


Abstract
Fragile X syndrome (FRAXA) is characterized at the molecular level by an expansion of a naturally occurring 5′-(CGG)n-3′ repeat in the promoter and 5′-untranslated region (5′-UTR) of the fragile X mental retardation (FMR1) gene on human chromosome Xq27.3. When expanded, this region is usually hypermethylated. Inactivation of the FMR1 promoter and absence of the FMR1 protein are the likely cause of the syndrome. By using the bisulfite protocol of the genomic sequencing method, we have determined the methylation patterns in this region on single chromosomes of healthy individuals and of selected premutation carriers and FRAXA patients. In control experiments with unmethylated or M-SssI-premethylated DNAs, this protocol has been ascertained to reliably detect all cytidines or 5-methylcytidines as unmethylated or methylated nucleotides, respectively. Analyses of the DNA from FRAXA patients reveal considerable variability in the lengths of the 5′-(CGG)n-3′ repeats and in the levels of methylation in the repeat and the 5′-UTR. In one patient (OEl) with high repeat length heterogeneity (n = 15 to >200), shorter repeats (n = 20–80) were methylated or unmethylated, longer repeats (n = 100–150) were often completely methylated, but one repeat with n = 160 proved to be completely unmethylated. This type of methylation mosaicism was observed in several FRAXA patients. In healthy females, methylated 5′-CG-3′ sequences were found in some repeats and 5′-UTRs, as expected for the sequences from one of the X chromosomes. The natural FMR1 promoter is methylation sensitive, as demonstrated by the loss of activity in transfection experiments using the unmethylated or M-SssI-premethylated FMR1 promoter fused to the luciferase gene as an activity indicator.
INTRODUCTION
Fragile X syndrome (FRAXA) is one of the most frequent genetic causes of mental retardation in humans and is associated with an inducible fragile chromosomal site at Xq27.3. Fragility is thought to be due to the expansion of a naturally occurring 5′-(CGG)n-3′ repeat in the promoter and 5′-region of the fragile X mental retardation 1 (FMR1) gene. The disease phenotype correlates with the absence of inactivating mutations in the FMR1 protein (1), a cytoplasmic protein which is found in neurons, in epithelial cells and in spermatogonia (2). In healthy individuals, the 5′-(CGG)n-3′ repeat is characterized by n = 6–50, in premutation carriers by 50 < n < 200 and in patients by n > 200 (3,4). In normal chromosomes, the 5′-(CGG)n-3′ repeats are interrupted by 5′-AGG-3′ interspersions. Loss of the latter, in particular at the 3′-ends of the repeats, seems to be linked to repeat instability (5–7).
Analyses using 5′-methyldeoxycytidine (5-mC)-sensitive restriction endonucleases, such as AclI (5′-AACGTT-3′), Fnu4HI (5′-GCNGC-3′), NruI (5′-TCGCGA-3′), EagI (5′-CGGCCG-3′) or BssHII (5′-GCGCGC-3′), have revealed that these sequences in the FMR1 promoter are methylated in FRAXA, but not in normal, individuals (8–11). Full repeat expansions in the absence of repeat methylation can be compatible with expression of the FMR1 protein in healthy individuals (12,13). Restriction enzyme analyses provide only limited insight into the complexity of DNA methylation patterns in the human and other genomes. The genomic sequencing method has been successfully used to address this problem in the upstream region of the FMR1 gene (11,14).
In previous work, we found the bisulfite protocol of the genomic sequencing method (15,16) to yield highly reproducible results (17–20). In the present investigation, considerable variability and methylation mosaicism have been found in the 5′-(CGG)n-3′ repeat and adjacent regions in fragile X males.
MATERIALS AND METHODS
Genomic sequencing of DNA from peripheral white blood cells by the bisulfite protocol
DNA was extracted from peripheral blood which was collected by venipuncture. The methylation status of the 5′-(CGG)n-3′ repeat was determined by the bisulfite protocol (15,16) of the genomic sequencing technique as described (17–20). Briefly, 5 µg of genomic DNA was alkali denatured in 0.3 M NaOH for 15 min at 37°C and for 2 min at 95°C. The DNA was then treated with sodium bisulfite. The bisulfite solution was prepared by dissolving 11.1 g of sodium bisulfite (Sigma) in 15 ml of degassed water, to which 1 ml of 40 mM hydrochinone was subsequently added. The solution was adjusted to pH 5.3 by adding 1.2 ml of 10 M NaOH. The denatured DNA solution (110 µl) was mixed with 1.5 ml of the bisulfite reagent, overlaid with mineral oil and incubated at 55°C for 4 h in a water bath in the dark. Subsequently, the DNA was purified using glass milk (Gene Clean II Kit; Bio 101 Inc.). Selected segments in the FMR1 sequence were amplified by PCR using appropriate oligodeoxyribonucleotide primers (Table 1 and see maps in Figs 1, 4 and 5).
Table 1. Oligodeoxyribonucleotide primers used in the amplification of DNA segments in the 5′-upstream region of the FMR1 genea.
| Primer | Sequence |
|---|---|
| P1 | 5′-AAACRTTCTAACCCTCRCRAAACAAATACRACC-3′ |
| P2 | 5′-CACCRCCCTTCAACCTTCCCRCCCTCCACCAAG-3′ |
| P3 | 5′-GTTYGTTTAGAGGGYGGTTTTTATYGGAAGTGAA-3′ |
| P4 | 5-GTYGTAYGTTTTTTGGTAGYGGYGTTTTYGT-3′ |
| P5 | 5′-ATTTCACTTCCRATAAAAAACCRCCTCTAAACRAAC-3′ |
| P6 | 5′-AAAACRCCRCTACCAAAAAACRTACRACAAC-3′ |
| P7 | 5′-TTTYGAGAGGTGGGTTGYGGGYGTTYGAGGTTTAG-3′ |
| P8 | 5′-TTTTATTTTTTTTTTAGTTTTGTTAGYGTYGGGAG-3′ |
aFor map locations of primers P1–P8 see Figure 1. Y = C or T; R = A or G.
Figure 1.

Map and nucleotide sequence of the 5′-upstream and promoter regions of the human FMR1 gene. The locations of primer oligodeoxyribonucleotides P1–P8 used in the genomic sequencing experiments (Table 1 and Figs 3–5) of the 5′-(CGG)n-3′ repeat and of the 5′-upstream region are listed. In the nucleotide sequence of the region 5′-upstream to the 5′-(CGG)n-3′ repeat, all 5′-CG-3′ dinucleotides are in bold print and numbered 1–49. Various restriction sites are indicated; HpaII (5′-CCGG-3′) sites are boxed. The underlined CT indicates a deviation from the published sequence.
Figure 4.

Methylation pattern in the DNA segment 5′-upstream of the 5′-(CGG)n-3′ repeat in the FMR1 upstream region. The DNA from FRAXA patients and normal female or male control individuals as indicated was analyzed by the genomic sequencing method. The map indicates the segment investigated and the locations of the nested primer sets P1–P4 used for PCR amplification. The numbers on the abscissa refer to the 5′-CG-3′ dinucleotides in the nucleotide sequence reproduced in Figure 1. Closed squares represent methylated, open squares unmethylated 5′-CG-3′ dinucleotides. Each horizontal row of squares displays the methylation pattern in one cloned molecule from that segment. Methylation-sensitive restriction sites are also designated.
Figure 5.

Methylation pattern in the 5′-upstream region of the FMR1 gene encompassing both the 5′-(CGG)n-3′ repeat and its 5′-upstream segment. The data are arranged as described in the legend to Figure 4, except that the P1, P2 and P7, P8 sets of nested primers have been used (see map). (a) Map of the region; (b) genomic sequencing data of FRAXA patients.
The PCR reaction products were then cloned into the pGEM-T® vector (Promega) and transfected into Escherichia coli strain XL1BlueMRF′ by standard methods (21). A number of clones were isolated, the DNA extracted and the nucleotide sequences determined (22) in an Applied Biosystems Model 377 DNA Sequencer. The bisulfite reaction converted all C residues to U residues and, after PCR amplification, the U residues were converted to T residues, whereas the 5-methyldeoxycytidine (5-mC) residues were refractory to this chemical conversion reaction. Thus, a C residue in the eventually determined nucleotide sequence proved the presence of a 5-mC residue in this position in the original genomic nucleotide sequence. All bona fide C residues scored as T residues in the final sequences. The sequence in each cloned molecule thus represents one X allele from male or two X alleles from female cells.
Determination of the repeat amplification
Genomic DNA was cleaved with EcoRI, HindIII, EagI or PstI restriction enzyme and the DNA fragments were separated by electrophoresis on 0.8% agarose gels, Southern blotted onto Boehringer Nylon-Plus membranes and hybridized to the 32P-labeled XhoI–PstI fragment of plasmid pE5.1 (4). The lengths of the amplified repeats were calculated based on length comparisons on the autoradiograms relative to size standards that were co-electrophoresed.
Western blot analyses of protein extracts isolated from peripheral white blood cells (PWBCs)
The lymphocyte fraction was isolated from whole blood of fragile X patients, premutation carriers or unaffected individuals by Ficoll gradient separation and lymphocyte proliferation was induced (23). For the preparation of crude cellular extracts, 5 × 108 cells were washed twice in phosphate-buffered saline (PBS) without Ca2+ and Mg2+, resuspended in 750 µl of extraction buffer [10 mM Na HEPES, 100 mM CaCl2, 300 mM KCl, 5 mM MgCl2, 0.05% Tween-20, 0.45% Triton-X 100, 1 mM dithiothreitol (DTT), 4 µg/ml aprotinin, 1 µg/ml leupeptin, pH 7.4] and incubated on ice for 20 min. After centrifugation for 30 min at 13 000 r.p.m. and 4°C, the supernatant was diluted with 750 µl of buffer (25 mM Na HEPES, 250 mM CaCl2, 50 mM KCl, 7.5 mM MgCl2, 1 mM DTT, 50% glycerol, 4 µg/ml aprotinin, 1 µg/ml leupeptin, pH 7.4), frozen in liquid nitrogen and stored at –80°C. For western blot analyses, 20 µg of crude cellular extract was fractionated on a 12% SDS–polyacrylamide gel (24), followed by electrotransfer to a polyvinylidene difluoride membrane (25). After treatment with 0.1% blocking reagent (Boehringer Mannheim, Mannheim, Germany), the membrane was incubated with the primary monoclonal antibody against FMR1 protein (clone IC3; Euromedex, Strasbourg, France), followed by incubation with the secondary antibody (anti-mouse Ig horseradish peroxidase conjugate). Signals were detected using the ECL-Plus system (Amersham) according to the manufacturer’s protocol. Rainbow colored protein molecular weight markers (Amersham) or SDS-7B (Sigma) were used as size standards.
In vitro methylation of the FMR1 promoter–luciferase gene construct
The FMR1 promoter segment from pE5.1 (4), nt 3–2819, was cloned in front of the luciferase gene in the vector pGL2-Basic (Promega, Madison, WI). In the 2537ΔRep construct, the FMR1 5′-region carrying a 5′-(CGG)n-3′ repeat was replaced by a segment without that repeat using synthetic oligodeoxyribonucleotides. Plasmid DNA was harvested from the methylation-negative E.coli strain JM110 (dam–, dcm–) (26), purified by CsCl density gradient centrifugation and methylated in vitro with M-HpaII or M-SssI DNA methyltransferase. Complete in vitro methylation was ascertained by cleavage of the plasmid DNA with HpaII, MspI or HhaI restriction enzyme and subsequent analysis by electrophoresis on a 5% polyacrylamide gel and Southern blot hybridization to the 32P-labeled construct. Mock-methylated DNA was prepared in the same reaction omitting S-adenosylmethionine as methyl donor (27), transfected and assayed as described. Methylated or mock-methylated plasmid DNA (2 pmol) was transfected into suspended human cells harvested from non-confluent cultures by electroporation in 4 mm cuvettes at 210 V, 960 µF in RPMI medium without glutamine and dye. Cells were grown in Dulbecco’s modified Eagle’s medium enriched with 10% fetal calf serum for 24 h before harvest. Cells were scraped off, washed and lysed in cell culture lysis reagent (Promega). Protein concentrations were determined by standard methods (28) and luciferase activities were measured in a luminometer (Lumat LB950m, Berthold), using assay buffers as recommended by the manufacturer.
RESULTS
Control experiments to assess the reliability of the bisulfite protocol for genomic sequencing experiments
The bisulfite protocol of the genomic sequencing technique has been applied to analyze the state of methylation in the 5′-upstream regions and in the 5′-(CGG)n-3′ repeats of the FMR1 gene in normal individuals, in female and male premutation carriers and in fragile X patients. Maps and nucleotide sequences of this DNA segment are presented in Figure 1 with all 5′-CG-3′ dinucleotides in bold print. Different sets of primers (P1–P8; see Table 1) were used for the PCR amplifications in the genomic sequencing experiments, and primer locations are shown in Figure 1.
In a series of control experiments, we have demonstrated that mock-methylated or 5′-CG-3′ M-SssI-premethylated sites in the FMR1 promoter region with a 5′-(CGG)16-3′ repeat cloned in plasmid pE5.1 (4) are unequivocally detected by the applied genomic sequencing protocol in the unmethylated or fully methylated state, respectively (data not shown). In mixtures of 1 genome equivalent to 5 µg of human DNA from control females with unmethylated control plasmid, unmethylated and methylated nucleotides can be reliably detected (data not shown).
We have also analyzed 1 µg of genomic DNA from a healthy male individual with a repeat length of 5′-(CGG)30-3′ in the unmethylated or in the 5′-CG-3′ (M-SssI) premethylated state. The results of the genomic sequencing analyses on these DNA preparations revealed unmethylated or completely 5′-CG-3′ methylated repeats, respectively (data not shown). Hence, previous results (17–20) and the present controls satisfy the requirements for a reliable method to detect methylated cytidines in genomic DNA. Lastly, the 5′-(CGG)n-3′ repeats have been investigated for their methylation status in the DNA from healthy control males (MM, JS and BG) and females (BS and LM). The primer pairs P5, P6 and P7, P8 were used for PCR amplification. In three male control individuals, the repeats are completely unmethylated. In two control females, many of the repeats analyzed are methylated, although possibly <50% (data not shown). In both male and female controls, the 5′-AGG-3′ interspersions are preserved.
Heterogeneity in repeat lengths in selected FRAXA patients as well as female and male premutation carriers
In families 1 (O) and 2 (F), mothers ON and FP are premutation carriers, their sons OEm and OEl and FPh, respectively, present with the phenotype of FRAXA. This diagnosis has been confirmed by the results of Southern blot analyses (Fig. 2). TK in family 3 is a male premutation carrier (Fig. 2c, I:2, and Table 2) whose two daughters also have the unexpanded or slightly expanded premutation (Fig. 2c, II:2 and II:3). The younger daughter (II:3) of TK has two sons, of whom the older one, the index patient (Fig. 2c, III:1), has FRAXA confirmed by molecular diagnosis while the younger one is healthy (data not shown). Figure 2a shows the pedigree of family 1 and the results of Southern DNA transfer hybridization analyses of the three family members and of an unrelated female control proband. The DNA samples from OEm and OEl demonstrate marked heterogeneity in the lengths of their repeats, as revealed by the HindIII and PstI restriction patterns (for a map see Fig. 1). The DNA from the mother ON exhibits one normal length allele of 28/29 repeats and one premutation allele varying in length between 64 and 93 repeats (Fig. 2a, ON-PstI lane) as compared with two normal alleles in the control female (closely spaced bands in the control PstI lane in Fig. 2a). The repeats in the two FRAXA brothers are highly methylated and hence not cleaved efficiently by EagI. The mother in family 2 (FP) carries a premutation of the 5′-(CGG)n-3′ repeat (Fig. 2b, PstI lane). In her son, FPh, the repeat is in the full mutation range (PstI lane) and is highly methylated (Fig. 2b, EagI lane). In the third family, the 5′-(CGG)n-3′ repeats in the male premutation carrier TK exhibit slightly polymorphic lengths between 82 and 90 (Table 2 and Fig. 2c).
Figure 2.


Restriction endonuclease analyses in the FMR1 promoter region in fragile X patients and in premutation and healthy individuals. (a–c) Pedigrees and restriction endonuclease patterns of two families with premutation mothers and fragile X sons and one family with grandparental transmission. The DNAs of healthy females [control (female)] were also analyzed. DNA (15 µg each) from PWBCs of family members as indicated were cleaved with the designated restriction endonucleases, the fragments were separated by electrophoresis in 0.8% agarose gels, blotted (29,30) to a positively charged nylon membrane and hybridized to the 32P-labeled XhoI–PstI probe depicted in (d). The sizes of the thus visualized PstI fragments best revealed the premutation (ON, FP, PH and TK) or full mutation (OEm, OEl and FPh) expansions of the 5′-(CGG)n-3′ repeats of the FMR1 promoter and 5′-upstream segment. Upon cleavage with EcoRI and the methylation-sensitive EagI (5′-CGGCCG-3′), DNA methylated at the underlined sites yielded high molecular mass fragments (OEm, OEl, FPh and TK). The molecular size marker lane (plasmid control) contained 100 pg each of a mixture of plasmid fragments generated by EcoRI, EcoRI + BamHI or PstI cleavage of plasmid pE5.1 (accession no. X61378). (d) Map of the pE5.1 plasmid. The XhoI–PstI fragment designated probe was used for the hybridization experiments (a–c). The locations of the size markers used in (a)–(c), with lengths of 5.2, 3.3 and 1.0 kb, are also indicated.
Table 2. Methylation mosaicism as revealed by genomic sequencing in individually cloned molecules of the 5′-(CGG)n-3′ repeat in the FMR1 promoter segment in probands as describeda.
| Proband | Sex | Repeat length (n) | Number of cloned PCR products sequenced | |||
|---|---|---|---|---|---|---|
| Total | Completely unmethylated | Partly methylatedb | Completely methylated | |||
| Family 1 ON (premutation) | Female | 28/29 | 38 | 25 | 7 | 6 |
| 64–93c | 6 | 0 | 4 | 2 | ||
| OEm (patient) | Male | 5–50 | 11 | 3 | 3 | 5 |
| 50–80 | 8 | 2 | 0 | 6 | ||
| 80–120 | 4 | 1 | 0 | 3 | ||
| 120–200d | 0 | 0 | 0 | 0 | ||
| OEl (patient) | Male | 5–50 | 6 | 0 | 0 | 6 |
| 50–80 | 29 | 10 | 0 | 19 | ||
| 80–120 | 16 | 2 | 0 | 14 | ||
| 120–200d | 8 | 1 | 0 | 7 | ||
| Family 2 FP (premutation) | Female | 19/20/21(~120)e | 24 | 8 | 10 | 6 |
| FPh (patient) | Male | 5–50 | 14 | 5 | 1 | 8 |
| 50–80 | 2 | 0 | 2 | 0 | ||
| 80–120 | 6 | 0 | 1 | 5 | ||
| 120–200e | 7 | 0 | 2 | 5 | ||
| TK (premutation) | Male | 82–90 | 5 | 5 | 0 | 0 |
| pE5.1 (plasmid control) | 16 | 25 | 0 | 0 | 25 | |
| 16 | 7 | 7 | 0 | 0 | ||
| BS (normal) | Female | 28/30 | 5 | 3 | 2 | 0 |
| 42/43 | 3 | 2 | 1 | 0 | ||
| ML (normal) | Female | 27/29 | 7 | 1 | 2 | 4 |
| 30 | 6 | 2 | 0 | 4 | ||
| MM (normal) | Male | 19/20 | 10 | 10 | 0 | 0 |
| JS (normal) | Male | 28/29 | 10 | 10 | 0 | 0 |
| BG (normal) | Male | 29/30 | 6 | 6 | 0 | 0 |
aFor each category, the table lists the number of cloned molecules for which the nucleotide sequence was determined after bisulfite treatment, PCR amplification and cloning into the pGEM-T vector. The repeat lengths in different probands were estimated by Southern transfer hybridization and by PCR. The actual sequence data with repeat lengths, state of methylation and 5′-AGG-3′ interspersions in the repeat are schematically shown in Figure 3.
bSome of the 5′-(CGG)n-3′ repeats were only partly methylated (see Fig. 3a–d).
cPremutation allele showed variations in repeat lengths.
dLonger repeats, though present, could not be analyzed by genomic sequencing due to limitations of the PCR which could not penetrate long 5′-(CGG)n-3′ DNA segments.
eThis value was derived from a Southern blot hybridization experiment.
Complex mosaic methylation patterns in expanded 5′-(CGG)-3′ repeats
Precise patterns of DNA methylation in the 5′-(CGG)n-3′ repeats in these FRAXA families, in healthy, premutation or fragile X individuals, have been determined using the bisulfite protocol of the genomic sequencing method. The results will be presented first for the 5′-(CGG)n-3′ repeat followed by those for the 5′-upstream segment of the FMR1 gene.
Methylation in the 5′-(CGG)n-3′ repeats
The 5′-(CGG)n-3′ repeats in patient OEl exhibit marked mosaicism in both repeat lengths and the extent of methylation in individually cloned repeat sequences (Fig. 3 and Table 2). Repeat lengths, 5′-AGG-3′ interspersions and methylation status of the repeats in patients OEl (Fig. 3a), OEm (Fig. 3b) and FPh (Fig. 3c) are presented. In some of the fully methylated repeats with n > 50, 5′-(AGG)-3′ interspersions are preserved in the 5′-segments of the repeats (Fig. 3b and c). Both short and longer repeats can be completely methylated or unmethylated. Repeats of normal lengths and repeats lacking 5′-(AGG)-3′ interspersions can be either unmethylated or methylated (Fig. 3). Most long repeats without 5′-(AGG)-3′ interruptions are methylated. Similarly, in family 2, patient FPh reveals considerable heterogeneity in the lengths of the 5′-(CGG)n-3′ repeats; some of the repeats are unmethylated while others only partly methylated (Fig. 3c). This heterogeneity in repeat lengths is not apparent in Southern transfer experiments (Fig. 2b).
Figure 3.



5′-(CGG)n-3′ repeat length and 5′-CG-3′ methylation mosaicism in three FRAXA patients. The results of the genomic sequencing experiments for all cloned PCR products are presented. Individual repeats are depicted as circles. Open circles, unmethylated 5′-(CGG)n-3′ repeats; closed circles, methylated repeats; inverted triangles, 5′-(AGG)-3′ interspersions. For PCR amplification the primer pairs P5, P6 and P7, P8 (Fig. 1) were used. (a) Patient OEl; (b) patient OEm; (c) patient FPh; (d) premutation carriers ON and FP.
Table 2 summarizes the results of all genomic sequencing experiments. In healthy female individuals (BS and ML), the 5′-(CGG)n-3′ repeats with normal lengths (n = 27–43) can be partly methylated, probably because of inactivation of one of the X chromosomes (Table 2). This inactivation may not be complete. The most remarkable methylation mosaicism exists in the two patient brothers OEl and OEm. There, even the longest repeat (n = 160), which has been genomically sequenced, is completely unmethylated (Fig. 3a). The very long repeats (n > 200) in fragile X patients unfortunately cannot be analyzed by the genomic sequencing technique, since there is an insurmountable length limit to 5′-CG-3′-rich sequences that can be amplified by PCR and stably propagated in E.coli (31,32). The repeats in the allele with a premutation expansion (n = 64–93) in the mother, ON, were completely methylated in two of six clones analyzed; four clones were partly methylated (Table 2 and Fig. 3d).
Short methylated repeats have also been found in fragile X patients, particularly in the two brothers of family 1 with a high degree of repeat length mosaicism. In normal and premutation individuals only limited variability in repeat lengths has been observed (33). In normal and premutation males (Table 2 and Fig. 3c, TK, and Fig. 4), the repeats are not methylated. In the FRAXA patients OEm and OEl both repeat lengths (Table 2 and Fig. 3) and the extent of repeat methylation varies considerably. With the exceptions shown in Figure 3a and b for some of the longer repeats with increasing repeat lengths, the degree of methylation in the repeats is augmented.
Methylation in the region 5′-upstream of the 5′-(CGG)n-3′ repeat
This DNA segment was investigated by the genomic sequencing method in two different sections. First, the region 5′-upstream of the 5′-(CGG)n-3′ repeats was subjected to genomic sequencing and the primer sets P1, P2 and P3, P4 were employed in PCR amplification (Fig. 4). In the FRAXA patients OEl and OEm, the DNA sequence 5′-upstream of the 5′-(CGG)n-3′ repeat is extensively methylated in all PCR-amplified products that have been sequenced (Fig. 4). In normal control females, the same DNA segment is methylated, whereas in normal males no methylation has been seen (Fig. 4).
PCR amplification of the long genome segment encompassed by the 5′-(CGG)n-3′ repeat and its 5′-upstream region proved difficult, since a 5′-CG-3′-rich sequence, possibly with a high degree of DNA methylation and a complex secondary structure in FRAXA patients, had to be copied by the polymerase system and stably propagated in E.coli upon cloning. In these experiments the primer sets P1, P2 and P7, P8 were used (map in Fig. 5a). The results reflect the selection of molecules that are not methylated and not of excessive length. In FRAXA patient FPh (Fig. 5b), only a few completely 5′-CG-3′ methylated molecules were amplified and detectable. In control or premutation males, this region showed no methylation, at least in the molecules amplified in our experiments (data not shown). This result further attests to the presence of unmethylated repeats and 5′-upstream regions in FRAXA patients and supports the notion of methylation mosaicism in these patients. Similar results on control females and premutation females (data not shown), in whom one expects to find the methylated alleles from the hypermethylated X chromosome, corroborate the interpretation of selection against the amplification of long, 5′-CG-3′-rich and methylated DNA segments by the procedure employed, since <50% of the repeats and even fewer molecules in the region 5′-upstream of the repeats have been found to be methylated.
Lack of FMR1 gene expression
Although some of the 5′-(CGG)n-3′ repeats and the 5′-upstream segment in patients OEl and OEm can be un- or hypomethylated, the FMR1 gene, which is controlled by the promoter carrying the repeats, is not detectably, though perhaps minimally, expressed in peripheral white blood cells (PWBCs). Lack of the FMR1 gene product (FMRP) is thought to be responsible for the fragile X phenotype. Western blotting detected no FMR1 protein in extracts of PWBCs from the two patients and another male full mutation carrier (Fig. 6), although trace amounts might be present. Extracts of PWBCs from the premutation carrier ON, from a control or another premutation individual exhibit normal levels of FMR1 protein. Apparently, methylated FMR1 promoter sequences do not permit efficient expression of this gene. Since unmethylated sequences in the FRAXA patients also do not seem to be expressed, it is likely that factors other than promoter methylation contribute to inactivation of the FMR1 promoter. Alternatively, the relative number of unmethylated sequences might be too low to allow production of detectable amounts of FMR1 protein, perhaps expressed only in a subset of cells. Here we have investigated expression of the FMR1 gene in PWBCs only, and have not evaluated FMR1 protein levels in other organs, particularly in the central nervous system during development.
Figure 6.
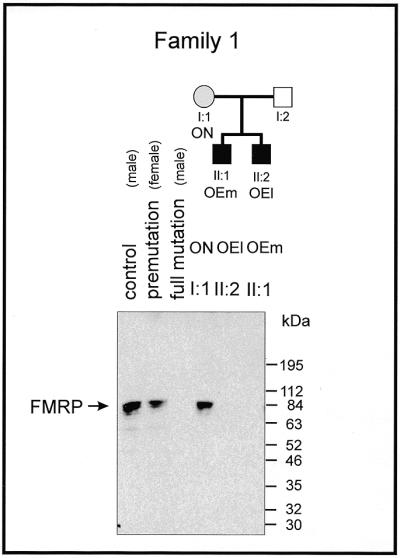
Lack of expression of the FMRP in PWBCs in the FRAXA patients OEm and OEl as determined by western blot analyses. Details are described in Materials and Methods.
Inactivation of the FMR1 promoter by in vitro 5′-(CG)-3′ premethylation
Sequence-specific promoter methylation in eukaryotes has been shown to silence promoter activity (34,35). Since methylation of the FMR1 promoter has been related to gene inactivation and the FRAXA syndrome, we have investigated whether the FMR1 promoter can be inactivated by in vitro premethylation of all of its 5′-CG-3′ sequences by M-SssI DNA methyltransferase. Several FMR1 promoter constructs of different lengths, with or without (2537ΔRep) the 5′-(CGG)n-3′ repeat, were fused to the luciferase gene as an activity indicator (Fig. 7). Upon transfection into human HeLa or 293 cells, the M-SssI-methylated FMR1 promoter–luciferase fusion constructs are almost completely inactivated, whereas unmethylated or mock-methylated constructs retain activity. M-HpaII (5′-CCGG-3′) methylation does not interfere with the activity of this promoter (Table 3). This construct carries a 2537 bp promoter and upstream sequences. A minimal promoter construct consisting of only 41 bp upstream of the transcriptional start site harboring the 5′-(CGG)n-3′ repeat has low activity and is also inactivated by M-SssI methylation. Does methylation of the 5′-(CGG)n-3′ repeat itself inactivate the FMR1 promoter? We have deleted the repeat in the 5′-UTR of the construct without affecting the flanking DNA sequences (2537ΔRep). This promoter construct has somewhat reduced activity. Efficient inhibition of this promoter construct by 5′-CG-3′ (M-SssI) methylation demonstrates that silencing by methylation does not depend on the presence of the 5′-(CGG)-3′ repeat (Table 3). Thus, at least in reconstruction experiments, the FMR1 promoter can be silenced by 5′-CG-3′ methylation, as reported earlier (36). It is unknown why the expanded 5′-(CGG)n-3′ repeats become de novo methylated. We have previously suggested the possibility that a repeat amplification might be recognized as foreign DNA (37).
Figure 7.

Fusion constructs between the luciferase gene and segments of different lengths from the FMR1 promoter.
Table 3. Activity of the FMR1 promoter upon in vitro M-SssI premethylationa.
| HeLa cells | 293 cells | ||||
|---|---|---|---|---|---|
| Length of promoter fragment (bp) | 2537 | 41 | 2537ΔRep | 2537 | 41 |
| Mock-methylated | 100%b | 9% | 100%c | 100%d | 30% |
| M-HpaII-methylated | 129% | 3% | 130% | 159% | 44% |
| M-SssI-methylated | 0.6% | 0.2% | 4% | 4% | 3% |
aExperimental details are described in Materials and Methods.
b18 580 r.l.u./mg protein/pmol plasmid.
c11 050 r.l.u./mg protein/pmol plasmid.
d29 500 r.l.u./mg protein/pmol plasmid.
DISCUSSION
In this study, we have investigated the endogenous methylation patterns of the 5′-upstream and the 5′-(CGG)-3′ repeat regions of the FMR1 gene. With respect to both its length and the extent of 5′-CG-3′ methylation, the 5′-(CGG)n-3′ repeat is mosaic in female premutation carriers and FRAXA patients (Table 2 and Figs 2–5). Similar observations have been reported by other laboratories (14,38–40). Even in FRAXA patients, we have observed shorter (normal or premutation length) and longer unmethylated repeats (Table 2 and Fig. 3). The data presented are consistent with the interpretation that in individual FRAXA patients some of the 5′-(CGG)n-3′ repeats in the FMR1 promoter are completely unmethylated while others can be partly or completely methylated. Although a part of these repeats appears to remain unmethylated, the FMR1 gene is not efficiently transcribed and the FRAXA phenotype develops. There is also variability in normal individuals. Repeat mosaicism in length and methylation is likely a consequence of the instability of the 5′-(CGG)n-3′ repeat. Particularly in the 3′-sections of the repeats, the 5′-AGG-3′ interspersions are always missing in FRAXA patients and 5′-AGG-3′ sequences are also often absent in the 5′-segments.
The data from numerous control experiments (not shown, but available on request), as well as previously reported analyses (17–20), have documented the reliability of the bisulfite protocol of the genomic sequencing method (15,16). In interpreting the data on repeat heterogeneity, we have to consider the fact that the genomic sequencing protocol employed utilizes PCR amplification and thus naturally selects for molecules with shorter repeats. PCR amplification and clone stability of more extended 5′-(CGG)n-3′ repeats impose severe technical limitations. Long repeats will be amplified in the PCR reaction less efficiently, but can occasionally be found, like the n = 160 repeat length clone observed without methylation (OEl, Table 2 and Fig. 3a).
The DNA segment 5′-upstream of the 5′-(CGG)n-3′ repeat is heavily methylated in FRAXA patients (Fig. 4), although not in all molecules (Fig. 5). It is technically difficult to amplify an expanded 5′-(CGG)n-3′ repeat jointly with the 5′-upstream DNA segment on the same molecule. The polymerase system apparently selects for the unmethylated molecules, which then appear over-represented. This conclusion is supported by a similar finding with the DNA of normal females (data not shown), in whom a high proportion of molecules are expected to be methylated due to the high level of methylation on one of the X chromosomes. Actually, however, fewer methylated molecules are found in control females. At present, it is unknown by what mechanism, if any, the expansion of a repeat is related to its methylation. It is conceivable that in some of the molecules with a highly expanded repeat it is recognized as foreign DNA and hence de novo methylated (37,41).
In western blot experiments, expression of the FMR1 protein in FRAXA patients has not been detected (Fig. 6). Due to the presence of unmethylated and/or normal length repeats in patients with a high degree of mosaicism (Table 2 and Fig. 5), a low level of FMR1 protein, difficult to detect by western blotting, might be produced in some of these patients. Reduction in the amount of FMR1 protein below a critical threshold would interfere with normal development and lead to the syndrome. On the other hand, FMR1 protein might in fact be completely absent, because factors other than DNA methylation or in addition to that genetic signal may have shut off expression of that gene. As shown earlier (36) in reconstruction experiments, we can demonstrate FMR1 gene expression to be sensitive to the methylation of all 5′-CG-3′ sequences in the 5′-region of that gene (Table 3). Of course, any discussion on the pathogenesis of FRAXA in the context of repeat mosaicism is subject to the limitation that all the molecular data are derived from only one cell type, namely PWBCs. Analyses by RT–PCR have revealed the absence of FMR1-specific RNA in PWBCs from FRAXA individuals (1,42).
However, the role of the repeat in promoter function is not yet clear. The results of the transfection experiments using FMR1 promoter–reporter gene constructs imply that methylation of the region 5′-upstream of the repeat seems to be more important than the methylation status of the repeat itself. This notion is supported by the finding of males with a normal phenotype but with an amplified repeat that is devoid of 5′-CG-3′ methylation in the 5′-upstream region of the FMR1 gene (13). Amplification of the repeat may serve as a start signal for the cellular DNA methyltransferase in a silencing reaction for modified DNA structures or foreign DNA sequences.
Acknowledgments
ACKNOWLEDGEMENTS
We are indebted to the patients and their families for collaborating in this study. We thank Ulf Pettersson (Uppsala Universitet, Uppsala, Sweden) for comments on the manuscript, Petra Böhm for expert editorial work and Gudrun Schell for DNA sequencing. This research was supported by the Center for Molecular Medicine, Köln, TV 13 (BMBF, Bonn) and by the Kaempgen Stiftung, Köln.
REFERENCES
- 1.Pieretti M., Zhang,F., Fu,Y.-H., Warren,S.T., Oostra,B.A., Caskey,C.T. and Nelson,D.L. (1991) Cell, 66, 817–822. [DOI] [PubMed] [Google Scholar]
- 2.Devys D., Lutz,Y., Rouyer,N., Bellocq,J.-P. and Mandel,J.-L. (1993) Nature Genet., 4, 335–340. [DOI] [PubMed] [Google Scholar]
- 3.Fu Y.-H., Kuhl,D.P.A., Pizzutti,A., Pieretti,M., Sutcliffe,J.S., Richards,S., Verkerk,A.J.M.H., Holden,J.J.A., Fenwick,R.G.,Jr, Warren,S.T. et al. (1991) Cell, 67, 1047–1058. [DOI] [PubMed] [Google Scholar]
- 4.Verkerk A.J.M.H., Pieretti,M., Sutcliffe,J.S., Fu,Y.-H., Kuhl,D.P.A., Pizutti,A., Reiner,O., Richards,S., Victoria,M.F., Zhang,F. et al. (1991) Cell, 65, 905–914. [DOI] [PubMed] [Google Scholar]
- 5.Eichler E.E., Holden,J.J.A., Popovich,B.W., Reiss,A.L., Snow,K., Thibodeau,S.N., Richards,C.S., Ward,P.A. and Nelson,D.L. (1994) Nature Genet., 8, 88–94. [DOI] [PubMed] [Google Scholar]
- 6.Eichler E.E., Hammond,H.A., Macpherson,J.N., Ward,P.A. and Nelson,D.L. (1995) Hum. Mol. Genet., 4, 2199–2208. [DOI] [PubMed] [Google Scholar]
- 7.Hirst M.C. and White,P.J. (1998) Nucleic Acids Res., 26, 2353–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oberlé I., Rousseau,F., Heitz,D., Kretz,C., Devys,D., Hanauer,A., Boué,J., Bertheas,M.F. and Mandel,J.L. (1991) Science, 252, 1097–1102. [DOI] [PubMed] [Google Scholar]
- 9.Hansen R.S., Gartler,S.M., Scott,C.R., Chen,S.-H. and Laird,C.D. (1992) Hum. Mol. Genet., 1, 571–578. [DOI] [PubMed] [Google Scholar]
- 10.Rousseau F., Heitz,D., Biancalana,V., Oberlé,I. and Mandel,J.L. (1992) Am. J. Med. Genet., 43, 197–207. [DOI] [PubMed] [Google Scholar]
- 11.Hornstra I.K., Nelson,D.L., Warren,S.T. and Yang,T.P. (1993) Hum. Mol. Genet., 2, 1659–1665. [DOI] [PubMed] [Google Scholar]
- 12.Hagerman R.J., Hull,C.E., Safanda,J.F., Carpenter,I., Staley,L.W., O’Connor,R.A., Seydel,C., Mazzocco,M.M.M., Snow,K., Thibodeau,S.N. et al. (1994) Am. J. Med. Genet., 51, 298–308. [DOI] [PubMed] [Google Scholar]
- 13.Smeets H.J.M., Smits,A.P.T., Verheij,C.E., Theelen,J.P.G., Willemsen,R., van de Burgt,I., Hoogeveen,A.T., Oosterwijk,J.C. and Oostra,B.A. (1995) Hum. Mol. Genet., 4, 2103–2108. [DOI] [PubMed] [Google Scholar]
- 14.Stöger R., Kajimura,T.M., Brown,W.T. and Laird,C.D. (1997) Hum. Mol. Genet., 6, 1791–1801. [DOI] [PubMed] [Google Scholar]
- 15.Frommer M., McDonald,L.E., Millar,D.S., Collis,C.M., Watt,F., Grigg,G.W., Molloy,P.L. and Paul,C.L. (1992) Proc. Natl Acad. Sci. USA, 89, 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark S.-J., Harrison,J., Paul,C.L. and Frommer,M. (1994) Nucleic Acids Res., 22, 2990–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeschnigk M., Schmitz,B., Dittrich,B., Buiting,K., Horsthemke,B. and Doerfler,W. (1997) Hum. Mol. Genet., 6, 387–395. [DOI] [PubMed] [Google Scholar]
- 18.Munnes M., Patrone,G., Schmitz,B., Romeo,G. and Doerfler,W. (1998) Oncogene, 17, 2573–2583. [DOI] [PubMed] [Google Scholar]
- 19.Remus R., Kämmer,C., Heller,H., Schmitz,B., Schell,G. and Doerfler,W. (1999) J. Virol., 73, 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schumacher A., Buiting,K., Zeschnigk,M., Doerfler,W. and Horsthemke,B. (1998) Nature Genet., 19, 324–325. [DOI] [PubMed] [Google Scholar]
- 21.Hanahan D. (1983) J. Mol. Biol., 166, 557–580. [DOI] [PubMed] [Google Scholar]
- 22.Sanger F., Nicklen,S. and Coulson,A.R. (1977) Proc. Natl Acad. Sci. USA, 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gillis S. and Watson,J. (1981) Immunol. Rev., 54, 81–109. [DOI] [PubMed] [Google Scholar]
- 24.Lämmli U.K. (1970) Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 25.Kyhse-Andersen J. (1984) J. Biochem. Biophys. Methods, 10, 203–209. [DOI] [PubMed] [Google Scholar]
- 26.Yanisch-Perron C., Vieira,J. and Messing,J. (1985) Gene, 33, 103–119. [DOI] [PubMed] [Google Scholar]
- 27.Langner K.-D., Vardimon,L., Renz,D. and Doerfler,W. (1984) Proc. Natl Acad. Sci. USA, 81, 2950–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradford M.M. (1976) Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 29.Southern E.M. (1975) J. Mol. Biol., 98, 503–517. [DOI] [PubMed] [Google Scholar]
- 30.Koetsier P.A., Schorr,J. and Doerfler,W. (1993) Biotechniques, 15, 260–262. [PubMed] [Google Scholar]
- 31.Bowater R.P., Jaworski,A., Larson,J.E., Parniewski,P. and Well,R.D. (1997) Nucleic Acids Res., 25, 2861–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iyer R.R. and Wells,R.D. (1999) J. Biol. Chem., 274, 3865–3877. [DOI] [PubMed] [Google Scholar]
- 33.Kunst C.B. and Warren,S.T. (1994) Cell, 77, 853–861. [DOI] [PubMed] [Google Scholar]
- 34.Doerfler W. (1983) Annu. Rev. Biochem., 52, 93–124. [DOI] [PubMed] [Google Scholar]
- 35.Munnes M. and Doerfler,W. (1997) Encyclopedia of Human Biology, Vol. 3. Academic Press, pp. 435–446.
- 36.Hwu W.-L., Lee,Y.-M., Lee,S.-C. and Wang,T.-R. (1993) Biochem. Biophys. Res. Commun., 193, 324–329. [DOI] [PubMed] [Google Scholar]
- 37.Behn-Krappa A. and Doerfler,W. (1994) Hum. Mutat., 3, 19–24. [DOI] [PubMed] [Google Scholar]
- 38.Tassone F., Hagerman,R.J., Gane,L.W. and Taylor,A.K. (1999) Am. J. Med. Genet., 84, 240–244. [PubMed] [Google Scholar]
- 39.Taylor A.K., Tassone,F., Dyer,P.N., Hersch,S.M., Harris,J.B., Greenough,W.T. and Hagerman,R.J. (1999) Am. J. Med. Genet., 84, 233–239. [PubMed] [Google Scholar]
- 40.Schmucker B. and Seidel,J. (1999) Am. J. Med. Genet., 84, 221–225. [PubMed] [Google Scholar]
- 41.Doerfler W. (1991) Biol. Chem. Hoppe-Seyler, 372, 557–564. [PubMed] [Google Scholar]
- 42.Feng Y., Lakkis,L., Devys,D. and Warren,S.T. (1995) Am. J. Hum. Genet., 56, 106–113. [PMC free article] [PubMed] [Google Scholar]