


Abstract
Methyl-CpG-binding protein 2 (MeCP2) contains a transcriptional repression domain (TRD), which can act by recruitment of a large transcriptional co-repressor complex containing histone deacetylases HDAC1 and 2. We demonstrate here that transient transcription from the SV40 enhancer/promoter or the SV40 promoter is strongly repressed in a histone deacetylase-independent manner, since repression is not alleviated by Trichostatin A (TSA). In a mutational analysis, repression depends on a conserved 30 residue sequence containing two clusters of basic amino acids. Mutation of the first of these clusters inhibits in vitro interaction between TRD and mSin3A. Furthermore, a subdomain of the TRD containing the conserved 30-residue sequence and 16 flanking amino acids was sufficient to compromise VP16-activated transcription. In summary, our results indicate an alternative, histone deacetylase-independent pathway of transcriptional repression by MeCP2.
INTRODUCTION
DNA methylation is essential for mammalian development and crucial for processes like imprinting, repression of gene activity and cancer. It has been long thought that the information encoded in the methylation of distinct sequences is transmitted by proteins, which specifically recognize methylated CpGs. The first reported member of this group of proteins is methyl-CpG-binding protein 2 (MeCP2) (1). It can bind to a single methylated CpG through a domain of 85 amino acids, termed the methyl-CpG-binding domain (MBD) (2). The protein is a highly abundant component in the heavily methylated pericentromeric heterochromatin of mouse chromosomes (1) and a transiently expressed MeCP2 fusion protein is targeted to methylated heterochromatin (3). The chicken homolog of MeCP2 was identified through the ability to bind nuclear matrix attachment regions (MARs), the putative anchorage sites of chromatin loop domains and named attachment region-binding protein (ARBP) (4). The chicken and the human protein can bind to MARs and methylated as well as unmethylated mouse satellite DNA in vitro and in vivo (1,5–7). The domain specifically recognizing MAR DNA (ARBD) comprises 125 amino acids and includes, in addition to the 85 amino acid MBD, sequence motifs known to bind in the minor groove of the DNA helix (6).
MeCP2 further contains a domain that represses transcription in vitro and in vivo, named the transcriptional repression domain (TRD) (8). A deletion mutational analysis localized the TRD to residues 207–310. Through this domain MeCP2 interacts with the co-repressor mSin3A, which in turn is part of a large co-repressor complex containing histone deacetylases HDAC1 and 2 (9,10). Repression by MeCP2 is (partially) relieved by Trichostatin A (TSA), a specific inhibitor of histone deacetylases. Thus it is thought that long-term silencing of methylated sequences is executed through the generation of a deacetylated inactive chromatin configuration.
However, several observations indicate that transcriptional repression by MeCP2 might also operate through an alternative mechanism. First, MeCP2 represses transcription from naked DNA in an in vitro system, which presumably does not reconstitute nucleosomes (8). Secondly, transcriptional repression of the human β-actin promoter is only partially relieved by Trichostatin A (TSA). Third, endogeneous genes silenced by CpG island methylation cannot be reactivated by TSA without prior partial demethylation of the DNA (11). Fourth, demethylation of the inactive promoter of the FMR1 gene in cells from fragile X patients resulted in association of acetylated histones H3 and H4 with FMR1 and transcriptional activation (12). TSA also led to acetylation of histones H4 and H3, but no detectable transcription. These findings led us to search for an alternative, histone deacetylase-independent pathway of transcriptional repression by MeCP2. We demonstrate that transient transcription from the SV40 enhancer/promoter or the SV40 promoter is strongly repressed in a TSA-insensitive manner. A mutational analysis revealed that repression depends on a conserved 30 residue sequence containing a cluster of critical basic amino acids.
MATERIALS AND METHODS
Plasmid constructs
Plasmids expressing fusion proteins of the Gal4 DNA-binding domain and the C-terminal half (amino acids 196–486) of MeCP2 (Gal4–TRD), amino acids 196–310 (Gal4–mTRD) or amino acids 311–486 (Gal4–CD) were constructed by ligation of the respective cDNA fragments of human MeCP2 into the vector pcDNA3-Gal4BD (a gift from J. Han). The control reporter vector contained the firefly luciferase gene driven by the SV40 enhancer/promoter (a gift from J. Bode) or by the SV40 promoter, which includes the three 21 bp repeats and the proximal AP-1 site (pGL3-Promoter; Promega). A sequence containing five Gal4 binding sites obtained by BamHI digestion of pFR-LUC (Stratagene) was inserted into the BamHI site of the control vector. Furthermore, the SV40 enhancer/promoter of the control vector and its Gal4 derivative was replaced by a 409 bp adenovirus type 5 major late (AdML) promoter fragment. Expression plasmids for GST fusion proteins were constructed by insertion of PCR-amplified cDNA fragments of MeCP2 into vector pGEX-2T. Mutation of segments A–F was carried out by PCR with appropriate primers. Double and quadruple mutations in segment D were generated using the QuikChange site-directed mutagenesis kit (Stratagene). To generate plasmids expressing the triple fusion proteins Gal4–mTRD–VP16 and Gal4–DE–VP16, sequences encoding mTRD and DE were amplified by PCR using primers encompassing KpnI sites. The products were cloned into the KpnI site separating the sequences for Gal4 and VP16 in plasmid pcDNA3-gal4-VP16. Constructs using amplified sequences and mutated plasmids were sequenced for confirmation.
Cell culture and transfection
Transient transfection of human HEK293 cells, an adenovirus-transformed embryonic kidney cell line (13), was performed by the calcium phosphate co-precipitation method. NIH 3T3 cells were transiently transfected using the Fugene 6 transfection kit (Roche). Cells were seeded in tissue culture dishes 35 mm in diameter and grown in Dulbecco’s modified Eagle’s medium (Life Technologies) supplemented with 10% fetal calf serum overnight before transfection. Firefly luciferase reporter plasmid (1.0 µg) and renilla luciferase reference plasmid pRL-TK (Promega) (0.05 µg) were co-transfected with 0.1 µg of effector plasmid. After overnight incubation, the medium and precipitates were removed and replaced with fresh medium containing 100 ng/ml TSA where indicated. Forty-eight hours after transfection, the cell monolayer was washed with phosphate-buffered saline, harvested by scraping and resuspended in PLB buffer (Promega). Firefly and renilla luciferase activities were assayed with the Dual-Luciferase Assay (Promega), following the instructions of the manufacturer, and firefly luciferase activities were standardized to renilla ones.
GST pull-down assay
Equal amounts (6 µg) of bacterially expressed GST and GST fusion proteins were immobilized on glutathione–Sepharose beads and then incubated for 2 h at 4°C with 60 µg of HEK293 nuclear extract (14) in buffer A of Nan et al. (9). Beads were washed five times with binding buffer and bound proteins were separated by SDS–PAGE and analyzed by western blotting using anti-mSin3A (K-20; Santa Cruz Biotechnology).
Microscopy
NIH 3T3 cells were fixed at 1% formaldehyde, permeabilized with cold methanol and then incubated for 20 min with anti-FLAG antibody M2 (Eastman Kodak Co.). After washing, slides were incubated for 20 min with fluorescein isothiocyanate-conjugated anti-mouse IgG antibody (Dianova) and 0.05% DAPI to detect anti-FLAG binding and DNA, respectively. Photomicrographs were taken by epifluorescence using a Leitz Orthoplan microscope with filter sets I2 and A.
RESULTS
The TRD of MeCP2 can repress transcription through a histone deacetylase-independent pathway
We sought to examine alternative pathways for the transcription-repressing activity of MeCP2. In transient transfection studies we used a reporter plasmid containing the SV40 enhancer/promoter (SV40) driving the luciferase gene. When Gal4 DNA-binding sites were cloned upstream of the SV40 enhancer and a fusion protein containing the DNA-binding domain of Gal4 and the C-terminal half of human MeCP2 (amino acids 196–486, including the TRD) (Gal4–TRD) was co-expressed in adenovirus-transformed HEK293 cells, SV40 enhancer/promoter activity was dramatically (on average 85%) inhibited (Fig. 1A). Surprisingly, the specific histone deacetylase inhibitor TSA failed to relieve this repression. Identical results were obtained (Fig. 1B) when the fusion protein contained the previously characterized TRD proper, here named miniTRD (mTRD, amino acids 196–310) (8). TSA was unable to alleviate the dramatic transcriptional repression of the SV40 enhancer/promoter by the miniTRD. The C-terminal 176 residues (C-terminal domain, CD) exhibit weak (on average 38%) inhibition of enhancer/promoter activity (Fig. 1C). Again, this repression was not alleviated by TSA. These results appear to indicate that repression of the SV40 enhancer/promoter by the TRD of MeCP2 acts through a novel, histone deacetylase-independent mechanism. When the SV40 enhancer/promoter was substituted by the adenovirus major late (AdML) promoter, the TRD in Gal4–TRD inhibited this promoter in HEK293 cells by only 25% and TSA was able to relieve this repression completely (Fig. 1D). This result is comparable to the previous observation that MeCP2 inhibits transcription from the human β-actin promoter in a TSA-sensitive manner (8). Taken together, the present and previous in vivo experiments indicate that the mode of action of the TRD of MeCP2 is dependent upon the promoter context. Within the same cell type (HEK293) two different pathways can be differentiated, a histone deacetylase-independent one and a histone deacetylase-dependent one.
Figure 1.

The TRD of MeCP2 can repress transcription through a histone deacetylase-independent pathway. Expression plasmids coded for fusion proteins between the Gal4 DNA-binding domain and the C-terminal half (TRD, amino acids 196–486) (A and D), the miniTRD (mTRD, amino acids 196–310) (B) or the C-terminal 176 residues (CD, amino acids 311–486) (C) of MeCP2. They were co-transfected with the reporter vector SV40 enhancer/promoter–luciferase, Gal4–SV40 enhancer/promoter–luciferase, AdML promoter–luciferase or Gal4–AdML promoter–luciferase into HEK293 cells. Where indicated, TSA was added after 24 h. Normalized relative luciferase activity (RLU) in the absence of Gal4 sites and TSA was set as 1.0. Results are representative of at least five separate experiments, each in triplicate (± SD).
To exclude the possibility that in adenovirus-transformed HEK293 cells transcriptional repression by the TRD is mediated by interference with any interaction of the viral oncoprotein E1A with cellular transcriptional activators or co-activators (see for example 15,16), we repeated the transfection experiments in NIH 3T3 cells. The TRD (amino acids 196–486) as well as the miniTRD (amino acids 196–310) repressed SV40 enhancer/promoter activity significantly (Fig. 2A and B). TSA failed to relieve repression by the miniTRD (Fig. 2B) and weakly alleviated repression by the TRD (Fig. 2A). This weak alleviation was likely due to a general transcription-enhancing effect of TSA, which is also observed in the control (reporter lacking a Gal4 recognition site). In summary, the histone deacetylase-independent pathway of repression by MeCP2 is cell type independent. To further investigate whether repression by TRD is dependent on the SV40 enhancer, we employed a reporter driven by the SV40 promoter solely containing the three 21 bp repeats (Sp1-binding region) and the promoter proximal AP-1-binding site, but lacking the remaining portion of the enhancer. The TRD and the miniTRD repressed this promoter as efficiently as the complete SV40 enhancer/promoter (Fig. 2C and D). TSA slightly increased the promoter activity in the controls as well as in the repressed state, again likely reflecting a general transcription-enhancing effect. Yet TSA did not abrogate the TRD-induced repression.
Figure 2.

Repression by TRD of the SV40 enhancer/promoter in NIH 3T3 cells and of the SV40 promoter in HEK293 cells. (A and B) Histone deacetylase-independent repression by TRD in NIH 3T3 cells. NIH 3T3 cells were transfected with reporter and expression constructs as in Figure 1A and B. (C and D) Repression of the SV40 promoter. HEK293 cells were transfected with the indicated expression plasmids and reporter constructs driven by the SV40 promoter containing the 21-bp repeats and the promoter proximal AP-1 site. Results are representative of at least three separate experiments, each in triplicate (± SD).
Mutational analysis of the TRD
The TRD of MeCP2 was previously localized to amino acids 207–310 (8). We performed a more detailed mutational analysis to determine the protein sequences important for each of the two suggested modes of repression, the histone deacetylase-independent one and the dependent one. In the Gal4–TRD fusion protein, the region amino acids 196–311 encompassing the TRD was mutated at six selected segments (A–F) of nine amino acids each by replacement through the FLAG-epitope sequence plus an additional valine (VDYKDDDDK). Figure 3A aligns the sequence of human MeCP2 from amino acids 196 to 318 to those of rat, Xenopus and chicken, excluding poly(Pro) (Pn) and poly(Gly) (Gn) runs of the chicken sequence that are likely generated by trinucleotide amplifications and do not occur within the mammalian and Xenopus MeCP2s (6). While the sequences of segments A and C diverge significantly, segments B and D–F are highly conserved from mammals to Xenopus. Since segment D is contained within the bipartite nuclear localization signal of MeCP2 (3), we first verified by cytological studies and western blotting analysis using an anti-FLAG antibody that all mutated fusion proteins accumulate in the nucleus and were expressed in equivalent amounts (Fig. 3B and western blotting data not shown). Measuring repression of SV40 enhancer/promoter activity, we found that the repressing activity of TRD wild-type (wt) was completely abrogated by mutation D and was greatly reduced by mutation E (Fig. 3C, left). This indicates that repression of the SV40 enhancer/promoter depends on a distinct amino acid sequence within the TRD of MeCP2. Repression of the AdML promoter was only weakly reduced by mutations D and E (Fig. 3C, right). In segment D a sequence of six amino acids, PKKRGK (amino acids 265–270), is highly conserved from human to Xenopus. We explored the role of this sequence by three double mutations, M1–M3, and a quadruple mutation, M4 (Fig. 4A). Change of the central KR267/268 to EE affected a decrease in the repressing activity of TRD-wt by 23.3 ± 7.0% (Fig. 4B). Statistical analysis by a parametric analysis (Student’s t-test) revealed that this decrease is significant (P < 0.05). Change of a total of four basic residues to acidic ones in mutation M4 (KRRK267/268/270/271EEEE) decreased the repressing activity of TRD by 55.3%. In summary, our mutational analysis localizes the TRD of MeCP2 to a conserved 30 residue region containing two clusters of basic amino acids. This region shows no significant similarity to the repression domains of Mad1 and Ume6 (17,18). The more detailed mutational analysis in segment D is in support of the notion that basic residues exert a critical role.
Figure 3.

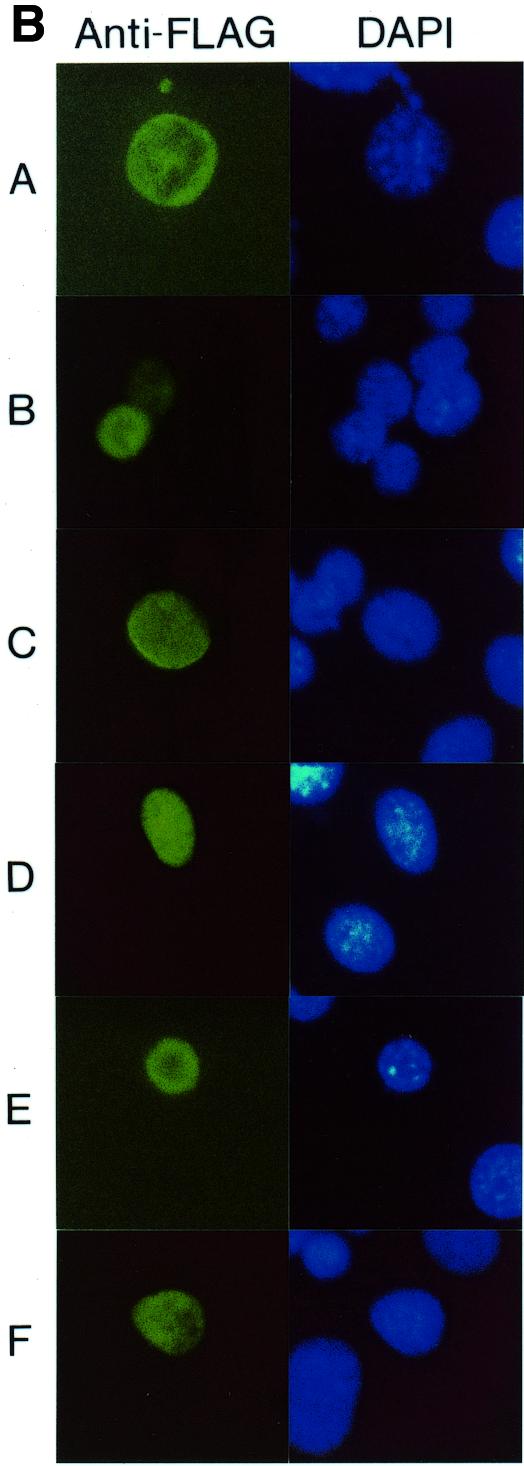

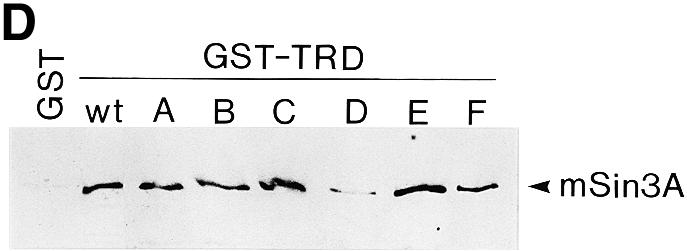
Mutational analysis of the TRD. (A) Sequence comparison of the TRDs from human (h), rat (r), Xenopus (x) and chicken (c) MeCP2 (accession nos Y12643, M94064, AF051768 and Y14166). Only the chicken sequence contains a run of glycine residues (Gn) and a poly(Pro) run (Pn), both of which are thought to be generated by trinucleotide amplifications (6). Gaps in the sequence are marked by dots. Six segments of nine amino acids each (A–F), indicated by bars above the human sequence, were mutated by replacement by the sequence VDYKDDDDK. The arrow indicates location of a nonsense mutation (R255X) in a Rett patient predicting a truncated protein lacking segments D and E (23). NLS, nuclear localization signal (3). (B) Nuclear localization of Gal4–TRD mutants A–F. Plasmids expressing Gal4–TRD mutants A–F were transfected into NIH 3T3 cells. Fixed slides were incubated with anti-FLAG antibody to detect Gal4–TRD mutant fusion proteins and stained for DNA with DAPI. (C) Mutational analysis of the trans-repressing activity of the TRD. The Gal4–SV40 enhancer/promoter–luciferase reporter (left) or the Gal4–AdML promoter–luciferase reporter (right) were transfected alone (–) or co-transfected with plasmids encoding Gal4–TRD wild-type (wt) or Gal4–TRD mutant A–F fusion proteins into HEK293 cells. Results represent means (± SD) of at least four independent experiments, each in triplicate. (D) In vitro binding of TRD mutants A–F to cellular mSin3A. In a GST pull-down assay, equal amounts of immobilized GST, GST–TRD wild-type (wt) or GST–TRD mutants A–F were incubated with nuclear HEK293 extracts. Bound proteins were analyzed by western blotting using anti-mSin3A antibody (K-20).
Figure 4.

Mutational analysis of segment D. (A) Change of conserved (bold) amino acids in segment D by mutations M1–M4. (B) The Gal4–SV40 enhancer/promoter–luciferase reporter was transfected alone (–) or co-transfected with plasmids encoding Gal4–TRD-wt or Gal4–TRD mutants M1–M4 into HEK293 cells. Results are representative of at least four separate experiments, each in triplicate (± SD).
Knowing that MeCP2 interacts with the co-repressor mSin3A (9,10), it was of interest to monitor the effects of mutations A–F on the ability of MeCP2 to bind mSin3A in GST pull-down assays. Nuclear extracts from HEK293 cells were incubated with equal amounts of either TRD-wt produced as a bacterial fusion to GST, mutants A–F of TRD as fusions to GST or GST alone and monitored for associated proteins by western blot analysis. Complexes of cellular mSin3A with GST–TRD-wt, but not the control GST protein, were easily detected (Fig. 3D), confirming previous results (9,10). Importantly, we found that cellular mSin3A interacted with GST–TRD mutants A–C, E and F, but weakly with GST–TRD mutant D (Fig. 3D). In four independent experiments, binding of mSin3A to TRD mutant D was reduced, on average, by 87.6% when compared with TRD-wt. We conclude that mutation D, which inactivates transcriptional repression by MeCP2 in vivo, inhibits in vitro interaction between MeCP2 and mSin3A.
We also attempted to explore whether segments D and E are sufficient to repress transcription. A 46 amino acid region (amino acids 249–294) containing both segments and fused to the Gal4 DNA-binding domain was unable to reduce transcription of the SV40 enhancer/promoter appreciably (data not shown). We therefore decided to monitor the effects of the TRD and its subdomains on VP16-activated transcription. The minimal promoter of plasmid pFR-LUC containing a TATA box and a Gal4 recognition sequence is inactive without tethered transcription factors but is efficiently transcribed when the VP16 activation domain is recruited to the promoter through a fused Gal4 DNA-binding domain (Table 1). When the minimal TRD (mTRD) was inserted into the middle position of the fusion protein, i.e. between the Gal4 DNA-binding domain and the VP16 activation domain, transcriptional activation was aboliswhed (Table 1). Significantly, the 46 amino acid region containing segments D and E also greatly reduced VP16-activated transcription. Thus, when directly bound to an activation domain, segments D and E are sufficient to severely compromise activated transcription.
Table 1. Repression of VP16-activated transcription by mTRD and TRD segment DE.
| Fusion protein | RLU ± SD | Percentage ± SD |
|---|---|---|
| Gal4 | 75 ± 40 | |
| Gal4–VP16 | 962 620 ± 306 030 | 100 ± 31.8 |
| Gal4–mTRD–VP16 | 9450 ± 5230 | 0.9 ± 0.5 |
| Gal4–DE–VP16 | 145 970 ± 72 450 | 15.1 ± 7.5 |
The pFR-LUC reporter (Stratagene) was co-transfected into HEK293 cells with plasmids expressing the indicated fusion proteins. Results are shown as means of three independent transfections, each in triplicate (± SD).
DISCUSSION
The significant feature of the present study is the identification of a novel, histone deacetylase-independent pathway for transcriptional repression by MeCP2. This pathway operates on the SV40 enhancer/promoter and the SV40 promoter in HEK293 cells, but not on the AdML promoter in the same cell type. The strong repression of the SV40 promoter by the TRD of MeCP2 is not relieved by TSA, a specific inhibitor of all known histone deacetylases (19). In contrast, the weak repression of the AdML promoter (in an otherwise identical reporter plasmid context) in the same cell type (HEK293 cells) was alleviated by TSA. Previous transfection studies demonstrated compelling evidence for a histone deacetylase-dependent pathway for transcriptional repression by the TRD of MeCP2 (9,10). Evidently, which pathway is chosen is dependent on the promoter context. The histone deacetylase-independent mode of repression is operative on the SV40 promoter and the histone deacetylase-dependent one on the AdML promoter. The partial relief by TSA of the transcriptional repression of the human β-actin promoter by MeCP2 may suggest that its trans-repressing activity can operate at this promoter through both pathways (9). The transcriptional repression activity of MeCP2 was originally identified in in vitro experiments in which naked DNA was incubated with MeCP2 and rat liver or HeLa nuclear extracts (8). Since it may be anticipated that nucleosomes were not assembled under the assay conditions, we speculate that transcriptional inhibition was achieved through the histone deacetylase-independent pathway. Furthermore, the histone deacetylase-independent pathway can at least partially explain the inability of TSA to activate endogeneous genes silenced by CpG island methylation or the FMR1 gene in cells from fragile X patients (11,12). MeCP2 is the first identified member of a family of nuclear proteins containing a methyl-CpG-binding domain as a common feature (20). Interestingly, another member of this family, MBD2, also possesses transcription-repressing activity and exerts this role on the human β-actin promoter through a histone deacetylase-independent mechanism (21). A paradigm for the ability of MeCP2 to repress transcription through a histone deacetylase-dependent and an independent pathway is the retinoblastoma protein, Rb (22). Rb can interact with histone deacetylases to repress transcription and secondly can directly repress transcription factors bound at the promoter, e.g. the ETS family member PU.1.
The TRD (amino acids 196–310), which operates through the histone deacetylase-independent mechanism, coincides with that effective through the histone deacetylase-dependent one (amino acids 207–310) (Fig. 1B; 9). Yet, some transcription-repressing activity also resides in the C-terminal 176 residues. In a regional mutational analysis we identified two neighboring sequences (D and E) whose mutation abrogated the transcription-repressing activity of MeCP2 at the SV40 enhancer/promoter. Mutation of the first (D) of these sequences inhibits in vitro interaction between MeCP2 and mSin3A. Furthermore, a 46 amino acid region containing sequences D and E was sufficient to compromise VP16-activated transcription. Sequence D harbors a cluster of basic amino acids conserved from human to Xenopus. A combined change of four basic residues to acidic ones within sequence D severely reduced trans-repressing activity. Thus the central sequences D and E within the TRD, including clustered basic residues, likely represent a functional core of the TRD. Recently it was reported that mutations of the MECP2 gene cause Rett syndrome, a progressive neurodevelopmental disorder (23). Among six mutations found, four affected the methyl-CpG-binding domain and two the TRD. Interestingly, one of the latter mutations is a nonsense mutation of codon 255 (marked by an arrow in Fig. 3A) that localizes immediately upstream of segment D, and presumably generates a truncated protein lacking segments D and E. This lends support to our conclusion that segments D and E are essential for transcriptional repression of the SV40 enhancer/promoter. In contrast, mutation of segments D and E only weakly relieved trans-repressing activity of the TRD on the AdML promoter. This would be consistent with redundant protein–protein interactions. Interactions involving region D might be replaceable by others to repress AdML promoter activity.
Our results suggest that the histone deacetylase-independent pathway involves mSin3A but not a TSA-inhibitable histone deacetylase. mSin3A contains two silencing domains, SD-A and SD-B (24): while domain SD-B overlaps with the HDAC interaction domain, it was recently found that domain SD-A complexes with HDAC7 (25). Furthermore, mSin3A interacts with the general transcription factor TFIIB (24). This interaction or a crosstalk to a yet to be intentified transcriptional activator or co-activator may be involved in the histone deacetylase-independent mode of repression by MeCP2.
Acknowledgments
ACKNOWLEDGEMENTS
We thank J. Bode, J. Han and F. Schnieders for gifts of plasmid DNA. This article is based on the doctoral theses of F. Yu and J. Thiesen in the Faculty of Biology, University of Hamburg. This work was supported by grants from the Deutsche Forschungsgemeinschaft to W.H.S.
REFERENCES
- 1.Lewis J.D., Meehan,R.R., Henzel,W.J., Maurer-Fogy,I., Jeppesen,P., Klein,F. and Bird,A. (1992) Cell, 69, 905–914. [DOI] [PubMed] [Google Scholar]
- 2.Nan X., Meehan,R.R. and Bird,A. (1993) Nucleic Acids Res., 21, 4886–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nan X., Tate,P., Li,E. and Bird,A. (1996) Mol. Cell. Biol., 16, 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.von Kries J.P., Buhrmester,H. and Strätling,W.H. (1991) Cell, 64, 123–135. [DOI] [PubMed] [Google Scholar]
- 5.Buhrmester H., von Kries,J.P. and Strätling,W.H. (1995) Biochemistry, 34, 4108–4117. [DOI] [PubMed] [Google Scholar]
- 6.Weitzel J.M., Buhrmester,H. and Strätling,W.H. (1997) Mol. Cell. Biol., 17, 5656–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strätling W.H. and Yu,F. (1999) Crit. Rev. Eukaryot. Gene Expr., 9, 311–318. [DOI] [PubMed] [Google Scholar]
- 8.Nan X., Campoy,F.J. and Bird,A. (1997) Cell, 88, 471–481. [DOI] [PubMed] [Google Scholar]
- 9.Nan X., Ng,H.-H., Johnson,C.A., Laherty,C.D., Turner,B.M., Eisenmann,R.N. and Bird,A. (1998) Nature, 393, 386–389. [DOI] [PubMed] [Google Scholar]
- 10.Jones P.L., Veenstra,G.J.C., Wade,P.A., Vermaak,D., Kass,S.U., Landsberger,N., Strouboulis,J. and Wolffe,A.P. (1998) Nature Genet., 19, 187–191. [DOI] [PubMed] [Google Scholar]
- 11.Cameron E.E., Bachman,K.E., Myöhänen,S., Herman,J.G. and Baylin,S.B. (1999) Nature Genet., 21, 103–107. [DOI] [PubMed] [Google Scholar]
- 12.Coffee B., Zhang,F., Warren,S.T. and Reines,D. (1999) Nature Genet., 22, 98–101. [DOI] [PubMed] [Google Scholar]
- 13.Graham F.L., Smiley,J., Russel,W.C. and Nairn,R. (1977) J. Gen. Virol., 36, 59–72. [DOI] [PubMed] [Google Scholar]
- 14.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eckner R., Ewen,M.E., Newsome,D., Gerdes,M., DeCaprio,J.A., Lawrence,J.B. and Livingston,D.M. (1994) Genes Dev., 8, 869–884. [DOI] [PubMed] [Google Scholar]
- 16.Yang X.-J., Ogryzko,V.V., Nishikawa,J., Howard,B.H. and Nakatani,Y. (1996) Nature, 382, 319–324. [DOI] [PubMed] [Google Scholar]
- 17.Eilers A.L., Billin,A.N., Liu,J. and Ayer,D.E. (1999) J. Biol. Chem., 274, 32750–32756. [DOI] [PubMed] [Google Scholar]
- 18.Kadosh D. and Struhl,K. (1997) Cell, 89, 365–371. [DOI] [PubMed] [Google Scholar]
- 19.Grozinger C.M., Hassig,C.A. and Schreiber,S.L. (1999) Proc. Natl Acad. Sci. USA, 96, 4868–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hendrich B. and Bird,A. (1998) Mol. Cell. Biol., 18, 6538–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng H.-H., Zhang,Y., Hendrich,B., Johnson,C.A., Turner,B.M., Erdjument-Bromage,H., Tempst,P., Reinberg,D. and Bird,A. (1999) Nature Genet., 23, 58–61. [DOI] [PubMed] [Google Scholar]
- 22.Luo R.X., Postigo,A.A. and Dean,D.C. (1998) Cell, 92, 463–473. [DOI] [PubMed] [Google Scholar]
- 23.Amir R.E., Van den Veyver,I.B.,Wan,M., Tran,C.Q., Francke,U. and Zoghbi,H.Y. (1999) Nature Genet., 23, 185–188. [DOI] [PubMed] [Google Scholar]
- 24.Wong C.-W. and Privalsky,M.L. (1998) Mol. Cell. Biol., 18, 5500–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kao H.-Y., Downes,M., Ordentlich,P. and Evans,R.M. (2000) Genes Dev., 14, 55–66. [PMC free article] [PubMed] [Google Scholar]