

Abstract
This review focuses on the synthesis and biological activity of flavones and their related flavonoidic compounds, namely flavonols and aurones. Among the biological activities of natural and synthetic flavones and aurones, their anticancer, antioxidant, and antimicrobial properties are highlighted and detailed in this review. Starting from the structures of natural flavones acting on multiple anticancer targets (myricetin, genkwanin, and other structurally related compounds), new flavone analogs were recently designed and evaluated for their anticancer activity. The most representative compounds and their anticancer activity are summarized in this review. Natural flavones recognized for their antimicrobial properties (baicalein, luteolin, quercetol, apigenin, kaempferol, tricin) have been recently derivatized or structurally modulated by chemical synthetic methods in order to obtain new effective antimicrobial flavonoidic derivatives with improved biological properties. The most promising antimicrobial agents are systematically highlighted in this review. The most applied method for the synthesis of flavones and aurones is based on the oxidative cyclization of o-hydroxychalcones. Depending on the reaction conditions and the structure of the precursor, in some cases, several cyclization products result simultaneously: flavones, flavanones, flavonols, and aurones. Based on the literature data and the results obtained by our research group, our aim is to highlight the most promising methods for the synthesis of flavones, as well as the synthetic routes for the other structurally related cyclization products, such as hydroxyflavones and aurones, while considering that, in practice, it is difficult to predict which is the main or exclusive cyclization product of o-hydroxychalcones under certain reaction conditions.
Keywords: chalcones, flavones, flavonols, aurones, anticancer activity, antimicrobial activity
1. Introduction
Flavonoids are a widely distributed group of natural polyphenolic compounds that are found in plants usually in glycosylated form and have been shown to possess a wide range of biological activities, including antioxidant, anti-inflammatory, antibacterial, antiviral, and anticancer properties, making them an attractive target for synthesis and further study.
Structurally, flavonoids are functional aromatic compounds constituted by a C6-C3-C6 structure. The bioprecursor of flavonoids is the amino acid L-phenylalanine, which is transformed into phenyl-propenoyl-S-CoA with the involvement of the phenylalanine ammonia-lyase enzyme. Enzymatic condensation of phenyl-propenoyl-S-CoA with three malonyl-S-CoA units, followed by cyclization, yields o-hydroxychalcones that are structurally 1,3-diarylpropen-1-ones [1].
The reactive α,β-unsaturated ketone structure and the presence of hydroxy groups in o-hydroxychalcones make their cyclization possible, resulting in different flavonoidic compounds. Similar to biochemical cyclization pathways, in organic synthesis, the cyclization of o-hydroxychalcones represents the most useful way to obtain compounds from aurones, flavanones, flavones, flavonols, and flavylium salts, as will be detailed in Section 3.
The outstanding biological potential of natural flavonoids has attracted interest in the medical field, meaning that many of their synthetic analogs are currently known as promising candidates in treatments for cancer; microbial, fungal, and viral infections; inflammatory diseases; and diabetes.
Among the flavonoidic compounds, flavones and flavonols are related by the fact that they possess the same basic skeleton, the 2-phenyl-chromen-4-one system [1]. Flavones represent one of the most studied sub-class of flavonoids due to their wide distribution in plants and their wide structural diversity.
Flavonols, also called hydroxyflavones, differ from flavones by the presence of a hydroxy group at position 3 in the chromen-4-one ring (C ring, Figure 1) [1]. Although they have very similar structures, natural flavonols are not formed from chalcones via flavones as intermediates but through another biochemical pathway, with the involvement of other enzymes, via flavanones. Flavanones are common bioprecursors for flavones and flavonols [2].
Figure 1.

General structures of flavones, flavonols, aurones, and their common precursors, the o-hydroxychalcones.
Aurones, 2-benzylidenebenzofuran-3(2H)-ones [2], also belong to the flavonoid class, being structural isomers of flavones (Figure 1). Even if aurones are less known compared to flavones, research on them has experienced significant development in recent years due to their promising therapeutic potential.
Because of their related structure, flavones, flavonols, and aurones have common properties, such as the interesting way they exert their antioxidant, anticancer, antimicrobial, and other pharmacological activities [2].
In recent years, various methods have been developed for synthesizing flavones and related compounds, mainly hydroxyflavones and aurones, including chemical, biochemical, enzymatic, and total synthesis. Chemical synthesis is based on different approaches involving the use of different precursors or reagents, with the earliest developed synthesis methodologies for flavones emanating from the late 1890s–1900s (the von Kostanecki methodology and von Auwers synthesis [3]).
This review aims to present the most relevant methods for flavone and aurone synthesis, starting with the earliest and concluding with the recent methods. We consider it important to specify some examples from the literature and from our own research in which aurones and hydroxyflavones were obtained via the cyclization of o-hydroxychalcones, considering that they are structurally related compounds and that sometimes it is difficult to predict which would be the main or exclusive reaction product under certain reaction conditions.
Although the synthesis methodologies were first applied for obtaining the basic skeleton of natural flavones or aurones (in which rings A and B are benzene rings), some of these methods were successfully extended to obtain new bioactive flavonoidic analogs of natural compounds or flavone/aurone hybrids through structural modulations at the level of the A, B, or C rings with the aim of obtaining new bioisosters with improved biological functions. All structural modulations retain the aromatic character of the A and B rings, which is essential for their biological activity.
The structural modulations made on the A ring of flavones and aurones involve grafting different electron-withdrawing (halogen atoms, cian, or nitro) or electron-donating substituents such as hydroxy, methoxy, or acyloxy groups. Bulkier substituents such as pyperidine, directly connected to the A ring or through a linker such as benzyloxy, benzylamino, isopentyloxy, or benzylaminomethylene, are also introduced at different positions of the A ring of some flavonoidic analogs with reported anticancer/antimicrobial activity, as exemplified below.
The structural modulations at the aromatic B ring include the introduction of various electron-withdrawing (halogen atoms, carbamoyl, or trifluoromethyl groups) or electron-donating substituents (hydroxy, methoxy, benzyloxy, alkyl, acylamino, alkylamino, dialkylamino, or other multifunctionalized residues such as amino acid residues). Other structural modifications are based on replacing the B ring with other pentaatomic or hexaatomic aromatic heterocycles such as thiazole, pyrazole, thiophene, and pyridine, alone or linked/condensed with other (hetero)aromatic rings in order to obtain extended π-conjugated aromatic systems such as 2-phenylthiazole, thiazolo [3,2-b][1,2,4]triazole, 1,3-diphenylpyrazole, 3-naphtyl-1-phenylpyrazole, and quinoline.
The structural changes at the level of the C ring of flavones and aurones have been less frequently investigated. The most frequently reported changes include the derivatization of the hydroxy group of hydroxyflavones via alkylation or acylation. Recent attempts to obtain azaaurones, compounds which contain nitrogen as a heteroatom in the C ring instead of oxygen, have been made. It was found that replacing the intracyclic oxygen of aurones with nitrogen is beneficial for selective cytotoxicity against some multidrug resistant cancer cells, such as the resistant cancer cell line P-glycoprotein-overexpressing human doxorubicin resistant uterine sarcoma cells (MES-SA/Dx5) [4].
2. Biological Activity of Flavones, Flavonols, and Aurones
2.1. Anticancer Activity
The antitumor activity of flavones is most often due to their ability to target certain key structures that lead to cell cycle arrest and the apoptosis of tumor cells. Thus, flavones can inhibit the specific enzymes responsible for tumorigenesis, which are normally involved in the regulation of the cell cycle but whose function is deregulated under pathological conditions, for example, protein kinase C (PKC) [5], cyclin-dependent kinases (CDK) [6], casein kinases (CK) [7], PIM-1 kinases [8], death-associated protein kinase 1 (DAPK-1), and tyrosine kinases [9]. Some flavones can inhibit the polymerization of tubulin, thus preventing the formation of microtubules [10]. All this leads to cell cycle arrest, most often in the G2/M phase. Flavones can also activate certain enzymes that cause tumor cell apoptosis, such as caspases [11,12].
Natural flavones such as apigenin and nobiletin can regulate the expression of important inflammatory signaling pathways, including nuclear factor erythroid 2-related factor 2 (Nrf2) and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). The antioxidant properties of several natural flavones are attributable to their ability to regulate the expression of Nrf2/heme oxygenase-1 (HO-1), which decreases free radical levels and oxidative stress [13]. Nuclear factor erythroid 2-related factor (Nrf2) can interact with the NF-κB signaling pathway to maintain cellular redox homeostasis during inflammatory states. As the NF-κB pathway activates the expression of genes implicated in inflammation that can lead to chronic inflammation, tumor development, or proliferation, the Nrf2 pathway displays important antioxidant roles, such as mediating the release of ROS (reactive oxygen species) induced by NF-κB or suppressing the transcription of NF-κB-dependent pro-inflammatory genes [14]. Thus, the activation of Nrf2 pathway will suppress the NF-κB pathway and reduce TNFα, IL-6, and IL-1β proinflammatory cytokine levels [14].
The potential of flavones to act on multiple anticancer targets or by synergic mechanisms of action allows them to be considered as key structures for the development of new multitarget-acting therapeutic agents.
In several cases, the anticancer activity of natural flavones and aurones is closely related to their antioxidant activity. Myricetin (Figure 2), a natural flavone with polyphenol structure, presents good antioxidant properties by acting as a scavenger for reactive oxygen species and by enhancing the activity of glutathione-S-transferase [15]. Myricetin also presents great antitumor properties by targeting key structures, leading to cell cycle arrest and apoptosis. Myricetin has been shown to inhibit several enzymes involved in cell cycle regulation whose functions were deregulated under pathological conditions, namely, PKC, CK2, PIM-1, and DAPK1 [16]. Myricetin promotes tumor cell apoptosis by modulating certain signaling pathways, including Bcl2 (B-cell lymphoma 2), NF-κB, MAPKs (mitogen-activated protein kinases), and the Wnt/β-catenin signaling pathway [16,17,18]. Recently, it was reported that myricetin inhibits interferon-γ-induced programmed death ligand-1 (PD-L1) and indoleamine 2,3-dioxygenase 1 (IDO1) expression in lung cancer cells via the regulation of the Janus kinase/signal transducer and activator of the JAK/STAT-IRF1 transcription pathway [19]. According to the authors of [19], in their study, Myricetin recovered the function of T cells in the lung cancer cells and Jurkat-PD-1 T cells. Myricetin restored the survival, proliferation, CD69 expression, and interleukin-2 (IL-2) secretion of Jurkat-PD-1 T cells suppressed by IFN-γ-treated lung cancer cells [19]. PD-L1 and ISO1 are two immune checkpoints responsible for the immune escape of tumors. Thus, as an inhibitor of IFN-γ-induced PD-L1 and ISO1, myricetin has potential applications in tumor immunotherapy.
Recent studies have shown that myricetin induces apoptosis and autophagy in human gastric cancer cells through the inhibition of the PI3K/Akt/mTOR pathway (phosphoinositide 3-kinase, PI3K/Protein kinase B, Akt/Mechanistic target of rapamycin, mTOR) [20]. The abnormal increase in the activity of the PI3K/Akt/mTOR pathway is associated with various malignancies; therefore, the modulation of this signaling pathway represents a new strategy, in particular in gastric cancer treatment [21].
Myricetin also proved to be effective in preventing mutagenesis induced by different carcinogenic compounds such as formaldehyde [22]. Myricetin alleviates the formaldehyde-enhanced Warburg effect in tumor cells through the inhibition of human hypoxia-inducible factor 1 subunit alpha (HIF-1α), an important target in lung and ovarian tumors [22].
Gu Ling et al. recently revealed that myricetin regulates the p38 MAPK pathway by targeting MAP Kinase Kinase 3 (MKK3) in non-small cell lung cancer cells (NSCLC) [23]. These results encourage future research on the development of new anticancer agents, MKK3 inhibitors, through the structural modulation of myricetin.
Genkwanin (Figure 2), another natural flavone with antioxidant properties, has demonstrated promising anticancer activity against a series of cancer cell lines, including human MCF-7 breast cancer (IC50 = 13.6 ± 0.3 µg/mL), HepG-2 human hepatocellular carcinoma (IC50 = 22.5 ± 0.3 µg/mL), and HCT-116 colon cancer (IC50 = 15.4 ± 0.5 µg/mL). Genkwanin is also able to reduce the migration, invasion, and proliferation of lung cancer cells by targeting the phosphoinositide 3-kinase (PI3K) and phospho-protein kinase B (AKT) signaling pathways [24]. Due to this mechanism, genkwanin represents an effective option for the treatment of cancer proliferation and metastasis. Because genkwanin presents low oral bioavailability, genkwanin nanosuspensions were prepared in order to improve its solubility and pharmacokinetic profile. Li Y et al. reported the therapeutic potential of genkwanin nanosuspensions as novel antitumor agents in breast carcinoma therapy [25].
Spiegel M. et al. established that, through the bond dissociation enthalpy (BDE) of the hydrogen atom transfer (HAT) mechanism, the antioxidant activity of flavones could be related to the presence of a hydroxy group located on the B ring, especially in position C4′, more than the A-ring substitution. Regarding flavonols, the presence of a hydroxy group in C3 is beneficial for their antioxidant activity. These positions present the lowest values of bond dissociation entalphy (BDE = 84.4 kcal/mol for C4′, in the case of luteolin, and BDE = 84.6 kcal/mol for C3, in the case of morin) [26].
The anticancer activity of flavones could be correlated with their antioxidant activity, but it is not a mandatory rule in all cases. Grigalius I. and Petrikaite V. studied the relationship between the anticancer and antioxidant activities of trihydroxyflavones. The antioxidant activity was evaluated by using the DPPH (2,2-diphenyl-1-picrylhydrazyl) method, and the anticancer activity was evaluated by using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) method, both of which were performed on three different types of human cancer cell lines: lung (A549), breast (MCF-7), and brain epithelium (U87). Based on the calculation of the Pearson coefficient (r), a moderate correlation was revealed between the two biological properties [27]. It was found that the substituents on the phenyl ring (B ring) are the most important for the antioxidant activity of trihydroxyflavones. Thus, the most potent antioxidants have the o-dihydroxy group (catechol) on the B ring and are involved in binding hydroxy, peroxyl, and peroxynitrile radicals [27] (Figure 2, compounds 3 and 4). However, hydroxyflavone 5 does not possess this structural feature, but it does present the best anticancer activity, thus, in this case, alluding to the existence of other mechanisms of action for anticancer activity besides the neutralizing effect of free radicals.
Figure 2.

Polyphenolic flavones with anticancer and antioxidant activity [24,27].
Zhao L. et al studied the structural elements of flavones capable of blocking different serine-threonine kinases involved in the cell cycle. Structure–activity relationship studies were conducted for PKC, CK-2, PIM-1 kinase, DAPK-1, and CDK. It was found that the hydroxy groups grafted on rings A, B, and C act as H-bond donors/acceptors in the interaction with PKC, PIM-1, DAPK-1, and CDK [28]. For the inhibition of CK-2, it was found that the presence of the halogen atoms Br and Cl at positions 6 and 8 of the A ring, respectively, and the hydroxy group only in position 4 of the B ring are beneficial. The carbonyl group located in position 4 of the chromen-4-one ring acts as a H-bond acceptor in the interaction with various amino acid residues from CK-2, CDK, and PIM-1. The benzene ring (B ring) interacts by π–π stacking with the phenylalanine residue Fen113 of CK-2, and through this mechanism, it also blocks the ATP binding site of these enzymes. The benzene ring (B ring) can also make van der Waals interactions with certain hydrophobic residues from CDK-9 and PIM-1, thus making additional contact with these enzymes without blocking the binding of ATP. It has also been observed that changing the position of the phenyl ring from 2 to 3, specific to isoflavones, leads to the loss of activity [28].
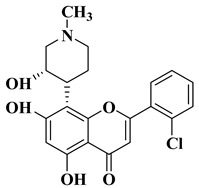
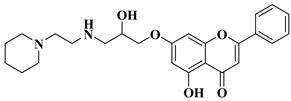
Flavopiridol (Table 1, line 1) is a semisynthetic flavone that is currently being used in clinical trials as an anticancer agent for the treatment of acute myeloid leukemia. This compound acts by inhibiting kinases CDK-1, -2, -4, -6, and -7, all of which are competitive with ATP. At the same time, flavopiridol significantly inhibits kinases CDK-9 (non-competitive with ATP) [9,28]. Flavopiridol also inhibits the activity of positive transcription elongation factor (P-TEFb), a cyclin-dependent kinase controlling elongation via RNA polymerase II [29].
The anticancer activity of flavopiridol is due to the presence of a chromone moiety that is bioisosteric with the purine ring of ATP and binds competitively to the ATP binding pocket of CDK. The benzene ring (ring B) provides additional contact with the enzyme, as it interacts with different regions than those occupied by ATP, participating in van der Waals-type interactions with other amino acid units [28]. Other important elements for the inhibition of kinase activity by flavopiridol are the hydroxy groups at C-7 and C-5, the carbonyl group at C-4, the nitrogen atom, and the hydroxy group from the piperidine, and all of these functional groups are involved in the formation of hydrogen bonds with CDK [28].
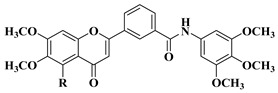
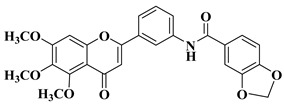
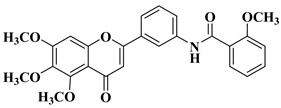
Starting from the structures of two natural products with anticancer activity with different mechanisms of action, 3,5,4′-trimethoxystilbene and 5,6,7-trimethoxyflavone, Hassan A.H. et al. synthesized new antiproliferative compounds by combining two pharmacophore moieties in the same molecule by replacing the vinylene residue in stilbene with the amide group [30]. The cytotoxic activities of the synthesized compounds against several cancer cell lines were determined at 10 μM doses in all cases. The structures of the most active compounds are presented in Table 1, lines 2–4.
Flavone–stylbene hybrids in which the nitrogen atom of the amide linker is attached to the flavone moiety proved to be more citotoxic than the corresponding compounds with the opposite amide linker configuration. Trimethoxylated flavone–stylbene hybrids showed superior activity compared to dimethoxylated flavone–stylbene hybrids on hematologic, colorectal, central nervous system, ovarian, renal, and breast cancer cell lines [30]. On lung cancer cell lines, the dimethoxylated derivatives were generally more active than the trimethoxylated ones. Most of the tested hybrid compounds showed selective activity, showing no cytotoxicity on normal cells. Their anticancer mechanism of action consists of inducing apoptosis and inhibiting cell proliferation [30].
Continuing their research, Hassan A.H. et al. synthesized a series of trimethoxyflavone-based aryl-amides, starting from the structures of already approved arylamide-type medicinal compounds (imatinib, masitinib) and replacing the bulky aromatic entity in their structure with 5,6,7-trimethoxyflavone and 5-hydroxy-6,7-dimethoxyflavone. The formation of the amide bond was carried out in the 3′ and 4′ positions on the B ring of the flavone using 3′-amino and 4′-amino precursors coupled with various acyl chlorides and 3′-carboxyl precursors condensed with aryl amines, respectively [31].
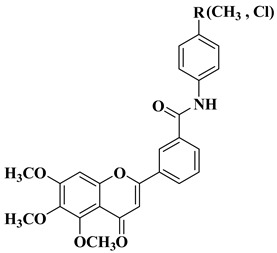
Two flavones presented good broad-spectrum anticancer activity by triggering cell cycle arrest in the G1 phase (Table 1, line 5). These compounds could represent hit compounds for the design of new, more potent inhibitors of STE20/GCK-IV kinase family members, including HGK, TNIK, and MINK1 kinases. It was found that the presence of the carbonyl of the amide linker attached to the flavone moiety is beneficial for the anticancer activity of the tested flavone-based aryl-amides. Reversing the attachment mode of the amide linker led to a significant decrease in anticancer activity [31].
A series of dimethoxyflavonols and trimethoxyflavonols derivatives were obtained via the alkylation of the hydroxy group at position 3 of the chromen-4-one ring (C ring) (Figure 3). The compounds were investigated for their anticancer activity on both androgen-sensitive (LNCaP) and androgen-insensitive (PC-3 and DU145) prostate cancer cell lines [32].
Figure 3.

3-O-substituted flavonols reported as anti-prostate cancer agents. Structure–activity relationship study [32].
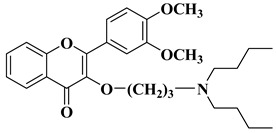
It was found that the alkylation of the hydroxy group in position 3 generally increased the antiproliferative activity of the compounds. The presence of an amino group linked to the hydroxy group at position 3 of the flavonols through a three- to five-carbon linker is beneficial for antiproliferative activity against the three human prostate cancer cell lines with tumor selectivity. N-methylpiperazin-1-yl, pyrrolidin-1-yl, and dibutyl amino groups proved to be beneficial, improving the anticancer activity of the tested compounds. The most promising derivative in terms of selectivity, anticancer activity, and bioavailability contains a dibutyl amino group linked to the oxygen at position 3 via a three-carbon linker (Table 1, line 6). The bioavailability of the tested compounds was superior to that of fisetin [32].
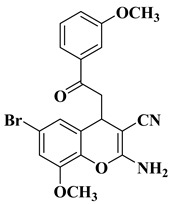
Starting from a series of differently substituted chalcones, Pontes et al. synthesized a series of chromene–chalcone hybrid compounds in order to test their anticancer activity on breast cancer cell lines. The most active compound is depicted in Table 1, line 7. The mechanism of action involves the inhibition of cell migration and induction of apoptosis, by determining cell cycle arrest in the G2/M phase. Moreover, this compound has been proved to alter tubulin polymerization, representing a promising new microtubule-destabilizing agent. It was found that the presence of the halogen atoms grafted on the basic skeleton of chromene–chalcone hybrids is beneficial to antitumor activity. Brominated compounds presented superior activity to chlorinated and fluorinated compounds. The evaluated compounds presented selective cytotoxicity on cancer cell lines compared to non-cancerous cell lines [10].
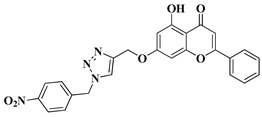
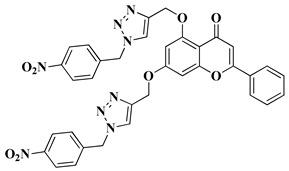
New hybrid compounds of flavones (chrysin and kaempferol) and substituted 1,2,3-triazoles were recently synthesized via the chemical derivatization of the hydroxyl groups of chrysin and kaempferol with functionalized 1,2,3-triazole compounds [33]. The antitumor activity of the obtained mono- and bis-coupled hybrids was evaluated in vitro on 60 cell lines of 9 common cancer types (NCI60) [33]. The hybrid compounds presenting the most significant antiproliferative effect are mentioned in Table 1, lines 8, 9.
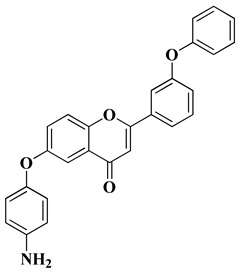
A series of new heterocyclic derivatives were recently synthesized via the functionalization of a flavone ring with an aminophenoxy moiety in different positions of the A ring and a phenoxy moiety in different positions of the B ring [34]. Their cytotoxicity was investigated in vitro against two human non-small cell lung cancer (NSCLC) cell lines (A549 and NCI-H1975). It was found that the presence of a 4-aminophenoxy group at the sixth position of the A ring and a terminal phenoxy group on the B ring is beneficial for cancer-selective cytotoxicity. A flavone derivative containing a phenoxy moiety at the C’3 position of the B ring and a p-aminophenoxy group at the sixth position of the A ring was the most effective, presenting micromolar IC50 values (for A549 and H1975) and a high selectivity index (SI > 10, Table 1, line 10). Further flow cytometric analyses showed that this compound induces apoptosis and cell cycle arrest in the G2/M phase through the up-regulation of p21 expression [34]. The absence of the phenoxy moiety on the B ring and the different position of the p-aminophenoxy moieties on the A ring decreased the efficacy and selectivity of aminophenoxy derivatives [34].
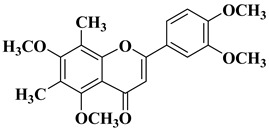
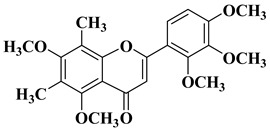
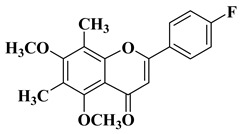
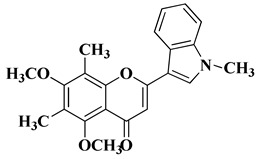
New C-dimethylated flavones were recently synthesized and evaluated for their anti-tubercular and anticancer activity [35]. In this study, four flavones presented anticancer activity against a human adenocarcinoma A549 cell line, with IC50 values between 39 and 48 μM (Table 1, lines 11–14). This study’s in silico docking simulations revealed that these four compounds present improved binding and interaction profiles against the epidermal growth factor receptor (EGFR) [35].
Other recently reported examples of synthetic flavones with antitumor activity are illustrated in Table 1, lines 15–20.
Natural and synthetic aurones possess a broad variety of biological activities, including antiproliferative activity against different cancer cell lines. The anticancer activity of aurones is due to their ability to interact with different key antitumor molecular targets, and examples of such interactions include the following: the inhibition of serine/threonine cyclin-dependent kinases (CDK 1 and 2) [36], the inhibition of topoisomerase IIα [37], the inhibition of sphingosine kinase (SphK) [38], and interfering with microtubule assembly [39]. In some cases, it was found that the anticancer activity of aurones is strongly related to their antioxidant activity [40].
Several aurones have been shown to modulate the activity of ATP-dependent efflux pumps such as P-glycoprotein [41] and breast cancer resistance protein (BCRP/ABCG2) [42]. Through this mechanism, aurones can potentiate the effect of simultaneously administered anticancer chemotherapeutics by blocking the multidrug resistance mechanisms of tumor cells.
Our research group synthesized a series of aurone analogs by replacing the B ring (phenyl) with the 2-arylthiazole system in order to obtain compounds with superior anticancer activity, considering the anticancer potential of thiazole derivatives. Two aurone analogs were active against cancer cell lines resistant to currently used chemotherapeutics, such as multidrug-resistant leukemia cell lines and breast cancer cell lines, and both showed cytotoxic activities that were superior to doxorubicin (Table 1, lines 21, 22) [43]. Other recently reported examples of synthetic aurones with antitumor activity are illustrated in Table 1, lines 23–31.
Table 1.
Synthetic analogs of flavones and aurones with antitumor properties.
| Entry | Chemical Structure | Cancer Cell Lines against the Tested Compounds Present Cytotoxic Activity | Ref. |
|---|---|---|---|
| 1 |
 Flavopiridol Flavopiridol |
|
[44] |
| 2 |
|
R = OCH3
|
[30] |
R = OH
| |||
| 3 |
|
|
[30] |
| 4 |
|
|
[30] |
| 5 |
|
|
[31] |
| 6 |
|
|
[32] |
| 7 |
|
|
[10] |
| 8 |
|
|
[33] |
| 9 |
|
|
[33] |
| 10 |
|
|
[34] |
| 11 |
|
|
[35] |
| 12 |
|
|
[35] |
| 13 |
|
|
[35] |
| 14 |
|
|
[35] |
| 15 |
|
|
[45] |
| 16 |
|
|
[46] |
| 17 |
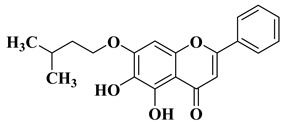
|
|
[11] |
| 18 |
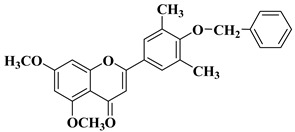
|
|
[47] |
| 19 |
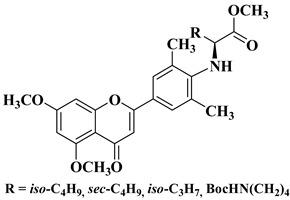
|
|
[47] |
| 20 |
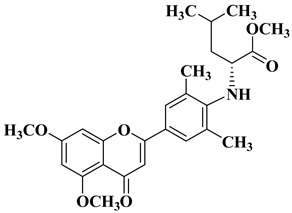
|
|
[47] |
| 21 |
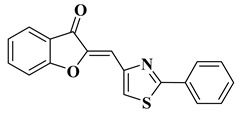
|
|
[43] |
| 22 |
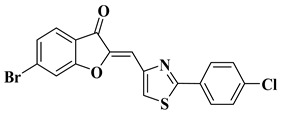
|
|
[43] |
| 23 |
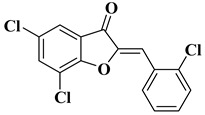
|
|
[48] |
| 24 |
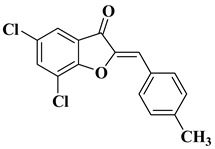
|
|
[48] |
| 25 |
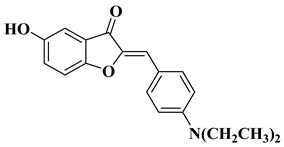
|
|
[49] |
| 26 |
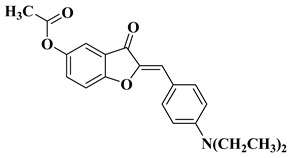
|
|
[49] |
| 27 |
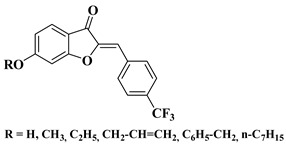
|
|
[50] |
| 28 |
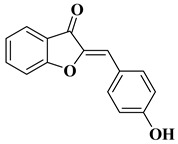
|
|
[51] |
| 29 |
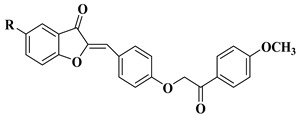
|
R = Cl: leukemia cell lines MOLT-4 (−17.79% mean growth percentage), and SR (−22.38% mean growth percentage). R = H: renal cancer cell line UO-31 (−44.36% mean growth percentage). The mean growth percentages were determined for five concentrations ranging from 10−4 to 10−8 M. |
[52] |
| 30 |
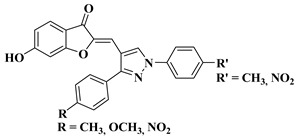
|
R′ = NO2 and R = NO2 (IC50 = 25.1 µM). |
[53] |
| 31 |
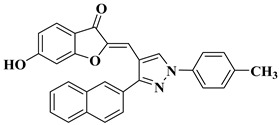
|
|
[53] |
2.2. Antibacterial and Antifungal Activity
Bacterial and fungal resistance to existing antibiotics is a worldwide health issue, particularly affecting the immunocompromised patients. Without effective antimicrobial agents, several medical procedures could endanger the lives of patients by increasing the risk of microbial infections. The basic structure of natural flavones and aurones have inspired researchers to develop new antimicrobial agents with improved bioavailability and antibacterial and antifungal properties.
Recently, it was reported that the natural flavone myricetin (Figure 2) presents anti biofilm activity against Staphylococcus aureus and attenuates osteomyelitis by inhibiting the Toll-like receptor-2 (TLR2)/mitogen-activated protein kinase (MAPK) pathway in experimental mice [54].
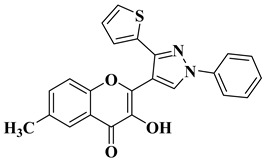
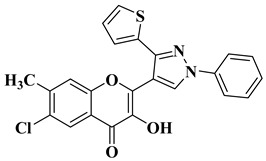
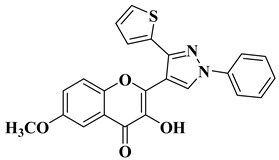
Ashok D. et al. synthesized new flavonol analogs bearing the extended heteroaromatic system 1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl instead of a phenyl ring (B ring) and containing various substituents on the chromone system. The synthesized flavonol derivatives were screened for their antimicrobial activity against several fungal strains (Aspergillus niger, Penicillium italicum, Fusarium oxysporum) and bacterial strains (Staphylococcus aureus, Pseudomonas aeruginosa, Escherichia coli, Bacillus subtilis). The inhibition zones (IZ) were determined at 50 μg/mL concentration for each compound in dimethyl sulfoxide (used as a solvent). Four of the tested compounds (Table 2, lines 1–4) show good antimicrobial activity and represent hit compounds for the design of new antifungal and/or antibacterial therapeutic agents [55].
In order to obtain new flavone analogs with antibacterial activity, Lv X.H. et al. synthesized a series of flavone Mannich base derivatives by applying the Mannich reaction between primary amines and using natural flavones as components with mobile hydrogen and formaldehyde as a carbonyl component. The natural flavones used as precursors were baicalein, luteolin, quercetol, apigenin, and kaempferol. Derivatization was performed at position 8 of the chromone moiety by applying the Mannich reaction [56]. The antibacterial activity of the obtained flavone Mannich bases was evaluated for two Gram-positive bacteria (S. aureus and Listeria monocytogenes) and two Gram-negative bacteria (E. coli and Salmonella gallinarium), and novobiocin and ciprofloxacin were used as standards. The structures of the most active compounds are shown in Table 2, lines 5, 6. Through performing in vitro experiments and in silico molecular docking studies, it was found that these compounds exhibit potent inhibition against topoisomerase II and topoisomerase IV isolated from E. coli [56].
New hydroxyflavone derivatives containing the dimethylamino group grafted at position 4 of the benzene ring (B ring) were synthesized and evaluated for their antifungal activity against Acremonium strictum, Penicillium expansum, and Aspergillus flavus. Four of the tested compounds presented very good antifungal activities against some of the tested fungal strains (Table 2, lines 7–10) [57].
New quinoline-based aurone analogs were synthesized and evaluated for their antibacterial, antifungal, and anti-biofilm activity. The compounds mentioned in Table 2, lines 11–13 presented the most significant antibacterial and antifungal activities, and some of them were also shown to be good anti-biofilm agents [58].
New C-dimethylated flavones were recently synthesized and evaluated for their anti-tubercular and anticancer activity [35]. Two dimethylated and dimethoxylated flavones bearing the fluoro and dimethylamino substituents in position 4 of the B ring were shown to have significant antibacterial activity against the H37Rv strain of replicating Mycobacterium tuberculosis, with sensitivity up to 6.25 µg/mL (Table 2, line 14).
2.3. Antiviral Activity
Viral infections represent a global health issue and have had many implications on public health throughout history, including the appearance of new mutant viral strains and the emergence of pandemics. Specific aspects of modernization, such as rapid air transit and urbanization, have accelerated the emergence and spread of viruses. Antiviral therapy is necessary when vaccination does not bring the expected results or in the case of infections for which vaccination has not been implemented. Flavones have also been included in the research of new molecules with antiviral potential, yielding some important results and positive prospects for the future.
According to a recently reported study, the natural flavone myricetin (Figure 2) possesses potency against SARS-CoV-2 infection through blocking viral-entry facilitators and suppressing inflammation through the RIPK1/NF-κB pathway [59]. Myricetin also inhibits SASR-CoV-2 infection and replication in Vero E6 cells (EC50 55.18 μM) [59]; these results suggest that this flavone represents a key structure for the design of new therapeutic agents against COVID-19.
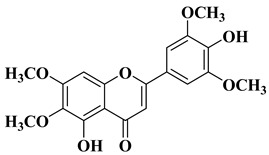
Regarding tricin, 4′,5,7-trihydroxy-3′,5′-dimethoxyflavone, a flavone derivative with activity against cytomegalovirus (CMV), Fujimoto K.J. et al. modulated its structure by grafting a fluorine atom on the chromen-4-one ring. Thus, two compounds were obtained—6-F-tricin and 7-F-tricin—and the antiviral activity of which was measured against cytomegalovirus replicated on embryonic lung cell cultures. Compared to ganciclovir, 6-F-tricin showed much stronger activity against cytomegalovirus (Table 2, line 15). Moreover, it was observed that 6-F-tricin did not produce cytotoxicity on the used embryonic cells. Substitution with fluorine is beneficial for increasing the affinity for target proteins (in this case, for CDK9, cyclin-dependent kinase 9) [60].
The antiviral potential of flavones has also been demonstrated against tropical diseases such as Chikungunya fever. Badavath V.N. et al. synthesized nineteen flavones in order to evaluate their antiviral activity against Chikungunya virus replication. Two compounds showed activity at concentrations below 1 µg/mL (Table 2, lines 16, 17). It was observed that the more potent compounds possess heterocycles (thiophen-2-yl and pyridyn-2-yl) in position 2 of the chromen-4-one ring instead of the benzene ring (B ring). Through conducting molecular docking studies, it was deduced that these compounds act by inhibiting the Chikungunya virus protease [61].
Table 2.
Synthetic analogs of flavones and aurones with antimicrobial (antibacterial/antifungal/antiviral) properties.
| Entry | Chemical Structure | Microbial Strains against the Tested Compounds Present Antimicrobial Activity | Ref. |
|---|---|---|---|
| 1 |
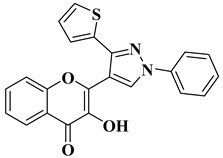
|
Antifungal activity (inhibition zone for 50 μg/mL solution): Aspergillus niger (IZ = 16 mm) Penicillium italicum (IZ = 20 mm) Fusarium oxysporum (IZ = 31 mm) |
[55] |
| 2 |
|
Antibacterial activity (inhibition zone for 50 μg/mL solution): Staphylococcus aureus (IZ = 31 mm) Pseudomonas aeruginosa (IZ = 11 mm) Escherichia coli (IZ = 30 mm) |
[55] |
| 3 |
|
Antibacterial activity (inhibition zone for 50 μg/mL solution): Staphylococcus aureus (IZ = 30 mm) Bacillus subtilis (IZ = 11 mm) Escherichia coli (IZ = 31 mm) Antifungal activity (inhibition zone for 50 μg/mL solution): Aspergillus niger (IZ = 13 mm) Penicillium italicum (IZ = 24 mm) Fusarium oxysporum (IZ = 25 mm) |
[55] |
| 4 |
|
Antibacterial activity (inhibition zone for 50 μg/mL solution): Staphylococcus aureus (IZ = 33 mm) Bacillus subtilis (IZ = 17 mm) Escherichia coli (IZ = 33 mm) Antifungal activity (inhibition zone for 50 μg/mL solution): Aspergillus niger (IZ = 14 mm) Penicillium italicum (IZ = 26 mm) Fusarium oxysporum (IZ = 27 mm) |
[55] |
| 5 |
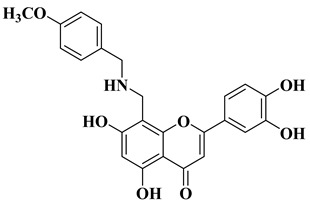
|
Antibacterial activity: Staphylococcus aureus (MIC = 2 mg/L) Escherichia coli (MIC = 4 mg/L) Salmonella gallinarum (MIC = 0.125 mg/L) |
[56] |
| 6 |
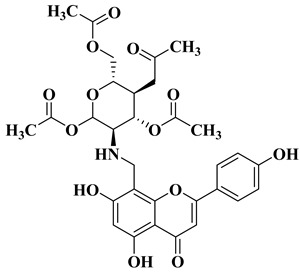
|
Antibacterial activity: Staphylococcus aureus (MIC = 1 mg/L) Escherichia coli (MIC = 2 mg/L) Salmonella gallinarum (MIC = 0.05 mg/L) Listeria monocytogenes (MIC = 0.5 mg/L) |
[56] |
| 7 |
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration): Acremonium strictum (81.33%; 100%) Penicillium expansum (60.87%; 100%) Aspergillus flavus (41.02%; 65.64%) |
[57] |
| 8 |
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration): Acremonium strictum (70%; 100%) Penicillium expansum (42.15%; 100%) Aspergillus flavus (6.41%; 46.15%) |
[57] |
| 9 |
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration): Acremonium strictum (76.88%; 100%) Aspergillus flavus (15.38%; 60.51%) |
[57] |
| 10 |
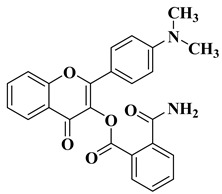
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration): Acremonium strictum (73.33%; 100%) |
[57] |
| 11 |
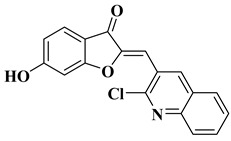
|
Antibacterial activity: Staphylococcus aureus (MIC = 1.25 mg/mL) Bacillus subtilis (MIC = 0.02 mg/mL) Mycobacterium smegmatis (MIC = 0.625 mg/mL) Antifungal activity: Fusarium oxysporum (MIC = 0.625 mg/mL) |
[58] |
| 12 |
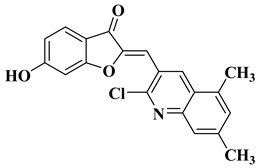
|
Antibacterial activity: Staphylococcus aureus (MIC = 2.5 mg/mL) Bacillus subtilis (MIC = 0.156 mg/mL) Mycobacterium smegmatis (MIC = 0.078 mg/mL) Anti biofilm and anti quorum sensing activity (100 μg/mL) Antifungal activity: Fusarium oxysporum (MIC = 0.313 mg/mL) Candida albicans (MIC = 0.078 mg/mL) |
[58] |
| 13 |
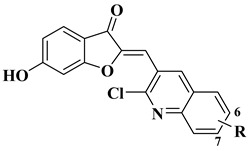
|
Antibacterial activity (R=6-OCH3, 7-Cl) Staphylococcus aureus (MIC = 1.25 mg/mL) Bacillus subtilis (MIC = 1.25 mg/mL) Klebsiella pneumoniae (MIC = 0.625 mg/mL) Anti biofilm activity (R=6-OCH3, 100 μg/mL) Antifungal activity (R=7-Cl): Candida albicans (MIC = 0.156 mg/mL) |
[58] |
| 14 |
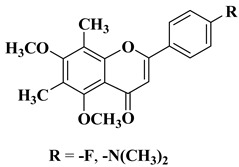
|
Antibacterial activity Mycobacterium tuberculosis H37Rv (MIC = 6.25 µg/mL) |
[35] |
| 15 |
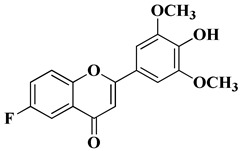
|
Antiviral activity Human cytomegalovirus (EC50 = 0.126 nM) |
[60] |
| 16 |
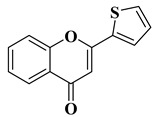
|
Antiviral activity Chikungunya Virus (IC50 = 0.44 µM) |
[61] |
| 17 |
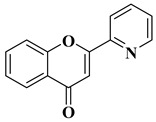
|
Antiviral activity Chikungunya Virus (IC50 = 0.45 µM) |
[61] |
3. Chemical Synthesis of Flavones
3.1. Von Kostanecki Method
Stanislaus von Kostanecki’s method was established in 1898–1899 and is considered one of the earliest methods for the synthesis of flavones. It uses o-hydroxyacetophenone (or o-acetoxyacetophenone) (6) and benzaldehyde (7) as precursors to form 2′-hydroxychalcone (or 2′-acetoxychalcone) (8) through Claisen–Schmidt condensation. In the next step, the obtained chalcone (8) is converted to flavone (9) through bromination followed by a dehydrobromination reaction in alkali alcoholic solution (Scheme 1).
Scheme 1.

von Kostanecki method for obtaining flavones.
The reaction pathway differs depending of the reaction conditions. According to von Kostanecki’s collaboration with Levi and Tambor, it is presumed that, instead of a chalcone, an aldol (11) between the two compounds forms; then, via cyclization, a flavanone (12) is formed. This flavanone is subsequently subjected to nuclear bromination with bromine in carbon disulfide, resulting in a 3-bromoflavanone (13) [62], and ultimately, the brominated intermediate suffers a dehydrobromination reaction, thus deriving a flavone (9) (Scheme 2) [63,64].
Scheme 2.

von Kostanecki method according to collaboration with Levi and Tambor.
However, according to von Kostanecki’s collaboration with Emilewicz and Tambor (also known as Emilewicz–von Kostanecki cyclization), the chalcone (14) is formed and then brominated, resulting in a chalcone dibromide (15). This brominated compound is cyclized through the elimination of one bromine atom, resulting in a 2-bromoflavanone (16) and, finally, the flavone (9) after eliminating the second atom (Scheme 3) [3,64].
Scheme 3.

Emilewicz–von Kostanecki cyclization reaction steps.
The proposed mechanism starts with the Claisen–Schmidt condensation of o-hydroxyacetophenone (10) with benzaldehyde (7), resulting in an o-hydroxychalcone (14). The nucleophilic species is represented by the intermediate (17), stabilized via conjugation, and formed via the deprotonation of acetophenone (10) in basic media. Through the bromination of the chalcone (14) on the C=C bond, a chalcone dibromide (15) is formed. Further dehydrobromination of 15 followed by cyclization of the intermediate 18 affords a 2-bromoflavanone intermediate (19). The intermediate 19 leads to the flavone 9 via the expulsion of Br− (Scheme 4) [3].
Scheme 4.

Emilewicz–von Kostanecki mechanism for the synthesis of flavones (adapted from Kshatriya et al.) [3].
The limitations of the von Kostanecki method are the possibility of nuclear bromination and the tendency to form 2-benzylidene-coumaran-3-ones (22) (benzalcoumaranones or aurones [65]) (Scheme 5) instead of flavones (21) when trying to synthesize natural flavones either with 5,7-disubstitution pattern, a 4′- or 5′-alkoxy substituent, or containing a phloroglucinol moiety. Better results can be obtained when methyl ether derivatives (20) are used as precursors [66,67,68]. Hutchins and Wheeler observed that treating the chalcone dibromides (20) with potassium cyanide in ethanolic solution or heating above their melting point will convert them into flavones (21) [68]. The same reagent converts aurones (22) back to flavones (21) (Scheme 5). The quantity of potassium cyanide influences the reaction’s outcome. In the case of 2-p-alkoxybenzylidenecoumaran-3-ones, refluxing with an excess of reagent will cause the ring expansion of the aurone, affording 4′-alkoxyflavones (21), while treating the chalcone dibromide (20) with an insufficient quantity of potassium cyanide produces 2-benzylidene-coumaran-3-one (aurone 22) instead of a flavone [69].
Scheme 5.

Hutchins and Wheeler method to convert chalcone dibromides and aurones to flavones using potassium cyanide in ethanol (image adapted from Fitzgerald et al.) [69].
The possible mechanism of obtaining flavones from aurones using ethanolic potassium cyanide starts with a nucleophilic attack by a cyanide anion on the methine carbon of the aurone (22), followed by hydrogen transfer and carbanion (24) formation. The carbanion’s electrons migrate, recreating the double bond and preparing the ring expansion. Conjugation and intramolecular nucleophilic attack by the newly formed oxygen anion (25) lead to the expulsion of the cyanide anion, a good nucleofuge, and ring closure (Scheme 6) [69].
Scheme 6.

Possible mechanism for converting aurones to flavones using ethanolic potassium cyanide (image adapted from Fitzgerald et al.) [69].
However, it has been established that aurone formation may be avoided by providing milder conditions for the dehydrohalogenation reaction [64]. Donnelly and Doran have observed that the quantity of flavone increases with a decrease in base concentration [70]. von Auwers and Anschutz have shown that 4′-alkoxyflavones can be obtained by performing the cyclization reaction in cold alcohol rather than hot alcohol, which generates aurones [64,71].
Zemplén and Bognár improved the von Kostanecki method and demonstrated that nuclear bromination could be avoided by submitting acetates of hydroxyflavanones to bromination in absolute chloroform and in the presence of UV light. The obtained intermediate was a 3-bromoflavanone that could be easily dehydrobrominated, thus forming a flavone. This method is suitable for obtaining 3-hydroxyflavones [62,66].
Improvements in the direct dehydrogenation of chalcones and flavanones were made by using selenium dioxide as an oxidative reagent [66,72].
A new method by von Kostanecki was established in 1904 in collaboration with Szabránski. This method is used for obtaining 3-hydroxyflavones (28) from flavanones (12) via isonitrosoflavanones. The flavanone is nitrosated with pentyl nitrite (26) and hydrochloric acid. The isonitrosoflavanone (27) is converted into 3-hydroxyflavone (28) via hydrolysis with diluted sulfuric acid (Scheme 7) [73].
Scheme 7.

von Kostanecki–Szabránski method for obtaining 3-hydroxyflavones.
3.2. Von Auwers–Müller Method
Karl von Auwers’s method was first established in 1908 in collaboration with Müller, and it consists of the conversion of aurones 29 into 3-hydroxyflavones 28 (Scheme 8) via bromination in chloroform, followed by the dehydrohalogenation of the intermediate (33) with ring rearrangement under the action of potassium hydroxide in ethanol solution (Scheme 9) [3,74].
Scheme 8.

The von Auwers–Müller method of obtaining 3-hydroxyflavones.
Scheme 9.

Original von Auwers synthesis using 5-methylcoumaranone [74].
Originally, the aurone used by von Auwers and Müller was 2-benzylidene-5-methylcoumaranone 32, obtained from 5-methylcoumaranone 31 (previously synthesized by Stoermer and Bartsch) [75]. Von Auwers and Müller synthesized coumaranone 31 starting from o-chloracetyl-p-cresol (30) (Scheme 9) [74]. This is supposed to lead to a cyclodehydrohalogenation via the action of sodium hydroxide in ethanol while heating. The obtained coumaranone 31, via condensation with benzaldehyde, is converted into the aurone 32, which can add bromine to the ethylene bond. The obtained dibromo derivative 33 is transformed into 3-hydroxyflavone 34 via dehydrobromination and recyclization under the action of potassium hydroxide in ethanol while heating [74].
The mechanism of von Auwers synthesis is presented in Scheme 10. It starts with the bromination of aurone on the double bond, resulting in a 1,2-dibrominated compound (35). The subsequent substitution of bromine via the nucleophilic attack of a hydroxide anion results in an α,β-unsaturated ketone (38), which yields hydroxyflavone through cyclodebromination [3].
Scheme 10.

Mechanism of von Auwers synthesis [3].
As much as this method tried to improve upon von Kostanecki’s one, it also had limited applicability for obtaining natural 3-hydroxyflavones. To improve the outcome of this method, they suggested chlorination instead of bromination. This resulted in a trichlorinated derivative that would convert into a chlorinated hydroxyflavone that has one chlorine atom in positions 5 or 7. However, any attempt to eliminate the last chlorine atom failed. 3-Hydroxyflavones could be obtained from aurones in better yields only if the splitting of the coumaranone ring would take place easier than the dehydrohalogenation. They concluded that the presence of chlorine, methoxy, and methyl groups in position 5 of the coumaranone ring facilitates the formation of 3-hydroxyflavones, while methoxy and methyl groups in meta position and two methoxy groups on the aldehyde make it more difficult [76,77].
3.3. Allan–Robinson Method
The Allan–Robinson method was first established in 1924, and it involves converting 2-hydroxyacetophenones to flavones via treatment with anhydrides of aromatic carboxylic acids and the sodium salts of the corresponding carboxylic acids while heating (Scheme 11). The first part of this procedure entails converting ω-methoxyresacetophenone (Scheme 11: R′ = OH, R = OCH3) into 7-hydroxy-3-methoxyflavone (41) using benzoic anhydride and sodium benzoate (Ar = C6H5). This method was further extended by using various 2-hydroxyacetophenone derivatives (40) and aromatic anhydrides as starting materials [78].
Scheme 11.

Allan–Robinson method for obtaining flavones.
In the first step of the reaction mechanism, the deprotonated enolic tautomer of 2-hydroxyacetophenone (42) performs a nucleophilic attack on the carbonyl group of the anhydride (43), affording a 1,3-diketone compound (o-hidroxydibenzoylmethane) (44). The basic conditions provided by the sodium salt transform the intermediate into a flavone via enolisation followed by intramolecular cyclocondensation (Scheme 12) [3].
Scheme 12.

General reaction mechanism of the Allan–Robinson method for the synthesis of flavones.
An alternative to the reaction conditions of this method was proposed by Wheeler. This modification implies that 2-hydroxyacetophenone (10) is turned into 2-benzoyloxyacetophenone (48) via treatment with benzoyl chloride and pyridine. The obtained acetophenone derivative (48) can be converted into a flavone (9) either in glycerol while heating or via treatment with KOH in pyridine, followed by the cyclization of the 1,3-diketone intermediate (44) with glacial acetic acid and concentrated sulfuric acid while heating (Scheme 13) [79].
Scheme 13.

Wheeler’s alternative reaction conditions for the synthesis of flavones [79].
Unlike the previous presented methods, this one can be used for synthesizing more complex structures, making the Allan–Robinson method suitable for obtaining natural flavones and 3-hydroxyflavones. It has been used for obtaining a large variety of natural compounds, including fisetin, quercitin [80], datiscetin [81], myricetin, methylgalangin [82], limocitrol, limocitrin, spinacitrin [83], kaempferol [84], axillarin [85], jaceidin [86], and hispidulin [8].
3.4. Baker–Verkataraman Method
This method was established after the individual work of Baker and Verkataraman in 1933. It is used to obtain flavones from o-acyloxyacetophenones (48). These precursors are first converted into o-hydroxydibenzoylmethane derivatives (44) via heating in benzene or toluene with anhydrous potassium carbonate. The o-hydroxydibenzoylmethane derivatives (44) are cyclized into the corresponding flavones (9) via treatment with cold concentrated sulfuric acid [3,87] (Scheme 14).
Scheme 14.

The Baker–Venkataraman method.
Verkataraman first used this method to obtain α-naphtoflavone from 2-acetyl-1-naphthyl benzoate via heating with sodium benzoate and benzoic anhydride [88,89]. Mahal and Verkataraman obtained the diketone derivative via treatment with NaNH2 in ether at room temperature. Further cyclization to the corresponding α-naphtoflavone was performed via treatment with concentrated sulfuric acid in ethanol at reflux [90].
According to the mechanism, this method starts with an intramolecular Claisen condensation between acetophenone and an ester group grafted in ortho position on the aromatic ring (an o-acyloxyacetophenone 48), which can also be interpreted as acyl group transfer. This is followed by cyclocondensation in acidic conditions via a 2-hydroxyflavanone intermediate (53) (Scheme 15) [3,91].
Scheme 15.

Baker found out that this method could yield 3-acylflavones (56) via the treatment of o-acyloxyacetophenone 48 with sodium salts of carboxylic acids. Instead of cyclocondensation, the o-hydroxydibenzoylmethane 44 can be acylated on the methylene carbon, affording a triacylmethane derivative (54). This intermediate is cyclized to 2-hydroxy-3-acylflavanone (55) and then dehydrated, affording the corresponding 3-acylflavone (56) (Scheme 16) [87].
Scheme 16.

Synthesis of 3-acylflavones.
However, the conventional method could not produce large yields of flavones [91]. Cramer and Elschnig discovered that the best catalyst is sodium ethoxide [92]. Ares et al. suggested using potassium tert-butoxide for the synthesis of the diketone intermediate, obtaining higher yields [93]. Jain et al. used benzoyl chloride in benzene under phase transfer-catalysis conditions with n-tetrabutylamonium hydrogen sulphate, obtaining o-hydroxydibenzoylmethane. Further treatment with p-toluenesulphonic acid yielded flavones with good results [94]. Modifying this method permits the synthesis of hydroxyflavones with phloroglucinol units via heating the 2-hydroxyacetophenones with aqueous 5% potassium carbonate followed by treatment with acetic acid [95]. Song and Ahn proposed the use of tetrabutylammonium fluoride as a phase transfer catalyst for the condensation of dibenzoylmethanes, also obtaining good yields [96]. Another useful reaction condition for cyclocondensation was discovered by Stanek and Stodulski, who used N-triflylphosphoramide, an organocatalyst which is active in mild reaction conditions [97]. Through using microwave irradiation, Pinto et al. managed to obtain 3-aroyl-5-hydroxyflavones from 2,6-diaroyloxyacetophenones [98]. Similar results were obtained with a shorter reaction time by using ethyl ammonium nitrate, a recyclable ionic liquid, under microwave irradiation [99].
Through the cyclization of dibenzoylmethanes with CuBr2, 3-bromoflavones are formed, and these can then be converted into 3-aminoflavones [100]. Other catalysts that are reportedly useful for converting dibenzoylmethanes into flavones include the following: FeCl3 in dichloromethane [101]; CuCl2 under microwave irradiation [102]; Cobalt(bis(salicylideniminato-3-propyl)hydroxide, a six coordinate cobalt Schiff base complex [103]; montmorillonite K 10 Clay under microwave irradiation (clay-catalyzed synthesis) [104]; amberlyst 15, a cation-exchange resin, under reflux in isopropyl alcohol [105]; solid supported catalysts like mesoporous titania/tungstophosphoric acid composites TiO2/H3PW12O40 at reflux [106], trifluoromethanesulfonic acid in toluene at reflux [107], Wells–Dawson heteropolyacid in toluene at reflux (or solvent-free) [108], molybdophosphoric and molybdosilicic Keggin heteropolyacids in acetonitrile at reflux [109], and silica gel supported NaHSO4 in toluene at reflux [110].
3.5. Algar–Flynn–Oyamada Method
This method represents the collaboration between Algar and Flynn and the individual work of Oyamada from 1934 to 1935. It can be used to obtain 3-hydroxyflavones (28) from o-hydroxychalcones (14) by means of hydrogen peroxide in aqueous sodium hydroxide solution and cooling [111]. Algar and Flynn used this method with hot potassium hydroxide alcoholic solution, and both achieved good yields (Scheme 17) [112].
Scheme 17.

The Algar–Flynn–Oyamada method.
The mechanism has experienced many alterations over time. At first, Algar and Flynn were not able to isolate the intermediates. They proposed the transitory formation of an ethylene peroxide in the first stage of oxidation [112]. Oyamada considered the existence of a flavanone intermediate (60) formed by the electrophilic attack of hydrogen peroxide on position 3 of the flavanone anion (59) [111,113]. Dean and Podimuang demonstrated that no epoxides were formed as intermediates for obtaining 3-hydroxyflavones (28). They argued that a β-position attack by the hydroperoxide anion would be difficult for phenolic chalcones due to the internal electronic inactivation (see mesomere structure 58) and the basic conditions that turn them into anions, facilitating the electrostatic repulsion of the hydrogen peroxide (Scheme 18) [114].
Scheme 18.

The mechanism of the Algar–Flynn–Oyamada method proposed by Dean and Podimuang.
Starting from o-hydroxychalcones, via epoxides as intermediates, two cyclization products can theoretically be formed, namely, aurones (63), if the attack takes place in the α position, and flavonols (65) (via flavanone 64), if the attack takes place in the β position (Scheme 19). When 6′-substituted-2′-hydroxychalcones (61) were used as precursors, it was found that the cyclization takes place preferentially via α attack, with the formation of aurones as major reaction products. This is due to the fact that the substituent grafted in the 6′ position of the chalcone displaces the keto group from the plane of the aromatic ring. This causes the steric inhibition of resonance from the 2’-oxygen anion and determines the activation of the α-carbon [77].
Scheme 19.

Algar–Flynn–Oyamada reaction products. Aurone formation happens in higher yields when a 6′-substituted chalcone is used.
Adams and Main argued that we should not rule out the idea that epoxides are precursors and intermediates in the formation of flavonols via β-attack. They demonstrated that treating an epoxide at various pHs in aqueous acetonitrile solution and room temperature led to small amounts of the β-cyclization product, which was a flavonol derivative [115,116].
Dean and Podimuang’s theory was also challenged by Serdiuk et al. They concluded that epoxides are indeed intermediates in this method by analyzing the thermodynamic characteristics of the intermediate reactions and finding out that the reactions of chalcones in anionic form with the hydroperoxide anion are energetically favorable [113]. However, Bhattacharyya and Hatua demonstrated through the density functional theory that epoxidation is unlikely because of the electrostatic interaction of the hydroperoxide anion with the conjugated double bond, although an epoxide intermediate could still be formed at high temperatures and converted into aurone, supporting Dean and Podimuang’s work [117].
The major limitation of the Algar–Flynn–Oyamada method is the fact that it cannot be applied for the synthesis of 3-hydroxyflavones via the cyclization of the corresponding 6′-substituted o-hydroxychalcones because, in this case, cyclization takes place preferentially via α-attack [118]. Also, besides 3-hydroxyflavones, aurones can be easily obtained through α-cyclization, and sometimes, even 2-benzyl-2-hydroxydihydrobenzofuran-3-ones (67) and 2-arylbenzofuran-3-carboxylic acids (68) are formed [116,119] (Scheme 19).
Oxidative cyclization with H2O2/NaOH or KOH of o-hydroxychalcones was applied by Li Xiang et al. for the synthesis of new 3-O-substituted flavonols as anti-prostate cancer agents [32]. Khdera H.A. et al. recently synthesized a series of flavonol derivatives with antifungal properties via the oxidative cyclization of o-hydroxychalcones with H2O2/KOH, followed by the further derivatization of the 3-OH group [57].
A couple of modifications for this method exist, namely, using Na2CO3 and H2O2 in methanol and water to obtain 5′-substituted-3-hydroxyflavones [120], phase transfer catalysis (tetrabutylammonium bromide, iodide, or hydrogensulphate; benzyltriphenylphosphonium chloride; ethyltriphenylphosphonium iodide; propyltriphenylphosphonium iodide or bromide) [121], and direct synthesis starting from acetophenone and aldehyde without isolating the chalcone intermediate (known as one-pot synthesis) [55,122,123,124,125].
Other modern improvements have been made for the Algar–Flynn–Oyamada reaction, such as performing the synthesis under microwave irradiation [55].
The oxidative cyclization of o-hydroxychalcones with H2O2/OH− was successfully extended by our research group for the synthesis of new analogs of hydroxyflavones containing the 2-phenylthiazole moiety instead of the phenyl B ring of the basic skeleton of natural flavones. In the first step, the thiazole o-hydroxy-heterochalcones (70) were obtained in 75–82% yields via the condensation of o-hydroxyacetophenone (10) with different 2-arylthiazol-4-yl carbaldehydes (69) (Scheme 20). Their epoxidation with hydrogen peroxide, followed by oxidative cyclization, afforded the corresponding 2-arylthiazole hydroxyflavones (71) in 65–71% yields [126].
Scheme 20.

Synthesis of new thiazole hydroxychromones via the oxidative cyclization of thiazole o-hydroxy chalcones [126].
The cyclization pathway of thiazole and thiazolo[3,2-b][1,2,4]triazole hetero-chalcones with hydrogen peroxide in basic media (NaOH) was investigated by V. Zaharia et al. Through treatment with the hydrogen peroxide of o-hydroxy-heterochalcones (72) in basic media, the corresponding hydroxyflavones (73) were obtained (Scheme 21a). The epoxyketones (75) were obtained in the same reaction conditions as the hydroxyflavones (73), starting from the unhydroxylated heterochalcones (74) as precursors (Scheme 21b) [127].
Scheme 21.

(a) Synthesis of thiazolo[3,2-b][1,2,4]triazole hydroxyflavones via the cyclization of o-hydroxychalcones. (b) Synthesis of thiazole and thiazolo[3,2-b][1,2,4]triazole epoxyketones [127].
3.6. Claisen–Schmidt Method
This method was established in 1962 and consists of two steps. The first step is Claisen–Schmidt condensation between an o-hydroxyacetophenone (10) and benzaldehyde derivative (7) in basic medium, which affords chalcones. The second step involves the oxidative cyclization of the obtained chalcones (14), which can be achieved using a large variety of conditions and catalysts [3] (Scheme 22).
Scheme 22.

The Claisen–Schmidt method.
The condensation mechanism begins with the formation of an anion of the acetophenone (76) under basic conditions. Through the nucleophilic attack of the anion (76) on the carbonyl group of benzaldehyde (7), followed by the elimination of H2O, the corresponding chalcones (14) are obtained [128] (Scheme 23).
Scheme 23.

The mechanism of Claisen–Schmidt condensation [128].
Cyclization can be realized by many methods, first starting with iodine in hot dimethyl sulfoxide (I2/DMSO, Δ). Patonay et al. observed that this method is suitable for a large variety of substituents, including electron-donating and electron-withdrawing groups and sensitive-to-oxidation and protecting groups. Thus, it can be considered a general method of cyclization for obtaining flavones from 2′-hydroxychalcones [129]. The mechanism involves the formation of an iodonium cation (79) via the interaction of I2 with the o-hydroxychalcone (14), followed by cyclization via the nucleophilic attack of the o-hydroxy group. Further elimination of hydroiodic acid affords the corresponding flavones (9). The solvent dimethyl sulfoxide (81) is important in this reaction because it acts as a co-oxidant and regenerates iodine [130] (Scheme 24).
Scheme 24.

Mechanism of the oxidative cyclization of o-hydroxychalcones with I2/DMSO. Role of DMSO in the regeneration of iodine (mechanism adapted from Masesane) [130].
Through using microwave irradiation, the reaction time is greatly reduced to approximatively three minutes [131]. However, this method is limited in the case of 2′-hydroxychalcones with a phloroglucinol moiety, affording complex mixtures. Hans and Grover extended the applicability of iodine as an oxidant agent by instead using sodium periodate in hot dimethyl sulfoxide (NaIO4/DMSO, Δ), managing to smoothly convert phloroglucinol-derived chalcones into the corresponding flavones [132] (Scheme 25).
Scheme 25.

Oxidative cyclization of o-hydroxychalcones using I2 or NaIO4 in DMSO [129,131,132].
In order to extend the general method of cyclization of o-hydroxychalcones with iodine in dimethyl sulfoxide, our research group investigated this method using a series of 2-arylthiazole o-hydroxychalcones as precursors (85). A similar chemical behavior was observed in this case, with the corresponding 2-arylthiazole flavones (86) being obtained with 32–55% yields (Scheme 26) [133].
Scheme 26.

Cyclization of 2-arylthiazole o-hydroxychalcones with iodine in dimethyl sulfoxide [133].
At present, the cyclization of o-hydroxychalcones using iodine in dimethyl sulfoxide still represents the principal method for the synthesis of flavones and their analogs with various structures. This method is still finding multiple applications because it generally allows for obtaining the target compounds with high yields, while the o-hydroxychalcones needed as precursors can be easily obtained via Claisen–Schmidt condensation [30,34,35,46,47,61].
Another method for the cyclization of o-hydroxychalcones was established by Litkei et al., who used iodosobenzene diacetate (phenyliodine(III) diacetate, PIDA), a hypervalent iodine reagent, which forms iodosylbenzene in situ [134] (Scheme 27). The same reagent was used for obtaining prenylated flavones, which are abundant in nature, from prenylated 2′-hydroxychalcones [135].
Scheme 27.

Oxidative cyclization using hypervalent iodine reagent [134].
The usage of ionic liquids for this oxidative cyclization was described in the literature. Du et al. developed a new method involving Cu(I) iodide, mediated by the ionic liquid [bmim][NTf2] (1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide, compound 88) at a low temperature. The reaction mechanism has not been fully elucidated, but the results so far reveal that a flavanone is formed intermediately. The flavanone seems to be dehydrogenated to the corresponding flavone in the same reaction conditions [136]. Lahyani and Trabelsi reported the oxidative cyclization of o-hydroxychalcones with iodine monochloride in dimethyl sulfoxide (ICl-DMSO) under ultrasound. This method has similar advantages to than of ultrasound processes, such as mild conditions, high yields, and eco-friendliness. The mechanism is similar to the one based on oxidative cyclization with I2-DMSO [137] (Scheme 28).
Scheme 28.

Oxidative cyclization of o-hydroxychalcones with CuI in ionic liquid [136] and ICl in DMSO, respectively, under ultrasound [137].
Heating o-hydroxychalcones with iodine in triethylene glycol is also a good and inexpensive method for their cyclization. While the underlying mechanism is not fully understood, the authors proposed a pathway that involves the iodination of chalcone, which affords chalcone diiodides (89), similar to the chalcone dihalides from the von Kostanecki’s method. Via the dehydrohalogenation of the intermediate (90), a 3-iodoflavanone (80) is formed, which yields the corresponding flavone (9) via β-elimination of a second hydroiodic acid molecule [138]. The use of iodine on silica gel (I2-SiO2) was reported by Babu et al. to provide favorable results and less harmful effects towards the environment [139]. Another solid supported catalyst, iodine on neutral alumina (I2-Al2O3), was reported by Sarda et al., providing short reaction times, simple conditions, and very good yields [140] (Scheme 29).
Scheme 29.

(a) Oxidative cyclization with iodine in triethylene glycol and solid supports (silica gel and alumina) [138,139,140]. (b) The proposed pathway for oxidation with molecular iodine (adapted from Miyake et al. [138]).
An alternative to the toxicity and corrosiveness of iodine was proposed by Kulkarni et al., who used ammonium iodide under exposure to air and solvent-free conditions, thus generating in situ iodine that acted as a cyclization agent [141] (Scheme 30).
Scheme 30.

Oxidative cyclization using NH4I in solvent-free conditions [141].
Selenium dioxide is another catalyst used for the cyclization of o-hydroxychalcones. It can be combined with various solvents, such as pentan-1-ol [142] (only providing low yields) [132], dioxane [143], and isoamyl alcohol with prolonged heating, to facilitate the formation of side products and low yields [144]. Dimethyl sulfoxide was also used with good yields [145], but in order to diminish the high toxicity of DMSO, Gupta et al. managed to use selenium dioxide and traces of solvent over silica gel under microwave irradiation, yielding very good results [146]. Similar to I2/DMSO, SeO2/DMSO is also problematic for chalcones with a phloroglucinol oxygenation pattern [132]. However, selenium dioxide is volatile and hazardous. Lamba and Makrandi proposed using sodium selenite (Na2SeO3), which is less volatile, and found out that it acts as a proper dehydrogenating agent in DMSO [147] (Scheme 31).
Scheme 31.

Oxidative cyclization of o-hydroxychalcones using selenium-based catalysts in different solvents and reaction conditions [142,143,144,145,146,147].
Palladium was also experimented on as a catalyst. Kasahara et al. used lithium chloropalladite (Li2PdCl4), palladium(II) acetate, and (CH3COO)2Pd to obtain flavones. The reaction was described as a phenoxypalladation, with the formation of the intermediates 91 and 92, followed by the elimination of palladium (II) hydride (HPdCl). This method also yielded small amounts of flavanone (12) [148] (Scheme 32).
Scheme 32.

Oxidative cyclization of o-hydroxychalcones with palladium(II) salts [148].
Cyclodehydrogenation with DDQ (2,3-dichloro-5,6-dicyano-p-benzoquinone) was proposed by Imafuku et al. Their method involves using dioxane as a solvent and results in a mixture of flavones, flavanones, and aurones in low yields [149]. Another agent that acts in a similar manner is nickel peroxide (NiO2) in dioxane, yielding a similar mixture. However, flavanones can be dehydrogenated to flavones under the action of NiO2 in benzene as a solvent [150] (Scheme 33).
Scheme 33.

Oxidative cyclization of o-hydroxychalcones using DDQ and nickel peroxide [149,150].
Disulfides are also a good choice for cyclodehydrogenation. Hoshino et al. used four disulfides, namely, dineopentyl disulfide, diisopentyl disulfide, dipentyl disulfide, and diphenyl disulfide, with the latter giving the best yields. The disadvantages of this method include the very high temperatures (260–290 °C) and very low yields when electron-withdrawing groups (NO2) are present [151] (Scheme 34).
Scheme 34.

Oxidative cyclization using organic disulfides.
Initially meant for obtaining quinolines from 2′-aminochalcones using FeCl3·6H2O in methanol, Kumar and Perumal applied the same method on 2′-hydroxychalcones and obtained flavones (96, X = O) with satisfactory results [152]. Similarly, Liu et al. used cerium(IV) sulphate tetrahydrate (Ce(SO4)2·4H2O) on silica gel to obtain flavones from 2′-hydroxychalcones at 100 °C, aza-flavones (96, X = NH), and aza-flavanones from 2′-aminochalcones (95, X = NH) [153] (Scheme 35).
Scheme 35.

Oxidative cyclization of o-hydroxychalcones using FeCl3 hexahydrate or Ce(SO4)2 tetrahydrate [152,153].
In addition to all the reagents previously mentioned, sodium perborate tetrahydrate (SPB, compound 97) was proposed by Ganguly et al. They observed that depending on the solvent, this method could yield different products, such as warm acetic acid and SPB in excess yielded flavones (9), while warm aqueous acetonitrile yielded flavanones. In the case of flavone formation, SPB and acetic acid generate peracetoxyboron anion species (101) that favorize the oxidative cyclization of o-hydroxychalcones [154,155] (Scheme 36).
Scheme 36.

(a) Oxidative cyclization of o-hydroxychalcones using sodium perborate tetrahydrate (SPB). (b) The mechanism for generating peracetoxyboron anionic species [154,155].
Other reagents for the oxidative cyclization include indium(III) halides (InCl3 and InBr3) on silica gel and in solvent-free conditions, which has been shown to provide higher yields when InBr3 is used [156] or when sodium tellurite is used in dimethyl sulfoxide and anhydrous conditions (Na2TeO3-DMSO) [157]. The reaction mechanism has not completely ben elucidated. The supposed reported pathway for the oxidative cyclization with indium halides involves the formation of a flavanone as an intermediate, which is dehydrogenated in the same reaction conditions to the corresponding flavone [156]. Oxalic acid in ethanol reflux, which was used by Zambare et al., has been shown to be a very useful and cheap method, with excellent yields over 90% [158] (Scheme 37).
Scheme 37.

Oxidative cyclization of o-hydroxychalcones with indium(III) halides [156], sodium tellurite in DMSO [157], and oxalic acid [158].
Photocyclization provides another way to perform this reaction. However, in one study, it yielded only flavanones and in low quantities [159]. By adding a heterocyclic N-oxide, pyrimido[5,4-g]pteridine N-oxide (102), Maki et al. managed to obtain flavones (9) but still only in unsatisfactory yields and in a mixture with flavanones (12). The photoreaction involves a single electron transfer (SET) process from chalcone (14) to N-oxide (102). Initially, the N-oxide is found in the oxygenated form, pyrimido [5,4-g]pteridin-2,4,6,8(1H,3H,7H,9H)-tetrone 5-oxide (102), and as the mixture forms, it gets deoxygenated to pyrimido [5,4-g]pteridine (109) [160]. Electrochemistry found applications in this reaction too. Saničanin and Tabaković cyclized 2′-hydroxychalcones by electrochemically generating a cation radical of tris-(4-bromophenyl)amine, which acted as a homogenous electron transfer reagent. This method creates a mixture of flavanones and flavones in moderate yields [161] (Scheme 38).
Scheme 38.

(a) Oxidative cyclization using photo- and electrochemistry [160,161]. (b) Mechanism of single-electron transfer process (adapted from Maki et al. [160]).
Through creating a totally non-hazardous medium, a different approach for the cyclization of o-hydroxychalcones was implemented by Tamuli et al. In this case, the catalyst consists of a mixture of two agro-food waste products—Musa sp. ‘Malbhog’ peel ash (MMPA) and Musa champa Hort. ex Hook. F. peel ash (MCPA)—which allowed for the cyclodehydrogenation of o-hydroxychalcones in solvent-free conditions and at room temperature [162] (Scheme 39).
Scheme 39.

Oxidative cyclization using banana peel ash in open air and solvent-free conditions (adapted from Tamuli et al. [162]).
Among the reported methods for the cyclization of o-hydroxychalcones, our research group investigated the most promising ones in order to obtain new flavonoid analogs containing the 2-arylthiazole moiety instead of the benzene ring B. Our aim was to also investigate the chemical behavior of 2-arylthiazole o-hydroxychalcones in the cyclization reactions.
The oxidative cyclization of the 2-arylthiazole o-hydroxychalcones (85) afforded various reaction products, depending on the oxidizing agent. Flavanones (112) were obtained with 40–55% yields via the cyclization of the corresponding 2-arylthiazole o-hydroxychalcones in acidic catalysis (H2SO4 conc. in ethanol) [133]. The cyclization of 2-arylthiazole o-hydroxychalcones in the presence of sodium acetate in methanol (used as a solvent) also afforded the corresponding flavanones (112) in good yields. The use of copper(II) acetate in dimethyl sulfoxyde resulted in a mixture of aurones (110) and the corresponding flavones (86) in an approximate 1:1 molar ratio [43]. Hydroxyflavones (111) were obtained via the cyclization of the same substrates with hydrogen peroxide in alkaline catalysis [43], and flavones (86) were obtained when iodine in dimethyl sulfoxide was used [43,133]. The cyclization with selenium dioxide in n-butanol led to a mixture of flavones (86) and hydroxyflavones (111) [43]. Cyclization with mercury(II) acetate in pyridine as a solvent afforded the corresponding Z-aurones (110) with 70–86% yields [43]. The cyclization products and the reactions conditions are summarized in Scheme 40.
Scheme 40.

Cyclization products obtained via the oxidative cyclization of 2-arylthiazole o-hydroxychalcones in different reaction conditions [43,133,163].
The cyclization of o-methoxylated chalcones bearing the 2-arylthiazole moiety (113) was further studied in similar reaction conditions. It was found that the reaction occurred differently depending on the oxidizing agent and the reaction conditions. Through treatment with iodine in dimethyl sulfoxide, at reflux, the corresponding flavones (86) were formed. This fact indicates that the demethylation of the methoxy group of chalcone occurred, resulting in the corresponding o-hydroxychalcone (85), which was further cyclized to the corresponding flavone [163]. Instead, when hydrogen peroxide in NaOH was used as a cyclization agent, the formation of the corresponding epoxides (114) was observed, which can be explained by the fact that the methoxy group is resistant in these reaction conditions; therefore, the cyclization to flavone cannot take place (Scheme 41) [163].
Scheme 41.

Cyclization products of 2-arylthiazole o-methoxychalcones [163].
3.7. Mentzer Method
This method involves synthesizing flavones based on the reaction between phenols (phenol, resorcinol, or phloroglucinol) and β-ketoesters (116) [164] (Scheme 42). It is based on the Pechmann reaction (used to obtain coumarins from phenol and ethyl acetoacetate) [165]. Mentzer et al. obtained flavones from resorcinol and ethyl 2-benzylacetoacetate by heating up the mixture for 48 h at 250 °C [166].
Scheme 42.

The Mentzer method for the synthesis of flavones.
The mechanism involves a nucleophilic attack by the phenolic compound (115) on the β-ketoester (116), resulting in an arenium ion (117). Under heating, the intermediate eliminates the alcohol and affords o-hydroxydibenzoylmethane (119), a compound which is also available in the Baker–Verkataraman method, which is cyclized into flavone (9) [164] (Scheme 43).
Scheme 43.

The mechanism of the Mentzer method (image adapted from Wang [164]).
Seijas et al. developed a solvent-free modification of this method that uses microwave irradiation instead of heating. The yields are good, and the time of reaction is significantly reduced [167] (Scheme 44).
Scheme 44.

A variation of the Mentzer method using microwave irradiation (image adapted from Seijas et al. [167]).
3.8. Suzuki–Miyaura Method
The Suzuki–Miyaura method is a cross-coupling reaction based on the insertion of palladium in sp2 hybridized C-X bonds and the usage of various organoboron precursors (124) under mild reaction conditions. In the case of flavones, 2-halogenochromones (123) are used as substrates; however, they are difficult to obtain [168] (Scheme 45).
Scheme 45.

The Suzuki–Miyaura method for the synthesis of flavones starting from 2-halogenochromones [168,169].
While this method mostly uses 2-bromo- or 2-iodochromones, Kraus and Gupta demonstrated that these precursors yield aurones rather than flavones. By using 2-chlorochromone, they managed to obtain flavones in proper yields (68–74%) [169].
Their synthesis starts with the esterification of a phenolic compound (125) and 3,3-dichloroacrylic acid (126), resulting in a dichloro acrylic ester (127). This compound undergoes Fries transposition, affording the intermediate 128. Further cycloelimination in basic conditions yields 2-chlorochromone (129). Finally, this is then coupled with arylboronic acids, which can replace the halogen atom from the chromone skeleton with the aryl rest of the boronic compound, thus affording a flavone (130) [169] (Scheme 46).
Scheme 46.

The pathway of the Suzuki–Miyaura method for obtaining substituted flavones (image adapted from Kraus and Gupta [169]).
The postulated mechanism of the Suzuki–Miyaura method involves a catalytic cycle initiated by the formation of the active Pd catalytic species Pd(PPh3)2 (132). Further oxidative insertion of palladium to the 2-halogenochromone (133) leads to the organopalladium intermediate 134 (chromon-2-yl-palladium(II) chloride). In the transmetalation step, the chlorine atom is transferred to the boronic compound 124, resulting in a chloroboronic acid (135) and chromon-2-yl-phenyl-palladium (136), which undergoes reductive elimination, yielding the flavone (9) and the active Pd catalyst Pd(PPh3)2 [170] (Scheme 47).
Scheme 47.

Postulated mechanism of the Suzuki–Miyaura method.
4. Chemical Synthesis of Aurones
Among the reported methods for the synthesis of aurones, the most applied are those based on the oxidative cyclization of o-hydroxychalcones. These reactions are mediated by transitional metal salts such as Hg(CH3COO)2 [40,48,171], CuBr2 [171,172], or Tl(NO3)3 [173], whose metal cation interacts with the double bond in chalcones, thus favoring the attack of the ortho hydroxy group on the alpha carbon (Scheme 48) [130].
Scheme 48.

Aurone synthesis via the cyclization of o-hydroxychalcones [171,172,173].
The cyclization of o-hydroxychalcones (137) with mercury(II) acetate afforded the best yields in aurones (138) when the reaction was performed in pyridine [40,48,171] or with dimethyl sulfoxide [174] as a solvent at reflux. In the case of cyclization with cupric bromide, aprotic polar solvents such as dimethyl sulfoxide [171] or N,N-dimethyl formamide [37,172] have been shown to be the optimal solvents.
In the case of thallium nitrate-mediated cyclization, reported data indicate that the reaction course depends on the nature of the substituents on the B ring; only the electron-withdrawing groups (chlorine, formyl, methoxycarbonyl, and nitro) grafted in the para position are favorable for cyclization to aurones. In other cases, mixtures of aurones and isoflavones, or exclusively isoflavones, are obtained [175].
Our research group synthesized a series of aurone analogs (140) containing the same 2-phenylthiazole aromatic system as the B ring in Z-configuration, achieving 70-86% yields via the cyclization of the corresponding o-hydroxychalcones (139) with mercury(II) acetate in pyridine at reflux (Scheme 49) [43].
Scheme 49.

Synthesis of aurone analogs containing the 2-phenylthiazole moiety [43].
Another chemical route towards aurones involves starting with substituted benzofuran-3-ones as precursors. Benzofuran-3-ones (143) are synthesized in two steps. The first step consists of the introduction of a chloroacetyl group on the aromatic ring of the phenols (141) by applying a Hoesch-type reaction (with 2-chloronitriles in the presence of anhydrous ZnCl2 and gaseous HCl) [176] or Friedel–Crafts acylation (with alfa-halogenated acyl chlorides in the presence of AlCl3) [49,177]. In the next step, an intramolecular Williamson-type reaction with ring closure is applied (Scheme 50) [49,176,177].
Scheme 50.

Synthesis of benzofuran-3-ones.
The condensation of benzofuran-3-ones (143) with aromatic aldehydes (144) affords the corresponding aurones 145 (Scheme 51). This step can be performed under acid catalysis with HCl [52] and HCl/CH3COOH [49,74], in basic catalysis with 50% KOH/Ethanol or KOH/Methanol [176,177,178] or NaOH/NaOCH3 [50], or with a Al2O3 catalyst [75]. A recently reported method for the condensation of benzofuran-3-ones with aromatic aldehydes uses an activated Ba(OH)2 catalyst in dimethyl sulfoxide as a solvent [53,58].
Scheme 51.

Synthesis of aurones via the condensation of aromatic aldehydes with benzofuran-3-ones.
Another reported method for the synthesis of aurones involves the cyclization of hydroxypropynylphenol derivatives (149) obtained via the alkynylation of salicylaldehyde derivatives (146) with lithium arylacetylures (147) (Scheme 52) [179,180].
Scheme 52.

Synthesis of aurones via hydroxypropynylphenol derivatives [179,180].
Harkat H. et al. performed the cyclization of hydroxypropynylphenol derivatives (149) in the presence of gold(I) chloride and potassium carbonate in acetonitrile. Further oxidation with MnO2 of the intermediate secondary alcohol (150) afforded the corresponding aurones 151 (Scheme 52) [179].
Li S. et al. proposed a variant of cyclization with AgNO3. Ag+ ions catalyze both the cyclocondensation reaction and the oxidation of the secondary alcohols to the corresponding aurones 151 (Scheme 52) [180].
5. Conclusions
From a pharmacological point of view, flavones, flavonols, and aurones are precious natural products that have inspired researchers over time to create new biologically active compounds for use as anticancer and anti-infectious agents with benzochromon-4-one or benzofuran-3-one structures.
The polyphenolic structure of natural flavones and aurones determines their low stability to oxidants in solution and also generally decreases their bioavailability after oral administration due to their low solubility, thus affecting their potential use as therapeutic agents. Consequently, the structural modulations on aromatic rings A and B that have been reported to aid the design of new flavone/aurone analogs and facilitate both the improvement of therapeutic properties and pharmacokinetic profiles and increases in stability must be taken into account.
The hybridization of the basic skeletons of flavones and aurones with other pharmacophore moieties has led to new compounds with superior pharmacological profiles compared to their natural analogs.
The most applied routes for the synthesis of flavones, hydroxyflavones, and aurones involve the cyclization of o-hydroxychalcones. As exemplified in Section 3, the cyclization pathway transpires differently depending on the reaction conditions, the catalysts used, and the nature and position of the substituents grafted on the aromatic moieties [118,127].
In order to obtain new biologically active compounds based on the flavone scaffold, two main directions can be outlined, offering future research perspectives. One research direction involves using natural flavones already recognized for their therapeutic potential as precursors. Through the alkylation of their OH groups [10,45] or by grafting different pharmacophore units on their aromatic rings [31], new flavonoid analogs or hybrid molecules such as Mannich bases [56] with improved biological functions can be obtained. Based on literature data regarding the discovery of new important pharmacophore moieties targeting tumor-associated structures or recognized for their antimicrobial properties, new multi-target-acting hybrid molecules with anticancer or antimicrobial potential could be developed using similar strategies that involve starting from the structures of natural flavones.
Another research direction with important future prospects in the development of new flavone analogs involves the synthesis of the 2-phenyl-chromen-4-one system via different procedures. The most applied and promising route consists of the cyclization of o-hydroxychalcones with I2/DMSO [30,46,47,61,133]. Although this method was implemented long time ago, this procedure is still largely exploited and extended on various structures because it generally allows one to obtain target compounds with high yields and because the o-hydroxychalcones needed for use as precursors are accessible and easily obtainable via Claisen–Schmidt condensation.
The oxidative cyclization of o-hydroxychalcones with H2O2 in basic media represents a promising route with future prospects for the synthesis of 3-hydroxyflavones. The cyclization pathway transpires differently depending on the nature and the position of the substituents grafted on the aromatic moieties, thus affording 3-hydroxyflavones or aurones [116,118,119]. Modern improvements have been made to this method, such as performing the synthesis under microwave irradiation [55].
Even if the presented methods for the synthesis of flavones and aurones generally refer to obtaining the basic skeleton for natural flavones and aurones, in which the aromatic moieties are benzene and benzochromon-4-one or benzofuran-3-one rings, these methods have also been successfully applied in obtaining other new synthetic analogs of flavones or aurones containing other aromatic moieties such as heteroaromatic systems [35,43,53,55,58,61]. In particular, flavone/aurone analogs bearing the heterocyclic moieties thiazole, pyrazole, thiophene, pyridine, and quinoline instead of ring B represent basic skeletons with future prospects for the design and development of new heterocyclic flavonoid analogs with anticancer and antimicrobial potential.
The results presented in this review regarding the biological activity of flavones and related compounds confirm that the general synthetic pathways towards flavones, flavonols, and aurones have great importance for future research on the development of new effective anticancer and antimicrobial agents.
Author Contributions
Writing—draft preparation, D.L. and D.U.; conceptualization and supervision, V.Z. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This research received no external funding.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Berim A., Gang D.R. Methoxylated flavones: Occurrence, importance, biosynthesis. Phytochem. Rev. 2016;15:363–390. doi: 10.1007/s11101-015-9426-0. [DOI] [Google Scholar]
- 2.Ververidis F., Trantas E., Douglas C., Vollmer G., Kretzschmar G., Panopoulos N. Biotechnology of flavonoids and other phenylpropanoid-derived natural products. Part I: Chemical diversity, impacts on plant biology and human health. Biotechnol. J. 2007;2:1214–1234. doi: 10.1002/biot.200700084. [DOI] [PubMed] [Google Scholar]
- 3.Kshatriya R., Jejurkar V.P., Saha S. In memory of Prof. Venkataraman: Recent advances in the synthetic methodologies of flavones. Tetrahedron. 2018;74:811–833. doi: 10.1016/j.tet.2017.12.052. [DOI] [Google Scholar]
- 4.Tóth S., Szepesi Á., Tran-Nguyen V.-K., Sarkadi B., Német K., Falson P., Di Pietro A., Szakács G., Boumendjel A. Synthesis and anticancer cytotoxicity of azaaurones overcoming multidrug resistance. Molecules. 2020;25:764. doi: 10.3390/molecules25030764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hou D.X., Kumamoto T. Flavonoids as protein kinase inhibitors for cancer chemoprevention: Direct binding and molecular modeling. Antioxid. Redox Signal. 2010;13:691–719. doi: 10.1089/ars.2009.2816. [DOI] [PubMed] [Google Scholar]
- 6.Polier G., Ding J., Konkimalla B.V., Eick D., Ribeiro N., Köhler R., Giaisi M., Efferth T., Desaubry L., Krammer P.H., et al. Wogonin and related natural flavones are inhibitors of CDK9 that induce apoptosis in cancer cells by transcriptional suppression of Mcl-1. Cell Death Dis. 2011;2:e182. doi: 10.1038/cddis.2011.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Golub A.G., Bdzhola V.G., Ostrynska O.V., Kyshenia I.V., Sapelkin V.M., Prykhod’ko A.O., Kukharenko O.P., Yarmoluk S.M. Discovery and characterization of synthetic 4′-hydroxyflavones—New CK2 inhibitors from flavone family. Bioorg. Med. Chem. 2013;21:6681–6689. doi: 10.1016/j.bmc.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 8.Chao S.W., Su M.Y., Chiou L.C., Chen L.C., Chang C.I., Huang W.J. Total Synthesis of Hispidulin and the Structural Basis for Its Inhibition of Proto-oncogene Kinase Pim-1. J. Nat. Prod. 2015;78:1969–1976. doi: 10.1021/acs.jnatprod.5b00324. [DOI] [PubMed] [Google Scholar]
- 9.Sedlacek H., Czech J., Naik R., Kaur G., Worland P., Losiewicz M., Parker B., Carlson B., Smith A., Senderowicz A., et al. Flavopiridol (L86 8275; NSC 649890), a new kinase inhibitor for tumor therapy. Int. J. Oncol. 1996;9:1143–1168. doi: 10.3892/ijo.9.6.1143. [DOI] [PubMed] [Google Scholar]
- 10.Pontes O., Costa M., Santos F., Sampaio-Marques B., Dias T., Ludovico P., Baltazar F., Proença F. Exploitation of new chalcones and 4H-chromenes as agents for cancer treatment. Eur. J. Med. Chem. 2018;157:101–114. doi: 10.1016/j.ejmech.2018.07.058. [DOI] [PubMed] [Google Scholar]
- 11.Monasterio A., Urdaci M., Pinchuk I., Moratalla N., Martínez I. Flavonoids induce apoptosis in human leukemia U937 cells through caspase-and caspase-calpain-dependent pathways. Nutr. Cancer. 2004;50:90–100. doi: 10.1207/s15327914nc5001_12. [DOI] [PubMed] [Google Scholar]
- 12.Moreira J., Ribeiro D., Silva P.M.A., Nazareth N., Monteiro M., Palmeira A., Saraiva L., Pinto M., Bousbaa H., Cidade H. New alkoxy flavone derivatives targeting caspases: Synthesis and antitumor activity evaluation. Molecules. 2019;24:129. doi: 10.3390/molecules24010129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kariagina A., Doseff A.I. Anti-Inflammatory Mechanisms of Dietary Flavones: Tapping into Nature to Control Chronic Inflammation in Obesity and Cancer. Int. J. Mol. Sci. 2022;23:15753. doi: 10.3390/ijms232415753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Occhiuto C.J., Moerland J.A., Leal A.S., Gallo K.A., Liby K.T. The Multi-Faceted Consequences of NRF2 Activation throughout Carcinogenesis. Mol. Cells. 2023;46:176–186. doi: 10.14348/molcells.2023.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ross J.A., Kasum C.M. Dietary flavonoids: Bioavailability, metabolic effects, and safety. Annu. Rev. Nutr. 2002;22:19–34. doi: 10.1146/annurev.nutr.22.111401.144957. [DOI] [PubMed] [Google Scholar]
- 16.Jiang M., Zhu M., Wang L., Yu S. Anti-tumor effects and associated molecular mechanisms of myricetin. Biomed. Pharmacother. 2019;120:109506. doi: 10.1016/j.biopha.2019.109506. [DOI] [PubMed] [Google Scholar]
- 17.Semwal D.K., Semwal R.B., Combrinck S., Viljoen A. Myricetin: A dietary molecule with diverse biological activities. Nutrients. 2016;8:90. doi: 10.3390/nu8020090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zang W., Wang T., Wang Y., Li M., Xuan X., Ma Y., Zhao G. Myricetin exerts anti-proliferative, anti-invasive, and pro-apoptotic effects on esophageal carcinoma EC9706 and KYSE30 cells via RSK2. Tumor Biol. 2014;35:12583–12592. doi: 10.1007/s13277-014-2579-4. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y.C., He X.L., Qi L., Shi W., Yuan L.-W., Huang M.-Y., Xu Y.-L., Chen X., Gu L., Zhang L.-L., et al. Myricetin inhibits interferon-γ-induced PD-L1 and IDO1 expression in lung cancer cells. Biochem. Pharmacol. 2022;197:114940. doi: 10.1016/j.bcp.2022.114940. [DOI] [PubMed] [Google Scholar]
- 20.Han S.-H., Lee J.-H., Woo J.-S., Jung G.-H., Jung S.-H., Han E.-J., Kim B., Cho S.-D., Nam J.-S., Che J.H., et al. Myricetin induces apoptosis and autophagy in human gastric cancer cells through inhibition of the PI3K/Akt/mTOR pathway. Heliyon. 2022;8:e09309. doi: 10.1016/j.heliyon.2022.e09309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh S.S., Yap W.N., Arfuso F., Kar S., Wang C., Cai W., Dharmarajan A.M., Sethi G., Kumar A.P. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: A reality for personalized medicine? World J. Gastroenterol. 2015;21:12261–12273. doi: 10.3748/wjg.v21.i43.12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li L., Ma H., Li D., Shu Q., Wang T., Song X., Xu H. Myricetin alleviates the formaldehyde-enhanced Warburg effect in tumor cells through inhibition of HIF-1α. Toxicology and Appl. Pharmacol. 2022;454:116246. doi: 10.1016/j.taap.2022.116246. [DOI] [PubMed] [Google Scholar]
- 23.Gu L., Li Z., Zhang X., Chen M., Zhang X. Identification of MAP Kinase Kinase 3 as a protein target of myricetin in non-small cell lung cancer cells. Biomed. Pharmacother. 2023;161:114460. doi: 10.1016/j.biopha.2023.114460. [DOI] [PubMed] [Google Scholar]
- 24.El Menyiy N., Aboulaghras S., Bakrim S., Moubachir R., Taha D., Khalid A., Abdalla A.N., Algarni A.S., Hermansyah A., Ming L.C., et al. Genkwanin: An emerging natural compound with multifaceted pharmacological effects. Biomed. Pharmacother. 2023;165:115159. doi: 10.1016/j.biopha.2023.115159. [DOI] [PubMed] [Google Scholar]
- 25.Li Y., Hong J., Li H., Qi X., Guo Y., Han M., Wang X. Genkwanin nanosuspensions: A novel and potential antitumor drug in breast carcinoma therapy. Drug Deliv. 2017;24:1491–1500. doi: 10.1080/10717544.2017.1384519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spiegel M., Andruniów T., Sroka Z. Flavones’ and Flavonols’ Antiradical Structure–Activity Relationship—A Quantum Chemical Study. Antioxidants. 2020;9:461. doi: 10.3390/antiox9060461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grigalius I., Petrikaite V. Relationship between antioxidant and anticancer activity of trihydroxyflavones. Molecules. 2017;22:2169. doi: 10.3390/molecules22122169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao L., Yuan X., Wang J., Feng Y., Ji F., Li Z., Bian J. A review on flavones targeting serine/threonine protein kinases for potential anticancer drugs. Bioorg. Med. Chem. 2019;27:677–685. doi: 10.1016/j.bmc.2019.01.027. [DOI] [PubMed] [Google Scholar]
- 29.Chao S.H., Price D.H. Flavopiridol Inactivates P-TEFb and Blocks Most RNA Polymerase II Transcription in Vivo. J. Biol. Chem. 2001;276:31793–31799. doi: 10.1074/jbc.M102306200. [DOI] [PubMed] [Google Scholar]
- 30.Hassan A.H., Choi E., Yoon Y.M., Lee K.W., Yoo S.Y., Cho M.C., Yang J.S., Kim H.I., Hong J.Y., Shin J.-S., et al. Natural products hybrids: 3,5,4′-Trimethoxystilbene-5,6,7-trimethoxyflavone chimeric analogs as potential cytotoxic agents against diverse human cancer cells. Eur. J. Med. Chem. 2019;161:559–580. doi: 10.1016/j.ejmech.2018.10.062. [DOI] [PubMed] [Google Scholar]
- 31.Hassan A.H.E., Lee K.T., Lee Y.S. Flavone-based arylamides as potential anticancers: Design, synthesis and in vitro cell-based/cell-free evaluations. Eur. J. Med. Chem. 2020;187:111965. doi: 10.1016/j.ejmech.2019.111965. [DOI] [PubMed] [Google Scholar]
- 32.Li X., Zhang C., Guo S., Rajaram P., Lee M., Chen G., Fong R., Gonzalez A., Zhang Q., Zheng S., et al. Structure-activity relationship and pharmacokinetic studies of 3-O-substitutedflavonols as anti-prostate cancer agents. Eur. J. Med. Chem. 2018;157:978–993. doi: 10.1016/j.ejmech.2018.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Németh-Rieder A., Keglevich P., Hunyadi A., Latif A.D., Zupkó I., Hazai L. Synthesis and In Vitro Anticancer Evaluation of Flavone—1,2,3-Triazole Hybrids. Molecules. 2023;28:626. doi: 10.3390/molecules28020626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mobbili G., Romaldi B., Sabbatini G., Amici A., Marcaccio M., Galeazzi R., Laudadio E., Armeni T., Minnelli C. Identification of Flavone Derivative Displaying a 4-Aminophenoxy Moiety as Potential Selective Anticancer Agent in NSCLC Tumor Cells. Molecules. 2023;28:3239. doi: 10.3390/molecules28073239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bollikolla H.B., Anandam R., Chinnam S., Varala R., Khandapu B.M.K., Kapavarapu R., Syed K.S., Dubasi N., Syed M.A. C-Dimethylated Flavones as Possible Potential Anti-Tubercular and Anticancer Agents. Chem. Biodivers. 2023;20:e202201201. doi: 10.1002/cbdv.202201201. [DOI] [PubMed] [Google Scholar]
- 36.Schoepfer J., Fretz H., Chaudhuri B., Muller L., Seeber E., Meijer L., Lozach O., Vangrevelinghe E., Furet P. Structure-based design and synthesis of 2-benzylidene-benzofuran-3-ones as flavopiridol mimics. J. Med. Chem. 2002;45:1741–1747. doi: 10.1021/jm0108348. [DOI] [PubMed] [Google Scholar]
- 37.Priyadarshani G., Nayak A., Amrutkar S.M., Das S., Guchhait S.K., Kundu C.N., Banerjee U.C. Scaffold-Hopping of Aurones: 2-Arylideneimidazo[1,2-a]pyridinones as Topoisomerase IIα-Inhibiting Anticancer Agents. ACS Med. Chem. Lett. 2016;7:1056–1061. doi: 10.1021/acsmedchemlett.6b00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.French K.J., Schrecengost R.S., Lee B.D., Zhuang Y., Smith S.N., Eberly J.L., Yun J., Smith C.D. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- 39.Lawrence N.J., Rennison D., McGown A.T., Hadfield J.A. The total synthesis of an aurone isolated from Uvaria hamiltonii: Aurones and flavones as anticancer agents. Bioorg. Med. Chem. Lett. 2003;13:3759–3763. doi: 10.1016/j.bmcl.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 40.Detsi A., Majdalani M., Kontogiorgis C.A., Hadjipavlou-Litina D., Kefalas P. Natural and synthetic 2′-hydroxy-chalcones and aurones: Synthesis, characterization and evaluation of the antioxidant and soybean lipoxygenase inhibitory activity. Bioorg. Med. Chem. 2009;17:8073–8085. doi: 10.1016/j.bmc.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 41.Hadjeri M., Barbier M., Ronot X., Mariotte A.M., Boumendjel A., Boutonnat J. Modulation of P-glycoprotein-mediated multidrug resistance by flavonoid derivatives and analogues. J. Med. Chem. 2003;46:2125–2131. doi: 10.1021/jm021099i. [DOI] [PubMed] [Google Scholar]
- 42.Sim H.M., Wu C.P., Ambudkar S.V., Go M.L. In vitro and in vivo modulation of ABCG2 by functionalized aurones and structurally related analogs. Biochem. Pharmacol. 2011;82:1562–1571. doi: 10.1016/j.bcp.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coman F.M., Mbaveng A.T., Marc G., Leonte D., Brém B., Vlase L., Imre S., Kuete V., Zaharia V. Heterocycles 47. Synthesis, Characterization and Biological Evaluation of some New Thiazole Aurones as Antiproliferative Agents. Farmacia. 2020;68:492–506. doi: 10.31925/farmacia.2020.3.15. [DOI] [Google Scholar]
- 44.Semenov I., Akyuz C., Roginskaya V., Chauhan D., Corey S.J. Growth inhibition and apoptosis of myeloma cells by the CDK inhibitor flavopiridol. Leuk. Res. 2002;26:271–280. doi: 10.1016/S0145-2126(01)00103-5. [DOI] [PubMed] [Google Scholar]
- 45.Jeon K.-H., Park S., Shin J.-H., Jung A.-R., Hwang S.-Y., Seo S.H., Jo H., Na Y., Kwon Y. Synthesis and evaluation of 7-(3-aminopropyloxy)-substituted flavone analogue as a topoisomerase IIα catalytic inhibitor and its sensitizing effect to enzalutamide in castration-resistant prostate cancer cells. Eur. J. Med. Chem. 2023;246:114999. doi: 10.1016/j.ejmech.2022.114999. [DOI] [PubMed] [Google Scholar]
- 46.Su L., Li W., Liu K., Wang Q. Synthesis and anti-proliferative activities of 5,6,7-trimethoxyflavones and their derivatives. Nat. Prod. Res. 2022;36:4070–4075. doi: 10.1080/14786419.2021.1961136. [DOI] [PubMed] [Google Scholar]
- 47.Zhang N., Yang J., Li K., Luo J., Yang S., Song J.-R., Chen C., Pan W.-D. Synthesis of flavone derivatives via N-amination and evaluation of their anticancer activities. Molecules. 2019;24:2723. doi: 10.3390/molecules24152723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elhadi A.A., Osman H., Iqbal M.A., Rajeswari S.K., Ahamed M.B.K., Majid A.M.A., Rosli M.M., Razak I.A., Majid A.S.A. Synthesis and structural elucidation of two new series of aurone derivatives as potent inhibitors against the proliferation of human cancer cells. Med. Chem. Res. 2015;24:3504–3515. doi: 10.1007/s00044-015-1400-2. [DOI] [Google Scholar]
- 49.Cheng H., Zhang L., Liu Y., Chen S., Cheng H., Lu X., Zheng Z., Zhou G.-C. Design, synthesis and discovery of 5-hydroxyaurone derivatives as growth inhibitors against HUVEC and some cancer cell lines. Eur. J. Med. Chem. 2010;45:5950–5957. doi: 10.1016/j.ejmech.2010.09.061. [DOI] [PubMed] [Google Scholar]
- 50.Zheng X., Wang H., Liu Y.-M., Yao X., Tong M., Wang Y.-H., Liao D.-F. Synthesis, Characterization, and Anticancer Effect of Trifluoromethylated Aurone Derivatives. J. Heterocycl. Chem. 2014;6:1098–1107. doi: 10.1002/jhet.1969. [DOI] [Google Scholar]
- 51.Uesawa Y., Sakagami H., Ikezoe N., Takao K., Kagaya H., Sugita Y. Quantitative structure-cytotoxicity relationship of aurones. Anticancer Res. 2017;37:6169–6176. doi: 10.21873/anticanres.12066. [DOI] [PubMed] [Google Scholar]
- 52.Demirayak S., Yurttas L., Gundogdu-Karaburun N., Karaburun A.C., Kayagil I. Synthesis and anticancer activity evaluation of new aurone derivatives. J. Enzym. Inhib. Med. Chem. 2015;30:816–825. doi: 10.3109/14756366.2014.976568. [DOI] [PubMed] [Google Scholar]
- 53.Lathwal E., Kumar S., Sahoo P.K., Ghosh S., Mahata S., Nasare V.D., Kumar S. Synthesis, cytotoxic evaluation and structure activity relationship of pyrazole hybrid aurones on gastric cancer (AGS) cell lines. Results Chem. 2022;4:100590. doi: 10.1016/j.rechem.2022.100590. [DOI] [Google Scholar]
- 54.Gao L., Tang Z., Li T., Wang J. Myricetin exerts anti-biofilm activity and attenuates osteomyelitis by inhibiting the TLR2/MAPK pathway in experimental mice. Microb. Pathog. 2023;182:106165. doi: 10.1016/j.micpath.2023.106165. [DOI] [PubMed] [Google Scholar]
- 55.Ashok D., Kifah M.A., Lakshmi B.V., Sarasija M., Adam S. Microwave-assisted one-pot synthesis of some new flavonols by modified Algar–Flynn–Oyamada reaction and their antimicrobial activity. Chem. Heterocycl. Compd. 2016;52:172–176. doi: 10.1007/s10593-016-1852-4. [DOI] [Google Scholar]
- 56.Lv X.H., Liu H., Ren Z.L., Wang W., Tang F., Cao H.Q. Design, synthesis and biological evaluation of novel flavone Mannich base derivatives as potential antibacterial agents. Mol. Divers. 2019;23:299–306. doi: 10.1007/s11030-018-9873-9. [DOI] [PubMed] [Google Scholar]
- 57.Khdera H.A., Saad S.Y., Moustapha A., Kandil F. Synthesis of new flavonoid derivatives based on 3-hydroxy-4′-dimethylamino flavone and study the activity of some of them as antifungal. Heliyon. 2022;8:e12062. doi: 10.1016/j.heliyon.2022.e12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar G., Lathwal E., Saroha B., Kumar S., Kumar S., Chauhan N.S., Kumar T. Synthesis and Biological Evaluation of Quinoline-Based Novel Aurones. ChemistrySelect. 2020;5:3539–3543. doi: 10.1002/slct.201904912. [DOI] [Google Scholar]
- 59.Pan H., He J., Yang Z., Yao X., Zhang H., Li R., Xiao Y., Zhao C., Jiang H., Liu Y., et al. Myricetin possesses the potency against SARS-CoV-2 infection through blocking viral-entry facilitators and suppressing inflammation in rats and mice. Phytomedicine. 2023;116:154858. doi: 10.1016/j.phymed.2023.154858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fujimoto K.J., Nema D., Ninomiya M., Koketsu M., Sadanari H., Takemoto M., Daikoku T., Murayama T. An in silico-designed flavone derivative, 6-fluoro-4′-hydroxy-3′,5′-dimetoxyflavone, has a greater anti-human cytomegalovirus effect than ganciclovir in infected cells. Antivir. Res. 2018;154:10–16. doi: 10.1016/j.antiviral.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 61.Badavath V.N., Jadav S., Pastorino B., de Lamballerie X., Sinha N.B., Jayaprakash V. Synthesis and Antiviral Activity of 2-aryl-4H-chromen-4-one Derivatives Against Chikungunya Virus. Lett. Drug Des. Discov. 2016;13:1019–1024. doi: 10.2174/1570180813666160711163349. [DOI] [Google Scholar]
- 62.Zemplén G., Bognár R. Umwandlung des Hesperetins in Diosmetin, des Hesperidins in Diosmin und des Isosakuranetins in Acacetin. Berichte Dtsch. Chem. Ges. 1943;76:452–457. doi: 10.1002/cber.19430760503. [DOI] [Google Scholar]
- 63.Kostanecki S.V., Levi R., Tambor J. Synthese des 2-Oxyflavons. Berichte Dtsch. Chem. Ges. 1899;32:326–332. doi: 10.1002/cber.18990320155. [DOI] [Google Scholar]
- 64.Donnelly J.A., Acton J.P., Donnelly D.J., Philbin E.M. Steric and Electronic Effects in the Emilewiczvon Kostanecki Cyclization of Chalcone Dihalides. Proc. R. Ir. Acad. B. 1983;83B:49–56. [Google Scholar]
- 65.Bate-Smith E.C., Geissman T.A. Benzalcoumaranones. Nature. 1951;167:688. doi: 10.1038/167688a0. [DOI] [Google Scholar]
- 66.Narasimhachari N., Seshadri T.R. A new synthesis of flavones. Proc. Indian Acad. Sci. Sect. A. 1949;30:151–162. doi: 10.1007/BF03049180. [DOI] [Google Scholar]
- 67.Donnelly D.J., Donnelly J.A., Murphy J.J., Philbin E.M., Wheeler T.S. Steric effects on the cyclisation of chalcone dibromides. Chem. Commun. 1966;12:351–352. doi: 10.1039/c19660000351. [DOI] [Google Scholar]
- 68.Hutchins W.A., Wheeler T.S. 17. Chalkones: A new synthesis of chrysin, apigenin, and luteolin. J. Chem. Soc. 1939;83:91. doi: 10.1039/jr9390000091. [DOI] [Google Scholar]
- 69.Fitzgerald D.M., O’Sullivan J.F., Philbin E.M., Wheeler T.S. Ring expansion of 2-benzylidenecoumaran-3-ones—A synthesis of flavones. J. Chem. Soc. 1955;860:860–862. doi: 10.1039/JR9550000860. [DOI] [Google Scholar]
- 70.Donnelly J.A., Doran H.J. Chalcone dihalides-VII. The course of the cyclization of 2′-hydroxy-6′-methoxyl derivatives. Tetrahedron. 1975;31:1565–1569. doi: 10.1016/0040-4020(75)87013-X. [DOI] [Google Scholar]
- 71.Donnelly J.A., Doran H.J. Chalcone dihalides-VI. Generalisation of their use in the synthesis of naturally occurring flavones. Tetrahedron Lett. 1974;15:4083–4084. doi: 10.1016/S0040-4039(01)92089-1. [DOI] [Google Scholar]
- 72.Mahal H.S., Rai H.S., Venkataraman K. Synthetical experiments in the chromone group. Part XVI. Chalkones and flavanones and their oxidation to flavones by means of selenium dioxide. J. Chem. Soc. 1935;866:866–868. doi: 10.1039/jr9350000866. [DOI] [Google Scholar]
- 73.Kostanecki V.S., Szabranski W. Synthese des Flavonols. Berichte Dtsch. Chem. Ges. 1904;37:2819–2820. doi: 10.1002/cber.19040370353. [DOI] [Google Scholar]
- 74.Auwers K., Müller K. Umwandlung von Benzal-cumaranonen in Flavonole. Berichte Dtsch. Chem. Ges. 1908;41:4233–4241. doi: 10.1002/cber.190804103137. [DOI] [Google Scholar]
- 75.Stoermer R., Bartsch F. Synthesen des Cumaranons (Ketocumarans) und seiner Homologen aus Phenoxyessigsäuren. Berichte Dtsch. Chem. Ges. 1900;33:3175–3181. doi: 10.1002/cber.19000330374. [DOI] [Google Scholar]
- 76.Auwers K.V., Pohl P. Über die Umwandlung von Benzalcumaranonen in Flavonole. Justus Liebig’s Ann. Chem. 1914;405:243–294. doi: 10.1002/jlac.19144050302. [DOI] [Google Scholar]
- 77.Auwers K.V. Zur Bildung von Flavonolen aus Benzal-cumaranonen. Berichte Dtsch. Chem. Ges. 1916;49:809–819. doi: 10.1002/cber.19160490188. [DOI] [Google Scholar]
- 78.Allan J., Robinson R. CCXC.—An accessible derivative of chromonol. J. Chem. Soc. 1924;125:2192–2195. doi: 10.1039/CT9242502192. [DOI] [Google Scholar]
- 79.Wheeler T.S. Flavone. Org. Synth. 1952;32:72. [Google Scholar]
- 80.Allan J., Robinson R. CCCIX.—A new synthesis of fisetin and of quercetin. J. Chem. Soc. 1926;129:2334–2336. doi: 10.1039/JR9262902334. [DOI] [Google Scholar]
- 81.Kalff J., Robinson R. CCLXIV.—A synthesis of datiscetin. J. Chem. Soc. Trans. 1925;127:1968–1973. doi: 10.1039/CT9252701968. [DOI] [Google Scholar]
- 82.Kalff J., Robinson R. XXVIII.—A synthesis of myricetin and of a galangin monomethyl ether occurring in galanga root. J. Chem. Soc. Trans. 1925;127:181–184. doi: 10.1039/CT9252700181. [DOI] [Google Scholar]
- 83.Dreyer D.L., Tabata S., Horowitz R.M. Flavonoids of citrus—VIII. Tetrahedron. 1964;20:2977–2983. doi: 10.1016/S0040-4020(01)98522-9. [DOI] [Google Scholar]
- 84.Robinson R., Shinoda J. CCLXV.—The synthesis of certain 2-styrylchromonol derivatives. J. Chem. Soc. Trans. 1925;127:1973–1980. doi: 10.1039/CT9252701973. [DOI] [Google Scholar]
- 85.Fukui K., Nakayama M., Horie T. Synthetic Studies of the Flavone Derivatives. IX. The Syntheses of Axillarin and Its Related Compounds. Bull. Chem. Soc. Jpn. 1969;42:1649–1652. doi: 10.1246/bcsj.42.1649. [DOI] [Google Scholar]
- 86.Fukui K., Matsumoto T., Nakamura S., Nakayama M., Horie T. Synthetic Studies of the Flavone Derivatives. VII. The Synthesis of Jaceidin. Bull. Chem. Soc. Jpn. 1968;41:1413–1417. doi: 10.1246/bcsj.41.1413. [DOI] [Google Scholar]
- 87.Baker W. Molecular Rearrangement of Some o-Acyloxyacetophenones. J. Chem. Soc. 1933;1933:1381–1389. doi: 10.1039/jr9330001381. [DOI] [Google Scholar]
- 88.Mahal H.S., Venkataraman K. A Synthesis of Flavones at Room Temperature. Curr. Sci. 1933;2:214–215. [Google Scholar]
- 89.Chadha T.C., Venkataraman K. 256. Synthetical experiments in the chromone group. Part VIII. Derivatives of o-hydroxy-,2:5-dihydroxy-, and 2:4:5-trihydroxy-acetophenone. J. Chem. Soc. 1933;1933:1073. doi: 10.1039/jr9330001073. [DOI] [Google Scholar]
- 90.Mahal H.S., Venkataraman K. 387. Synthetical experiments in the chromone group. Part XIV. The action of sodamide on 1-acyloxy-2-acetonaphthones. J. Chem. Soc. 1934;1934:1767. doi: 10.1039/jr9340001767. [DOI] [Google Scholar]
- 91.Ameen D., Snape T.J. Mechanism and application of Baker-Venkataraman O→C acyl migration reactions. Synthesis. 2015;47:141–158. doi: 10.1055/s-0034-1379498. [DOI] [Google Scholar]
- 92.Cramer F., Elschnig G.H. Über Einschluß-verbindungen, IX. Mitteil.: Die blauen Jodverbindungen der Flavone. Chem. Ber. 1956;89:1–12. doi: 10.1002/cber.19560890102. [DOI] [Google Scholar]
- 93.Ares J.J., Outt P.E., Kakodkar S.V., Buss R.C., Geiger J.C. A Convenient Large-Scale Synthesis of 5-Methoxyflavone and Its Application to Analog Preparation. J. Org. Chem. 1993;58:7903–7905. doi: 10.1021/jo00079a041. [DOI] [Google Scholar]
- 94.Jain P.K., Makrandi J.K., Grover S.K. A Facile Baker-Venkataraman Synthesis of Flavones using Phase Transfer Catalysis. Synthesis. 1982;1982:221–222. doi: 10.1055/s-1982-29755. [DOI] [Google Scholar]
- 95.Saxena S., Makrandi J., Grover S. Synthesis of 5- and/or 7-Hydroxyflavones using A Modified Phase Transfer-Catalysed Baker-Venkataraman Transformation. Synthesis. 1985;1985:697. doi: 10.1055/s-1985-31319. [DOI] [Google Scholar]
- 96.Song G.-Y., Ahn B.-Z. Synthesis of dibenzoylmethanes as intermediates for flavone synthesis by a modified Baker-Venkataraman rearrangement. Arch. Pharm. Res. 1994;17:434–437. doi: 10.1007/BF02979121. [DOI] [PubMed] [Google Scholar]
- 97.Stanek F., Stodulski M. Mild and efficient organocatalytic method for the synthesis of flavones. Tetrahedron Lett. 2016;57:3841–3843. doi: 10.1016/j.tetlet.2016.07.042. [DOI] [Google Scholar]
- 98.Pinto D.C.G.A., Silva A.M.S., Cavaleiro J.A.S. Baker-Venkataraman rearrangement under microwave irradiation: A new strategy for the synthesis of 3-aroyl-5-hydroxyflavones. Synthesis. 2007;12:1897–1900. doi: 10.1055/s-2007-984525. [DOI] [Google Scholar]
- 99.Sarda S.R., Pathan M.Y., Paike V.V., Pachmase P.R., Jadhav W.N., Pawar R.P. A facile synthesis of flavones using recyclable ionic liquid under microwave irradiation. Arkivoc. 2006;2006:43–48. doi: 10.3998/ark.5550190.0007.g05. [DOI] [Google Scholar]
- 100.Miyake H., Nishino S., Nishimura A., Sasaki M. New synthesis of 3-bromoflavones via bromination of 1-(2-hydroxyphenyl)-3- arylpropane-1,3-dione by CuBr2, and conversion into 3-aminoflavones. Chem. Lett. 2007;36:522–523. doi: 10.1246/cl.2007.522. [DOI] [Google Scholar]
- 101.Zubaidha P.K., Hashmi A.M., Bhosale R.S. FeCl3 catalyzed dehydrative cyclisation of 1, 3-(diaryl diketones) to flavones. Heterocycl. Commun. 2005;11:97–100. doi: 10.1515/HC.2005.11.1.97. [DOI] [Google Scholar]
- 102.Kabalka G.W., Mereddy A.R. Microwave-assisted synthesis of functionalized flavones and chromones. Tetrahedron Lett. 2005;46:6315–6317. doi: 10.1016/j.tetlet.2005.07.038. [DOI] [Google Scholar]
- 103.Nishinaga A., Ando H., Maruyama K., Mashino T. A New Metal Complex Promoted System for Highly Selective Synthesis of 4 H -Chromen-4-ones (Chromones) Synthesis. 1992;1992:839–841. doi: 10.1055/s-1992-26241. [DOI] [Google Scholar]
- 104.Varma R.S., Saini R.K., Kumar D. An Expeditious Synthesis of Flavones on Montmorillonite K 10 Clay with Microwaves. J. Chem. Res. Synop. 1998;6:348–349. doi: 10.1039/a709146j. [DOI] [Google Scholar]
- 105.Hoshino Y., Takeno N. A Facile Preparation of Flavones Using Nonaqueous Cation-Exchange Resin. Bull. Chem. Soc. Jpn. 1987;60:1919–1920. doi: 10.1246/bcsj.60.1919. [DOI] [Google Scholar]
- 106.Pérez M.E., Ruiz D.M., Autino J.C., Blanco M.N., Pizzio L.R., Romanelli G.P. Mesoporous titania/tungstophosphoric acid composites: Suitable synthesis of flavones. J. Porous Mater. 2013;20:1433–1440. doi: 10.1007/s10934-013-9729-8. [DOI] [Google Scholar]
- 107.Bennardi D.O., Romanelli G.P., Autino J.C., Pizzio L.R. Supported trifluoromethanesulfonic acid as catalyst in the synthesis of flavone and chromone derivatives. Appl. Catal. A Gen. 2007;324:62–68. doi: 10.1016/j.apcata.2007.03.009. [DOI] [Google Scholar]
- 108.Jorge L., Autino C. Synthesis of substituted flavones and chromones using a Wells-Dawson heteropolyacid as catalyst. Arkivoc. 2008;11:123. [Google Scholar]
- 109.Bennardi D.O., Romanelli G.P., Jios J.L., Vázquez P.G., Cáceres C.V., Autino C. Synthesis of substituted flavones and arylchromones using ρ and si keggin heteropolyacids as catalysts. Heterocycl. Commun. 2007;13:77–82. doi: 10.1515/HC.2007.13.1.77. [DOI] [Google Scholar]
- 110.Kucukislamoglu M., Nebioglu M., Zengin M., Arslan M., Yayli N. An environmentally benign synthesis of flavones from 1,3-diketones using silica gel supported NaHSO4 catalyst. J. Chem. Res. 2005;9:556–557. doi: 10.3184/030823405774308790. [DOI] [Google Scholar]
- 111.Oyamada T. A new general method for the synthesis of the derivates of flavonol. Bull. Chem. Soc. Jpn. 1935;10:182–186. doi: 10.1246/bcsj.10.182. [DOI] [Google Scholar]
- 112.Algar J., Flynn J. A New Method for the Synthesis of Flavonols. Proc. R. Ir. Acad. B. 1934;42:1–8. [Google Scholar]
- 113.Serdiuk I.E., Roshal A.D., Błażejowski J. Quantum-chemical analysis of the Algar-Flynn-Oyamada reaction mechanism. Chem. Heterocycl. Compd. 2014;50:396–403. doi: 10.1007/s10593-014-1487-2. [DOI] [Google Scholar]
- 114.Dean F.M., Podimuang V. 737. The course of the Algar–Flynn–Oyamada (A.F.O.) reaction. J. Chem. Soc. 1965;3978:3978–3987. doi: 10.1039/JR9650003978. [DOI] [Google Scholar]
- 115.Adams C.J., Main L. Synthesis of 2′-hydroxychalcone epoxides. Tetrahedron. 1991;47:4959–4978. doi: 10.1016/S0040-4020(01)80960-1. [DOI] [Google Scholar]
- 116.Bennett M., Burke A.J., Ivo O’Sullivan W. Aspects of the Algar-Flynn-Oyamada (AFO) reaction. Tetrahedron. 1996;52:7163–7178. doi: 10.1016/0040-4020(96)00334-1. [DOI] [Google Scholar]
- 117.Bhattacharyya S., Hatua K. Computational insight of the mechanism of Algar-Flynn-Oyamada (AFO) reaction. RSC Adv. 2014;4:18702–18709. doi: 10.1039/c3ra46623j. [DOI] [Google Scholar]
- 118.Fougerousse A., Gonzalez E., Brouillard R. A convenient method for synthesizing 2-aryl-3-hydroxy-4-oxo-4H-1- benzopyrans or flavonols. J. Org. Chem. 2000;65:583–586. doi: 10.1021/jo990735q. [DOI] [PubMed] [Google Scholar]
- 119.Cummins B., Donnelly D., Eades J., Fletcher H., Cinnéide F., Philbin E., Swirski J., Wheeler T., Wilson R. Oxidation of chalcones (AFO reaction) Tetrahedron. 1963;19:499–512. doi: 10.1016/S0040-4020(01)98539-4. [DOI] [Google Scholar]
- 120.Shen X., Zhou Q., Xiong W., Pu W., Zhang W., Zhang G., Wang C. Synthesis of 5-subsituted flavonols via the Algar-Flynn-Oyamada (AFO) reaction: The mechanistic implication. Tetrahedron. 2017;73:4822–4829. doi: 10.1016/j.tet.2017.06.064. [DOI] [Google Scholar]
- 121.Nhu D., Hawkins B.C., Burns C.J. Phase transfer catalysis extends the scope of the Algar-Flynn-Oyamada synthesis of 3-Hydroxyflavones. Aust. J. Chem. 2015;68:1102–1107. doi: 10.1071/CH14620. [DOI] [Google Scholar]
- 122.Jain A.C., Gupta S.M., Sharma A. Synthesis of 2,2-Dimethyl-2 H-pyran-fused Flavonols Using the Modified Algar-Flynn-Oyamada Reaction. Bull. Chem. Soc. Jpn. 1983;56:1267–1268. doi: 10.1246/bcsj.56.1267. [DOI] [Google Scholar]
- 123.Dharia J.R., Johnson K.F., Schlenoff J.B. Synthesis and Characterization of Wavelength-Shifting Monomers and Polymers Based on 3-Hydroxyflavone. Macromolecules. 1994;27:5167–5172. doi: 10.1021/ma00096a046. [DOI] [Google Scholar]
- 124.Tong J., Liu C., Wang B. Improved Synthesis of Icaritin and Total Synthesis of β-Anhydroicaritin. Chem. Res. Chin. Univ. 2019;35:616–620. doi: 10.1007/s40242-019-9012-x. [DOI] [Google Scholar]
- 125.Ashraf J., Mughal E.U., Sadiq A., Bibi M., Naeem N., Ali A., Massadaq A., Fatima N., Javid A., Zafar M.N., et al. Exploring 3-hydroxyflavone scaffolds as mushroom tyrosinase inhibitors: Synthesis, X-ray crystallography, antimicrobial, fluorescence behaviour, structure-activity relationship and molecular modelling studies. J. Biomol. Struct. Dyn. 2020;39:7107–7122. doi: 10.1080/07391102.2020.1805364. [DOI] [PubMed] [Google Scholar]
- 126.Simiti I., Zaharia V., Mager S., Horn M., Koteles-Popa T. Heterocyclen 67. Mitt.: Darstellung und charakterisierung einiger 2-(2-aryl-thiazol-4-yl)-3-hydroxy-chromone. Arch. Pharm. 1991;324:913–915. doi: 10.1002/ardp.2503241117. [DOI] [Google Scholar]
- 127.Zaharia V., Imre S., Palibroda N. Heterocycles. Obtaining and physico-chemical characterization of some thiazolo and thiazolo [3,2-b][1,2,4] triazolic hydroxy-heterochalcones. Rev. Chim. 2009;4:391–397. [Google Scholar]
- 128.Enchev V., Mehandzhiyski A.Y. Computational insight on the chalcone formation mechanism by the Claisen–Schmidt reaction. Int. J. Quantum Chem. 2017;117:e25365. doi: 10.1002/qua.25365. [DOI] [Google Scholar]
- 129.Patonay T., Cavaleiro J.A.S., Lévai A., Silva A.M.S. Dehydrogenation by iodine-dimethylsulfoxide system: A general route to susbtituted chromones and thiochromones. Heterocycl. Commun. 1997;3:223–229. doi: 10.1515/HC.1997.3.3.223. [DOI] [Google Scholar]
- 130.Masesane I.B. A comprehensive review of the oxidative cyclisation of 2′-hydroxychalcones to aurones and flavones. Int. J. Chem. Stud. 2015;3:53–59. [Google Scholar]
- 131.Menezes M.J., Manjrekar S., Pai V., Patre R.E., Tilve S.G. A facile microwave assisted synthesis of flavones. Indian J. Chem Sect. B Org. Med. Chem. 2009;48:1311–1314. [Google Scholar]
- 132.Hans N., Grover S.K. An efficient conversion of 2′-hydroxychalcones to flavones. Synth. Commun. 1993;23:1021–1023. doi: 10.1080/00397919308013299. [DOI] [Google Scholar]
- 133.Constantinescu T., Leonte D., Bencze L.C., Vlase L., Imre S., Hanganu D., Zaharia V. Heterocycles 43. Synthesis, characterization and antioxidant activity of some thiazole hydroxychalcones and their flavonoidic derivatives. Farmacia. 2018;66:663–673. doi: 10.31925/farmacia.2018.4.16. [DOI] [Google Scholar]
- 134.Litkei G., Gulácsi K., Antus S., Blaskó G. Cyclodehydrogenation of 2′-hydroxychalcones with hypervalent iodine reagent: A new synthesis of flavones. Liebigs Ann. 1995;1995:1711–1715. doi: 10.1002/jlac.1995199509240. [DOI] [Google Scholar]
- 135.Gulácsi K., Litkei G., Antus S., Gunda T.E. A short and facile synthetic route to prenylated flavones. Cyclodehydrogenation of prenylated 2′-hydroxychalcones by a hypervalent iodine reagent. Tetrahedron. 1998;54:13867–13876. doi: 10.1016/S0040-4020(98)00853-9. [DOI] [Google Scholar]
- 136.Du Z., Ng H., Zhang K., Zeng H., Wang J. Ionic liquid mediated Cu-catalyzed cascade oxa-Michael-oxidation: Efficient synthesis of flavones under mild reaction conditions. Org. Biomol. Chem. 2011;9:6930–6933. doi: 10.1039/c1ob06209c. [DOI] [PubMed] [Google Scholar]
- 137.Lahyani A., Trabelsi M. Ultrasonic-assisted synthesis of flavones by oxidative cyclization of 2′-hydroxychalcones using iodine monochloride. Ultrason. Sonochem. 2016;31:626–630. doi: 10.1016/j.ultsonch.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 138.Miyake H., Takizawa E., Sasaki M. Syntheses of flavones via the iodine-mediated oxidative cyclization of 1,3-diphenylprop-2-en-1-ones. Bull. Chem. Soc. Jpn. 2003;76:835–836. doi: 10.1246/bcsj.76.835. [DOI] [Google Scholar]
- 139.Rajesh Babu K., Vijaya Kumar K., Vijaya M., Madhavarao V. A novel solid supported synthesis of flavones. Int. J. Pharm. Technol. 2012;4:3943–3950. [Google Scholar]
- 140.Sarda S.R., Jadhav W.N., Pawar R.P. I2-Al2O3: A suitable heterogeneous catalyst for the synthesis of flavones under microwave irradiation. Int. J. ChemTech Res. 2009;1:539–543. [Google Scholar]
- 141.Kulkarni P.S., Kondhare D.D., Varala R., Zubaidha P.K. Cyclization of 2′-hydroxychalcones to flavones using ammonium iodide as an iodine source—An eco-friendly approach. J. Serbian Chem. Soc. 2013;78:909–916. doi: 10.2298/JSC120901119K. [DOI] [Google Scholar]
- 142.Lee H.H., Tan C.H. 496. Syntheses of flavones from Lindera lucida. J. Chem. Soc. 1965;1965:2743–2749. doi: 10.1039/jr9650002743. [DOI] [Google Scholar]
- 143.Zwaagstra M.E., Timmerman H., van de Stolpe A.C., de Kanter F.J., Tamura M., Wada Y., Zhang M.Q. Synthesis and structure—Activity relationships of carboxyflavones as structurally rigid CysLT1 (LTD4) receptor antagonists. J. Med. Chem. 1998;41:1428–1438. doi: 10.1021/jm970179x. [DOI] [PubMed] [Google Scholar]
- 144.Akama T., Shida Y., Sugaya T., Ishida H., Gomi K., Kasai M. Novel 5-aminoflavone derivatives as specific antitumor agents in breast cancer. J. Med. Chem. 1996;39:3461–3469. doi: 10.1021/jm950938g. [DOI] [PubMed] [Google Scholar]
- 145.Makrandi J.K., Seema S. ChemInform Abstract: An Efficient Procedure for Cyclization of 2′-Hydroxychalcones into Flavones. ChemInform. 1990;21:181. doi: 10.1002/chin.199010181. [DOI] [Google Scholar]
- 146.Gupta M., Paul S., Gupta R., Loupy A. A rapid method for the cyclization of 2′-hydroxychalcones into flavones. Org. Prep. Proced. Int. 2000;32:280–283. doi: 10.1080/00304940009355926. [DOI] [Google Scholar]
- 147.Lamba M., Makrandi J.K. Sodium selenite-dimethylsulfoxide: A highly efficient reagent for dehydrogenation. J. Chem. Res. 2008;4:225–226. doi: 10.3184/030823408X313591. [DOI] [Google Scholar]
- 148.Kasahara A., Izumi T., Ooshima M. A New Method of Preparing Flavones. Bull. Chem. Soc. Jpn. 1974;47:2526–2528. doi: 10.1246/bcsj.47.2526. [DOI] [Google Scholar]
- 149.Imafuku K., Honda M., Mcomie J.F. Cyclodehydrogenation of 2′-Hydroxychalcones with DDQ: A Simple Route for Flavones and Aurones. Synthesis. 1987;1987:199–201. doi: 10.1055/s-1987-27891. [DOI] [Google Scholar]
- 150.Mallik U.K., Saha M.M., Mallik A.K. ChemInform Abstract: Cyclodehydrogenation of 2′-Hydroxychalcones and Dehydrogenation of Flavanones Using Nickel Peroxide. Abstract 182. ChemInform. 1990;21:116. doi: 10.1002/chin.199009182. [DOI] [Google Scholar]
- 151.Hoshino Y., Oohinata T., Takeno N. The Direct Preparation of Flavones from 2′-Hydroxychalcones Using Disulfides. Bull. Chem. Soc. Jpn. 1986;59:2351–2352. doi: 10.1246/bcsj.59.2351. [DOI] [Google Scholar]
- 152.Hemanth Kumar K., Perumal P.T. A novel one-pot oxidative cyclization of 2′-amino and 2′-hydroxychalcones employing FeCl3•6H2O-methanol. Synthesis of 4-alkoxy-2-aryl-quinolines and flavones. Tetrahedron. 2007;63:9531–9535. doi: 10.1016/j.tet.2007.06.051. [DOI] [Google Scholar]
- 153.Liu R., Zhang Y., Xu K., Tan G. Silica-gel-supported Ce(SO4)2•4H2O-mediated cyclization of 2′-amino and 2′-hydroxychalcones under solvent-free conditions. Synth. Commun. 2017;47:1–9. doi: 10.1080/00397911.2016.1230217. [DOI] [Google Scholar]
- 154.Ganguly N.C., Chandra S., Barik S.K. Sodium perborate tetrahydrate-mediated transformations of 2′-hydroxychalcones to flavanones, flavones, and 3′,5′-diiodoflavone under mild, environmentally friendly conditions. Synth. Commun. 2013;43:1351–1361. doi: 10.1080/00397911.2011.633734. [DOI] [Google Scholar]
- 155.McKillop A., Kemp D. Further functional group oxidations using sodium perborate. Tetrahedron. 1989;45:3299–3306. doi: 10.1016/S0040-4020(01)81008-5. [DOI] [Google Scholar]
- 156.Ahmed N., Ali H., Van Lier J.E. Silica gel supported InBr3 and InCl3: New catalysts for the facile and rapid oxidation of 2′-hydroxychalcones and flavanones to their corresponding flavones under solvent free conditions. Tetrahedron Lett. 2005;46:253–256. doi: 10.1016/j.tetlet.2004.11.062. [DOI] [Google Scholar]
- 157.Kumar S., Sharma D. Oxidative cyclisation of 2′-hydroxychalcones using sodium tellurite: Synthesis of flavones. Orient. J. Chem. 2011;27:761–763. [Google Scholar]
- 158.Zambare A.S., Sangshetti J.N., Kokare N.D., Shinde D.B. Development of mild and efficient method for synthesis of substituted flavones using oxalic acid catalyst. Chin. Chem. Lett. 2009;20:171–174. doi: 10.1016/j.cclet.2008.10.042. [DOI] [Google Scholar]
- 159.Matsushima R., Kageyama H. Photochemical cyclization of 2′-hydroxychalcones. J. Chem. Soc. Perkin Trans. II. 1985;53:743–748. doi: 10.1039/P29850000743. [DOI] [Google Scholar]
- 160.Maki Y., Shimada K., Sako M., Hirota K. Photo-oxidative cyclisation of 2′-hydroxychalcones leading to flavones induced by heterocycle-oxides: High efficiency of pybimido[54-]pteridine -oxide for the photochemical dehydrogenation. Tetrahedron. 1988;44:3187–3194. doi: 10.1016/S0040-4020(01)85950-0. [DOI] [Google Scholar]
- 161.Saničanin Z., Tabaković I. Electrochemical transformations of 2-hydroxychalcones into flavanoids. Tetrahedron Lett. 1986;27:407–408. doi: 10.1016/S0040-4039(00)84031-9. [DOI] [Google Scholar]
- 162.Tamuli K.J., Sahoo R.K., Bordoloi M. A Biocatalytic Green Alternative to Existing Hazardous Reaction Medium: Synthesis of Chalcone and Flavone Derivatives via Claisen-Schmidt Reaction at Room Temperature. New J. Chem. 2020;44:20956–20965. doi: 10.1039/D0NJ03839C. [DOI] [Google Scholar]
- 163.Nana F., Kuete V., Zaharia V., Ngameni B., Sandjo L.P. Synthesis of Functionalized 1-Aryl-3-phenylthiazolylpropanoids and Their Potential as Anticancer Agents. ChemistrySelect. 2020;5:7675–7678. doi: 10.1002/slct.202002060. [DOI] [Google Scholar]
- 164.Wang Z. Mentzer Pyrone Synthesis. In: Wang Z., editor. Compr Org Name React Reagents. John Wiley & Sons, Inc.; New York, NY, USA: 2010. pp. 1901–1904. [Google Scholar]
- 165.Von Pechmann H., Duisberg C. Ueber die Verbindungen der Phenole mit Acetessigäther. Berichte Dtsch. Chem. Ges. 1883;16:2119–2128. doi: 10.1002/cber.188301602117. [DOI] [Google Scholar]
- 166.Mentzer C., Molho D., Vercier P. Sur un nouveau mode de condensation desters beta-cetoniques et de phenols en chromones. Comptes Rendus Hebd. Seances L Acad. Sci. 1951;232:1488–1490. [Google Scholar]
- 167.Seijas J.A., Vázquez-Tato M.P., Carballido-Reboredo R. Solvent-Free Synthesis of Functionalized Flavones under Microwave Irradiation. J. Org. Chem. 2005;70:2855–2858. doi: 10.1021/jo048685z. [DOI] [PubMed] [Google Scholar]
- 168.Selepe M.A., Van Heerden F.R. Application of the Suzuki-Miyaura reaction in the synthesis of flavonoids. Molecules. 2013;18:4739–4765. doi: 10.3390/molecules18044739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kraus G.A., Gupta V. Divergent approach to flavones and aurones via dihaloacrylic acids. Unexpected dependence on the halogen atom. Org. Lett. 2010;12:5278–5280. doi: 10.1021/ol1023294. [DOI] [PubMed] [Google Scholar]
- 170.Corbet J.P., Mignani G. Selected patented cross-coupling reaction technologies. Chem. Rev. 2006;106:2651–2710. doi: 10.1021/cr0505268. [DOI] [PubMed] [Google Scholar]
- 171.Agrawal N.N., Soni P.A. A new process for the synthesis of aurones by using mercury (II) acetate in pyridine and cupric bromide in dimethyl sulfoxide. Indian J. Chem Sect. B Org. Med. Chem. 2006;45:1301–1303. doi: 10.1002/chin.200639120. [DOI] [Google Scholar]
- 172.Ameta K.L., Rathore N.S., Kumar B., Malaga M.E.S., Manuela Verastegui P., Gilman R.H., Verma B.L. Synthesis and Trypanocidal Evaluation of Some Novel 2-(Substituted Benzylidene)-5,7-Dibromo-6-Hydroxy-1-Benzofuran-3(2H)-Ones. Int. J. Org. Chem. 2012;2:295–301. doi: 10.4236/ijoc.2012.223040. [DOI] [Google Scholar]
- 173.Thakkar K., Cushman M. A Novel Oxidative Cyclization of 2′-Hydroxychalcones to 4,5-Dialkoxyaurones by Thallium(III) Nitrate. J. Org. Chem. 1995;60:6499–6510. doi: 10.1021/jo00125a041. [DOI] [Google Scholar]
- 174.Khan M.S.Y., Mueed M.A. Scope of mercuric acetate oxidation of chalcones and the antibacterial activity of resulting aurones. Ind. J. Chem. 2004;43:1794–1797. doi: 10.1002/chin.200449099. [DOI] [Google Scholar]
- 175.Thanigaimalai P., Yang H.M., Sharma V.K., Kim Y., Jung S.H. The scope of thallium nitrate oxidative cyclization of chalcones; synthesis and evaluation of isoflavone and aurone analogs for their inhibitory activity against interleukin-5. Bioorg. Med. Chem. 2010;18:4441–4445. doi: 10.1016/j.bmc.2010.04.075. [DOI] [PubMed] [Google Scholar]
- 176.Meguellati A., Ahmed-Belkacem A., Yi W., Haudecoeur R., Crouillère M., Brillet R., Pawlotsky J.-M., Boumendjel A., Peuchmaur M. B-ring modified aurones as promising allosteric inhibitors of hepatitis C virus RNA-dependent RNA polymerase. Eur. J. Med. Chem. 2014;80:579–592. doi: 10.1016/j.ejmech.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 177.Li Y., Qiang X., Luo L., Yang X., Xiao G., Liu Q., Ai J., Tan Z., Deng Y. Aurone Mannich base derivatives as promising multifunctional agents with acetylcholinesterase inhibition, anti-β-amyloid aggragation and neuroprotective properties for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017;126:762–775. doi: 10.1016/j.ejmech.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 178.Sutton C.L., Taylor Z.E., Farone M.B., Handy S.T. Antifungal activity of substituted aurones. Bioorg. Med. Chem. Lett. 2017;27:901–903. doi: 10.1016/j.bmcl.2017.01.012. [DOI] [PubMed] [Google Scholar]
- 179.Harkat H., Blanc A., Weibel J.-M., Pale P. Versatile and Expeditious Synthesis of Aurones via Au I -Catalyzed Cyclization. J. Org. Chem. 2008;73:1620–1623. doi: 10.1021/jo702197b. [DOI] [PubMed] [Google Scholar]
- 180.Li S., Jin F., Viji M., Jo H., Sim J., Kim H.S., Lee H., Jung J.-K. A novel cyclization/oxidation strategy for a two-step synthesis of (Z)-aurone. Tetrahedron Lett. 2017;58:1417–1420. doi: 10.1016/j.tetlet.2017.02.074. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.