


Abstract
XRCC1 (X-ray cross-complementing group 1) is a DNA repair protein that forms complexes with DNA polymerase β (β-Pol), DNA ligase III and poly-ADP-ribose polymerase in the repair of DNA single strand breaks. The domains in XRCC1 have been determined, and characterization of the domain–domain interaction in the XRCC1-β-Pol complex has provided information on the specificity and mechanism of binding. The domain structure of XRCC1, determined using limited proteolysis, was found to include an N-terminal domain (NTD), a central BRCT-I (breast cancer susceptibility protein-1) domain and a C-terminal BRCT-II domain. The BRCT-I–linker–BRCT-II C-terminal fragment and the linker–BRCT-II C-terminal fragment were relatively stable to proteolysis suggestive of a non-random conformation of the linker. A predicted inner domain was found not to be stable to proteolysis. Using cross-linking experiments, XRCC1 was found to bind intact β-Pol and the β-Pol 31 kDa domain. The XRCC1-NTD1–183 (residues 1–183) was found to bind β-Pol, the β-Pol 31 kDa domain and the β-Pol C-terminal palm-thumb (residues 140–335), and the interaction was further localized to XRCC1-NTD1–157 (residues 1–157). The XRCC1-NTD1–183-β-Pol 31 kDa domain complex was stable at high salt (1 M NaCl) indicative of a hydrophobic contribution. Using a yeast two-hybrid screen, polypeptides expressed from two XRCC1 constructs, which included residues 36–355 and residues 1–159, were found to interact with β-Pol, the β-Pol 31 kDa domain, and the β-Pol C-terminal thumb-only domain polypeptides expressed from the respective β-Pol constructs. Neither the XRCC1-NTD1–159, nor the XRCC136–355 polypeptide was found to interact with a β-Pol thumbless polypeptide. A third XRCC1 polypeptide (residues 75–212) showed no interaction with β-Pol. In quantitative gel filtration and analytical ultracentrifugation experiments, the XRCC1-NTD1–183 was found to bind β-Pol and its 31 kDa domain in a 1:1 complex with high affinity (Kd of 0.4–2.4 µM). The combined results indicate a thumb-domain specific 1:1 interaction between the XRCC1-NTD1–159 and β-Pol that is of an affinity comparable to other binding interactions involving β-Pol.
INTRODUCTION
XRCC1 (X-ray cross-complementing group 1) is a DNA repair protein that in mammals is required for efficient repair of DNA damage caused by ionizing radiation or DNA methylating agents (1). In cells lacking XRCC1 activity, aberrant levels of sister chromatid exchange have been observed during mitosis (1). Transfection of a vector carrying the gene for human XRCC1 into hamster cell lines deficient in XRCC1 activity results in complementation and restoration of normal DNA damage repair function (1). In transgenic mice, XRCC1(–/–) knockouts or DNA polymerase β (β-Pol)(–/–) knockouts are embryonic lethal, indicating an essential role for these interacting proteins (2,3).
XRCC1 has been considered a component of base excision repair (BER) (recently reviewed in 4). XRCC1 mediates DNA repair by forming DNA repair complexes consisting of XRCC1, β-Pol, poly-ADP-ribose polymerase (PARP, preferentially in the ADP ribosylated form), and DNA ligase III (5–7). XRCC1 and DNA ligase III are associated in vivo, and expression of active DNA ligase III requires the co-expression of native XRCC1 (8). The N-terminal domain (NTD) of XRCC1 (XRCC1-NTD1–183) interacts with β-Pol (5,6,9). Additionally, the XRCC1-NTD1–183 interacts with DNA containing a single-strand break (9).
The BER pathway consists of steps that include excision of the abasic site nucleotide by the deoxyribose-5-phosphate (dRP) lyase activity that is carried by the β-Pol NTD (10,11), single nucleotide gap-filling DNA synthesis and DNA ligation (12,13). Many thousands of abasic sites are generated in cells each day by either purine hydrolysis or by a group of damage specific DNA glycosylases. Incision by AP endonuclease (APE) results in a single-strand break containing the 5′-dRP group. The XRCC1/PARP complex has been described as providing a nick sensing function in BER (6). XRCC1 may function in ‘short patch’ BER by recruiting ligation machinery to the site of DNA repair (5,6,14). A comparison of BER products in cell-free extracts from wild type and xrcc1 mutant CHO cell lines has shown that XRCC1 may suppress strand displacement synthesis (5), or alternatively, enhance single-nucleotide insertion ligation (15). β-Pol has a well-defined domain architecture first shown by limited proteolysis (16–18) and later apparent from X-ray crystal structures (19). The three-dimensional structure of β-Pol is formed from the C-terminal polymerase domain with fingers, palm and thumb sub-domains (19) and the N-terminal 8 kDa DNA binding domain (19,20). The three-dimensional NMR structure of the XRCC1-NTD1–183 has been reported together with the single-strand break binding activity (9). Mapping of the β-Pol and DNA binding sites showed that the structure of the XRCC1-NTD1–183 is well-suited for interaction with the inside curvature of 90° bent DNA and with polypeptide segments in the β-Pol palm and thumb in a β-Pol–gapped DNA complex (9).
While long-patch BER also generates single-strand breaks, it is not clear what role, if any, XRCC1 might serve in long-patch BER. In long-patch BER, 2–13 nt are incorporated by a DNA polymerase (reviewed in 21), and a flap endonuclease (FEN1) is required for removal of the flap structure generated by strand displacement synthesis (22). In cell-free extracts, long-patch BER is conducted by β-Pol (23–25). A BER regulatory role for PARP, another XRCC1 partner, has been suggested (26), and PARP may provide single-strand break recognition function (14). PARP binds specifically to single-strand DNA breaks (27) and induces a V-like bend in DNA (28). Upon binding, PARP poly-ADP ribosylates itself and nearby proteins causing dissociation of the poly-ADP-ribosylated proteins from the damaged DNA. Whether there is a competition or cooperation of β-Pol and/or XRCC1 with PARP for damaged sites is not yet known. It has been suggested that PARP could relax the nucleosome structures and recruit the BER complex through interaction with XRCC1 (29,30).
To date, structural characterization of domains shows that XRCC1 is formed by the independently folded NTD (residues 1–151) (9) and the C-terminal BRCT-II (breast cancer susceptibility protein-1 homology C-terminal) domain (residues 538–633) (31). A central BRCT domain, BRCT-I (residues 315–403), was predicted on the basis of sequence homology (32,33). BRCT-II was found to interact with a C-terminal BRCT domain of DNA ligase III (34), and BRCT-I was found to bind to PARP (7). On PARP, binding was mapped to a BRCT domain of PARP, as well as to a region N-terminal to the BRCT domain. Nearly half of the XRCC1 sequence is formed by two additional segments, one unclassified segment that separates the XRCC1-NTD1–151 from the BRCT-I domain and a second segment that separates the BRCT domains.
In the present study, we have addressed the biochemical properties of the XRCC1–β-Pol interaction in order to understand the specificity and mechanism of binding. We have characterized the XRCC1-NTD, BRCT-I and BRCT-II domains in XRCC1 by use of limited proteolysis methods. The polypeptide segment separating the NTD from BRCT-I was found not to be stable to proteolysis, while the segment separating the BRCT domains was found to be relatively stable to proteolysis. We have studied the domain specific interaction between N-terminal regions of XRCC1 and domains in β-Pol by in vitro cross-linking of purified components, yeast two-hybrid methods, quantitative gel filtration and sedimentation equilibrium. We show evidence for a specific interaction between the β-Pol catalytic domain and the XRCC1-NTD with cross-linking localizing the interaction to the β-Pol palm–thumb domain and a yeast two-hybrid analysis localizing the interaction to the β-Pol thumb. The binding affinities and stoichiometry of the interactions have been characterized.
MATERIALS AND METHODS
Escherichia coli expression plasmids and purification of proteins
Full-length XRCC1 with a C-terminal His6 tag was expressed from the plasmid pET16BXH (14) kindly provided by Dr Keith W. Caldecott (School of Biological Sciences, University of Manchester, Manchester, UK). BL21(DE3) cells containing the plasmid were grown in LB medium to OD600 of 0.9 and induced with 1 mM IPTG for 3 h. After centrifugation, pellets were resuspended in buffer containing 10 mM Na phosphate, pH 7.9, 400 mM NaCl, 2 mM β-mercaptoethanol (β-ME), 1 mM phenylmethylsulfonyl fluoride (PMSF) and sonicated. XRCC1 was purified from the crude extract by selective ammonium sulfate precipitation. XRCC1 was soluble at 30% saturation and precipitated at 50% saturation. The protein was further purified on a TALON His tag affinity column (Clontech Laboratories, Palo Alto, CA), according to the manufacturer’s instructions. Finally, gel filtration was performed using a Sephadex G-75 column in the same buffer as used for cell lysis. The final purity was >90%, as estimated from acrylamide gels stained with Coomassie blue.
The plasmid pET23a-X183, overexpressing the first 183 residues of human XRCC1, was constructed by sub-cloning the corresponding fragment from XRCC1-δ184–633 (5) into NdeI–BamHI sites in the pET23a vector (Novagen, Madison, WI) using the polymerase chain reaction (PCR). The presence of the gene for XRCC1-NTD1–183 was confirmed by restriction digestion and sequencing. The isolated plasmid DNA was transformed into the BL21(DE3) pLysS E.coli strain for overexpression. Cultures in LB medium were grown to OD600 of 0.9 and induced with 1 mM IPTG for 3 h. After centrifugation, pellets were resuspended in lysis buffer (25 mM Tris–HCl, pH 7.5, 400 mM NaCl, 1 mM EDTA, 2 mM β-ME, 1 mM PMSF) and sonicated. XRCC1-NTD1–183 was purified from the crude extract by selective ammonium sulfate precipitation. XRCC1-NTD1–183 was largely soluble at 50% and precipitated at 77% saturation. Gel filtration was performed using a Sephadex G-50 column in the above-described buffer. The final purity was >95%, as estimated from acrylamide gels stained with Coomassie blue.
Full-length recombinant human β-Pol and the recombinant rat β-Pol 31 and 8 kDa domains, and 16 kDa fragment (consisting of the 8 kDa domain and the fingers sub-domain of the 31 kDa domain) were purified as described previously (18). Human and rat β-Pol are 96% identical. The 27 kDa C-terminal fragment of β-Pol (consisting of the palm and thumb sub-domains of the 31 kDa domain) was generated by cleavage of the 31 kDa domain (0.6 mg/ml) with trypsin (10 µg/ml) for 20 min at room temperature in 20 mM Tris–HCl, pH 7.0, 100 mM NaCl and contained some residual intact 31 kDa domain. Reactions were stopped by addition of the inhibitor, 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF) to 3 mM final concentration.
Domain characterization of XRCC1
A solution containing XRCC1 (0.2 mg/ml) in 20 mM Na phosphate buffer, pH 7.0, 100 mM NaCl or XRCC1-NTD1–183 (1 mg/ml) in 5 mM Tris–HCl, pH 7.5, 250 mM NaCl, was incubated with trypsin (0.2 or 1 µg/ml, respectively) at 25°C and aliquots were taken at 5, 10, 20, 30, 40, 50 and 60 min. The proteolysis reaction was stopped by addition of AEBSF to a concentration of 0.1 mM and boiling in SDS sample buffer. The samples were characterized by SDS–PAGE using 10–20% gradient Tris–HCl (for XRCC1) or 16.5% Tris–Tricine (for XRCC1-NTD1–183) acrylamide gels. The separated protein fragments were electro-transferred onto Immobilon PSQ membrane (Millipore, Bedford, MA) using CAPS buffer, pH 11, at room temperature for 100 min at 40 V. The membranes were stained with Coomassie blue and bands of interest were cut out. N-terminal microsequencing was performed on an ABI/Perkin Elmer Procise 494 protein sequencer in the Laboratory for Protein Microsequencing, University of Massachusetts Medical Center, Shrewsbury, MA.
Sequence comparisons
Sequence alignments were performed with MAP (35) and manually modified based on alignments from BLAST search (33). Highlighting of conserved positions in the alignment was done using BOXSHADE (Kay Hofmann and Michael D. Baron, ISREC Bioinformatics group, Lausanne, Switzerland). The PHD program (36,37) was used for secondary structure prediction.
Glutaraldehyde cross-linking
Proteins were mixed and incubated in 20 mM Na-phosphate buffer, pH 7.0, 100 mM NaCl for 20 min at 25° C before addition of glutaraldehyde. Cross-linking was performed by addition of glutaraldehyde (4 mM) and incubation for 30 min at 25°C, unless otherwise stated. The reactions were stopped by boiling in SDS sample buffer and samples were run on 15% or on 10–20% gradient acrylamide gels. Gels were stained with Coomassie blue.
Yeast two-hybrid constructs
The β-Pol two-hybrid constructs used in this study were prepared from a full-length human β-Pol cDNA as restriction fragments. Adapters were used as needed for in-frame insertion relative to the GAL4 activation domain (AD) encoded in the pACT2 yeast two-hybrid vector plasmid (Clontech). The β-Pol 31 kDa domain construct codes for Arg102 to Glu335 and was prepared by insertion of an XhoI restriction fragment of β-Pol. The β-Pol thumbless construct codes for Met1 to Asp251 and was prepared by insertion of a NcoI–EcoRV restriction fragment of β-Pol and contains a vector encoded stop codon. The β-Pol thumb-only construct codes for Asp251 to Glu335 and was prepared by insertion of a EcoRV–XhoI restriction fragment of β-Pol. Full-length β-Pol and N-terminal 8 kDa domain were also prepared as in-frame inserts into pACT2. The β-Pol NTD construct codes for Met1 to Arg102.
The XRCC1 two-hybrid constructs used in this study were prepared from a full-length human XRCC1 cDNA as restriction fragments (adapters were used as needed) or as PCR product inserts in-frame relative to the GAL4 binding domain (BD) in the pAS2-1 yeast two-hybrid vector plasmid (Clontech). The XRCC136–355 construct codes for Ala36 to Arg355 and was prepared by insertion of a StyI–SmaI fragment of XRCC1 and contains a vector encoded stop codon. The XRCC175–212 construct codes for Ser75 to Ala212 and was prepared by insertion of a PvuII fragment of XRCC1. The XRCC1-NTD1–159 construct was prepared as a PCR product and codes for Met1 to Glu159. All of the constructs were confirmed by sequencing.
Two-hybrid analysis
The yeast media used to determine nutritional requirements in the two-hybrid selections were prepared in the facility of NIEHS following established recipes. Chemical reagents for transformation were obtained from Sigma and the yeast plasmid vectors and host cells were obtained from Clontech.
Before testing for protein interactions, each construct was tested for background HIS3 expression on defined media without histidine. No growth on media lacking histidine was observed for any of the constructs transformed into yeast strain CG1945. The CG1945 yeast strain carries the HIS3 and LacZ reporter genes under control of a tightly regulated GAL4 responsive element. Transformation was confirmed by reversion to the trp+ or leu+ phenotype for XRCC1 BD or β-Pol AD constructs, respectively.
Protein interactions were tested by selection for his+ revertants following co-transformation of yeast strain CG1945 with XRCC1 BD constructs and β-Pol AD constructs. Co-transformed cells were plated on dropout media containing 25 mM His3 inhibitor 3AT (3-amino-1,2,4-triazole) and lacking Trp, Leu and His (DO3). The preparation of competent cells and transformations were performed by the LiCl method as described in the Matchmaker GAL4 Two-Hybrid User Manual (Clontech PT3061-1). Since the growth phase was found to affect transformation efficiency more than any other factor, transformation utilized cells that were collected immediately after the culture had doubled twice following dilution. Starting with a saturated culture diluted to OD600 of 0.125; cells were collected when the OD600 was at least 0.50, but <0.55 to avoid crowding. Transformation reactions were split and added in equal amount to DO2 (media lacking Trp and Leu) and DO3 selection plates, grown at 30°C and photographed after 5 days. All protein interactions detected by nutrition selection were confirmed by β-galactosidase assays performed as described in the Matchmaker User Manual.
Gel filtration high-pressure liquid chromatography (HPLC)
XRCC1-NTD1–183 and β-Pol 31 kDa domain were mixed at 1:1 molar ratio with concentrations ranging from 0.5 to 200 µM. The samples (20 µl) were applied to a 300 mm Bio-Sil SEC 125 column and eluted at 1 ml/min in 50 mM Na phosphate buffer, pH 7.0, 150 mM NaCl, 5 mM DTT. The buffer was kept under constant helium purge to prevent DTT oxidation. Detection was performed at 280 nm. Molecular mass standards (Bio-Rad Laboratories, Hercules, CA) of 158, 44, 17 and 1.35 kDa were used to plot a standard curve of log (molecular mass) versus elution time. XRCC1-NTD1–183 and the 31 kDa domain migrated as an average molecular mass complex throughout the entire range of concentrations, whereas the 31 kDa domain alone (5–20 µM) and XRCC1-NTD1–183 alone (12.5–125 µM) migrated as monomers with apparent molecular masses (Mapp) of 35.5 and 26 kDa, respectively. Mapp of the XRCC1-NTD1–183–β-Pol 31 kDa domain complex was plotted as a function of the concentration, and the Kd was fitted as described previously (38). The weighted average molecular mass (Mavg) was calculated on the basis of the mole fractions of the contributing species using equation 1:
Mavg = a 58.5 + b 35.5 + b 26 1
where 58.5, 35.5 and 26 are the fitted Mapp of the complex, the observed Mapp for the free 31 kDa domain and the observed Mapp for the free XRCC1-NTD1–183, respectively, and a and b are the mole fractions of the complex and the free species, respectively, for all species within the same volume. a and b were calculated using equations 2 and 3:
a = (x – y)/[(x – y) + 2y] 2
b = y/[(x – y) + 2y] 3
where x is the total concentration of XRCC1-NTD1–183, which is equal to the total concentration of the 31 kDa domain, and y is the concentration of the free species at equilibrium. y was calculated by solving the quadratic equation 4:
y2 + Kd y – Kd x = 0 4
which is derived from the equilibrium expression for Kd as shown by equation 5:
Kd = y2/(x – y) 5
The Kd was fitted using Microsoft Excel (Microsoft Corp.), and the curve was generated using KaleidaGraph (Abelbeck Software).
Analytical ultracentrifugation
The associative behavior, in solution, of the XRCC1-NTD1–183 with intact β-Pol and with its two complementary domains (the 31 kDa C-terminal and the 8 kDa NTDs) were studied in the analytical ultracentrifuge by sedimentation equilibrium. Four experiments were performed to investigate first, the self-associative behavior of XRCC1-NTD1–183, and second, its heterogeneous interactions with β-Pol and with its 8 and 31 kDa domains. The experiments were run simultaneously using an eight-hole rotor at a speed of 18 000 r.p.m. in a Beckman-XLA analytical ultracentrifuge. Scans were taken at 280 nm after equilibrium had been reached at two different temperatures, 4 and 20°C. The proteins were prepared in a 25 mM HEPES, pH 7.5, 50 mM KCl, 1.5 mM DTT, 1 mM EDTA buffer except for the 31 kDa β-Pol domain preparations for which an additional 100 mM KCl was necessary to avoid precipitation. An initial experiment in the absence of DTT from the buffer, displayed significant dimerization of XRCC1-NTD1–183, presumably due to formation of irreversible disulfide bonds. DTT (1.5 mM) was added to the buffer in the final experiment in order to competitively reverse such covalently formed homodimers. Two centrifuge cells were loaded with 180 µl columns of XRCC1-NTD1–183 that had concentrations with observed absorbances of 0.2 and 0.3 at 280 nm. A single cell was loaded with a 1:1 mixture of the XRCC1-NTD1–183 with the 8 kDa N-terminal domain of β-Pol with an observed absorbance of ~0.22 at 280 nm. Two additional cells were loaded with 1:1 molar ratio mixtures of XRCC1-NTD1–183 and intact β-Pol at concentrations with observed absorbances of 0.15 and 0.25, respectively. The remaining two cells were loaded with 1:1 molar ratio mixtures of XRCC1-NTD1–183 and the 31 kDa domain of β-Pol at concentrations with observed absorbances of 0.15 and 0.25.
Analytical ultracentrifugation data analysis
Although the XRCC1-NTD1–183 dimer has a mass that is very close to the mass of intact β-Pol, the amount of XRCC1-NTD1–183 dimer is small, and the β-Pol oligomerization weak. Thus, a stronger heterogeneous interaction should be readily observable. A number of equilibrium mathematical models were fitted globally to the data from the two cells; the most complex (and comprehensive) model was as shown in equation 6:

where the subscripts and superscripts X and B refer to XRCC1-NTD1–183 and β-Pol, respectively, and lnK is the natural logarithm of the association constant of the species indicated by its superscript and of molar ratios indicated by its subscript. M is the molar mass of the respective protein, and the buoyancy and the effects of the centrifugal field are represented in the term Ai, which is described by equation 7
Ai = (1 – ρ vi)ω2/2RT 7
where vi is the compositional partial specific volume of the solute molecule i at the temperature T, ρ is the specific mass of the buffer at that temperature, ω is the rotational speed in rad/s, R is the gas constant and T is the absolute temperature. ɛ is the baseline offset correction for the finite absorbance of the buffer. The (possibly virtual) association constant for dimerization of XRCC1-NTD1–183 estimated above was used as was the corresponding association constants for the oligomerization of β-Pol published recently (39). Estimation of the remaining association constants, concentrations and baseline offset correction parameters was achieved by curve fitting the collected data using the above model and a number of reduced variants of the same and the method recently described (39). All computations were performed on a Pentium PC using the software package MLAB (Civilized Software, Bethesda, MD).
RESULTS
Domain characterization of XRCC1
Limited proteolysis of XRCC1 yielded products with Mapp of ~16, 23, 33 and 45 kDa that were stable for digestion periods of up to 60 min (Fig. 1A). The proteolytically obtained protein fragments were identified by sequence analysis of the first five N-terminal residues. Sequence analysis of the 16 kDa fragment indicated that the N-terminal residue was Ala311 (Fig. 1B). The size of the 16 kDa fragment together with the finding that another cleavage site in XRCC1 was observed between Lys436 and Thr437 to yield a 33 kDa fragment (discussed below) suggests that the C-terminus of the 16 kDa is Lys436 (Fig. 1B). Thus, these data indicate that the low molecular weight 16 kDa product is the proteolytically generated BRCT-I domain.
Figure 1.
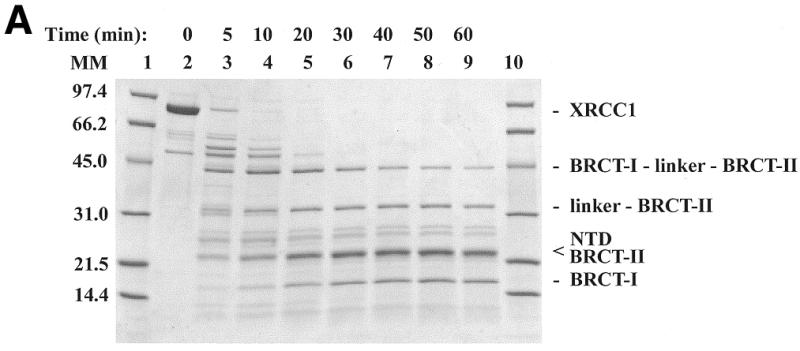

Limited tryptic digestion of XRCC1. (A) SDS–PAGE analysis of the tryptic digestion using 0.2 mg/ml of XRCC1 and 0.2 µg/ml of trypsin. Lanes 1 and 10, molecular mass markers; lane 2, undigested purified XRCC1; and lanes 3–9, digestion times as indicated. The identity of the tryptic fragments is indicated on the right. (B) Alignment of XRCC1 sequences from human (huXRCC1, SwissProt accession no. P18887), mouse (muXRCC1, SwissProt accession no. Q60596), hamster (haXRCC1, GenBank accession no. AAC40038) and Drosophila (drXRCC1, GenBank accession no. AAD33589). Identities in at least three sequences are highlighted in cyan and similarities are highlighted in green. The numbering over the sequences is based on huXRCC1. Insertions with respect to huXRCC1 in the other sequences are not shown and their positions are indicated with pairs of lower case letters. The conserved regions are indicated above the sequence. The known secondary structure elements for NTD and BRCT-II are marked with a solid line and the secondary structure elements for BRCT-I, predicted based on homology to BRCT-II, are marked with a dotted line. The start and expected end of a tryptic fragment (A) are indicated with │-> and ->│, respectively.
The band that migrated at ~23 kDa was found to contain two proteolytic products, that were identified as the XRCC1-NTD and the C-terminal BRCT-II domain. In the 40 min proteolysis time point the XRCC1-NTD and the C-terminal BRCT-II were in an ~2:1 molar ratio. Sequence analysis indicated that the N-terminal residue in the proteolytically generated XRCC1-NTD was Pro 2 (NTD) and the presumed C-terminal residue was Lys164 or Lys169 (Fig. 1B). The N-terminal residue in the proteolytically generated BRCT-II domain was Val495. Limited proteolysis of the cloned XRCC1-NTD1–183, consisting of residues 1–183, similarly yielded a 23 kDa fragment resulting from the loss of ~2 kDa (data not shown). These results for the intact XRCC1 and for the 1–183 fragment are consistent with cleavage at Lys164 or Lys169.
Sequence analysis of the 33 kDa fragment (Fig. 1A) indicated that the N-terminal residue was Thr437. An apparent mass of 33 kDa and the N-terminal sequencing data identify this product as the linker–BRCT-II fragment. Analysis of the 45 kDa fragment indicated that the N-terminal residue was Ala311, which corresponds to a tryptic cleavage site that is four residues N-terminal to the predicted N-terminus of BRCT-I domain (32,33). An apparent mass of 45 kDa and the N-terminal sequencing data identify this product as the BRCT-I–linker–BRCT-II C-terminal fragment. In addition to these products, multiple bands migrating at 55–65 kDa are apparent at 5 and 10 min proteolysis periods. Several of these bands correspond to impurities that co-purify with the His-tagged XRCC1, and are indicative of C-terminal His-tagged fragments that have resulted from proteolytic removal of the NTD, and possible inner domain (ID) fragments, from the remainder of the protein. Accordingly, mass spectrometer analysis of XRCC1, proteolyzed for 10 min, showed four identified low molecular weight polypeptides corresponding to tryptic fragments within the predicted ID domain (data not shown).
XRCC1 sequence comparisons
There are four XRCC1 sequences in the databases, which include the sequences from human (huXRCC1) (1), mouse (muXRCC1) (40), hamster (haXRCC1) (41) and Drosophila (drXRCC1) (42,43) (Fig. 1B). The three mammalian proteins have an identity of >80%. The NTD (1–151), BRCT-I (315–403), and BRCT-II (538–633) show >90% sequence identity. The segments between the NTD and BRCT-1 and the segment between the BRCT domains consist of polar Pro, Ser and Arg/Lys-rich regions and are only ~60% identical for the mammalian proteins. There are, however, two regions within these segments with >80% identity (Fig. 1B). Residues 158–250 (conserved region 1) have significant hydrophobicity and are predicted by PHD (36,37) to have secondary structure suggestive of folding to form an ID. This predicted domain, however, is unstable to proteolysis. Conserved region 2 (residues 469–526) is shorter and more polar, with only one helix predicted by the PHD program and is not likely to form a folded domain.
The drXRCC1 shows limited homology to the mammalian XRCC1 sequences. It contains the NTD and BRCT-I domains with >40% identity. It lacks BRCT-II and has an ~100 residue insertion between the predicted ID and BRCT-I. In addition, the predicted ID shows 27% identity, but a BLAST search with residues 152–250 of huXRCC1 does not independently find the weakly homologous Drosophila sequence. Homology searches for the linker regions between the BRCT domains showed conserved region 2, residues 469–526, with 37% identity from drXRCC1 to huXRCC1. The region 2 conservation in drXRCC1 suggests that its function is not related to BRCT-II.
Cross-linking analysis of XRCC1-NTD1–183 binding to β-Pol and β-Pol domains
We performed glutaraldehyde cross-linking experiments to determine whether specific domains of β-Pol preferentially interact with XRCC1-NTD1–183. In the SDS–PAGE analysis, the 31 kDa domain of β-Pol was found to efficiently cross-link to XRCC1-NTD1–183, as judged from the appearance and intensity of a band migrating at the expected molecular mass (~50 kDa) of the XRCC1-NTD1–183–β-Pol 31 kDa domain complex (Fig. 2A, lanes 9–11). No such band was observed on addition of glutaraldehyde to either of the individual proteins, further indicating that the 50 kDa band is the heterodimer. For XRCC1-NTD1–183 alone, incubation with glutaraldehyde under the same conditions, including the same cross-linker concentration, yielded only minor cross-linking to itself, as detected by a faint band at ~40 kDa (Fig. 2A, lanes 3 and 4). Glutaraldehyde modification of XRCC1-NTD1–183 was found to result in a second band of near equal intensity below the band corresponding to the untreated protein indicating an intramolecular cross-link. Together, these results established that a 1:1 complex of the XRCC1-NTD and the β-Pol 31 kDa domain was present in the 50 kDa band, which was clearly of higher molecular mass than the molecular mass of the minor 40 kDa XRCC1-NTD1–183 self-crosslinked species.
Figure 2.

Glutaraldehyde (GA) cross-linking of XRCC1-NTD1–183 designated as X(1–183) with full-length β-Pol and β-Pol domains. β-Pol domains included the N-terminal 8 kDa domain and the C-terminal 31 kDa domain. The 27 kDa palm–thumb domain is a C-terminal proteolysis product derived from the 31 kDa domain. The 16 kDa domain includes the 8 kDa domain and the fingers domain of the 31 kDa domain. Cross-linking was performed for 30 min, unless otherwise stated. The concentrations of the proteins were as follows: X(1–183), 20 µM; β-Pol, 4 µM; β-Pol 31 kDa domain (31K), 10 µM; β-Pol 27 kDa fragment (27K), <10 µM (Materials and Methods); β-Pol 16 kDa fragment (16K), 40 µM; β-Pol 8 kDa domain (8K), 40 µM. The reactions were stopped by boiling in SDS sample buffer and the proteins were separated on 15% acrylamide gels. Gels were stained with Coomassie blue. (A) Lane 1, molecular mass markers; lane 2, X(1–183); lane 3, X(1–183) + GA, 10 min; lane 4, X(1–183) + GA, 30 min; lane 5, 31K; lane 6, 31K + GA, 10 min; lane 7, 31K + GA, 30 min; lane 8, X(1–183) + 31K; lane 9, X(1–183) + 31K + GA, 10 min; lane 10, X(1–183) + 31K + GA, 20 min; lane 11, X(1–183) + 31K + GA, 30 min. (Note: the band of free X(1–183) doubles upon modification by GA.) (B) Lane 1, X(1–183) + β-Pol; lane 2, X(1–183) + β-Pol + GA; lane 3, β-Pol + GA; lane 4, molecular mass markers; lane 5, X(1–183) + 27K; lane 6, X(1–183) + 27K + GA; lane 7, 27K + GA. (C) Lane 1, molecular mass markers; lane 2, X(1–183) +16K; lane 3, X(1–183) + 16 K + GA; lane 4, 16K + GA; lane 5, X(1–183) + 8K; lane 6, X(1–183) + 8K + GA; lane 7, 8K + GA. (D) Effect of salt on cross-linking of XRCC1-NTD1–183 [X(1–183)] with β-Pol 31 kDa domain (31K). Lanes 1 and 8, molecular mass markers; lane 2, X(1–183) + 31K; lanes 3–7, X(1–183) + 31K + GA in the following salt concentrations: 0 mM NaCl (lane 3); 100 mM NaCl (lane 4); 500 mM NaCl (lane 5); 1 M NaCl (lane 6); 2 M NaCl (lane 7).
XRCC1-NTD1–183 cross-linked efficiently to full-length β-Pol (Fig. 2B, lane 2). Higher molecular mass cross-linked species were observed for XRCC1-NTD1–183 cross-linking to β-Pol, in addition to the primary 1:1 cross-linked species. However, as shown by sedimentation equilibrium ultracentrifugation (later section), the heterodimer is the predominant species. Similarly, full-length XRCC1 cross-linked to β-Pol and to the β-Pol 31 kDa domain, as judged from the disappearance of the free proteins and formation of cross-linked species in the presence of glutaraldehyde (data not shown). Minor self cross-linking of XRCC1 alone was observed.
To further map the region(s) of β-Pol interacting with XRCC1-NTD1–183, we used the 27 kDa C-terminal fragment of β-Pol corresponding to the palm and thumb. Nearly one-half of the 27 kDa fragment in the mixture was cross-linked to XRCC1-NTD1–183 (Fig. 2B, lane 6). In the control, a small amount of the β-Pol 27 kDa homodimer was observed that migrated close to the XRCC1-NTD1–183–β-Pol 27 kDa heterodimer. The intensity of the crosslinked heterodimer band was significantly higher in comparison to the β-Pol 27 kDa homodimer in the control lane. The role of the C-terminal 27 kDa region of β-Pol in XRCC1-NTD binding was further addressed using the yeast two-hybrid screen (see below). In the β-Pol-XRCC1-NTD1–183 and the β-Pol 27 kDa domain–XRCC1-NTD1–183 lanes, higher molecular mass cross-linked products were observed. The oligomeric cross-linked products migrated at higher masses than the self cross-linked β-Pol and the self cross-linked 27 kDa fragment, and this likely reflects aggregates of the heterodimeric complexes. No significant cross-linking of XRCC1-NTD1–183 to the 8 kDa domain and the 16 kDa fragment, consisting of the 8 kDa domain and the fingers sub-domain of the 31 kDa domain, was observed (Fig. 2C, lanes 6 and 3). In controls, self cross-linking was observed with the 16 kDa fragment (Fig. 2C, lane 4). Minor self cross-linking of the β-Pol 8 kDa domain was observed (Fig. 2C, lane 7). Finally, in separate experiments, a further truncated polypeptide, XRCC1-NTD1–157, was shown to cross-link to both the β-Pol 31 kDa domain and to the overexpressed and purified β-Pol palm–thumb domain (data not shown).
The sensitivity of the cross-linking reaction to increasing salt concentration was studied in order to determine the nature of the interaction. The complex between XRCC1-NTD1–183 and the 31 kDa domain was resistant to an increase in salt concentration with little detectable change in cross-linking between 100 and 500 mM NaCl and only a slight decrease in cross-linking at 1 and 2 M NaCl (Fig. 2D). The finding that the complex is stable at high salt concentrations shows that the interaction has an appreciable hydrophobic contribution. Such salt resistance is consistent with specific contacts as opposed to non-specific ionic contacts. Taken together, the cross-linking results are indicative of an interaction between XRCC1-NTD1–183 and the 31 kDa domain of β-Pol and more specifically, with the thumb and/or palm domains.
Yeast two-hybrid analysis of XRCC1 N-terminal segment interaction with β-Pol and β-Pol domains
We further examined the XRCC1 interaction with β-Pol and β-Pol domains with a yeast two-hybrid screen using DNA constructs that expressed parts of the N-terminal region of XRCC1 as fusions to GAL4 BD and DNA constructs that expressed domains of β-Pol as fusions to GAL4 DNA AD. The full-length β-Pol was tested for interaction with the XRCC136–355, XRCC175–212 and the XRCC1-NTD1–159 polypeptides. Co-transformation of the full-length β-Pol construct with either the XRCC136–355 or XRCC1-NTD1–159 constructs resulted in the appearance of yeast colonies on DO3 plates that were only slightly delayed relative to co-transformation controls grown on DO2 plates. No growth of cells co-transformed with β-Pol/XRCC175–212 was observed on DO3 plates.
In order to map the site of the two-hybrid interaction to domains of β-Pol, XRCC136–355 and XRCC1-NTD1–159 constructs were tested with constructs expressing the β-Pol 31 kDa domain, the β-Pol 8 kDa domain, β-Pol thumbless and β-Pol thumb-only. Interaction of XRCC136–355 and XRCC1-NTD1–159 with the β-Pol 31 kDa domain (Fig. 3A) was not significantly different from the interaction with full-length β-Pol for growth rate, the effect of the HIS3 inhibitor 3AT, or β-galactosidase activity. No growth on DO3 plates was observed for CG1945 cells co-transformed with XRCC136–355/8 kDa domain or XRCC1-NTD1–159/8 kDa domain constructs. The interactions are summarized in Figure 3B. To further localize the region of XRCC1-NTD interaction on β-Pol, a β-Pol thumbless construct was co-transformed with the XRCC1-NTD1–159 and the XRCC136–355 constructs separately (Fig. 3A). Similarly, the β-Pol thumb-only construct was co-transformed with the XRCC1-NTD1–159 and the XRCC136–355 constructs separately. The thumb domain of β-Pol showed affinity for the XRCC1-NTD in both of the XRCC1 constructs tested as growth on DO3 plates was only slightly inhibited relative to growth on DO2 transformation control plates (Fig. 3A). Consistent with an interaction between XRCC1 and the β-Pol thumb domain, either the XRCC1-NTD1–159 or the XRCC136–355 construct when co-transformed with the β-Pol thumbless construct showed inhibited growth on DO3 plates (Fig. 3A). The slight growth that was observed could be the result of a low affinity secondary interaction between XRCC1 and β-Pol fingers or palm domain. Figure 3B and C summarizes the yeast two-hybrid interactions determined for XRCC1 and β-Pol.
Figure 3.
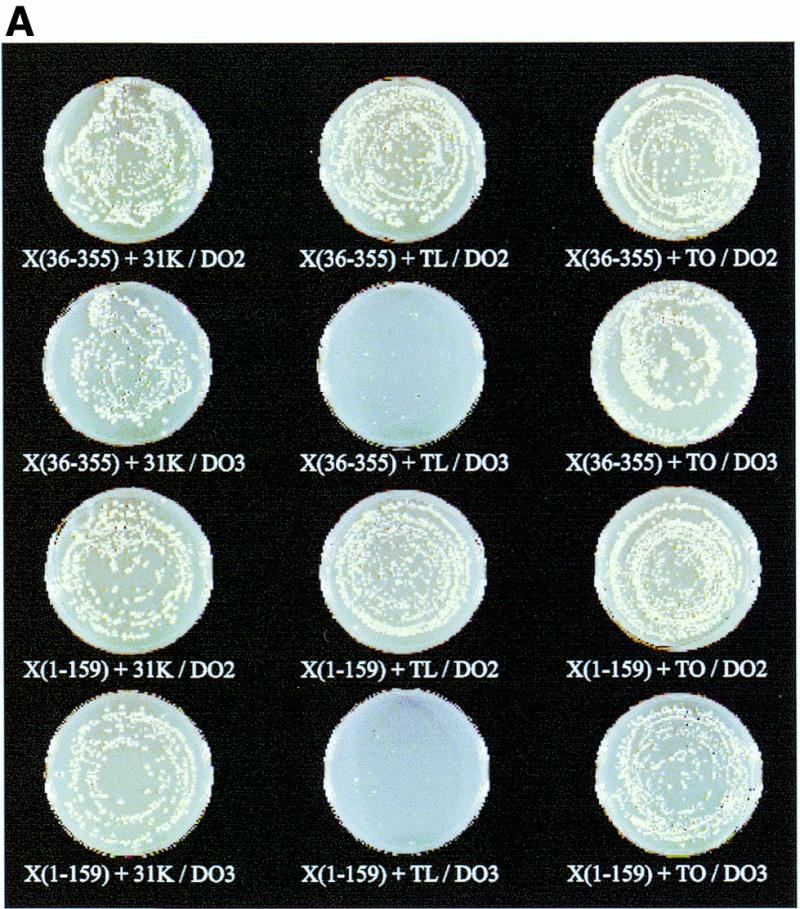
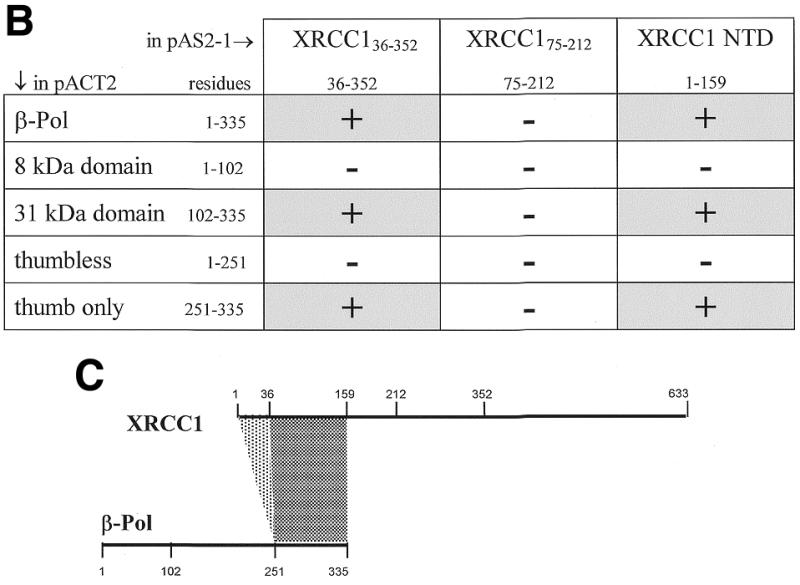
Yeast two-hybrid interactions between regions of XRCC1 and β-Pol. (A) Yeast CG1945 cells co-transformed with the indicated XRCC1 BD and β-Pol AD constructs and grown on DO2 and DO3 plates. Yeast colonies of at least 1 mm were visible 3 days after co-transformation. The designations are as follows: XRCC136–355 [X(36–355)]; XRCC1-NTD1–159 [X(1–159)]; β-Pol 31 kDa domain, Arg102 to the C-terminal Glu335 (31K); β-Pol thumbless, Met1 to Asp251 (TL); β-Pol thumb-only, Asp251 to the C-terminal Glu335 (TO). DO2 is defined media lacking Trp and Leu. Visible colonies on DO2 plates indicate transformation with the BD and AD two-hybrid vector plasmids. DO3 is defined media lacking Trp, Leu and His with HIS3 inhibitor 3AT added to a final concentration of 25 mM. Visible colonies on DO3 plates report protein interactions. (B) Summary of the interactions observed between the XRCC1 BD and β-Pol AD expressed proteins. (C) Summary of the polypeptide segments that displayed a yeast two-hybrid interaction.
Gel filtration analysis of XRCC1-NTD1–183 binding to the β-Pol 31 kDa domain
In order to establish the binding affinity and stoichiometry of the XRCC1-NTD1–183 for the β-Pol 31 kDa domain, a gel filtration experiment was performed. In the gel filtration HPLC experiment, XRCC1-NTD1–183 and the β-Pol 31 kDa domain, at an equimolar ratio, migrated as an average molecular mass complex throughout the concentration range tested (from 0.5 to 200 µM) (Fig. 4). The average molecular mass is due to a rapid equilibrium between the free states of the two proteins and the complex with respect to the separation time. At the highest concentrations tested (200 µM), the XRCC1-NTD1–183–31 kDa domain complex migrated at an Mapp of 56 kDa. On decreasing the concentration of the constituent proteins, the migration was found to be slowed, resulting in an average molecular mass of the complex at the lowest concentrations (0.5 µM) of 37.5 kDa. This result can be explained by a shift in the equilibrium toward the free species. The Mapp of the complex as a function of the concentration of XRCC1-NTD1–183 and β-Pol 31 kDa domain (at an equimolar ratio) was fit to a dissociation equilibrium assuming that all species present contribute to the observed molecular mass as shown in equation 1. The Kd of the XRCC1-NTD1–183–31 kDa domain complex in 50 mM Na-phosphate, pH 7.0, 150 mM NaCl was determined to be 0.43 ± 0.05 µM (Fig. 4). The calculation of the Kd assumes no dilution in the column and, therefore, represents an upper limit of the Kd, which could be within the range of 0.08–0.5 µM. Control experiments showed that the mobility of XRCC1-NTD1–183 did not change between 12.5 and 125 µM (not shown). The mobility of the 31 kDa domain of β-Pol (31K) did not change between 5 and 20 µM (not shown). 31K alone and XRCC1-NTD1–183 alone migrated as monomers with Mapp of 35.5 and 26 kDa, respectively. Thus, the two proteins alone are monomeric in the concentration range used. XRCC1-NTD1–183 bound to full-length β-Pol when mixed at 20 µM concentrations as judged from the following observations: (i) β-Pol showed a greatly slowed migration indicating interaction with the column resin via the highly positively charged 8 kDa domain under the low salt conditions, and (ii) β-Pol and XRCC1-NTD1–183 together were both greatly slowed in their migration due to β-Pol interaction with the column resin and XRCC1-NTD1–183 interaction with β-Pol (data not shown). No apparent binding between XRCC1-NTD1–183 and the 8 kDa domain of β-Pol was seen at an 80 µM (i.e., 4-fold higher) concentration as judged by the following result. The 8 kDa domain showed a greatly slowed migration, while XRCC1-NTD1–183 migrated at its characteristic Mapp of 26 kDa (data not shown).
Figure 4.

Determination of the Kd (0.43 µM) for the binding of XRCC1-NTD1–183 to the 31 kDa domain of β-Pol (31K) by gel filtration HPLC. The proteins were mixed at 1:1 molar ratio with concentrations ranging from 0.5 to 200 µM. XRCC1-NTD1–183 and 31K migrated as a single chromatographic peak throughout the entire range of concentrations and the complex displayed a concentration-dependent average molecular mass. The apparent molecular mass of the complex, XRCC11–183–31K, calculated from the retention time was plotted as a function of the concentration. The Kd of 0.43 ± 0.05 µM was fitted as described in Materials and Methods.
Analytical ultracentrifugation analysis of XRCC1-NTD1–183 binding to β-Pol and β-Pol domains
To further characterize the binding equilibria for XRCC1-NTD1–183 interaction with β-Pol and with β-Pol domains, we analyzed these interactions using analytical ultracentrifugation. As a first step, any possible self-association of XRCC1-NTD1–183 alone was characterized. The addition of DTT (1.5 mM) significantly reduced the formation of irreversible dimers through disulfide bonds. Data analysis, however, indicated the presence of small amount of dimer in the solution. When the data were fitted for a monomer, it was found that the mean molecular mass of the protein species in the solution slightly exceeded the monomeric mass. The data were then fit by assuming a monomer–dimer reversible association or by assuming the presence of two non-interacting species with molecular masses equal to and twice that of the monomer. The statistical fit was excellent in both cases and indicated the presence of ~5% dimer. The hypothetical reversible interaction would be rather weak with Kd equal to ~0.4 and 0.3 mM at temperatures 4 and 20° C, respectively.
The mean molecular mass of the mixture of XRCC1-NTD1–183 with the β-Pol 8 kDa domain was found to lie between the two protein masses but no 1:1 heterogeneous complex of the two proteins could be detected. The best fit was achieved by assuming three non-interacting species: the XRCC1-NTD1–183 monomer and its dimer and the 8 kDa domain of β-Pol. The previous finding that the 8 kDa domain of the β-Pol did not self-associate (39) simplified the analysis.
It has recently been reported that intact β-Pol has the tendency to oligomerize forming oligomers of various sizes (39). The association has been fit by a weak indefinite isodesmic association, meaning that the addition of each monomer to the oligomeric chain is energetically identical. Therefore, the protein species in the equilibrated solution could include intact β-Pol oligomers of all sizes, XRCC1-NTD1–183 monomer and dimer along with possible heterogeneous complexes. In each experiment, a heterogeneous complex of XRCC1-NTD1–183 and β-Pol in a 1:1 molar ratio, was strongly present. No X2B2 complex (Materials and Methods) could be detected at either temperature and inclusion of the isodesmic association of β-Pol or of the XRCC1-NTD1–183 dimer in the analysis model was interchangeable with little change in the final results. The association constant for the 1:1 complex of XRCC1-NTD1–183 lies between 2 × 106 and 3.4 × 106 M–1 (i.e., Kd = 0.3–0.5 µM) at the temperatures examined, with good fits at 4°C (Fig. 5A) and 20°C. This affinity is much stronger than the β-Pol self association or the XRCC1-NTD1–183 virtual association. Similar results were obtained for XRCC1-NTD with the 31 kDa domain of β-Pol using the same models as with intact β-Pol (Fig. 5B). As with intact β-Pol, there was heterogeneous association for XRCC1-NTD1–183 and the 31 kDa domain but the association was slightly weaker than with intact β-Pol. At 4°C the association constant was 0.15 × 106 M–1 or Kd = 6.7 µM, and at 20°C the values were 0.4 × 106 M–1 and 2.4 µM, respectively. The slight differences in the Kd (0.4 versus 2.4 µM) of the XRCC1–31 kDa domain complex determined by gel filtration and analytical ultracentrifugation experiments likely result from differences in temperatures, buffer conditions and pH in the two experiments. The results indicate binding specificity by XRCC1-NTD1–183 for the 31 kDa domain of β-Pol. The 8 kDa domain of the intact enzyme provides only a small degree of additional binding free energy.
Figure 5.

Analytical ultracentrifugation analysis of the interaction of XRCC1-NTD1–183 with full-length β-Pol and its 31 kDa domain. (A) Global curve-fitting of transmitted light intensity data at 4°C from two centrifuge cells loaded with different total concentrations of XRCC1-NTD1–183 and full-length β-Pol, in a 1:1 molar ratio, using the mathematical model given in equation 6 (Materials and Methods). The top two curves represent the reference channels and the bottom two the corresponding sample channels. The data points (open squares) correspond to the cells loaded with concentrations of ~9 µM of each protein while data points shown by stars correspond to the cells loaded with 16 µM of each protein. For each sample concentration, the sample and reference data curves and the fitting error plots have the same symbol. The quality of fit is excellent as can be seen in the inset, which shows the fitting errors for the two sample channels. (B) Curve fitting of transmitted light intensity data from a single centrifuge cell containing XRCC1-NTD1–183 and the β-Pol 31 kDa domain, in a 1:1 ratio, at 20°C, using the mathematical model given in equation 6. The concentration for each protein was ~15 µM. The data points for the reference channel are shown as (open squares), and the data points for the sample channel are shown by triangles. The quality of fit is excellent as can be seen in the inset, which shows the fitting errors. The top error plot represents the sample and the bottom the reference channel. The latter was fitted using cubic splines.
DISCUSSION
Biochemical and sequence analysis of the domain structure of XRCC1 indicated that XRCC1 is formed by an NTD (XRCC1-NTD), a central BRCT domain (BRCT-I) and a C-terminal BRCT domain (BRCT-II). A predicted fourth domain (XRCC1 ID) formed by the conserved residues 158–250 adjacent to the NTD was found not to be stable to proteolysis. The XRCC1-NTD, with a low pI of 6.1, binds single-strand break DNA (9). A possible role of the adjoining sequence (residues 158–310), which includes the predicted ID, might be facilitation of DNA binding, since this segment has a high pI (10.7) and overall positive charge. In the predicted ID, a human polymorphic mutation (R194W) is seen at a conserved Arg in mammals making it of interest in terms of cancer susceptibility (44). Interestingly, Arg194 (positively charged) aligns with a Trp (hydrophobic) in Drosophila, while Ile195 (hydrophobic), which is conserved in mammals, aligns with a Lys (positively charged) in Drosophila.
Unexpectedly, the region between BRCT-I and BRCT-II was found to be relatively resistant to proteolysis, suggesting that this segment of the protein is not totally flexible or exposed (Fig. 1A). Instead, the resistance of this segment to proteolysis, even though it contains Arg and Lys residues for tryptic cleavage, suggests that it associates with one or both of the BRCT domains of XRCC1. Approximately half of the linker segment between the BRCT domains was found to be conserved from Drosophila to humans (Fig. 1B). Since the BRCT-II domain is absent from Drosophila, this 60 residue segment (conserved region II) may have an important role that is functionally distinct from BRCT-II. Moreover, the absence of BRCT-II in Drosophila XRCC1 may make BRCT-I a more likely candidate for association with the conserved region II. Residues 487–500 of conserved region II were predicted using PHD (36,37) to form a helix.
Previously, the XRCC1 BRCT-II was shown to bind to the C-terminal BRCT domain of DNA ligase III (34). Additionally, XRCC1 BRCT-I has been shown to bind to the BRCT domain of PARP in conjunction with a region N-terminal to the PARP BRCT domain (7). No such mapping studies have been previously reported for the interaction between the XRCC1-NTD and domains of β-Pol. We have found that XRCC1-NTD interacts with the C-terminal 31 kDa domain of β-Pol and not with the 8 kDa domain. The interaction has a substantial hydrophobic contribution as judged by the negligible effect of increasing salt on binding. The dissociation constant (Kd of 0.4–2.4 µM) of the complex formed by XRCC1-NTD and either β-Pol or its 31 kDa domain indicates that the interaction is physiologically relevant and is consistent with the finding that XRCC1 is co-immunoprecipitated with β-Pol, DNA ligase III and PARP from HeLa cell extracts (6).
We have found that residues in XRCC1 that are C-terminal to Glu159 (two-hybrid screen) or Glu157 (cross-linking) were not required for β-Pol binding. Importantly, using the yeast two-hybrid screen, we localized the interaction of the XRCC1-NTD to the thumb of β-Pol. Less significant contributions by the β-Pol palm domain to XRCC1-NTD interaction were not ruled out. Primary thumb contacts as well as possible palm contacts are consistent with a previously proposed model for XRCC1-NTD interaction with β-Pol (9). As predicted from the XRCC1-NTD structure, the yeast two-hybrid screen did not show an interaction between XRCC175–212 and β-Pol, since the XRCC175–212 expression fragment would be missing strands βABD on the five-stranded βABGDE sheet and strand βC on the three-stranded βFCH sheet, in addition to other secondary structural elements that together are required for the central β-sandwich fold of the XRCC1-NTD (9). Interestingly, the XRCC1-NTD could be stripped of the N-terminal 35 residues and retain interaction with β-Pol when expressed as a fusion construct. The N-terminal 35 residues form an extended segment, two strands and a helix that pack on one side of the domain opposite to the side of β-Pol interaction. Such a deletion apparently allows the folding of the β-sandwich formed by the βBGDE (missing βA) and the βFCH sheets as well as the packing of the secondary structures that include α2, α3 and the βB′G′ two-stranded sheet.
The XRCC1-NTD–β-Pol interaction (Kd = 400 ± 100 nM) is slightly weaker than the XRCC1-NTD interaction with duplex DNA containing a single-strand gap (half maximal binding at ~60 nM) or with a β-Pol single-nucleotide gap complex (9). The XRCC1-NTD binding affinity for β-Pol is approximately 100 times weaker than the binding affinity of gapped DNA for β-Pol (13) but is comparable to the binding affinity of dNTP substrates for a β-Pol-gapped DNA complex (45). XRCC1 is the only protein known to interact with the catalytic domain of β-Pol. The interaction likely involves contacts with both gapped DNA and loops in the thumb and possibly palm domains of β-Pol. This mechanism of interaction is supported by the results from this study. An alternate pathway for DNA repair via β-Pol utilizes DNA ligase I for ligation (46). Unlike the XRCC1-NTD interaction, DNA ligase I was found to interact with the 8 kDa domain and the contacts involved several charged residues on the 8 kDa domain as assessed by mutational analysis (39). In this interaction a ligase I trimer associates with three β-Pol monomers to form a hexameric complex. At 32°C, the Kd for β-Pol 8 kDa domain interaction with DNA ligase I was calculated to be ~10 µM. These differing interactions by XRCC1/DNA ligase III and DNA ligase I with β-Pol will likely relate to unique BER pathways but do not suggest competitive interactions with β-Pol.
The binding constant for the β-Pol–XRCC1 complex should be considered in view of the ordered BER binding events during DNA repair and the mechanism by which XRCC1 is likely to participate. The binding constant and previous studies suggest that XRCC1 will associate with β-Pol in vivo and preferentially interact with β-Pol bound at sites of DNA damage. XRCC1 is associated with DNA ligase III in vivo through interaction between C-terminal BRCT domains on each of these proteins, and mutations in XRCC1 have been shown to decrease DNA ligase III activity in vivo. PARP interacts with the central BRCT-I domain of XRCC1. Whether PARP remains associated with XRCC1 during all steps in single-strand break repair remains to be determined. APE facilitates β-Pol interaction at an abasic site after incision of abasic site DNA to form a strand break, but APE does not associate with β-Pol in the absence of DNA (39,47). A recent structure shows the interaction of APE1 with bent DNA containing an abasic site analog (48). Interestingly, like β-Pol, the APE is found to make contacts on the outside of the bend (i.e., the side opposite to XRCC1-NTD interaction). The DNA bend in these BER complexes is likely of importance in the ordered steps of the BER pathway (49).
The XRCC1-NTD binds single-strand break DNA in conjunction with β-Pol and contributes to the overall stability of a β-Pol single-nucleotide gap DNA complex. The interaction of the XRCC1-NTD with the thumb is of significance, since the β-Pol thumb is a required domain in polymerase catalysis (functioning analogously to the fingers in the Pol I family of enzymes). Furthermore, motion of the β-Pol thumb with respect to the palm is likely to be important in polymerase catalysis and fidelity (50). Such motion during catalysis could be affected by XRCC1-NTD interaction.
Acknowledgments
ACKNOWLEDGEMENT
This research was supported by a Grant GM52738 from the National Institutes of Health (to G.P.M.).
REFERENCES
- 1.Thompson L.H., Brookman,K.W., Jones,N.J., Allen,S.A. and Carrano,A.V. (1990) Mol. Cell. Biol., 10, 6160–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tebbs R.S., Flannery,M.L., Meneses,J.J., Hartmann,A., Tucker,J.D., Thompson,L.H., Cleaver,J.E. and Pedersen,R.A. (1999) Dev. Biol., 208, 513–529. [DOI] [PubMed] [Google Scholar]
- 3.Sobol R.W., Horton,J.K., Kuhn,R., Gu,H., Singhal,R.K., Prasad,R., Rajewsky,K. and Wilson,S.H. (1996) Nature, 379, 183–186. [DOI] [PubMed] [Google Scholar]
- 4.Thompson L.H. and West,M.G. (2000) Mutat. Res., 459, 1–18. [DOI] [PubMed] [Google Scholar]
- 5.Kubota Y., Nash,R.A., Klungland,A., Schar,P., Barnes,D.E. and Lindahl,T. (1996) EMBO J., 15, 6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 6.Caldecott K.W., Aoufouchi,S., Johnson,P. and Shall,S. (1996) Nucleic Acids Res., 24, 4387–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masson M., Niedergang,C., Schreiber,V., Muller,S., Menissier-de Murcia,J. and de Murcia,G. (1998) Mol. Cell. Biol., 18, 3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caldecott K.W., McKeown,C.K., Tucker,J.D., Ljungquist,S. and Thompson,L.H. (1994) Mol. Cell. Biol., 14, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marintchev A., Mullen,M.A., Maciejewski,M.W., Pan,B., Gryk,M.R. and Mullen,G.P. (1999) Nature Struct. Biol., 6, 884–893. [DOI] [PubMed] [Google Scholar]
- 10.Matsumoto Y. and Kim,K. (1995) Science, 269, 699–702. [DOI] [PubMed] [Google Scholar]
- 11.Mullen G.P. and Wilson,S.H. (1997) In Hickson,I.D. (ed.), Base Excision Repair of DNA Damage. Landes Bioscience, Austin, TX, pp. 121–135.
- 12.Singhal R.K. and Wilson,S.H. (1993) J. Biol. Chem., 268, 15906–15911. [PubMed] [Google Scholar]
- 13.Prasad R., Beard,W.A. and Wilson,S.H. (1994) J. Biol. Chem., 269, 18096–18101. [PubMed] [Google Scholar]
- 14.Caldecott K.W., Tucker,J.D., Stanker,L.H. and Thompson,L.H. (1995) Nucleic Acids Res., 23, 4836–4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cappelli E., Taylor,R., Cevasco,M., Abbondandolo,A., Caldecott,K. and Frosina,G. (1997) J. Biol. Chem., 272, 23970–23975. [DOI] [PubMed] [Google Scholar]
- 16.Kumar A., Widen,S.G., Williams,K.R., Kedar,P., Karpel,R.L. and Wilson,S.H. (1990) J. Biol. Chem., 265, 2124–2131. [PubMed] [Google Scholar]
- 17.Casas-Finet J.R., Kumar,A., Karpel,R.L. and Wilson,S.H. (1992) Biochemistry, 31, 10272–10280. [DOI] [PubMed] [Google Scholar]
- 18.Beard W.A. and Wilson,S.H. (1995) Methods Enzymol., 262, 98–107. [DOI] [PubMed] [Google Scholar]
- 19.Sawaya M.R., Pelletier,H., Kumar,A., Wilson,S.H. and Kraut,J. (1994) Science, 264, 1930–1935. [DOI] [PubMed] [Google Scholar]
- 20.Liu D., DeRose,E.F., Prasad,R., Wilson,S.H. and Mullen,G.P. (1994) Biochemistry, 33, 9537–9545. [DOI] [PubMed] [Google Scholar]
- 21.Wilson S.H. (1998) Mutat. Res., 407, 203–215. [DOI] [PubMed] [Google Scholar]
- 22.Kim K., Biade,S. and Matsumoto,Y. (1998) J. Biol. Chem., 273, 8842–8848. [DOI] [PubMed] [Google Scholar]
- 23.Klungland A. and Lindahl,T. (1997) EMBO J., 16, 3341–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dianov G.L., Prasad,R., Wilson,S.H. and Bohr,V.A. (1999) J. Biol. Chem., 274, 13741–13743. [DOI] [PubMed] [Google Scholar]
- 25.Horton J.K., Prasad,R., Hou,E. and Wilson,S.H. (2000) J. Biol. Chem., 275, 2211–2218. [DOI] [PubMed] [Google Scholar]
- 26.Althaus F.R. (1997) In Hickson,I.D. (ed.), Base Excision Repair of DNA Damage. Landes Bioscience, Austin, TX, pp. 169–181.
- 27.Gradwohl G., Menissier de Murcia,J., Molinete,M., Simonin,F. and de Murcia,G. (1989) Nucleic Acids Res., 17, 7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le Cam E., Fack,F., Menissier-de Murcia,J., Cognet,J.A., Barbin,A., Sarantoglou,V., Revet,B., Delain,E. and de Murcia,G. (1994) J. Mol. Biol., 235, 1062–1071. [DOI] [PubMed] [Google Scholar]
- 29.Wilson D.M. III and Thompson,L.H. (1997) Proc. Natl Acad. Sci. USA, 94, 12754–12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trucco C., Rolli,V., Oliver,F.J., Flatter,E., Masson,M., Dantzer,F., Niedergang,C., Dutrillaux,B., Menissier-de Murcia,J. and de Murcia,G. (1999) Mol. Cell. Biochem., 193, 53–60. [PubMed] [Google Scholar]
- 31.Zhang X., Morera,S., Bates,P.A., Whitehead,P.C., Coffer,A.I., Hainbucher,K., Nash,R.A., Sternberg,M.J., Lindahl,T. and Freemont,P.S. (1998) EMBO J., 17, 6404–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bork P., Hofmann,K., Bucher,P., Neuwald,A.F., Altschul,S.F. and Koonin,E.V. (1997) FASEB J., 11, 68–76. [PubMed] [Google Scholar]
- 33.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nash R.A., Caldecott,K.W., Barnes,D.E. and Lindahl,T. (1997) Biochemistry, 36, 5207–5211. [DOI] [PubMed] [Google Scholar]
- 35.Huang X. (1994) Comput. Appl. Biosci., 10, 227–235. [DOI] [PubMed] [Google Scholar]
- 36.Rost B. and Sander,C. (1993) J. Mol. Biol., 232, 584–599. [DOI] [PubMed] [Google Scholar]
- 37.Rost B. and Sander,C. (1994) Proteins, 19, 55–72. [DOI] [PubMed] [Google Scholar]
- 38.Liu T., Liu,D., DeRose,E.F. and Mullen,G.P. (1993) J. Biol. Chem., 268, 16309–16315. [PubMed] [Google Scholar]
- 39.Dimitriadis E.K., Prasad,R., Vaske,M.K., Chen,L., Tomkinson,A.E., Lewis,M.S. and Wilson,S.H. (1998) J. Biol. Chem., 273, 20540–20550. [DOI] [PubMed] [Google Scholar]
- 40.Brookman K.W., Tebbs,R.S., Allen,S.A., Tucker,J.D., Swiger,R.R., Lamerdin,J.E., Carrano,A.V. and Thompson,L.H. (1994) Genomics, 22, 180–188. [DOI] [PubMed] [Google Scholar]
- 41.Shen M.R., Zdzienicka,M.Z., Mohrenweiser,H., Thompson,L.H. and Thelen,M.P. (1998) Nucleic Acids Res., 26, 1032–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor R.M. (1998) EMBL accession no. CAB45234.
- 43.Brodsky M.H., Rubin,G.M. and Tsang,G. (1999) GenBank accession no. AAD33589.
- 44.Shen M.R., Jones,I.M. and Mohrenweiser,H. (1998) Cancer Res., 58, 604–608. [PubMed] [Google Scholar]
- 45.Beard W.A., Osheroff,W.P., Prasad,R., Sawaya,M.R., Jaju,M., Wood,T.G., Kraut,J., Kunkel,T.A. and Wilson,S.H. (1996) J. Biol. Chem., 271, 12141–12144. [DOI] [PubMed] [Google Scholar]
- 46.Prasad R., Singhal,R.K., Srivastava,D.K., Molina,J.T., Tomkinson,A.E. and Wilson,S.H. (1996) J. Biol. Chem., 271, 16000–16007. [DOI] [PubMed] [Google Scholar]
- 47.Bennet R.A.O., Wilson,D.M.,III, Wong,D. and Demple,B. (1997) Proc. Natl Acad. Sci. USA, 94, 7166–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mol C.D., Izumi,T., Mitra,S. and Tainer,J.A. (2000) Nature, 403, 451–456. [DOI] [PubMed] [Google Scholar]
- 49.Wilson S.H. and Kunkel,T.A. (2000) Nature Struct. Biol., 7, 176–178. [DOI] [PubMed] [Google Scholar]
- 50.Sawaya M.R., Prasad,R., Wilson,S.H., Kraut,J. and Pelletier,H. (1997) Biochemistry, 36, 11205–11215. [DOI] [PubMed] [Google Scholar]