

Abstract
Chirality is important in drug discovery since stereoselective drugs can ameliorate therapeutic difficulties including adverse toxicity and poor pharmacokinetic profiles. The human kinome, a major druggable, enzyme class, has encountered a push for chiral drug development. As a consequence, kinase inhibitors have been exploited to treat a wide range of diseases. However, many kinase inhibitors are planar and overlap in chemical space, which leads to selectivity and toxicity issues. By exploring chirality within the kinome, a new iteration of kinase inhibitors are being developed to better exploit the three-dimensional nature of the kinase active site. Exploration into novel chemical space, in turn, will also improve drug solubility and pharmacokinetic profiles. This perspective explores the role of chirality to improve kinome druggability and will serve as a resource for pioneering kinase inhibitor development to address current therapeutic needs.
Graphical Abstract
1. Introduction
Stereochemistry has a vital role in pharmacology by dictating pharmacokinetic and pharmacodynamic properties of a drug molecule.1–2 The enantiomers of a chiral drug may have identical physical and chemical properties but, in a chiral environment, enantiomeric effects can be significantly different.3 Hypothetically, an active enantiomer will perfectly fit into the receptor binding site to produce a pharmacological effect whereas the inactive enantiomer (distomer) does not and will not generate any active response.4–5 Moreover, the distomer may interact at a site elsewhere and cause an unwanted or adverse effect as a consequence of its pharmacodynamics eg. Thalidomide, a drug used for morning sickness in the late 1950’s, caused teratogenic effects in offspring (phocomelia).6–9 Later, it was discovered that only the S-isomer could cross the blood-placental barrier and was responsible for teratogenicity.10 Considering the possible detrimental effects of the distomer, the FDA provided guidelines to assure stereoisomeric composition of a drug and pharmacokinetics of specific enantiomers.11–12
In certain drugs, such as atropisomers,13 a chiral center does not exist but hindrance around a rotatable bond generates axial chirality. Compounds with axial chirality exhibit similar properties to that of traditional enantiomers, which can significantly impact drug properties. Recently, drug design and synthetic campaigns have been carried out to address the issue of atropisomers in pharmaceutical development. Since axial chirality can give rise to a single, stereochemically stable drug with improved efficacy and safety, atropisomers are being developed in a similar fashion to that of enantiomers to enhance drug properties.14–15
Chiral drug discovery has gained much attention in the human kinome as a method to improve drug properties of kinase inhibitors. As is known, the kinome represents promising drug targets because of their pivotal roles in cellular signalling.16 Kinase inhibitors are divided between type I (ATP competitive), II (ATP non-competitive), and III (allosteric) and are typically planar, non-chiral structures.17, 18 An emerging paradigm to improve kinome selectivity is to explore the three-dimensional nature of the kinase active site by generating chiral inhibitors. Researchers have investigated this strategy to successfully address potency and selectivity issues.19 For example, FDA approved drugs crizotinib and lorlatinib were developed by exploiting chirality to improve potency, selectivity, and pharmacokinetics.20–21 Additionally, the FDA approved drug tofacitinib was selectively developed for JAK3 by evaluating selectivity of diastereomers.22 In general, chirality can be engineered into kinase inhibitors to improve drug properties. Various approved enantio-specific kinase inhibitors are listed in Table 1.
Table 1:
Various approved chiral kinase inhibitors
| Drug Name | Tradename | Approval | Developer | Target | Indication |
|---|---|---|---|---|---|
|
| |||||
| Sirolimus* | RAPAMUNE® | 199923 | Wyeth-Ayerst Research | FKBP12/mTOR | Immunosuppressant |
| Temsirolimus* | TORISEL® | 200724 | Wyeth Pharmaceuticals | FKBP12/mTOR | Advanced RCCa |
| Everolimus* | AFINITOR® | 2009 | Novartis | FKBP12/mTOR | Immunosuppressant |
| Crizotinib | XALKORI® | 2011 | Pfizer | ALK, c-MET, HGFR | NSCLC |
| Ruxolitinib | JAKAFI® | 2011 | Incyte | JAK1, JAK2 | Myelofibrosis |
| Tofacitinib | XELJANZ® | 2012 | Pfizer | JAK2, JAK3 | RAb |
| Afatinib | GILOTRIF® | 2013 | Boehringer Ingelheim | EGFR, HER2 | NSCLCc |
| Inbrutinib | IMBRUVICA® | 2013 | Pharmacyclics | BTK | CLLd, MCLe |
| Idelalisib | ZYDELIG® | 2014 | Gilead | PI3Kδ | Haematological cancers |
| Cobimetinib | COTELLIC® | 201525 | Exelixis and Genentech | MEK1/2 | Melanoma |
| Midostaurin* | RYDAPT® | 201726 | Novartis | FLT3 | AMLf |
| Netarsudil | RHOPRESSA® | 201827 | Aerie Pharm. | Rho Kinase | Glaucoma |
| Acalabrutinib | CALQUENCE® | 201728 | Acerta Pharm | BTK | MCLe |
| Encorafenib | BRAFTOVI® | 201829 | Array BioPharma | B-RafV600E/K | Melanoma |
| Larotrectinib | VITRAKVI® | 201830 | Bayer | NTRK | NTRK positive solid tumors |
Natural-product derived.
RCC, renal cell carcinoma.
RA, rheumatoid arthritis.
NSCLC, non-small cell lung cancer.
CLL, chronic lymphocytic leukemia.
MCL, mantle cell lymphoma.
AML, acute myeloid leukemia.
In this perspective, we consider the role of chirality in the discovery of safer, more efficacious kinase inhibitors to treat chronic diseases with an emphasis on malignancies. A comprehensive overview is provided outlining the discovery of clinically approved chiral kinase inhibitors or inhibitors under clinical development. This perspective serves as a resource for pioneering kinase inhibitor discovery to address new therapeutic needs through enantio-specific approaches.
2. Chiral Kinase Inhibitors
2.1. ALK and c-MET Receptor Tyrosine Kinases
Receptor tyrosine kinases (RTKs) are transmembrane receptors activated by growth factors, cytokines, or hormones. They transfer the γ-phosphate of ATP to a tyrosine residue of a downstream signaling partner. RTKs play fundamental roles in cellular processes such as proliferation, migration, metabolism, differentiation, and survival.31 Uncontrolled RTK activities are associated with various types of cancers and numerous small molecules targeting RKTs have been approved for the treatment of cancers.32 In particular, hepatocyte growth factor receptor (HGFR) and anaplastic lymphoma kinase (ALK) have been targeted for their roles in non-small cell lung cancer.33–34
HGFR, also known as c-MET, has participating roles in normal cellular processes with subsequent signaling from RTKs. Consequently, aberrant c-MET signaling is correlated with poor prognosis and metastatic progression in a number of major cancers.35–37 ALK, belonging to the insulin receptor superfamily, was first discovered as a NPM-ALK fusion protein in anaplastic lymphoma.38 ALK chromosomal alterations have been observed in anaplastic large cell lymphoma (50%–60%), inflammatory myofibroblastic tumors (27%), and non-small cell lung cancer (NSCLC) (4%–7%). Hence, both c-MET and ALK underwent extensive targeting strategies for the treatment of cancer.39
2.1.1. FDA-Approved RTK Drugs
A. Crizotinib (Aug 2011, XALKORI®, Pfizer)
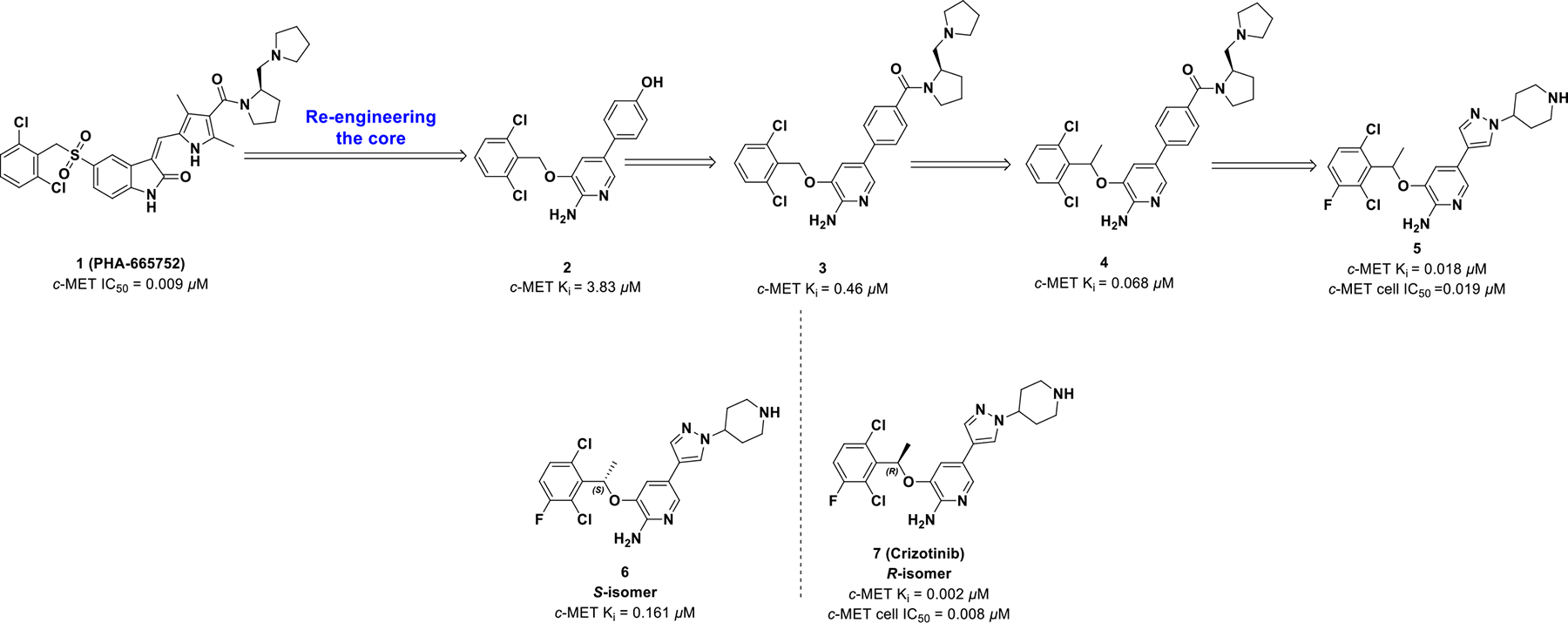
In 2011, crizotinib (7, PF-02341066) became the first FDA approved chiral ALK kinase inhibitor for the treatment of NSCLC.40 Crizotinib demonstrates concentration-dependent inhibition of ALK and c-MET and has antitumor activity in mice bearing tumor xenografts with c-MET and EML4- or NPM-ALK fusion proteins.41 Although originally approved for ALK fusion oncogenes, crizotinib is also a potent inhibitor of c-MET and ROS1. Crizotinib was discovered through lead-based optimization, which focused on improving selectivity and pharmacokinetic properties of lead candidates by exploiting chirality.
The discovery of crizotinib initiated with the identification of compound 1 (PHA-665752) (Scheme 1), which is a c-MET inhibitor with cellular potency (IC50 = 0.009 μM in GTL-16 cell line) and moderate selectivity (>50-fold for c-MET compared to other kinases).42 However, poor pharmacokinetic properties of 1 led to an optimization campaign to improve drug properties as well as kinase selectivity. The co-crystal structure43 of compound 1 with c-MET revealed inefficient interaction within the ATP pocket so the warhead was re-engineered to 4-(6-amino-5-((2,6-dichlorobenzyl)oxy)pyridin-3-yl)phenol (2), which led to an increase in receptor affinity (Ki = 3.83 μM). Scaffold 2 was further optimized to improve solubility to afford 3 (Ki = 0.46 μM).
Scheme 1.
Drug design of compound 7.
Further modification of the linkage between the 2-aminopyridine core and the phenyl ring was investigated due to a small lipophilic pocket adjacent to the binding location of the 3-benzyloxy linker. Addition of a small α-methyl group enhanced the binding efficiency of 4 (Ki = 0.068 μM, IC50 = 0.14 μM) along with lipophilicity (LipE Ki = 4.20; LipE IC50 = 3.86). Also, the α-methyl group improved pharmacokinetics by impairing benzylic oxidation and created a chiral center. Finally, the combination of 2,6-dichloro, 3-fluoro, and α-methyl into compound 5 provided the greatest inhibition against c-MET and the highest LipE values (enzymatic LipE = 4.82, cell LipE = 4.60; Ki = 0.012 μM, cell IC50 = 0.020 μM).
Aforementioned, the addition of the α-methyl introduced a chiral center (5), and the two enantiomers of compound 5 were synthesized and assessed. The R-enantiomer (7) exhibited better potency than the racemate (5) and S-isomer (6), which can be attributed to improved solubility, binding affinity, and assay variation. Hence, compound 7 efficiently binds c-MET resulting in improved cell-based ligand efficacy (LE) and LipE values with blocked benzylic oxidation.
The co-crystal structure of compound 720 bound to nonphosphorylated c-MET was solved (Figure 1; left panel). 2-Aminopyridine is involved in two hydrogen bonds with the hinge region (Pro1158 and Met1160). The chiral methyl group interacts with the c-MET A-loop, which rigidifies the benzyl group and occupies a small lipophilic pocket between Val1092 and Leu1157 (Figure 1; right panel). Moreover, the chiral benzyl group orients in a π-π interaction with Tyr1230. It can be concluded from structural analysis that the R-enantiomer preferentially interacts with Val1092, Leu1157, and Try1230; the S-enantiomer, however, would sterically clash with the c-MET back pocket and justifies the 100-fold loss in activity. Therefore, by exploiting chirality, R-enantiomer 7 was developed with enhanced enzymatic activity and improved pharmacokinetic profiles over the S-enantiomer 6.
Figure 1.
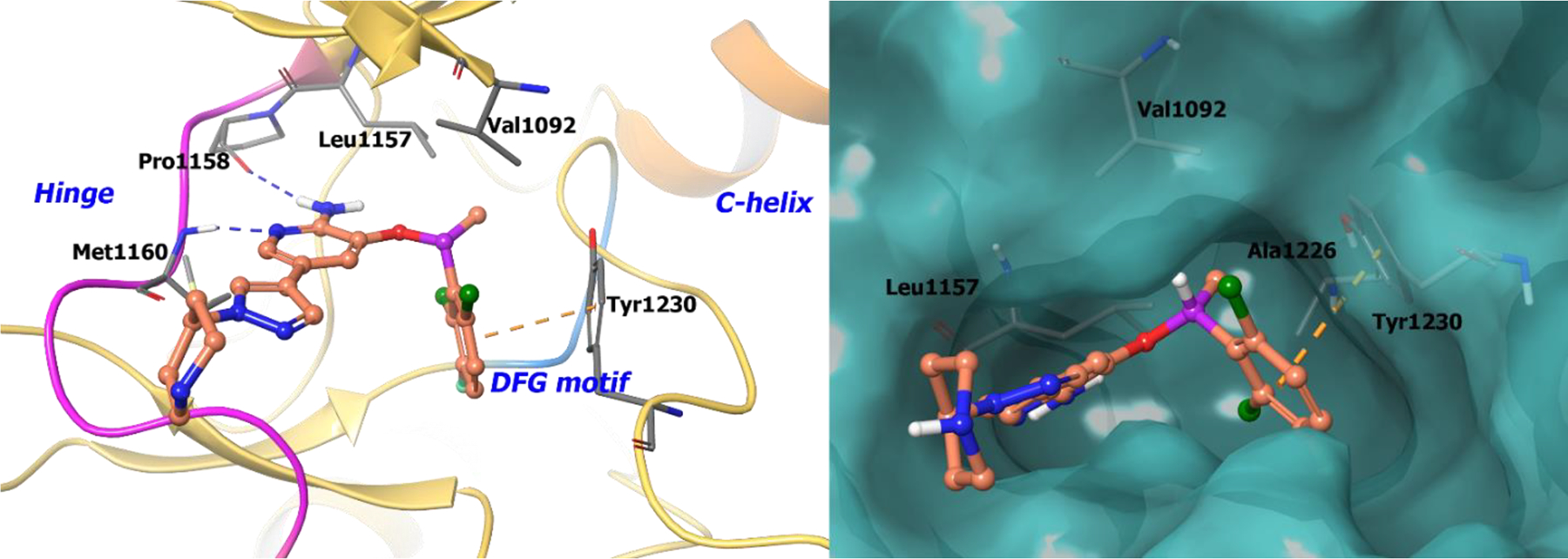
The co-crystal structure of compound 7 bound to c-MET (PDB 2WGJ, 2.0 Å). Left Panel: Interaction of compound 7 with the protein. The protein is depicted as yellow ribbons, and hydrogen bonds are illustrated with blue dashed lines; Right panel: The compound 7 binding site in c-MET. The surface of the protein is depicted in blue. compound 7 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
B. Lorlatinib (Nov 2018; LORBRENA®, Pfizer)
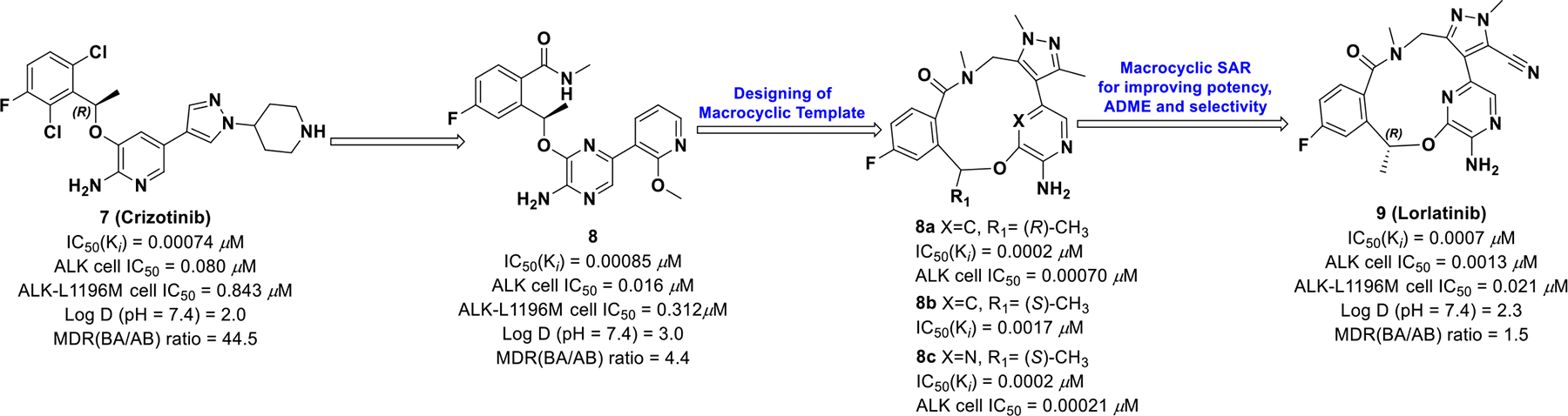
Due to poor blood brain barrier permeability of crizotinib, modifications were made to the molecule to better target ALK-positive NSCLC brain metastasis (Scheme 2).44–45 It was hypothesized that simultaneous improvement in cellular potency and multidrug resistance (MDR) BA/AB ratios (<2.5) could be achieved if crizotinib was redesigned with a restricted conformation and removal of the highly basic amine.21 The co-crystal structure of compound 8 bound to ALK highlighted an acyclic U-shaped pose with the fluorophenyl and heteroaromatic tail in proximity to the binding site. This suggested that novel macrocyclic design templates, with restricted conformations, could retain potency while achieving desirable central nervous system (CNS) ADME properties.
Scheme 2.
Drug design of compound 9.
A series of 12 to 14-membered macrocycles (8a-c) were tested and 8a exhibited an improvement in potency (ALK Ki = 0.0002 μM, ALK-L1196M Ki = 0.00029 μM) against both wild-type and mutant ALK compared with the S-enantiomer (8b) (ALK Ki = 0.0017 μM, ALK-L1196M Ki = 0.0021 μM). This stereochemical preference of the R-isomer accounts for a 210-fold improvement compared to the S-enantiomer. Additional modifications were performed to obtain the required combination of potency, ADME, and CNS availability, which provided compound 9 (lorlatinib). Interestingly, 9 was found to exhibit atropisomerism via a “ring-flip”. But, due to a high energy barrier, only a single diastereomer is physiologically relevant.46
Selectivity of the macrocycles for ALK over the highly conserved TrkB kinase was investigated because of the importance of TrkB in CNS homeostasis.47 It was found that the cyano group of compound 9 (lortatinib) was highly selective for the ALK mutation L1196M.48–49 The chiral α-methyl on compound 9 orients the florobenzene into a lipophilic pocket formed by Leu1256, Cys1255 and Gly1269 (Figure 2; right panel). Consequently, in TrkB, the cyano moiety of 9 is oriented to Tyr635. This interaction causes unfavorable desolvation energies between the nitrile and Tyr635 resulting in 40-fold selectivity for ALK over TrkB. The co-crystal structure of 8c bound to ALK depicts key hydrogen bonding of the NH2 and ring nitrogen of the 2-aminopyrazine core with hinge residues Glu1197 and Met1199 (Figure 2; left panel).
Figure 2.
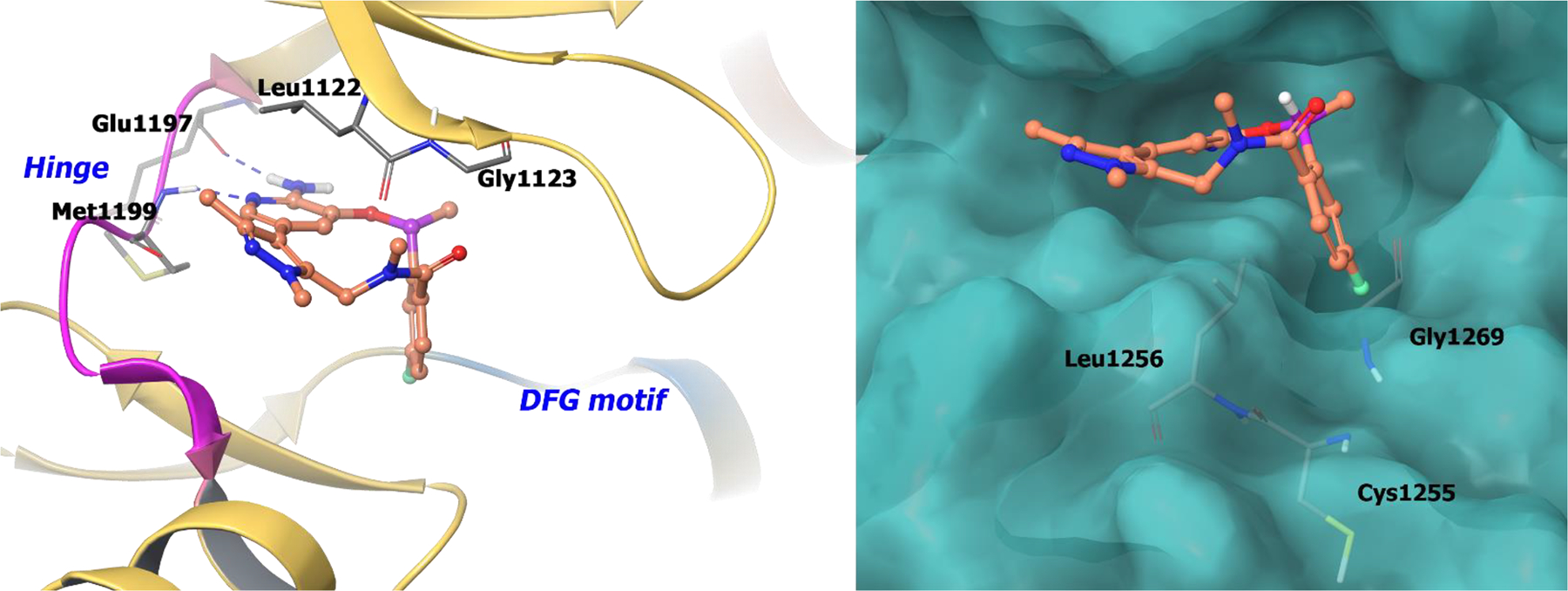
The co-crystal structure of compound 9 bound to ALK (PDB 4CMU, 1.8 Å). Left panel: Interaction of compound 9 with the protein. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Right panel: The compound 9 binding site in ALK, protein is shown as surface. Compound 9 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
The macrocycles were examined for CNS availability by analyzing MDR BA/AB efflux. Compound 9 exhibited low efflux potential, low clearance, and improved potency against wild-type and ALK mutants compared to crizotinib. On November 2, 2018, the FDA granted accelerated approval of compound 9 for ALK-positive NSCLC with metastatic disease.50 By exploiting chirality and macrocyclic atropisomerism, compound 9 achieved enhanced CNS penetration and improved kinome selectivity.
2.1.2. RTK Inhibitors Under Clinical Development
Although crizotinib provides upfront efficacy, patients frequently develop resistance to treatment because of ALK mutations (L1196M, G1269A, S1206Y, C1156Y, F1174L, L1152R and 1151Tins). Therefore, crizotinib was reengineered and optimized for ALK mutations by using a combination of LipE analysis and structure-based drug design.51 Comparing the crizotinib-ALK co-crystal structure to its apo form suggested a smaller C2 group would relax the Gly1269 carbonyl and enhance ALK mutant potency. Hence, strategies focused on optimizing the 2,6-dichloro-3-fluorophenyl group as well as limiting structural bulk at the pyrazolopiperidine tail (Scheme 3).
Scheme 3.
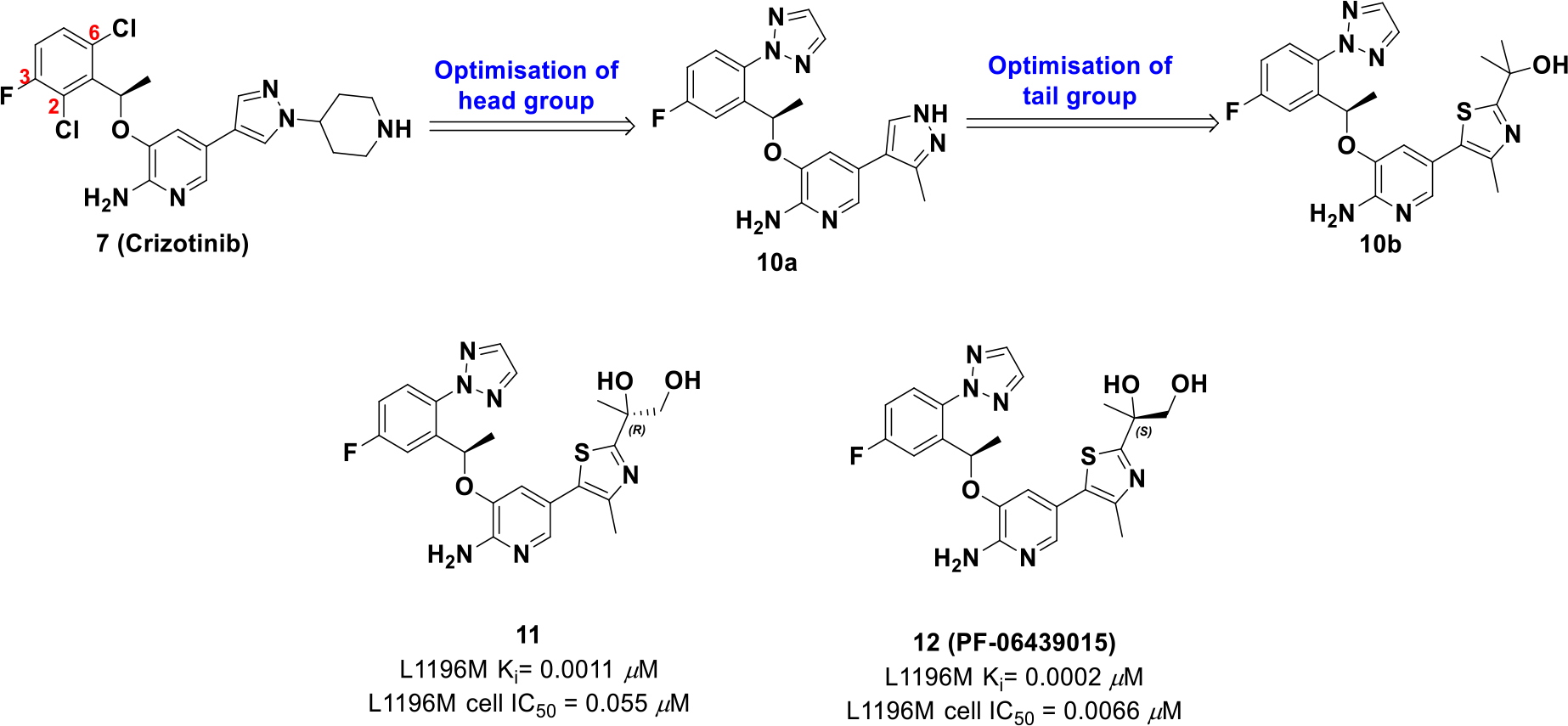
Drug design of compound 12
Optimization efforts uncovered that a triazole on C6 and a thiazole with a tertiary alcohol (10b) afforded the best potency and lipophilic values (L1196M cell IC50 = 0.027 μM, log D = 3.6). The alcohol formed a hydrogen bond with the carbonyl group of Asp1203 and structural analysis suggested another alcohol in proximity to Asp1203 could improve binding site interactions. The resulting R and S enantiomers with a primary alcohol were synthesized, amongst which the S enantiomer 12 (PF-06439015) exhibited an improved LipE value and an increase in cellular potency compared to crizotinib. Importantly, compound 12 also exhibited activity against ALK mutations L1196M and G1269A of which crizotinib is not active.
The co-crystal structure of compound 12 with wild-type ALK illustrates that the hydroxy group formed two hydrogen-bonds with Asp1203 (Figure 3). Moreover, introduction of the additional hydroxy group reduces lipophilicity but maintains membrane permeability because of intramolecular hydrogen bonding. Interestingly, the R-enantiomer of the chiral alcohol (11) demonstrated a reduced LipE value suggesting that an optimal chiral alignment is essential for improved drug properties.
Figure 3.
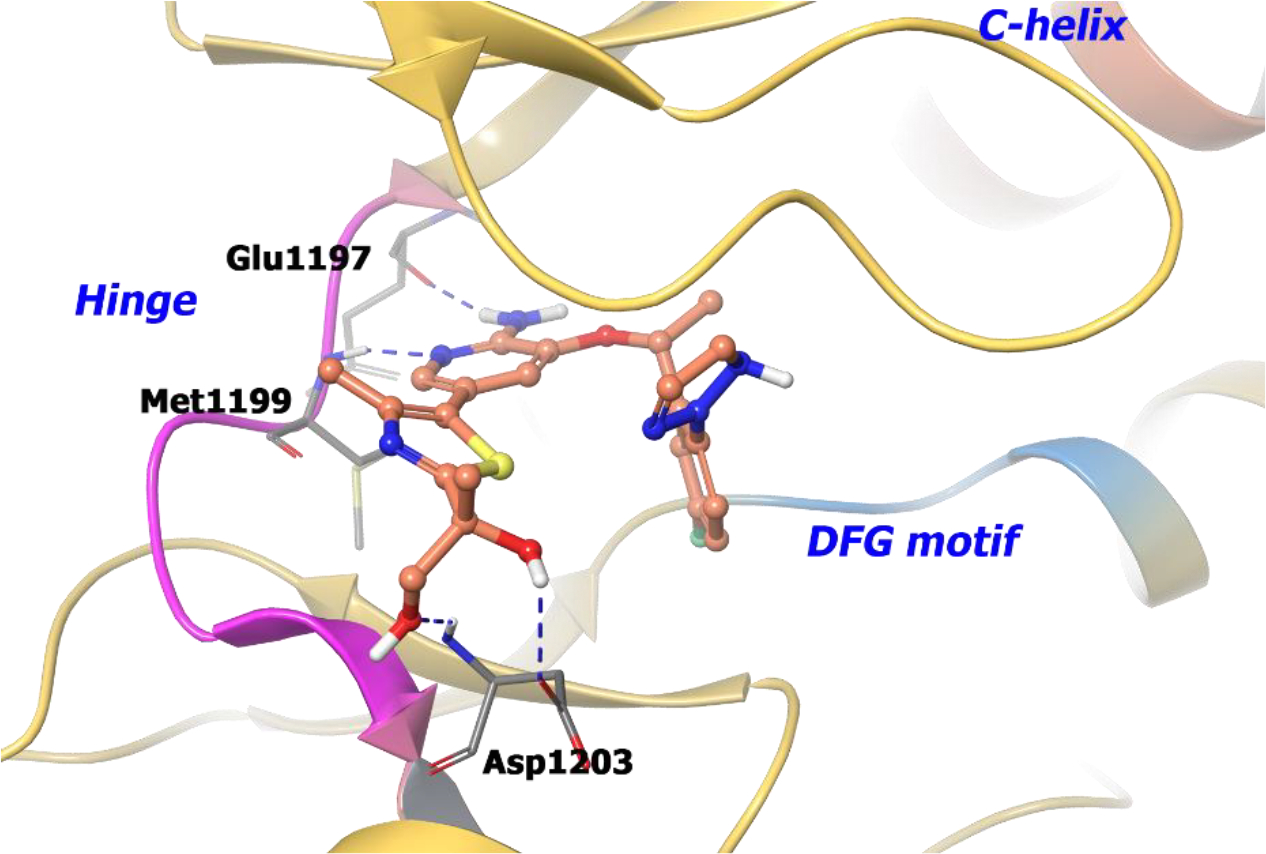
The co-crystal structure of compound 12 bound to ALK L1196M protein (PDB 4CD0, 2.23 Å). Interaction of compound 12 into the binding site of the protein. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound 12 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
Harmange et al discovered a series of triazolo-pyridines/pyridazines52 exhibiting cellular and enzymatic potency against c-MET.53 The U-shaped inhibitors adopt a unique c-MET binding mode and presented with exciting selectivity properties over other kinases. The first series was based on O-linked (13) and N-linked triazolopyridazine (14), but both suffered from poor solubility and rapid clearance in a patient-derived xenograft (PDX) model.54 These limitations were addressed with the generation of compound 15. The combination of a PEG-like ethoxy solubilizing group, a fluorine to block aromatic oxidation, and an R α-methyl to block benzylic oxidation furnished the best combination of potency and pharmacokinetic properties. However, efficacy was not optimal in PDX models (Scheme 4).55 Further improvements were investigated to improve drug properties, which resulted in compound 16 (AMG 337). Compound 16 was predicted to form intramolecular hydrogen bonds to reinforce the rigidity of the U-shaped binding mode.56 Compound 16 exhibited an improved half-life, high oral bioavailability (~65%), and strong inhibition of c-MET over a 12 h period. Compound 16 was also evaluated in a c-MET dependent xenograft model and exhibited an ED50 of 0.3 mg/kg.
Scheme 4.
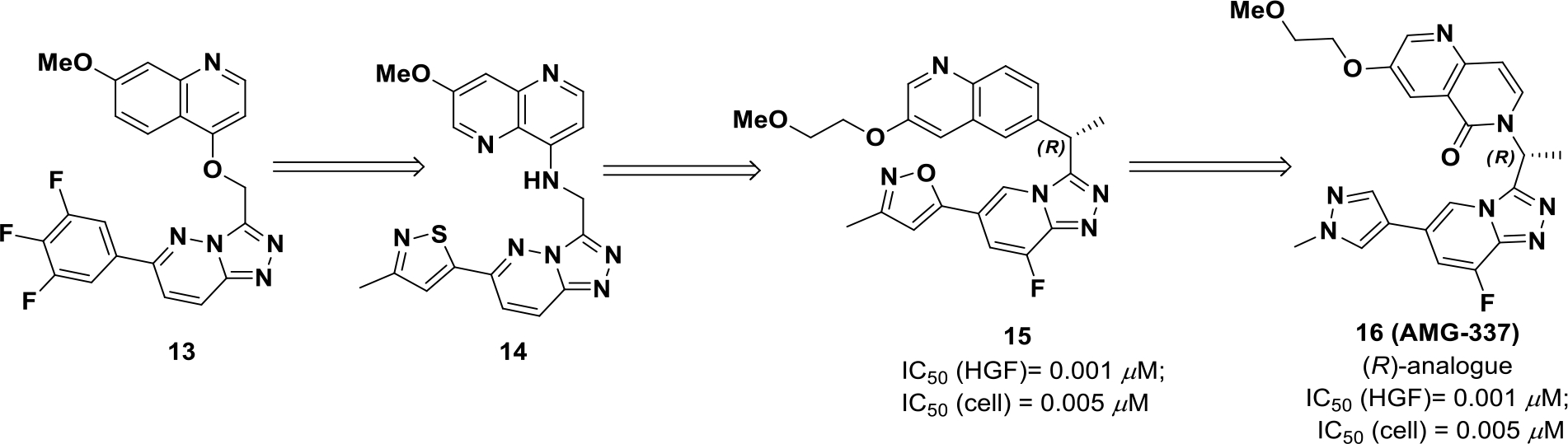
Drug design of compound 16
Boezio et al. produced a co-crystal structure of compound 16 with unphosphorylated c-MET56 and confirmed that the inhibitor adopts the predicted U-shaped binding mode. The chiral benzylic methyl forces the compound in a stable U-shaped binding mode around Met1211 (Figure 4, left panel), which would cause a steric clash with the S-enantiomer. The naphthyridinone nitrogen hydrogen bonds with the hinge at Met1160, whereas the nitrogen of the fluorotriazolopyridine engages the amide backbone of Asp1222. Furthermore, Tyr1230 and the fluorotriazolopyridine ring system exhibit face-to-face π-π stacking. The bulky fluorotriazolopyridine orients towards the solvent front, which is unlikely for the S-analogue because of geometric constraints in the c-MET back pocket. Interestingly, the R-configuration is encapsulated in a small, lipophilic pocket generated by Val1092, Leu1157, and Lys1110 (Figure 4, right panel), but the S-enantiomer is unable to interact in the same pocket. Also, the fluorine atom on fluorotriazolopyridine is confined to a small cleft perpendicular to the amide backbone of Asn1209. Compound 16 was selected as a clinical candidate and is currently in phase II clinical trials for c-MET amplified gastric/esophageal adenocarcinoma and other solid tumors (NCT02016534).
Figure 4.
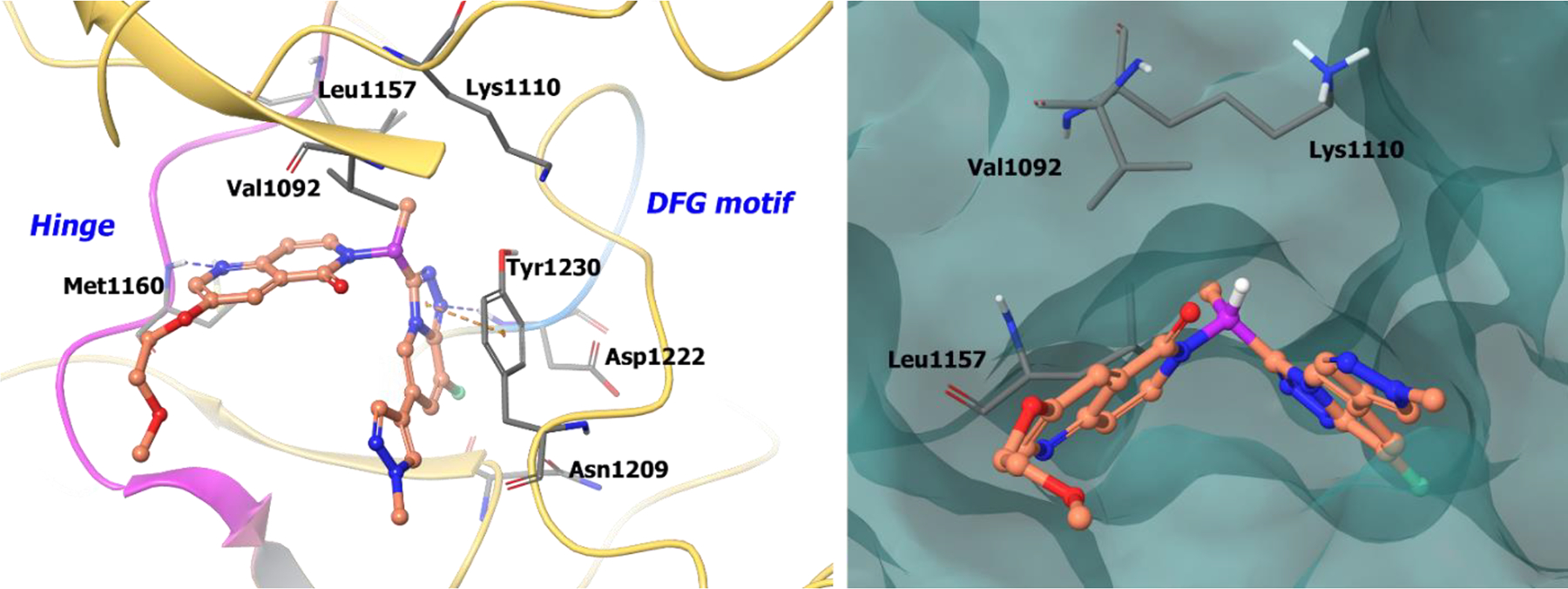
The co-crystal structure of compound 16 bound to c-MET (PDB 5EYD, 1.85 Å). Left panel: Interaction of compound 16 with the protein. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Right panel: Compound 16 binding site in c-MET, protein is shown as surface. Compound 16 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
Katz et al.57 uncovered inhibitor 17 while screening a Merck library. Follow on studies identified 17 as a potent ATP competitive inhibitor of c-MET (IC50 = 0.031 μM). The tricyclic heptanone core is a unique kinase warhead; therefore, optimization efforts focused on the phenyl and sulfonamide regions while keeping the warhead fixed. Aryl and heteroaryl derivatives were tested at the 3-position, amongst which the N-methyl pyrazolyl analogue 18 displayed a 27-fold increase in potency (IC50 = 0.004 μM). To improve solubility, alterations were examined on the 7-position sulfonamide with analogues such as vinyl, phenyl, imidazolyl, trifluoromethyl, and tert-butyl. Further modifying the pyrazole substituent and diversifying the sulfonamide moiety led to free amine 18a (c-MET IC50 = 0.003 μM) and monomethyl amine 18b (c-MET IC50 = 0.002 μM), which were subjected to metabolic profiling. Metabolic studies revealed that rapid dealkylation of the sulfonamide was the main metabolic path. To block dealkylation, cyclic analogues were generated, which exhibited optimal cell-based activities and pharmacokinetics. The R-dioxanyl cyclic analogue 20 (MK-2461), displayed the best cell potency (GTL-16 IC50 = 0.056 μM,) and excellent rat pharmacokinetics (Clp = 10 mL/min/kg, t1/2 = 1.3 h).
A piperidine analogue of compound 20, 18c (c-MET IC50 = 0.0026 μM), was co-crystallized with c-MET and adopts a similar pose as compound 20. The analogue forms a hydrogen bond with Met1160 at the hinge region, and the substituent on the pyrazole moiety extends into the solvent (Figure 5). Other key interactions include a hydrogen bond with Asp1222 and a hydrogen bond between the (R)-1,4-dioxane moiety and Arg1086. The sulfonamide moiety is buried near the catalytic lysine and the glycine rich loop. The stereospecific alignment of (R)-1,4-dioxane allows a water mediated hydrogen bond between the most proximal dioxanyl oxygen and the carbonyl oxygen of the central core. As a result, the tricyclic core adopts a conformation slightly out of plane. Hence, inclusion of the chiral (R)-analogue generates a unique binding mode for compound 20 in c-Met and effectively interacts with the solvent improving pharmacokinetic properties. Compound 20 has progressed into clinical studies and is the first candidate to enter phase II clinical trials from the tricyclic series (NCT00518739).
Figure 5.
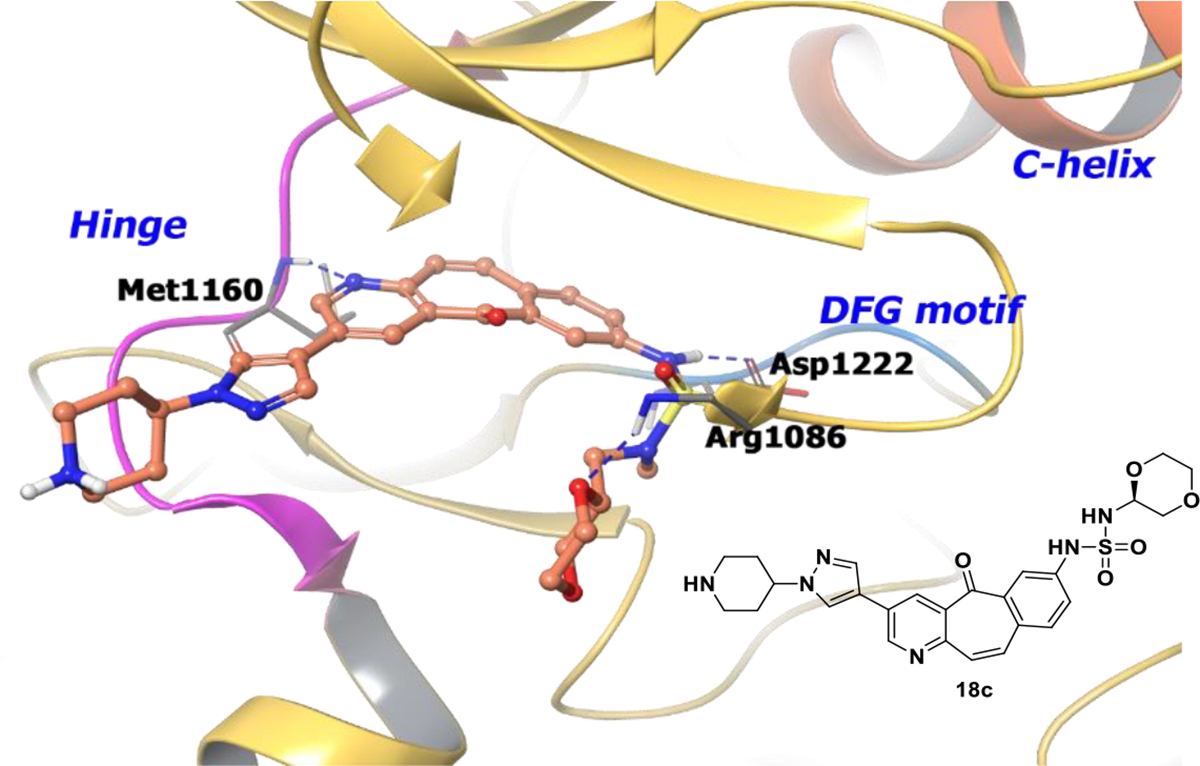
The co-crystal structure of analogue 18c bound to c-MET (PDB 3R7O, 2.3 Å). Interaction of analogue 18c at the binding site of the protein. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound 18c atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
Insulin-like growth factor (IGF) receptor is another RTK that has been targeted with chiral molecules.58 The IGF system is comprised of three receptors (IR, IGF-1R, IGF-2R), three ligands (insulin, IGF-1, IGF-2), and six 3-indole-D-glycerol-3’-phosphate (IGP) binding proteins (IGFBP1–6). The IGF-1R downstream signaling pathway is activated by insulin receptor (IR) substrates-1–4 and Src-homology collagen proteins as adapter molecules. Activation of the receptor triggers both PI3K/Akt and Ras/Raf pathways, thereby controlling apoptosis. Several studies have highlighted the key roles IGF-1R plays in cancer cell proliferation and metastasis. Over-expression of IGF-1R has been associated with colorectal and breast cancer. 59 In addition, increased levels of IGF-1 have been associated with prostate cancer and pre-menopausal breast cancer.60–61 The two main approaches to control IGF-1R signaling include monoclonal antibody therapy and small molecule inhibition.62
BMS-754807 (23) is a pyrrolo-triazine based IGF-1R inhibitor exhibiting good clinical results.66 For development, structural modifications were performed on compound 21 (Scheme 7). The core of the inhibitor was developed into pyrrolo-triazine with cyclopropyl substitution on pyrazole 22 providing further scope of structure-activity relationship (SAR) studies on the C2 amine.
Scheme 7.
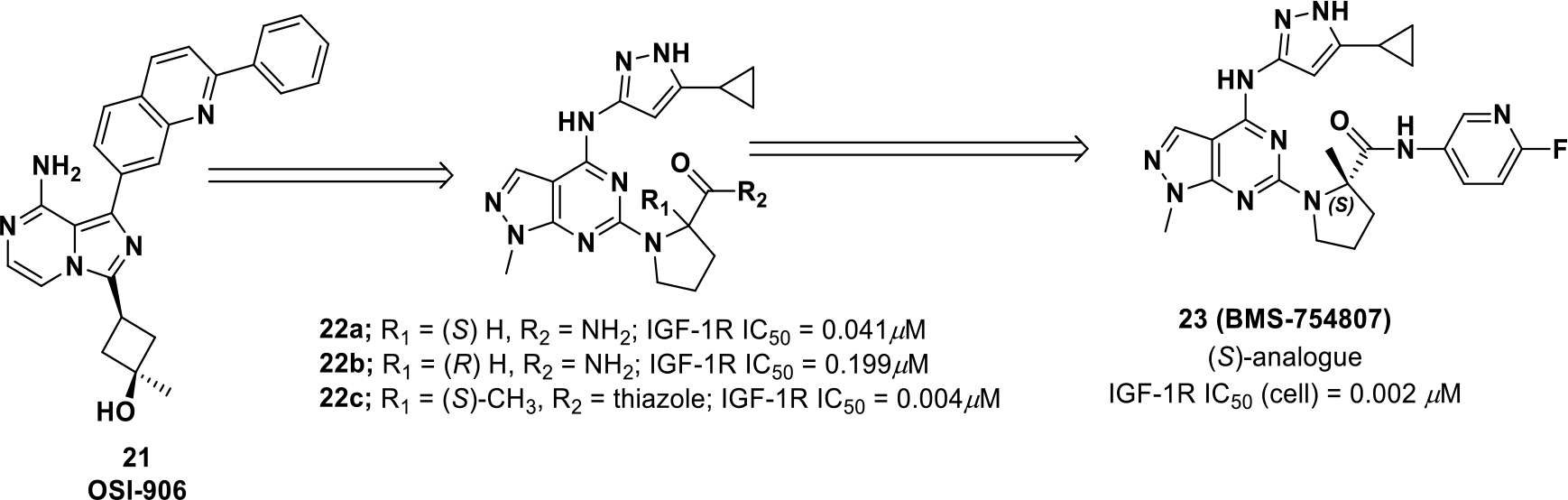
Drug design of compound 23
The primary amide (22a) was found to be potent against IGF-1R but also exhibited CDK2E inhibition. The S- amide (22) was found to be five-fold more potent than the R-amide (22b). Other substituents, such as phenyl, thiazole, and pyridine, showed improved potency, but the (S)-2-methyl analogues, with heterocyclic amides (22c), maintained potency with better selectivity. By optimizing the pyridyl substitution, analogue 23 was discovered and had an optimal combination of both potency (IC50 = 0.002 μM). It exhibited lower toxicity and good pharmacokinetic properties.
The co-crystal structure of compound 23 with IGF-1R demonstrates a donor/acceptor/donor hydrogen bonding motif with Met1052, Leu1051, and Glu1050 at the hinge. Moreover, the pyridyl ring nitrogen is involved in a hydrogen bond with the backbone of Asp1123. In the S-enantiomer, the cyclopropyl substituent on pyrazole could accommodate the gatekeeper (Met1049) and the fluoropyridyl amide engages the back pocket. The less active R-enantiomer would have a steric clash between the fluoropryidyl ring and the Gly-rich loop of the kinase. Because of the favorable interactions, the S-isomer (23) exhibits better potency compared to the R-isomer (Figure 6).66
Figure 6.
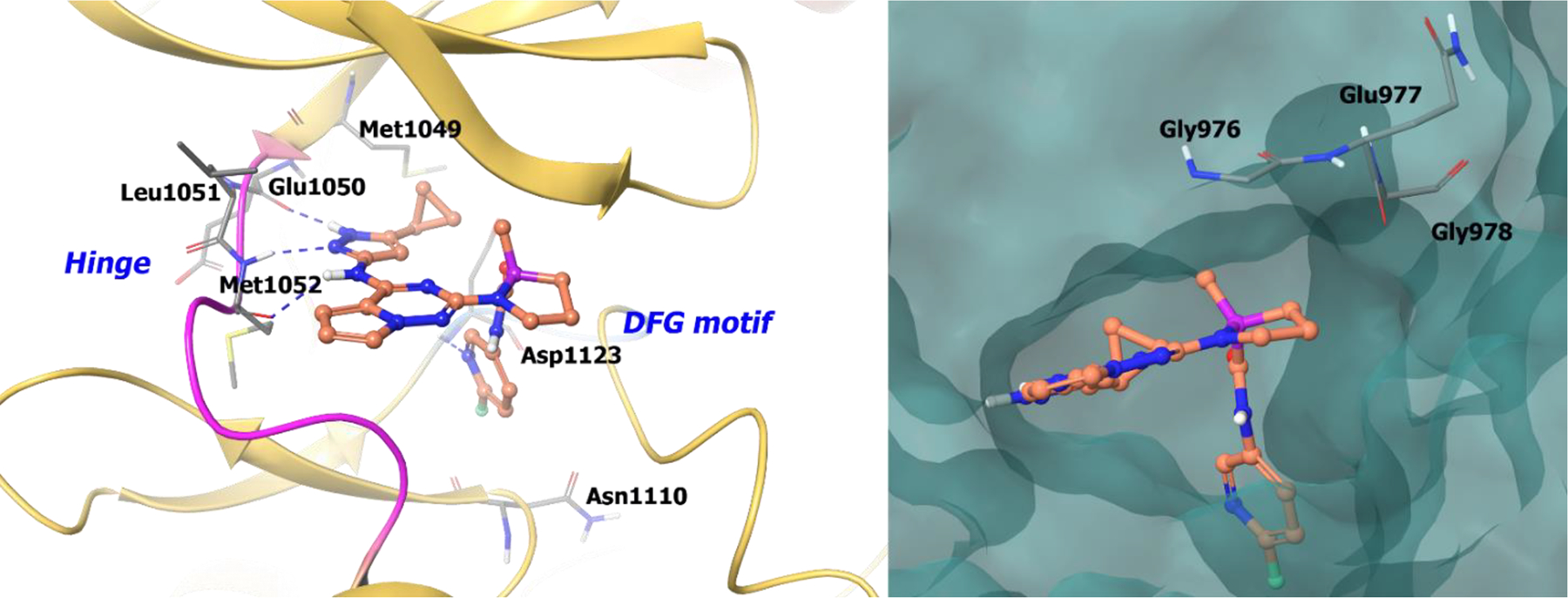
The co-crystal structure of compound 23 bound to IGF-1R (PDB 3I81, 2.08 Å). Left Panel: Interaction of compound 23 with the protein. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Right panel: The binding site of compound 23 in IGFR-1R. The surface of the protein is depicted in blue. Compound 23 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
Additional chiral RTK inhibitors (Scheme 8) are being evaluated in clinical studies including LY2874455, AZD4547, and BMS-582664. Compound 24 (LY2874455) is a novel pan-FGFR inhibitor and is in phase I clinical trials for solid tumors (NCT01212107).67 Compound 25 (BMS-582664) is a prodrug for dual inhibition of VEGFR-2/FGFR-1 and is in phase II clinical trials for hepatocellular carcinoma.69 Clearly, chirality is an important chemical addition to target RTKs, which can improve potency, selectivity, and pharmacokinetic profiles and can result in improved target inhibition.
Scheme 8.
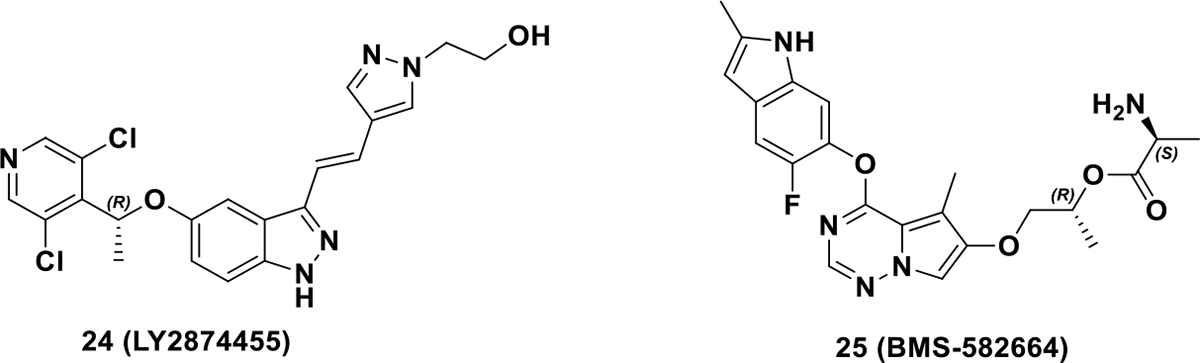
Chemical structures of chiral RTK inhibitors 24 and 25.
2.2. Non-Receptor Tyrosine Kinase Inhibitors
Non-receptor tyrosine kinases (nRTKs) are a subgroup of tyrosine kinases with the same primary function as RTKs except that they are solubilized, cytoplasmic enzymes. NRTK phosphorylation is associated with activation of T-cell receptors (TCR), B-cell receptors (BCR), IL-2 receptors (IL-2R), Ig, and prolactin receptors, thereby contributing to the immune system and immune response.70 Also, oncogenic nRTK mutations (e.g. BCR-ABL) are prevalent in a variety of hematological malignancies. Several small molecule nRTK inhibitors have been investigated for both cancer and immune disorders.71–73
2.2.1. FDA-Approved Non-RTK Drugs
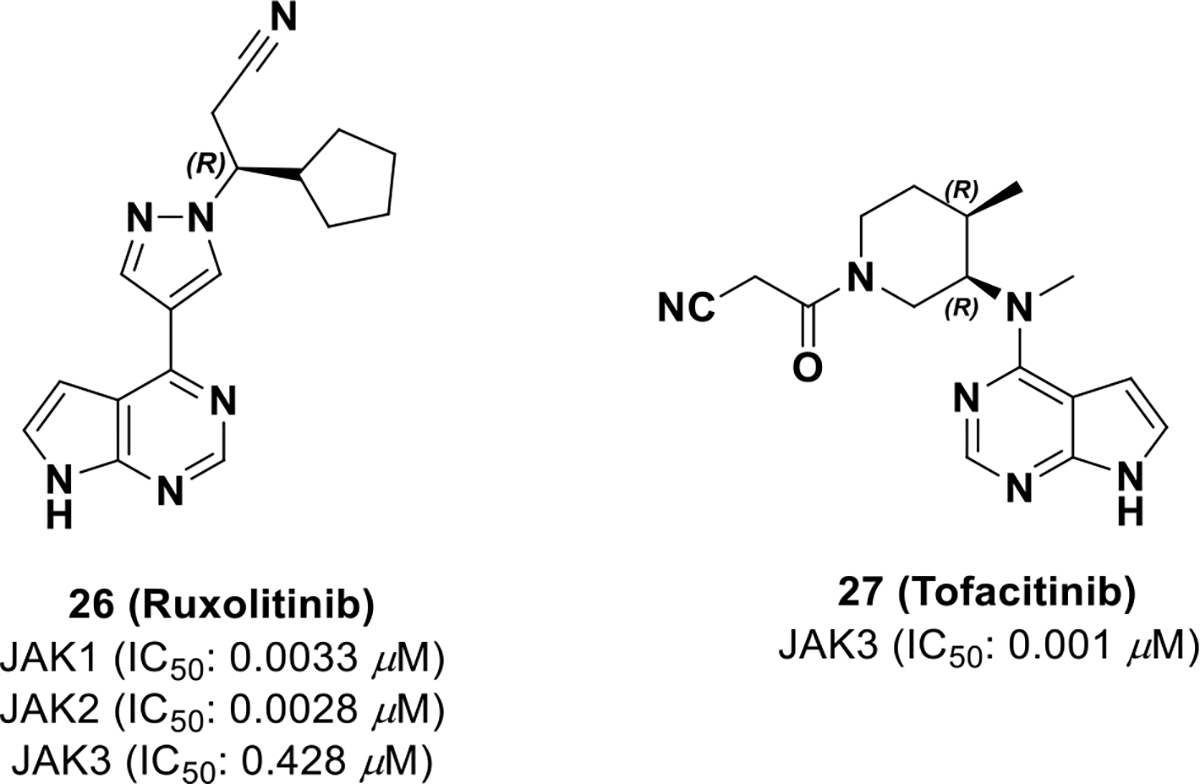
A. Ruxolitinib (Nov. 2011, JAKAFI®, Incyte) and tofacitinib (Nov. 2012, XELJANZ®, Pfizer)
Ruxolitinib and tofacitinib are the first-generation Janus kinase (JAK) inhibitors approved for primary myelofibrosis and immunological diseases (Scheme 9).74–75 The JAK family is comprised of JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). The JAK pathway is activated upon binding of a cytokine to a cytokine receptor leading to the phosphorylation of JAK. In its active, phosphorylated form, JAK signals through STAT, which translocates to the nucleus and regulates gene transcription and cellular function.76–77
Scheme 9.
FDA approved chiral JAK inhibitors
Ruxolitinib (26, INCB018424)78 was the first FDA approved JAK inhibitor for the treatment of myelofibrosis, and is selective for JAK1/JAK2 over JAK3/TYK2 (NCT01164163). To date, the preclinical drug discovery and development campaign of ruxolitinib has not been disclosed. There are reports of resistance to ruxolitinib, but the exact mechanism is still unknown and is likely patient dependent.79 Resistance could be due to on-target, JAK2 mutations or from mutations in the heterodimerization domain between JAK2 and JAK1.78
Tofacitinib (27, CP-690550) received regulatory approval in 2012 for the treatment of severe rheumatoid arthritis. It has also been developed for other disorders such as psoriasis, psoriatic arthritis, ankylosing spondylitis, ulcerative colitis, and Crohn’s disease. 80 Because tofacitinib is approved to treat chronic, non-lethal disease, the side effect profile should be minimal. In the discovery of tofacitinib, chirality was exploited for JAK3 selectivity, which resulted in low off-target liabilities and a well-tolerated side effect profile.
Tofacitinib was discovered through high-throughput screening, which uncovered a series of pyrrolo[2,3-d]pyrimidines (28) (Scheme 10).22 The pyrrolopyrimidine warhead was hypothesized to bind the hinge of JAK3, and different heterocyclic substitutions were examined, but all reduced potency. To reduce bulk at the binding site, the tricyclic ring system was truncated to N-methylcycloalkyl, which increased potency (29). Substitutions were further explored on the cyclohexane ring and it was found that the introduction of 2’,5’-dimethyl groups increased activity (30). Compound 30 is a complex mixture of 8 isomers so stereospecific isomers were synthesized and tested. It was found that a natural product-like terpenoid derivative (31) provided optimal activity but required further optimization because of the exposed alkene and stereochemical complexity. To improve drug-like properties, piperidine-based analogues were explored, which was hypothesized to provide access the JAK3 activation loop. Through the analysis of piperidine analogues, a cyanoacetamide substituent was found to provide the best combination of potency and selectivity resulting in tofacitinib (27).81–82
Scheme 10.
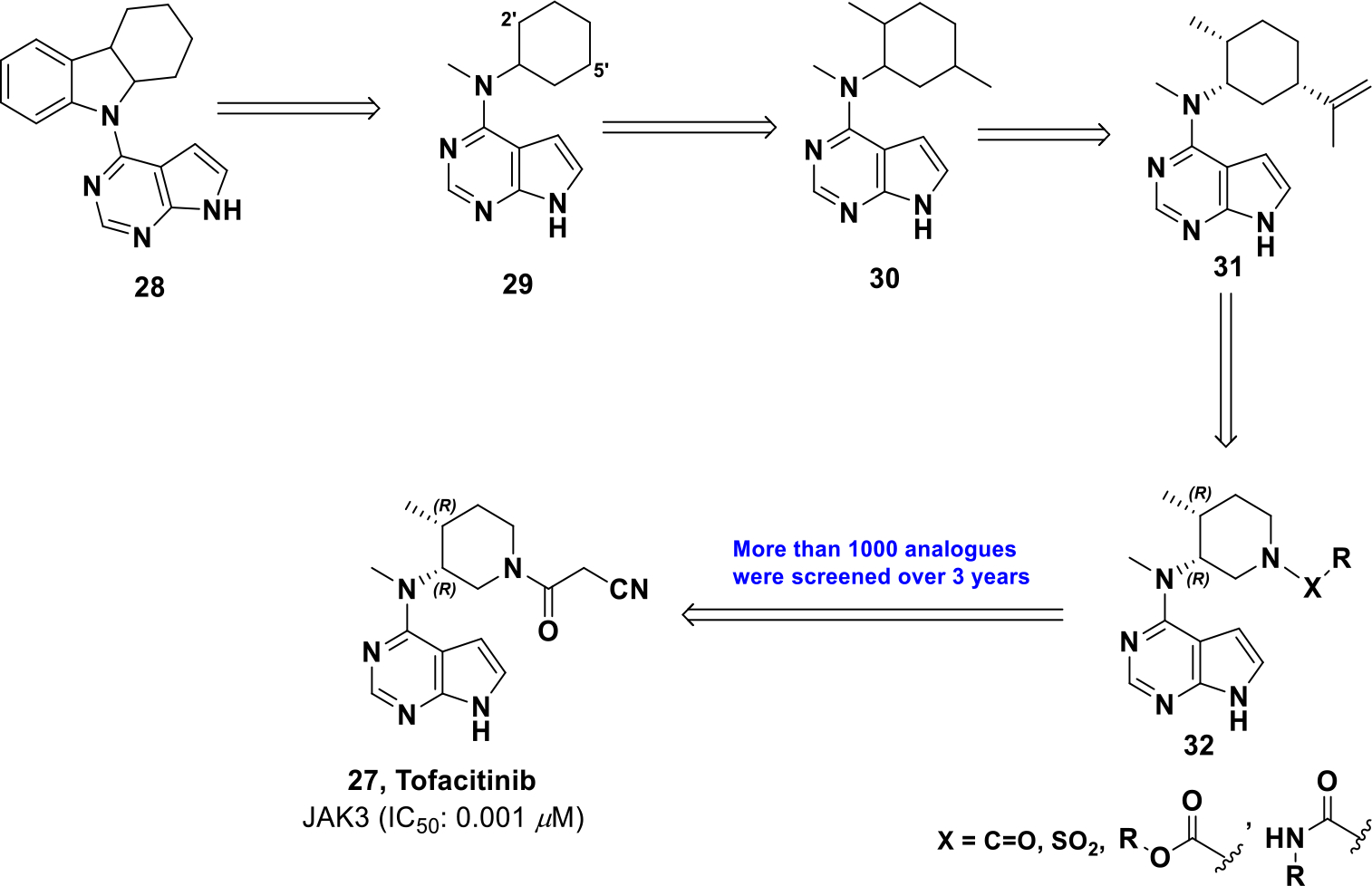
Drug development of compound 27
Compound 27 was docked into the JAK3 binding pocket and demonstrated that the 7-deazapurine warhead hydrogen bonds to the JAK3 hinge (Figure 7). In addition, the nitrile group hydrogen bonds with Arg953 at the opening of the JAK3 cleft. This docking pose in JAK3 illustrates that the 4R-methyl group is equatorial while the 3R-base moiety is axial, suggesting stereo-specific JAK3 binding.
Figure 7.
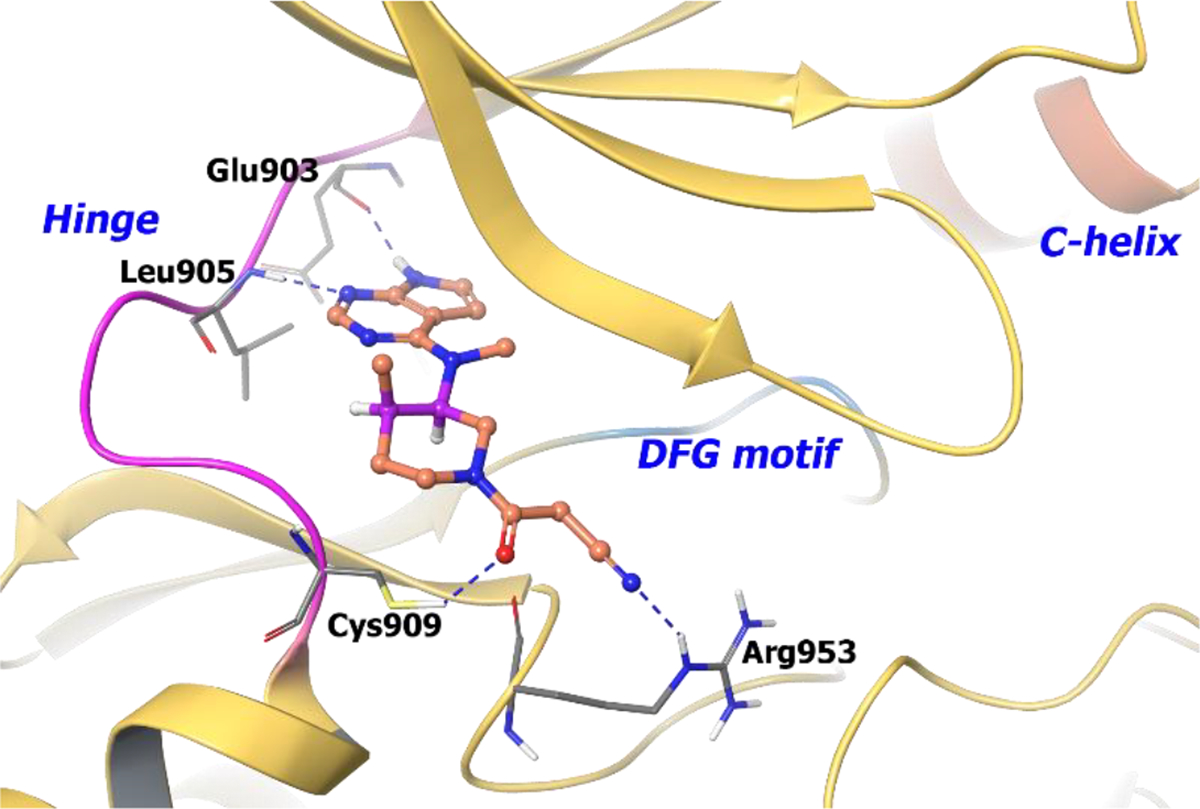
The binding of compound 27 into JAK3 active site (PDB 1YVJ, 2.55 Å). Interaction of compound 27 at the binding site of protein. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Compound 27 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
To investigate the stereo-specificity of compound 27 for JAK3, its three stereoisomers were synthesized and subjected to cell-based assays, molecular modeling studies, and target profiling. JAK3 and JAK2/TYK2 serve as a shared receptor for selected cytokines and is downregulated by STAT5 and STAT4 phosphorylation, respectively. Hence, the degree of STAT5 and STAT4 phosphorylation was evaluated via immunoblotting with each stereoisomer and only compound 27 (Scheme 11) could affect STAT5 phosphorylation at the concentration of 0.5 μM and no clear inhibition of STAT4 phosphorylation was observed.
Scheme 11.
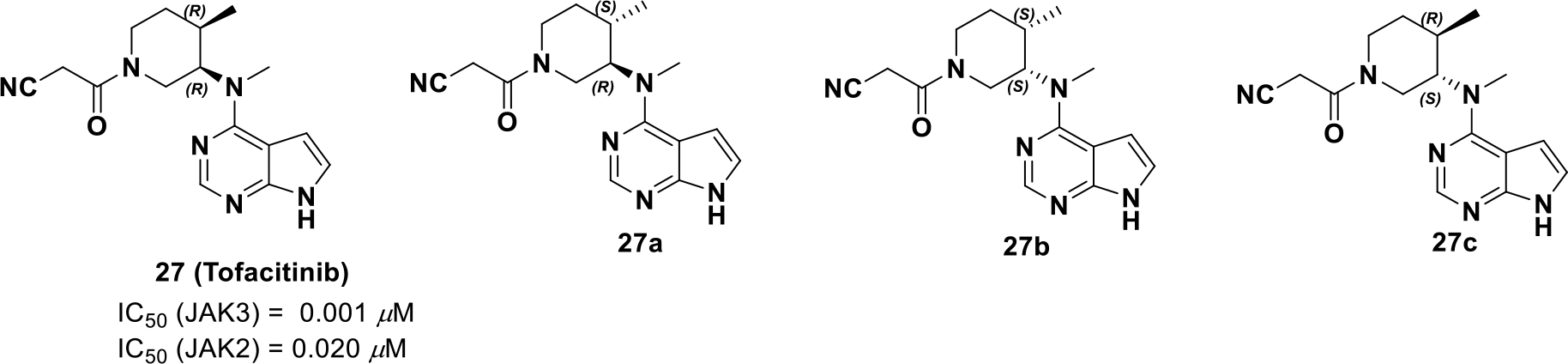
Four stereoisomers of tofacitinib
Next, the selectivity profile over other kinases was determined and all four isomers were JAK selective, but compound 27 was more selective for JAK3 (Kd = 0.0075 μM for JAK3).83 Further, the stereoisomers were docked into the JAK3 catalytic cleft.84 The lowest energy conformations of the unbound compounds 27 and 27a-c were super-imposed with their respective best docked poses (Figure 8). It was found that only compound 27 (3R, 4R) binds to JAK3 in a conformation that best resembles its minimum energy conformation, whereas the other isomers, 27a (3R, 4S) and 27b (3S, 4R), resemble an unstable half chair and a twisted boat conformation. The (3S, 4S) isomer 27c assumes a chair conformation in JAK3 but the axial and equatorial substitutions on the cyclohexane are high energy, respectively. Therefore, by exploiting chirality, tofacitinib was developed as a selective JAK3 inhibitor that is well tolerated for non-lethal indications of inflammation.
Figure 8.
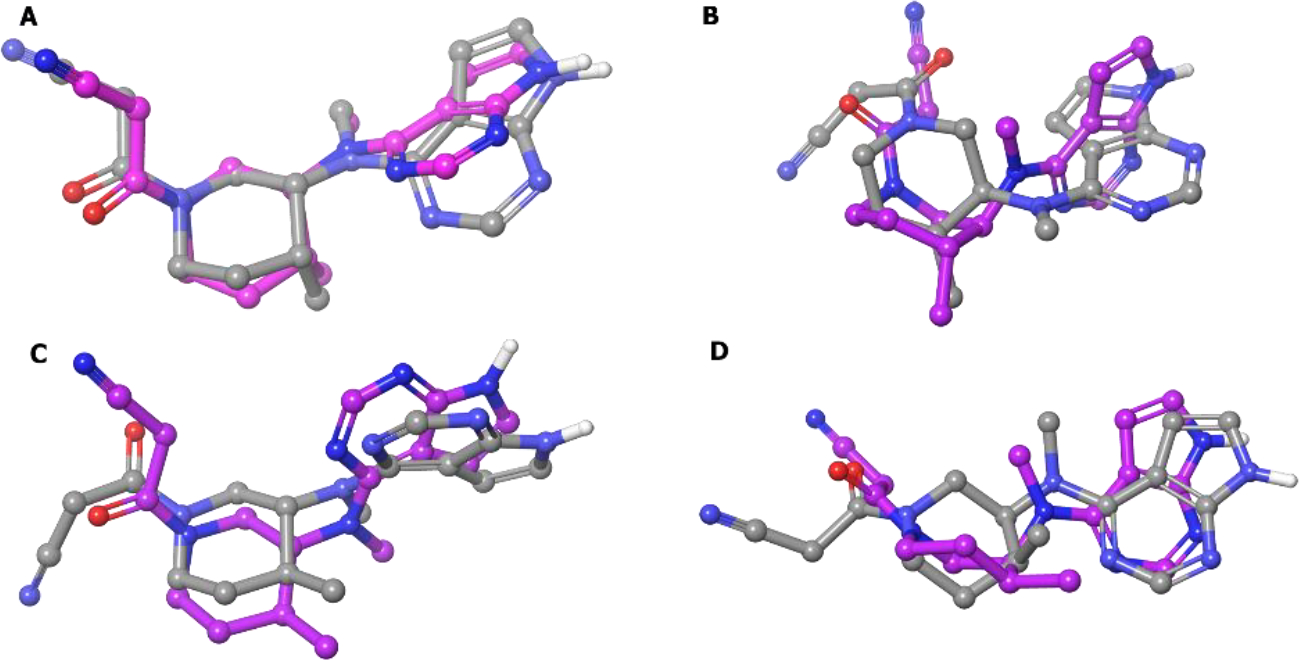
Superimposition of the six-membered ring of the lowest energy conformation (MM2 energy minimization) of compounds A:[27 (R,R)]; B:[27a (R,S)]; C:[27b (S,R)]; D:[27c (S,S)] (colored by atom type) and their respective best docked poses (colored in pink).
2.2.2. Non-RTK Inhibitors in Clinical Development
Proliferation and differentiation of T-cells or B-cells is induced by cytokines or chemokines via the JAK/STAT pathway.85 Amongst the four members of the JAK family, JAK1 is activated by γ-common chain cytokines, IFNγ, IL-6, and other gp130 cytokines.86 Whereas TYK2 plays a significant role in cytokine signaling from IL-dependent type I interferons and IL-12 and IL-23. Thus, the idea of dual inhibition of JAK1 and TYK2 with selectivity over JAK2/JAK3 (minimizing erythropoietin signaling pathway) was envisioned.87
To identify JAK1/TYK2 dual inhibitors, a series of constrained 2,4-diamino pyrimidines were investigated (compounds 33a-d) (Scheme 12).88 A piperazine ring was found optimal at the 4-position of 2,4-diamino pyrimidine and the ring could tolerate a methylene bridge, which generated two enantiomers. The preferential S,S-enantiomer 33a exhibited better activity (TYK2 IC50 = 0.023 μM) compared to R,R-enantiomer 33b (TYK2 IC50 = 2.690 μM). The SAR was expanded to include more bridged diamines, which provided compound 34 with a 3,8-diazabicyclo[3.2.1]octane ring, N-methyl pyrazole, enhanced potency, and high LipE. Next, keeping the N-methyl pyrazole constant, a series of acylated derivatives, amides, and ureas were screened and uncovered that 1,1-difluoro-cyclopropane was very effective at this position (compounds 35a-c). The eutomer S-1,1-difluoro cyclopropane 36a (PF-06700841) provided a more balanced, potent JAK1/TYK2 profile (TYK2 and JAK1 IC50 = 0.023 and 0.017 μM) compared to the R-enantiomer 36b (TYK2 and JAK1 IC50 = 0.702 μM and 0.842 μM).
Scheme 12.
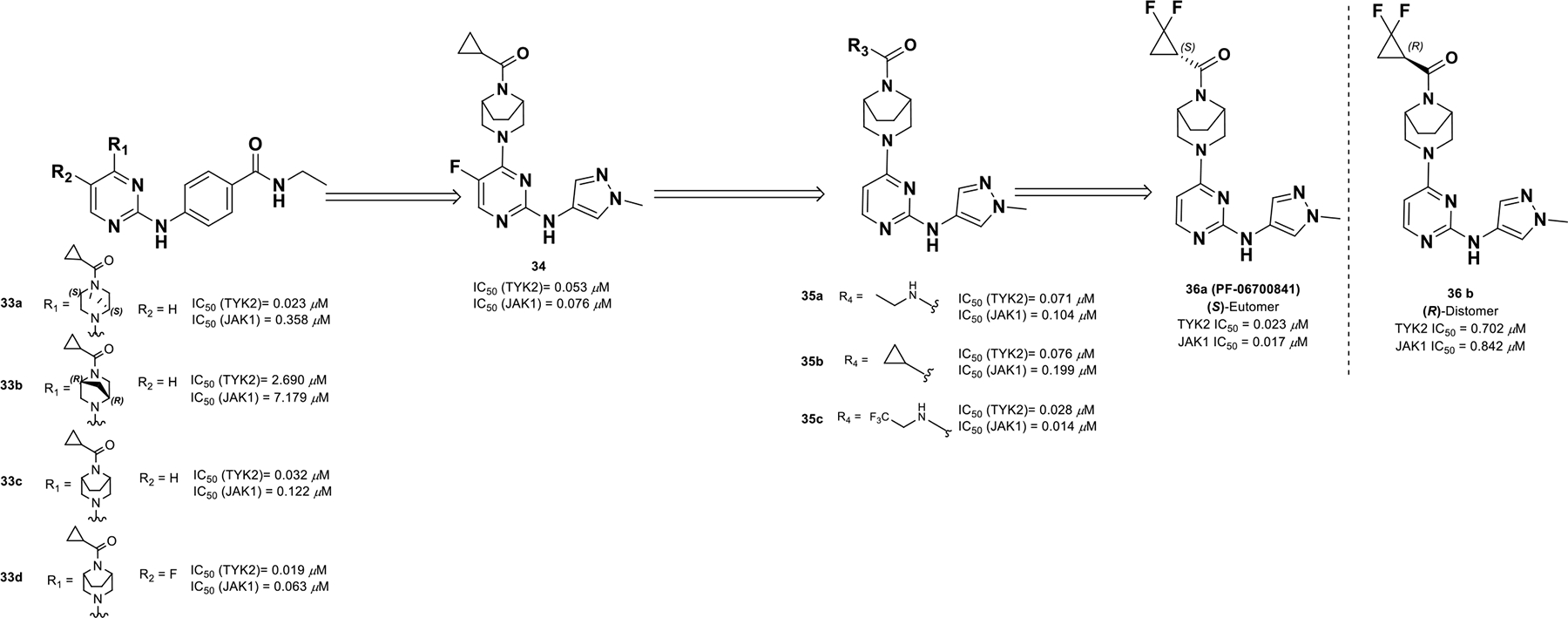
Discovery of compound 36a profile.
The co-crystal structure of compound 36a with TYK2 shows the formation of two hydrogen bonds at the hinge region, whereas the ethylene bridge of the [3.2.1]-diazabicycle protrudes into a hydrophobic pocket between Gly1040 and Leu1030. The difluoromethylene of the cyclopropyl amide orients towards the P-loop of the ATP binding site composed of Gly909 and Lys910 (Figure 9). With the (R)-distomer (36b), the difluoromethylene group orients into a negative electrostatic-potential region formed by side chain residues Asp1041 and Asn1028. This explains the remarkable 40-fold potency difference between the S and R isomers with respect to TYK2 binding. Compound 36a also binds JAK1 in a similar manner, justifying the similar potencies against both enzymes.
Figure 9:
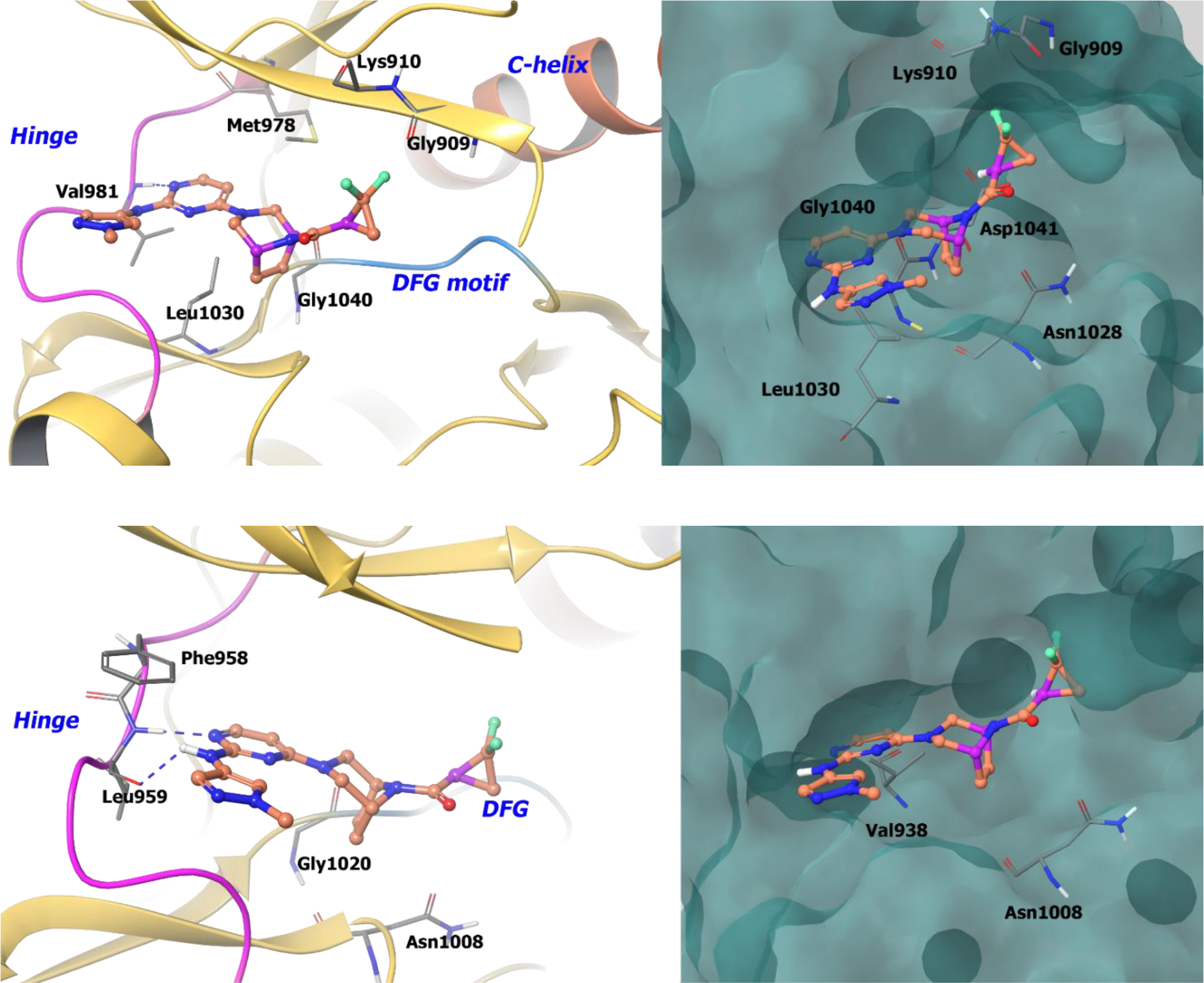
A. The co-crystal structure of compound 36a bound to TYK2 (PDB 6DBM, 2.36 Å). Left Panel: Interaction of compound 36a with TYK2, the protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Right panel: The binding site of compound 36a in TYK2. The surface of the protein is depicted in blue. B. The co-crystal structure of compound 36a bound to JAK1 (PDB 6DBN, 2.48 Å). Left Panel: Interaction of compound 36a with JAK1. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Right panel: The compound 36a binding site in JAK1. The surface of the protein is depicted in blue. Compound 36a atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
Compound 36a was progressed through a variety of pre-clinical testing and exhibited an excellent off-target polypharmacology profile and an ADME profile consistent with once-daily dosing. Compound 36a is currently in phase II clinical trials for psoriasis (NCT02969018). Clearly, the application of chirality in the development of compound 36a enabled enhanced target selectivity and improved drug properties.
Bruton’s tyrosine kinase (BTK) is another nRTK and plays a key role in BCR-mediated activation and development. Phosphorylated BTK activates downstream NFAT and NFĸB pathways, resulting in proliferation, cytokine production, and expression of stimulatory molecules. As a result, inhibiting BTK regulates BCR signaling and Fcγ and Fcε pathways, thereby ameliorating autoimmune diseases such as rheumatoid arthritis (RA), lupus, and multiple sclerosis etc.89–91
After much effort in identifying a reversible BTK inhibitor, compound 37 was discovered as a promising lead.92 SAR exploration led to compound 38 (BMS-935177) (Scheme 13) with reasonable potency against BTK (IC50 = 0.003 μM),93 but the compound presented with a very narrow therapeutic window (< 20-fold). Researchers believed the toxicity was a result of four inter-converting atropisomers with different selectivity and toxicity profiles. Hence, it was envisioned that trapping a single atropisomer, with simultaneous improvement in potency and selectivity, could enhance drug safety. For this reason, another carbonyl was introduced into the quinazolinone ring generating derivative 39 that provided better pharmacokinetic properties. The two enantiomers were separated and the (S)-isomer was found to be more potent against human whole blood cells (BTK IC50 = 0.0023 ± 0.0012 μM). Next, a second point of chirality was introduced by locking carbazole C4 through the addition of a C3 chloro substituent, which was found to be 1900-fold selective for BTK over JAK2 with improved potency. Anticipating improvement in kinase selectivity and the overall PK profile, SARs were expanded to an additional point of chirality on tetrahydrocarbazoles 40.
Scheme 13.
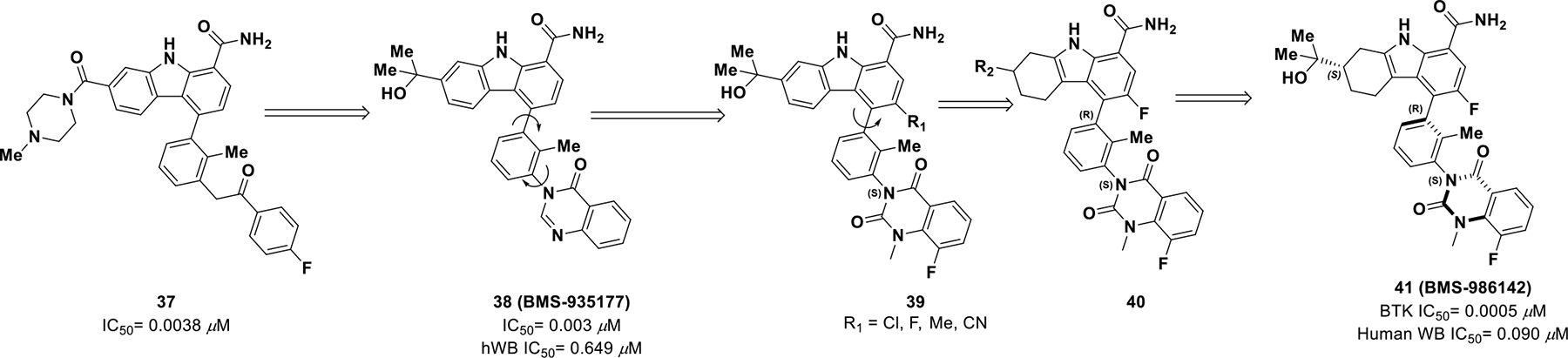
Drug discovery of compound 41
Ultimately, adding (S)-dimethyl carbinol on tetrahydrocarbazoles and locking the carbazole C4 in the bioactive (R)-analogue by a fluoro substituent exhibited good potency (BTK IC50 = 0.0005 μM and hWB IC50 = 0.090 μM) and pharmacokinetic properties (AUC = 39 μM*h). The optimized analogue 41 (BMS-986142) inhibited only five TEC family kinases with less than 100-fold selectivity for BTK, exhibited a good pharmacokinetic profile in mice (with <4% formation of the main des-methylquinazoline dione metabolite) and a clean liability profile.
Compound 41 was co-crystalized with BTK94 and revealed that the tetrahydrocarbazole NH and the carboxamide carbonyl form two hydrogen bonds at the hinge region (Figure 10). The C2 dimethyl carbinol is directed towards the solvent front and does not exhibit any specific interaction. The (R)-configuration of the C5 phenyl linker projects the methyl group into a hydrophobic pocket adjacent to Cys481. The orthogonal nature of the phenyl linker orients the (S)-quinazolinedione into the active site. In the (S)-configuration, the methyl group would be pushed out of the hydrophobic pocket causing an increase in entropy. Compound 41 demonstrated a desirable safety profile in multiple species and completed its phase II trials recently for moderate to severe rheumatoid arthritis (NCT02638948).The drug discovery campaign of compound 41 illustrates the use of axial chirality leading to a single, stable atropisomeric compound with enhanced potency and selectivity profiles.
Figure 10.
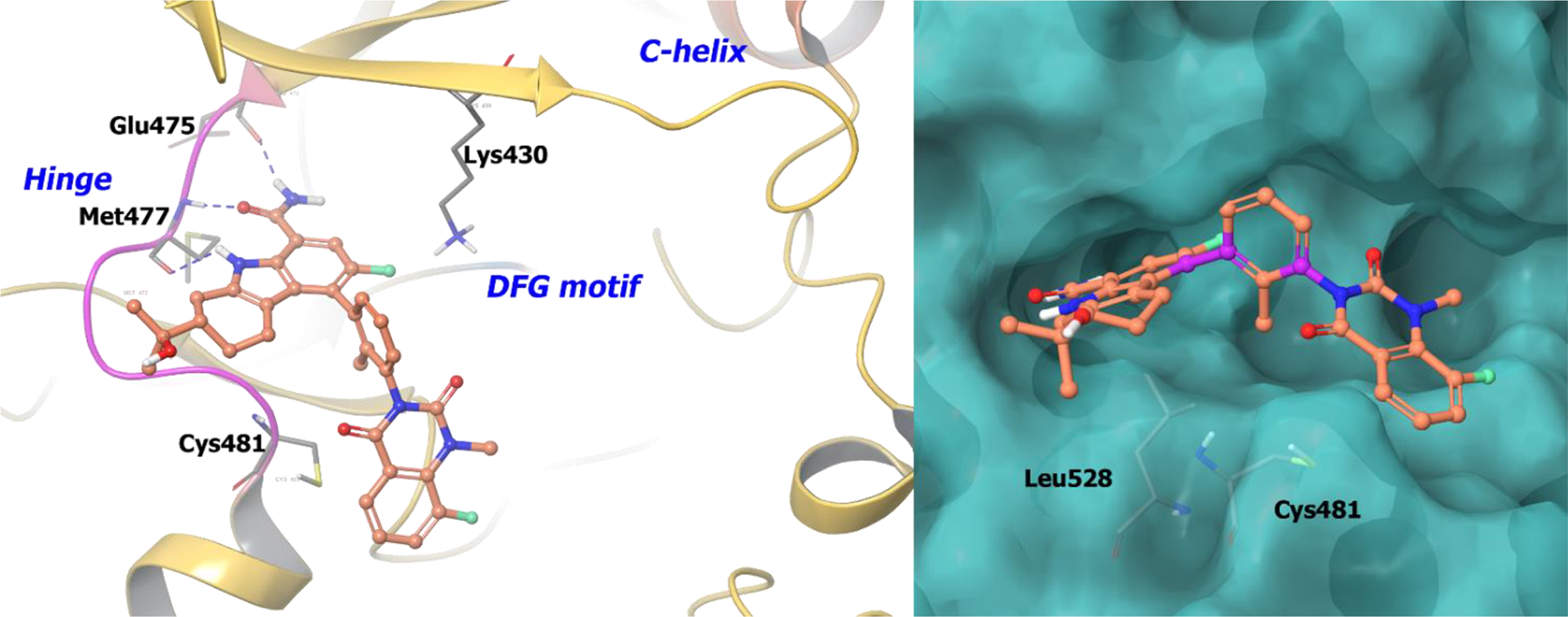
The co-crystal structure of compound 41 bound to BTK (PDB 5T18, 1.5 Å). Left Panel: Interaction of compound 41 with the protein. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Right panel: The binding site of compound 41, The surface of the protein is depicted in blue. compound 41 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
With continued interest in discovering BTK inhibitors, compound 42 (CGI-1746)95 was developed as a highly selective and potent BTK inhibitor with excellent selectivity but suffered from poor ADME properties. Pharmacokinetic optimization of compound 42 focused on two main regions (Scheme 14). 96 The first region was analogued with moieties to engage the solvent exposed pocket (Region 1, H2 Pocket). The second region was explored by varying substitution on the phenyl ring or replacing it with alternate arenes or heterocycles (Region 2, H3 Pocket). Adding polarity to the phenyl ring by substituting different heterocyclic arenes caused a decrease in binding affinity. Finally, a hydrobenzothiophene substituent at the H3 pocket and dimethyl-3-oxopiperazin-2-yl group on the H2 region optimized potency and pharmacokinetic properties. The (R)-isomer of substituent 44 (IC50 = 0.006 μM; CL = 4.4 mL/min/Kg; F = 35%) provided a 3-fold improvement in potency and clearance from its enantiomeric (S)-isomer (IC50 = 0.02 μM; CL = 11 mL/min/Kg; F = 41%). Additionally, compound 44 (GDC-0834) was found to have excellent selectivity against a panel of 331 kinases and minimal off-target receptor activity.
Scheme 14.
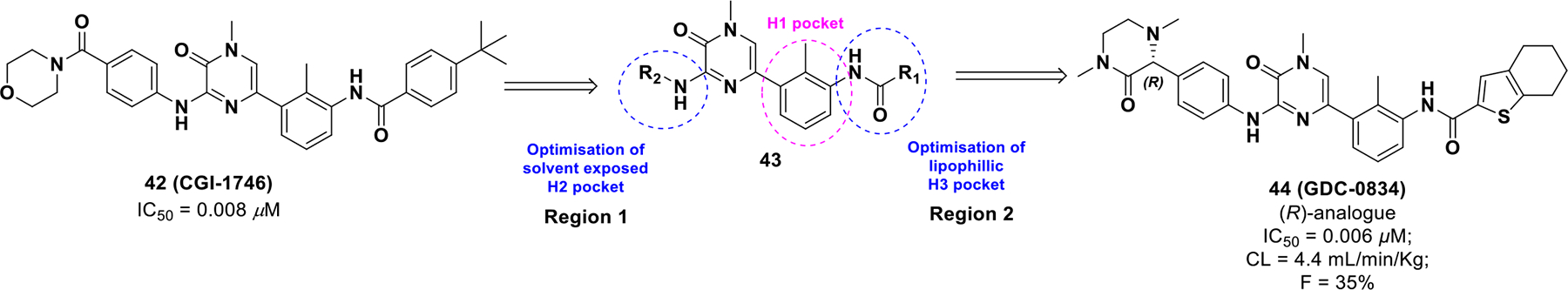
Drug discovery of compound 44
The co-crystal structure of compound 44 in complex with BTK revealed that the tetrahydrobenzothiophene moiety engages the hydrophobic pocket created by Leu542, Val546, Ser543, and Tyr551 of the activation loop (Figure 11). The amide linking the central aryl to the tetrahydro benzothiophene forms a hydrogen bond to Lys430. The pyrazinone binds Met477 at the hinge while the dimethyl-3-oxopiperazin-2-yl extends into the solvent front. In the discovery of compound 44, chirality was used to improve pharmacokinetic properties and receptor potency.
Figure 11.
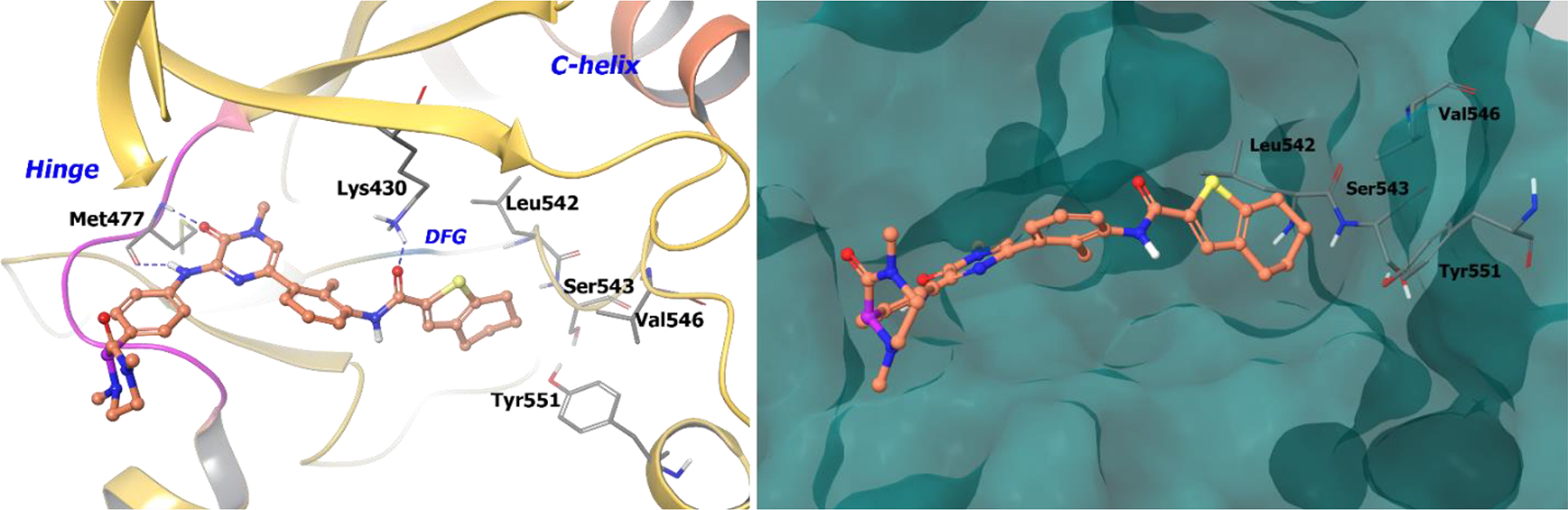
The co-crystal structure of compound 44 bound to BTK (PDB 4OTF, 1.95 Å). Left Panel: Interaction of compound 44 with the protein. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Right panel: The binding site of compound 44 in BTK. The surface of the protein is depicted in blue. compound 44 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
2.3. Serine/Threonine Kinase Inhibitors
The human kinome consist of more than 500 human kinases of which 300 are serine/threonine kinases (STK).97 As with any kinase, the prime function of an STK is to phosphorylate an amino acid hydroxy group (serine or threonine), thereby turning on or off a pathway. STKs are regulators of cell proliferation, apoptosis, cell differentiation, and embryonic development, and have relevance in numerous forms of disease. STKs can be receptors, which are categorized as type I (including activin like receptors [ALKs], TGFβR1 [transforming growth factor beta receptor 1]), type II (ActR2, ActR2B, MISR2, BMPR1A, TGFβR2), and type III (TGFβR3), or solubilized, cytosolic proteins such as protein kinase A, B (Akt kinase), and C.98 Other major classes of STKs include second messenger-dependent protein kinases (cAMP kinase, cGMP kinase, Ca2+/calmodulin kinase, Protein kinase C), MAP kinases (ERKs, JNKs or SAPKs), MAPKinase-regulating kinases (MEKs, SEKs, Raf, MEK kinases), CDKs, CDK-regulating kinases (CAK, CAK kinase), GRKs, RSKs and casein kinases.99 STKs also include dual specificity kinases that act on both tyrosine as well as serine/threonine residues, such as MEKs. Additionally, STKs are activated in signal transduction pathways triggered by many RTKs and other receptors.100 Another class of serine-threonine kinases include cyclin-dependent kinases (CDKs), known for cell cycle progression. Hence, CDK inhibitors exhibit therapeutic relevance for several diseases including cancer, diabetes, renal, neurodegenerative, and infectious diseases.
2.3.1. FDA-Orphan Drug Designation
CDK inhibitors have undergone clinical development with no disclosed explanation for incorporating chirality into the clinical candidate. Alvocidib (45), a flavonoid CDK9 inhibitor, was granted with FDA orphan drug designation in 2014 for AML (acute myeloid leukemia).101 Among other CDK inhibitors, seliciclib (46, R-roscovitine, CYCLACEL®) is a CDK-2,7,9 inhibitor under phase II development102 whereas dinaciclib (47), a CDK-2,7,9 inhibitor, is in phase II clinical trials for advanced breast cancer and NSCLC (NCT00732810).103
Cobimetinib (48, COTELLIC®, GDC-0973, XL518), a MEK1/2 inhibitor, obtained orphan drug status for malignant melanoma exhibiting BRAFV600 mutations in 2014.104 Compound 48 is a carboxamide-based, selective MEK inhibitor (100-fold selective over 100 other serine–threonine and tyrosine kinases)105 and exhibits higher efficacy in BRAF mutated tumors because of its strong MEK inhibition.12
2.3.2. Additional STK Inhibitors under Clinical Development
Lee et al. (2017)106 initiated a drug discovery campaign against IRAK4 (Interleukin-1-receptor associated kinase 4) for the treatment of inflammation. IRAK4 is activated by the cytokine receptor interleukin-1-receptor in response to binding of interleukin-1 (IL-1). In addition, IRAK4 is involved in the innate and adaptive immune system and is expressed in T- and B-lymphocytes. Therefore, IRAK4 is an attractive target for treating inflammatory and autoimmune diseases. To initiate development, Lee et al. completed a fragment based screening using the Pfizer Global Fragment Initiative library. 10 best hits were co-crystallized with IRAK4. Co-crystal structure analysis suggests that exploration of the three-dimensional space within the ATP pocket could simultaneously improve potency, pharmacological and pharmaceutical properties. Binding of the carboxamide 49 to IRAK4 suggested switching the core to a more polar heterocyclic system, such as quinoline or isoquinoline, would improve potency. Compound 50 was generated and the resulting protein-ligand complex (Scheme 16) suggested interactions at the base of the binding pocket could be optimized. The piperidine was replaced with a five-membered lactam ring generating compound 51. The (S)-isomer (51a) was found to be significantly more potent than the (R)-isomer (51b). The increase in potency of the (S)-isomer stems from its stereochemistry that enables the lactam ring to efficiently hydrogen bond to Ala315 and Asn316. In addition, the carbonyl accepts hydrogen bonds from Ser328 and an interaction between the isoquinoline nitrogen and Asp272 via a water molecule.
Scheme 16.
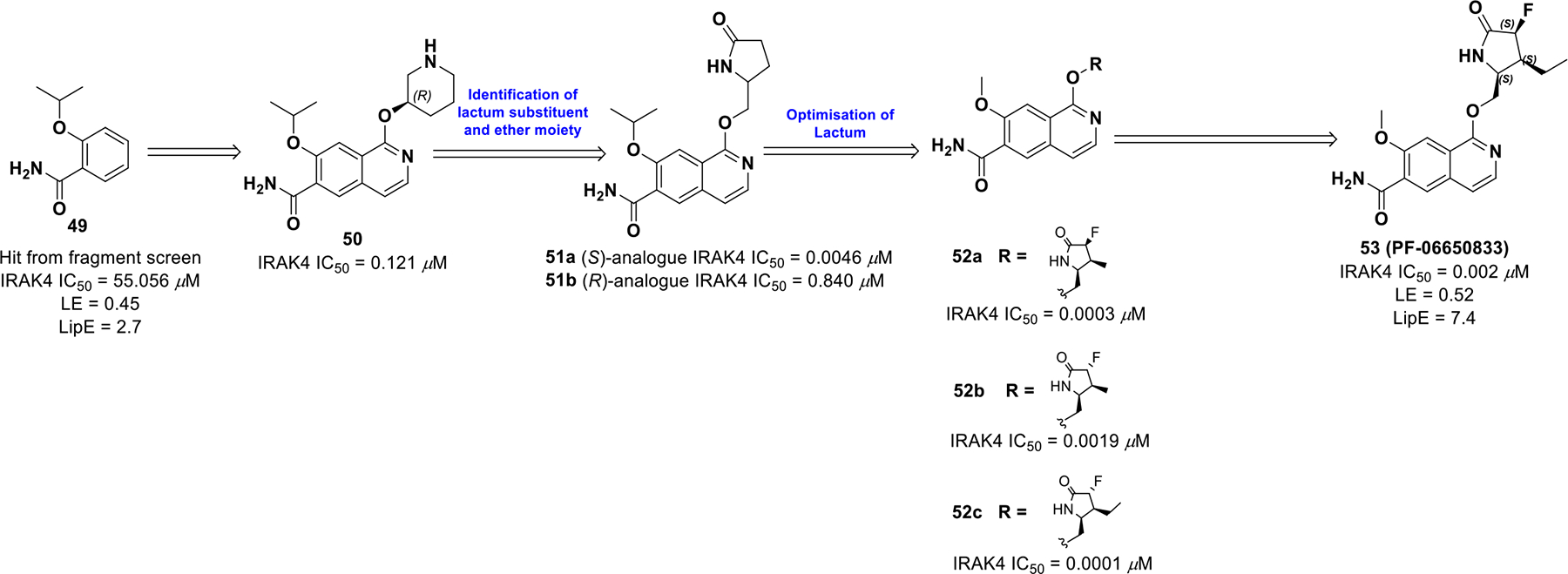
Drug discovery of compound 53
Compound 51a achieved a good pharmacokinetic profile (CL = 23 L/hr/Kg, T1/2 = 1.2 h, F = 57% in rat), which suggested oral administration was feasible for dosing. Unfortunately, allometric scaling to determine the human dose led to termination of the compound. To improve pharmacokinetics, compound 51 was further modified at the lactam position, thereby reducing the planarity of the molecule. A fluoro substituent with syn stereochemistry (52a) to the ether linker provided better potency than the anti-isomer (52b). However, α-syn fluorine substituent and a β-3-ethyl substituent led to compound 53 (PF-0665083) with improved potency (IRAK4 IC50 = 0.001 μM, PBMC IC50 = 0.0024 μM) and optimized ADME properties (LE = 0.52, LipE = 7.4). The potency rise can be attributed to enhanced hydrogen bonding capability of the lactam ring conferred by appropriate positioning of the fluoro substituent.
The co-crystal structure of 53 bound to IRAK4 is shown in Figure 12, illustrating key interactions. The terminal, primary amide group forms two hydrogen bonds with Val263 and Met265. The cyclic lactam is involved in a hydrogen bond with the bottom pocket of IRAK4. The (S)-stereochemistry orients the lactam amide into a favorable position to take part in hydrogen bonding with Ala315. Moreover, the ethyl group on the cyclic amide is encapsulated in a lipophilic pocket formed by Gly195, Lys213 and Val261. Hence, introduction of 3 points of chirality into 53 reduced planarity, which improved binding site interactions, solubility, and pharmacokinetics.
Figure 12.
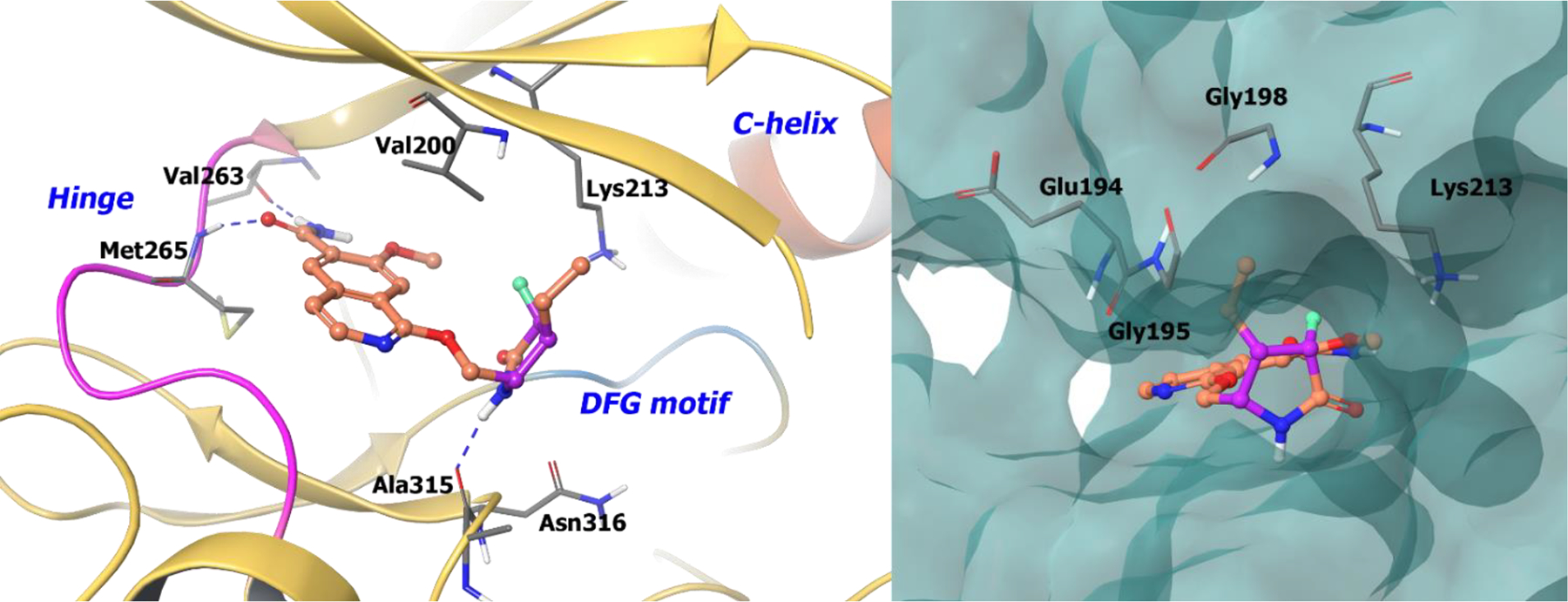
The co-crystal structure of compound 53 bound to IRAK4 (PDB 5UIU, 2.02 Å). Left Panel: Interaction of compound 53 with the protein. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Right panel: The binding site of compound 53 in IRAK4. The surface of the protein is depicted in blue. compound 53 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
Another serine/threonine kinase closely related to the IRAK family is receptor interacting protein 1 (RIP1). RIP1 is involved in cell death/growth signaling circuits and is affected by exposure to several stress signals such as inflammatory cytokines, infections, and genotoxic stress.107 Functions of RIP1 include T-cell homeostasis, activation of NF-κB, and activation of mitogen activated protein kinases (MAPKs), such as p38 MAPK, JNK, and ERK.108 It was also found that RIP1 plays a significant role in various downstream pathways of the death receptors TNFR1, FasL, TRAIL, and Toll-like receptors. Thus, obstructing this pathway may be therapeutically relevant for various inflammatory diseases.109
Initial RIP1 inhibitors suffered from poor pharmacodynamics and pharmacokinetic properties. To identify a more viable RIP1 inhibitor, a GSK DNA-encoded library was screened via high-throughput screening, which uncovered compound 54a (GSK-481), a more selective RIP1 benzoxazepinone pharmacophore. The binding interactions of the (S)-analogue 54a with the protein is of significance as the (R)-analogue 54b is not active. Therefore, chirality is integral for RIP1 activity.110
Replacing the heteroatom in the benzoxepinone with -N, -S, -CH2, -NMe provided high RIP1 in vitro potency (Scheme 17). Altering the size of the benzoxazepine or removal of the benzo function yielded inactive analogues as the benzoxazepinone moiety fits tightly into the RIP1 pocket. Heteroaryl substitutions on the aryl ring at 7,8-positions suffered from low solubility. For example, oxazole (56a) and imidazole (56e) were found to be more active than their isomers, whereas thiazole (56b) exhibited lower activity. Also, N-benzyl-1,2,3-triazole (56g) displayed better rat oral exposure than its 1,2,4-triazole (56f) analogue, but, the 3-benzyl-1,2,4-triazole isomer provided the best combination of in vitro potency, lipophilicity (logD 3.8), and rat oral exposure (AUC0−∞ 2.3 μg/h/mL at a dosage of 2 mg/kg). All other tetrazole, phenyl, or pyridine analogues suffered from high lipophilicity and low solubility. Thus, compound 57 (GSK-2982772) was progressed for further development and has entered phase IIa clinical trials for ulcerative colitis. (NCT02903966)
Scheme 17.
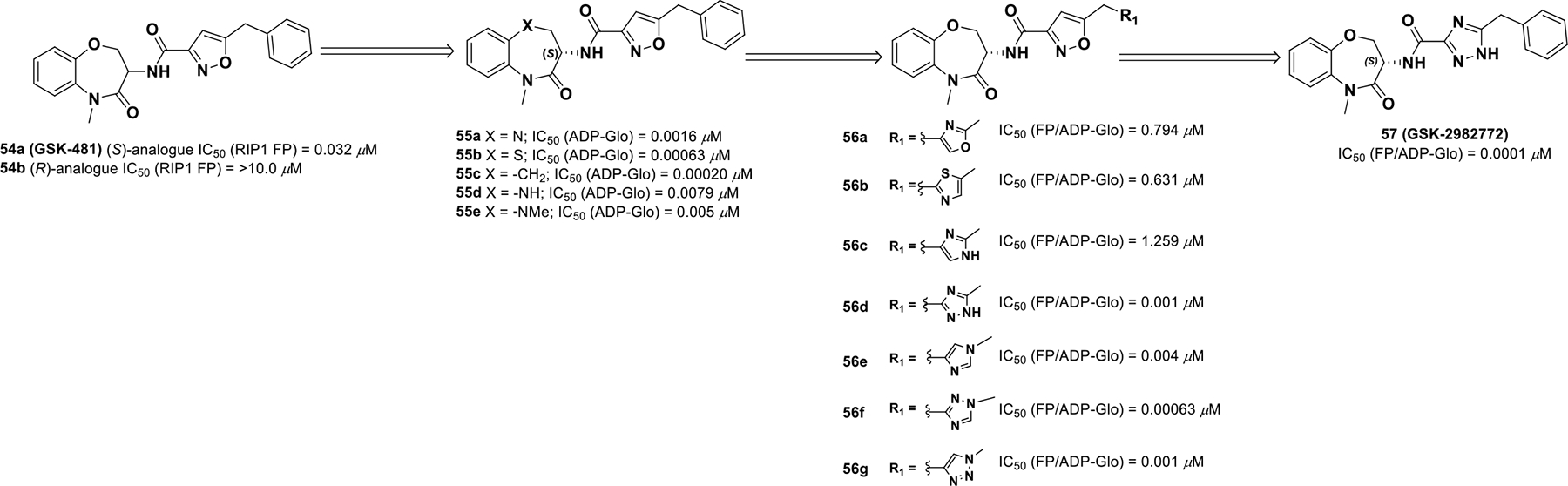
Drug discovery of compound 57
The co-crystal structure of benzoxazepinone 57 bound to RIP1 demonstrates 111 the binding of the seven-membered ring to an allosteric site between the N-terminal and C-terminal domains without any interaction at the hinge region (Figure 13). The triazole and benzyl groups occupy an allosteric lipophilic pocket at the back of the ATP binding site. The benzoxazepinone ring resides in a pocket formed by two β-strands defined by Leu90-Val91-Met92 and Ile43-Met44-Lys45. The amide carbonyl linker makes a direct hydrogen-bond with the backbone of Asp156. The (S)-stereochemistry of the molecule allows the benzyl triazole amide access into a hydrophobic tunnel, while the (R)-analogue is not able to access this tunnel and is virtually inactive (> 10,000-fold potency). Hence, any change in chirality or conformation of the seven membered ring is not tolerated.
Figure 13.
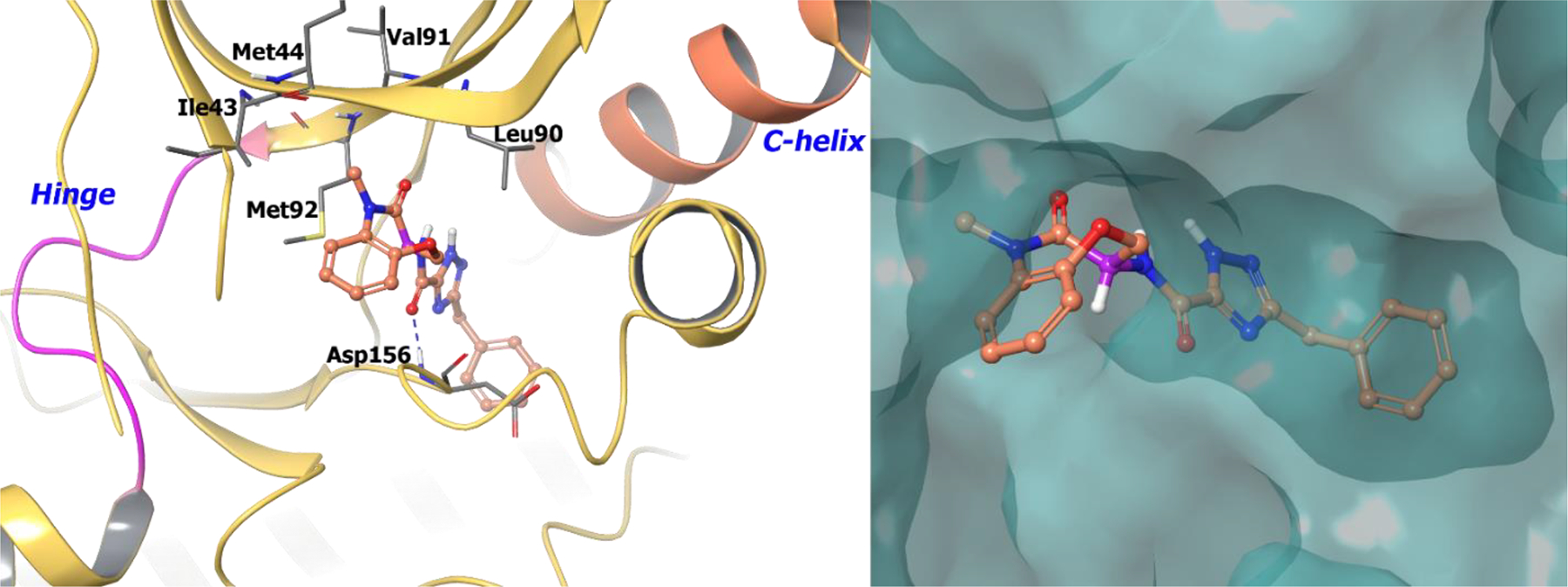
The co-crystal structure of compound 57 bound to RIP1 (PDB 5TX5, 2.56 Å). Left Panel: Interaction of compound 61 with the protein. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Right panel: The binding site of compound 57 in RIP1, The protein surface is depicted in blue. Compound 57 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
Polo-like kinases (PLKs), which are serine/threonine kinases, are well-validated cell cycle targets with vital roles in mitosis, spindle formation, chromosome segregation, and cytokinesis.112 They include PLK1, PLK2 (SNK), PLK3 (PRK/FNK) and PLK4 (SAK), among which PLK1 is the most studied in oncology with an established correlation between PLK1 expression and cancer prognosis.113 As such, there have been numerous campaigns targeting PLKs for antineoplastic drug development.
The PLK1 inhibitor 58 exhibited strong activity against PLK1, but was also active on PLK3 and lacked desirable drug properties warranting further clinical studies. Compound 58 was further optimized to improve potency and pharmacokinetic properties (Scheme 18). Incorporating a methyl group at the benzylic carbon displayed an improvement in cell potency (40-fold). The (R)-analogue (59a) exhibited approximately 40-fold improvement in growth inhibition of HCT116 (colorectal carcinoma) over the (S)-analogue (59b).114 Further, improvement in solubility was considered by incorporating of more polar groups. By analyzing the binding of 59a in the active site of PLK1, it was found that the 6-position of benzimidazole orients towards the solvent and could tolerate polar groups.
Scheme 18.
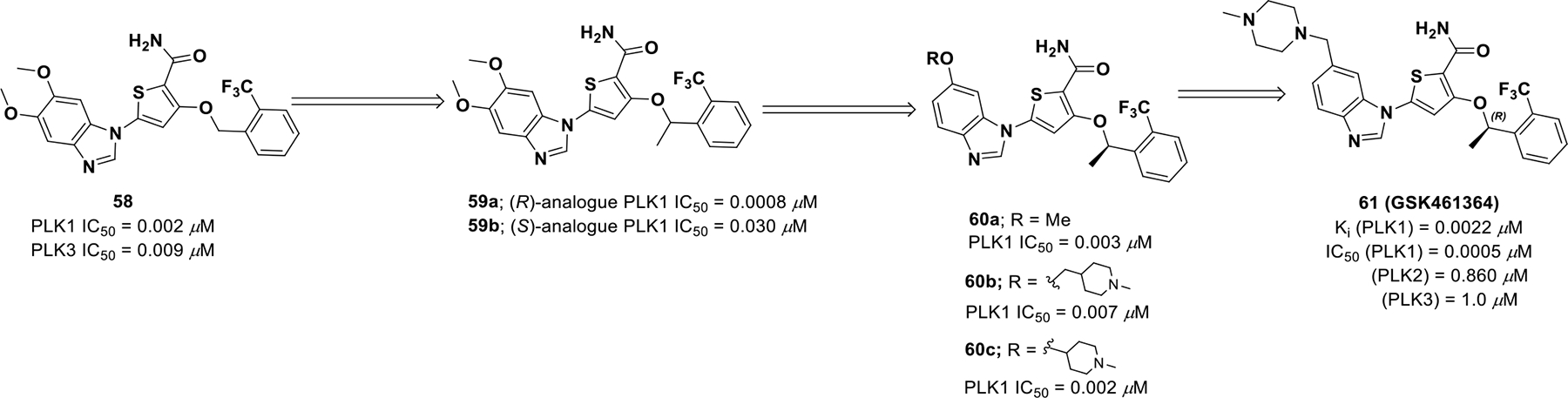
Drug discovery of compound 61
Methyl ether analogs at the 6-position of benzimidazole (60a) displayed high protein binding but poor solubility. Potency was further refined by eliminating the methylene spacer between the oxygen atom and the piperidine ring (60b). The basic amine analogs displayed an excellent combination of potency, solubility, and reduced protein binding with selectivity over PLK2/PLK3. Homology modeling of compound 60c bound to the ATP-binding site of PLK1 displayed a hydrogen bond interaction between Glu140 and the piperidine nitrogen. Whereas in PLK3 and PLK2, Glu140 is replaced by a histidine, whose interaction with the charged amine is not favorable, resulting in reduced potency. Subsequently, more cyclic amines were screened to obtain suitable potency, selectivity, and solubility, which resulted in the discovery of compound 61 (GSK461364). Compound 61 is currently under evaluation in phase I clinical trials for non-Hodgkin’s lymphoma (NCT00536835).
2.3. Lipid kinase Inhibitors
2.3.1. FDA-Approved Lipid Kinase Drugs
Lipid kinases are responsible for phosphorylation of lipids in the cell, which in turn change the reactivity and localization of lipids causing signal transduction.115 For instance, Sphingosine kinase, a lipid kinase, catalyzes the conversion of sphingosine to sphingosine-1-phosphate (S1P) whereas phosphatidylinositol kinases generate phosphatidylinositol 3,4-bisphosphate (PI(3,4)P2), phosphatidylinositol 3,4,5-trisphosphate (PIP3), and phosphatidylinositol 3-phosphate (PI3P).116 This family of kinases include phosphoinositide 3-kinases (PI3Ks), phosphatidylinositol-4-phosphate 3-kinase, and phosphatidylinositol-4,5-bisphosphate 3-kinase.117 PI3Ks are lipid kinases that phosphorylate the 3’ position of inositol in phosphatidylinositol, which functions as a secondary messenger that regulates proliferation, motility, and differentiation. PI3Ks are further classified into three classes, among which class I plays a critical role in regulating PIP3 levels in the cell. PIP3 is an essential cellular mediator involved in activation of downstream signaling cascades, which is generated through the phosphorylation of PIP2.118 A lack of regulation in PIP3 levels, such as impairment in PTEN function, is found in numerous, aggressive cancers.119
A. Idelalisib (Jul. 2014, ZYDELIG®, Gilead)
In July 2014, idelalisib (62, GS-1101, CAL-101, IC489666) received FDA approval for treatment of relapsed chronic lymphocytic leukemia, follicular B-cell non-Hodgkin’s lymphoma, and small lymphocytic lymphoma.120 Idelalisib is the first-in-class small molecule PI3K inhibitor, selectively targeting PI3Kδ. PI3Kδ is a class I PI3K comprised of p110δ as a catalytic subunit and p85 as a regulatory subunit. PI3Kδ expression is restricted to leukocytes making it an excellent therapeutic target to selectively impair the PI3K-AKT pathway in hematopoietic cells.121–123 Idelalisib is similar to 63 (IC87114), which was discovered by the ICOS corporation and has been studied comprehensively as a PI3Kδ inhibitor.124 Idelalisib has an IC50 for PI3Kδ of 0.019 μM and exhibits better potency and metabolic stability than compound 63 (IC87114) (Scheme 19).125,126 Further, idelalisib is selective for PI3Kδ with relatively weak inhibition of PI3Kα, PI3Kβ, and PI3Kγ isoforms (IC50 = 8.6, 4.0, and 2.10 μM, respectively).127,128
Scheme 19.
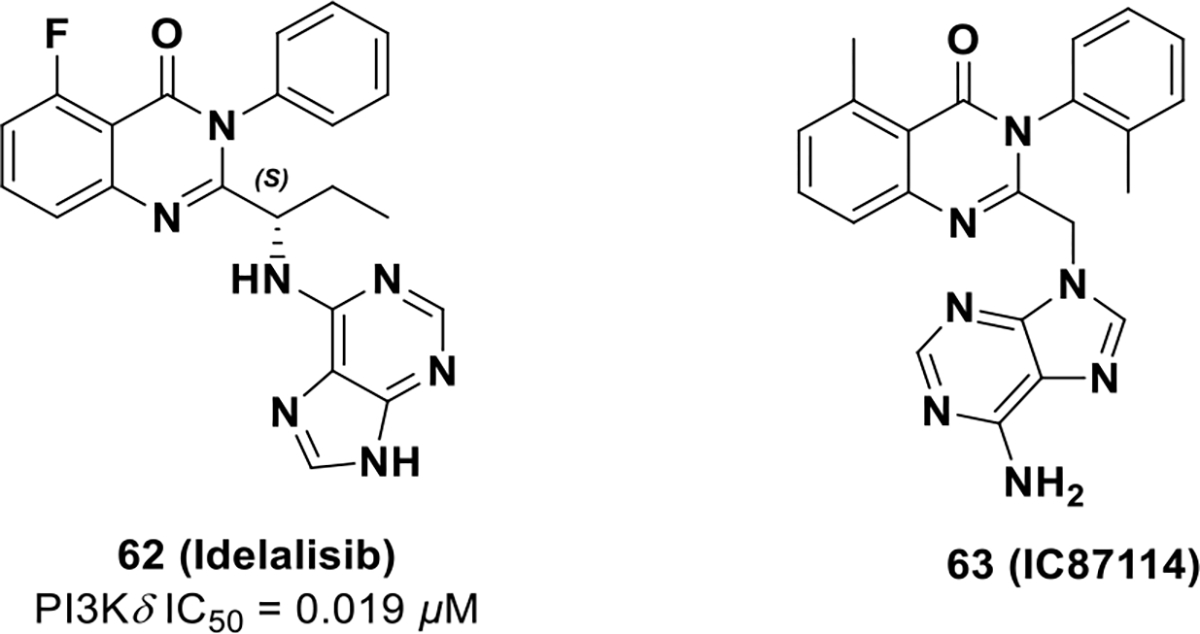
FDA approved drug 62 and its chemotype 63
Idelalisib interacts with PI3Kδ hinge residues Val828 and Glu826 via the N3 and N9 nitrogens of the purine group, respectively (Figure 14). The (S)-chiral carbon directs the ethyl group of idelalisib into the hydrophobic pocket of PI3Kδ formed by Ile910, Met900, and Met752. The ATP-binding pocket of PI3Kδ undergoes a conformational change (from the apo-enzyme PDB: 2WXR) to accommodate the fluoro quinazolinone ring in a hydrophobic pocket (the specificity pocket) made up of Met752, Trp760, and Lys708 on the top of the active site.129,130 It is hypothesized the energy required to open the specificity pocket defines the selectivity of idelalisib for PI3Kδ over other isoforms. Further, because of geometric constraints in the selectivity pocket, the R-enantiomer of idelalisib does not fit properly in the PI3Kδ active site. The chiral attributes of idelalisib, as well as low energy binding for PI3Kδ, enable selectivity in the PI3K family. The selectivity, in turn, improves toxicity profiles and drug properties of idelalisib.
Figure 14.
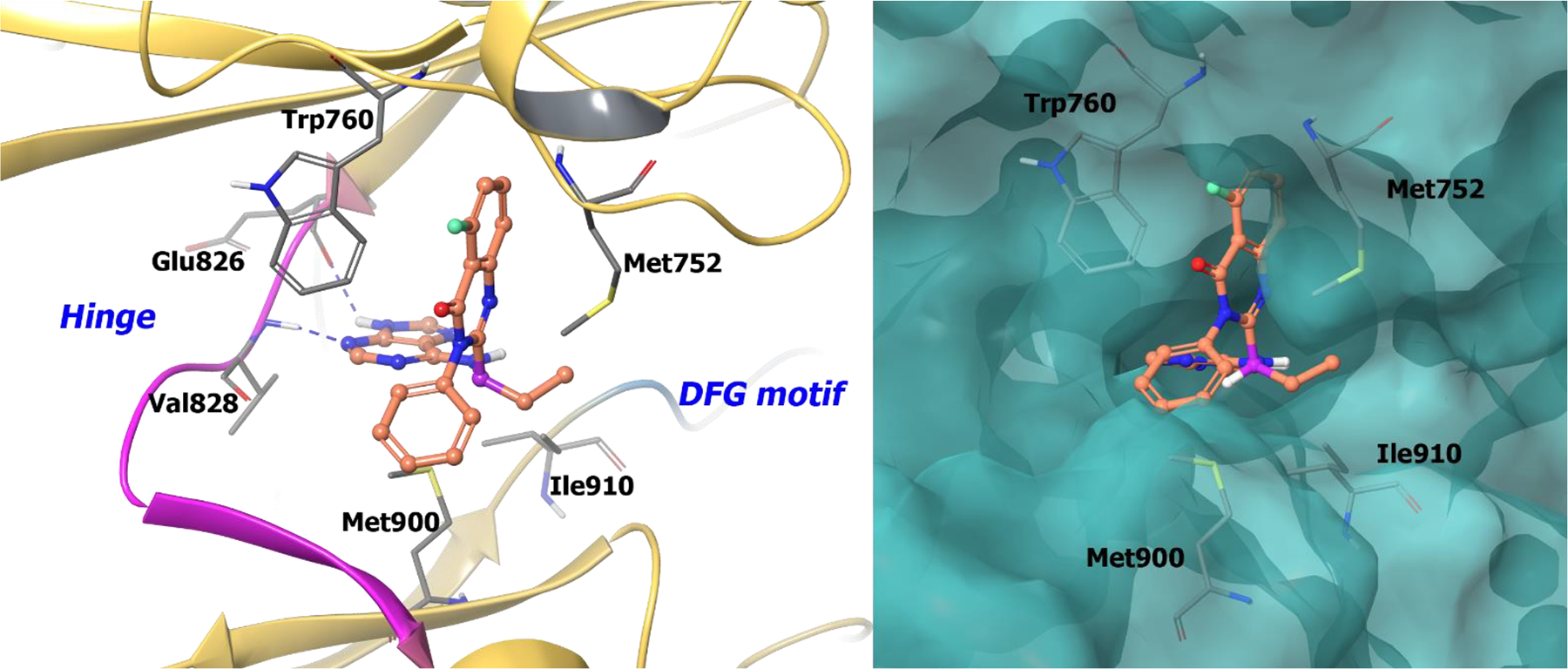
The co-crystal structure of compound 62 bound to PI3Kδ (PDB 4XE0, 2.35 Å). Left Panel: Interaction of compound 62 with the protein. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Right panel: The binding site of compound 62 in PI3Kδ. The protein surface is depicted in blue. Compound 62 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
In 2012, Amgen disclosed compound 64, an inhibitor of Class I PI3Ks with activity in biochemical (Ki values of 0.012, 0.005, 0.002 and 0.004 μM for PI3Kα, PI3Kβ, PI3Kδ, and PI3Kγ, respectively) and cellular (U87 MG IC50 = 0.016 μM) assays (Scheme 20). The compound exhibited promising in vivo activity, but exhibited rapid clearance (CL = 2.5 L/h/kg) with a very short mean residence time (MRT = 1.6 h for both); hence, it did not have appropriate drug-like properties.131 Upon further investigation, it was found that the metabolites were the product of oxidative metabolism, including O-demethylation (65a) and benzylic oxidation (65b). To reduce metabolism of 64, further optimization was completed, which resulted in compound 67a (AMG 511).131
Scheme 20.
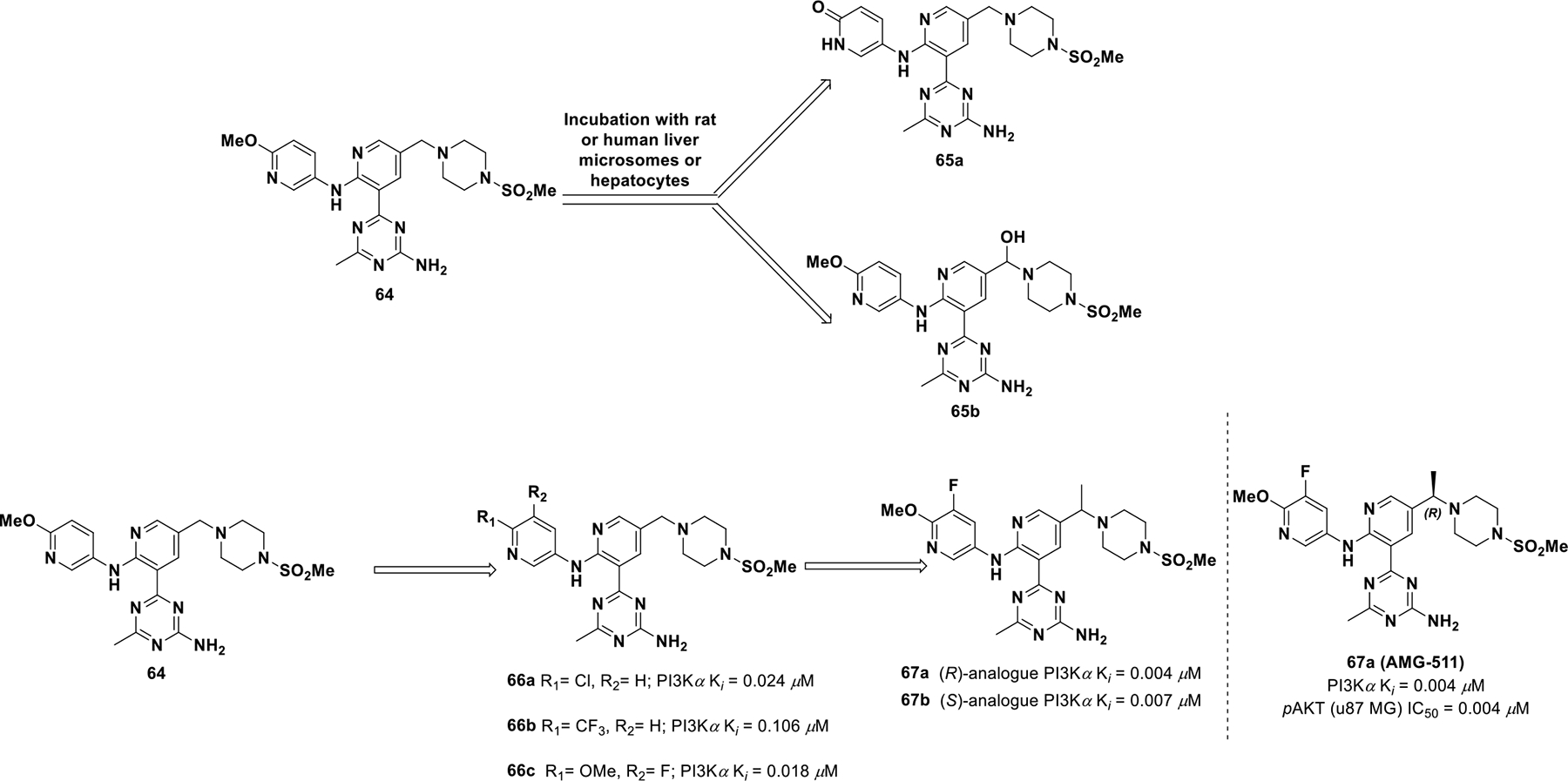
Drug discovery of compound 67a
To improve drug properties of 64, Norman et al. explored altering electron density of the terminal pyridine ring.131 Introducing a fluorine atom at the C3 position of the pyridine ring furnished compound 66c (Ki for PI3Kδ = 0.006 μM and Ki for PI3Kα, PI3Kβ, PI3Kγ are 0.018, 0.008, 0.021 μM respectively) and reduced O-demethylation but did not improve clearance. Introduction of a benzylic methyl decreased PI3Kδ selectivity either with or without substitution at the piperazine position. There was no significant stereo-chemical preference for PI3K inhibition, but pharmacokinetic properties improved with one isomer. When exploring rat microsomal stability, the (R)-Methyl benzylic isomer (67a; IC50 = 0.004 μM for U87 MG) was more stable than the (S)-Methylpiperizine isomer (67b; IC50 = 0.007 μM for U87 MG). Therefore, compound 67a was selected for clinical investigation.
The co-crystal structure of 67a bound to PI3Kγ showed the aminotriazine of compound 67a forms two hydrogen bonds with Val882 at the hinge region (Figure 15). The methyl substituent on the triazine occupies a small hydrophobic pocket found only in PI3Ks (near Tyr867), which is responsible for kinase selectivity. Both the methoxy oxygen and the 3-fluoro substituent exhibit a favorable interaction with Lys833 within the affinity pocket. The nitrogen in the pyridine central core forms a network of water-mediated hydrogen bonds with Asp964. The benzylic (R)-methyl group efficiently occupies a hydrophobic pocket at the floor of the enzyme formed by Thr887, Ile963, and Asp950. This directs the methylsulfonamide piperidine moiety to engage in hydrogen bonds with Lys802 and Ala805.
Figure 15.
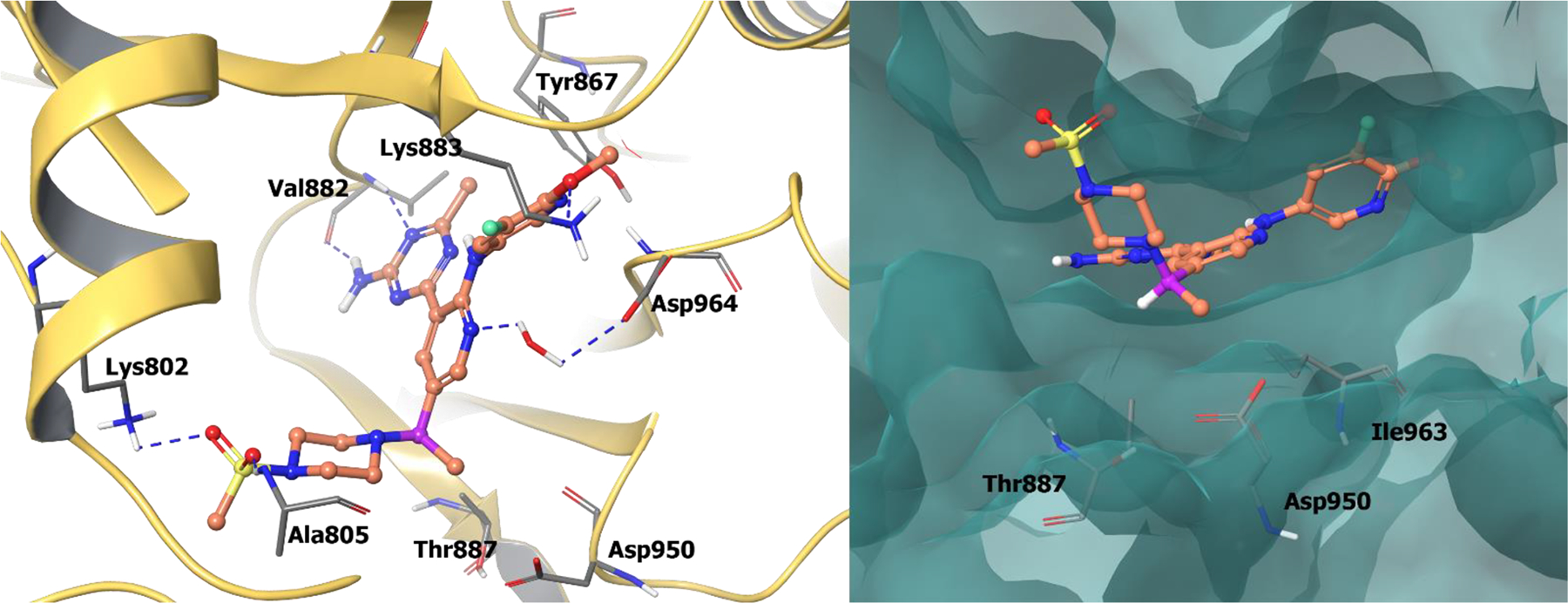
The co-crystal structure of compound 67a bound to PI3Kγ (PDB 4FLH, 2.6 Å). Left Panel: Interaction of compound 67a with the protein. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Right panel: The binding site of compound 67a in PI3Kγ. The protein surface is depicted in blue. Compound 67a atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
Compound 67a was subjected to a kinase panel against phosphatidylinositol 3-kinase-related kinases and exhibited good selectivity against mTOR (Kd > 10.0 μM), hVPS34 (Kd > 9.0 μM) and DNA-PK (Kd > 10.0 μM). From the series, compound 67a exhibited the highest stability in rat microsomes (CLint = 20 μL/min/mg) and also the lowest in-vivo clearance (CL = 0.4 L/kg/h) with a high mean residence time of ~2–5h. Compound 67a also possessed high oral bioavailability (F = 60%) commensurate with a large oral exposure (AUC = 5.0 μMh). Furthermore, compound 67a was also shown to potently block the PI3K pathway in an in vivo model and exhibited a dose-dependent decrease in AKT phosphorylation with an EC50 of 228 ng/mL. In a U87 MG xenograft model, compound 67a effectively inhibited tumor growth with an ED50 of 0.6 mg/Kg. Hence, by introducing a chiral methyl at the benzylic position, compound 67a achieved enhanced metabolic stability, which improved clearance and mean residence time.
Scientists at Amgen discovered another PI3K drug candidate, compound 71 (AMG 319), which completed phase I/II studies against relapsed or refractory lymphoid malignancies. Compound 71 was also examined for the treatment of human papillomavirus (HPV) and head and neck squamous cell carcinoma (HNSCC). However, clinical investigation of compound 71 was terminated due to safety concerns (NCT02540928).132
Compound 71 was discovered through modeling known PI3K inhibitors from the literature, which uncovered lead compound 68 (Scheme 21). Compound 68 served as a validated starting point since the compound achieved an IC50 of 0.24 μM against PI3Kδ. However, the thioether of 68 was sensitive to in vivo oxidation.133 SAR exploration revealed that an ether (69b) or amino (69c) linker improved potency against PI3K and also improved selectivity for PI3Kδ over PI3Kγ. However, the ether and amino analogs were still poorly soluble and exhibited microsomal instability. In order to minimize benzylic oxidation, a methyl was introduced at the α-position to both ether and amine linkers (70a-b). Microsomal stability did not improve yet enzymatic potency against PI3Kδ did improve (70a) (PI3Kδ IC50 = 0.071 μM). The introduction of a methyl at this position limits free rotation and rigidified the structure in a low energy state, which may mimic the binding state. Upon further analysis, it was observed that the (S)-methyl isomer (70a) (IC50 = 0.071 μM) was significantly preferred over the (R)-methyl isomer (70b) (IC50 = 2.6 μM) due to steric interference. Further SAR exploration resulted in identification of compound 71, which involved removal of the metabolically labile methyl, deactivating the quinoline with a fluorine, and substituting 2-aryl for 2-pyridyl. Compound 71 exhibited decent pharmacokinetic properties and solubility (PI3Kδ IC50 = 0.018 μM; PI3Kβ IC50 = 2.7 μM; PI3Kα IC50 = 33 μM; PI3Kγ IC50 = 0.85 μM, sol. (PBS) 146 mg/μL, rat PK CL = 0.34 L/h/kg; F = 54%).
Scheme 21.
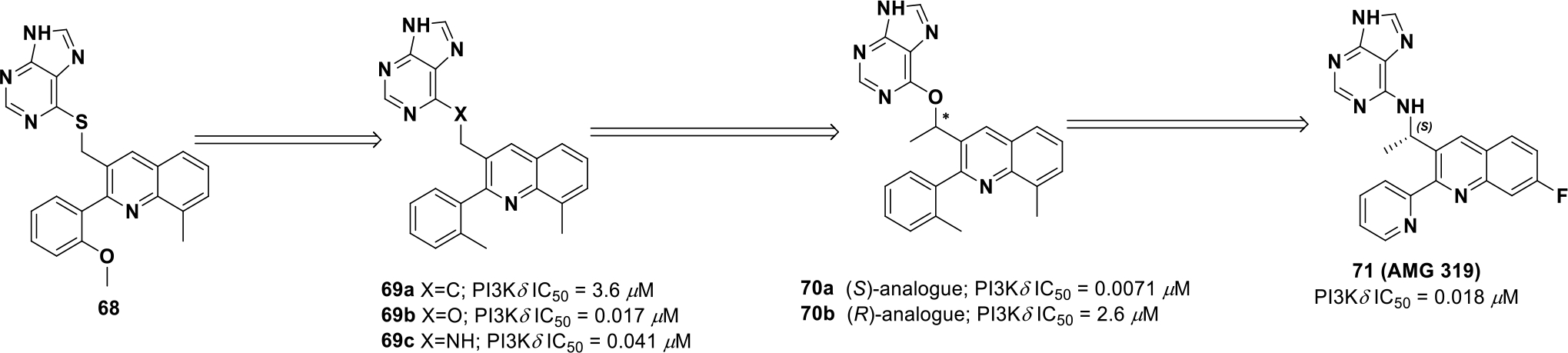
Drug discovery of compound 71
Compound 71 has been co-crystalized in the PI3Kγ active site. The 7-fluoroquinoline lies in the inducible specificity pocket between Trp812 and Met804, and the pyridine orients towards a hydrophobic pocket comprised of Val882 and Lys890. The chiral (S)-methyl group on the benzylic position efficiently occupies a hydrophobic pocket adjacent to the hinge region formed by Thr887, Ile963, and Asp905 (Figure 16). The purine core binds to the hinge region at Val882 and Glu880. The (R)-enantiomer is unable to form similar interactions in PI3Kγ and therefore has substantially less activity. Therefore, by exploiting chirality, (S)-AMG 319 exhibited improved selectivity, solubility, and pharmacokinetic properties.
Figure 16.
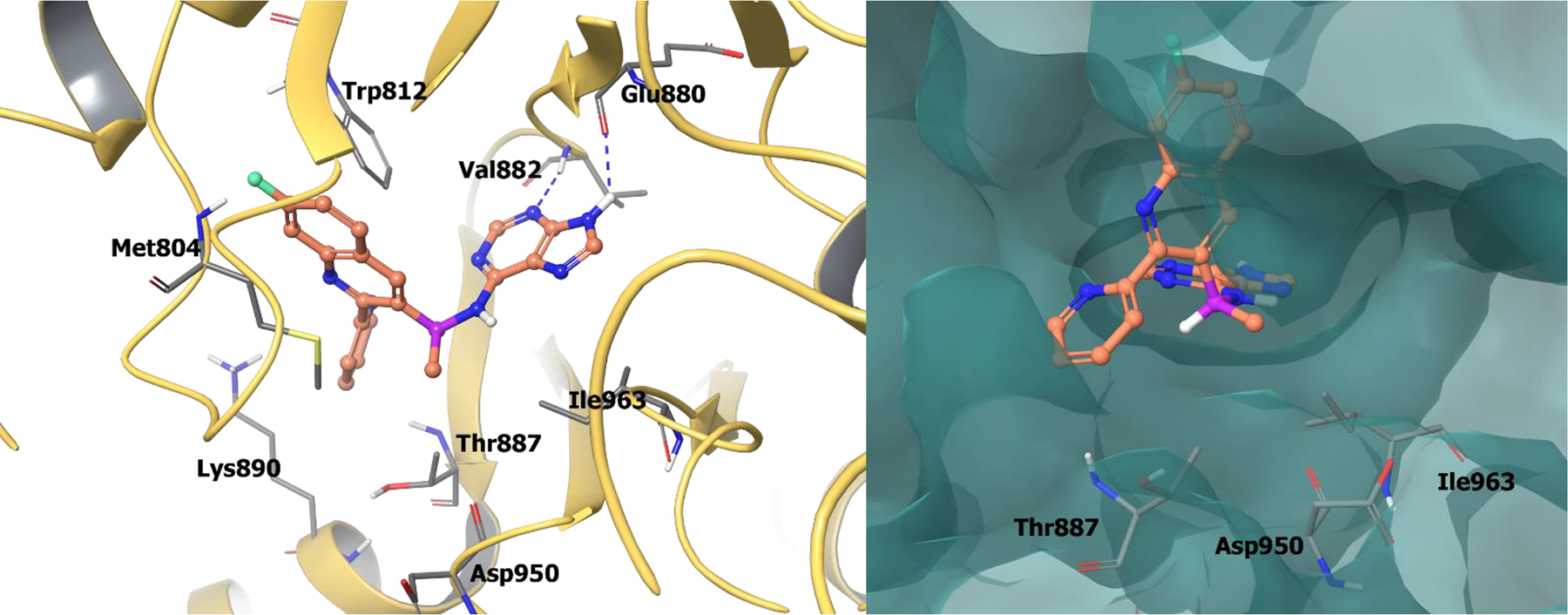
The co-crystal structure of compound 71 bound to PI3Kγ (PDB 4WWN, 2.7 Å). Left Panel: Interaction of compound 71 with the protein. The protein is depicted as yellow ribbons, and hydrogen bonds are illustrated with blue dashed lines; Right panel: The compound 71 binding site in PI3Kγ. The surface of the protein is depicted in blue. Compound 71 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
A series of 4H-pyrido[1,2-a]pyrimidin-4-ones, illustrated by compound 72 (TGX-221) (PI3Kβ IC50 = 0.022 μM), were reported as PI3Kβ inhibitors. Compound 72 has a benzylic, chiral center but previous investigations were completed with the racemic mixture. Chirality of compound 72 was later resolved, and it was observed that the R-enantiomer (73a) (PI3Kβ IC50 = 0.006 μM) was 100-fold more potent than the S-enantiomer (73b) (PI3Kβ IC50 = 0.80 μM). This suggests that chirality plays a crucial role in receptor binding and was further investigated to obtain insight about the receptor/ligand complex. Compound 72 was modeled in a homology model of PI3Kβ and identified that morpholine interacts with Val854 at the hinge and the pyrido-pyrimidinone core interacts in the central pocket formed by Met926 and Ile residues 803, 851, and 936. The carbonyl group engages the back-pocket Tyr839 via a hydrogen bond network. Also, the aniline ring of compound 73a (TGX-221-R) is believed to induce a conformational switch in the P-loop at the top of the ATP binding site, forming a specificity pocket as observed with other PI3K inhibitors. Because of the energetics of PI3Kβ, induction of the specificity pocket by 73a is selective for the β-isoform over others.134,–135
Further efforts by AstraZeneca resulted in the discovery of another PI3Kβ inhibitor, AZD6482 (74a) (Scheme 22).135 Similar to 73a, compound 74a (PI3Kβ IC50 = 0.04 μM) exhibited 200 times more activity than the (S)-enantiomer (74b) (PI3Kβ IC50 = 2.3 μM). 74a was evaluated for anti-platelet effects via ADP-induced impedance aggregometry and exhibited efficacy in humans. 137
Scheme 22.
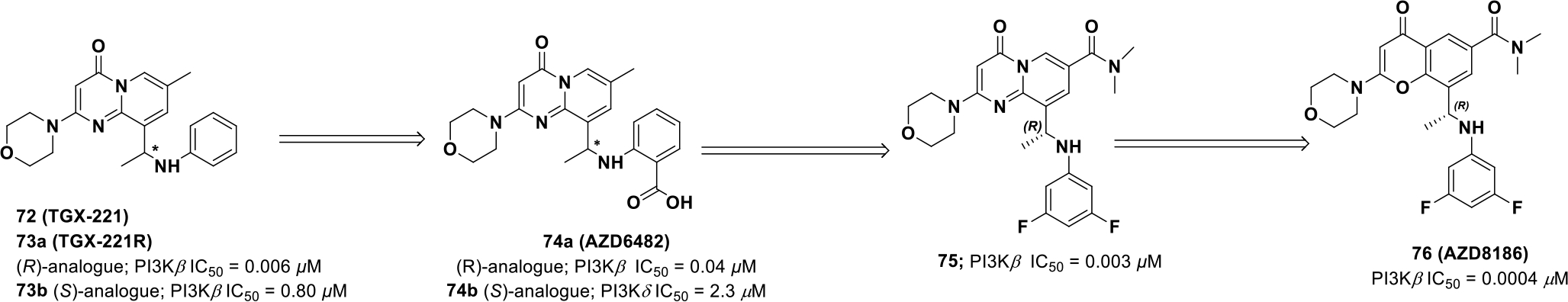
Drug discovery of compound 74a and 76.
Barlaam et al. sought to identify an orally available PI3Kβ inhibitor and utilized compounds 73a and 74a. For this investigation, the researchers considered removing the carboxylic acid on 74a, believing the acid was responsible for poor oral exposure. With the objective of balancing lipophilicity and permeability, the researchers identified the R-isomer (75), with a difluoro substituted aniline and a tertiary amide on the core. Compound (R)-75 had average metabolic stability and exhibited good cell potency (IC50 = 0.003 μM), 31% oral bioavailability, and a clearance of 82 mL/min/kg. However, due to solubility issues (R)-75 was terminated.136
The same research group sought to further optimize (R)-75 with the intention of generating a more hydrophilic molecule with improved solubility and metabolic stability. By modifying (R)-75, AZD8186 (76) was discovered that has a similar difluoro substituted aniline and a tertiary amide on a different core. Compound 76 exhibited a significant increase in both potency and selectivity (PI3Kβ IC50 = 0.0004 μM; PI3Kα IC50 = 0.035 μM; PI3Kδ IC50 = 0.012 μM; PI3Kγ IC50 = 0.675 μM, in cell, MDA-MB-468 pAKT IC50 = 0.003 μM) than compound 75. Further, through oral administration, 76 blocked Akt phosphorylation in PTEN-deficient PC3 xenografts. With moderate permeability (Caco-2 Papp 8.0×10−6 cm/s), high metabolic stability, and potency, 76 was progressed into clinical studies for patients with advanced castration-resistant prostate cancer (CRPC), squamous NSCLC, triple negative breast cancer (TNBC), and PTEN-deficient/mutated or PIK3CB mutated/amplified advanced, solid malignancies (NCT03218826).138
In an attempt to develop a PI3Kδ inhibitor that could be administered via inhalation to treat NSCLC, Erra et al. developed LAS195319 (78), which displayed less side effects than idelalisib (Scheme 23). 139 Modifications to 63 were performed to reduce systemic side effects by deliberately increasing clearance. These efforts resulted in compound 78, which exhibited improved lung retention and potency against PI3Kδ (PI3Kδ IC50 = 0.0005 μM, PI3Kα IC50 = 1.9 μM, PI3Kβ IC50 = 0.01 μM, PI3Kγ IC50 = 0.036 μM, M-CSF-induced AKT in THP-1 cells IC50 = 0.027 μM, rat PK AUC = 171 ng/h/mL, F = 1%).140 Erra et al. developed another pyrrolotriazine based inhibitor by adding a methyl group on the benzylic carbon to minimize benzylic oxidation on IC87114 (Scheme 23). Additional modifications uncovered compound 80 (LAS191954), which entered clinical development for the treatment of pemphigus (PI3Kδ IC50 = 0.026 μM, PI3Kα IC50 = 8.2 μM, PI3Kβ IC50 = 94.0 μM, PI3Kγ IC50 = 72.0 μM, M-CSF-induced AKT in THP-1 cells IC50 = 0.078 μM, rat PK CL = 1.4mL/min/Kg, F = 98%).141
Scheme 23.
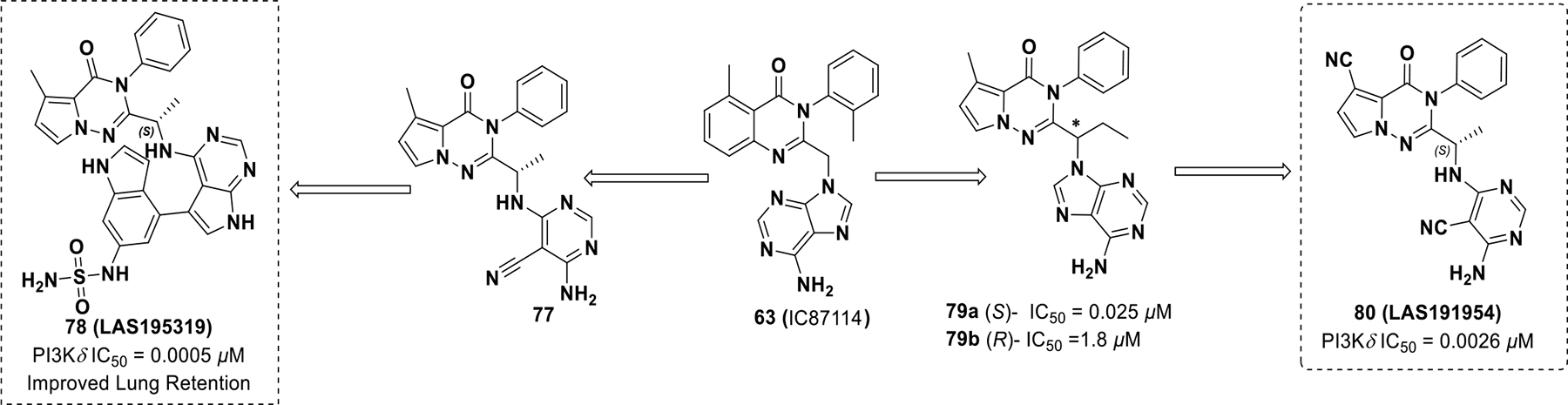
Drug discovery of compound 78 and 80
The co-crystal structure of compound 81 (an analogue of 78) bound to PI3Kδ revealed that the molecule adopts a chirality-induced conformation. The pyrrolopyrimidine forms hydrogen bonds to Glu826 and Val828 at the hinge. As a result, the pyrrolotriazinone moiety is sandwiched orthogonally into the specificity pocket between Trp760 and Met752 (Figure 17). Also, the sulfone forms a hydrogen bond with the side chain of Lys779, and the phenol group hydrogen bonds to Asp787.
Figure 17.
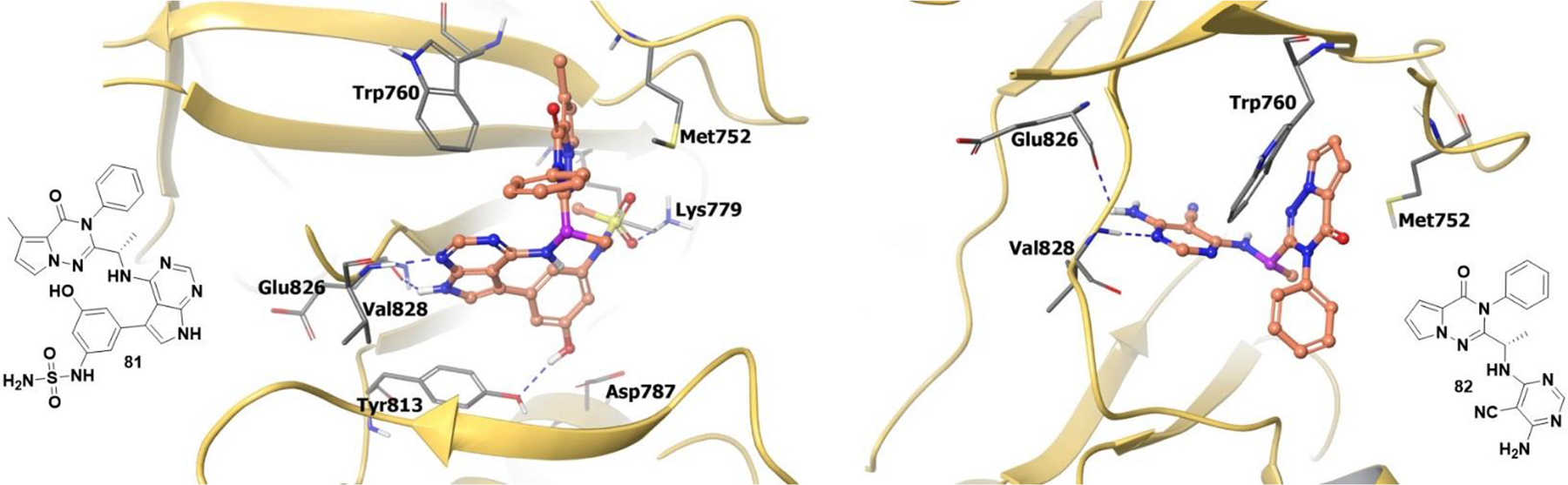
Left Panel: The co-crystal structure of analogue 81 bound to catalytic site of PI3Kδ (PDB 6G6W, 2.72 Å). Right Panel: The co-crystal structure of analogue 82 bound to catalytic site of PI3Kδ (PDB 5M6U, 2.85 Å). The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Analogue 81 and 82 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet.
Compound 82 (an analogue of 80) occupies the prototypical binding pose in PI3Kδ where the pyrrolotriazinone moiety enters the hydrophobic specificity pocket between Trp760 and Met752; the cyanopyrimidine binds to Val828 and Glu826 at the hinge (Figure 17). The R-enantiomer of analogue 82 cannot enter the specificity pocket with the correct geometry and would therefore form undesirable steric clashes in the binding pocket.
Additional inhibitors of the PI3K family are under clinical development. This includes umbralisib (83), a selective PI3Kδ inhibitor in phase II clinical trials for chronic lymphocytic leukemia with resistance to BTK inhibitors or prior PI3Kδ inhibitor therapy (NCT03801525) (Scheme 24).141–142 This also includes duvelisib (84), a dual PI3Kδ and PI3Kγ inhibitor in clinical trials for advanced hematologic malignancies and relapsed or refractory peripheral T-cell lymphoma (NCT01476657, NCT03372057), and tenalisib (85), another dual PI3Kδ and PI3Kγ inhibitor in phase II clinical trials for relapsed/refractory indolent non-Hodgkin’s lymphoma and T-cell lymphoma (NCT03711578). The additional PI3K inhibitors are chiral and the S-enantiomers are active. The S-chiral configuration permits appropriate geometric alignment in the selectivity pocket.
Scheme 24.
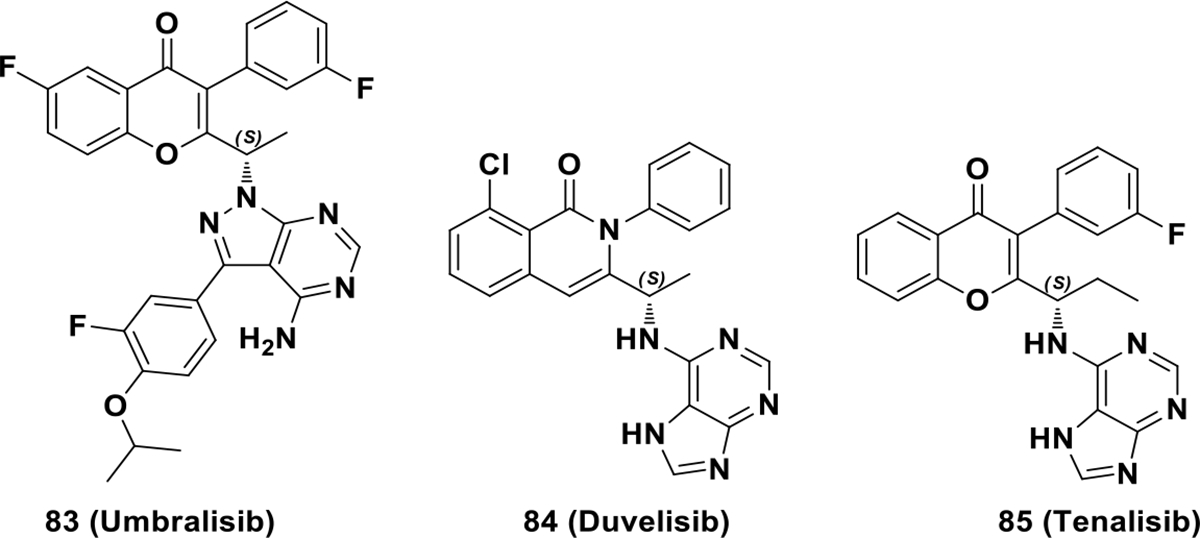
Chemical structures of PI3K clinical candidates 83, 84 and 85
2.5. Irreversible Chiral Kinase Inhibitors
Irreversible kinase inhibitors form covalent bonds with the target kinase.143 This occurs via nucleophilic attack by a cysteine residue on an electrophilic center of the irreversible inhibitor. The typical electrophilic centers used for this purpose include acrylamides, vinyl sulphonates, quinones, alkynyl amides, propargylic acid derivatives, α-halo ketones, thiocyanates, epoxides, etc. Covalent inhibition serves as a platform to fine tune selectivity and affinity for the target kinase.144 Kinase classes explored with covalent inhibitors include EGFR, Her-2, Her-4, the Tec family (BMX, BTK, ITK, TEC and TXK), and one member of the Src family (BLK).145 Among clinically approved covalent candidates, two chiral molecules have been approved by the FDA, afatinib and ibrutinib.
2.5.1. FDA-Approved Irreversible Kinase Inhibitors
A. Afatinib (Jul 2013, GILOTRIF®, Boehringer Ingelheim Corp)
In 2009, afatinib (88, BIBW-2992) was the first quinazoline-based, irreversible kinase inhibitor approved by the FDA for treatment of NSCLC. Compound 88 covalently binds to Cys797 (EGFR) and Cys805 (HER2) and exhibits potent phosphorylation inhibition on both EGFR (IC50 = 0.005 μM) and HER2 (IC50 = 0.014 μM) compared to other irreversible kinase inhibitors (EKB-569 and HKI-272) (Scheme 25).146,147
Scheme 25.
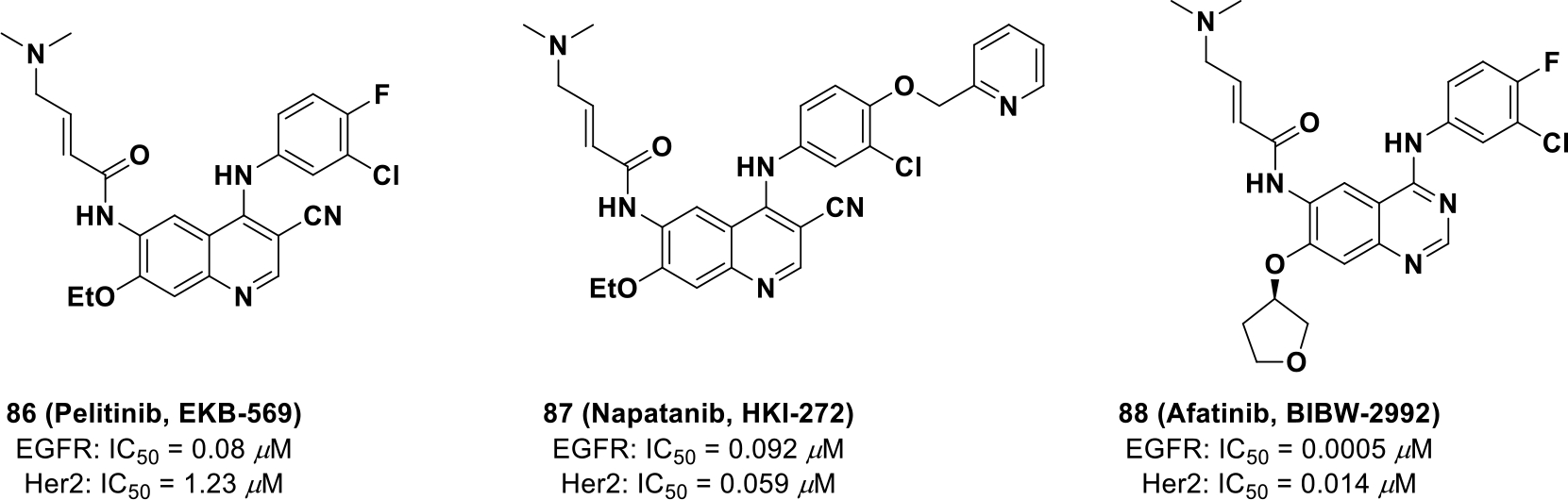
Chemical structures of irreversible EGFR and Her2 inhibitors 86, 87 and 88
The drug discovery campaign for compound 88 has not been disclosed, but its co-crystal structure with EGFR illustrates a key covalent bond formation between Cys797 and the electrophilic center of afatinib along with a hydrogen bond with Met793 at the hinge region (Figure 19). The specific role of the chiral tetrahydrofuran ring has not described but, since that region is in the solvent, it can be inferred the chirality does not influence the substrate-ligand complex.148 Instead, the chiral tetrahydrofuran ring likely influences pharmacokinetic properties of compound 88. Metabolic studies reveal minimal cytochrome P450 (CYP) metabolism, while the majority of 88 is excreted as a covalent adduct with plasma proteins.149 Also, compound 88 exhibits a low renal elimination rate (5%).149
Figure 19.
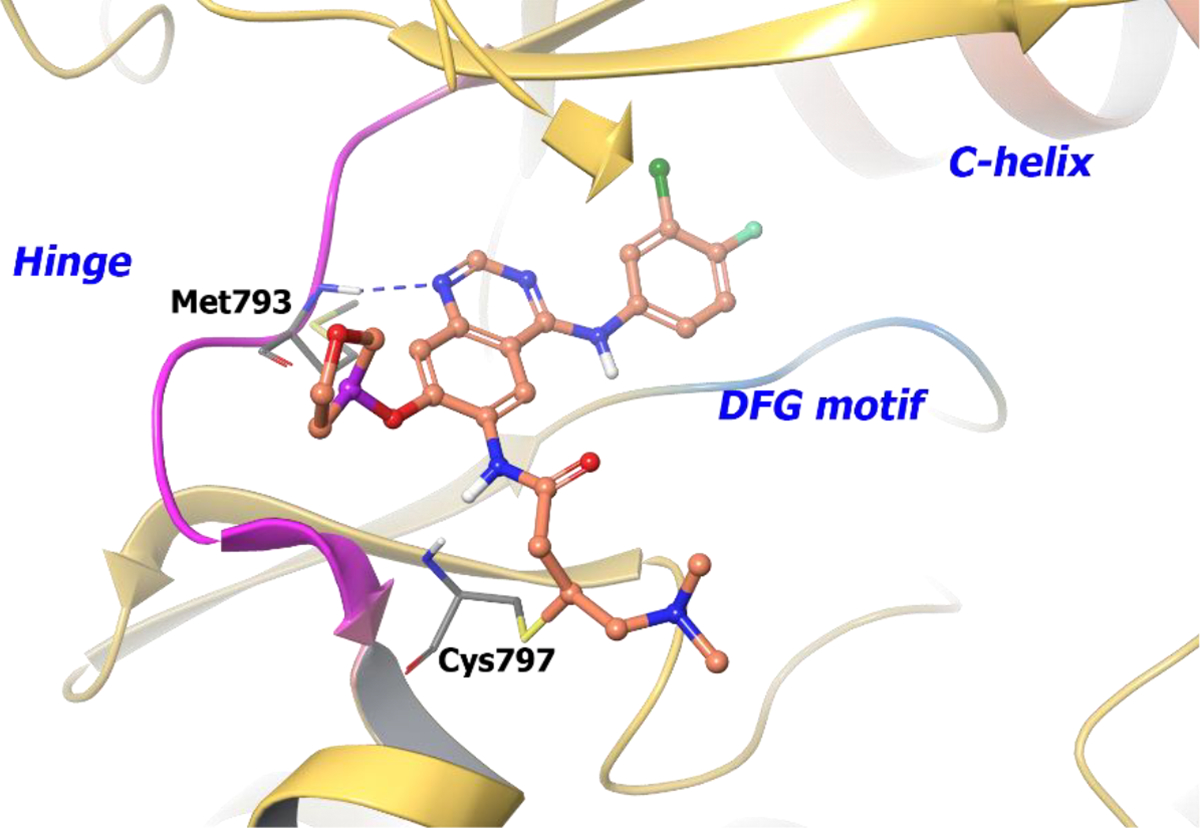
The co-crystal structure of compound 88 bound to EGFRT790M mutant (PDB 4G5P, 3.17 Å). The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound 88 atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet..
B. Ibrutinib (Nov 2013, IMBRUVICA®, Pharmacyclics)
Ibrutinib (90b, PCI-32765) is another irreversible inhibitor approved for B-cell non-Hodgkin’s lymphoma. To identify compound 90b, a library was screened against the BTK kinase and compound 89 (PCI-29732) was identified as a hit compound (BTK IC50 = 0.082 μM) (Scheme 26).150
Scheme 26.
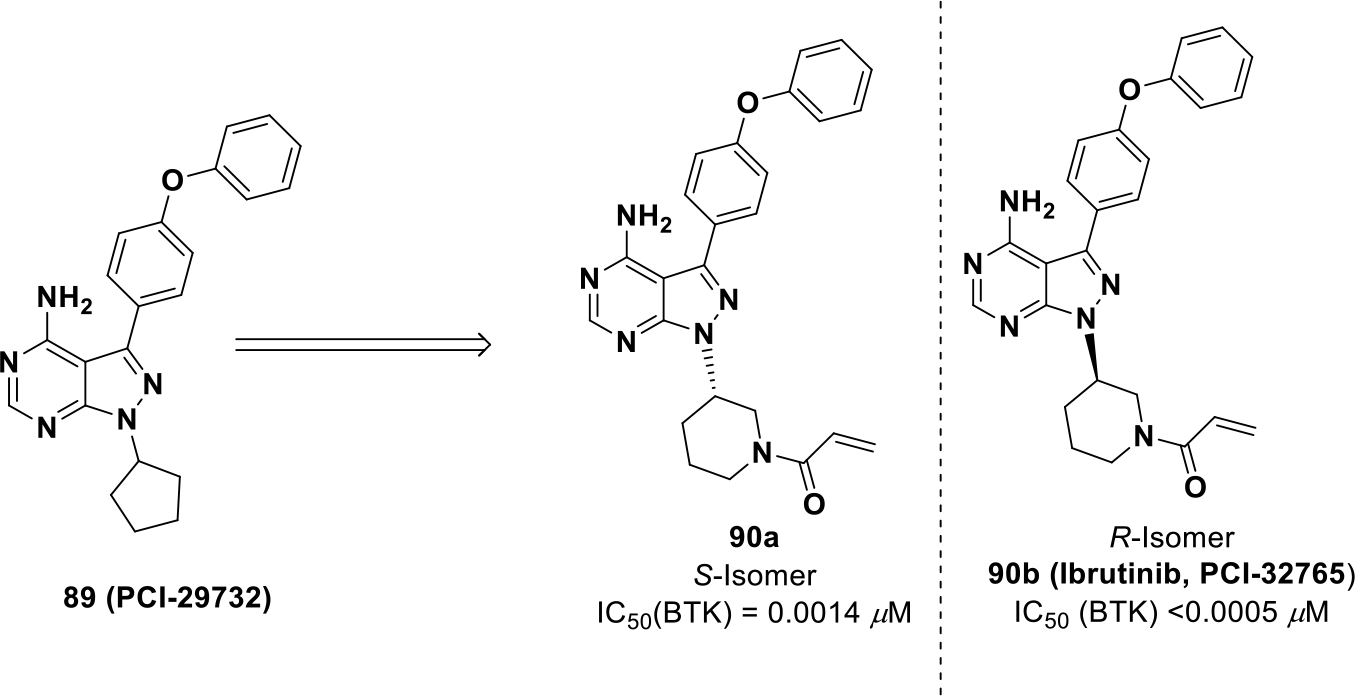
Drug development of compound 90b
A series of molecules were synthesized bearing different electrophilic centers to engage the conserved cysteine residue in BTK (Cys481). On the electrophilic center, adding a trans methyl group to the vinyl group reduced potency whereas addition of a tertiary amine improved potency. Investigation into piperidine and pyrrolidine based Michael acceptors was completed, and it was found that a piperidine based Michael acceptor with absolute (R)-configuration provided optimal potency (IC50 = 0.0005 μM).151
Drug-receptor interaction studies were completed by modeling compound 90b in BTK. The 4-amino group hydrogen bonds to Thr474 (gatekeeper) and Glu475, whereas the N-H of the pyrazolo[3,4-d]pyrimidine core forms a hydrogen bond with Met477 at the hinge (Figure 20). The thiol group of Cys481 covalently binds to the electrophilic center of compound 90b. Chirality of the piperidine ring places compound 90b in an orientation appropriate for covalent interaction with Cys481 and optimizes hydrogen bonding and hydrophobic interactions within the protein.
Figure 20.
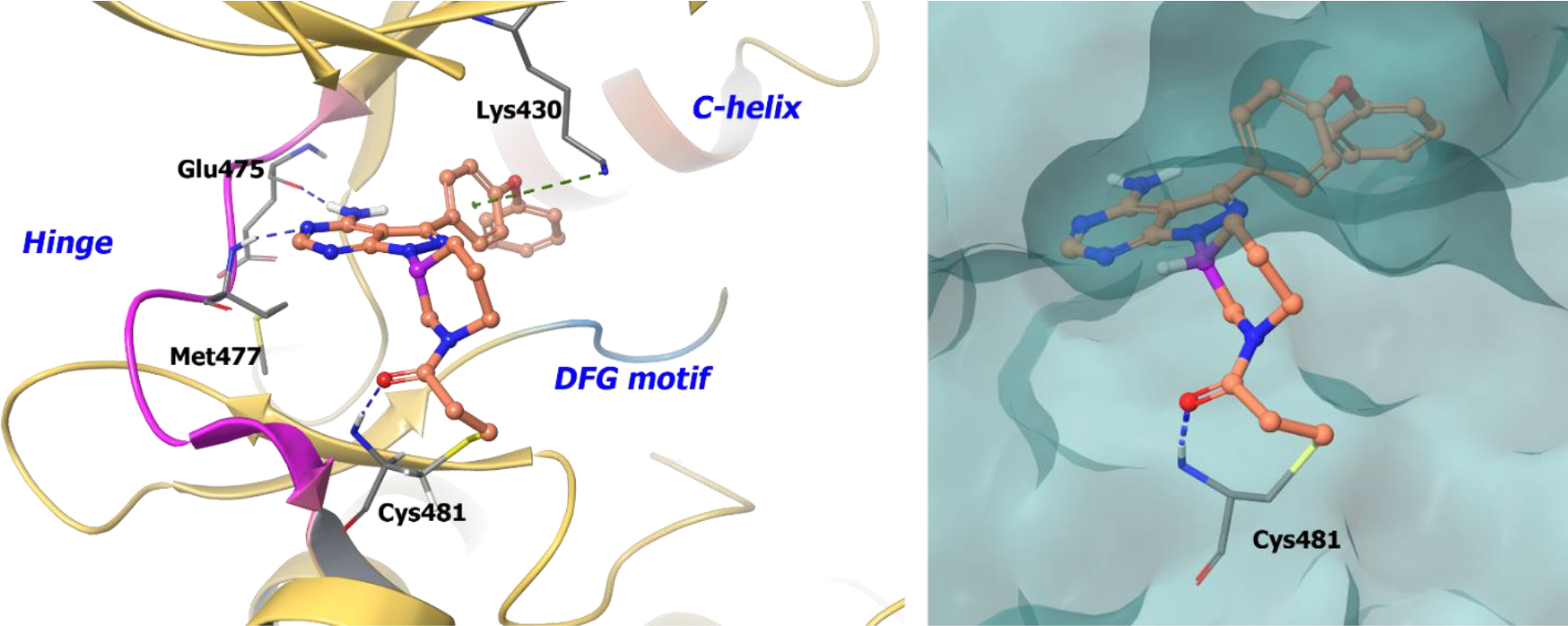
Compound 90b docked into the ATP binding pocket of EGFR (PDB 5FBN, 3.17 Å). Left Panel: Interaction of compound 90b with the protein. The protein is depicted as yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines; Right panel: The binding site of compound 90b in EGFR. The protein surface is depicted in blue. Compound 90b atoms are colored as follows: carbon, red-orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, jade; hydrogen, white; the chiral carbon is highlighted in violet..
Although ibrutinib is effective against B-cell malignancies, the inhibitor has numerous off-target activities due to irreversible binding to other kinases [e.g., EGFR, tyrosine kinase expressed in hepatocellular carcinoma (TEC), interleukin-2-inducible T-cell kinase (ITK), and T-cell X chromosome kinase (TXK)]. This led to the development of more selective, second-generation, irreversible BTK inhibitors tirabrutinib152 (91, ONO/GS-4059) and acalabrutinib (92, ACP-196, CALQUENCE®, AstraZeneca Pharmaceuticals Inc.) (Scheme 27).153 Compound 92 was developed for selectivity while maintaining an efficacious, irreversible profile against BTK. It received FDA approval in 2017 for treatment of adult patients with relapsed mantle cell lymphoma (MCL).25
Scheme 27:
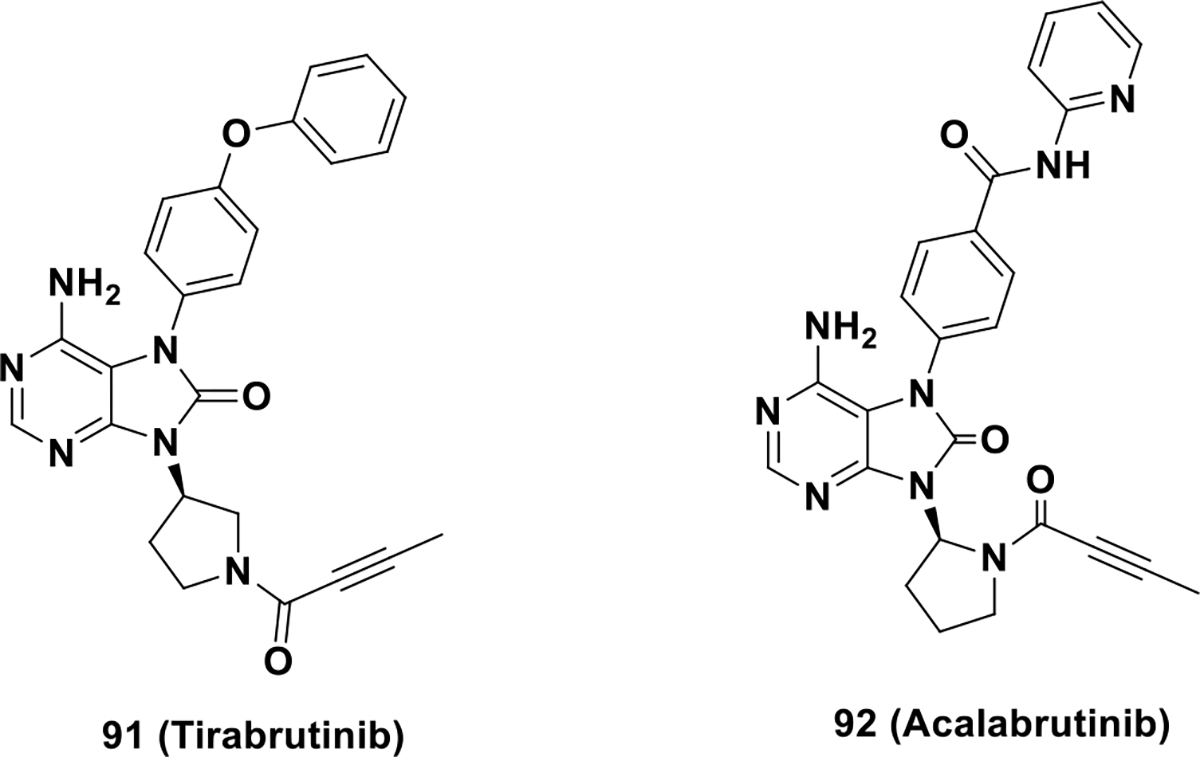
Second generation irreversible BTK inhibitors 91 and 92.
It should be noted that the JAK inhibitors 26 and 27 as described above are also irreversible kinase inhibitors.
3. Conclusion and Future Perspectives
Chirality is a prominent attribute in the biological world. Many organic molecules, including sugars and most natural amino acids, are chiral. Furthermore, all biomolecules within the central dogma of biology are chiral. Incorporating chirality into drug discovery is an important technique to better engage biological targets with enhanced drug properties. During drug discovery, the majority of failures are due to a lack of either efficacy or safety, which can be attributed to poor ADME properties of the drug.154 Consequently, a balance of optimized pharmacokinetic and pharmacodynamic parameters maximizes the safety and efficacy of a drug candidate. Chirality has the potential to remedy both challenges of drug optimization by exploiting the three dimensional nature of biology. As such, chiral small molecules are emerging as an attractive clinical advantage in drug discovery.
In kinase drug discovery, most approved inhibitors are planar and have no stereocenters. Although planar structures are efficacious, some drug discovery efforts within the kinome are currently focused on the generation of kinase inhibitors with stereocenters. These efforts emphasize chirality as a property to improve pharmacokinetics and selectivity within the kinome. Solubility is also a major liability in drug development, especially with planar drug structures. Since most kinase inhibitors mimic adenine, the structures are planar and aromatic and exhibit intramolecular π-π stacking interaction that limits their aqueous solubility. Hence, incorporation of a chiral center can reduce π-π stacking interaction, thus improving drug solubility and absorption. Axial chirality is also being explored within the kinome by generating atropisomeric structures. Dynamics of such axial systems permits fine-tuning of receptor/ligand interactions. Further, chirality provides a new dimension to access new chemical space increasing novelty in structure and thereby accelerating drug discovery.
Understanding the importance of chirality in interdisciplinary areas of drug development may contribute to major progress in kinase inhibitor development. Crizotinib, the first FDA approved chiral kinase inhibitor for NSCLC, supports the importance of chirality in kinase drug discovery. On similar lines, the recent approval of lorlatinib for the treatment of NSCLC has further galvanized the importance of chirality in kinase research. The recent major advancements in new asymmetric synthetic methodologies and enantiomeric separation techniques encourage the effort of chiral drug development. Other evolving medicinal chemistry practices, such as kinome profiling and X-ray crystallography, are providing better insight into the role of chirality in kinase receptor engagement and receptor selectivity. Hence, developing chiral kinase inhibitors can help enhance druggability within the kinome. It is expected that chirality-driven, drug discovery campaigns will promote the development of kinase inhibitors with improved selectivity, potency, and drug properties. A surge of chiral kinase inhibitors are under clinical investigation and many have been approved in recent years. Enhancing druggability by implementing chirality-focused drug discovery will expand structural diversity when targeting the kinome. This, in turn, will uncover new chemotypes with augmented pharmacokinetic and pharmacodynamic profiles for improved druggability within the human kinome.
Scheme 5.
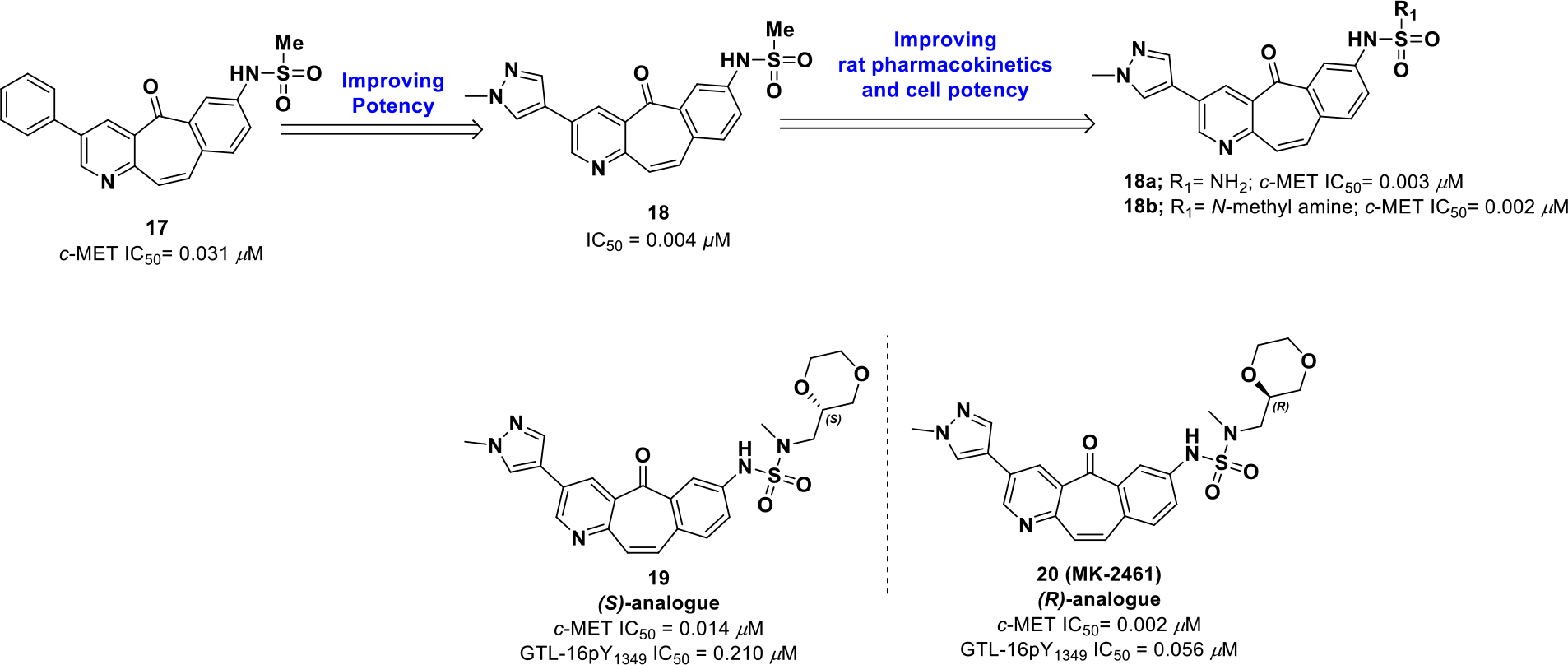
Drug design of compound 18
Scheme 15:
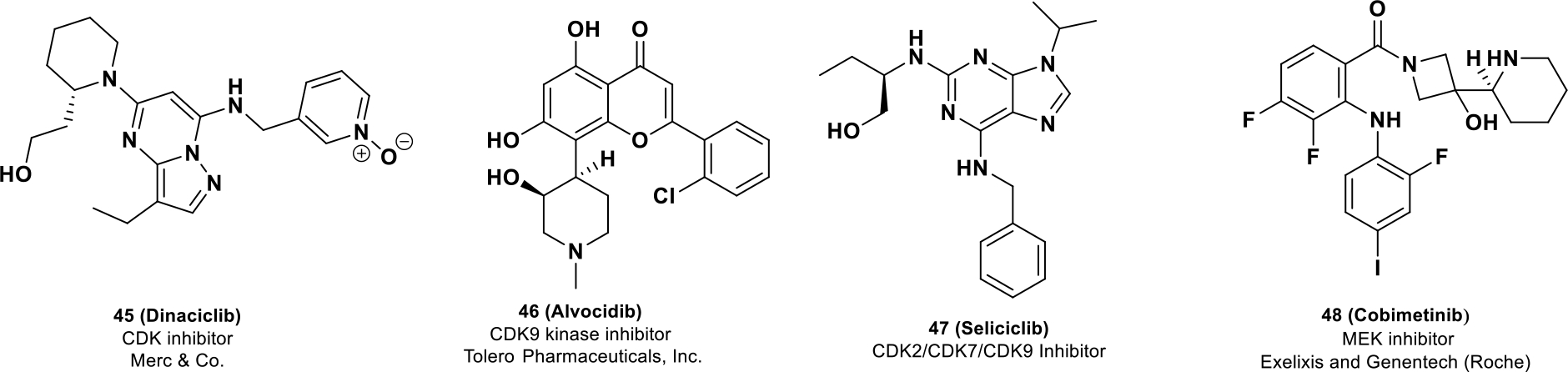
Chemical structures of approved CDK and MEK inhibitors
Acknowledgement:
HL was supported by the grants (NIH 1R01CA194094 and 1R01CA197178)
List of Non-standard Abbreviations
- BTK
Bruton’s tyrosine kinase
- BCR
B-cell receptors
- BLK
B Lymphocyte Kinase
- CDKs
Cyclin-dependent kinases
- CL
Blood Clearance
- CRPC
Castration-resistant prostate cancer
- ERKs
Extracellular signal–regulated kinases
- FLT3
FMS like tyrosine kinase 3
- GSK
Glaxo smith Kline
- HER2
Human epidermal growth factor receptor 2
- ITK
Interleukin-2-inducible T-cell kinase
- IFNγ
Interferon gamma
- IGF
Insulin-like growth factor-1 receptor
- IL-2R
IL-2 receptors
- IRAK4
Interleukin-1-receptor associated kinase
- ITK
Interleukin-2-inducible T-cell kinase
- JAK
Janus Kinase
- JNKs
c-Jun N-terminal kinases
- MAPKs
Mitogen activated protein kinases
- MDA-MB
M.D. Anderson Metastasis Breast cancer
- MDR
Multi drug resistance
- NFAT
Nuclear factor of activated T-cell
- PBMC
Peripheral blood monomorphonuclear cells
- PDX
Patient-derived xenograft
- PI(3,4)P2
Phosphatidylinositol 3,4-bisphosphate
- PI3K
Phosphoinositide 3-kinase
- PI3P
Phosphatidylinositol 3-phosphate
- PIP3
Phosphatidylinositol 3,4,5-trisphosphate
- PLKs
Polo-like kinases
- PTEN
Phosphatase and tensin homolog
- RIP1
Receptor interacting protein 1
- S1P
Sphingosine-1-phosphate
- STAT
Signal transducer and activator of transcription
- STK
Serine threonine Kinase
- TCR
T-cell receptors
- TGFβR1
Transforming growth factor beta receptor 1
- TNBC
Triple negative breast cancer
Biographies
1. Debasmita Saha received her Ph.D. in organic synthesis from Indian Institute of Technology, Roorkee, India. She also worked as an International Research Scholar at KU Leuven, Belgium followed by a post-doctoral assignment there. Currently, she is a post-doctoral fellow at University of Arkansas for Medical Sciences working in the area of kinase drug discovery. Her research interests include the design and synthesis of novel organic therapeutic frameworks and medicinal chemistry.
2. Anupreet Kharbanda is currently a Graduate student in the Department of Pharmaceutical Sciences, University of Arkansas for Medical Sciences. She obtained her bachelor’s in chemistry from University of Delhi, and master’s in chemistry from Indian Institute of Technology, Roorkee, India. Her current research centers on developing small molecule for targeting the tumor microenvironment with TGFβ Inhibitor, using synthetic medicinal chemistry and in-vitro assays.
3. Wei Yan received his Ph.D. from East China University of Science and Technology, Shanghai, China, and worked jointly at Shanghai Institute of Materia Medica, Chinese Academy of Sciences. Following, he worked at WuXi AppTec as a process chemistry scientist. He is currently a postdoctoral researcher at the University of Arkansas for Medical Sciences. His expertise is in medicinal chemistry and process chemistry for pilot plant manufacture of APIs.
4. Naga Rajiv Lakkaniga is currently a Ph.D. candidate in the Department of Pharmaceutical Sciences, University of Arkansas for Medical Sciences. He received a Bachelors in Pharmacy and Master of Pharmacy in Pharmaceutical Chemistry from Birla Institute of Technology and Science, Pilani, India. His current research is focused on discovering novel small molecule kinase inhibitors for anticancer therapy using synthetic medicinal chemistry and employing computational methods for lead optimization and conformational study of protein kinases.
5. Brendan Frett is an Assistant Professor of Pharmaceutical Sciences in the College of Pharmacy at the University of Arkansas for Medical Sciences. He received his Ph.D. degree from the University of Arizona, where he codiscovered a clinical candidate in IND studies. He has successfully transferred academic-based discoveries to pharmaceutical companies for clinical development. He is interested in pursuing translational research projects, where research completed in his laboratory can directly help patients.
6. Hong-yu Li is a Professor of Medicinal Chemistry at the University of Arkansas for Medical Sciences (UAMS). He is also an Arkansas Research Alliance (ARA) Scholar, the Helen Adams & ARA endowed chair in drug discovery, and codirector for the Therapeutics Science Program, Winthrop P Rockefeller Cancer Institute. He received his Ph.D. degree from the University of Tokyo and did postdoctoral training at Columbia University and Harvard University. He previously worked at Eli Lilly and the University of Arizona where he focused on oncology drug discovery. His current research interests are in chemical biology and drug discovery, especially for oncology related targets and phenotypes. In his lab at UAMS, a robust oncology pipeline is under development exploiting single agent polypharmacology and synergistic medicinal chemistry approaches.
Footnotes
The authors declare no competing financial interest.
References
- 1.Francotte E; Lindne W Chirality in drug research. WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007. [Google Scholar]
- 2.Waldeck B Three-dimensional pharmacology, a subject ranging from ignorance to overstatements. Pharmacol. and Tox. 2003, 93, 203–210. [DOI] [PubMed] [Google Scholar]
- 3.Agranat I; Caner H; Caldwell J Putting chirality to work: the strategy of chiral switches Nat. Rev. Drug Discov. 2002, 1, 753–68. [DOI] [PubMed] [Google Scholar]
- 4.Burke D; Henderson DJ Chirality: a blueprint for the future. Br. J. Anaesth. 2002, 88, 563–576. [DOI] [PubMed] [Google Scholar]
- 5.McConathy J; Owens MJ Stereochemistry in drug action. Prim. Care Companion J. Clin. Psychiatry 2003, 5, 70–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eriksson T Pharmacokinetics of the enantiomers of thalidomide. Malmö University Hospital: Malmö, 1997. [Google Scholar]
- 7.Eriksson T; Björkman S; Höglund P Clinical pharmacology of thalidomide. Eur. J. Clin. Pharmacol. 2001, 57, 365–376. [DOI] [PubMed] [Google Scholar]
- 8.Eriksson T; Björkman S; Roth B; Björk H; Höglund P Hydroxylated metabolites of thalidomide: formation in-vitro and in-vivo in man. J. Pharm. Pharmacol. 1998, 50, 1409–1416. [DOI] [PubMed] [Google Scholar]
- 9.Meyring M; Mühlbacher J; Messer K; Kastner-Pustet N; Bringmann G; Mannschreck A; Blaschke G In vitro biotransformation of (R)-and (S)-thalidomide: application of circular dichroism spectroscopy to the stereochemical characterization of the hydroxylated etabolites. Anal. Chem. 2002, 74, 3726–3735. [DOI] [PubMed] [Google Scholar]
- 10.Vargesson N Thalidomide-induced teratogenesis: history and mechanisms. Birth Defects Res. C. Embryo Today: Reviews 2015, 105, 140–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Development of new stereoisomeric drugs. https://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/guidances/ucm122883.htm (accessed Jan 19, 2019).
- 12.Caner H; Groner E; Levy L; Agranat I Trends in the development of chiral drugs. Drug Discov. Today 2004, 9, 105–110. [DOI] [PubMed] [Google Scholar]
- 13.a) Toenjes ST; Gustafson JL. Atropisomerism in medicinal chemistry: challenges and opportunities. Fut. Med. Chem. 2018, 10, 409–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Clayden J; Moran WJ; Edwards PJ; LaPlante SR. The challenge of atropisomerism in drug discovery. Angew. Chem. Int. Ed. Engl. 2009, 48, 6398–6401; [DOI] [PubMed] [Google Scholar]; b) Barrett KT; Metrano AJ; Rablen PR; Miller SJ. Spontaneous transfer of chirality in an atropisomerically enriched two-axis system. Nature 2014, 509, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laplante SR; L DF; Fandrick KR; Fandrick DR; Hucke O; Kemper R; Miller SP; Edwards PJ. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem. 2011, 54, 7005–7022. [DOI] [PubMed] [Google Scholar]
- 16.Wu P; Nielsen TE; Clausen MH Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov. Today 2016, 21, 5–10. [DOI] [PubMed] [Google Scholar]
- 17.Wu P; Nielsen TE; Clausen MH FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [DOI] [PubMed] [Google Scholar]
- 18.Davis MI; Hunt JP; Herrgard S; Ciceri P; Wodicka LM; Pallares G; Hocker M; Treiber DK; Zarrinkar PP Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- 19.Brooks H, W.; Guida C, W.; Daniel G, K. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 2011, 11, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui JJ; Tran-Dube M; Shen H; Nambu M; Kung PP; Pairish M; Jia L; Meng J; Funk L; Botrous I; McTigue M; Grodsky N; Ryan K; Padrique E; Alton G; Timofeevski S; Yamazaki S; Li Q; Zou H; Christensen J; Mroczkowski B; Bender S; Kania RS; Edwards MP Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [DOI] [PubMed] [Google Scholar]
- 21.Johnson TW; Richardson PF; Bailey S; Brooun A; Burke BJ; Collins MR; Cui JJ; Deal JG; Deng YL; Dinh D; Engstrom LD; He M; Hoffman J; Hoffman RL; Huang Q; Kania RS; Kath JC; Lam H; Lam JL; Le PT; Lingardo L; Liu W; McTigue M; Palmer CL; Sach NW; Smeal T; Smith GL; Stewart AE; Timofeevski S; Zhu H; Zhu J; Zou HY; Edwards MP Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J. Med. Chem. 2014, 57, 4720–4744. [DOI] [PubMed] [Google Scholar]
- 22.Flanagan ME; Blumenkopf TA; Brissette WH; Brown MF; Casavant JM; Shang-Poa C; Doty JL; Elliott EA; Fisher MB; Hines M; Kent C; Kudlacz EM; Lillie BM; Magnuson KS; McCurdy SP; Munchhof MJ; Perry BD; Sawyer PS; Strelevitz TJ; Subramanyam C; Sun J; Whipple DA; Changelian PS Discovery of CP-690,550: a potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J. Med. Chem. 2010, 53, 8468–8484. [DOI] [PubMed] [Google Scholar]
- 23.Drug approval package: Rapamune (sirolimus) oral solution. https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/21083A.cfm (accessed Aug 30, 2018).
- 24.Kwitkowski VE; Prowell TM; Ibrahim A; Farrell AT; Justice R; Mitchell SS; Sridhara R; Pazdur R FDA approval summary: temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 2010, 15, 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drug trials Snapshots: COTELLIC. https://www.fda.gov/Drugs/InformationOnDrugs/ucm478351.htm (accessed November 10, 2018).
- 26.Midostaurin. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm555756.htm (accessed Oct 28, 2018).
- 27.Drug Trials Snapshots: RHOPRESSA. https://www.fda.gov/Drugs/InformationOnDrugs/ucm591430.htm (accessed Dec 05, 2018).
- 28.FDA grants accelerated approval to acalabrutinib for mantle cell lymphoma. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm583106.htm (accessed Dec 31, 2018).
- 29.FDA approves encorafenib and binimetinib in combination for unresectable or metastatic melanoma with BRAF mutations. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm611981.htm (accessed Jan 27, 2019).
- 30.FDA approves larotrectinib for solid tumors with NTRK gene fusions. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm626720.htm (accessed Dec 28, 2018).
- 31.Lemmon MA; Schlessinger J Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gschwind A; Fischer OM; Ullrich A The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [DOI] [PubMed] [Google Scholar]
- 33.Cheng HL; Trink B; Tzai TS; Liu HS; Chan SH; Ho CL; Sidransky D; Chow NH Overexpression of c-met as a prognostic indicator for transitional cell carcinoma of the urinary bladder: a comparison with p53 nuclear accumulation. J. Clin. Oncol. 2002, 20, 1544–1550. [DOI] [PubMed] [Google Scholar]
- 34.Morris SW; Kirstein MN; Valentine MB; Dittmer KG; Shapiro DN; Saltman DL; Look AT Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [DOI] [PubMed] [Google Scholar]
- 35.Maulik G; Shrikhande A; Kijima T; Ma PC; Morrison PT; Salgia R Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth F. R. 2002, 13, 41–59. [DOI] [PubMed] [Google Scholar]
- 36.Ma PC; Maulik G; Christensen J; Salgia R c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003, 22, 309–325. [DOI] [PubMed] [Google Scholar]
- 37.Knudsen BS; Vande Woude G Showering c-MET-dependent cancers with drugs. Curr. Opin. Genet. Dev. 2008, 18, 87–96. [DOI] [PubMed] [Google Scholar]
- 38.Morris S; Kirstein M; Valentine M; Dittmer K; Shapiro D; Saltman D; Look A Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [DOI] [PubMed] [Google Scholar]
- 39.Christensen JG; Zou HY; Arango ME; Li Q; Lee JH; McDonnell SR; Yamazaki S; Alton GR; Mroczkowski B; Los G Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer. Ther. 2007, 6, 3314–3322. [DOI] [PubMed] [Google Scholar]
- 40.FDA Approves Crizotinib Capsules. https://www.fda.gov (accessed Jan. 30, 2019).
- 41.Zou HY; Li Q; Lee JH; Arango ME; McDonnell SR; Yamazaki S; Koudriakova TB; Alton G; Cui JJ; Kung PP; Nambu MD; Los G; Bender SL; Mroczkowski B; Christensen JG An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007, 67, 4408–4417. [DOI] [PubMed] [Google Scholar]
- 42.Cui JR, Y.; Liang C; Sun L; Wei CC; Tang PC. Preparation of 5-aralkylsulfonyl-3-(pyrrol-2-ylmethylidene)-2-indolinone derivatives as kinase inhibitors. WO2002096361, 2002. [Google Scholar]
- 43.Schiering N; Knapp S; Marconi M; Flocco MM; Cui J; Perego R; Rusconi L; Cristiani C Crystal structure of the tyrosine kinase domain of the hepatocyte growth factor receptor c-Met and its complex with the microbial alkaloid K-252a. Proc. Natl. Acad. Sci. U S A 2003, 100, 12654–12659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen TD; DeAngelis LM Brain metastases. Neurol. Clin. 2007, 25, 1173–1192. [DOI] [PubMed] [Google Scholar]
- 45.Katayama R; Khan TM; Benes C; Lifshits E; Ebi H; Rivera VM; Shakespeare WC; Iafrate AJ; Engelman JA; Shaw AT Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. U S A 2011, 108, 7535–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elleraas J; Ewanicki J; Johnson TW; Sach NW; Collins MR; Richardson PF Conformational studies and atropisomerism kinetics of the ALK clinical candidate lorlatinib (PF-06463922) and desmethyl congeners. Angew. Chem. Int. Ed. Engl. 2016, 55, 3590–3595. [DOI] [PubMed] [Google Scholar]
- 47.Bertrand T; Kothe M; Liu J; Dupuy A; Rak A; Berne PF; Davis S; Gladysheva T; Valtre C; Crenne JY; Mathieu M The crystal structures of TrkA and TrkB suggest key regions for achieving selective inhibition. J. Mol. Biol. 2012, 423, 439–453. [DOI] [PubMed] [Google Scholar]
- 48.He H; Lyons KA; Shen X; Yao Z; Bleasby K; Chan G; Hafey M; Li X; Xu S; Salituro GM; Cohen LH; Tang W Utility of unbound plasma drug levels and P-glycoprotein transport data in prediction of central nervous system exposure. Xenobiotica 2009, 39, 687–693. [DOI] [PubMed] [Google Scholar]
- 49.Kodaira H; Kusuhara H; Fujita T; Ushiki J; Fuse E; Sugiyama Y Quantitative evaluation of the impact of active efflux by P-glycoprotein and breast cancer resistance protein at the blood-brain barrier on the predictability of the unbound concentrations of drugs in the brain using cerebrospinal fluid concentration as a surrogate. J. Pharmacol. Exp. Ther. 2011, 339, 935–944. [DOI] [PubMed] [Google Scholar]
- 50.FDA approves lorlatinib for second- or third-line treatment of ALK-positive metastatic NSCLC. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm625027.htm (accessed Feb 14, 2019).
- 51.Huang Q; Johnson TW; Bailey S; Brooun A; Bunker KD; Burke BJ; Collins MR; Cook AS; Cui JJ; Dack KN; Deal JG; Deng YL; Dinh D; Engstrom LD; He M; Hoffman J; Hoffman RL; Johnson PS; Kania RS; Lam H; Lam JL; Le PT; Li Q; Lingardo L; Liu W; Lu MW; McTigue M; Palmer CL; Richardson PF; Sach NW; Shen H; Smeal T; Smith GL; Stewart AE; Timofeevski S; Tsaparikos K; Wang H; Zhu H; Zhu J; Zou HY; Edwards MP Design of potent and selective inhibitors to overcome clinical anaplastic lymphoma kinase mutations resistant to crizotinib. J. Med. Chem. 2014, 57, 1170–1187. [DOI] [PubMed] [Google Scholar]
- 52.Albrecht BK; Harmange JC; Bauer D; Berry L; Bode C; Boezio AA; Chen A; Choquette D; Dussault I; Fridrich C; Hirai S; Hoffman D; Larrow JF; Kaplan-Lefko P; Lin J; Lohman J; Long AM; Moriguchi J; O’Connor A; Potashman MH; Reese M; Rex K; Siegmund A; Shah K; Shimanovich R; Springer SK; Teffera Y; Yang Y; Zhang Y; Bellon SF Discovery and optimization of triazolopyridazines as potent and selective inhibitors of the c-Met kinase. J. Med. Chem. 2008, 51, 2879–2882. [DOI] [PubMed] [Google Scholar]
- 53.Boezio AA; Berry L; Albrecht BK; Bauer D; Bellon SF; Bode C; Chen A; Choquette D; Dussault I; Fang M; Hirai S; Kaplan-Lefko P; Larrow JF; Lin MH; Lohman J; Potashman MH; Qu Y; Rex K; Santostefano M; Shah K; Shimanovich R; Springer SK; Teffera Y; Yang Y; Zhang Y; Harmange JC Discovery and optimization of potent and selective triazolopyridazine series of c-Met inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 6307–6312. [DOI] [PubMed] [Google Scholar]
- 54.Bode CM; Boezio AA; Albrecht BK; Bellon SF; Berry L; Broome MA; Choquette D; Dussault I; Lewis RT; Lin MH; Rex K; Whittington DA; Yang Y; Harmange JC Discovery and optimization of a potent and selective triazolopyridinone series of c-Met inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 4089–4093. [DOI] [PubMed] [Google Scholar]
- 55.Peterson EA; Teffera Y; Albrecht BK; Bauer D; Bellon SF; Boezio A; Boezio C; Broome MA; Choquette D; Copeland KW; Dussault I; Lewis R; Lin MH; Lohman J; Liu J; Potashman M; Rex K; Shimanovich R; Whittington DA; Vaida KR; Harmange JC Discovery of potent and selective 8-fluorotriazolopyridine c-Met inhibitors. J. Med. Chem. 2015, 58, 2417–2430. [DOI] [PubMed] [Google Scholar]
- 56.Boezio AA; Copeland KW; Rex K; B KA; Bauer D; Bellon SF; Boezio C; Broome MA; Choquette D; Coxon A; Dussault I; Hirai S; Lewis R; Lin MH; Lohman J; Liu J; Peterson EA; Potashman M; Shimanovich R; Teffera Y; Whittington DA; Vaida KR; Harmange JC Discovery of (R)-6-(1-(8-fluoro-6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)-1,6-naphthyridin-5(6H)-one (AMG 337), a potent and selective inhibitor of MET with high unbound target coverage and robust in vivo antitumor activity. J. Med. Chem. 2016, 59, 2328–2342. [DOI] [PubMed] [Google Scholar]
- 57.Katz JD; Jewell JP; Guerin DJ; Lim J; Dinsmore CJ; Deshmukh SV; Pan BS; Marshall CG; Lu W; Altman MD; Dahlberg WK; Davis L; Falcone D; Gabarda AE; Hang G; Hatch H; Holmes R; Kunii K; Lumb KJ; Lutterbach B; Mathvink R; Nazef N; Patel SB; Qu X; Reilly JF; Rickert KW; Rosenstein C; Soisson SM; Spencer KB; Szewczak AA; Walker D; Wang W; Young J; Zeng Q Discovery of a 5H-benzo[4,5]cyclohepta[1,2-b]pyridin-5-one (MK-2461) inhibitor of c-Met kinase for the treatment of cancer. J. Med. Chem. 2011, 54, 4092–4108. [DOI] [PubMed] [Google Scholar]
- 58.Li R; Pourpak A; Morris SW Inhibition of the insulin-like growth factor-1 receptor (IGF1R) tyrosine kinase as a novel cancer therapy approach. J. Med. Chem. 2009, 52, 4981–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Samani AA; Yakar S; LeRoith D; Brodt P The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr. Rev. 2007, 28, 20–47. [DOI] [PubMed] [Google Scholar]
- 60.Chan JM; Stampfer MJ; Giovannucci E; Gann PH; Ma J; Wilkinson P; Hennekens CH; Pollak M Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science 1998, 279, 563–566. [DOI] [PubMed] [Google Scholar]
- 61.Perks CM; Holly JM The insulin-like growth factor (IGF) family and breast cancer. Breast Dis. 2003, 18, 45–60. [DOI] [PubMed] [Google Scholar]
- 62.Chen C; Zhu Y-F; Liu X-J; Lu Z-X; Xie Q; Ling N Discovery of a series of nonpeptide small molecules that inhibit the binding of insulin-like growth factor (IGF) to IGF-binding proteins. J. Med. Chem. 2001, 44, 4001–4010. [DOI] [PubMed] [Google Scholar]
- 63.Jin M; Gokhale PC; Cooke A; Foreman K; Buck E; May EW; Feng L; Bittner MA; Kadalbajoo M; Landfair D; Siu KW; Stolz KM; Werner DS; Laufer RS; Li AH; Dong H; Steinig AG; Kleinberg A; Yao Y; Pachter JA; Wild R; Mulvihill MJ Discovery of an orally efficacious imidazo[5,1-f][1,2,4]triazine dual inhibitor of IGF-1R and IR. ACS Med. Chem. Lett. 2010, 1, 510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jin M; Kleinberg A; Cooke A; Gokhale PC; Foreman K; Dong H; Siu KW; Bittner MA; Mulvihill KM; Yao Y; Landfair D; O’Connor M; Mak G; Pachter JA; Wild R; Rosenfeld-Franklin M; Ji Q; Mulvihill MJ Potent and selective cyclohexyl-derived imidazopyrazine insulin-like growth factor 1 receptor inhibitors with in vivo efficacy. Bioorg. Med. Chem. Lett. 2011, 21, 1176–1180. [DOI] [PubMed] [Google Scholar]
- 65.Mulvihill MJ; Cooke A; Rosenfeld-Franklin M; Buck E; Foreman K; Landfair D; O’Connor M; Pirritt C; Sun Y; Yao Y; Arnold LD; Gibson NW; Ji QS Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med. Chem. 2009, 1, 1153–1171. [DOI] [PubMed] [Google Scholar]
- 66.Wittman MD; Carboni JM; Yang Z; Lee FY; Antman M; Attar R; Balimane P; Chang C; Chen C; Discenza L; Frennesson D; Gottardis MM; Greer A; Hurlburt W; Johnson W; Langley DR; Li A; Li J; Liu P; Mastalerz H; Mathur A; Menard K; Patel K; Sack J; Sang X; Saulnier M; Smith D; Stefanski K; Trainor G; Velaparthi U; Zhang G; Zimmermann K; Vyas DM Discovery of a 2,4-disubstituted pyrrolo[1,2-f][1,2,4]triazine inhibitor (BMS-754807) of insulin-like growth factor receptor (IGF-1R) kinase in clinical development. J. Med. Chem. 2009, 52, 7360–7363. [DOI] [PubMed] [Google Scholar]
- 67.Zhao GS; Li WY; Chen DH; Henry JR; Li HY; Chen ZG; Zia-Ebrahimi M; Bloem L; Zhai Y; Huss K; Peng SB; McCann DJ A novel, selective inhibitor of fibroblast growth factor receptors that shows a potent broad spectrum of antitumor activity in several tumor xenograft models. Mol. Cancer Ther. 2011, 10, 2200–2210. [DOI] [PubMed] [Google Scholar]
- 68.Gavine PR; Mooney L; Kilgour E; Thomas AP; Al-Kadhimi K; Beck S; Rooney C; Coleman T; Baker D; Mellor MJ; Brooks AN; Klinowska T AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012, 72, 2045–2056. [DOI] [PubMed] [Google Scholar]
- 69.Cai ZW; Zhang Y; Borzilleri RM; Qian L; Barbosa S; Wei D; Zheng X; Wu L; Fan J; Shi Z; Wautlet BS; Mortillo S; Jeyaseelan R Sr.; Kukral DW; Kamath A; Marathe P; D’Arienzo C; Derbin G; Barrish JC; Robl JA; Hunt JT; Lombardo LJ; Fargnoli J; Bhide RS Discovery of brivanib alaninate ((S)-((R)-1-(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-5-methylpyrrolo[2,1-f][1,2,4] triazin-6-yloxy)propan-2-yl)2-aminopropanoate), a novel prodrug of dual vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1 kinase inhibitor (BMS-540215). J. Med. Chem. 2008, 51, 1976–1980. [DOI] [PubMed] [Google Scholar]
- 70.Weiss A; Littman DR Signal transduction by lymphocyte antigen receptors. Cell 1994, 76, 263–274. [DOI] [PubMed] [Google Scholar]
- 71.Kontzias A; Kotlyar A; Laurence A; Changelian P; O’Shea JJ Jakinibs: a new class of kinase inhibitors in cancer and autoimmune disease. Curr. Opin. Pharmacol. 2012, 12, 464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pesu M; Laurence A; Kishore N; Zwillich SH; Chan G; O’Shea JJ Therapeutic targeting of Janus kinases. Immunol. Rev. 2008, 223, 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Laurence A; Pesu M; Silvennoinen O; O’Shea J JAK kinases in health and disease: an update. Open Rheumatol. J. 2012, 6, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.O’Shea JJ; Schwartz DM; Villarino AV; Gadina M; McInnes IB; Laurence A The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Furumoto Y; Gadina M The arrival of JAK inhibitors: advancing the treatment of immune and hematologic disorders. BioDrugs 2013, 27, 431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ghoreschi K; Laurence A; O’Shea JJ Janus kinases in immune cell signaling. Immunol. Rev. 2009, 228, 273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.O’Shea JJ; Laurence A; McInnes IB Back to the future: oral targeted therapy for RA and other autoimmune diseases. Nat. Rev. Rheumatol. 2013, 9, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mesa RA; Yasothan U; Kirkpatrick P Ruxolitinib. Nat. Rev. Drug. Discov. 2012, 11, 103–104. [DOI] [PubMed] [Google Scholar]
- 79.Andreoli A; Verger E; Robin M; Raffoux E; Zini JM; Rousselot P; Socie G; Rea D; Parquet N; Giraudier S; Chomienne C; Cassinat B; Kiladjian JJ Clinical resistance to ruxolitinib is more frequent in patients without MPN-associated mutations and is rarely due to mutations in the JAK2 kinase drug-binding domain. Blood 2013, 122, 1591–1591. [Google Scholar]
- 80.FDA approves xeljanz for rheumatoid arthritis. https://www.drugs.com/newdrugs/fda-approves-xeljanz-rheumatoid-arthritis-3558.html (accessed Jan 12, 2019).
- 81.Clark JD; Flanagan ME; Telliez JB Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J. Med. Chem. 2014, 57, 5023–5038. [DOI] [PubMed] [Google Scholar]
- 82.Jiang JJJ; Wang XY; Zhang Y; Jin Y; Lin J Advances in the inhibitors of janus kinase. Med. Chem. 2014, 4, 540–548. [Google Scholar]
- 83.Jiang JK; Ghoreschi K; Deflorian F; Chen Z; Perreira M; Pesu M; Smith J; Nguyen DT; Liu EH; Leister W; Costanzi S; O’Shea JJ; Thomas CJ Examining the chirality, conformation and selective kinase inhibition of 3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-y l)-3-oxopropanenitrile (CP-690,550). J. Med. Chem. 2008, 51, 8012–8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boggon TJ; Li Y; Manley PW; Eck MJ Crystal structure of the Jak3 kinase domain in complex with a staurosporine analog. Blood 2005, 106, 996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O’Sullivan LA; Liongue C; Lewis RS; Stephenson SE; Ward AC Cytokine receptor signaling through the JAK-STAT-SOCS pathway in disease. Mol. Immunol. 2007, 44, 2497–2506. [DOI] [PubMed] [Google Scholar]
- 86.Baker SJ; Rane SG; Reddy EP Hematopoietic cytokine receptor signaling. Oncogene 2007, 26, 6724–6737. [DOI] [PubMed] [Google Scholar]
- 87.Menet CJ A dual inhibition, a better solution: development of a JAK1/TYK2 inhibitor. J. Med. Chem. 2018, 61, 8594–8596. [DOI] [PubMed] [Google Scholar]
- 88.Fensome A; Ambler CM; Arnold E; Banker ME; Brown MF; Chrencik J; Clark JD; Dowty ME; Efremov IV; Flick A; Gerstenberger BS; Gopalsamy A; Hayward MM; Hegen M; Hollingshead BD; Jussif J; Knafels JD; Limburg DC; Lin D; Lin TH; Pierce BS; Saiah E; Sharma R; Symanowicz PT; Telliez JB; Trujillo JI; Vajdos FF; Vincent F; Wan ZK; Xing L; Yang X; Yang X; Zhang L Dual inhibition of TYK2 and JAK1 for the treatment of autoimmune diseases: discovery of ((S)-2,2-difluorocyclopropyl)((1R,5S)-3-(2-((1-methyl-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octan-8-yl)methanone (PF-06700841). J. Med. Chem. 2018, 61, 8597–8612. [DOI] [PubMed] [Google Scholar]
- 89.Mohamed AJ; Yu L; Backesjo CM; Vargas L; Faryal R; Aints A; Christensson B; Berglof A; Vihinen M; Nore BF; Smith CI Bruton’s tyrosine kinase (BTK): function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 2009, 228, 58–73. [DOI] [PubMed] [Google Scholar]
- 90.Mohamed AJ; Nore BF; Christensson B; Smith CIE Signalling of bruton’s tyrosine kinase, BTK. Scand. J. Immun. 1999, 49, 113–118. [DOI] [PubMed] [Google Scholar]
- 91.Takata M A role for Bruton’s tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J. Exp. Med. 1996, 184, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu Q; Batt DG; Lippy JS; Surti N; Tebben AJ; Muckelbauer JK; Chen L, An Y, Chang C; Pokross M; Yang Z; Wang H; Burke JR; Carter PH; Tino JA Design and synthesis of carbazole carboxamides as promising inhibitors of Bruton’s tyrosine kinase (BTK) and Janus kinase 2 (JAK2) Bioorg. Med. Chem. Lett. 2015, 25, 4265–4269. [DOI] [PubMed] [Google Scholar]
- 93.De Lucca GV; Shi Q; Liu Q; Batt DG; Beaudoin Bertrand M; Rampulla R; Mathur A; Discenza L; D’Arienzo C; Dai J; Obermeier M; Vickery R; Zhang Y; Yang Z; Marathe P; Tebben AJ; Muckelbauer JK; Chang CJ; Zhang H; Gillooly K; Taylor T; Pattoli MA; Skala S; Kukral DW; McIntyre KW; Salter-Cid L; Fura A; Burke JR; Barrish JC; Carter PH; Tino JA Small molecule reversible inhibitors of bruton’s tyrosine kinase (BTK): structure-activity relationships leading to the identification of 7-(2-hydroxypropan-2-yl)-4-[2-methyl-3-(4-oxo-3,4-dihydroquinazolin-3-yl)phenyl]-9H-carbazole-1-carboxamide (BMS-935177). J. Med. Chem. 2016, 59, 7915–7935. [DOI] [PubMed] [Google Scholar]
- 94.Watterson SH; De Lucca GV; Shi Q; Langevine CM; Liu Q; Batt DG; Beaudoin Bertrand M; Gong H; Dai J; Yip S; Li P; Sun D; Wu DR; Wang C; Zhang Y; Traeger SC; Pattoli MA; Skala S; Cheng L; Obermeier MT; Vickery R; Discenza LN; D’Arienzo CJ; Zhang Y; Heimrich E; Gillooly KM; Taylor TL; Pulicicchio C; McIntyre KW; Galella MA; Tebben AJ; Muckelbauer JK; Chang C; Rampulla R; Mathur A; Salter-Cid L; Barrish JC; Carter PH; Fura A; Burke JR; Tino JA Discovery of 6-fluoro-5-(R)-(3-(S)-(8-fluoro-1-methyl-2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-1H-carbazole-8-carboxamide (BMS-986142): a reversible inhibitor of bruton’s tyrosine kinase (BTK) conformationally constrained by two locked atropisomers. J. Med. Chem. 2016, 59, 9173–9200. [DOI] [PubMed] [Google Scholar]
- 95.Di Paolo JA; Huang T; Balazs M; Barbosa J; Barck KH; Bravo BJ; Carano RA; Darrow J; Davies DR; DeForge LE; Diehl L; Ferrando R; Gallion SL; Giannetti AM; Gribling P; Hurez V; Hymowitz SG; Jones R; Kropf JE; Lee WP; Maciejewski PM; Mitchell SA; Rong H; Staker BL; Whitney JA; Yeh S; Young WB; Yu C; Zhang J; Reif K; Currie KS Specific BTK inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat. Chem. Biol. 2011, 7, 41–50. [DOI] [PubMed] [Google Scholar]
- 96.Young WB; Barbosa J; Blomgren P; Bremer MC; Crawford JJ; Dambach D; Gallion S; Hymowitz SG; Kropf JE; Lee SH; Liu L; Lubach JW; Macaluso J; Maciejewski P; Maurer B; Mitchell SA; Ortwine DF; Di Paolo J; Reif K; Scheerens H; Schmitt A; Sowell CG; Wang X; Wong H; Xiong JM; Xu J; Zhao Z; Currie KS Potent and selective Bruton’s tyrosine kinase inhibitors: discovery of GDC-0834. Bioorg. Med. Chem. Lett. 2015, 25, 1333–1337. [DOI] [PubMed] [Google Scholar]
- 97.Alexander SP; Christopoulos A; Davenport AP; Kelly E; Marrion NV; Peters JA; Faccenda E; Harding SD; Pawson AJ; Sharman JL; Southan C; Davies JA The concise guide to pharmacology 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 2017, 174 Suppl 1, S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Boulton TG; Nye SH; Robbins DJ; Ip NY; Radzlejewska E; Morgenbesser SD; DePinho RA; Panayotatos N; Cobb MH; Yancopoulos GD ERKs: A family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 1991, 65, 663–675. [DOI] [PubMed] [Google Scholar]
- 99.Nestler EJ; Greengard P Protein serine-threonine kinases. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects.;Siegel GJ; B.W A; Albers RW, Eds.; Lippincott-Raven: Philadelphia, 1999. [Google Scholar]
- 100.Shen B; Manley JL Pelle kinase is activated by autophosphorylation during Toll signaling in Drosophila. Development 2002, 129, 1925–1933. [DOI] [PubMed] [Google Scholar]
- 101.Losiewicz MD; Carlson BA; Kaur G; Sausville EA; Worland PJ Potent inhibition of CDC2 kinase activity by the flavonoid L86–8275. Biochem. Biophys. Res. Commun. 1994, 201, 589–595. [DOI] [PubMed] [Google Scholar]
- 102.Meijer LB, A.; Mulner O; Chong JP; Blow JJ; Inagaki N; Inagaki M; Delcros JG Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases CDC2, CDK2 and CDK5. FEBS 1997, 243, 527. [DOI] [PubMed] [Google Scholar]
- 103.Parry D; Guzi T; Shanahan F; Davis N; Prabhavalkar D; Wiswell D; Seghezzi W; Paruch K; Dwyer MP; Doll R; Nomeir A; Windsor W; Fischmann T; Wang Y; Oft M; Chen T; Kirschmeier P; Lees EM Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer. Ther. 2010, 9, 2344–2353. [DOI] [PubMed] [Google Scholar]
- 104.Larkin J; Ascierto PA; Dreno B; Atkinson V; Liszkay G; Maio M; Mandala M; Demidov L; Stroyakovskiy D; Thomas L; de la Cruz-Merino L; Dutriaux C; Garbe C; Sovak MA; Chang I; Choong N; Hack SP; McArthur GA; Ribas A Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [DOI] [PubMed] [Google Scholar]
- 105.Andrlova H; Zeiser R; Meiss F Cobimetinib (GDC-0973, XL518). Recent Results Cancer Res. 2018, 211, 177–186. [DOI] [PubMed] [Google Scholar]
- 106.Lee KL; Ambler CM; Anderson DR; Boscoe BP; Bree AG; Brodfuehrer JI; Chang JS; Choi C; Chung S; Curran KJ; Day JE; Dehnhardt CM; Dower K; Drozda SE; Frisbie RK; Gavrin LK; Goldberg JA; Han S; Hegen M; Hepworth D; Hope HR; Kamtekar S; Kilty IC; Lee A; Lin LL; Lovering FE; Lowe MD; Mathias JP; Morgan HM; Murphy EA; Papaioannou N; Patny A; Pierce BS; Rao VR; Saiah E; Samardjiev IJ; Samas BM; Shen MWH; Shin JH; Soutter HH; Strohbach JW; Symanowicz PT; Thomason JR; Trzupek JD; Vargas R; Vincent F; Yan J; Zapf CW; Wright SW Discovery of clinical candidate 1-{[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}−7-methoxyisoquinoli ne-6-carboxamide (PF-06650833), a potent, selective inhibitor of interleukin-1 receptor associated kinase 4 (IRAK4), by fragment-based drug design. J. Med. Chem. 2017, 60, 5521–5542. [DOI] [PubMed] [Google Scholar]
- 107.Festjens N; Vanden Berghe T; Cornelis S; Vandenabeele P RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 2007, 14, 400–410. [DOI] [PubMed] [Google Scholar]
- 108.Kelliher MA; Grimm S; Ishida Y; Kuo F; Stanger BZ; Leder P The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 1998, 8, 297–303. [DOI] [PubMed] [Google Scholar]
- 109.Micheau O; Tschopp J Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [DOI] [PubMed] [Google Scholar]
- 110.Harris PA; Berger SB; Jeong JU; Nagilla R; Bandyopadhyay D; Campobasso N; Capriotti CA; Cox JA; Dare L; Dong X; Eidam PM; Finger JN; Hoffman SJ; Kang J; Kasparcova V; King BW; Lehr R; Lan Y; Leister LK; Lich JD; MacDonald TT; Miller NA; Ouellette MT; Pao CS; Rahman A; Reilly MA; Rendina AR; Rivera EJ; Schaeffer MC; Sehon CA; Singhaus RR; Sun HH; Swift BA; Totoritis RD; Vossenkamper A; Ward P; Wisnoski DD; Zhang D; Marquis RW; Gough PJ and Bertin J Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [DOI] [PubMed] [Google Scholar]
- 111.Xie T; Peng W; Liu Y; Yan C; Maki J; Degterev A; Yuan J; Shi Y Structural basis of RIP1 inhibition by necrostatins. Structure 2013, 21, 493–499. [DOI] [PubMed] [Google Scholar]
- 112.Zitouni S; Nabais C; Jana SC; Guerrero A; Bettencourt-Dias M Polo-like kinases: structural variations lead to multiple functions. Nat. Rev. Mol. Cell. Biol. 2014, 15, 433–452. [DOI] [PubMed] [Google Scholar]
- 113.Lee SY; Jang C; Lee KA Polo-like kinases (plks), a key regulator of cell cycle and new potential target for cancer therapy. Dev. Reprod. 2014, 18, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Emmitte KA; Adjabeng GM; Andrews CW; Alberti JG; Bambal R; Chamberlain SD; Davis-Ward RG; Dickson HD; Hassler DF; Hornberger KR; Jackson JR; Kuntz KW; Lansing TJ; Mook RA Jr.; Nailor KE; Pobanz MA; Smith SC; Sung CM; Cheung M Design of potent thiophene inhibitors of polo-like kinase 1 with improved solubility and reduced protein binding. Bioorg. Med. Chem. Lett. 2009, 19, 1694–1697. [DOI] [PubMed] [Google Scholar]
- 115.Toker A; Cantley LC Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature 1997, 387, 673–676. [DOI] [PubMed] [Google Scholar]
- 116.Fruman DA; Rommel C PI3K and cancer: lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jean S; Kiger AA Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 2014, 127, 923–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhao W; Qiu Y; Kong D Class I phosphatidylinositol 3-kinase inhibitors for cancer therapy. Acta Pharm. Sin. B 2017, 7, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chalhoub N; Baker SJ PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.FDA Approves Zydelig (idelalisib) for CLL and Lymphoma. https://www.drugs.com/newdrugs/fda-approves-zydelig-idelalisib-cll-lymphoma-4056.html (accessed Aug 23, 2018).
- 121.Polak R; Buitenhuis M The PI3K/PKB signaling module as key regulator of hematopoiesis: implications for therapeutic strategies in leukemia. Blood 2012, 119, 911–923. [DOI] [PubMed] [Google Scholar]
- 122.Martelli AM; Evangelisti C; Chiarini F; Grimaldi C; McCubrey JA The emerging role of the phosphatidylinositol 3-kinase/ akt/mammalian target of rapamycin signaling network in cancer stem cell biology. Cancers 2010, 2, 1576–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Meadows SA; Vega F; Kashishian A; Johnson D; Diehl V; Miller LL; Younes A; Lannutti BJ PI3Kdelta inhibitor, GS-1101 (CAL-101), attenuates pathway signaling, induces apoptosis, and overcomes signals from the microenvironment in cellular models of hodgkin lymphoma. Blood 2012, 119, 1897–1900. [DOI] [PubMed] [Google Scholar]
- 124.Sadhu C; Masinovsky B; Dick K; Sowell CG; Staunton DE Essential role of phosphoinositide 3-kinase in neutrophil directional movement. J. Immunol. 2003, 170, 2647–2654. [DOI] [PubMed] [Google Scholar]
- 125.Berndt A; Miller S; Williams O; Le DD; Houseman BT; Pacold JI; Gorrec F; Hon WC; Liu Y; Rommel C; Gaillard P; Ruckle T; Schwarz MK; Shokat KM; Shaw JP; Williams RL The p110 delta structure: mechanisms for selectivity and potency of new PI(3)K inhibitors. Nat. Chem. Biol. 2010, 6, 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yang Q; Modi P; Newcomb T; Queva C; Gandhi V Idelalisib: first-in-class PI3K delta inhibitor for the treatment of chronic lymphocytic leukemia, small lymphocytic leukemia, and follicular lymphoma. Clin. Cancer Res. 2015, 21, 1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Do B; Mace M; Rexwinkle A Idelalisib for treatment of B-cell malignancies. Am. J. Health Syst. Pharm. 2016, 73, 547–555. [DOI] [PubMed] [Google Scholar]
- 128.Lannutti BJ; Meadows SA; Herman SE; Kashishian A; Steiner B; Johnson AJ; Byrd JC; Tyner JW; Loriaux MM; Deininger M; Druker BJ; Puri KD; Ulrich RG; Giese NA CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 2011, 117, 591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Somoza JR; Koditek D; Villasenor AG; Novikov N; Wong MH; Liclican A; Xing W; Lagpacan L; Wang R; Schultz BE; Papalia GA; Samuel D; Lad L; McGrath ME Structural, biochemical, and biophysical characterization of idelalisib binding to phosphoinositide 3-kinase delta. J. Biol. Chem. 2015, 290, 8439–8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Smith AL; D’Angelo ND; Bo YY; Booker SK; Cee VJ; Herberich B; Hong FT; Jackson CL; Lanman BA; Liu L; Nishimura N; Pettus LH; Reed AB; Tadesse S; Tamayo NA; Wurz RP; Yang K; Andrews KL; Whittington DA; McCarter JD; Miguel TS; Zalameda L; Jiang J; Subramanian R; Mullady EL; Caenepeel S; Freeman DJ; Wang L; Zhang N; Wu T; Hughes PE; Norman MH Structure-based design of a novel series of potent, selective inhibitors of the class I phosphatidylinositol 3-kinases. J. Med. Chem. 2012, 55, 5188–5219. [DOI] [PubMed] [Google Scholar]
- 131.Norman MH; Andrews KL; Bo YY; Booker SK; Caenepeel S; Cee VJ; D’Angelo ND; Freeman DJ; Herberich BJ; Hong FT; Jackson CL; Jiang J; Lanman BA; Liu L; McCarter JD; Mullady EL; Nishimura N; Pettus LH; Reed AB; Miguel TS; Smith AL; Stec MM; Tadesse S; Tasker A; Aidasani D; Zhu X; Subramanian R; Tamayo NA; Wang L; Whittington DA; Wu B; Wu T; Wurz RP; Yang K; Zalameda L; Zhang N; Hughes PE Selective class I phosphoinositide 3-kinase inhibitors: optimization of a series of pyridyltriazines leading to the identification of a clinical candidate, AMG 511. J. Med. Chem. 2012, 55, 7796–7816. [DOI] [PubMed] [Google Scholar]
- 132.AMG 319 in HPV Positive and Negative HNSCC. https://clinicaltrials.gov/ct2/show/NCT02540928 (accessed Oct 18, 2018).
- 133.Cushing TD; Hao X; Shin Y; Andrews K; Brown M; Cardozo M; Chen Y; Duquette J; Fisher B; Gonzalez-Lopez de Turiso F; He X; Henne KR; Hu YL; Hungate R; Johnson MG; Kelly RC; Lucas B; McCarter JD; McGee LR; Medina JC; San Miguel T; Mohn D; Pattaropong V; Pettus LH; Reichelt A; Rzasa RM; Seganish J; Tasker AS; Wahl RC; Wannberg S; Whittington DA; Whoriskey J; Yu G; Zalameda L; Zhang D; Metz DP Discovery and in vivo evaluation of (S)-N-(1-(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine (AMG319) and related PI3Kdelta inhibitors for inflammation and autoimmune disease. J. Med. Chem. 2015, 58, 480–511. [DOI] [PubMed] [Google Scholar]
- 134.Lin H; Erhard K; Hardwicke MA; Luengo JI; Mack JF; McSurdy-Freed J; Plant R; Raha K; Rominger CM; Sanchez RM; Schaber MD; Schulz MJ; Spengler MD; Tedesco R; Xie R; Zeng JJ; Rivero RA Synthesis and structure-activity relationships of imidazo[1,2-a]pyrimidin-5(1H)-ones as a novel series of beta isoform selective phosphatidylinositol 3-kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 2230–2234. [DOI] [PubMed] [Google Scholar]
- 135.Knight ZA; Gonzalez B; Feldman ME; Zunder ER; Goldenberg DD; Williams O; Loewith R; Stokoe D; Balla A; Toth B; Balla T; Weiss WA; Williams RL; Shokat KM A pharmacological map of the PI3K family defines a role for p110alpha in insulin signaling. Cell 2006, 125, 733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Barlaam B; Cosulich S; Degorce S; Fitzek M; Giordanetto F; Green S; Inghardt T; Hennequin L; Hancox U; Lambert-van der Brempt C; Morgentin R; Pass S; Ple P; Saleh T; Ward L Discovery of 9-(1-anilinoethyl)-2-morpholino-4-oxo-pyrido[1,2-a]pyrimidine-7-carboxamides as PI3Kbeta/delta inhibitors for the treatment of PTEN-deficient tumours. Bioorg. Med. Chem. Lett. 2014, 24, 3928–3935. [DOI] [PubMed] [Google Scholar]
- 137.Nylander S; Kull B; Bjorkman JA; Ulvinge JC; Oakes N; Emanuelsson BM; Andersson M; Skarby T; Inghardt T; Fjellstrom O; Gustafsson D Human target validation of phosphoinositide 3-kinase (PI3K)beta: effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kbeta inhibitor. J. Thromb. Haemost. 2012, 10, 2127–2136. [DOI] [PubMed] [Google Scholar]
- 138.Barlaam B; Cosulich S; Degorce S; Fitzek M; Green S; Hancox U; Lambert-van der Brempt C; Lohmann JJ; Maudet M; Morgentin R; Pasquet MJ; Peru A; Ple P; Saleh T; Vautier M; Walker M; Ward L; Warin N Discovery of (R)-8-(1-(3,5-difluorophenylamino)ethyl)-N,N-dimethyl-2-morpholino-4-oxo-4H-chrom ene-6-carboxamide (AZD8186): a potent and selective inhibitor of PI3Kbeta and PI3Kdelta for the treatment of PTEN-deficient cancers. J. Med. Chem. 2015, 58, 943–962. [DOI] [PubMed] [Google Scholar]
- 139.Williams O; Houseman BT; Kunkel EJ; Aizenstein B; Hoffman R; Knight ZA; Shokat KM Discovery of dual inhibitors of the immune cell PI3Ks p110delta and p110gamma: a prototype for new anti-inflammatory drugs. Chem. Biol. 2010, 17, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Erra M; Taltavull J; Bernal FJ; Caturla JF; Carrascal M; Pages L; Mir M; Espinosa S; Gracia J; Dominguez M; Sabate M; Paris S; Maldonado M; Hernandez B; Bravo M; Calama E; Miralpeix M; Lehner MD; Calbet M Discovery of a novel inhaled PI3Kdelta inhibitor for the treatment of respiratory diseases. J. Med. Chem. 2018, 61, 9551–9567. [DOI] [PubMed] [Google Scholar]
- 141.Erra M; Taltavull J; Greco A; Bernal FJ; Caturla JF; Gracia J; Dominguez M; Sabate M; Paris S; Soria S; Hernandez B; Armengol C; Cabedo J; Bravo M; Calama E; Miralpeix M; Lehner MD Discovery of a potent, selective, and orally available PI3Kdelta inhibitor for the treatment of inflammatory diseases. ACS Med. Chem. Lett. 2017, 8, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Garces AE; Stocks MJ Class 1 PI3K clinical candidates and recent inhibitor design strategies: a medicinal chemistry perspective. J. Med. Chem.2019, 62, 4815–4850. [DOI] [PubMed] [Google Scholar]
- 143.Zhao Z; Bourne PE Progress with covalent small-molecule kinase inhibitors. Drug Discov. Today 2018, 23, 727–735. [DOI] [PubMed] [Google Scholar]
- 144.Liu Q; Sabnis Y; Zhao Z; Zhang T; Buhrlage SJ; Jones LH; Gray NS Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hur W; Velentza A; Kim S; Flatauer L; Jiang X; Valente D; Mason DE; Suzuki M; Larson B; Zhang J; Zagorska A; Didonato M; Nagle A; Warmuth M; Balk SP; Peters EC; Gray NS Clinical stage EGFR inhibitors irreversibly alkylate Bmx kinase. Bioorg. Med. Chem. Lett. 2008, 18, 5916–5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bordoni RE Afatinib (BIBW-2992): a novel dual EGFR/HER2neu inhibitor with promising activity in non-small-cell lung cancer. Therapy 2011, 8, 15–22. [Google Scholar]
- 147.Hirsh V Afatinib (BIBW 2992) development in non-small-cell lung cancer. Future Oncol. 2011, 7, 817–825. [DOI] [PubMed] [Google Scholar]
- 148.Solca F; Dahl G; Zoephel A; Bader G; Sanderson M; Klein C; Kraemer O; Himmelsbach F; Haaksma E; Adolf GR Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [DOI] [PubMed] [Google Scholar]
- 149.Stopfer P; Marzin K; Narjes H; Gansser D; Shahidi M; Uttereuther-Fischer M; Ebner T Afatinib pharmacokinetics and metabolism after oral administration to healthy male volunteers. Cancer Chemother. Pharmacol. 2012, 69, 1051–1061. [DOI] [PubMed] [Google Scholar]
- 150.Pan Z; Scheerens H; Li SJ; Schultz BE; Sprengeler PA; Burrill LC; Mendonca RV; Sweeney MD; Scott KC; Grothaus PG; Jeffery DA; Spoerke JM; Honigberg LA; Young PR; Dalrymple SA; Palmer JT Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. Chem. Med. Chem. 2007, 2, 58–61. [DOI] [PubMed] [Google Scholar]
- 151.Roskoski R Jr. Ibrutinib inhibition of bruton protein-tyrosine kinase (BTK) in the treatment of B cell neoplasms. Pharmacol. Res. 2016, 113, 395–408. [DOI] [PubMed] [Google Scholar]
- 152.Walter HS; Rule SA; Dyer MJ; Karlin L; Jones C; Cazin B; Quittet P; Shah N; Hutchinson CV; Honda H; Duffy K; Birkett J; Jamieson V; Courtenay-Luck N; Yoshizawa T; Sharpe J; Ohno T; Abe S; Nishimura A; Cartron G; Morschhauser F; Fegan C; Salles G A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood 2016, 127, 411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Byrd JC; Harrington B; O’Brien S; Jones JA; Schuh A; Devereux S; Chaves J; Wierda WG; Awan FT; Brown JR; Hillmen P; Stephens DM; Ghia P; Barrientos JC; Pagel JM; Woyach J; Johnson D; Huang J; Wang X; Kaptein A; Lannutti BJ; Covey T; Fardis M; McGreivy J; Hamdy A; Rothbaum W; Izumi R; Diacovo TG; Johnson AJ; Furman RR Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2016, 374, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Harrison RK Phase II and phase III failures: 2013–2015. Nat. Rev. Drug Discov. 2016, 15, 817–818. [DOI] [PubMed] [Google Scholar]