


Abstract
Synaptosome cybrids were used to confirm the presence of heteroplasmic mtDNA sequence variants in the human brain. Synaptosomes contain one to several mitochondria, and when fused to mtDNA-deficient (ρ°) mouse or human cell lines result in viable cybrid cell lines. The brain origin of mouse synaptosome cybrid mtDNAs was confirmed using sequence polymorphisms in the mtDNA COIII, ND3 and tRNAArg genes. The brain origin of the human synaptosome cybrids was confirmed using a rare mtDNA MboI polymorphism. Fusion of synaptosomes from the brain of a 35-year-old woman resulted in 71 synaptosome cybrids. Sequencing the mtDNA control region of these cybrid clones revealed differences in the number of Cs in a poly C track between nucleotide pairs (nps) 301 and 309. Three percent of the cybrid clones had mtDNAs with 10 Cs, 76% had nine, 18% had eight and 3% had seven Cs. Comparable results were obtained by PCR amplification, cloning and sequencing of mtDNA control regions directly from the patient’s brain tissue, but not when the control region was amplified and cloned from a synaptosome cybrid homoplasmic for a mtDNA with nine Cs. Thus, we have clonally recovered mtDNA control region length variants from an adult human brain without recourse to PCR, and established the variant mtDNAs within living cultured cells. This confirms that some mtDNA heteroplasmy can exist in human neurons, and provides the opportunity to study its functional significance.
INTRODUCTION
It has been proposed that somatic mtDNA mutations accumulate in post-mitotic tissues with age and may be an important factor in aging and the progression of neuro-degenerative diseases (1). A variety of studies in humans have reported the age-related accumulation of both mtDNA deletion (2–9) and base substitution (10–14) mutations. In one extensive study, base substitution and small insertion–deletion mutations were detected in the control regions of 35–40% of the mtDNAs from human brains. The most common variants were small insertion–deletion mutations in homopolymeric stretches of nucleotides at nucleotide pairs (nps) 71 (G) and 309 (C). Within the stretch of Cs between nps 301 and 309, 30% of the mtDNAs were reported to have lost one C, giving nine Cs as opposed to the predominant 10 Cs. Moreover, the extent of this mutability increased with age as there was a 7.7-fold increase in variation in 96- and 99-year-old brains compared to a 28-year-old brain. By contrast, somatic mtDNA variation was not found in a 300-np region of coding sequence surrounding the tRNALeu(UUR) gene (13).
One concern about such studies of somatic mtDNA variation is that the mtDNA is PCR-amplified from the tissue, prior to testing for heteroplasmic mutations. PCR amplification using thermal stable DNA polymerases can be error prone and thus can introduce mutations during amplification. To circumvent this concern, we have developed a somatic cell genetic procedure for the isolation and cloning of neuronal mtDNAs from mouse and human brain. Synaptosomes containing brain mitochondria are fused to human and mouse cells which lack mtDNA (ρ° cells) (15).
Synaptosomes are derived from neuronal synaptic boutons which pinch off during the homogenization of brain tissue to form membrane-bound cytoplasmic particles which include mitochondria. When fused to a ρ° cell, the brain mtDNAs are replicated and amplified by the normal DNA replication system. Hence, they are faithfully reproduced and maintained in a biologically active state.
In mice, mtDNAs have been recovered from the brain by fusion of brain homogenates to ρ° cells and some cybrids reported to contain trace amounts of deleted mtDNA, detected by PCR amplification (16). However, no stable cybrids harboring mutant mtDNAs from the donor mice were obtained.
In the present study, we report the recovery of neuronal mtDNAs from both mouse and human brains, and show that 24% of the cybrid mtDNAs derived from the human brain differed in the length of the control region poly C track between nps 301 and 309. This confirms that at least some mtDNA heteroplasmy does exist in the brain, and provides the opportunity for the characterization of its biochemical significance.
MATERIALS AND METHODS
Cell lines and culture conditions
A mouse ρ° cell line was produced by exposure of LM(TK–) cells to ethidium bromide (EtBr) as described previously for avian (17,18) and human (19) cells. Four ρ° LM(TK–) clones were isolated by exposure of 5 × 106 cells in a single flask to 250 ng/ml EtBr in DMEM containing 4.5 mg/ml glucose, 10% fetal calf serum (FCS) and 50 µg/ml uridine for 28 days. An aliquot of the cells was cloned at 104 cells/dish in the same medium containing 100 ng/ml of EtBr. After an additional 9 weeks (13 weeks after onset of EtBr treatment), five clones were ring-isolated and tested for uridine dependency. All five clones were found to be uridine and pyruvate dependent, as has been shown for human ρ° cells (19). Southern blot analysis confirmed that these clones lacked detectable mtDNA. The population doubling rate of the ρ° LM(TK–) cells was ∼72 h, substantially longer than that to the parental cell line of ∼10 h. The ρ° cells were more fusiform than their mtDNA-containing (ρ+) counter parts, and fusion of the ρ° clones with enucleated mouse RAG cells and selection in uridine-free medium containing 30 µg/ml of bromodeoxyuridine (BrdU) gave rise to cybrids at a frequency of ∼10–4. These cybrids had the same growth rate and morphology as the parental LM(TK–) line. The ρ° derivative of the human 143B-TK– cell line has been described previously (20).
Synaptosome isolation
Mouse studies. Synaptosomes were isolated by a rapid Percoll step-gradient method (21). For mice, whole brains were removed from C57BL/6NNia males (NIA, Jefferson, AZ) using sterile technique and transferred immediately to chilled SED isolation medium (0.32 M sucrose, 1 mM EDTA and 0.25 mM DTT, adjusted to pH 7.4 with dilute HCl). After cooling for 10 min, the tissue was minced with scissors and homogenized in 9 vol SED using 10 strokes each of loose and tight pestles in a Dounce homogenizer. The 10% homogenate was centrifuged at 1000 g for 10 min to remove nuclei and unbroken cells and the supernatant diluted 1:1 with SED. Two milliliters of the supernatant was layered onto each of six gradients consisting of 2 ml 23, 15, 10 and 3% Percoll, prepared in SED. The gradients were centrifuged at 32 500 g for 5 min (RC5B with SS-34 rotor, 20 000 r.p.m., 4°C) excluding acceleration and braking time. The fractions at the 23/15% Percoll interface were aspirated and combined, diluted 10-fold with 0.3 M mannitol (pH 7.4) and centrifuged at 15 000 g for 15 min. The loose pellet was then used for electrofusion with ρ° cells.
Human studies. Three samples of fresh human brain cortex, ranging from 0.2 to 1.5 g, were obtained from patients undergoing surgery for tumor removal. Macroscopically normal appearing tissue was separated by the surgeon from around the central tumor mass, and processed for synaptosomes fusion. In another three experiments, post-mortem cortical samples were obtained at 7–13 h after death.
Fusion and selection of cybrids
The washed synaptosome fraction was combined with 2 × 106 ρ° cells, and washed once with 0.3 M mannitol (pH 7). The mixture was brought to 0.6 ml in the same media, and subjected to electrofusion (22). Cybrids were selected in uridine-free DMEM containing 5% dialysed fetal bovine serum and 50 µg/ml BrdU. Cybrid clones were visible after 6–8 days and were isolated after 10–14 days.
Sequencing of mouse mtDNAs
Genomic DNA was extracted from C57BL/6NNia brain, LM(TK–) cells and mouse synaptosome cybrids using proteinase K digestion followed by phenol–chloroform extraction and ethanol precipitation. Regions of the mtDNA sequenced were PCR amplified using the mtDNA-specific primers listed in the Appendix. The fragments were purified by Centrion 100 separators (Amicon, Beverly, MA), and subjected to cycle sequencing using AmpliTaq FS DNA polymerase with fluorescent dye-terminator chemistry using the manufacturer’s recommended protocol (Prism Ready Reaction Cycle Sequencing Kit, Applied Biosystems, Foster City, CA) and an ABI Prism 377 DNA Sequencer.
To identify mtDNA sequence polymorphisms, a total of 6.7 kb of the C57BL/6NNia mtDNA was sequenced encompassing the control region, part of the ND1 gene, as well as the COIII, ND3, ND4L, ND4 and ND5 genes. These sequences were compared to the published mouse LA9 mtDNA sequence (23), and the LM(TK–) and LA9 (Emory) mouse mtDNA sequences (Table 1). Interestingly, all three mtDNAs proved to be different, even though both mouse LA9 (24) and LM(TK–) (24) were derived from the same mouse L929 cells (25). Primary sequence differences between CS7BL/6NNia mouse and LM(TK–) mtDNAs included a G→A transition at np 9348, a T→C transition at np 9461 and insertion of two As in the tRNAArg gene between nps 9818 and 9825. These variants were then used to analyze the cybrid mtDNAs.
Table 1. Inheritance of mtDNA sequence variants in mouse synaptosome cybrids.
| Nucleotide position | Gene | LA9 mtDNAa reference sequence | LM(TK–) n = 1 | C57BL/6NNia n = 2 | Cybrids n = 63 |
|---|---|---|---|---|---|
| 9348 | COIII | A | A | G | G |
| 9461 | ND3 | C | C | T | T |
| 9818–9825 | TRNAArg | 8A | 10A | 8A | 8A |
The number of A nucleotides found in the homopolymeric stretch of As from np 9818–9825 is indicated.
aSee (23).
To screen for de novo mtDNA mutations in the mouse synaptosome cybrids, a 1261-np fragment encompassing the entire mouse mtDNA control region was PCR-amplified using primers from nps 15184 to 150. Seven internal primer pairs were used to produce overlapping fragments for direct cycle sequencing.
Sequence analysis of human mtDNA control regions in synaptosome cybrid clones
Genomic DNA was extracted from each human synaptosome cybrid. A 1795-np region of the mtDNA from nps 15838 to 1064 was PCR-amplified and the entire control region (1121 bp) was sequenced by cycle sequencing using overlapping primers. Both strands were sequenced for most of the target sequence and ambiguities were resolved by additional sequence analysis using independent primers (see Appendix). The entire control region mtDNA sequence was determined for 71 independent cybrid clones.
Cloning of human brain and synaptosome cybrid derived mtDNAs
A 394-bp fragment of the mtDNA from nps 242 to 636 was PCR-amplified from a frozen aliquot of the brain homogenate of the 35-year-old female subject, the same homogenate used for synaptosome isolation. This region encompassed the variable poly C track from np 301 to 309. The resulting DNA fragments were cloned into the vector pCR2.1 and transformed into INVαF′ cells (TA Cloning kit, Invitrogen, Carlsbad, CA). White colonies were isolated, the mtDNA fragment PCR-amplified and the sequence determined using the same primers employed to amplify the 394-bp fragment. A total of 102 brain mtDNAs, cloned into plasmid pCR21, were sequenced.
As a control experiment to determine if PCR amplification generated increased mtDNA control region sequence variation, the mtDNA of one cybrid clone harboring nine Cs between nps 301 and 309 was PCR amplified and cloned in pCR2.1. The resulting DNA clones were sequenced and compared. A total of 148 of these ‘control’ cybrid clones were sequenced in the np 301–309 region.
Primer extension assay
A 528-np mtDNA fragment from nps 242 to 770, which encompassed the variable poly C track of the control region, was PCR-amplified from brain DNA of the 35-year-old female subject as well as from selected of her synaptosome cybrids and the DNA purified by gel-extraction (Qiagen, Valencia, CA). The number of Cs in the poly C track was determined by primer extension reactions using a 32P-labeled oligonucleotide corresponding to nps 340–321 (5′-GTGTTTAAGTGGTGTGGCCA-3′), which extended to within 12 np of the poly C region. Each reaction mixture comprised 100 ng of purified PCR product; 3.2 pmol of primer; 2.5 U of thermosequenase (Amersham Pharmacia Biotech, Piscataway, NJ); 0.3 mM dNTP-mix containing dATP, dGTP, dCTP; and 3 mM ddTTP, the latter terminating the extension at the base following the poly C track. The products of the primer extension were purified using Centri-Sep columns (Princeton Separations, Adelphia, NJ) and separated on a 7 M urea, 8% polyacrylamide gel. The gel was electrophoresed for 2 h and the primer extension products revealed by exposure to X-ray film for 5–8 h. Three 32P-labeled oligonucleotides, ranging from 37 to 40 nps were used as size standards.
RESULTS
Synaptosomes as viable mitochondrial DNA donors in mouse cybrids
Electron micrographs of synaptosomes isolated from a mouse brain revealed a homogeneous fraction of membrane-bound vesicles encapsulating one to several morphologically normal mitochondria (Fig. 1). These synaptosomes were fused to the LM(TK–) ρ° cell line, LMEB4, and the cybrids selected in medium containing BrdU but lacking uridine. When synaptosomes were isolated from freshly killed mouse brains that were chilled rapidly in SED medium, the yields of cybrids were consistently high, ∼200 clones per 2 × 106 ρ° cells or ∼1 per 104 ρ° cells. This high yield was also obtained when the brain was removed immediately after death and kept on ice in SED for 4 h before synaptosome isolation. However, delaying the removal of the brain, with the carcass kept at 4°C for 4 or 24 h, resulted in a drastic (100-fold) reduction in cybrid frequencies.
Figure 1.
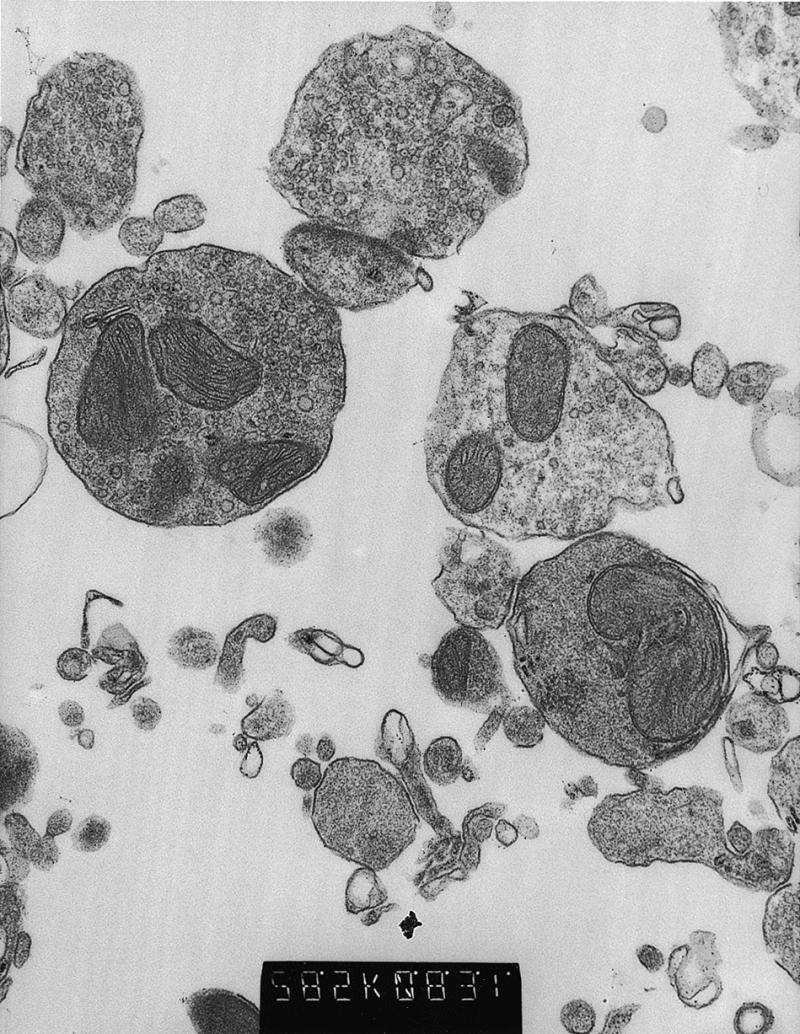
Electron micrograph of a synaptosome fraction from a mouse brain. Intact synaptosomes predominate, with one to several intact mitochondrial profiles in most. This fraction was isolated from a 6-week-old animal where the carcass was stored at 4°C for 4 h before processing. Magnification = ×28 500.
The brain origin of the mouse synaptosome cybrid mtDNAs was confirmed by testing for the nucleotide polymorphisms at nps 9348 and 9461 and for the number of As in the poly A track from nps 9818 to 9825, which differ between the C57BL/6NNia brain donor mtDNA and the LM(TK–) recipient mtDNA. A total of 63 synaptosome cybrids were tested for these markers including 18 cybrids from a 1.5-month-old mouse brain and 45 cybrids from a 34-month-old mouse brain. All of the synaptosome cybrids were found to have the variants of the C57BL/6NNia mtDNA, not those characteristic of LM(TK–) mtDNA (Table 1). Sequencing the control regions of the cybrids revealed no additional sequence variants. Hence, in the mouse synaptosome cybrids, the LM(TK–) ρ° cells are invariably repopulated with the C57Bl/6NNia brain mtDNAs confirming that mouse mtDNAs can be recovered from brain tissue by cybrid fusion.
MtDNA heteroplasmy demonstrated in human brains by synaptosome fusions
Human synaptosome cybrids were prepared from synaptosomes isolated from freshly obtained surgical specimens and fused to 143B-TK– ρ° cells, with cybrids selected in medium containing BrdU but lacking uridine. In one experiment using 0.2 g of frontal cortex from a 35-year-old woman, ∼200 clones were obtained or ∼1 per 104 ρ° cells. However, in two other experiments using 1.1 or 1.5 g of brain, only five and three clones were obtained, respectively. Three attempts to isolate synaptosome cybrids using post-mortem samples resulted in only one clone.
The brain origin of the mtDNAs in the human synaptosome cybrids was confirmed using an MboI restriction fragment length polymorphism (RFLP) at np 15397 (Fig. 2). In a PCR fragment encompassing nps 15005–15701, the human brain donor mtDNA lacked this MboI site and gave a 234-np MboI fragment, while the mtDNA of the parental 143B-TK– cells had this site, such that the 234-np fragment is cleaved into 195- and 39-np fragments. Three independent synaptosome cybrid clones were examined and found to harbor only mtDNAs derived from the brain (Fig. 2). Hence, human synaptosome cybrids are also repopulated with brain mtDNAs.
Figure 2.
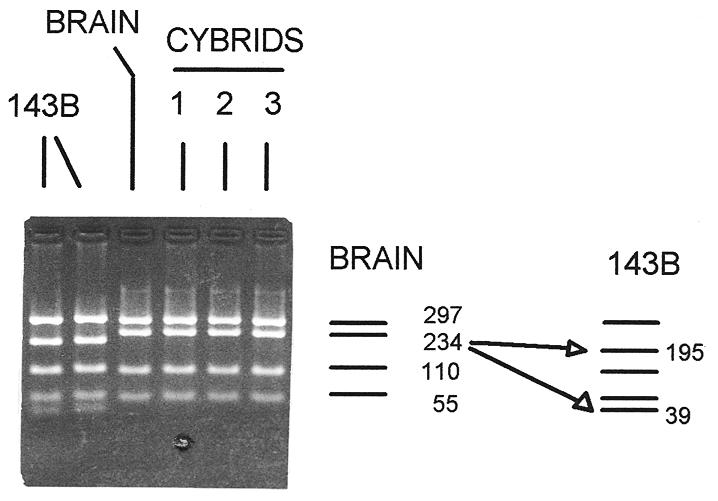
The brain origin of human synaptosome cybrids mtDNAs. The mtDNA region from nps 15005 to 15701 was PCR-amplified, digested with MboI and the restriction fragments electrophoresed in a 3% agarose gel. The restriction pattern of the brain and cybrid clones differs from that of the 143B-TK– cell mtDNA due to a nucleotide polymorphism at np 15397, an A→G transition. Most human mtDNAs lack this site and give a 234 np fragment while 143B-TK– mtDNAs have the site and give 195 and 39 np fragments (indicated on the right). The presence of the np 15397 site in the brain and cybrid mtDNAs confirms that the cybrids mtDNAs were derived from the brain tissue. The two lanes for the 143B-TK– cell line show the parental line (left) and a sub-clone which subsequently gave rise to the ρ° clone (right). This mtDNA PCR product could not be amplified from the DNA of the 143B-TK– ρ° clone.
Synaptosome clones reveal heteroplasmy in the human brain mtDNA
Sequence analysis of the mtDNA control regions of 71 synaptosome cybrids derived from the brain of the 35-year-old woman revealed clonal variation in the number of Cs in the poly C track between nps 301 and 309. Of the 71 synaptosome cybrids, most (76%) harbored mtDNAs with nine Cs, two Cs more than the Cambridge reference sequence (26). Since the length of this poly C region is known to vary between individuals (27), the nine C genotype is likely to represent this woman’s germline sequence. The remaining 24% of the cybrid clones differed in the number of Cs, with 3% having 10 Cs (three more than the reference sequence, +3), 18% having eight Cs (+1) and 3% having seven Cs (+0).
The distribution in the number of Cs at nps 301–309 was shown to reflect the pre-existing mtDNA sequence variation in the brain by PCR amplification, cloning and sequencing of the mtDNA control region directly from the subject’s brain DNA. This revealed essentially the same distribution of poly C length variants as the cybrids with 73% having nine, 4% having 10, 19% having eight and 4% having seven Cs. To confirm that this distribution of Cs was not caused by PCR or cloning artifacts, this same region was PCR amplified and cloned from a single synaptosome cybrid clone which harbored only the modal nine C mtDNA sequence. This resulted in only 4% of variant clones, with 1% having 10, 96% having nine and 3% having eight Cs (Table 2).
Table 2. Variation in the length of a poly C track in human synaptosome cybrids and brain.
| No. Cs between np 301 and 309 | Synapasome cybrids | Brain clones | Control clones | ||||
|---|---|---|---|---|---|---|---|
| No. clones | (%) | No. clones | (%) | No. clones | (%) | ||
| 7 | (+0)a | 2 | (2.8) | 4 | (3.9) | 0 | |
| 8 | (+1)a | 13 | (18.2) | 19 | (18.6) | 5 | (3.4) |
| 9 | (+2)a | 54 | (76.1) | 74 | (72.5) | 142 | (95.9) |
| 10 | (+3)a | 2 | (2.8) | 4 | (3.9) | 1 | (0.7) |
| 11 | (+4)a | 0 | (0) | 1 | (1.0) | 0 | (0) |
aRelative to the Cambridge reference sequence (26).
The length variation of the np 301–309 poly C track between the different synaptosome cybrid clones was confirmed by primer extension analysis. A primer was annealed adjacent to the run of Cs, and extended across the Cs to the first A, where extension was terminated by incorporation of a ddTTP. The length of the resulting fragments was then determined by gel electrophoresis. The primer extension assay revealed that each cybrid clone harbored one predominant mtDNA, with a np 301–309 poly C track of either seven Cs (0), eight Cs (+1), nine Cs (+2), or 10 Cs (+3) (Fig. 3). Thus, each cybrid clone gave the primer extension fragment length predicted for that clone’s mtDNA sequence.
Figure 3.
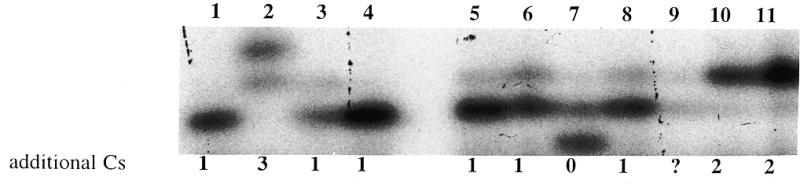
Confirmation of the variation in length of the np 301–309 poly C track in human synaptosome cybrids by primer extension. Lanes 1–11 represent the primer extension products from the mtDNAs of individual synaptosome cybrid clones derived from the brain of a 35-year-old female. Numbers below the figure represent the number of additional Cs contained in a clone relative to the Cambridge sequence. The germline mtDNA of the brain donor probably contained nine Cs (two more than the Cambridge sequence or +2) as shown in the cybrids in lanes 10 and 11.
DISCUSSION
Synaptosome cybrids have permitted us to recover somatic mtDNA mutations from fresh human brain tissue without PCR amplification. Among the synaptosome cybrids isolated from the brain tissue of a 35-year-old female, we found that 24% differed in the number of Cs between nps 301 and 309, relative to the predominant and thus likely germline number of nine Cs. Our result is consistent with a published paper that reported that ∼30% of the mtDNA control region clones obtained by PCR amplification, cloning and sequencing directly from human brain DNA had variants in the number of Cs between nps 301 and 309 (13). Thus heteroplasmy of mtDNA control region length variants in the brain has been confirmed by both synaptosome cybrids and PCR amplification.
While this study proves that mtDNA sequence variation does exist in the human brain, the level was surprisingly low. Besides the np 301–309 length polymorphism, no other control region sequence variants were identified in the 71 human and 63 mouse synaptosome cybrids examined. This stands in striking contrast to the 5.6 × 10–4 frequency of control region mtDNA nucleotide variants identified in the brains of older subjects (13), and to the prevalence of specific nucleotide variants found in the mtDNA control regions of fibroblasts derived from older humans (14), both analyzed by PCR amplification, denature gradient gel electrophoresis, cloning and sequencing. Neither the T414C variant found in over half of the fibroblasts from subjects over 64 (14), nor the T146C and T195C variants found in the fibroblasts of a centenarian (14) and the brain of a 96-year-old subject (13) were observed in any of our synaptosome cybrids. However, as our human brain synaptosomes were derived from a 35-year-old donor, it is clear that more extensive synaptosome cybrid studies will be required to accurately assess the frequency and nature of somatic mtDNA mutations which accumulate in human and mammalian brain with age.
In an attempt to maximize the sampling of synaptic bouton mtDNAs, we examined several parameters relating to brain sample collection and processing. We found that while synaptosome cybrids were readily obtained from fresh brain, it proved difficult to isolate viable synaptosomes from post-mortem brain specimens. This was confirmed using the mouse system which showed a sharp decline in cybrid frequency when mouse brains were recovered even 4 h post-mortem. If, however, the brain tissue was immediately removed and placed in ice-cold isolation medium, successful synaptosome isolation and fusion could be achieved up to 4 h after brain removal.
In a recent report (16), mouse ρ° cells were fused to a post-nuclear pellet (17 000 g × 20 min) from mouse brain homogenate and the fusions promoted using polyethylene glycol. The cybrid yield of this study was ∼100-fold lower than ours. The higher cybrid yields using our methods could be due to use of a homogenous synaptosome fraction and/or use of electrofusion instead of PEG.
A great advantage of the synaptosome method for assessing the extent of somatic mtDNA variation in brain is that it permits the clonal propagation of the mutant mtDNAs in cultured cells. This permits the mtDNAs to be maintained in their biological active form making it feasible to characterize the biochemical consequences of the somatic mtDNA mutations.
Thus, synaptosome cybrids provide us a unique opportunity to determine both the quantitative and functional consequences of the somatic mtDNA mutations which accumulate with age in the mammalian and human brain.
APPENDIX
PCR amplification conditions
Amplification of mouse mtDNA was achieved using an initial denaturation at 94°C for 1 min and 30 cycles of 94°C denaturation for 30 s followed by annealing at 55–65°C for 45 s and extension at 72°C for 45–120 s.
Amplification of human mtDNA was achieved using a denaturation temperature of 94°C for 1 min and 35 cycles of 94°C denaturation for 45 s followed by annealing at 49–55°C for 45 s and extension at 72°C for 45–120 s.
For sequencing, either the fragment primers or the above mentioned internal primers were used.
A. Primers used for amplification of mouse synaptosome cybrid mtDNA.
| Primer pair | Primer coordinates | Primer sequence |
|---|---|---|
| (5′→3′) | ||
| 1 | 15184–15216 | 5′-CTACTTCTTGAGTACATAAATTTACATAGTAC-3′ |
| 150–125 | 5′-TAAGGGATTTTACACCGGTCTATGGA-3′ | |
| 2 | 14877–14898 | 5′-CCAGACATACTAGGAGACCCA-3′ |
| 150–125 | 5′-TAAGGGATTTTACACCGGTCTATGGA-3′ | |
| 3 | 2716–2790 | 5′-CCTTGTTCCCAGAGGTTCAAATCCT-3′ |
| 4033–4009 | 5′-AATTGCTAGTAGGCTGAATTCTAGG-3′ | |
| 4 | 8861–8880 | 5′-ATTCATCGTCTCGGAAGTAT-3′ |
| 11840–11821 | 5′-GTACAGTGGGAAGTTGATGT-3′ | |
| 5 | 11697–11706 | 5′-TAGTAATCCATTGGTCTTAGGAACCAA-3′ |
| 14207–14183 | 5′-GGCAGGTAGGTCAATGAATGAGTGG-3′ | |
| 6 | 8861–8880 | 5′-ATTCATCGTCTCGGAAGTAT-3′ |
| 10381–10362 | 5′-GGCAGTAATCAGGCTGTTAA-3′ |
B. Primers used for sequencing mouse synaptosome cybrid mtDNA.
| Primer | Primer coordinates | Primer sequence |
|---|---|---|
| (5′→3′) | ||
| 1 | 15431–15459 | 5′-CATAGTACAACAGTACATTTATGTATATC-3′ |
| 2 | 15771–15791 | 5′-ACTTTATCAGACATCTGGTTC-3′ |
| 3 | 16285–16257 | 5′-TTATTGCGTAATAGAGTATGATTAGAGTT-3′ |
| 4 | 13853–13834 | 5′-CTGCTATAGCTACTGAGGAA-3′ |
| 5 | 15630–15611 | 5′-GACTGTATGGTGTATGTCAG-3′ |
| 6 | 15407–15438 | 5′-CTACTTCTTGAGTAATAAATTTACATAGTAC-3′) |
| 7 | 15630–15611 | 5′-GACTGTATGGTGTATGTCAG-3′ |
| 8 | 15771–15791 | 5′-ACTTTATCAGACATCTGGTTC-3′ |
| 9 | 222–201 | 5′-TGTGGCTAGGCAAGGTGTCTTA-3′) |
| 10 | 3351–3370 | 5′-ACAGAAGGAGAATCAGAATT-3′ |
| 11 | 3500–3481 | 5′-ATTGATATAGTATAGGGGTC-3′ |
| 12 | 9415–9442 | 5′-CTGACTTCCAATTAGTAGATTCTGAATA-3′ |
| 13 | 11586–11561 | 5′-TCCTGTTGTCAGATTCACAGTCTAAT-3′ |
| 14 | 10261–10280 | 5′-TTAGTTTAACCAGCCTAACA-3′ |
| 15 | 8950–8931 | 5′-CAGCCTCCTAGATCATGTGT-3′ |
| 16 | 9561–9538 | 5′-GATTTGCTTCTGAGTACAGATTTA-3′ |
| 17 | 9203–9224 | 5′-GGCTACTGGATTCCATGGACTC-3′ |
| 18 | 9571–9590 | 5′-GCGGATTCGACCCTACAAGC-3′ |
| 19 | 9943–9920 | 5′-AGCGAAATATAAGTGTCCCTAGA-3′ |
| 20 | 9415–9442 | 5′-CTGACTTCCAATTAGTAGATTCTGAATA-3′ |
| 21 | 14520–14545 | 5′-TTTATAGGCTACGTCCTTCCATGAGG-3′ |
| 22 | 12251–12232 | 5′-GATTGCTTGTAGGGCTGCAG-3′ |
| 23 | 12804–12785 | 5′-GGATGTCTTGTTCGTCTGCC-3′ |
| 24 | 13183–13159 | 5′-GGTATTGTGAGGACTGGAATGCTGG-3′ |
| 25 | 13641–13616 | 5′-GCGGCAATATATAGTTGTGCTACTTG-3′ |
C. Primers used for amplification of human synaptosome cybrid mtDNA.
| Primer pair | Primer coordinates | Primer sequence |
|---|---|---|
| (5′→3′) | ||
| 1 | 15838–15857 | 5′-CCTAATACCAACTATCTCCC-3′ |
| 1064–1045 | 5′-GTCTTAGCTATTGTGTGTTC-3′ |
D. Primers used for sequencing of human synaptosome cybrid mtDNA.
| Primer | Primer coordinates | Primer sequence |
|---|---|---|
| (5′→3′) | ||
| 1 | 240–221 | 5′-TATTATTATGTCCTACAAGC-3′ |
| 2 | 1–20 | 5′-GATCACAGGTCTATCACCCT-3′ |
| 3 | 636–617 | 5′-TGATGTGAGCCCGTCTAAAC-3′ |
| 4 | 429–408 | 5′-CTGTTAAAAGTGCATACCGCCA-3′ |
| 5 | 242–261 | 5′-CAATTGAATGTCTGCACAGC-3′ |
| 6 | 16225–16244 | 5′-AACTATCACACATCAACTG-3′ |
| 7 | 16501–16843 | 5′-ATGTCGGATACAGTTCAC-3′ |
| 8 | 16547–16527 | 5′-GGAACGTGTGGGCTATTTAGG-3′ |
E. Primers used for production of human brain mtDNA bacterial clones.
| Template fragment for cloning: | ||
|---|---|---|
| Primer pair | Primer coordinates | Primer sequence |
| (5′→3′) | ||
| 1 | 242–261 | 5′-CAATTGAATGTCTGCACAGC-3′ |
| 636–617 | 5′-TGATGTGAGCCCGTCTAAAAC-3′ | |
Same primers were used for mtDNA sequence analysis of bacterial clones.
Acknowledgments
ACKNOWLEDGEMENTS
The authors would like to express their appreciation for the technical assistance of Stephanie Letellier and Shawn Levy. The work was supported by NIH grants AG13154, HL45572 and NS21328 awarded to D.C.W.
REFERENCES
- 1.Wallace D.C. (1992) Annu. Rev. Biochem., 61, 1175–1212. [DOI] [PubMed] [Google Scholar]
- 2.Corral-Debrinski M., Horton,T., Lott,M.T., Shoffner,J.M., Beal,M.F. and Wallace,D.C. (1992) Nature Genet., 2, 324–329. [DOI] [PubMed] [Google Scholar]
- 3.Corral-Debrinski M., Shoffner,J.M., Lott,M.T. and Wallace,D.C. (1992) Mutat. Res., 275, 169–180. [DOI] [PubMed] [Google Scholar]
- 4.Cortopassi G.A. and Arnheim,N. (1990) Nucleic Acids Res., 18, 6927–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cortopassi G.A., Shibata,D., Soong,N.W. and Arnheim,N. (1992) Proc. Natl Acad. Sci. USA, 89, 7370–7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayakawa M., Sugiyama,S., Hattori,K., Takasawa,M. and Ozawa,T. (1993) Mol. Cell. Biochem., 119, 95–103. [DOI] [PubMed] [Google Scholar]
- 7.Simonetti S., Chen,X., DiMauro,S. and Schon,E.A. (1992) Biochim. Biophys. Acta, 1180, 113–122. [DOI] [PubMed] [Google Scholar]
- 8.Soong N.W., Hinton,D.R., Cortopassi,G. and Arnheim,N. (1992) Nature Genet., 2, 318–323. [DOI] [PubMed] [Google Scholar]
- 9.Melov S., Shoffner,J.M., Kaufman,A. and Wallace,D.C. (1995) Nucleic Acids Res., 23, 4122–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munscher C., Muller-Hocker,J. and Kadenbach,B. (1993) Biol. Chem. Hoppe Seyler, 374, 1099–1104. [DOI] [PubMed] [Google Scholar]
- 11.Munscher C., Rieger,T., Muller-Hocker,J. and Kadenbach,B. (1993) FEBS Lett., 317, 27–30. [DOI] [PubMed] [Google Scholar]
- 12.Rieger T., Munscher,C., Seibel,P., Muller-Hocker,J. and Kadenbach,B. (1993) Methods in Molecular and Cellular Biology, Vol. 4, pp. 121–127.
- 13.Jazin E.E., Cavelier,L., Eriksson,I., Oreland,L. and Gyllensten,U. (1996) Proc. Natl Acad. Sci. USA, 93, 12382–12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michikawa Y., Mazzucchelli,F., Bresolin,N., Scarlato,G. and Attardi,G. (1999) Science, 286, 774–779. [DOI] [PubMed] [Google Scholar]
- 15.Trounce I., Neill,S. and Wallace,D.C. (1995) Am. J. Hum. Genet., 57, A252. [Google Scholar]
- 16.Inoue K., Ito,S., Takai,D., Soejima,A., Shisa,H., LePecq,J.B., Segal-Bendirdjian,E., Kagawa,Y. and Hayashi,J.I. (1997) J. Biol. Chem., 272, 15510–15515. [DOI] [PubMed] [Google Scholar]
- 17.Desjardins P., de Muys,J.M. and Morais,R. (1986) Somatic Cell Genet., 12, 133–139. [Google Scholar]
- 18.Morais R., Desjardins,P., Turmel,C. and Zinkewich-Peotti,K. (1988) In Vitro Cell. Dev. Biol., 24, 649–658. [DOI] [PubMed] [Google Scholar]
- 19.King M.P. and Attardi,G. (1989) Science, 246, 500–503. [DOI] [PubMed] [Google Scholar]
- 20.Trounce I., Neill,S. and Wallace,D.C. (1994) Proc. Natl Acad. Sci. USA, 91, 8334–8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunkley P.R., Heath,J.W., Harrison,S.M., Jarvie,P.E., Glenfield,P.J. and Rostas,J.A. (1988) Brain Res., 441, 59–71. [DOI] [PubMed] [Google Scholar]
- 22.Trounce I.A., Kim,Y.L., Jun,A.S. and Wallace,D.C. (1996) Methods Enzymol., 264, 484–509. [DOI] [PubMed] [Google Scholar]
- 23.Bibb M.J., Van Etten,R.A., Wright,C.T., Walberg,M.W. and Clayton,D.A. (1981) Cell, 26, 167–180. [DOI] [PubMed] [Google Scholar]
- 24.Littlefield J.W. (1964) Nature, 203, 1142–1144. [DOI] [PubMed] [Google Scholar]
- 25.Kit S., Dubbs,D.R., Pielarski,L.J. and Hsu,T.S. (1963) Exp. Cell Res., 31, 297–312. [DOI] [PubMed] [Google Scholar]
- 26.Anderson S., Bankier,A.T., Barrell,B.G., de Bruijn,M.H., Coulson,A.R., Drouin,J., Eperon,I.C., Nierlich,D.P., Roe,B.A., Sanger,F., et al. (1981) Nature, 290, 457–465. [DOI] [PubMed] [Google Scholar]
- 27.Wallace D.C., Lott,M.T., Brown,M.D., Huoponen,K. and Torroni,A. (1995) In Cuticchia,A.J. (ed.), Human Gene Mapping 1994, A Compendium. The Johns Hopkins University Press, Baltimore, pp. 910–954.