

Abstract
A series of novel 3-cyanopyridone/pyrazoline hybrids (21–30) exhibiting dual inhibition against EGFR and BRAFV600E has been developed. The synthesized target compounds were tested in vitro against four cancer cell lines. Compounds 28 and 30 demonstrated remarkable antiproliferative activity, boasting GI50 values of 27 nM and 25 nM, respectively. These hybrids exhibited dual inhibitory effects on both EGFR and BRAFV600E pathways. Compounds 28 and 30, akin to Erlotinib, displayed promising anticancer potential. Compound 30 emerged as the most potent inhibitor against cancer cell proliferation and BRAFV600E. Notably, both compounds 28 and 30 induced apoptosis by elevating levels of caspase-3 and -8 and Bax, while downregulating the antiapoptotic Bcl2 protein. Molecular docking studies confirmed the potential of compounds 28 and 30 to act as dual EGFR/BRAFV600E inhibitors. Furthermore, in silico ADMET prediction indicated that most synthesized 3-cyanopyridone/pyrazoline hybrids exhibit low toxicity and minimal adverse effects.
Keywords: pyridine, pyrazoline, synthesis, anticancer, apoptosis, docking
1. Introduction
Cancer is a leading cause of death [1,2]. Cancer has the ability to invade and harm healthy tissues and disrupt functions; metastasis worsens treatment prospects [3,4]. Early detection, prevention, and enhanced treatment are vital to combat cancer’s impact on individuals and society [5,6]. Chemotherapy damages rapidly dividing cells like bone marrow, digestive tract, and hair follicle cells, causing side effects like hair loss, nausea, fatigue, and infection vulnerability [7]. Targeted treatments offer personalized, less harmful cancer care than traditional chemotherapy [8,9,10]. Tailored to cancer type and molecular traits, they enable precise and personalized therapy.
The EGFR (Epidermal Growth Factor Receptor) is crucial in cancer due to its frequent overexpression or mutations [11,12]. EGFR targeting is prominent in cancer treatment, with TKIs and monoclonal antibodies designed to disrupt signaling and reduce proliferation [13,14]. These targeted therapies have shown clinical success in several cancer types, including lung, colorectal, and head and neck cancer [15,16,17]. EGFR-targeted therapy faces resistance through diverse mechanisms, underscoring the need for ongoing research to enhance its effectiveness and counter resistance [18]. BRAF is pivotal in cancer treatment, especially for melanoma and colorectal cancer, driven by frequent V600E mutations promoting tumor growth [19,20]. BRAF inhibitors target mutated BRAF, halting its aberrant function and suppressing cancer cell growth [21]. Combining BRAF inhibitors with EGFR-targeted drugs shows promise in enhancing patient outcomes and tackling resistance [22,23]. Simultaneous EGFR and BRAF inhibition exhibits potential for increased efficacy and overcoming resistance [24,25]. Targeting EGFR and BRAF concurrently in preclinical studies shows synergistic antitumor effects [26,27].
Pyridine derivatives are organic compounds containing a pyridine ring, which is a six-membered aromatic ring with one nitrogen atom [28]. Pyridine derivatives hold medicinal value and versatile synthetic roles due to their heterocyclic nature [29]. Cyanopyridine derivatives have shown potential as antimicrobial agents, antibiotics [30], analgesics [31], and anticancer agents [32,33]. The anticancer activities of these compounds have attracted significant interest due to their potential to target various biological entities such as tubulin [34], HDAC [35], and PIM-1 Kinase [36,37]. Vemurafenib (Figure 1), marketed as Zelboraf®, is an FDA-approved pyridine derivative designed to treat advanced melanoma. It is a selective inhibitor of the V600E-mutated BRAF kinase [38,39,40].
Figure 1.

Structures of vemurafenib, Ibrutinib, compound I, and II.
In a recent publication [41], we described the antiproliferative activity of a novel series of cyanopyridine compounds as dual EGFR/BRAFV600E inhibitors. Compound I (Figure 1), in particular, was discovered to be the most active derivative, with antiproliferative action and the ability to inhibit both EGFR and BRAFV600E. Compound I exhibits a noteworthy IC50 value (0.80 µM) against Panc-1 cancer, surpassing doxorubicin (1.00 µM). Compound I displays comparable EGFR and BRAFV600E inhibition (IC50: 89 nM and 65 nM), like Erlotinib (IC50: 60 nM and 80 nM). Compound I, on the other hand, induces G0/G1 cell cycle arrest while also triggering apoptosis.
Moreover, the pyrazoline scaffold is a nitrogen-containing five-membered heterocyclic structure. Pyrazoline is a dihydropyrazole derivative with a ring-based double bond and neighboring nitrogen atoms. Pyrazoline ring cyclization via Michael addition occurs with chalcones and hydrazine monohydrate under basic conditions [42,43]. Due to its ease of synthesis and notable pharmacological and biological activities, especially concerning its anticancer properties [44,45], the pyrazoline ring has emerged as a pivotal scaffold in various heterocycles and pharmacologically active compounds [46,47]. The pyrazoline scaffold has been utilized to develop several approved drugs [48]. Ibrutinib, exemplifying exceptional anticancer activity, features a fused pyrazoline ring (Figure 1). Ibrutinib, a tyrosine kinase inhibitor, treats mantle cell lymphoma and chronic lymphocytic leukemia [49]. Pyrazoline derivatives have been documented to exhibit various pharmacological activities, including anti-diabetic [50], anti-cancer [44], anticonvulsant [51], and antidepressant [52] properties.
We recently reported the design, synthesis, and antiproliferative activity of compound II (Figure 1), a pyrazoline derivative, as a dual EGFR and BRAFV600E inhibitor [53]. Compound II showed an IC50 of 1.00 µM against A-549 cancer, outperforming doxorubicin (IC50: 1.40 µM). Compound II was effective against BRAFV600E, with an IC50 value of 93 nM, whereas Erlotinib had an IC50 of 60 nM. Furthermore, Compound II had an IC50 value of 81 nM, matching that of the EGFR inhibitor erlotinib. Pyrazoline II shows potential as a dual EGFR/BRAFV600E inhibitor with antiproliferative efficacy.
Building on our prior anticancer research (compounds I and II), we designed and synthesized compact pyridone-2-one–pyrazoline hybrids 21–30 (Figure 2). Newly created hybrids were assessed for antiproliferative effectiveness on four human cancer cell lines. Highly active compounds were then evaluated in vitro for EGFR/BRAFV600E inhibition and apoptotic effectiveness. An ADMET analysis was performed to evaluate the drug-likeness and toxicity of the novel hybrids. Molecular docking explored the binding affinities of the new pyridine/pyrazoline hybrids at EGFR and BRAFV600E active sites.
Figure 2.
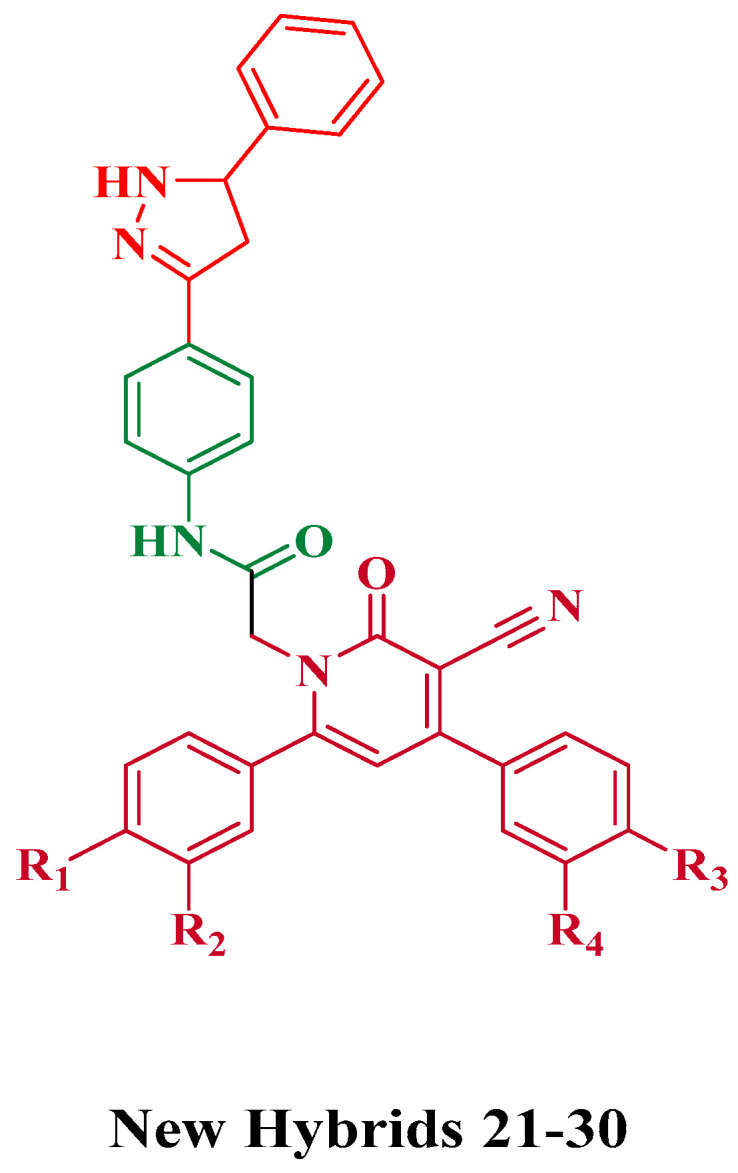
Structures of the new hybrids 21–30.
2. Results and Discussion
2.1. Chemistry
The synthetic procedures for the intermediates 4a–f, 7a–c, and the target 3-cyanopyridone–pyrazoline novel hybrids 21–30 are described in Scheme 1, Scheme 2 and Scheme 3. Shown in Scheme 1, compounds 3a–f were synthesized through a base-catalyzed Claisen–Schmidt condensation of 4-aminoacetophenone 1 with substituted benzaldehyde derivatives 2a–f [54]. Chalcones 3a–f were reacted with bromoacetyl bromide in a potassium carbonate solution in dichloromethane to produce the acetylated chalcones 4a–f [53].
Scheme 1.

Synthesis of acetylated chalcone derivatives 4a–f.
Scheme 2.

Synthesis of the intermediate compounds 7a–c.
Scheme 3.

Synthesis of 3-cyanopyridone–pyrazoline hybrids 21–30.
The key intermediates, 3-cyano-4,6-bis(phenyl)-pyridones 7a–c, were efficiently synthesized using a one-pot four-component reaction without any solvent. Equimolar amounts of the appropriately substituted benzaldehydes 2b, 2c, or 2f; the substituted acetophenones 5a, 5b, or 5c; ethyl cyanoacetate; and ammonium acetate were directly stirred at 110 °C for 10–15 min, resulting in the formation of the target compounds in high yields (Scheme 2) [55]. The sequential two-step reaction, which involved condensing benzaldehydes with acetophenones and then treating the resulting chalcones with ethyl cyanoacetate and excess ammonium acetate, resulted in a lower yield and a more time-consuming process. This observation highlights the advantage of the one-pot, four-component reaction used in this study, which provided a more efficient and time-saving method for synthesizing the desired compounds [32].
In this study, the synthesis of N-alkylated hybrids 8–20 was achieved by alkylating 3-cyanopyridones 7a–c with acetylated chalcones 4a–f. The reaction was performed using the sodium salt of the precursor cyanopyridones and conducted in a polar aprotic solvent, specifically dimethyl sulfoxide (DMSO), under inert conditions. This approach aimed to enhance the yield of N-alkylated hybrids 8–20. Importantly, this method offered the convenience of not requiring chromatography to separate O-alkylated isomer by-products with low yields. Subsequently, the final 3-cyanopyridone–pyrazoline hybrids 21–30 were obtained by refluxing the N-alkylated cyanopyridones 8–20 with hydrazine monohydrate in absolute ethanol for 12 h. This synthetic pathway is depicted in Scheme 3.
Compound 24, a 3-cyano-4,6-(methoxyphenyl)-pyridone–pyrazoline hybrid, was subjected to 1H NMR spectroscopic analysis. The pyrazoline ring displayed an AMX pattern for three protons (HA, HM, and HX), which manifested as a doublet of a doublet at δ: 2.78 ppm (J values of 16.20 and 11.1 Hz) for HA. However, the HM proton of the pyrazoline ring was split into a doublet of doublet signal at δ: 3.39 ppm (J values of 16.23 and 3.09 Hz), while the HX protons appeared as a doublet of doublets at a higher downfield shift at δ 4.76 ppm (J values of 11.02 and 3.06 Hz), as illustrated in Figure 3.
Figure 3.

AMX pattern representation of compound 24.
In the 1H NMR analysis, several signals were observed for compound 24, confirming its structure. Two singlet signals at δ 3.74 and 3.72 ppm corresponded to the (4-phenyl-4-OCH3) and (6-phenyl-4-OCH3) groups, respectively, with three proton integrations each. The linker’s methylene protons (N-CH2-CO) were assigned a singlet signal at δ 5.16 ppm, indicating two proton integration. Another singlet signal at δ 7.77 ppm represented the pyridine-C5–H proton with one proton integration. The amide proton NH was identified by a singlet signal at δ 10.51 ppm.
Confirmation of the final structure was further supported by DEPTQ 13C NMR spectroscopy. Two signals at δ 55.88 and 55.83 ppm were assigned to the (4-phenyl-4-OCH3) and (6-phenyl-4-OCH3) groups, respectively. The methylene carbon in the pyrazoline nucleus was observed at δ 40.83 ppm, while the methine carbon of the pyrazoline ring showed a chemical shift at δ 64.00 ppm. The methylene carbon in the linker exhibited a signal at δ 66.05 ppm. Signals at δ 163.81 and 166.78 ppm indicated the presence of a pyridone carbonyl and an amide carbonyl, respectively. The remaining carbons displayed the expected chemical shifts.
2.2. Biological Evaluation
2.2.1. Evaluation of Cell Viability
The viability of new compounds 21–30 was examined using the human mammary gland epithelial (MCF-10A) cell line [56]. The cell viability of compounds 21–30 was assessed using the MTT assay after four days of incubation on MCF-10A cells. Table 1 shows that none of the compounds tested were cytotoxic, and all hybrids had cell viability at 50 µM of more than 88%.
Table 1.
Comparative analysis of antiproliferative IC50 values for Compounds 21–30 and Erlotinib across different cancer cell lines.
| Comp. | Cell Viability % | Antiproliferative Activity IC50 ± SEM (nM) | ||||
|---|---|---|---|---|---|---|
| A-549 | MCF-7 | Panc-1 | HT-29 | Average IC50 (GI50) |
||
| 21 | 90 | 26 ± 2 | 30 ± 3 | 28 ± 2 | 28 ± 2 | 28 |
| 22 | 89 | 32 ± 3 | 35 ± 3 | 34 ± 3 | 32 ± 3 | 33 |
| 23 | 91 | 29 ± 2 | 34 ± 3 | 32± 3 | 32 ± 3 | 32 |
| 24 | 90 | 27 ± 2 | 31 ± 3 | 29± 2 | 28 ± 2 | 29 |
| 25 | 88 | 40 ± 3 | 45 ± 4 | 42 ± 4 | 42 ± 4 | 42 |
| 26 | 92 | 35 ± 3 | 40 ± 4 | 36 ± 3 | 36 ± 3 | 37 |
| 27 | 88 | 36 ± 3 | 42 ± 4 | 38 ± 3 | 38 ± 3 | 38 |
| 28 | 89 | 25 ± 2 | 30 ± 3 | 26 ± 2 | 26 ± 2 | 27 |
| 29 | 91 | 27 ± 2 | 31 ± 3 | 30 ± 3 | 30 ± 3 | 30 |
| 30 | 90 | 23 ± 2 | 28 ± 2 | 24 ± 2 | 24 ± 2 | 25 |
| Erlotinib | ND | 30 ± 3 | 40 ± 3 | 30 ± 3 | 30 ± 3 | 33 |
ND: Not Determined.
2.2.2. Evaluation of Antiproliferative Activity
Using Erlotinib as a control, an MTT assay was utilized to assess the antiproliferative effect of hybrids 21–30 versus four human cancer cell lines: a colon cancer (HT-29) cell line, a pancreatic cancer (Panc-1) cell line, a lung cancer (A-549) cell line, and a breast cancer (MCF-7) cell line [57]. The median inhibitory concentration (IC50) and GI50 [58] (average IC50) against the four cancer cell lines are shown in Table 1.
In general, the examined hybrids 21–30 revealed potent antiproliferative activity with GI50 values ranging from 25 nM to 42 nM versus the tested four cancer cell lines, in comparison to the standard Erlotinib, which had a GI50 value of 33 nM. Compounds 21, 24, 28, 29, and 30 were the most potent five derivatives, with GI50 values ranging from 25 nM to 30 nM, making them more potent than Erlotinib (GI50 = 33 nM).
Compound 30 (R = 3,4-diOCH3, R1 = R2 = R3 = R4 = OCH3) was the most potent derivative of all newly synthesized hybrids 21–30, with a GI50 value of 25 nM, which is 1.3-fold more potent than the reference Erlotinib (GI50 = 33 nM). The 3,4-dimethoxyphenyl moiety of the pyrazole ring appears to be crucial for activity, with a drop in the number of methoxy groups related to a decrease in antiproliferative activity. For example, compound 28 (R = 4-OCH3, R1 = R2 = R3 = R4 = OCH3) ranked second in activity with a GI50 value of 27 nM, and compound 22 (R = H, R1 = R2 = R3 = R4 = OCH3), with a GI50 value of 33 nM, was 1.3-fold less potent than compound 30, demonstrating the importance of the methoxy group number on the phenyl moiety at the pyrazole fifth position. Activity increased in the order 3,4-diOCH3 > 4-OCH3 > H.
Another important factor influencing the antiproliferative activity of the novel hybrids is the type of substituents on both 4, 6-diphenyl moieties of the pyridone ring. Compounds 24 (R = 3,4-diOCH3, R1 = R3 = R4 = OCH3, R2 = H) and 29 (R = 3,4-diOCH3, R1 = R3 = Cl, R2 = R4 = H) revealed GI50 values of 29 nM and 30 nM, respectively, being less potent than compound 30 but still more potent than the reference Erlotinib (GI50 = 33 nM).
Moreover, compound 23 (R = 4-Cl, R1 = R3 = Cl, R2 = R4 = H) revealed a GI50 value of 32 nM, which was less powerful than compound 29 (R = 3,4-diOCH3, R1 = R3 = Cl, R2 = R4 = H) (GI50 = 30 nM). Moreover, compounds 25 (R = 4-OCH3, R1 = R3 = Cl, R2 = R4 = H), 26 (R = 2,4-diCH3, R1 = R3 = Cl, R2 = R4 = H), and 27 (R = 2,4-diCl, R1 = R3 = Cl, R2 = R4 = H) were the least potent derivatives with GI50 values of 42 nM, 37 nM, and 38 nM, respectively, being less potent than compound 29, providing more support for the importance of the dimethoxy groups of the phenyl moiety at the pyrazole fifth position.
2.2.3. Evaluation of EGFR Inhibitory Activity
The five most effective antiproliferative compounds (21, 24, and 28–30) were evaluated for inhibition of EGFR as a possible target for their antiproliferative action [59]. Table 2 displays the results as IC50 values versus Erlotinib as a reference drug.
Table 2.
Determination of inhibitory concentration (IC50) for Compounds 21, 24, 28–30 against Epidermal Growth Factor Receptor (EGFR) and BRAFV600E oncogene.
| Compd. | EGFR Inhibition IC50 ± SEM (nM) |
BRAFV600E Inhibition IC50 ± SEM (nM) |
|---|---|---|
| 21 | 72 ± 5 | 73 ± 6 |
| 24 | 73 ± 5 | 77 ± 6 |
| 28 | 70 ± 5 | 69 ± 6 |
| 29 | 75 ± 5 | 80 ± 7 |
| 30 | 68 ± 4 | 65 ± 5 |
| Erlotinib | 80 ± 5 | ± 5 |
The results showed that the investigated hybrids 21, 24, and 28–30 had significant EGFR inhibitory effects, with IC50 values ranging from 68 nM to 75 nM, outperforming the reference Erlotinib (IC50 = 80 nM). Moreover, the results of an EGFR inhibitory assay were consistent with those of the antiproliferative assay, in which the most potent antiproliferative derivatives, compounds 28 (R = 4-OCH3, R1 = R2 = R3 = R4 = OCH3) and 30 (R = 3,4-diOCH3, R1 = R2 = R3 = R4 = OCH3), were the most potent EGFR inhibitors, with IC50 values of 70 ± 5 nM and 68 ± 5 nM, respectively, being 1.2-fold more potent than the reference Erlotinib (IC50 = 80 ± 5). These findings revealed that the studied compounds 21, 24, and 28–30 had significant EGFR inhibitory action and are potential antiproliferative agents.
2.2.4. Evaluation of BRAFV600E Inhibitory Activity
Hybrids 21, 24, and 28–30 were studied further as potential BRAFV600E inhibitors. Table 2 shows the IC50 values compared to Erlotinib, employed as a control [60]. According to Table 2, the examined hybrids displayed a promising BRAFV600E suppressive activity, with IC50 values ranging from 65 to 80 nM. In all cases, the examined derivatives were less potent than Erlotinib (IC50 = 60 nM). Compounds 28 and 30, the most potent derivatives in the antiproliferative and EGFR suppressive assays, were likewise the most effective derivatives as anti-BRAFV600E, with IC50 values of 65 ± 5 nM and 69 ± 6 nM, respectively. These data indicate that compounds 28 and 30 exhibit substantial antiproliferative action as dual EGFR/BRAFV600E inhibitors, hinting that additional structural modifications may be necessary to develop a more potent lead molecule for future development.
2.2.5. Valuation of Apoptotic Activity
One approach to treating cancer is regulating or terminating the uncontrolled multiplication of cancer cells. Using the cell’s natural dying process is an extremely effective method. Apoptosis evasion is a characteristic of cancer and is not specific to the etiology or type of cancer; thus, targeting apoptosis is beneficial for many types of cancer. Many anticancer drugs target various stages in both the intrinsic and extrinsic pathways [61,62,63]. Compounds 21, 28, and 30, the most effective derivatives in all in vitro studies, were examined for their ability to trigger the apoptosis cascade and reveal their proapoptotic potential.
2.2.6. Caspase 3 Activation Assay
Caspases are essential for the induction and maintenance of apoptosis. Caspase-3 is an important caspase that cleaves several cell proteins, causing apoptosis [64,65]. Compounds 21, 28, and 30 were investigated as caspase-3 activators against the human epithelial cancer cell line (A-594) [66], and the results are shown in Table 3.
Table 3.
Caspase-3 and -8, Bax, and Bcl-2 levels for compounds 21, 28, and 30 and Staurosporine on a human epithelial cancer cell line (A-594).
| Compd. No. | Caspase-3 | Caspase-8 | Bax | Bcl-2 | ||||
|---|---|---|---|---|---|---|---|---|
| Conc (Pg/mL) | Fold Change | Conc (ng/mL) | Fold Change | Conc (Pg/mL) | Fold Change | Conc (ng/mL) | Fold Reduction | |
| 21 | 530 ± 5 | 8 | ND | ND | ND | ND | ND | ND |
| 28 | 595 ± 5 | 9 | 2.25 | 25 | 325 | 36 | 0.80 | 6 |
| 30 | 710 ± 6 | 11 | 2.50 | 28 | 345 | 38 | 0.65 | 8 |
| Staurosporine | 465 ± 4 | 7 | 1.85 | 21 | 288 | 32 | 1.00 | 5 |
| Control | 65 | 1 | 0.09 | 1 | 9 | 1 | 5.00 | 1 |
ND: Not Determined.
Compounds 21, 28, and 30 demonstrated promising caspase-3 protein overexpression levels of 530 ± 5, 590 ± 5, and 710 ± 6 pg/mL, respectively. Compared to untreated control cells, they increased the protein caspase-3 in the A-594 cancer cell line by about 8-, 9-, and 11-fold. Compounds 21, 28, and 30 were more active than standard staurosporine, which had a caspase-3 overexpression level of 465 ± 4 pg/mL. Compound 30, the most effective antiproliferative agent, was once again the most active caspase-3 activator. These findings indicate the apoptotic potential of the studied compounds, which could explain their antiproliferative effect.
2.2.7. Caspase-8, Bax, and Bcl-2 Levels Assay
Compounds 28 and 30 were studied further for their influence on caspase-8, Bax, and antiapoptotic Bacl-2 levels against the A-594 cancel cell line using staurosporine as a control. Results are shown in Table 3. Caspase-8 overexpression was found to be highest in compound 30 (2.50 ng/mL), followed by compound 28 (2.25 ng/mL) and the reference staurosporine (1.85 ng/mL). Compared to the untreated control cell, compounds 28 and 30 elevated caspase-8 levels by 25- and 28-fold, respectively.
Compared to untreated A-594 cancer cells, compounds 28 and 30 induced Bax 36- and 38-fold (325 pg/mL and 345 pg/mL, respectively), more than staurosporine (288 pg/mL, a 32-fold induction). Finally, compared to staurosporine, compounds 28 and 30 triggered equipotent down-regulation of anti-apoptotic Bcl-2 protein levels in the A-594 cell line. These findings imply that compounds 28 and 30 serve as caspase-3 and -8 and Bax activators and down-regulators of the anti-apoptotic Bcl-2, and can be categorized as apoptotic inducers.
2.3. In Silico Studies
2.3.1. Docking Study
Utilizing in silico molecular docking simulation models, we tested compounds 28 and 30 against the EGFR and BRAFV600E [67] proteins. The objective was to determine the binding affinity and elucidate the inhibition mechanisms of the most potent compounds (28, 30) with potential cellular targets within this class. The results yielded a promising outlook throughout the assessment of cellular macromolecules employed in this investigation. Employing the Discovery Studio program, we conducted molecular docking studies involving crystal structures to investigate the binding modes of the EGFR (PDB ID: 1M17) [68] and BRAFV600E (BRAFm; PDB ID: 3OG7) [69].
The docking model involving the co-crystallized ligand (Erlotinib) positioned within the EGFR active site, which exhibited a docking score (S) of -7.7 kcal/mol and an RMSD of 1.33 Å upon re-docking within the same site. With this established and dependable docking model, we investigated the potential binding interactions of compounds 28 and 30 within the EGFR active site.
Through an analysis of the optimal docking pose, observed in Figure 4, for hybrids 28 and 30, a noteworthy pattern emerged, wherein the hybrids established a consistent hydrogen bond connection with the critical EGFR amino acid Met769. This hydrogen bond interaction was also observed between the Erlotinib pyrimidine nitrogen and Met769, as illustrated in Figure 4. Furthermore, compound 30 displayed an additional set of hydrogen bonding interactions with HIS 781, LEU 694, GLY 772, and GLN 767, in conjunction with pi–sigma interactions involving the amino acid residue CYS 773 (as depicted in Figure 4). These findings harmonize with the outcomes of the in vitro EGFR inhibition assay.
Figure 4.

Three-dimensional design of compounds 28 and 30 within the active site of EGFR (PDB ID: 1M17) showing the binding pocket molecular surface occupied completely by novel hybrids.
In this context, the most potent hybrids, 28 and 30, underwent a subsequent docking procedure within the active site of BRAFm. Upon inspecting the 2D representation of the molecular docking poses for compounds 28 and 30, as illustrated in Figure 5, a favorable alignment within the BRAFm active site was discernible, accompanied by an array of bonding interactions.
Figure 5.

Three-dimensional binding interaction diagrams of 28, 30, and Erlotinib within the active site of BRAFV600E (BRAFm; PDB ID: 3OG7).
Furthermore, upon probing the interactions between compound 28 and the BRAF active site, a typical hydrogen bond interaction with GLN 530, reminiscent of Erlotinib, was observed(Figure 5). Conversely, compound 30 exhibited a significant array of vital interactions with CYS 532, SER 536, and ASP 594. Additionally, the two methoxy groups of the pyridone moiety formed supplementary interactions with GLY 534 and GLN 461. Notably, these findings harmonize coherently with the in vitro BRAF inhibition assay outcomes.
2.3.2. ADMET Studies
Given the compelling in vitro and silico docking outcomes, we undertook supplementary ADMET studies for the synthesized compounds. This decision was driven by the desire to further enhance our understanding of these pivotal activities [70]. In the ADMET investigations, Erlotinib served as the established reference compound. Utilizing Discovery Studio 4.0, we predicted the ADMET descriptors for all the compounds. The anticipated descriptors are provided in Table 4 and Figure 6. Each hybrid compound exhibited a modest predicted level of intestinal absorption (absorption level = 2), positioning them as promising candidates for localized treatment of gastrointestinal tumors or potential candidates for intravenous administration. Most of these novel hybrids demonstrated a low aqueous solubility (ADME aqueous solubility level = 1), indicating a dependency on pH for solubility. The solubility improved as the pH decreases and ionization occurs. Additionally, the creation of hydrochloride salts presents an auxiliary avenue to enhance the solubility of these hybrids.
Table 4.
Comprehensive prediction of the absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles of synthesized 3-cyanopyridone–pyrazoline hybrids 21–30.
| Comp. ID | PSA | PPB a | Absorption Level b | CYP2D6 Prediction c | BBB Level d | Solubility Level e | AlogP98 |
|---|---|---|---|---|---|---|---|
| 21 | 97.832 | Yes | 2 | No | 4 | 1 | 6.356 |
| 22 | 133.553 | No | 2 | No | 4 | 2 | 4.962 |
| 23 | 97.832 | Yes | 3 | No | 4 | 1 | 7.021 |
| 24 | 142.483 | No | 2 | No | 4 | 3 | 4.945 |
| 25 | 106.763 | Yes | 2 | No | 4 | 1 | 6.34 |
| 26 | 97.832 | Yes | 3 | No | 4 | 1 | 7.329 |
| 27 | 97.832 | Yes | 3 | No | 4 | 1 | 7.685 |
| 28 | 142.483 | No | 2 | No | 4 | 2 | 4.945 |
| 29 | 115.693 | Yes | 2 | No | 4 | 1 | 6.323 |
| 30 | 151.413 | Yes | 3 | No | 4 | 3 | 4.929 |
| Erlotinib | 71.052 | Yes | 0 | No | 1 | 2 | 4.309 |
a PPB, plasma protein binding, FALSE means less than 90%, TRUE means >90%. b Absorption level, 0 = good, 1 = moderate, 2 = poor, 3 = very poor. c CYP2D6, cytochrome P2D6, TRUE = inhibitor, FALSE = non inhibitor. d BBB level, blood–brain barrier level, 0 = very high, 1 = high, 2 = medium, 3 = low, 4 = very low. e Solubility level, 1 = very low, 2 = low, 3 = good, 4 = optimal.
Figure 6.

The predicted ADMET study of 3-cyanopyridone–pyrazoline hybrids 21–30.
In the ADMET assessment, all the newly synthesized hybrids were situated at a blood–brain barrier (BBB) level of 4, effectively preventing their penetration across the BBB. Notably, drug bioavailability was linked with the fundamental property of the 2D polar surface area (ADMET 2D PSA). Employing the calculated 2D polar surface area (PSA 2D) and atom-based Log P98 (A log P98) properties, the outcomes were visualized in the form of a 2D ADMET plot (Figure 6). Notably, molecules possessing a PSA of >145 generally exhibit low bioavailability and passive absorption characteristics [71]. Employing a 2D chemical structure as input, the model for cytochrome P450 2D6 (CYP2D6) predicts the inhibition potential of the CYP2D6 enzyme. The liver enzyme CYP2D6 plays a pivotal role in the metabolism of numerous substrates, contributing significantly to most drug–drug interaction scenarios [72]. Consequently, an experiment to assess CYP2D6 inhibition is imperative within the regulatory protocols employed during drug discovery and development [73]. Every assessed hybrid compound was predicted to exhibit non-inhibitory behavior towards CYP2D6. Consequently, the likelihood of inducing liver dysfunction after administering these hybrids is minimal.
The plasma protein binding model aids in determining whether a substance will exhibit strong binding (>90% bound) to blood carrier proteins. A notable binding to plasma proteins (>90%) was anticipated for most hybrid compounds, as outlined in Table 4.
2.3.3. In Silico Toxicity Predictions
Toxicity prediction was performed for the synthesized compounds using the constructed and validated models within the Discovery Studio software [BIOVIA Corp V16.1.0.15350] [74]. The rodent carcinogenicity test conducted by the FDA assesses the potential of a chemical structure to induce cancer in rats. The rat maximum tolerated dose (MTD) prediction estimates the maximum dose at which a chemical substance can be administered to rats without causing adverse effects [75]. During toxicity assessments for a chemical compound, the rat oral LD50 prediction is employed to anticipate the rat acute median lethal dose (LD50) following oral administration [76]. Within the framework of the Draize test, ocular irritancy analysis is employed to ascertain the potential of a specific compound to induce ocular irritation and to gauge the extent of the irritation severity [77]. In rabbit-based assessments, skin irritancy investigations determine the likelihood of a substance causing skin irritation and the degree of severity it might induce. Most compounds exhibited low toxicity and demonstrated minimal adverse effects according to in silico assessments, as presented in Table 5.
Table 5.
Extensive prediction and analysis of toxicity properties for synthesized hybrid compounds (21–30).
| Comp. ID | FDA Rodent Carcinogenicity (Mouse, Female) |
Rat Maximum Tolerated Dose (Feed) a |
Rat Oral LD50 a |
Ocular Irritancy |
Skin Irritancy |
|---|---|---|---|---|---|
| 21 | Non-Carcinogen | 0.0760 | 1.27741 | Mild | Non-Irritant |
| 22 | Non-Carcinogen | 0.0312 | 8.48863 | Mild | Non-Irritant |
| 23 | Non-Carcinogen | 0.0597 | 1.67537 | Moderate-Severe | Non-Irritant |
| 24 | Non-Carcinogen | 0.0293 | 14.0883 | Mild | Non-Irritant |
| 25 | Non-Carcinogen | 0.0351 | 0.932102 | Mild | Non-Irritant |
| 26 | Non-Carcinogen | 0.0582 | 4.49508 | Mild | Non-Irritant |
| 27 | Non-Carcinogen | 0.0468 | 2.05463 | Mild | Non-Irritant |
| 28 | Non-Carcinogen | 0.0293 | 14.0883 | Mild | Non-Irritant |
| 29 | Non-Carcinogen | 0.0329 | 5.06156 | Mild | Non-Irritant |
| 30 | Non-Carcinogen | 0.0274 | 12.7428 | Mild | Non-Irritant |
| Erlotinib | Non-Carcinogen | 0.0828 | 0.662169 | Mild | Non-Irritant |
a Unit: g/kg body weight.
Furthermore, all the tested hybrids were predicted to possess non-carcinogenic properties, as initially determined by the FDA rodent carcinogenicity assessment. The evaluated compounds displayed rat oral LD50 values of 0.932 to 14.088 mg/kg body weight/day, similar to the Erlotinib value (0.662 mg/kg body weight/day).
Moreover, the predictive models indicated that all hybrids are expected to induce no or mild irritation in the cases of skin and ocular irritancy.
To summarize, the undertaken ADMET investigations within this study give vital insights into the newly developed hybrids’ potential effectiveness, safety, and pharmacokinetic behavior. The knowledge gleaned from these assessments holds paramount significance in steering the drug discovery and development trajectory. This wealth of information aids in pinpointing promising drug candidates worthy of further evaluation and advancement in the testing and developmental phases.
2.4. Structure–Activity Relationship (SAR)
Based on the observed results, the structure–activity relationship of our novel pyridone/pyrazoline hybrids (21–30) is as follows:
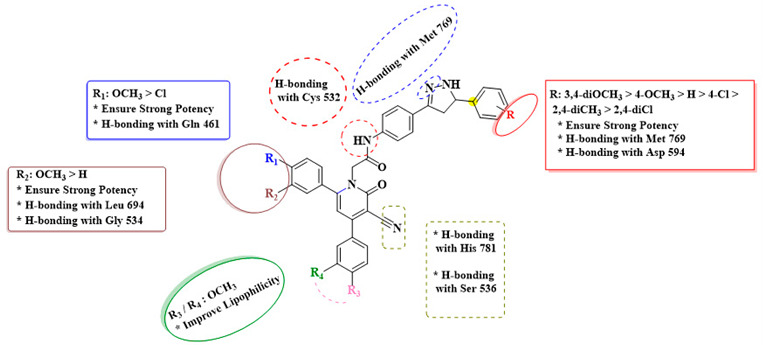
In summary, the presence and arrangement of substituents on the pyrazole and pyridone moieties appear to influence the potency of the new pyridone–pyrazoline hybrids. The involvement of specific functional groups, such as dimethoxy groups, and their positions are critical in defining the antiproliferative efficacy of these molecules.
3. Materials and Methods
3.1. Chemistry
General Details: See Supplementary Materials File.
Acetylated chalcones 4a-f, 3-cyanopyridones 7a-c, and 3-cyanopyridones-chalcones 8–20 were synthesized using previously reported procedures [55].
General Procedure for the Synthesis of Compounds 21–30
In a 50 mL round-bottom flask, a mixture of the appropriate 3-cyanopyridone-chalcone 8–20 (1.32 mmol) and hydrazine monohydrate (0.2 mL, 8.92 mmol) in 30 mL of absolute ethanol was heated under reflux for 12 h. The resulting mixture was cooled to room temperature and filtered through a Buchner funnel to collect the solid. The desired solid was washed with cold ethanol to remove impurities, affording the target 3-cyanopyridone/pyrazoline novel hybrids 21–30.
2-(3-Cyano-4,6-bis(4-chlorophenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-phenyl-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (21). White crystals (0.71 g, 82% yield); m.p 273–274 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.30 (1H, s, O=C-NH), 8.30 (2H, d, J = 8.50 Hz, Ar-H), 8.21 (1H, s, pyrazoline NH), 7.77 (3H, m, Ar-H), 7.73–7.69 (5H, m, Ar-H), 7.64 (2H, d, J = 8.50 Hz, Ar-H), 7.60–7.55 (5H, m, Ar-H), 7.36 (1H, s, Ar-H), 5.42 (2H, s, NCH2), 4.85–4.83 (1H, m, pyrazoline H), 3.44 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.84 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 167.17, 164.82, 157.12, 156.35, 154.68, 153.64, 147.69, 138.06, 136.75, 136.21, 136.03, 135.61, 131.27, 131.00, 130.88, 130.53, 129.77, 129.52, 129.42, 129.25, 129.14, 128.87, 127.11, 112.77, 109.67, 92.86, 66.33, 64.10, 40.82; Anal. Calcd. For C35H25Cl2N5O2 (618.52): C, 67.97; H, 4.07; N, 11.32. Found: C, 67.66; H, 4.11; N, 11.45.
2-(3-Cyano-4,6-bis(3,4-dimethoxyphenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-phenyl-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (22). White crystals (0.81 g, 86% yield); m.p 262–263 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.50 (1H, s, O=C-NH), 7.85–7.81 (2H, m, pyrazoline NH + Ar-H), 7.70 (1H, s, Ar-H), 7.62 (2H, d, J = 8.50 Hz, Ar-H), 7.57 (2H, d, J = 8.50 Hz, Ar-H), 7.49 (1H, s, Ar-H), 7.33–7.39 (6H, m, Ar-H), 7.26 (1H, s, Ar-H), 7.18 (1H, d, J = 9.00 Hz, Ar-H), 6.96 (1H, d, J = 9.00 Hz, Ar-H), 5.19 (2H, s, NCH2), 4.81 (1H, dd, J = 11.02 and 3.06 Hz, pyrazoline H), 3.88 (3H, s, OCH3), 3.86 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.63 (3H, s, OCH3), 3.43 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.80 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.79, 163.77, 157.10, 156.56, 151.61, 150.87, 149.32, 149.18, 148.86, 143.54, 139.03, 129.43, 128.98, 128.86, 128.56, 127.58, 127.10, 126.53, 121.99, 121.38, 119.52, 116.30, 113.73, 112.75, 112.26, 111.98, 110.70, 91.30, 65.97, 64.10, 56.20, 56.18, 56.08, 55.82, 41.14. Anal. Calcd. For C39H35N5O6 (669.74): C, 69.94; H, 5.27; N, 10.46. Found: C, 70.03; H, 5.16; N, 10.39.
2-(3-Cyano-4,6-bis(4-chlorophenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (23). White powder (0.81 g, 89% yield); m.p 286–287 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.56 (1H, s, O=C-NH), 8.24 (2H, d, J = 8.50 Hz, Ar-H), 7.91 (1H, s, pyrazoline NH), 7.81 (2H, d, J = 8.50 Hz, Ar-H), 7.70–7.60 (8H, m, Ar-H), 7.85–7.81 (5 H, m, Ar-H), 5.20 (2H, s, NCH2), 4.81–4.85 (1H, m, pyrazoline H), 3.45 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.79 (1H, dd, J = 11.23 and 3.03 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.57, 163.68, 156.20, 155.94, 148.99, 142.59, 138.93, 136.23, 135.73, 135.51, 134.85, 132.03, 131.09, 129.72, 129.44, 129.31, 129.01, 128.93, 128.80, 126.61, 119.71, 115.45, 114.80, 93.08, 66.26, 63.33, 41.10; Anal. Calcd. For C35H24Cl3N5O2 (652.96): C, 64.38; H, 3.70; N, 10.73. Found: C, 64.46; H, 3.82; N, 10.59.
2-(3-Cyano-4-(3,4-dimethoxyphenyl)-6-(4-methoxyphenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (24). White crystals (0.86 g, 88% yield); m.p 244–245 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.50 (1H, s, O=C-NH), 8.16 (2H, d, J = 8.5 Hz, Ar-H), 7.77 (1H, s, pyrazoline NH), 7.58–7.66 (4H, m, Ar-H), 7.34–7.41 (3H, m, Ar-H + pyridine-C5–H), 7.16 (1H, d, J = 8.5 Hz, Ar-H), 6.88–6.99 (5H, m, Ar-H), 5.16 (2H, s, NCH2), 4.78 (1H, dd, J = 11.02 and 3.06 Hz, pyrazoline H), 3.87 (3H, s, OCH3), 3.86 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.74 (3H, s, OCH3), 3.72 (3H, s, OCH3), 3.39 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.81 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.78, 163.81, 161.85, 157.01, 156.62, 150.90, 149.20, 149.13, 148.41, 138.92, 135.77, 129.69, 129.28, 129.10, 128.51, 126.51, 121.99, 119.65, 119.09, 116.27, 114.61, 113.49, 112.68, 112.23, 112.16, 112.12, 110.84, 91.22, 66.05, 64.00, 56.18, 56.16, 56.03, 55.88, 55.83, 40.83. Anal. Calcd. For C40H37N5O7 (699.76): C, 68.66; H, 5.33; N, 10.01. Found: C, 68.58; H, 5.21; N, 10.12.
2-(3-Cyano-4,6-bis(4-chlorophenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (25). White powder (0.83 g, 92% yield); m.p 256–257 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.53 (1H, s, O=C-NH), 8.22 (2H, d, J = 8.50 Hz, Ar-H), 7.89 (1H, s, pyrazoline NH), 7.80 (2H, d, J = 8.50 Hz, Ar-H), 7.58–7.69 (6H, m, Ar-H), 7.46–7.40 (3H, m, Ar-H), 7.28 (2H, d, J = 8.50 Hz, Ar-H), 6.89 (2H, d, J = 8.50 Hz, Ar-H), 5.20 (2H, s, NCH2), 4.73–4.78 (1H, m, pyrazoline H), 3.79-385 (1H, m, pyrazoline H), 3.72 (3H, s, OCH3), 2.77 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.74, 163.20, 155.79, 155.38, 135.83, 135.36, 134.99, 134.32, 130.57, 129.19, 128.97, 128.82, 128.55, 127.78, 127.16, 126.49, 126.12, 125.70, 119.34, 115.00, 114.22, 113.78, 113.45, 92.59, 65.83, 63.26, 55.54, 39.85; Anal. Calcd. For C36H27Cl2N5O3 (648.54): C, 66.67; H, 4.20; N, 10.80. Found: C, 66.75; H, 4.36; N, 10.73.
2-(3-Cyano-4,6-bis(4-chlorophenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(2,4-dimethylphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (26). White crystals (0.78 g, 86% yield); m.p 269–270 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.52 (1H, s, O=C-NH), 8.21 (2H, d, J = 8.50 Hz, Ar-H), 7.90 (1H, s, pyrazoline NH), 7.81–7.79 (3H, m, Ar-H), 7.69–7.58 (6H, Ar-H), 7.45 (2H, d, J = 8.50 Hz, Ar-H), 7.29 (1H, d, J = 8.50 Hz, Ar-H), 6.96 (2H, d, J = 8.50 Hz, Ar-H), 5.20 (2H, s, NCH2), 4.92 (1H, dd, J = 11.02 and 3.06 Hz, pyrazoline H), 3.44 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.65 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H), 2.28 (3H, s, CH3), 2.22 (3H, s, CH3); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.54, 163.70, 156.21, 155.94, 148.44, 138.78, 138.54, 136.23, 136.16, 135.73, 135.53, 135.26, 134.87, 131.37, 131.11, 129.74, 129.50, 129.31, 126.92, 126.51, 126.06, 119.69, 119.63, 115.46, 114.83, 93.09, 66.27, 60.74, 40.47, 21.01, 19.45; Anal. Calcd. For C37H29Cl2N5O2 (646.57): C, 68.73; H, 4.52; N, 10.83. Found: C, 68.88; H, 4.46; N, 10.74.
2-(3-Cyano-4,6-bis(4-chlorophenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(2,4-dichlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (27). White powder (0.78 g, 91% yield); m.p 291–292 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.52 (1H, s, O=C-NH), 8.22 (2H, d, J = 8.50 Hz, Ar-H), 7.91 (1H, s, pyrazoline NH), 7.82 (2H, d, J = 8.50 Hz, Ar-H), 7.70–7.57 (9H, m, Ar-H), 7.47–7.43 (3H, m, Ar-H), 5.20 (2H, s, NCH2), 5.05 (1H, dd, J = 11.02 and 3.06 Hz, pyrazoline H), 3.58 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.73 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.77, 163.21, 155.72, 155.44, 139.68, 138.59, 135.75, 135.28, 135.02, 134.35, 132.71, 132.31, 131.64, 130.62, 130.08, 129.24, 128.96, 128.82, 128.20, 127.75, 127.57, 126.24, 119.21, 114.93, 114.33, 92.61, 65.79, 59.88, 40.15; Anal. Calcd. For C35H23Cl4N5O2 (687.40): C, 61.16; H, 3.37; N, 10.19. Found: C, 61.23; H, 3.43; N, 10.25.
2-(3-Cyano-4,6-bis(3,4-dimethoxyphenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (28). White powder (0.81 g, 83% yield); m.p 256–257 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.53 (1H, s, O=C-NH), 8.17 (2H, d, J = 8.5 Hz, Ar-H), 7.77 (1H, s, pyrazoline NH), 7.65 (2H, d, J = 8.50 Hz, Ar-H), 7.60 (2H, d, J = 8.50 Hz, Ar-H), 7.41 (1H, s, Ar-H), 7.36 (2H, d, J = 8.5 Hz, Ar-H), 7.17 (1H, s, Ar-H), 6.99–6.88 (5H, m, Ar-H), 5.16 (2H, s, NCH2), 4.76 (1H, dd, J = 11.02 and 3.06 Hz, pyrazoline H), 3.87 (3H, s, OCH3), 3.86 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.74 (3H, s, OCH3), 3.73 (3H, s, OCH3), 3.44 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.81 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.99, 163.36, 161.47, 156.62, 156.27, 153.09, 150.53, 148.82, 143.48, 142.79, 129.47, 129.27, 128.86, 128.62, 128.10, 127.37, 121.86, 121.62, 118.93, 115.84, 114.22, 113.18, 112.30, 111.86, 111.36, 110.05, 92.02, 90.84, 65.67, 62.13, 55.86, 55.83, 55.81, 55.78, 55.44, 38.89. Anal. Calcd. For C40H37N5O7 (699.76): C, 68.66; H, 5.33; N, 10.01. Found: C, 68.58; H, 5.26; N, 10.09.
2-(3-Cyano-4,6-bis(4-chlorophenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (29). White crystals (0.79 g, 83% yield); m.p 243–245 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.53 (1H, s, O=C-NH), 8.23 (2H, d, J = 8.50 Hz, Ar-H), 7.91 (1H, s, pyrazoline NH), 7.82 (2H, d, J = 8.50 Hz, Ar-H), 7.70–7.58 (7H, m, Ar-H),), 7.47–7.40 (3H, m, Ar-H), 6.99 (1H, s, Ar-H), 6.89 (1H, d, J = 8.50 Hz, Ar-H), 5.20 (2H, s, NCH2), 4.76 (1H, dd, J = 11.02 and 3.06 Hz, pyrazoline H), 3.74 (3H, s, OCH3), 3.72 (3H, s, OCH3), 3.37 (1H, m, pyrazoline H), 2.80 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.09, 163.25, 155.74, 155.41, 148.73, 148.62, 147.98, 138.38, 135.76, 135.30, 135.02, 134.34, 130.60, 129.23, 128.95, 128.81, 128.55, 126.07, 119.27, 118.83, 118.64, 114.96, 114.29, 111.70, 110.41, 92.60, 65.81, 63.57, 55.56, 55.42, 40.64. Anal. Calcd. For C37H29Cl2N5O4 (678.57): C, 65.49; H, 4.31; N, 10.32. Found: C, 65.57; H, 4.44; N, 10.40.
2-(3-Cyano-4,6-bis(3,4-dimethoxyphenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (30). White powder (0.89 g, 87% yield); m.p 233–234 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.48 (1H, s, O=C-NH), 7.81–7.57 (8H, m, pyrazoline NH + 7 Ar-H), 7.36 (2H, d, J = 8.50 Hz, Ar-H), 7.17 (1H, d, J = 8.50 Hz, Ar-H), 6.88–6.99 (4H, m, Ar-H), 5.19 (2H, s, NCH2), 4.76-4.73 (1H, m, pyrazoline H), 3.87 (3H, s, OCH3), 3.85 (3H, s, OCH3), 3.78 (3H, s, OCH3), 3.74 (3H, s, OCH3), 3.72 (3H, s, OCH3), 3.63 (3H, s, OCH3), 2.81 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.42, 163.36, 156.67, 156.08, 151.18, 150.46, 148.91, 148.75, 148.02, 138.60, 135.33, 129.02, 128.67, 128.13, 126.09, 121.58, 120.91, 119.14, 118.96, 115.92, 113.23, 112.29, 111.97, 111.50, 110.44, 110.27, 90.86, 65.57, 63.63, 55.74, 55.71, 55.61, 55.58, 55.46, 55.39, 40.66..Anal. Calcd. For C41H39N5O8 (729.79): C, 67.48; H, 5.39; N, 9.60.
3.2. Biology
3.2.1. Cell Viability Assay
The human mammary gland epithelial (MCF-10A) normal cell line was utilized to investigate the viability of new derivatives 21–30 [56]. For more information, see Supplementary Materials Files.
3.2.2. Antiproliferative Assay
An MTT assay was used to assess the antiproliferative activity of 21–30 against four human cancer cell lines: a colon cancer (HT-29) cell line, a pancreatic cancer (Panc-1) cell line, a lung cancer (A-549) cell line, and a breast cancer (MCF-7) cell line, using Erlotinib as the control, see Supplementary Materials Files)
3.2.3. EGFR Inhibitory Assay
The five most active antiproliferative compounds (21, 24, and 28–30) were evaluated for inhibition of EGFR as a possible target for their antiproliferative action [59]. See Supplementary Materials Files.
3.2.4. BRAFV600E Inhibitory Assay
Hybrids 21, 24, and 28–30 were studied further as potential BRAFV600E inhibitors. Table 2 shows the IC50 values compared to Erlotinib, employed as a control [60]. See Supplementary Materials Files.
3.2.5. Apoptotic Markers Assays
Compounds 21, 28, and 30 were investigated as caspase-3, caspase-8, Bax activators, and Bcl-2 down-regulators against the human epithelial cancer cell line (A-594) [66]. See Supplementary Materials Files.
3.2.6. Docking Study
For the molecular docking study, we utilized the computational software BIOVIA I Discovery Studio 2016, provided by manufacturers located in San Diego, California. The registration addresses and copyright details are as follows: Copyright 2015, Dassault Systèmes BIOVIA Corp V16.1.0.15350. The chosen proteins underwent preparation for docking analysis via the Protein Preparation Wizard [78]. Ligands were then mapped onto a three-dimensional model and subjected to energy minimization using LigPrep. To enhance potential binding, a receptor grid was generated for the selected binding site using the Receptor Grid Generation Tool. Ultimately, the Glide tool was utilized to assess both docking scores and various binding modes for the ligands.
3.2.7. In Silico ADMET Analysis
ADMET studies were performed using BIOVIA I Discovery Studio 2016 [79]. The chemical structures of all compounds were imported, and ADMET descriptors were predicted using integrated models, including Lipinski’s Rule of Five and assessments of absorption, distribution, metabolism, excretion, and toxicity. The obtained results were analyzed to ascertain the drug-likeness and safety profiles of the compounds.
4. Conclusions
In this study, novel 3-cyanopyridone/pyrazoline derivatives were synthesized as potential dual-targeting antiproliferative agents. Compounds 28 and 30 exhibited remarkable antiproliferative activity, surpassing Erlotinib, with notable potency against the EGFR and BRAFV600E pathways. The induction of apoptosis in MCF-7 cells was linked to the upregulation of caspase-3 and an altered Bax/Bcl-2 gene ratio, thereby emphasizing the potential mechanism of compounds 28 and 30. Molecular docking affirmed the strong inhibition of compound 30 against both EGFR and BRAFV600E. ADMET analysis indicated favorable safety profiles for most compounds. Further in vitro and in vivo studies, along with chemical optimizations, are warranted to enhance efficacy. Overall, this research introduces promising dual inhibitors (28 and 30) against EGFR/BRAFV600E pathways, laying the groundwork for advanced antiproliferative agents.
Acknowledgments
The authors acknowledge the support by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2023R3), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT -Publication Fund of the Karlsruhe Institute of Technology.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28186586/s1.
Author Contributions
E.A.M.B., A.M.H. and M.A.-A.: Supervision, conceptualization, editing and revision. B.G.M.Y., H.A.A.-Z.: methodology, formal analysis, writing, editing and revision. S.B.: formal analysis, writing and editing. M.H.: Docking Study., L.H.A.-W.: Funding Acquisition, editing and revision. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data will be provided upon request.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples will be available upon request form the authors.
Funding Statement
This work was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2023R3), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Fitzmaurice C., Dicker D., Pain A., Hamavid H., Moradi-Lakeh M., MacIntyre M.F., Allen C., Hansen G., Woodbrook R., Wolfe C. The global burden of cancer 2013. JAMA Oncol. 2015;1:505–527. doi: 10.1001/jamaoncol.2015.0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mattiuzzi C., Lippi G. Current cancer epidemiology. J. Epidemiol. Glob. Health. 2019;9:217. doi: 10.2991/jegh.k.191008.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abbott M., Ustoyev Y. Seminars in Oncology Nursing. Elsevier; Amsterdam, The Netherlands: 2019. Cancer and the immune system: The history and background of immunotherapy; p. 150923. [DOI] [PubMed] [Google Scholar]
- 4.Sheikh A., Md S., Kesharwani P. Rgd engineered dendrimer nanotherapeutic as an emerging targeted approach in cancer therapy. J. Control. Release. 2021;340:221–242. doi: 10.1016/j.jconrel.2021.10.028. [DOI] [PubMed] [Google Scholar]
- 5.Shah S.C., Kayamba V., Peek R.M., Jr., Heimburger D. Cancer control in low-and middle-income countries: Is it time to consider screening? J. Glob. Oncol. 2019;5:1–8. doi: 10.1200/JGO.18.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balata H., Fong K.M., Hendriks L.E., Lam S., Ostroff J.S., Peled N., Wu N., Aggarwal C. Prevention and early detection for nsclc: Advances in thoracic oncology 2018. J. Thorac. Oncol. 2019;14:1513–1527. doi: 10.1016/j.jtho.2019.06.011. [DOI] [PubMed] [Google Scholar]
- 7.Amjad M.T., Chidharla A., Kasi A. Cancer Chemotherapy. [(accessed on 5 September 2023)]. Available online: https://europepmc.org/article/nbk/nbk564367.
- 8.Mosaa Z.A., Al-Majidi M., Bader A.T., Boulhaoua M., Ahmad I. Bioconjugates: A new class of therapeutics for cancer treatment. J. Univ. Babylon Pure Appl. Sci. 2023;31:161–175. doi: 10.29196/jubpas.v31i2.4670. [DOI] [Google Scholar]
- 9.Gomaa H.A., Shaker M.E., Alzarea S.I., Hendawy O., Mohamed F.A., Gouda A.M., Ali A.T., Morcoss M.M., Abdelrahman M.H., Trembleau L. Optimization and sar investigation of novel 2, 3-dihydropyrazino [1, 2-a] indole-1, 4-dione derivatives as egfr and brafv600e dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg. Chem. 2022;120:105616. doi: 10.1016/j.bioorg.2022.105616. [DOI] [PubMed] [Google Scholar]
- 10.Epstein J.B., Thariat J., Bensadoun R.J., Barasch A., Murphy B.A., Kolnick L., Popplewell L., Maghami E. Oral complications of cancer and cancer therapy: From cancer treatment to survivorship. CA A Cancer J. Clin. 2012;62:400–422. doi: 10.3322/caac.21157. [DOI] [PubMed] [Google Scholar]
- 11.London M., Gallo E. Epidermal growth factor receptor (egfr) involvement in epithelial-derived cancers and its current antibody-based immunotherapies. Cell Biol. Int. 2020;44:1267–1282. doi: 10.1002/cbin.11340. [DOI] [PubMed] [Google Scholar]
- 12.Gan H.K., Cvrljevic A.N., Johns T.G. The epidermal growth factor receptor variant iii (egfr v iii): Where wild things are altered. FEBS J. 2013;280:5350–5370. doi: 10.1111/febs.12393. [DOI] [PubMed] [Google Scholar]
- 13.Liang Y., Zhang T., Zhang J. Natural tyrosine kinase inhibitors acting on the epidermal growth factor receptor: Their relevance for cancer therapy. Pharmacol. Res. 2020;161:105164. doi: 10.1016/j.phrs.2020.105164. [DOI] [PubMed] [Google Scholar]
- 14.Shi K., Wang G., Pei J., Zhang J., Wang J., Ouyang L., Wang Y., Li W. Emerging strategies to overcome resistance to third-generation egfr inhibitors. J. Hematol. Oncol. 2022;15:1–44. doi: 10.1186/s13045-022-01311-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reda M., Ngamcherdtrakul W., Gu S., Bejan D.S., Siriwon N., Gray J.W., Yantasee W. Plk1 and egfr targeted nanoparticle as a radiation sensitizer for non-small cell lung cancer. Cancer Lett. 2019;467:9–18. doi: 10.1016/j.canlet.2019.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chong C.R., Jänne P.A. The quest to overcome resistance to egfr-targeted therapies in cancer. Nat. Med. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gold K.A., Lee H.Y., Kim E.S. Targeted therapies in squamous cell carcinoma of the head and neck. Cancer: Interdiscip. Int. J. Am. Cancer Soc. 2009;115:922–935. doi: 10.1002/cncr.24123. [DOI] [PubMed] [Google Scholar]
- 18.Dong R.-F., Zhu M.-L., Liu M.-M., Xu Y.-T., Yuan L.-L., Bian J., Xia Y.-Z., Kong L.-Y. Egfr mutation mediates resistance to egfr tyrosine kinase inhibitors in nsclc: From molecular mechanisms to clinical research. Pharmacol. Res. 2021;167:105583. doi: 10.1016/j.phrs.2021.105583. [DOI] [PubMed] [Google Scholar]
- 19.Crispo F., Notarangelo T., Pietrafesa M., Lettini G., Storto G., Sgambato A., Maddalena F., Landriscina M. Braf inhibitors in thyroid cancer: Clinical impact, mechanisms of resistance and future perspectives. Cancers. 2019;11:1388. doi: 10.3390/cancers11091388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Youssif B.G., Gouda A.M., Moustafa A.H., Abdelhamid A.A., Gomaa H.A., Kamal I., Marzouk A.A. Design and synthesis of new triarylimidazole derivatives as dual inhibitors of brafv600e/p38α with potential antiproliferative activity. J. Mol. Struct. 2022;1253:132218. doi: 10.1016/j.molstruc.2021.132218. [DOI] [Google Scholar]
- 21.Mohassab A.M., Hassan H.A., Abdelhamid D., Gouda A.M., Youssif B.G., Tateishi H., Fujita M., Otsuka M., Abdel-Aziz M. Design and synthesis of novel quinoline/chalcone/1, 2, 4-triazole hybrids as potent antiproliferative agent targeting egfr and brafv600e kinases. Bioorg. Chem. 2021;106:104510. doi: 10.1016/j.bioorg.2020.104510. [DOI] [PubMed] [Google Scholar]
- 22.Notarangelo T., Sisinni L., Condelli V., Landriscina M. Dual egfr and braf blockade overcomes resistance to vemurafenib in braf mutated thyroid carcinoma cells. Cancer Cell Int. 2017;17:1–9. doi: 10.1186/s12935-017-0457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Emburgh B.O., Sartore-Bianchi A., Di Nicolantonio F., Siena S., Bardelli A. Acquired resistance to egfr-targeted therapies in colorectal cancer. Mol. Oncol. 2014;8:1084–1094. doi: 10.1016/j.molonc.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tandon R., Kapoor S., Vali S., Senthil V., Nithya D., Venkataramanan R., Sharma A., Talwadkar A., Ray A., Bhatnagar P.K. Dual epidermal growth factor receptor (egfr)/insulin-like growth factor-1 receptor (igf-1r) inhibitor: A novel approach for overcoming resistance in anticancer treatment. Eur. J. Pharmacol. 2011;667:56–65. doi: 10.1016/j.ejphar.2011.04.066. [DOI] [PubMed] [Google Scholar]
- 25.Mao M., Tian F., Mariadason J.M., Tsao C.C., Lemos R., Jr., Dayyani F., Gopal Y.V., Jiang Z.-Q., Wistuba I.I., Tang X.M. Resistance to braf inhibition in braf-mutant colon cancer can be overcome with pi3k inhibition or demethylating agents. Clin. Cancer Res. 2013;19:657–667. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan L., Zhang J., Wang Y., Wang X., Wang Y., Zhang Z., Shuai W., Wang G., Chen J., Wang C. Development of dual inhibitors targeting epidermal growth factor receptor in cancer therapy. J. Med. Chem. 2022;65:5149–5183. doi: 10.1021/acs.jmedchem.1c01714. [DOI] [PubMed] [Google Scholar]
- 27.Del Curatolo A., Conciatori F., Cesta Incani U., Bazzichetto C., Falcone I., Corbo V., D’Agosto S., Eramo A., Sette G., Sperduti I. Therapeutic potential of combined braf/mek blockade in braf-wild type preclinical tumor models. J. Exp. Clin. Cancer Res. 2018;37:1–14. doi: 10.1186/s13046-018-0820-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Begunov R., Sokolov A. Biological activity of condensed pyridine derivatives with a bridgehead nitrogen atom. Pharm. Chem. J. 2023;56:1553–1567. doi: 10.1007/s11094-023-02827-y. [DOI] [Google Scholar]
- 29.Teague S.J. Synthesis of heavily substituted 2-aminopyridines by displacement of a 6-methylsulfinyl group. J. Org. Chem. 2008;73:9765–9766. doi: 10.1021/jo801303v. [DOI] [PubMed] [Google Scholar]
- 30.Mamedov I., Naghiyev F., Maharramov A., Uwangue O., Farewell A., Sunnerhagen P., Erdelyi M. Antibacterial activity of 2-amino-3-cyanopyridine derivatives. Mendeleev Commun. 2020;30:498–499. doi: 10.1016/j.mencom.2020.07.031. [DOI] [Google Scholar]
- 31.Al-Omar M.A., Amr A.E.G.E., Al-Salahi R.A. Anti-inflammatory, analgesic, anticonvulsant and antiparkinsonian activities of some pyridine derivatives using 2,6-disubstituted isonicotinic acid hydrazides. Arch. Pharm. 2010;343:648–656. doi: 10.1002/ardp.201000088. [DOI] [PubMed] [Google Scholar]
- 32.Ismail M.M., Farrag A.M., Harras M.F., Ibrahim M.H., Mehany A.B. Apoptosis: A target for anticancer therapy with novel cyanopyridines. Bioorg. Chem. 2020;94:103481. doi: 10.1016/j.bioorg.2019.103481. [DOI] [PubMed] [Google Scholar]
- 33.Kotb E.R., El-Hashash M., Salama M.A., Kalf H.S., Abdel Wahed N.A. Synthesis and reactions of some novel nicotinonitrile derivatives for anticancer and antimicrobial evaluation. Acta Chim. Slov. 2009;56:908–919. [Google Scholar]
- 34.Ryad N., My A.-S., Ismail M.M., El Meligie S. Design, synthesis and screening of 4,6-diaryl pyridine and pyrimidine derivatives as potential cytotoxic molecules. Chem. Pharm. Bull. 2018;66:939–952. doi: 10.1248/cpb.c18-00269. [DOI] [PubMed] [Google Scholar]
- 35.Bass A.K., Nageeb E.-S.M., El-Zoghbi M.S., Mohamed M.F., Badr M., Abuo-Rahma G.E.-D.A. Utilization of cyanopyridine in design and synthesis of first-in-class anticancer dual acting pim-1 kinase/hdac inhibitors. Bioorg. Chem. 2022;119:105564. doi: 10.1016/j.bioorg.2021.105564. [DOI] [PubMed] [Google Scholar]
- 36.Abdelaziz M.E., El-Miligy M.M., Fahmy S.M., Mahran M.A., Hazzaa A.A. Design, synthesis and docking study of pyridine and thieno [2, 3-b] pyridine derivatives as anticancer pim-1 kinase inhibitors. Bioorg. Chem. 2018;80:674–692. doi: 10.1016/j.bioorg.2018.07.024. [DOI] [PubMed] [Google Scholar]
- 37.Abouzid K.A., Al-Ansary G.H., El-Naggar A.M. Eco-friendly synthesis of novel cyanopyridine derivatives and their anticancer and pim-1 kinase inhibitory activities. Eur. J. Med. Chem. 2017;134:357–365. doi: 10.1016/j.ejmech.2017.04.024. [DOI] [PubMed] [Google Scholar]
- 38.Tarazi H., El-Gamal M.I., Oh C.-H. Discovery of highly potent v600e-b-raf kinase inhibitors: Molecular modeling study. Bioorg. Med. Chem. 2019;27:655–663. doi: 10.1016/j.bmc.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 39.Ayala-Aguilera C.C., Valero T., Lorente-Macías Á., Baillache D.J., Croke S., Unciti-Broceta A. Small molecule kinase inhibitor drugs (1995–2021): Medical indication, pharmacology, and synthesis. J. Med. Chem. 2021;65:1047–1131. doi: 10.1021/acs.jmedchem.1c00963. [DOI] [PubMed] [Google Scholar]
- 40.Motati D.R., Amaradhi R., Ganesh T. Azaindole therapeutic agents. Bioorg. Med. Chem. 2020;28:115830. doi: 10.1016/j.bmc.2020.115830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abou-Zied H.A., Beshr E.A., Gomaa H.A., Mostafa Y.A., Youssif B.G., Hayallah A.M., Abdel-Aziz M. Discovery of new cyanopyridine/chalcone hybrids as dual inhibitors of egfr/brafv600e with promising antiproliferative properties. Arch. Der Pharm. 2023;356:2200464. doi: 10.1002/ardp.202200464. [DOI] [PubMed] [Google Scholar]
- 42.Farooq S., Ngaini Z. One-pot and two-pot synthesis of chalcone based mono and bis-pyrazolines. Tetrahedron Lett. 2020;61:151416. doi: 10.1016/j.tetlet.2019.151416. [DOI] [Google Scholar]
- 43.Yamali C., Gul H.I., Kazaz C., Levent S., Gulcin I. Synthesis, structure elucidation, and in vitro pharmacological evaluation of novel polyfluoro substituted pyrazoline type sulfonamides as multi-target agents for inhibition of acetylcholinesterase and carbonic anhydrase i and ii enzymes. Bioorg. Chem. 2020;96:103627. doi: 10.1016/j.bioorg.2020.103627. [DOI] [PubMed] [Google Scholar]
- 44.Matiadis D., Sagnou M. Pyrazoline hybrids as promising anticancer agents: An up-to-date overview. Int. J. Mol. Sci. 2020;21:5507. doi: 10.3390/ijms21155507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ansari A., Ali A., Asif M. Biologically active pyrazole derivatives. New J. Chem. 2017;41:16–41. doi: 10.1039/C6NJ03181A. [DOI] [Google Scholar]
- 46.Eid N.M., George R.F. Facile synthesis of some pyrazoline-based compounds with promising anti-inflammatory activity. Future Med. Chem. 2018;10:183–199. doi: 10.4155/fmc-2017-0144. [DOI] [PubMed] [Google Scholar]
- 47.Saleh N.M., El-Gazzar M.G., Aly H.M., Othman R.A. Novel anticancer fused pyrazole derivatives as egfr and vegfr-2 dual tk inhibitors. Front. Chem. 2020;7:917. doi: 10.3389/fchem.2019.00917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garnock-Jones K.P. Eltrombopag: A review of its use in treatment-refractory chronic primary immune thrombocytopenia. Drugs. 2011;71:1333–1353. doi: 10.2165/11207390-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 49.Shanafelt T.D., Wang X.V., Kay N.E., Hanson C.A., O’Brien S., Barrientos J., Jelinek D.F., Braggio E., Leis J.F., Zhang C.C. Ibrutinib–rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N. Engl. J. Med. 2019;381:432–443. doi: 10.1056/NEJMoa1817073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ibraheem F., Ahmad M., Ashfaq U.A., Aslam S., Ali Khan Z., Sultan S. Synthesis, molecular docking and anti-diabetic studies of novel benzimidazole-pyrazoline hybrid molecules. Pak. J. Pharm. Sci. 2020;33:847–855. [PubMed] [Google Scholar]
- 51.Beyhan N., Kocyigit-Kaymakcioglu B., Gümrü S., Aricioglu F. Synthesis and anticonvulsant activity of some 2-pyrazolines derived from chalcones. Arab. J. Chem. 2017;10:S2073–S2081. doi: 10.1016/j.arabjc.2013.07.037. [DOI] [Google Scholar]
- 52.Revanasiddappa B., Jisha M., Kumar M.V., Kumar H. Synthesis, antibacterial and antifungal evlaution of novel pyrazoline derivatives. Dhaka Univ. J. Pharm. Sci. 2018;17:221–226. doi: 10.3329/dujps.v17i2.39179. [DOI] [Google Scholar]
- 53.Al-Wahaibi L.H., Abou-Zied H.A., Beshr E.A.M., Youssif B.G.M., Hayallah A.M., Abdel-Aziz M. Design, synthesis, antiproliferative actions, and dft studies of new bis–pyrazoline derivatives as dual egfr/brafv600e inhibitors. Int. J. Mol. Sci. 2023;24:9104. doi: 10.3390/ijms24109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abou-Zied H.A., Youssif B.G., Mohamed M.F., Hayallah A.M., Abdel-Aziz M. Egfr inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019;89:102997. doi: 10.1016/j.bioorg.2019.102997. [DOI] [PubMed] [Google Scholar]
- 55.Al-Wahaibi L.H., Mahmoud M.A., Mostafa Y.A., Raslan A.E., Youssif B.G. Novel piperine-carboximidamide hybrids: Design, synthesis, and antiproliferative activity via a multi-targeted inhibitory pathway. J. Enzym. Inhib. Med. Chem. 2023;38:376–386. doi: 10.1080/14756366.2022.2151593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Al-Wahaibi L.H., Mostafa Y.A., Abdelrahman M.H., El-Bahrawy A.H., Trembleau L., Youssif B.G. Synthesis and biological evaluation of indole-2-carboxamides with potent apoptotic antiproliferative activity as egfr/cdk2 dual inhibitors. Pharmaceuticals. 2022;15:1006. doi: 10.3390/ph15081006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Al-Wahaibi L.H., Mohammed A.F., Abdel Rahman F.E.-Z.S., Abdelrahman M.H., Gu X., Trembleau L., Youssif B.G. Design, synthesis, apoptotic, and antiproliferative effects of 5-chloro-3-(2-methoxyvinyl)-indole-2-carboxamides and pyrido [3, 4-b] indol-1-ones as potent egfrwt/egfrt790m inhibitors. J. Enzym. Inhib. Med. Chem. 2023;38:2218602. doi: 10.1080/14756366.2023.2218602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DeRosa T.F. Chapter XXV—Proliferative disorders. In: DeRosa T.F., editor. Significant Pharmaceuticals Reported in Us Patents. Elsevier Science B.V.; Amsterdam, The Netherlands: 2007. pp. 497–608. [Google Scholar]
- 59.Maghraby M.T.E., Salem O.I., Youssif B.G., Sheha M.M. Design, synthesis, and modelling study of new 1, 2, 3-triazole/chalcone hybrids with antiproliferative action as epidermal growth factor receptor inhibitors. Chem. Biol. Drug Des. 2023;101:749–759. doi: 10.1111/cbdd.14178. [DOI] [PubMed] [Google Scholar]
- 60.Al-Wahaibi L.H., Mohammed A.F., Abdelrahman M.H., Trembleau L., Youssif B.G. Design, synthesis, and antiproliferative activity of new 5-chloro-indole-2-carboxylate and pyrrolo [3, 4-b] indol-3-one derivatives as potent inhibitors of egfrt790m/brafv600e pathways. Molecules. 2023;28:1269. doi: 10.3390/molecules28031269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Y., Zhu X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 2017;6:1–8. doi: 10.1186/s40035-017-0092-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Villa-Pulgarin J.A., Gajate C., Botet J., Jimenez A., Justies N., Varela-M R.E., Cuesta-Marban A., Müller I., Modolell M., Revuelta J.L. Mitochondria and lipid raft-located fof1-atp synthase as major therapeutic targets in the antileishmanial and anticancer activities of ether lipid edelfosine. PLoS Negl. Trop. Dis. 2017;11:e0005805. doi: 10.1371/journal.pntd.0005805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bao H., Zhang Q., Zhu Z., Xu H., Ding F., Wang M., Du S., Du Y., Yan Z. Bhx, a novel pyrazoline derivative, inhibits breast cancer cell invasion by reversing the epithelial-mesenchymal transition and down-regulating wnt/β-catenin signalling. Sci. Rep. 2017;7:9153. doi: 10.1038/s41598-017-09655-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin S. Caspases: Executioners of apoptosis. Pathobiol. Hum. Dis. 2014;2014:145–152. [Google Scholar]
- 65.Wall D.M., McCormick B.A. Bacterial secreted effectors and caspase-3 interactions. Cell. Microbiol. 2014;16:1746–1756. doi: 10.1111/cmi.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Youssif B.G., Mohamed A.M., Osman E.E.A., Abou-Ghadir O.F., Elnaggar D.H., Abdelrahman M.H., Treamblu L., Gomaa H.A. 5-chlorobenzofuran-2-carboxamides: From allosteric cb1 modulators to potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019;177:1–11. doi: 10.1016/j.ejmech.2019.05.040. [DOI] [PubMed] [Google Scholar]
- 67.Hafliger E., Boccaccino A., Lapeyre-Prost A., Perret A., Gallois C., Antista M., Pilla L., Lecomte T., Scartozzi M., Soularue E. Encorafenib plus cetuximab treatment in braf v600e-mutated metastatic colorectal cancer patients pre-treated with an anti-egfr: An ageo-gono case series. Eur. J. Cancer. 2022;168:34–40. doi: 10.1016/j.ejca.2022.03.011. [DOI] [PubMed] [Google Scholar]
- 68.Bhat M.A., Tüzün B., Alsaif N.A., Khan A.A., Naglah A.M. Synthesis, characterization, molecular modeling against egfr target and adme/t analysis of novel purine derivatives of sulfonamides. J. Mol. Struct. 2022;1257:132600. doi: 10.1016/j.molstruc.2022.132600. [DOI] [Google Scholar]
- 69.Umar A.B., Uzairu A., Shallangwa G.A., Uba S. Qsar modelling and molecular docking studies for anti-cancer compounds against melanoma cell line sk-mel-2. Heliyon. 2020;6:e03640. doi: 10.1016/j.heliyon.2020.e03640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu Z., Lei T., Shen C., Wang Z., Cao D., Hou T. Admet evaluation in drug discovery. 19. Reliable prediction of human cytochrome p450 inhibition using artificial intelligence approaches. J. Chem. Inf. Model. 2019;59:4587–4601. doi: 10.1021/acs.jcim.9b00801. [DOI] [PubMed] [Google Scholar]
- 71.Abd El-Sattar N.E., Badawy E.H., AbdEl-Hady W.H., Abo-Alkasem M.I., Mandour A.A., Ismail N.S. Design and synthesis of new cdk2 inhibitors containing thiazolone and thiazolthione scafold with apoptotic activity. Chem. Pharm. Bull. 2021;69:106–117. doi: 10.1248/cpb.c20-00714. [DOI] [PubMed] [Google Scholar]
- 72.Rai H., Barik A., Singh Y.P., Suresh A., Singh L., Singh G., Nayak U.Y., Dubey V.K., Modi G. Molecular docking, binding mode analysis, molecular dynamics, and prediction of admet/toxicity properties of selective potential antiviral agents against sars-cov-2 main protease: An effort toward drug repurposing to combat covid-19. Mol. Divers. 2021;25:1905–1927. doi: 10.1007/s11030-021-10188-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roy P.P., Roy K. Qsar studies of cyp2d6 inhibitor aryloxypropanolamines using 2d and 3d descriptors. Chem. Biol. Drug Des. 2009;73:442–455. doi: 10.1111/j.1747-0285.2009.00791.x. [DOI] [PubMed] [Google Scholar]
- 74.Xia X., Maliski E.G., Gallant P., Rogers D. Classification of kinase inhibitors using a bayesian model. J. Med. Chem. 2004;47:4463–4470. doi: 10.1021/jm0303195. [DOI] [PubMed] [Google Scholar]
- 75.Goodrnan G., Wilson R. Comparison of the dependence of the td50 on maximum tolerated dose for mutagens and nonmutagens. Risk Anal. 1992;12:525–533. doi: 10.1111/j.1539-6924.1992.tb00709.x. [DOI] [PubMed] [Google Scholar]
- 76.Gonella Diaza R., Manganelli S., Esposito A., Roncaglioni A., Manganaro A., Benfenati E. Comparison of in silico tools for evaluating rat oral acute toxicity. SAR QSAR Environ. Res. 2015;26:1–27. doi: 10.1080/1062936X.2014.977819. [DOI] [PubMed] [Google Scholar]
- 77.Wilhelmus K.R. The draize eye test. Surv. Ophthalmol. 2001;45:493–515. doi: 10.1016/S0039-6257(01)00211-9. [DOI] [PubMed] [Google Scholar]
- 78.Ibrahim T.S., Bokhtia R.M., Al-Mahmoudy A.M., Taher E.S., AlAwadh M.A., Elagawany M., Abdel-Aal E.H., Panda S., Gouda A.M., Asfour H.Z. Design, synthesis and biological evaluation of novel 5-((substituted quinolin-3-yl/1-naphthyl) methylene)-3-substituted imidazolidin-2, 4-dione as hiv-1 fusion inhibitors. Bioorg. Chem. 2020;99:103782. doi: 10.1016/j.bioorg.2020.103782. [DOI] [PubMed] [Google Scholar]
- 79.Gomaa H.A., El-Sherief H.A., Hussein S., Gouda A.M., Salem O.I., Alharbi K.S., Hayallah A.M., Youssif B.G. Novel 1, 2, 4-triazole derivatives as apoptotic inducers targeting p53: Synthesis and antiproliferative activity. Bioorg. Chem. 2020;105:104369. doi: 10.1016/j.bioorg.2020.104369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data will be provided upon request.