


Abstract
The transactivator Staf, which contains seven contiguous zinc fingers of the C2-H2 type, exerts its effects on gene expression by binding to specific targets in vertebrate small nuclear RNA (snRNA) and snRNA-type gene promoters. Here, we have investigated the interaction of the Staf zinc finger domain with the optimal Xenopus selenocysteine tRNA (xtRNASec) and human U6 snRNA (hU6) Staf motifs. Generation of a series of polypeptides containing increasing numbers of Staf zinc fingers tested in binding assays, by interference techniques and by binding site selection served to elucidate the mode of interaction between the zinc fingers and the Staf motifs. Our results provide strong evidence that zinc fingers 3–6 represent the minimal zinc finger region for high affinity binding to Staf motifs. Furthermore, we show that the binding of Staf is achieved through a broad spectrum of close contacts between zinc fingers 1–6 and xtRNASec or optimal sites or between zinc fingers 3–6 and the hU6 site. Extensive DNA major groove contacts contribute to the interaction with Staf that associates more closely with the non-template than with the template strand. Based on these findings and the structural information provided by the solved structures of other zinc finger–DNA complexes, we propose a model for the interaction between Staf zinc fingers and the xtRNASec, optimal and hU6 sites.
INTRODUCTION
The Cys2/His2 (C2-H2) zinc finger motif, first discovered in Xenopus laevis transcription factor IIIA (TFIIIA) (1), represents an important class of eukaryotic DNA-binding motifs. To date, more than 300 different cDNA sequences have been found to encode the classical zinc finger motif and many of their products have been shown to play central roles in developmental processes and general transcription (2–4). Typically, more than one zinc finger forms a cluster within the DNA-binding domain of transcription factors. The fingers are separated by H–C links that are short spacers of seven amino acids linking the last histidine of one zinc finger to the first cysteine of the next (5). The structure of individual zinc fingers of the C2-H2 type has been solved, showing that the zinc finger motif is an independent fold consisting of a small β-sheet and an α-helix stabilized in a compact structure by the bound zinc (6,7). The understanding as to how zinc fingers interact with the DNA has largely been gained from the crystal structures of DNA–protein complexes (8–16). These structures show that, in general, zinc fingers interact with the DNA in a very similar fashion. A run of fingers wraps around the DNA and the N-terminal portion of the α-helix from each finger projects into the major groove. Each zinc finger primarily makes contacts to a three base subsite on one DNA strand using residues –1, 2, 3 and 6 of the α-helix (numbering with respect to the start of the α-helix). Typically, residues –1, 3 and 6 of the α-helix interact with residues at the 3′, middle and 5′ positions of a 3 bp subsite, respectively. Residues at position 2 of the α-helix can contact bases on the opposite strand, 3′ to a 3 bp subsite.
The zinc finger protein Staf, originally identified in X.laevis as the transcriptional activator of the tRNASec gene (17,18), is also involved in transcriptional activation of snRNA and snRNA-type genes, some of which are transcribed by RNA polymerase II and others by RNA polymerase III (19). ZNF76 and ZNF143 are two human homologs of Staf, ZNF143 being the ortholog while ZNF76 is a DNA-binding protein related to Staf and ZNF143 (20). In the central part, Staf contains seven contiguous zinc fingers of the C2-H2 type (18). The first six are of the C-X4-C-X3-F/Y-X5-L-X2-H-X3-H type (X stands for any amino acid), except that the leucine residue (L) is not found in the fourth and fifth fingers, where it is replaced by arginine (R) and tyrosine (Y) residues, respectively (Fig. 1). The seventh, however, is of the C-X2-C-X3-Y-X5-L-X2-H-X4-H type. The H–C link is highly conserved, giving rise to the consensus TGE(K/R)PYX, but the GE sequence is not found between the sixth and seventh fingers. Comparative footprinting analyses, performed either with the entire protein or with the zinc finger domain only, established that the seven tandemly repeated zinc fingers contain the DNA-binding domain of Staf (18). In a recent study, binding site selection allowed us to identify the 21 bp ATTACCCATAATGCATYGCGG sequence as the high affinity Staf consensus sequence (21). Sequence comparisons of the known Staf binding sites with the consensus sequence derived from binding site selection revealed a high degree of sequence divergence. This is well illustrated by the Xenopus tRNASec site, which lacks the 5′-part of the consensus sequence, and by the absence of the 3′-part in the human U6 site (19). Very recently, we demonstrated that not all of the seven zinc fingers are required for binding to Staf motifs. Zinc finger (Zf) 1 exhibits a flexible requirement since it contacts the DNA of X.laevis tRNASec but not at the hU6 Staf motifs (21). This flexibility promotes maximization of transcriptional activation from xtRNASec and hU6 promoters (22). The non-utilization of Zf 1 at the hU6 promoter enables the simultaneous binding of Staf and Oct-1 to their cognate DNA motifs, Oct-1 being another factor involved in transcriptional activation of the hU6 gene (22).
Figure 1.


Binding of wild-type and truncated Staf zinc finger domains to the optimal Staf-binding site and to the Staf motifs in the human snRNA U6 (hU6 site) and in the X.laevis tRNASec (xtRNASec site) promoters. (A) Amino acid sequence of residues 255–476 displaying the sequence alignments of the seven zinc fingers (18). Gaps (<) have been introduced at two locations to maximize the match. Cysteines, histidines and invariant hydrophobic residues are depicted in bold. Open and solid triangles indicate the N- and the C-termini of the various Staf zinc finger domains expressed as GST fusion proteins in E.coli, respectively. (B) Relative binding efficiencies of wild-type and truncated Staf zinc finger domains for the optimal, hU6 and xtRNASec sites. The histogram plots the amount of probe bound to the truncated proteins, relative to that obtained with the wild-type zinc finger domain Zf 1–7. Binding reactions contained 2 nM DNA and 60 nM fusion protein. The results of one representative experiment for each protein and probe are shown. Two other independent determinations gave similar results. Sequence comparisons between the optimal, hU6 and xtRNASec Staf-binding sites used in this study are shown in the upper right pannel. Nucleotide identities between the different elements are boxed.
In the current study, the DNA-binding properties of portions of the Staf zinc finger domain were studied by various techniques, also allowing identification of binding sites for individual zinc fingers on the optimal, xtRNASec and hU6 sites. These results provide new insights into the interaction of Staf zinc fingers with Staf DNA motifs.
MATERIALS AND METHODS
Plasmid constructs
Plasmids pBS(+)-optimal Staf binding site, pGST-Zf 1–7, pGST-Zf 1–6, pGST-Zf 1–5 and pGST-Zf 2–7 were described by Schaub et al. (21). Plasmids pGST-Zf 1–3, pGST-Zf 2–4, pGST-Zf 3–5, pGST-Zf 4–6, pGST-Zf 5–7, pGST-Zf 1–4, pGST-Zf 2–5, pGST-Zf 3–6, pGST-Zf 4–7, pGST-Zf 2–6 and pGST-Zf 3–7 were obtained by insertion into BamHI/EcoRI-cleaved pGEX-3X of DNA fragments corresponding to Staf amino acids 257–351, 294–381, 324–411, 354–446, 384–475, 257–381, 294–411, 324–446, 354–475, 294–446 and 324–475, respectively (18).
Preparation of wild-type and truncated zinc finger domains
Wild-type and truncated zinc finger domains fused in-frame to the glutathione S-transferase (GST) gene were produced as described previously (19,21). Protein concentrations were determined using the Bradford microassay (Bio-Rad) with bovine serum albumin as the standard.
DNA binding assays
Probes were prepared as follows. The non-template and template strands of the human U6 (positions –357 to –171) and Xenopus tRNASec (positions –280 to –102) genes were 5′-end-labeled by PCR amplification using distal and proximal 32P-labeled primers. The non-template and template strands of the 94 bp optimal probe were 5′-end-labeled by PCR amplification of plasmid pBS(+)-optimal Staf binding site, using distal and proximal 32P-labeled primers. The distal and proximal primers were complementary to positions 911–931 and 880–900 of pBS(+), respectively. For the semi-quantitative binding analysis of wild-type and truncated Staf zinc finger domains to optimal, xtRNASec and hU6 sites, gel retardation assays were performed in a total volume of 10 µl, in the presence of 20 fmol (1.3 × 105 d.p.m.) of labeled Staf-binding sites and 0.3–0.6 pmol of wild-type or truncated zinc finger domains. Complexes were formed in 10 mM HEPES–NaOH pH 7.5, 1 mM dithiothreitol, 5 mM MgCl2, 50 mM KCl, 20 µM ZnCl2, 5% glycerol, 0.1% NP-40. Other conditions were as described in Schaub et al. (21). After gel electrophoresis, the intensities of free and bound DNA bands were quantitated with a Fuji Bioimage Analyzer BAS 2000. A binding value of 100 was arbitrarily assigned to the amount of complex formed between the wild-type Staf zinc finger domain and the Staf-binding sites. The binding efficiencies of the truncated zinc finger domains were expressed as a percentage of that obtained with the wild-type zinc finger domain.
Chemical modifications of DNA
The labeled probes were partially methylated with dimethylsulfate (DMS) as described previously (23,24). Thymine-specific modification with KMnO4 was performed according to Lee et al. (24) and Truss et al. (25). Depyrimidation of DNA with hydrazine was done as described (26). The probes were partially carbethoxylated by diethylpyrocarbonate (DEPC) at adenine residues according to Sturm et al. (27). Hydroxyl radical cleavage reactions were carried out as described previously (21,28).
Interference of binding of the Staf zinc finger domain
Binding reactions of the GST-fused wild-type Staf zinc fingers, in buffer conditions as described above, were done in the presence of premodified DNA fragments (4 × 105 d.p.m.) and the amount of protein required to retard ~50% of the probe. Bound and free probes were separated on non-denaturating gels. The DNA fragments were eluted and strand scission occurred at the positions modified by DMS, DEPC, KMnO4 or hydrazine by heating for 30 min at 90°C in 100 µl of 1 M piperidine. The cleaved fragments were separated on sequencing gels.
Binding site selection
Binding site selection was performed as described previously (19,21).
RESULTS
Mapping of Staf Zf 3–6 as the minimal region for high affinity binding to Staf-binding sites
To identify the minimal Staf DNA-binding domain, we generated three series of GST-fused polypeptides that contained precise deletions of zinc fingers, starting either from the C-terminus, the N-terminus or both termini of the zinc finger domain. The end points of the polypeptides are shown in Figure 1A. Each polypeptide was purified from Escherichia coli lysates by affinity chromatography and tested for binding activity to the optimal Staf-binding site and to the Staf-binding sites in the X.laevis tRNASec (xtRNASec site) and human U6 snRNA (hU6 site) promoters (19,21). The sequence of the optimal binding site identified by binding site selection and those of the xtRNASec and hU6 sites are shown in Figure 1B. Only 33% sequence identity was found between the three sites and 47% between the xtRNASec and hU6 sites. Binding activities were assayed by electrophoresis on non-denaturating gels. The relative efficiency with which wild-type and truncated Staf zinc finger domains bound to different Staf-binding sites was estimated by comparing the amounts of shifted complexes formed with the different labeled sites and three different protein concentrations. Figure 1B provides a graphical representation of the data for the complete set of zinc finger polypeptides, at one protein concentration and with the three Staf-binding sites. The complexes were all specific because they could be competed by an excess of the unlabeled optimal Staf-binding site, but not of the mutated xtRNASec Staf-binding site (data not shown). The effects of the C-terminal deletions will be described first. Zf 1–6 exhibited a binding efficiency to the optimal and xtRNASec probes similar to that of the entire Staf zinc finger domain (Zf 1–7), but only 61% of the wild-type value to the hU6 site. Zf 1–5 bound significantly to the optimal (28%) and xtRNASec (20%) sites, but not to the hU6 site. Zf 1–3 did not bind to any of the sites, nor did Zf 1–4, with the exception of a 5% residual value, to the optimal site. Next, the N-terminal deletions were evaluated. Zf 2–7 exhibited wild-type binding efficiencies for the optimal and hU6 sites, but only 48% for the xtRNASec site. With Zf 3–7, the binding efficiency decreased to ~70% for the optimal and hU6 sites and 28% for the xtRNASec site. A drop to 20% was observed for the optimal and hU6 sites with Zf 4–7, which did not bind to the xtRNASec site at all. Zf 5–7, containing only the last three C-terminal zinc fingers, did not bind to any of the sites. In a previous work (21), we showed that Zf 1 and Zf 7 were not required for high affinity binding. To determine the minimal set of zinc fingers that would retain high binding affinity, we generated a series of C- and N-terminal deletions in Zf 2–7 and Zf 1–6 (two-termini deletions in Fig. 1B). Only Zf 2–6 and Zf 3–6 exhibited significant binding efficiencies to the three sites (Fig. 1B). Zf 2–6 bound with values of 82, 68 and 41% to the optimal, hU6 and xtRNASec sites, respectively. For Zf 3–6 we observed a binding efficiency of ~55% to the optimal and hU6 sites and 22% to the xtRNASec site. Zf 2–4 and Zf 4–6 were unable to bind the Staf motifs and Zf 2–5 bound, with a very low efficiency (7%), only to the optimal binding site. Finally, a deletion of the C-terminal finger in Zf 3–6, to generate Zf 3–5, abolished binding to all Staf motifs. Altogether, our findings concluded that Zf 3–6 represents the minimal Staf zinc finger subdomain retaining specific and significant DNA binding affinity to the xtRNASec, hU6 and optimal sites.
Staf–purine contacts on the xtRNASec, hU6 and optimal Staf-binding sites
To determine the guanine and adenine residues important for binding of Staf to the DNA, methylation and carbethoxylation interference experiments were carried out with the entire GST-fused Staf zinc finger domain. DEPC reacts predominantly at the N7 position of adenines (29,30). DMS methylates N7-G residues in the major groove and, at a much slower rate, N3-A residues in the minor groove. Figure 2A and B shows the results of the methylation and carbethoxylation interference experiments, with compilation of the data in Figure 3C; the 21 bp optimal Staf-binding site (21) stands as the numbering reference. Figure 2A shows that methylation of G9 and G20 in the non-template (compare lanes 2 and 3) and G10 in the template strand (compare lanes 5 and 6) abolished binding of Staf to the xtRNASec site. The same effect was obtained in the hU6 site by methylation of G10 in the non-template strand and G5 and G7 in the template strand (Fig. 2A, compare lanes 8 and 9 and lanes 11 and 12). Carbethoxylation of A8 and A11 in the non-template strand of the three Staf-binding sites completely abolished the binding capacity for Staf (Fig. 2B, compare lanes 2 and 3, 8 and 9 and 14 and 15). Similarly, complete interference was induced by DEPC modification of A12 in the xtRNASec template strand, A4 in the hU6 template strand and A1 and A10 in the non-template strands of the hU6 and optimal sites, respectively (Fig. 2B, compare lanes 5 and 6, 11 and 12, 8 and 9, and 14 and 15). In addition to the contacts listed above, partial guanine methylation and adenine carbethoxylation interferences were observed at other residues in each strand of the xtRNASec and optimal sites and in the template strand of the hU6 site (Fig. 2). These results indicate that the interaction of Staf with the different binding sites is achieved through a broad spectrum of close contacts between the zinc finger domains and A/G residues. The purines are not confined to a core sequence but rather distributed along the entire Staf-binding sites. Since DMS modifies N7-G and DEPC N7-A, our data convincingly point to the involvement of the N7 positions of the bases cited above in specific contacts through the major groove.
Figure 2.
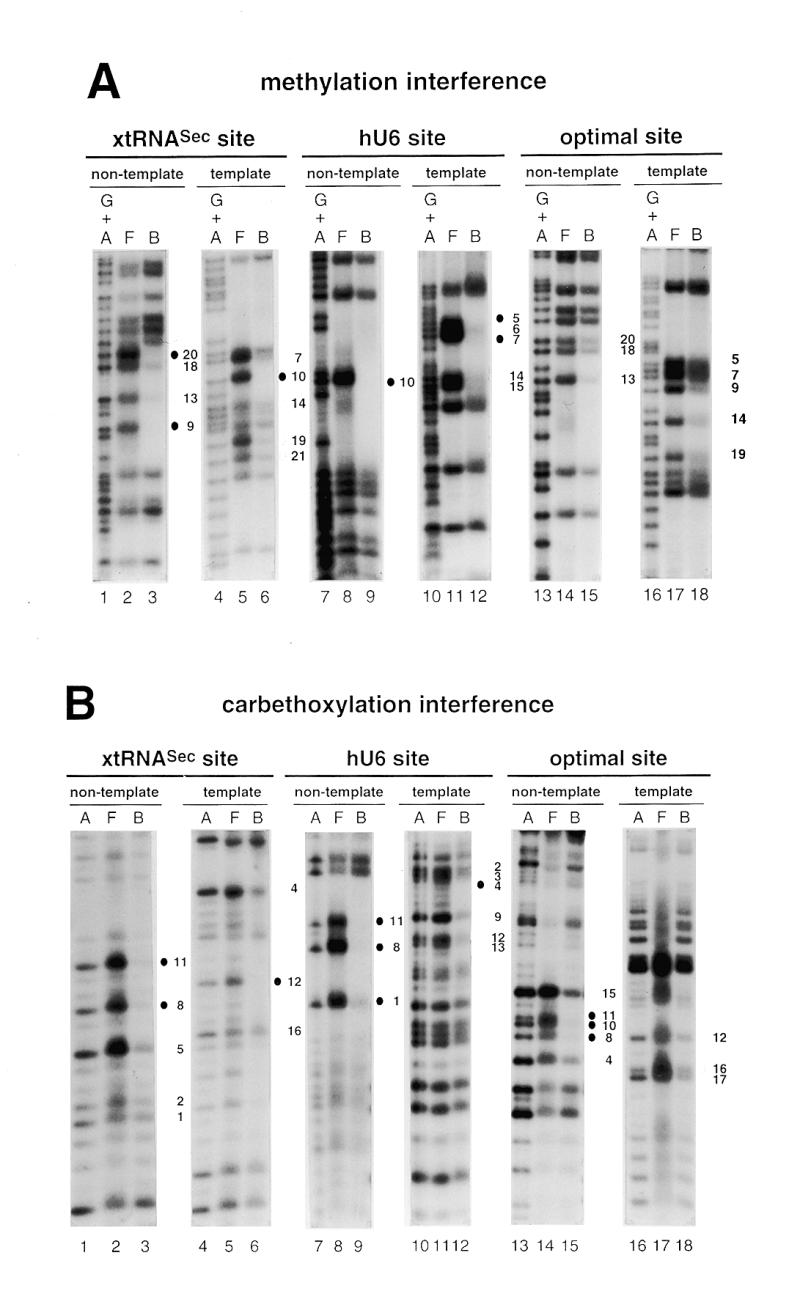

Interference with Staf binding by methylation, carbethoxylation, KMnO4 modification and depyrimidation of DNA fragments containing the xtRNASec, hU6 or optimal Staf-binding sites. The DNA fragments were 32P-labeled at their 5′-ends on the template or non-template strands, partially methylated (A) or carbethoxylated (B) and subjected to interference analysis. DMS modification and treatment of the modified DNA were performed under conditions allowing cleavage at only the modified guanine bases. Lanes A and G + A, sequencing reactions; lanes F and B, free and bound DNAs. Bases whose modifications interfered with Staf binding are shown by dark (total interference) or gray (partial interference) circles. Bases are numbered according to the Staf consensus binding site (21). (C) Summary of the interference experiments. The full and partial interference effects of guanine methylation, adenine carbethoxylation and pyrimidine removal on Staf binding are represented by dark and gray squares, respectively. The full and partial interference effects on the binding of Staf by KMnO4 modification of thymines are depicted by dark and gray triangles, respectively. Open squares and triangles show positions where base modifications or removal did not interfere with Staf binding; nd, not determined.
Figure 3.
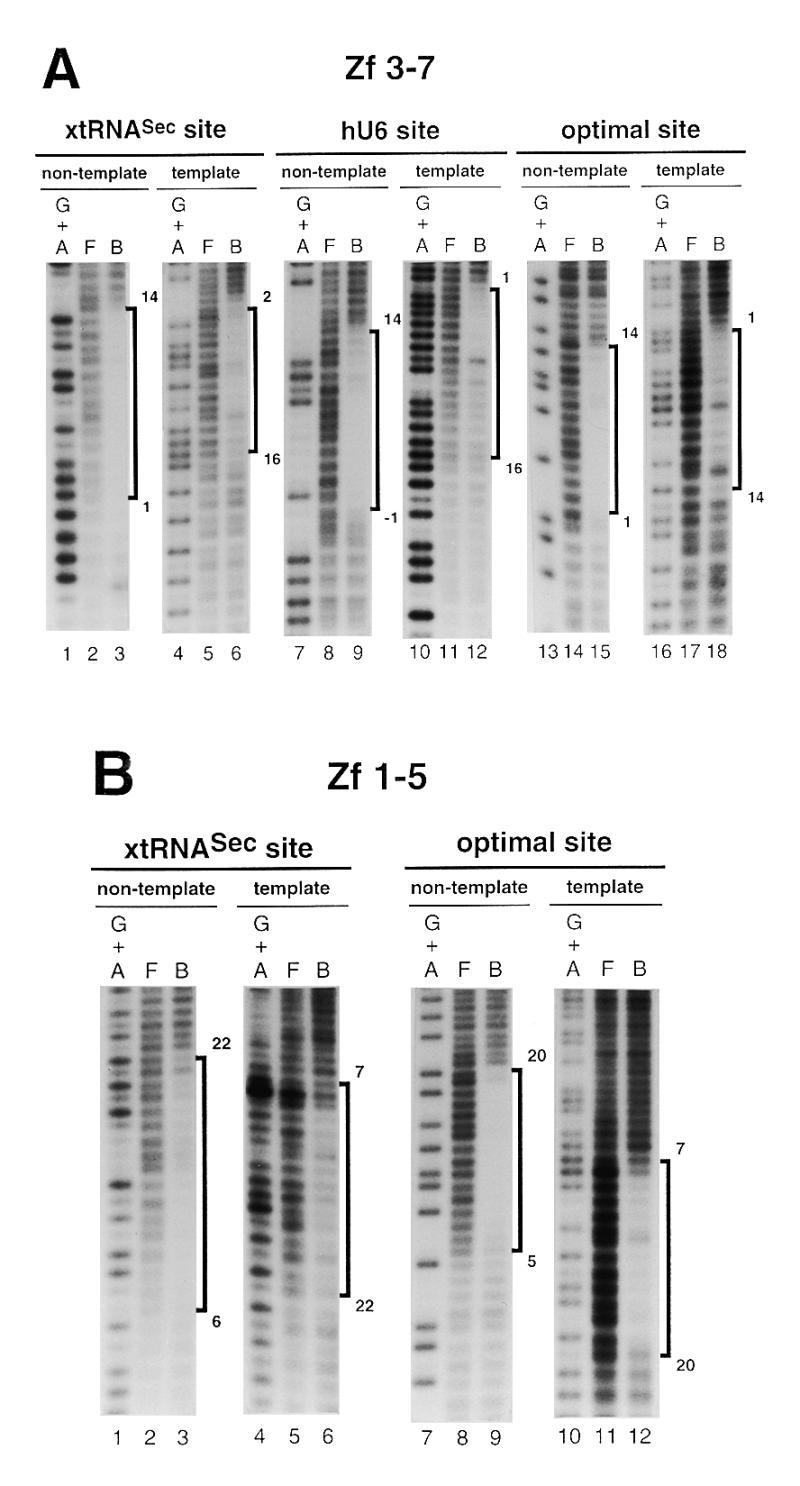

Hydroxyl radical interference patterns obtained with Staf Zf 1–5 and Zf 3–7 on the xtRNASec, hU6 and optimal sites. The 5′-end-labeled non-template or template strands containing the xtRNASec, hU6 and optimal sites were subjected to hydroxyl radical cleavage. Gapped DNAs were incubated separately with GST-fused Zf 1–5 and Zf 3–7. (A) Missing nucleoside interference patterns obtained on the xtRNASec, hU6 and optimal sites with Zf 3–7 and (B) with Zf 1–5 on the xtRNASec and optimal sites. Lanes G + A, F and B as in Figure 2. (C) Schematic representation of the results with comparison to previous results obtained with Zf 1–7, Zf 1–6 and Zf 2–7 (21,22). Regions of interference are boxed; dark boxes, strongest interference; hatched boxes, moderate interference; open boxes, weakest interference. The base pairs in the Staf-binding site are numbered –1 to 22, starting at the 5′-end of the non-template strand, with reference to the consensus binding site derived by in vitro selection (21).
Staf–pyrimidine contacts on the xtRNASec, hU6 and optimal Staf-binding sites
The interaction between Staf and thymine residues was analyzed using thymidine-specific modification with KMnO4 (25,31). A compilation of the data is shown in Figure 2C. Several positions interfered completely with the binding of Staf: T5 in the xtRNASec and T11 in the hU6 template sites; T4 and T16 in the xtRNASec, T2–T4 and T12 in the hU6 and T12 in the optimal non-template sites. In addition to these strong contacts, modification of T1 and T2 in the template strand of the xtRNASec site, T13 in the non-template strand of the hU6 site and T1–T3, T16 and T17 in the non-template strand and T11 and T15 in the template strand of the optimal site moderately interfered with binding of Staf. The results of the KMnO4 interference assay were compared with those obtained from the missing pyrimidine contact assay. In the latter procedure, pyrimidine residues in the DNA are removed by hydrazine treatment to determine their participation in protein binding (26). When applied to Staf, this method revealed contacts on the two strands essentially at those same thymine residues detected in the KMnO4 interference assay. Additional contacts at T9 in the non-template strand of the hU6 site and T12 in the non-template and T11 in the template strands of the xtRNASec site were also observed (Fig. 2C). In contrast to the KMnO4 modification, no interference was detected by removal of T1 and T2 in the template strand of the xtRNASec site and T1 and T17 in the non-template strand of the optimal site. Staf binding was not inhibited by modification or removal of the highly conserved T8 in the template strand of almost all the identified Staf-binding sites (19,21).
The missing pyrimidine contact assay could also detect interactions between Staf and cytosine residues. On the non-template strand, removal of C6 and C7 in the xtRNASec site and C5–C7 in the hU6 site totally interfered with binding of Staf (Fig. 2C). Partial interference was observed with the removal of C9, C10, C14, C18 and C19 in the xtRNASec site, C14 in the hU6 site and C5–C7, C14 and C18 in the optimal site (Fig. 2C). Thus, as observed for purines, contacts with thymines and cytosines were not restricted to a limited region of the Staf-binding sites. Rather, it appeared that a broad spectrum of base contacts are required for the binding of Staf. Given the more pronounced interference pattern on the non-template strand, it can be concluded that Staf associates more closely with the non-template than with the template strand. Comparison of the Staf–purine and Staf–pyrimidine contacts with the xtRNASec, optimal and hU6 sites revealed that the interference pattern is more limited in the hU6 site: chemical modification of residues 16–20 on both strands interfered with the binding of Staf to the xtRNASec and optimal sites but did not alter it to the hU6 site, suggesting a perhaps different set of zinc fingers binding to the hU6 site.
Mapping of the Staf zinc finger-binding sites
In an attempt to characterize the binding sites of the Staf zinc fingers, missing nucleoside interference experiments were carried out with hydroxyl radicals, using truncated Staf zinc finger domains. Zf 1–5 contained deletions of the last two C-terminal zinc fingers, while Zf 3–7 lacked the first two N-terminal zinc fingers. The DNA fragments containing the xtRNASec, hU6 and optimal sites were subjected to a mild treatment with hydroxyl radicals, incubated with the Zf 1–5 and Zf 3–7 proteins and analyzed as above. Figure 3A and B displays the pattern of the resulting DNA fragments. Figure 3C compiles the data in comparison with previous results obtained with the entire Staf zinc finger domain Zf 1–7 and the truncated zinc finger domains Zf 1–6 and Zf 2–7 (21). Removal of any nucleoside from positions 1 to 14 (xtRNASec site and optimal site) and –1 to 14 (hU6 site) in the non-template strand and from 2 to 16 (xtRNASec site), 1 to 16 (hU6 site) and 1 to 14 (optimal site) in the template strands interfered with binding of Zf 3–7. The interference patterns obtained for binding of Zf 3–7 to the xtRNASec and optimal sites were very different in their 3′-parts from those found for Zf 1–7 binding. Likewise, on the non-template strand a reduction in the interference pattern of seven (position 21 being excluded) and six nucleosides was observed for the xtRNASec and optimal sites, respectively. A surprising result was observed in the 3′-part of the non-template strand of the xtRNASec and optimal sites. Deletion of zinc fingers 1 and 2 in Zf 3–7 provoked a reduction in the interference pattern equivalent to that observed with the single deletion of zinc finger 1 in Zf 2–7. This was quite different from what we obtained with the hU6 site, where the interference pattern for the binding of Zf 3–7 was very similar to that found for Zf 1–7 and Zf 2–7. In the 3′-part of the hU6 site, the size of the interference pattern obtained with Zf 3–7 was reduced by one nucleoside only (position 15 in the non-template strand) with respect to that obtained with Zf 1–7 and Zf 2–7.
With Zf 1–5, missing nucleoside interference assays were performed only with the xtRNASec and optimal sites since Zf 1–5 failed to bind to the hU6 site (this work; 21). Removal of any nucleoside from positions 6 to 20 and 22 (xtRNASec site) and 5 to 20 (optimal site) in the non-template strand and from 7 to 22 (xtRNASec site) and 7 to 20 (optimal site) in the template strand interfered with binding of Zf 1–5 (Fig. 3B and C). Comparison of the Zf 1–5 interference pattern with those obtained with Zf 1–6 and Zf 1–7 revealed that removal of Zf 6 and Zf 7 reduced the size of the interference pattern in the 5′-part of the Staf elements: the reduction was four and five nucleosides for the optimal and xtRNASec sites, respectively, whether on the non-template or template strands. This contrasts with the removal of Zf 7 in Zf 1–6. Comparison of the Zf 1–5 and Zf 3–7 interference patterns suggested that the binding of Zf 3–Zf 5 required residues from positions 6 to 14 (xtRNASec site) and 5 to 14 (optimal site) in the non-template strand and from 7 to 16 (xtRNASec site) and 7 to 14 (optimal site) in the template strand. Furthermore, comparison of the Zf 1–7, Zf 1–6 and Zf 1–5 interference patterns suggested that residues from positions 1 to 4 (optimal site) and 1 to 5 (xtRNASec site) in the non-template strand and from 1 to 6 (optimal site) and 2 to 6 (xtRNASec) in the template strand are required for the binding of Zf 6. Finally, inspection of the Zf 1–7 and Zf 3–7 interference patterns indicated that residues 15–20 and 22 (xtRNASec site) and 15–20 (optimal site) in the non-template strand and residues 17–22 (xtRNASec site) and 16–20 (optimal site) in the template strand are necessary for binding of Zf 1 and Zf 2.
We next analyzed the recognition properties of Zf 3–7 by using this polypeptide, under stringent conditions, to select binding sites from an oligonucleotide pool of random sequences. The PCR products from the sixth round of amplification were cloned and 57 representative DNAs were sequenced. A comparison of the frequencies of each nucleotide at the 20 tabulated positions shows that the fusion protein Zf 3–7 yielded the 14 bp consensus sequence ATTACCCATAATNC, which overlaps with positions 1–14 in the 21 and 15 bp consensus obtained with the entire zinc finger domain Zf 1–7 and the fusion protein Zf 2–7 (21). For residues 15–19 the frequencies of each nucleotide selected by Zf 3–7 were entirely different from those observed with Zf 1–7 and Zf 2–7 (21). Zf 3–7 led to a significant decrease in the frequencies of the bases selected at positions 12–14 compared to Zf 1–7 and Zf 2–7 (21).
DISCUSSION
In an earlier work, we showed that most of the binding energy of Staf to the DNA arises from the interaction of Zf 2–Zf 6 (21). In this report, we have confirmed the previous results and established that the contribution of the individual Zf 2–Zf 6 to the DNA binding affinity of Staf appears different. Mutant Zf 3–6 lacking Zf 1, Zf 2 and Zf 7, bound specifically and with high affinity to the optimal, xtRNASec and hU6 sites. Thus, Zf 3–6 represents the minimal Staf zinc finger subdomain that retained specific and high binding affinity to the three tested motifs. In addition, Zf 1–5 bound to the optimal and xtRNASec sites but not to the hU6 site; Zf 4–7 interacted at the hU6 and optimal sites but was unable to recognize the xtRNASec site. This indicates a greater contribution of Zf 3 to the binding affinity for the xtRNASec site and of Zf 6 to that for the hU6 site.
Staf–purine and Staf–pyrimidine contacts to the xtRNASec, hU6 and optimal sites were analyzed with the entire Staf zinc finger domain by various chemical interference techniques. Our results revealed that the binding of Staf is achieved through a broad spectrum of close contacts between the zinc finger domain and residues not confined to a core sequence but rather distributed along the entire Staf-binding sites. Also arising from this study, our data established that the Staf–DNA complexes are canonical zinc finger–DNA complexes with extensive DNA major groove contacts. Furthermore, given the more pronounced interference pattern on the non-template strand, Staf probably associates more closely with the non-template than with the template strand.
Sequence comparisons of the optimal, xtRNASec and hU6 sites with the natural and in vitro selected sites detected the presence on the non-template strand of invariant C6 and C7 and highly conserved C5, A8, A11, T12 and C14 residues (19,21). Chemical interference analyses indicated that the three residues A8, A11 and T12, out of the seven listed above, are crucial for complex formation with the xtRNASec, hU6 and optimal sites. C6 and C7 are strictly required for the binding of Staf to the xtRNASec and hU6 sites, but binding to the three sites was only moderately impaired by the removal of C14. On the template strand, the requirement for the invariant and highly conserved residues was more relaxed and variable between sites. Notwithstanding, conservation of these residues strongly suggests that the zinc finger–DNA contacts in which they participate contribute to the specificity and affinity of binding. Surprisingly, however, chemical and missing interference analyses disclosed a crucial role of non-conserved residues for binding of Staf to the three tested binding sites. This is well illustrated by the variable base pair at position 10 occurring as C-G in the xtRNASec, G-C in the hU6 and A-T in the optimal site (Fig. 2C). G10 methylation in the template strand of the xtRNASec site and in the non-template strand of the hU6 site as well as A10 carbethoxylation in that same strand of the optimal site totally interfered with binding of Staf. This very likely reflects flexible DNA–zinc finger interactions involved in the affinity of binding.
Comparison of our previous missing nucleoside interference assays with Zf 1–7, Zf 2–7 and Zf 1–6 (21) to those with Zf 1–5 and Zf 3–7 in this study led to the discovery of interesting features concerning the binding sites for the individual zinc fingers. For example, the interference pattern for the binding of Zf 3–7 to the hU6 site resembled that for Zf 1–7 and Zf 2–7, strongly suggesting that Zf 2 does not interact with the DNA at the hU6 site, in much the same way as for Zf 1 (21,22). On the xtRNASec and optimal sites, the Zf 3–7 and Zf 2–7 interference patterns derived from missing nucleoside assays were identical to each other but shorter than with Zf 1–7. We can thus propose that the identical Zf 2–7 and Zf 3–7 interference patterns on the xtRNASec and optimal sites can be accounted for by the lack of interaction of Zf 2 with the DNA, in the absence of Zf 1. We previously showed that the GCG sequence at positions 18–20 was critical to Zf 1 binding to the xtRNASec site (22). In this study, methylation interference experiments with the xtRNASec site revealed that G20 on the non-template strand is crucial for Zf 1–7 binding, strongly suggesting that Zf 1 contacts G20 in the non-template strand of the xtRNASec site. Comparison of the Zf 1–5 and Zf 3–7 missing nucleoside interference patterns established that in the non-template strand subsites CCAGCATGC at positions 6–14 in the xtRNASec site and CCCAGCATGC at positions 5–14 in the optimal site were required for binding of Staf. In these subsites, the CCA sequence (positions 6–8) is invariant and C14 is highly conserved. We propose that C14 establishes base-specific contacts with Zf 3 and that the CCA sequence contacts Zf 5. The possible involvement of C14 in the interaction with Zf 3 is in agreement with the binding site selection performed with Zf 3–7 where the 14 bp selected sequence contained predominantly a C residue at position 14.
Figure 4 hypothesizes possible contacts of Zf 1–Zf 6 to the xtRNASec, hU6 and optimal sites. The Staf–base contacts were predicted based on the solved structures of other zinc finger protein–DNA complexes (8–16,32), tentative recognition codes (33–36) and our chemical interference analysis. In a zinc finger, the histidine residue at position 3 in the α-helix shows a strong preference for binding to a guanine residue located in the middle position of a 3 bp subsite (8,12–15). Paralleling this established interaction, we proposed an interaction between His3 in the α-helix of Zf 1 and G20 as the starting point for building the model with the xtRNASec and optimal sites. Assuming that Zf 1–Zf 6 contact the DNA at 3 bp subsites, the interaction involving G20 defines the CGC sequence (positions 19–21) as the 3 bp subsite for binding of Zf 1 to the xtRNASec and optimal sites. The CGC triplet thus provided the register allowing identification of the remaining 3 bp subsites interacting with Zf 2–Zf 6. Five additional Staf–DNA interactions in the template strands of the three Staf-binding sites could also be predicted from the solved structures of zinc finger–DNA complexes: Asp3 in Zf 3 with C14 in the xtRNASec, optimal and hU6 sites; Lys6 in Zf 3 with G13 in the xtRNASec and optimal sites; Asn3 in Zf 5 with A8 in the xtRNASec, optimal and hU6 sites; Ser3 in Zf 6 with A5 in the xtRNASec site; Ser2 in Zf 6 with G7′ in the three Staf binding sites. From the tentative recognition codes (33–36), three other interactions could be predicted: Lys6 in the α-helix of Zf 3 with T13 in the hU6 site; Thr–1 of the α-helix with T12 (which anchors Zf 4 to the xtRNASec, optimal and hU6 sites); Lys6 in the α-helix of Zf 6 with T4 in the xtRNASec and hU6 sites. In the model, we assumed that all of the six zinc fingers interact with the DNA, each finger recognizing a 3 bp subsite. One would then expect Staf to interact with an 18 bp sequence and not the 21 bp sequence identified by binding site selection and interference experiments. A tentative explanation for this apparent paradox was found in the structures of the TFIIIA–DNA and GLI–DNA complexes. The C-terminal zinc finger involved in major groove interaction, Zf 3 in TFIIIA and Zf 5 in GLI, binds to an extended subsite (10,12–14). It may well be that Zf 6 in Staf, being the C-terminal zinc finger contacting the DNA, could also interact with more than 3 bp. Alternatively, it is possible that the presence of the AT base pairs in the 5′-part of the Staf-binding sites induces a peculiar DNA conformation that indirectly influences the binding of Staf.
Figure 4.

Models for the binding of Staf to the xtRNASec, optimal and hU6 Staf-binding sites. C2-H2 Zf 1–Zf 6 are represented with the two invariant cysteines, histidines and the bound zinc (hatched). The amino acids at positions –1, 2, 3 and 6 in the zinc finger α-helix that are crucial for making base contacts in the solved structures of zinc finger–DNA complexes are indicated. The DNA helix represents the Staf-binding sites, with bases on the non-template (nt) strand numbered 1–21 according to the consensus Staf-binding site (21) and 1′–21′ on the template strand. Horizontal lines define triplet subsites. Solid and dotted arrows indicate the predicted base contacts based on the solved structures and tentative recognition codes, respectively. Only base pairs involved in the putative zinc finger–DNA contacts are indicated; gray boxes, contacts with the three sites; hatched boxes, contacts with two sites; dark boxes, contact with one site. The relevant sites are indicated on the right.
Our model proposes that a limited number of residues at positions –1, 2, 3 and 6 of the recognition helix interact with bases in the Staf-binding sites. This suggests that the Staf–DNA complexes contain novel interaction principles absent in the known repertoire of base–amino acids contacts in zinc finger–DNA complexes. Further structural studies are needed to identify them in a more detailed fashion.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to C. Loegler for excellent technical assistance. This work was supported by grants from the Association pour la Recherche sur le Cancer (ARC). M.S. was awarded fellowships from the Ministère de l’Education Nationale, de la Recherche et de la Technologie and from the ARC.
REFERENCES
- 1.Miller J., McLachlan,A.D. and Klug,A. (1985) EMBO J., 4, 1609–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg U.B., Schröder,C., Preiss,A., Kienlin,A., Cote,S., Riede,I. and Jäckle,H. (1986) Nature, 319, 336–339. [Google Scholar]
- 3.Tautz D., Lehmann,R., Schnürch,H., Schuh,R., Seifert,E., Kienlin,A., Jones,K. and Jäckle,H. (1987) Nature, 327, 383–389. [Google Scholar]
- 4.Turner J. and Crossley,M. (1999) Trends Biochem. Sci., 24, 236–240. [DOI] [PubMed] [Google Scholar]
- 5.Schuh R., Aicher,W., Gaul,U., Côté,S., Preiss,A., Maier,D., Seifert,E., Nauber,U., Schröder,C., Kemler,R. and Jäckle,H. (1986) Cell, 47, 1025–1032. [DOI] [PubMed] [Google Scholar]
- 6.Parraga G., Horvath,S.J., Eisen,A., Taylor,W.E., Hood,L., Young,E.T. and Klevit,R.E. (1988) Science, 241, 1489–1492. [DOI] [PubMed] [Google Scholar]
- 7.Lee M.S., Gippert,G.P., Soman,K.V., Case,D.A. and Wright,P.E. (1989) Science, 245, 635–637 [DOI] [PubMed] [Google Scholar]
- 8.Pavletich N.P. and Pabo,C.O. (1991) Science, 252, 809–817. [DOI] [PubMed] [Google Scholar]
- 9.Fairall L., Schwabe,J.W.R., Chapman,L., Finch,J.T. and Rhodes,D. (1993) Nature, 366, 483–487. [DOI] [PubMed] [Google Scholar]
- 10.Pavletich N.P. and Pabo,C.O. (1993) Science, 261, 1701–1707. [DOI] [PubMed] [Google Scholar]
- 11.Houbaviy H.B., Usheva,A., Shenk,T. and Burley,S.K. (1996) Proc. Natl Acad. Sci. USA, 93, 135777–13582. [Google Scholar]
- 12.Foster M.P., Wuttke,D.S., Radhakrishnan,I., Case,D.A., Gottesfeld,J.M. and Whright,P.E. (1997) Nature Struct. Biol., 4, 605–608. [DOI] [PubMed] [Google Scholar]
- 13.Wuttke D.S., Foster,M.P., Case,D.A., Gottesfeld,J.M. and Wright,P.E. (1997) J. Mol. Biol., 273, 183–206. [DOI] [PubMed] [Google Scholar]
- 14.Nolte R.T., Conlin,R.M., Harrison,S.C. and Brown,R.S. (1998) Proc. Natl Acad. Sci. USA, 95, 2938–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elrod-Erickson M., Rould,M.A., Nekludova,L. and Pabo,C.O. (1996) Structure, 4, 1171–1180. [DOI] [PubMed] [Google Scholar]
- 16.Kim C.A. and Berg,J.M. (1996) Nature Struct. Biol., 3, 940–945. [DOI] [PubMed] [Google Scholar]
- 17.Myslinski E., Krol,A. and Carbon,P. (1992) Nucleic Acids Res., 20, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuster C., Myslinski,E., Krol,A. and Carbon,P. (1995) EMBO J., 14, 3777–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaub M., Myslinski,E., Schuster,C., Krol,A. and Carbon,P. (1997) EMBO J., 16, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myslinski E., Krol,A. and Carbon,P. (1998) J. Biol. Chem., 273, 21998–22006. [DOI] [PubMed] [Google Scholar]
- 21.Schaub M., Krol,A. and Carbon,P. (1999) J. Biol. Chem., 274, 24241–24249. [DOI] [PubMed] [Google Scholar]
- 22.Schaub M., Myslinski,E., Krol,A. and Carbon,P. (1999) J. Biol. Chem., 274, 25042–25050. [DOI] [PubMed] [Google Scholar]
- 23.Siebenlist U. and Gilbert,W. (1980) Proc. Natl Acad. Sci. USA, 77, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee D.K., Horikoshi,M. and Roeder,R.G. (1991) Cell, 67, 1241–1250. [DOI] [PubMed] [Google Scholar]
- 25.Truss M., Chalapakis,G. and Beato,M. (1990) Proc. Natl Acad. Sci. USA, 87, 7180–7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brunelle A. and Schleif,R.F. (1987) Proc. Natl Acad. Sci. USA, 84, 6673–6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sturm R., Baumrucker,T., Franza,B.R.,Jr and Herr,W. (1987) Genes Dev., 1, 1147–1160. [DOI] [PubMed] [Google Scholar]
- 28.Hayes J.J. and Tullius,T.D. (1989) Biochemistry, 28, 9521–9527. [DOI] [PubMed] [Google Scholar]
- 29.Leonard N.J., McDonald,J.J., Henderson,R.E.L. and Reichmann,M.E. (1971) Biochemistry, 10, 3335–3342. [DOI] [PubMed] [Google Scholar]
- 30.Herr W. (1985) Proc. Natl Acad. Sci. USA, 82, 8009–8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubin C.M. and Schmid,C.W. (1980) Nucleic Acids Res., 8, 4613–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Omichinski J.G., Pedone,P.V., Felsenfeld,G., Gronenborn,A.M. and Clore,G.M. (1997) Nature Struct. Biol., 4, 122–132. [DOI] [PubMed] [Google Scholar]
- 33.Choo Y. and Klug,A. (1994) Proc. Natl Acad. Sci. USA, 91, 11163–11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choo Y. and Klug,A. (1994) Proc. Natl Acad. Sci. USA, 91, 11168–11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choo Y. and Klug,A. (1997) Curr. Opin. Struct. Biol., 7, 117–125. [DOI] [PubMed] [Google Scholar]
- 36.Wolfe S.A., Greisman,H.A., Ramm,E.I. and Pabo,C.O. (1999) J. Mol. Biol., 285, 1917–1934. [DOI] [PubMed] [Google Scholar]