


Abstract
A restriction map of the 2.8-Mb genome of the unicellular eukaryote Encephalitozoon cuniculi (phylum Microspora), a mammal-infecting intracellular parasite, has been constructed using two restriction enzymes with 6 bp recognition sites (BssHII and MluI). The fragments resulting from either single digestions of the whole molecular karyotype or double digestions of 11 individual chromosomes have been separated by two-dimensional pulsed field gel electrophoresis (2D-PFGE) procedures. The average distance between successive restriction sites is ~19 kb. The terminal regions of the chromosomes show a common pattern covering ~15 kb and including one 16S–23S rDNA unit. Results of hybridisation and molecular combing experiments indicate a palindromic-like orientation of the two subtelomeric rDNA copies on each chromosome. We have also located 67 DNA markers (clones from a partial E.cuniculi genomic library) by hybridisation to restriction fragments. Partial or complete sequencing has revealed homologies with known protein-coding genes for 32 of these clones. Evidence for two homologous chromosomes III, with a size difference (3 kb) related to a subtelomeric deletion/insertion event, argues for diploidy of E.cuniculi. The physical map should be useful for both the whole genome sequencing project and studies on genome plasticity of this widespread parasite.
INTRODUCTION
As with the other members of the phylum Microspora, Encephalitozoon cuniculi is an amitochondrial unicellular eukaryote and an obligate intracellular parasite. Infestation relies on the explosive extrusion of the long polar tube, initially coiled within the microsporidian spore and insuring the inoculation of the sporoplasm into the host cell. Intracellular development is divided into two major phases: the merogony, which involves the multiplication of naked forms (meronts), and the sporogony. During this second phase, the meronts transform into sporonts characterised by a dense cell wall and the division products of sporonts (sporoblasts) differentiate into small spores with a more complex wall and several specific internal structures collectively termed the extrusion or invasion apparatus. Most microsporidian species infect animals and some of them, including E.cuniculi, are recognised as being responsible for opportunistic infections in AIDS patients (1). The serological prevalence has been estimated to be ~8% in the European population (2).
Current knowledge of the organisation of the microsporidian nuclear genome mainly derives from karyotyping studies based on pulsed field gel electrophoresis (PFGE) techniques. Rather extensive inter-species variations in chromosome size and number and in haploid genome size have been reported. For example, in the fish microsporidian Glugea atherinae, the molecular karyotype comprises 16 chromosomal bands ranging between 420 and 2.7 Mb, and the genome size should be close to 19.5 Mb (3). The smallest microsporidian chromosome has only 139 kb in the insect parasite Vavraia oncoperae and different chromosome distributions account for low genome sizes in two Nosema species: 7.4 Mb in Nosema costelytrae and 5.3 Mb in Nosema locustae (4). Three Encephalitozoon species, able to infect man, possess extremely reduced genomes, down to 2.3 Mb in Encephalitozoon intestinalis (5). In the case of the most widely studied species E.cuniculi, 11 chromosomal bands range between 217 and 315 kb and the genome size has been estimated to be 2.9 Mb (6). About 100 anonymous DNA probes from a partial genomic library have been assigned to the E.cuniculi chromosomes (7) and rDNA units have been shown to be located on all these chromosomes (8). The microsporidian rDNA unit lacks ITS2 (no free 5.8S rRNA) and the sizes of the two corresponding rRNA (16S and 23S) are of prokaryotic type (9,10, see also Fig. 4A).
Figure 4.

Chromosomal localisation of rDNA units in E.cuniculi. (A) Schematic representation of the rDNA locus and positions of the oligonucleotides designed for long-PCR amplifications (small horizontal arrowheads) of which the combined products provide 8- and 11-kb probes. The position of probes for 16S coding region and 3′ part of the 23S coding region are also indicated. Note that the transcription unit lacks ITS2; the 5.8S sequence (white box) is associated with the 23S 5′ end. (B) FISH images illustrating the subtelomeric localisation of isolated rDNA units (green signals) on E.cuniculi combed chromosomes. The upper image (×100 objective) shows a chromosomal fragment including one rDNA signal. The letters assigned to the three different signals for statistical analysis are indicated on the scheme. The lower image (×40 objective) illustrates an entire chromosome harbouring two subterminal rDNA signals. Bar, 10 µm = 20 kb.
Growing amounts of sequencing data are becoming available for E.cuniculi. A rather high density of intron-less protein-coding genes has been suggested by an analysis of a 4.3-kb region of the chromosome I (11). However, a small intron (28 bp) has been identified in a ribosomal protein gene, indicating the occurrence of cis-splicing processes in microsporidia (12). The isolation of a gene encoding a homologue of mitochondrial HSP70 supports a secondary loss of mitochondria during the evolution of these parasites (13–15). Sequencing of the single rDNA internal transcribed spacer (ITS1) and karyotyping of E.cuniculi isolates from various mammalian hosts have provided evidence for three different strains (I, II and III) and genomic heterogeneity of two strains (16,17).
In order to help in the understanding of the physiology of microsporidian parasites and in the comparison with other eukaryotic genomes, a project of sequencing of the complete E.cuniculi genome has begun in collaboration with Génoscope (Evry, France). As this genome is very small, the strategy used consists in the in silico assembly of the sequences obtained from random genomic clones. No ordered library is required. However, in silico joining can be facilitated by independent chromosomal assignment of anchor markers. Standard genetic techniques cannot be applied to E.cuniculi because no sexual phase has been detected. Taking into consideration the restricted chromosomal size range of this organism and the requirement of tools for genomic analyses and intra-species genomic variability studies, we undertook the construction of a physical genome map of E.cuniculi. We adopted strategies based on two-dimensional (2D)-PFGE separation of restriction fragments arising either from a single digestion of the whole molecular karyotype (KARD-PFGE, for karyotype and restriction display by 2D-PFGE; Fig. 1) or from a double digestion of isolated chromosomal bands (DDIC-PFGE; Fig. 2). These mapping procedures, which derive from those successfully applied to bacterial genomes (18,19), are unbiased by repetitive DNA or highly recombinant DNA fragments. A better visualisation of the restriction fragments in the second dimension was provided by an original radioactive labelling procedure of the chromosomal DNA (20). This paper describes the establishment of a BssHII and MluI restriction map of the E.cuniculi chromosomes and the placing of several sequence-tagged sites (STSs). Molecular combing was also applied to investigate the location of rDNA units.
Figure 1.
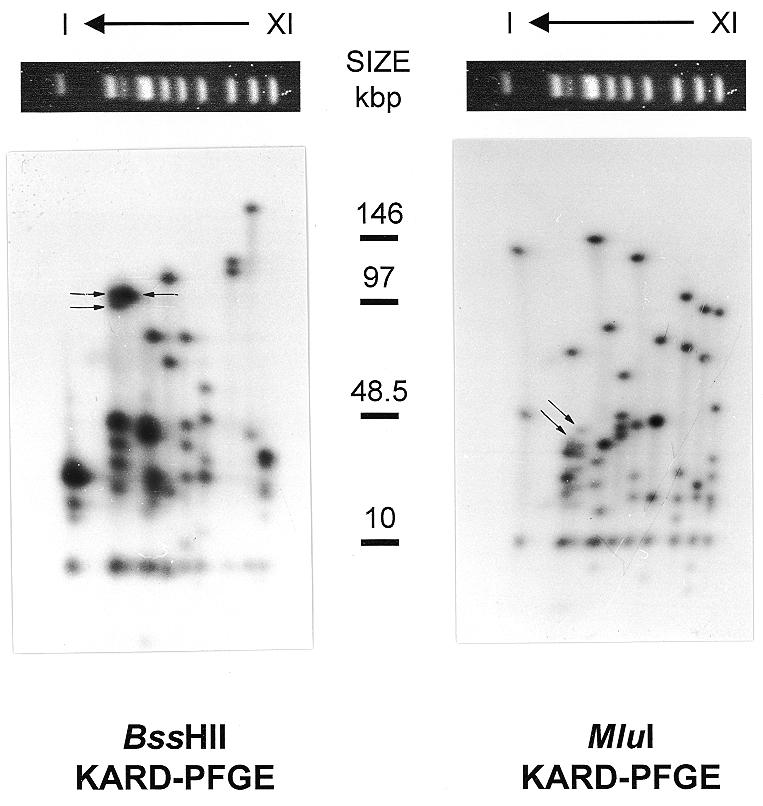
KARD-PFGE of the E.cuniculi genome. Autoradiographs of radiolabelled chromosomal DNA are shown after resolution of the molecular karyotype (at the top of the 2D-gel) and in-gel digestion with BssHII (left part) or MluI (right part) followed by a second orthogonal PFGE. Chromosome order is indicated above, the size of standards (in kb) in the middle. The two restriction fragments derived from chromosomes IIIa and IIIb, which can be differentiated by their size, are indicated by arrows. The decreasing intensity of spots in the right part of the lower region of BssHII KARD autoradiograph is due to a heterogeneous drying of the gel.
Figure 2.
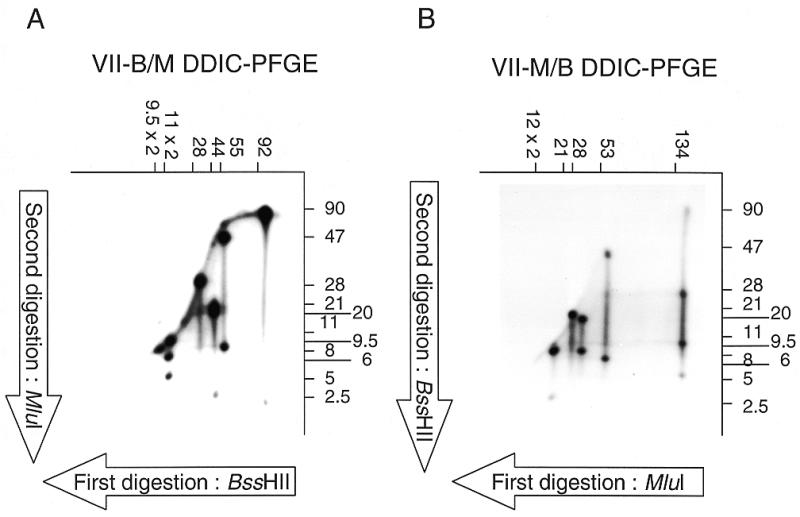
DDIC-PFGE of the chromosome VII of E.cuniculi. After isolation of the chromosome VII from the molecular karyotype, chromosomal DNA was digested by BssHII (A) or MluI (B), and radiolabelled. A second PFGE was performed leading to the separation of fragments (sizes in kb, above the autoradiograph). A second digestion was then performed with MluI (A) or BssHII (B), and the fragments separated with a third PFGE, orthogonal to the second (sizes in kb, on the right of each autoradiograph). Autoradiographs on (A) and (B) are therefore called VII B/M DDIC-PFGE and VII M/B DDIC-PFGE, respectively.
The sequences of the STSs (FASTA format) are available at http:\\www.protistes.univ-bpclermont.fr/fr/parasito/genome , as well as the principle of the KARD- and DDIC-PFGE.
MATERIALS AND METHODS
Cell culture
A mouse E.cuniculi isolate (strain I, karyotype form A) was kindly provided by Professor E. U. Canning (Imperial College of Science, London, UK). Parasites were grown at 37°C on MDCK (Madin Darby canine kidney) cells (21).
KARD-PFGE and DDIC-PFGE
Purification of E.cuniculi spores and preparation of agarose blocks for PFGE analyses were performed as described by Biderre et al. (6). Contour-clamped homogeneous electric field PFGE in a Gene Navigator™ (Pharmacia Biotech Europe, Saclay, France) system was performed at 12°C, using 1.3% chromosomal grade agarose (SeaKEM GTG, FMC Bioproducts, Rockland, ME) in 0.5× TBE (45 mM Tris base, 45 mM boric acid, 1 mM EDTA) and 16.5-s pulses for 38 h at 200 V. When desired, gels were stained with ethidium bromide and photographed under UV. Detailed 2D-PFGE protocols are described elsewhere (20). Briefly, for KARD-PFGE, a 1-mm wide agarose band was cut from the electrophoretic karyotype (first dimension step) and equilibrated six times in 1× High-Salt restriction buffer (50 mM Tris–HCl at pH 7.8, 100 mM NaCl, 10 mM MgCl2, 1 mM DTT and 0.1 mg/ml BSA). DNA was digested overnight with 100 U of MluI or BssHII (New England Biolabs, Beverly, MA). Radioactive labelling was then performed in the same buffer, using 5 µCi of [α-32P]dCTP, 5 U of Klenow fragment of the DNA polymerase (USB, Cleveland, OH), at 30°C for 3 h. Restriction fragments were PFGE-separated at 8°C in 1.2% agarose gel (0.5× TBE), with 4-s pulses for 11.5 h then 16-s pulses for 7.5 h at 230 V (second dimension step). The DDIC-PFGE procedure consisted in the 2D-PFGE analysis of restriction fragments derived from two successive digestions of individual chromosomal DNA bands. As a first step, BssHII or MluI digestion, radioactive labelling and separation of restriction fragments were performed as described for KARD-PFGE. Following digestion of a first-dimension gel slice with the other enzyme, the third PFGE was carried out at 8°C in 1.2% agarose gel (0.15× TBE), with 0.7-s pulses for 6 h at 450 V. For visualising radioactive DNA, gels were vacuum-dried for 1.5 h with a Gel Slab Dryer (Model 224, Bio-Rad, Ivry, France), and placed against a Biomax™ MS film (Kodak, Rochester, NY) between two intensifying screens at –80°C.
Hybridisation
DNA probes were synthesised using a PCR procedure (22) with minor modifications. Inserts in pUC 18 vectors (Pharmacia) were amplified and labelled with Universal and Reverse primers (respectively GTTTTCCCAGTCACGAC and AACAGCTATGACCATG) with 10 µCi of [α-32P]dCTP, 25 µM d[A,G,T]TP and 2 mM MgCl2 after 30 cycles of amplification comprising 30 s at 94°C, 30 s at 53°C and 2 min at 72°C. The exception was for the synthesis of the 16S rDNA probe (Fig. 4) where the primers used were those described by Delbac and Vivarès (23).
Radiolabelled probes were hybridised to PFGE-separated restriction fragments after transfer onto nylon membranes or directly in dried gels. Capillary transfer with 5× SSC buffer onto Hybond™ N(+) nylon membranes (Amersham, Saclay, France) was performed after the following treatments: 0.25 M HCl for 15 min, 0.4 M NaOH/1.5 M NaCl for 20 min and 1.5 M Tris–HCl, pH 7.0/1.5 M NaCl for 20 min (24,25). Membranes were baked for 2 h at 80°C. For in-gel hybridisation, the gel was treated with 0.4 M NaOH/0.15 M NaCl for 20 min then 1.5 M Tris–HCl, pH 7.0/0.15 M NaCl for 20 min and dried for 55 min at 55°C in a Slab Gel Dryer SGD 4050 (Savant, Bioblock, Strasbourg, France) (26). All hybridisations were performed at 65°C for 18 h in 5× SSPE, 1% SDS and 5× Denhardt’s (25). Membranes or gels were washed three times in 0.1× SSPE, 0.1% SDS, at 65°C for 30 min each, before exposition to Biomax‘ MS film (Kodak).
Molecular combing
Intact sporal DNA of E.cuniculi was extracted from agarose plugs. Contaminating spore wall components and precipitated Percoll‘ that might interfere with combing were eliminated through a short-duration preparative PFGE in 0.9% low-melting point (LMP) agarose gel (1× TAE buffer, 12°C, 200 V, 60-s pulse for 4 h). Agarose slices containing unstained DNA were cut then kept at 4°C in 0.5 M EDTA. DNA extraction and combing procedures were as described by Michalet et al. (27).
For fluorescence in situ hybridisation (FISH) experiments, we used 8- and 11-kb E.cuniculi rDNA probes corresponding to a mixture of two and three long-PCR products, respectively. Amplification was performed using the Long-Template PCR kit (Roche Molecular Biochemicals, Meylan, France) and the following oligonucleotide primer pairs: Fw1 (CTGGGGCAGTAGGGAGCTCTTTTCG) and Rv1 (CACAGACAGGGCTCAGGAGAGGTTC); Fw2 (GGAAGCGAAGGCTGTGCTCTTGGAC) and Rv2 (GGTGATCCTCTGTCAACGCAAGGGG); Fw3 (CCCCTTGCGTTGACAGAGGATCACC) and Rv3 (TCAAGAGCCGAGACCAAACAGACCC) (Fig. 4A). The preparation of a whole-chromosome probe consisted in the isolation of the largest chromosome (XI) from an LMP-agarose preparative gel (PFGE conditions as for karyotype). The agarose slice including this chromosome was melted and diluted one-fifth. Random PCR (28,29) was performed with this solution. Labelling of probes and FISH were carried out as previously described (27). Images were captured with a Charge-Coupled Device (CCD) camera (SITE 68 LX, DAGE MTI) mounted on an epifluorescence microscope Axiovert 135 (Zeiss, Oberkochen, Germany), with ×100, ×63 or ×40 objective. Size measurements and statistical analysis were performed with the Cartographix software (© X. Michalet). Since the ratio between the amounts of rDNA and chromosome XI probes was very high (~50), yellow signals resulting from the possible superimposition of red and green colours in rDNA regions were rarely observed.
Sequencing
Plasmid inserts were sequenced by the dideoxynucleotide chain-termination procedure (30) using the ABI Prism‘ Dye Terminator Cycle Sequencing kit according to the recommendations of the manufacturer (Perkin Elmer, Norwalk, CT). Thermocycling was performed on the GeneAmp‘ PCR system 2400, and electrophoresis on the ABI Prism‘ 377 Sequencer (Perkin Elmer).
Computational analysis
In silico restriction analysis of E.cuniculi DNA and gel readings were treated using the STADEN package (31) available from the French Molecular Biology Server INFOBIOGEN. Homologies with known nucleic acids sequences and proteins were determined with public databases using BLAST programs (32). Analysis of the chromosome I sequence of the Saccharomyces cerevisiae genome was achieved on the World Wide Web using the S.cerevisiae Genome Database (SGD) provided by Stanford University (http://genome-www.stanford.edu/Saccharomyces/ ).
RESULTS
Restriction map of the E.cuniculi genome
The construction of a physical map of the E.cuniculi genome was performed with an isolate belonging to strain I, regularly used in our previous investigations and then selected for the whole genome sequencing project. Numbering of the chromosomes was derived from the study of Biderre et al. (6), with a slight modification accounting for the differential migration of two homologous chromosomes (IIIa and IIIb) shown in the present work.
The choice of restriction endonucleases was made following in silico identification of restriction sites over ~25 kb of known E.cuniculi sequences, mainly represented by unique sequences of the chromosome I (11) and interspersed rDNA repeats (8). One 8-bp and 14 6-bp recognition site enzymes, predicted to cut less than three times in known sequences, were tested in KARD-PFGE experiments. The 8-bp recognition site enzyme PmeI, cutting only four times in the E.cuniculi genome (once in chromosomes VI and X, twice in chromosome VIII), was not used further. Of the 6-bp recognition site enzymes, 12 (Acc65I, BstEII, DraI, HpaI, NheI, PvuI, SalI, SnaBI, SpeI, SspI, XbaI and VspI) gave numerous fragments per chromosome and were also excluded. We retained the enzymes BssHII and MluI, which recognised the GCGCGC and the ACGCGT sequences, respectively, and these provided sizeable fragments for all chromosomes (Fig. 1). The smallest fragments seen on overexposed autoradiographs migrated at ~3 kb. A total of 139 sites (69 BssHII and 70 MluI) were identified. It should be stressed that all the chromosomes have in common a 9.5-kb BssHII fragment and a 12-kb MluI fragment, for which autoradiograph image analyses indicated that they are present twice in every chromosome. The repartition and the size of the restriction fragments led to new estimates of the sizes of the 11 different chromosomes, based on the sums of respective restriction fragments (reported in Fig. 3). The haploid genome size should then be slightly beyond 2.8 Mb. The 139 restriction sites identified correspond to 150 restriction fragments for 11 chromosomes. If we assume a random distribution of these sites throughout the genome, the mean interval between two successive sites should be 2822/150 = ~19 kb. In fact, the distance varies rather largely with the chromosome considered: from 28 kb in chromosome IX to 14 kb in chromosome XI (Fig. 3).
Figure 3.

Restriction map of the E.cuniculi genome. The 11 chromosomes are represented horizontally. Chromosome size and mean distance between two successive MluI or BssHII sites are indicated on the left. MluI sites (M) are marked above each chromosome and BssHII sites (B) below. Numbers between successive sites stand for the size interval in kb, as determined by KARD-PFGE. Vertical bars represent sites placed with DDIC-PFGE (solid lines) and rDNA hybridisation experiments with partially (dotted lines) or fully digested DNA (dotted lines topped by a black circle) on KARD-PFGE gels. Uncertain sites are represented with a cross. Horizontal arrows depict the rDNA units placed above when transcribed from the Watson strand or below when transcribed from the Crick strand. Only the longest chromosome III (IIIb) is shown; a 3-kb deletion in the 49-kb MluI fragment (vertical arrow) is characteristic of its homologue.
In order to place the BssHII and MluI restriction sites on each chromosome, we developed a mapping procedure applied to individual chromosomes (DDIC-PFGE) that can be viewed as the counterpart of 2D-PFGE in bacterial genomics (33). A prerequisite is the excision of chromosomal DNA bands from the molecular karyotype. Figure 2 shows autoradiographs obtained for the chromosome VII. The construction of the restriction map involved the swinging between data from the two sequentially different double digestions. For example, we deduce from the KARD-PFGE experiments that chromosome VII is characterised by eight BssHII restriction fragments (two of 9.5 kb, two of 11 kb and the others of 28, 44, 55 and 92 kb), and six MluI restriction fragments (two of 12 kb and the others of 21, 28, 53 and 134 kb). The 134-kb MluI fragment can be used to initiate the map of the chromosome VII. As indicated by M/B DDIC-PFGE (Fig. 2B), this fragment is divided into four BssHII restriction fragments (5, 11, 28 and 90 kb). The 11- and 28-kb fragments are internal since their sizes are compatible with data derived from BssHII-KARD-PFGE. Moreover, one of the two 11-kb fragments and the 28-kb BssHII fragments have no MluI site as judged by B/M DDIC-PFGE (Fig. 2A). Thus, the two other BssHII fragments (5 and 90 kb) must be placed at the extremities of the 134-kb MluI fragment. These fragments can be allocated to larger ones in the B/M profile (a 92-kb and the other 11-kb BssHII fragments). MluI digestion of this 11-kb fragment yields the above-mentioned 5-kb fragment and a 6-kb one that also belongs to a 53-kb MluI fragment. This indicates that the 134- and 53-kb MluI fragments are adjacent. Since BssHII digestion of the 53-kb fragment gives a 47-kb fragment also found in a 55-kb BssHII fragment, we deduce that the 11-kb BssHII fragment is adjacent to the 55-kb one. The smaller product (8 kb) derived from the 55-kb fragment also belongs to a 28-kb MluI fragment. The remaining fragment of 28 – 8 = 20 kb corresponds to one of the three MluI digestion products of a 44-kb BssHII fragment (21, 20 and 2.5 kb). Thus, this 44-kb fragment is also adjacent to the 55-kb BssHII fragment. MluI-KARD-PFGE shows a 21-kb fragment that is devoid of any BssHII site and can therefore be joined to the 28-kb MluI fragment. The deduced orders are 44–55–11–(28+11)–92 for the BssHII sites (one uncertain site location within a 39-kb region) and 21–28–53–134 for the MluI sites. The location of the two 9.5-kb BssHII and two 12-kb MluI fragments can be determined. The latter are digested by BssHII in 9.5- and 2.5-kb fragments (Fig. 2B) and the BssHII fragments that we have placed at extreme positions (44 and 92 kb) yield a 2.5-kb fragment after MluI digestion (Fig. 2A). Our interpretation is that each of the two 9.5-kb BssHII fragments occupies each end of chromosome VII, the most extreme BssHII and MluI sites being 2.5 kb apart. Finally, the following orders are obtained: 9.5–44–55–11–(28+11)–92–9.5 for the BssHII sites and 12–21–28–53–134–12 for the MluI sites.
The restriction map of the E.cuniculi genome deduced from DDIC-PFGE analyses is shown in Figure 3. Of the 139 restriction sites, 23 have uncertain localisations (eight BssHII and 15 MluI sites). Data from experiments involving hybridisations of fully or partially digested DNA eliminated the doubt for 13 of these sites (see below). A striking feature is that all the E.cuniculi chromosomes exhibit the same restriction pattern near the telomeric regions, characterised by one BssHII site and one MluI site located at 9.5 and 12 kb from each chromosomal end, respectively.
Subtelomeric distribution of the rDNA units
In E.cuniculi, the rDNA copies are scattered over all the chromosomes in a non-tandemly arranged organisation (8). The question was whether this unusual dispersion of the rDNA units is related to a common chromosomal location or not. The published rDNA sequence contains one MluI site at the end of the 23S coding region and one BssHII site in the 3′ untranscribed region, 2.5 kb apart. Thus, we may assume that each 2.5-kb MluI–BssHII fragment placed close to each telomeric region belongs to an rDNA copy. To test this hypothesis, molecular combing of the entire genome of E.cuniculi was performed. The DNA molecules stretched on glass were subsequently hybridised with whole-chromosome (XI) and rDNA fluorescent probes, respectively labelled with Texas Red and fluorescein. Due to the small size of the expected rDNA signal, we conducted all our analyses on images viewed with the ×100 objective. Because chromosomal breakage generally occurred, the most frequent images were short DNA stretches comprising a single green segment (rDNA, signal B) flanked by two red segments of unequal length (non-rDNA chromosomal regions, signals A and C), as illustrated by the upper photograph in Figure 4B. Statistical analysis of such images was performed in two independent experiments and data are shown in Table 1. Control of the surfaces with λ DNA provided a normal elongation factor (2 kb/µm). We first used an 8-kb rDNA probe (Fig. 4A). The size distribution for the signal A strongly deviates from the normality with a plurimodal curve and a mean of 22.2 kb [standard deviation (SD) = 38 kb]. We can explain this as a result from cross-hybridisation of chromosome XI probe with other chromosomes (Discussion) and random DNA fragmentation. In contrast, normal distributions with identical low standard deviations (1.2 kb) and mean sizes (~7 kb) are obtained for the lengths of the B and C signals. The mean size of the signal B is in agreement with the size of the rDNA probe used. The low variability for the signal C supports the hypothesis that this signal may be characteristic of each chromosomal extremity. Observations with a ×40 objective revealed a few images that may be interpreted as unbroken chromosomes bearing two telomere-associated rDNA copies (Fig. 4B, lower photograph). To determine the orientation of the rDNA unit, hybridisation of combed genomic DNA was performed with an 11-kb rDNA probe that more extends in the 3′ flanking region (Fig. 4A). Significant shortening of signal C suggests that the 3′ rDNA end is closer to the telomere than the 5′ rDNA end. The two rDNA units of each chromosome should then be divergently transcribed.
Table 1. Statistical analysis of molecular combing data.
| Signal | N | 8-kb rDNA probe | N | 11-kb rDNA probe | ||
|---|---|---|---|---|---|---|
| Mean SD | 95% C.I. | Mean SD | 95% C.I. | |||
| A | 185 | 11.1 19.0 | – | 61 | 11.8 9.5 | – |
| B | 206 | 3.5 0.6 | 0.04 | 77 | 5.3 1.0 | 0.22 |
| C | 192 | 3.4 0.6 | 0.04 | 57 | 2.4 0.7 | 0.18 |
One chromosome-XI probe labelled with Texas Red and two fluorescein-labelled rDNA probes of different sizes were used in FISH experiments. The 11-kb probe spanned both the rDNA unit and 3′ flanking region (Fig. 5A). All values are in µm (elongation factor: 2 kb/µm). The 95% confidence interval (95% C.I.) was calculated only for signals B and C, with normal size distributions. The difference between the mean signal C sizes for the two experiments is highly significant (Student’s t-test: t = 9.77, P < 0.001). N, number of measurements; SD, standard deviation.
We also hybridised rDNA probes to KARD-PFGE dried gels. The BssHII autoradiograph shows two rDNA signals per chromosome after hybridisation with either a 3′ rDNA probe spanning the end of the 23S region (left part of Fig. 5A) or a 16S probe (data not shown). Two exceptions were for the chromosomes I and III exhibiting a single signal but with a double intensity. The 3′ rDNA probe hybridised to the 12-kb MluI bands of all the chromosomes (right part of Fig. 5A). This confirms that each chromosome contains two divergent subtelomeric rDNA units.
Figure 5.

rDNA mapping by hybridisation on KARD-PFGE. BssHII- and MluI-KARD-PFGE dried gels were hybridised with either a 3′ rDNA probe (A) or a 16S rDNA probe (B) (see Fig. 4A for localisation of the probes). Data confirm the subtelomeric position of the rDNA unit on all the chromosomes and provide information about the MluI fragments adjacent to the 12-kb terminal fragments. Marker size is indicated in kb.
The use of the 16S rDNA probe allowed us to determine the MluI fragments within the BssHII fragments bearing the rDNA unit, for the chromosomes III, VIII, IX and X (Fig. 5B). The five newly placed MluI sites are symbolised with a vertical dotted line topped by a black circle in Figure 3. Partially digested DNA was also fractionated by KARD-PFGE then hybridised with the 16S rDNA probe (data not shown), leading to the localisation of five extra BssHII sites and three extra MluI sites (vertical dotted lines in Fig. 3). Thus, uncertainties remain for ~7% of the sites (three BssHII sites and seven MluI sites).
Mapping of STSs
Probes corresponding to five unique E.cuniculi genes isolated in our laboratory (12,34; C.Biderre, personal communication), were hybridised to BssHII- and MluI-KARD-PFGE gels. We also performed hybridisations with 62 anonymous chromosome-specific probes originating from a partial genomic library (7). They now correspond to 32 fully sequenced clones (average size: 650 bp) and 30 partially sequenced clones (average size: 950 bp). The localisation data is in agreement with the restriction map.
It should be stressed that nine of the 62 probes were known to recognise the chromosomal bands II and/or III. As these two bands are not easily resolvable (see Fig. 1 for example), we investigated their specificity by KARD-PFGE. Five clones were shown to be specifically associated with the chromosome II profile (c033, c048, c057, c124 and c140). The four others gave two signals of similar intensity corresponding to differentially migrated restriction fragments. The slow-migrating fragment belongs to the chromosome III KARD-PFGE profile while the fast-migrating one (size difference: 3 kb) is included within the chromosome II KARD-PFGE profile. This argues for two homologous chromosomes III of different sizes, that we termed IIIa (230 kb) and IIIb (233 kb), the smallest one (IIIa) co-migrating with the chromosome II. That the ethidium bromide-stained band II is three times more intense than the band III is in agreement with this. The chromosome IIIa may be viewed as the result of a 3-kb deletion in one of the two 107-kb BssHII fragments and in the 49-kb MluI fragment (indicated by an arrow in Fig. 1). Hybridisation data with c112 and c128 also support this interpretation (data not shown).
Four clones were found to be associated with two or three chromosomes. The clone c126, assigned to both chromosomes V and VII, has homology with a GTP-binding protein and is located relatively far from the extremities. The three others (c137, c147 and c123) have no significant homology with known sequences and are co-localised on a MluI restriction fragment of 21 kb in both chromosomes VII, X and XI. This suggests that three chromosomes share a similar terminal region, comprising the 12-kb MluI fragment common to the 11 chromosomes plus the 21-kb MluI fragment with internal identical sequences.
Four probes associated with all the chromosomes were also studied. Two clones (c030 and c198) hybridised the 9.5-kb BssHII- and 12-kb MluI-fragments and therefore appear as near-telomeric probes, while the two others (c059 and c150) recognised the same fragments as a 16S rDNA probe did (Fig. 5). No significant coding function is found. The richness in simple DNA repeats is suggestive of minisatellites.
DISCUSSION
For the first time, a physical and genetic genome map of an intracellular parasite (E.cuniculi) classified within the eukaryotic phylum Microspora has been obtained. The mapping involved the use of PFGE techniques and molecular combing. After selection of two restriction enzymes on the basis of well-resolved KARD-PFGE patterns, a nearly complete restriction map of the E.cuniculi genome was derived from DDIC-PFGE data. DNA hybridisations enabled us to map various STSs and previously ambiguous restriction sites. To our knowledge, 2D-PFGE has been previously applied to a eukaryotic genome only through a study of genetic markers for several chromosomes of Plasmodium falciparum (35). Restriction mapping has several advantages over other mapping approaches. First, there is no bias due to cloning artefacts. Secondly, phenotypic markers and the occurrence of meiotic recombination in the life cycle of the organism are not necessary, unlike genetic mapping. Thirdly, the procedure is rapid and does not require a large amount of genomic DNA.
Each of the two 6-bp cutters chosen to elaborate the map, BssHII and MluI, gives an average spacing of 40 kb. These enzymes have been previously described as rare cutters and used for physical maps of various bacteria: spirochetes (36), cyanobacteria (37), Gram-postive bacteria (38). An in silico digest of the smallest yeast chromosome sequences provides cutting frequencies similar to E.cuniculi. This low frequency might also be explained by a DNA modification to which both BssHII and MluI are sensitive, e.g. cytosine methylation as in higher eukaryotes. If DNA methylation occurs in E.cuniculi, differences in the methylation pattern of two homologous chromosomes should lead to differentially migrated fragments in KARD-PFGE gels. This has not been observed. The average resolution of the map should be sufficient to assist the ordering of sequences from the genome project as for further studies about inter- and intra-strain genome variability.
The restriction mapping and localisation of STSs has confirmed that the E.cuniculi haploid genome comprises 11 different chromosomes, as previously inferred from the electrophoretic karyotype (6) and DNA probe hybridisations (7). The calculated chromosome sizes show an apparent reduction of 5–10%. Although the sizes of chromosomal fragments are more easily assessed than whole chromosomes, we cannot exclude that this may be an underestimation due to the lack of detection of very small fragments (<1 kb). We tentatively suggest that some relationship exists between the chromosomes VII, X and XI on the basis of common sequences associated with the 21-kb MluI fragment just upstream from one of the two extreme 12-kb fragments. In Figure 3, these chromosomes are represented with the 21-kb fragment placed on the left.
Encephalitozoon cuniculi lacks sexual cycle and has been previously considered as a diploid organism (6,17). The KARD-PFGE technique combined with chromosome-specific probe hybridisation has been useful for revealing two homologous chromosomes III with a size difference of 3 kb, related to a deletion/insertion just upstream from one subtelomeric rDNA unit and entailing the co-migration of chromosomes II and IIIa. The assignment of some probes to both chromosomes II and III in a previous karyotyping study (7) is therefore erroneous. The mutational event should be relatively ancient, given that the karyotype and KARD-PFGE profiles were unchanged over investigation periods of 5 and 2 years, respectively.
Molecular combing has been successfully applied to the DNA from various organisms: λ phage, Escherichia coli, S.cerevisiae and human (27,39). The purpose of fiber-FISH on combed DNA has thus far been the physical mapping of loci and some genetic diagnostics e.g., the detection of micro-deletions (27,40). We have shown the suitability of molecular combing to map small genome structure through the visualisation of the subtelomeric position of the rDNA units in E.cuniculi. The chromosomal repartition of rDNA units varies from only one chromosome in Nosema bombycis (41) to all chromosomes in E.cuniculi (8). Tandem rDNA repeats have been reported in Nosema apis (42), which contrasts with the E.cuniculi case. A low number of non-tandemly organized rDNA genes are commonly found in the nuclear genome of apicomplexan parasites such as Plasmodium (43) or Cryptosporidium (44) and of the fungal parasite Pneumocystis (45). The rDNA units of some chromosomes of the diplomonad Giardia lamblia, a gut parasitic flagellate also characterised by an amitochondrial status and a small haploid genome (<12 Mb) (46), are subtelomeric but organised in tandem arrays (47). In fact, the E.cuniculi rDNA chromosomal arrangement resembles that of the free palindromic rDNA molecules found in slime moulds (48) and in the macronucleus of the ciliate Tetrahymena (49). However, such palindromes do not represent a general feature of the chromosomal organisation in these organisms.
The case of the genome of the nucleomorph in chlorarachniophytes should be also considered (reviewed in 50). These amoeboflagellates indeed possess a green alga endosymbiont, of which the vestigial nucleus (nucleomorph) harbours a tiny genome (380 kb) divided into three chromosomes with an rDNA unit at each end but with centripetal orientation (51). As subtelomeric rDNA exists in non-closely related eukaryotes, it is tempting to think that the positioning of the rDNA units near the chromosomal ends reflects more the occurrence of DNA rearrangements during the vegetative life of organisms prone to an important genome reduction. This genomic plasticity may be somewhat similar to that found in Giardia, in which the rearrangements depend on a common breakpoint upstream from the subtelomeric rDNA repeats (reviewed in 52). Probes hybridising to all the E.cuniculi chromosomes represent non-coding DNA sequences surrounding the subtelomeric rDNA units. A detailed discussion of the potential genes localised in internal regions is beyond the scope of this article. Some of them are of interest for further physiological studies, e.g. an aquaporin gene supporting the hypothesis of a role of aquaporin in the rapid entry of water into the spore during germination (53).
Taken together, our data enable us to propose a first model of the general organisation of an E.cuniculi chromosome. Identical or nearly identical terminal domains from one chromosome to another flank the variable core region containing most of the genes. Each terminal domain is occupied by one transcriptionally active rDNA unit and some repeated DNA sequences. The size of a terminal domain may be minimally estimated to be 15 kb (from the beginning of the rDNA unit to the chromosomal end). However, the longest conserved region may extend to ~30 kb, as suggested by the two MluI fragments common to chromosomes VII, X and XI as well as by hybridisation data with 16S rDNA probe applied to HpaI- and SspI-KARD-PFGE gels (data not shown). It would be of interest to further investigate intra- and/or interchromosomal mechanisms underlying sequence homogenisation of subtelomeric domains in the Encephalitozoon genome.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank C. White and P. Peyret for critical reading of the manuscript, F. Delbac and F. Duffieux for the gift of the probes, B. Chebance and R. Guerry for technical assistance. J.-F.B. would like to thank C. Biderre for the gift of the E.cuniculi partial genomic library and the CPSF probe before publication, M. Barbazanges and T. Bourbon for technical assistance. J.-F.B. is very grateful to E. Peyretaillade for critical comments and helpful discussion. E.C. is grateful to the staff of the laboratory ‘Biophysique de l’ADN’ for welcome technical assistance in molecular combing.
REFERENCES
- 1.Desportes-Livage I. (1996) Parasite, 3, 107–103. [DOI] [PubMed] [Google Scholar]
- 2.Van Gool T., Vetter,J.C.M., Weinmayr,B., Van Dam,A., Derouin,F. and Dankert,J. (1997) J. Infect. Dis., 175, 1020–1024. [DOI] [PubMed] [Google Scholar]
- 3.Biderre C., Pages,M., Méténier,G., David,D., Bata,J., Prensier,G. and Vivarès,C.P. (1994) C.R. Acad. Sci. III, 317, 399–404. [PubMed] [Google Scholar]
- 4.Malone L.A. and McIvor,C.A. (1995) J. Invertebr. Pathol., 65, 269–273. [DOI] [PubMed] [Google Scholar]
- 5.Biderre C., Canning,E.U., Méténier,G. and Vivarès,C.P. (1999) Eur. J. Protistol., 35, 194–196. [Google Scholar]
- 6.Biderre C., Pages,M., Méténier,G., Canning,E.U. and Vivarès,C.P. (1995) Mol. Biochem. Parasitol., 74, 229–231. [DOI] [PubMed] [Google Scholar]
- 7.Biderre C., Duffieux,F., Peyretaillade,E., Glaser,P., Peyret,P., Danchin,A., Pages,M., Méténier,G. and Vivarès,C.P. (1997) Gene, 191, 39–45. [DOI] [PubMed] [Google Scholar]
- 8.Peyretaillade E., Biderre,C., Peyret,P., Duffieux,F., Méténier,G., Gouy,M., Michot,B. and Vivarès,C.P. (1998) Nucleic Acids Res., 26, 3513–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vossbrinck C.R. and Woese,C.R. (1987) Nature, 6059, 287–288. [DOI] [PubMed] [Google Scholar]
- 10.Curgy J.J., Vavra,J. and Vivarès,C.P. (1980) Biol. Cell, 38, 49–52. [Google Scholar]
- 11.Duffieux F., Peyret,P., Roe,B.E. and Vivarès,C.P. (1998) Microb. Comp. Genomics, 3, 1–11. [DOI] [PubMed] [Google Scholar]
- 12.Biderre C., Méténier,G. and Vivarès,C.P. (1998) Mol. Biochem. Parasitol., 94, 283–286. [DOI] [PubMed] [Google Scholar]
- 13.Germot A., Philippe,H. and Le Guyader,H. (1997) Mol. Biochem. Parasitol., 87, 159–168. [DOI] [PubMed] [Google Scholar]
- 14.Hirt R.P., Healy,B., Vossbrinck,C.R., Canning,E.U. and Embley,T.M. (1997) Curr. Biol., 12, 995–998. [DOI] [PubMed] [Google Scholar]
- 15.Peyretaillade E., Broussolle,V., Peyret,P., Méténier,G., Gouy,M. and Vivarès,C.P. (1998) Mol. Biol. Evol., 15, 683–689. [DOI] [PubMed] [Google Scholar]
- 16.Didier E.S., Vossbrinck,C.R., Baker,M.D., Rogers,L.B., Bertucci,D.C. and Shadduck,J.A. (1995) Parasitology, 111, 411–421. [DOI] [PubMed] [Google Scholar]
- 17.Biderre C., Mathis,A., Deplazes,P., Weber,R., Méténier,G. and Vivarès,C.P. (1999) Parasitology, 118, 439–445. [DOI] [PubMed] [Google Scholar]
- 18.Römling U., Grothues,D., Heuer,T. and Tümmler,B. (1992) Electrophoresis, 13, 626–631. [DOI] [PubMed] [Google Scholar]
- 19.Cornillot E., Croux,C. and Soucaille,P. (1997) J. Bacteriol., 179, 7426–7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brugère J.-F., Cornillot,E., Méténier,G. and Vivarès,C.P. (2000) Nucleic Acids Res., 28, e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beauvais B., Sarfati,C., Challier,S. and Derouin,F. (1994) Antimicrob. Agents Chemother., 38, 2440–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schowalter D.B. and Sommer,S.S. (1989) Anal. Biochem., 177, 90–94. [DOI] [PubMed] [Google Scholar]
- 23.Delbac F. and Vivarès,C.P. (1997) J. Eukaryot. Microbiol., 44, 75S. [DOI] [PubMed] [Google Scholar]
- 24.Southern E.M. (1975) J. Mol. Biol., 98, 503. [DOI] [PubMed] [Google Scholar]
- 25.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 26.Tsao S.G., Brunk,C.F. and Pearlman,R.E. (1983) Anal. Biochem., 131, 365–372. [DOI] [PubMed] [Google Scholar]
- 27.Michalet X., Ekong,R., Fougerousse,F., Rousseaux,S., Schurra,C., Hornigold,N., Van Slegtenhorst,M., Wolfe,J., Povey,S., Beckmann,J.S. and Bensimon,A. (1997) Science, 277, 1518–1523. [DOI] [PubMed] [Google Scholar]
- 28.Froussard P.R. (1993) PCR Methods Appl., 2, 185–190. [DOI] [PubMed] [Google Scholar]
- 29.Grothues D., Cantor,C.R. and Smith,C.L. (1993) Nucleic Acids Res., 21, 1321–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanger F., Nicklen,S. and Coulson,A.R. (1977) Proc. Natl Acad. Sci. USA, 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Staden R. (1996) Mol. Biotechnol., 5, 233–241. [DOI] [PubMed] [Google Scholar]
- 32.Altschul S.F., Madden,T.L., Schäffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 17, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Römling U. and Tümmler,B. (1991) Nucleic Acids Res., 19, 3199–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delbac F., Peyret,P., Méténier,G., David,D., Danchin,A. and Vivarès,C.P. (1998) Mol. Microbiol., 29, 825–834. [DOI] [PubMed] [Google Scholar]
- 35.Hernandez-Rivas R. and Scherf,A. (1997) Mem. Inst. Oswaldo Cruz, 92, 815–819. [DOI] [PubMed] [Google Scholar]
- 36.Casjens S. and Huang,W.M. (1993) Mol. Microbiol., 8, 967–980. [DOI] [PubMed] [Google Scholar]
- 37.Bancroft I., Wolk,C.P. and Oren,E.V. (1989) J. Bacteriol., 171, 5940–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Canard B. and Cole,S.T. (1989) Proc. Natl Acad. Sci. USA, 86, 6676–6680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bensimon A., Simon,A., Chiffaudel,A., Croquette,V., Heslot,F. and Bensimon,D. (1994) Science, 265, 2096–2098. [DOI] [PubMed] [Google Scholar]
- 40.Weier H.-U., Wang,M., Mullikin,J.C., Zhu,Y., Cheng,J.F., Gruelich,K.M., Bensimon,A. and Gray,J.W. (1995) Hum. Mol. Genet., 10, 1903–1910. [DOI] [PubMed] [Google Scholar]
- 41.Kawakami Y., Inoue,T., Ito,K., Kitamizu,K., Hanawa,C., Ando,T., Iwano,H. and Ishihara,R. (1994) J. Invertebr. Pathol., 64, 147–148. [DOI] [PubMed] [Google Scholar]
- 42.Gatehouse H.S. and Malone,L.A. (1998) J. Invertebr. Pathol., 71, 97–105. [DOI] [PubMed] [Google Scholar]
- 43.Langsley G., Hyde,J.E., Goman,M. and Scaife,J.G. (1983) Nucleic Acids Res., 11, 8703–8717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le Blancq S.M., Khramtsov,N.V., Zamani,F., Upton,S.J. and Wu,T.W. (1997) Mol. Biochem. Parasitol., 90, 463–478. [DOI] [PubMed] [Google Scholar]
- 45.Stringer J.R. and Cushion,M.T. (1998) FEMS Immunol. Med. Microbiol., 22, 15–26. [DOI] [PubMed] [Google Scholar]
- 46.Fan J.B., Korman,S.H., Cantor,C.R. and Smith,C.L. (1991) Nucleic Acids Res., 19, 1905–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adam R.D., Nash,T.E. and Wellems,T.E. (1991) Mol. Cell. Biol., 11, 3326–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emery H.S. and Weiner,A.M. (1981) Cell, 26, 411–419. [DOI] [PubMed] [Google Scholar]
- 49.Karrer K.M. and Gall,J.G. (1976) J. Mol. Biol., 104, 421–453. [DOI] [PubMed] [Google Scholar]
- 50.Gilson P.R. and McFadden,G.I. (1997) Bioessays, 19, 167–173. [DOI] [PubMed] [Google Scholar]
- 51.Gilson P.R. and McFadden,G.I. (1995) Chromosoma, 103, 635–641. [DOI] [PubMed] [Google Scholar]
- 52.Upcroft P. and Upcroft,J.A. (1999) Protist, 150, 17–23. [DOI] [PubMed] [Google Scholar]
- 53.Frixione E., Ruiz,L., Cerbon,J. and Undeen,A.H. (1997) J. Eukaryot. Microbiol., 44, 109–116. [DOI] [PubMed] [Google Scholar]