

ABSTRACT
Nascent nucleocapsids of herpesviruses acquire a primary envelope during their nuclear export by budding through the inner nuclear membrane into the perinuclear space between the inner and outer nuclear membranes. This process is mediated by a conserved viral heterodimeric complex designated the nuclear egress complex, which consists of the nuclear matrix protein and the nuclear membrane protein. In addition to its essential roles during nuclear egress, the nuclear matrix protein has been shown to interact with intracellular signaling pathway molecules including NF-κB and IFN-β to affect viral or cellular gene expression. The human herpesvirus 6A (HHV-6A) U37 gene encodes a nuclear matrix protein, the role of which has not been analyzed. Here, we show that HHV-6A U37 activates the heat shock element promoter and induces the accumulation of the molecular chaperone Hsp90. Mechanistically, HHV-6A U37 interacts with heat shock transcription factor 1 (HSF1) and induces its phosphorylation at Ser-326. We report that pharmacological inhibition of HSF1, Hsp70, or Hsp90 decreases viral protein accumulation and viral replication. Taken together, our results lead us to propose a model in which HHV-6A U37 activates the heat shock response to support viral gene expression and replication.
IMPORTANCE
Human herpesvirus 6A (HHV-6A) is a dsDNA virus belonging to the Roseolovirus genus within the Betaherpesvirinae subfamily. It is frequently found in patients with neuroinflammatory disease, although its pathogenetic role, if any, awaits elucidation. The heat shock response is important for cell survival under stressful conditions that disrupt homeostasis. Our results indicate that HHV-6A U37 activates the heat shock element promoter and leads to the accumulation of heat shock proteins. Next, we show that the heat shock response is important for viral replication. Overall, our findings provide new insights into the function of HHV-6A U37 in host cell signaling and identify potential cellular targets involved in HHV-6A pathogenesis and replication.
KEYWORDS: herpesviruses, HHV-6, heat shock response, HSP, nuclear egress
INTRODUCTION
There are nine species of human herpesviruses which cause life-long persistent infection. Herpesviruses replicate their genomes and package them into capsids within the host cell nucleus (1 – 3). These capsids must translocate from the nucleus to the cytoplasm through a process designated nuclear egress. In brief, the nascent nucleocapsids bud at the inner nuclear membrane (INM) to form primary virions in the perinuclear space, after which their envelopes fuse with the outer nuclear membrane (ONM), releasing the nucleocapsids into the cytoplasm where they mature further (1 – 3).
The viral-encoded nuclear egress complex (NEC) is composed of two viral proteins and plays a crucial role in the nucleocytoplasmic transport of newly-assembled nucleocapsids (1 – 3). In the case of herpes simplex virus 1 (HSV-1), these proteins are designated UL31 and UL34, while in the case of human cytomegalovirus (HCMV), they are designated UL50 and UL53. The latter is a membrane protein targeted to both the INM and the ONM (4, 5), whereas the former is a nuclear matrix protein that is held in close apposition to the inner and outer surfaces of the INM and ONM through its interaction with UL34 (4, 5). Conversely, INM localization of UL34 is enhanced in the presence of UL31 (4). Crystal structures of NEC from HSV-1 and the other Herpesviridae reveal the highly conserved features of this complex (6 – 13) and their NECs are considered to have an intrinsic ability to vesiculate membranes through formation of hexagonal lattices (12, 14, 15). Furthermore, HSV-1 NECs recruit capsids into vesicles (16 – 19) and mediate scission of the INM to finalize envelopment through host scission ESCRT-III machinery (20 – 23).
In addition to its role in nuclear egress, the nuclear matrix protein UL31 affects intracellular signaling. First, HSV-1 UL31 can activate the NF-κB signaling pathway to promote the expression of viral proteins (24). Second, herpes simplex virus 2 (HSV-2) UL31 can be recruited to sites of DNA damage through poly (ADP-ribose) (PAR), although the details have not been fully elucidated (25). Finally, HSV-1 UL31 blocks interferon (IFN)-β production via its interaction with signaling molecules (26). These reports suggest that UL31 and its homolog proteins may possibly contribute to the regulation of viral and/or host gene expression prior to their essential role in nuclear egress. However, whether these functions of UL31 homologs are conserved among Herpesviridae has not been elucidated.
Human herpesvirus 6A (HHV-6A) is a ubiquitous virus belonging to the Roseolovirus genus within the Betaherpesvirinae subfamily (27 – 31). HHV-6A is frequently found in patients with neuroinflammatory diseases such as multiple sclerosis and Hashimoto’s thyroiditis, although whether primary infection with HHV-6A has a causal role in these or other illnesses has not yet been determined (32, 33). The HHV-6A homologs of UL31 and UL34 are designated U37 and U34, respectively; their roles have not been well-studied. Although the structures of the NEC are conserved in the members of all subfamilies of Herpesviridae, the amino acid sequences of the UL31 homologs are diverse and are clustered into individual subfamilies and genera (Fig. 1). The amino acid sequence at the N-terminal region of the UL31 homologs is poorly conserved compared to the region of UL31 homologs which form heterodimers with UL34 homologs (Fig. 1B).
Fig 1.

Conservation of nuclear matrix proteins in Herpesviridae. (A) Amino acid sequences of nuclear matrix proteins from nine human herpesviruses were aligned using multiple-sequence comparisons, and phylogeny was constructed using a neighbor-joining tree without distance corrections and scaled for equal branch lengths (Genetycs ver. 2.2.5). Shaded boxes indicate herpesvirus subfamilies, which group closely to established phylogenetic trees. The dotted box indicates genus Roseolovirus. HSV-1 UL31 (GenBank accession no. GU734771.1), HSV-2 UL31 (GenBank accession no. Z86099.2), VZV ORF27 (GenBank accession no. CAA27910.1), HCMV UL53 (GenBank accession no. NC_006273.2), HHV-6A U37 (GenBank accession no. NC_001664.4), HHV-6B U37 (GenBank accession no. AB021506.1), HHV-7 U37 (GenBank accession no. AAC40751.1), EBV BFLF2 (GenBank accession no. AJ507799.2 https://www.ncbi.nlm.nih.gov/nuccore/AJ507799.2) and KSHV ORF69 (GenBank accession no. AF148805.2). (B) Alignment of amino acid sequences of HHV-6A U37 homologs in members of three subfamilies of the Herpesviridae. The residues conserved in at least two herpesviruses are shaded. Secondary structural elements determined in HSV-1 UL31 (PDB: 4ZXS) are shown as blocks for α-helices and arrows for β-sheets. The region of HSV-1 UL31 (PDB: 4ZXS) which is the solved structure of a UL31/UL34 complex is indicated by the dashed line.
Here, we focus on the role of the HHV-6A nuclear matrix protein U37 in intracellular signaling. The heat shock element (HSE) promoter is activated in cells expressing exogenous HHV-6A U37. We demonstrate that heat shock transcriptional factor 1 (HSF1) is phosphorylated and responsible for the activation of the HSE in the presence of HHV-6A U37. This depends on the N-terminal region of HHV-6A U37. Consistent with this, heat shock proteins (HSPs) were up-regulated in HHV-6A-infected cells and viral replication was impaired by inhibitors of HSF1 or HSPs. Collectively, the results of our study reveal an additional role of HHV-6A U37 for intracellular signaling and highlight the importance of molecular chaperones during HHV-6A infection.
RESULTS
HHV-6A U37 activates the HSE promoter
Previous work had revealed that UL31 of HSV-1 and HSV-2 can modulate intracellular signaling pathways (24 – 26). Despite the conserved structures of the NEC among herpesviruses, the amino acid sequences of the UL31 homologs are diverse (Fig. 1). We thus determined whether U37, the nuclear matrix protein encoded in the HHV-6A genome, would elicit similar effects. HEK293T cells were co-transfected with Flag-tagged HHV-6A U37 (Flag-HHV-6A U37) and a series of firefly luciferase reporter plasmids harboring the IFN-βpromoter (IFN-β-luc), the NF-κB-responsive element (NF-κB-luc), the cAMP response element (cAMP-luc), or the heat shock element (HSE-luc). As shown in Fig. 2, ectopic expression of Flag-HHV-6A U37 did not have any effect on IFN-β-luc, NF-κB-luc, or cAMP-luc either in the presence or absence of the individual stimuli. However, ectopic expression of Flag-HHV-6A U37 significantly stimulated the activity of HSE-luc (Fig. 2A and B). Of note, heat shock increased the HSE-luc activity in the presence or absence of HHV-6A U37, suggesting that heat shock and HHV-6A U37 synergistically activate the HSE promoter.
Fig 2.

Exogenous expression of HHV-6A U37 activates the HSE promoter. (A and D) HEK293T cells were co-transfected with either IFN-β-luc (A), CRE-luc (B), NF-κB-luc (C), or HSE-luc (D) together with pRL-CMV as an internal control plasmid, along with either an empty plasmid (control) or plasmid expressing Flag-HHV-6A U37. For IFN-β promoter activation (A), mitochondrial antiviral signaling (MAVS) expression plasmid was co-transfected in addition. For CRE promoter activation (B) or NF-κB promoter activation (C), cells were treated with 10 µM colforsin or 10 µM TNF-α, respectively, at 8 hours post-transfection and incubated for another 12 hours. Luciferase activity was measured 24 hours post-transfection. For HSE promoter activation (D), at 18 hours post-transfection, cells were left untreated, or exposed to heat shock at 43°C for 30 minutes followed by 5 hours recovery before measuring the luciferase activity. (E) HEK293T cells were transfected with Flag-HHV-6A U37 expression plasmid or empty plasmid (control). At 24 hours post-transfection, the cells were analyzed by immunoblotting. Data are shown as means of three independent experiments ± standard error (*P < 0.05, unpaired Student’s t test).
In general, exposure to 5%–10°C above the optimal growth temperature leads to a heat shock response which is characterized by activated transcription of HSP genes orchestrated by a family of transcription factors designated heat shock factors (HSFs). The heat shock response causes the elevation of HSPs, such as Hsp27, Hsp40, Hsp70, and Hsp90, most of which are molecular chaperones that function to prevent protein misfolding and aggregation (34, 35). In infected cells, HSPs are known to be important for the precise function of viral proteins (36). To investigate whether HHV-6A U37 increases expression of HSPs, HEK293T cells were transfected with the Flag-HHV-6A U37 expression plasmid. As shown in Fig. 3, Flag-HHV-6A U37 localized to the nucleus in a manner similar to its homolog proteins reported earlier (4, 7). Flag-HHV-6A U37 co-localized with Hsp90 in the nucleus. Furthermore, accumulation of Hsp90 was significantly increased in Flag-HHV-6A U37-expressing cells compared to the mock transfected cells. These results suggest that HHV-6A specifically activates the cellular heat shock response system.
Fig 3.

Exogenous expression of HHV-6A U37 leads to accumulation of Hsp90. (A) HEK293T cells were transfected with Flag-HHV-6A U37 expression plasmid or empty plasmid (control). At 48 hours post-transfection, cells were fixed and observed by confocal microscopy. Bars, 10 µm. (B) The mean fluorescence intensity (MFI) of Hsp90 in the cells (>6 cells per sample) described in (A) was quantified. Data are shown as means of three independent experiments ± standard error (P < 0.05, unpaired Student’s t test). The mean value in the control cells was normalized to 100% relative MFI.
HSE is not activated by HHV-6A U34/U37 complexes or by HCMV UL53
The homolog proteins of HHV-6A U37 in other herpesviruses (such as HSV-1 UL31 or HCMV UL53) form heterodimers with the other NEC components such as HSV-1 UL34 or HCMV UL50 to localize at the nuclear rim in infected cells [although a fraction of HSV-1 UL31 or HCMV UL53 can be detected in the nucleus possibly as monomers (37 – 40)]. We next analyzed the effect of HHV-6A U37 in the presence of HHV-6A U34, the homolog of HSV UL34, and HCMV UL50. HEK293T cells were transfected with the expression plasmids for Flag-HHV-6A U37 or HHV-6A U34-Strep, or the co-expression plasmid for HHV-6A U34-Strep/Flag-HHV-6A U37. The HHV-6A U34-Strep/Flag-U37 co-expression plasmid encodes the fusion protein of HHV-6A U34-Strep, P2A self-cleaving peptides, and HHV-6A Flag-U37 to produce U34-Strep-P2A and Flag-U37 separately. At 48 hours after transfection, the cells were analyzed by immunoblotting and fluorescence assays. As expected, Flag-U37 and/or U34-Strep was detected in the cells transfected with these plasmids (Fig. 4A). Furthermore, Flag-HHV-6A U37 relocalized to the nuclear rim in the presence of HHV-6A U34-Strep. Of note, HHV-6A U34-Strep was detected as cytoplasmic membranous structures but relocalized to the nuclear rim in the presence of Flag-HHV-6A U37 (Fig. 4B). However, expression of Flag-HHV-6A U37 did not stimulate HSE-luc in the presence of HHV-6A U34-Strep (Fig. 4C). Because structural analysis of the NECs from HSV-1, Epstein-Bar virus (EBV), and HCMV had revealed that they form a hexagonal lattice through inter-molecular interactions (6, 12, 13, 15), the element in HHV-6A U37 responsible for HSE stimulation was possibly masked after formation of the NEC.
Fig 4.

HHV-6A U37 does not activate HSE in the presence of U34. (A and B) HEK293T cells were transfected with HHV-6A U34-Strep or Flag-HHV-6A U37 expression plasmids, or HHV-6A U34-Strep/Flag-HHV-6A U37 co-expression plasmids. At 24 hours post-transfection, cells were collected and analyzed by immunoblotting (A) or fixed and visualized by confocal microscopy (B). Bars, 10 µm. (C) HEK293T cells were co-transfected with HSE-luc together with pRL-CMV as an internal control plasmid, along with either an empty plasmid or plasmid expressing HHV-6A U34-Strep, Flag-HHV-6A U37, or Strep-HHV-6A U34-Flag-HHV-6A U37. Luciferase activity was measured 24 hours post-transfection. Data are shown as means of three independent experiments ± standard error (**P < 0.01, Tukey’s test).
To investigate the region in HHV-6A U37 responsible for HSE stimulation, we constructed a series of truncated HHV-6A U37s (Fig. 5A) and analyzed their effects on HSE-luc. As shown in Fig. 5B and C, U37 1-200 and U37 1–150, in which the C-terminal region of HHV-6A U37 was deleted, still enhanced HSE-luc to similar levels as wild-type U37. In contrast, HHV-6A U37 90–264, in which the N-terminal region of HHV-6A U37 was deleted, was expressed properly but failed to activate the HSE (Fig. 5B and C). These results indicate that the N-terminal region of HHV-6A U37 is important for activation of the HSE.
Fig 5.

N-terminal of HHV-6A U37 is important for HSE promoter activation. (A) Schematic representation of Flag-HHV-6A U37 with different deletions. Dotted lines indicate deleted domains. (B) HEK293T cells were co-transfected with HSE-luc together with pRL-CMV as an internal control plasmid, along with either an empty plasmid or plasmid expressing different truncated versions of Flag-HHV-6A U37. Luciferase activity was measured 24 hours post-transfection. Fold-changes of luciferase activity relative to control are shown as means of six independent experiments with standard error (***P < 0.001, Tukey’s test). (C) HEK293T cells were transfected with the series of Flag-HHV-6A U37 expression plasmids. At 24 hours post-transfection, cells were analyzed by immunoblotting.
Next, we aimed to determine whether the effect of HHV-6A U37 is conserved among betaherpesviruses. As shown in Fig. 6, ectopic expression of HCMV UL53, which is the homolog of HHV-6A UL37, did not increase HSE-luc activity in the presence or absence of the other NEC component HCMV UL53. These results indicate that the ability of HHV-6A U37 to stimulate the HSE is not conserved in the other herpesviruses.
Fig 6.
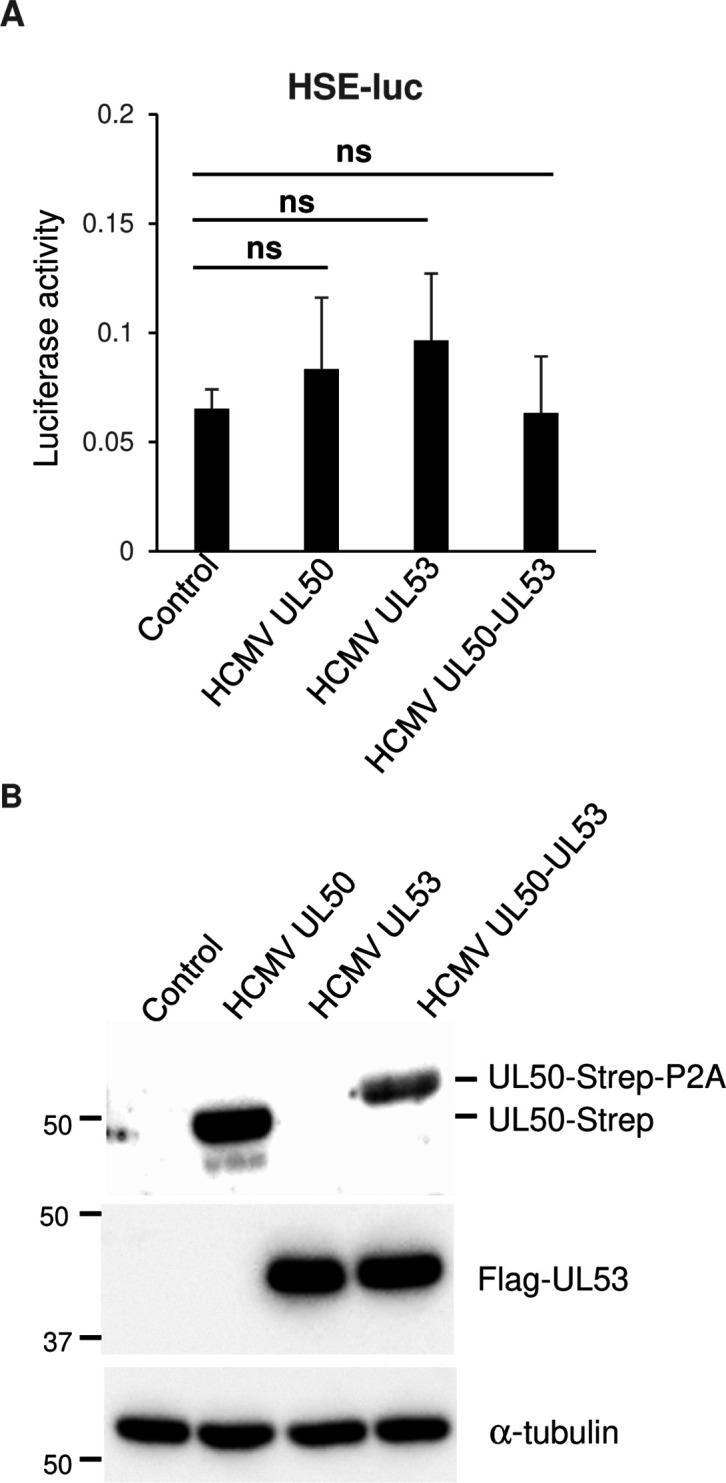
HCMV UL53 has no effect on HSE. (A) HEK293T cells were co-transfected with HSE-luc together with pRL-CMV as an internal control plasmid, along with either an empty plasmid or plasmid expressing HCMV UL50-Strep, Flag-HCMV UL53, or HCMV UL50-Strep-Flag-HCMV UL53. Luciferase activity was measured 24 hours post-transfection. The data are shown as means of six independent experiments and standard error (Tukey’s test). (B) HEK293T cells were transfected with HCMV UL50-Strep, Flag-HCMV UL53, or HCMV UL50-Strep-Flag-HCMV UL53 expression plasmid. At 24 hours post-transfection, cells were analyzed by immunoblotting.
The role of HSF1 in activation of the HSE promoter by HHV-6A U37
HSF1 is the best-studied member of the HSF family. Under unstressed conditions, HSF1 exists as a monomer and its transcriptional activity is suppressed. When cells are stressed, HSF1 undergoes several activating post-translational modifications and forms a transcriptionally active trimer that accumulates in the nucleus and binds to HSEs that are found in the upstream regulatory regions of the HSP genes (41). Among these modifications, stress-inducible phosphorylation of Ser-326 (S326) in the regulatory domain contributes to the transactivation function of HSF1 and is used as a surrogate marker for its activation. To investigate the mechanism of HSE activation by HHV-6A U37, we analyzed the role of HSF1. To this end, first, HEK293T cells were transfected with the expression plasmid for Flag-HSF1 in the presence or absence of plasmids expressing Strep-HHV-6A U37. The phosphorylation of HSF1 at S326 was enhanced in cells transfected with the Strep-HHV-6A U37 expression plasmid compared to control, whereas the accumulation of Flag-HSF1 was not affected (Fig. 7A and B). This finding suggests that HHV-6A U37 mediates activation of HSF1. Unexpectedly, Strep-HHV-6A U37 was reduced in cells expressing HSF1 but could be stabilized in the presence of MG132 (Fig. 7A). This might be because HHV-6A U37 is degraded through the proteasomal system in HSF1-expressing cells by some unidentified mechanism, as reflected by the finding that treatment with the proteasome inhibitor MG132 enhanced the accumulation of Strep-HHV-6A U37 in these cells (Fig. 7C).
Fig 7.

HHV-6A U37 increases HSF1 phosphorylation. (A) HEK293T cells were co-transfected with Flag-HSF1 either with a control empty plasmid or Strep-HHV-6A U37 expression plasmid in the presence or absence of 100 nM MG132 for 16 hours. At 24 hours post-transfection, cells were analyzed by immunoblotting. (B and C) The intensity of HSF1 pS326 in each lane normalized against Flag-HSF1 (B) or the intensity of Strep-U37 in each lane normalized against α-tubulin (C) is shown as the mean of six independent experiments with standard error (*P < 0.05; **P < 0.01, unpaired Student’s t test).
Second, fluorescence assays were performed to investigate whether the two proteins co-localized in the cells. HEK293T cells were transfected with the Flag-HSF1 expression plasmid in the presence or absence of the Strep-HHV-6A U37 expression plasmid, and then incubated with medium containing MG132. As expected, Flag-HSF1 co-localized with Strep-HHV-6A U37 in the nucleus (Fig. 8A).
Fig 8.

HHV-6A U37 interacts with HSF1. (A) HEK293T cells were co-transfected with Flag-HSF1 expression plasmid in the presence or absence of the Strep-HHV-6A U37 expression plasmid. At 8 hours post-transfection, the cells were treated with 100 nM MG132. At 24 hours post-transfection, cells were fixed and visualized by confocal microscopy. Bars, 10 µm. (B) HEK293T cells were co-transfected with the indicated combination of NanoBit plasmids. At 24 hours post-transfection, luminescence was detected. Data are shown as means of three independent experiments with standard error (*P < 0.05, unpaired Student’s t test).
Third, interactions between HHV-6A U37 and HSF1 were investigated by the NanoBiT assay, which enables measurement of protein-protein interactions in cells. In this assay, the luciferase is split into two subunits, LgBiT and SmBiT, which do not fluoresce when separated, but luciferase enzyme activity is reconstituted when they are in close proximity. By fusing two proteins with LgBiT and SmBiT, respectively, interactions between them brings the two BiT subunits together and leads to luminescence signals. Because interactions between U37 and U34 homologs or HSF1 and Hsp90 are well characterized (4, 42, 43), we used these combinations as positive controls. Sm-BiT fused to HaloTag (SmBit-HALO), which is unlikely to interact with HHV-6A U37, was used as a negative control. As expected, co-expression of HHV-6A U37-LgBit and HHV-6A U34-SmBit or Hsp90-LgBit and HSF1-SmBit led to luminescence enhancement (Fig. 8B). As shown in Fig. 8B, co-expression of HHV-6A U37-LgBit and HSF1-SmBit significantly increased luminescence compared to HHV-6A U37-LgBit and SmBit-HALO, suggesting that HHV-6A U37 interacts with HSF1.
Because HHV-6A U37 interacts with and activates HSF1, we analyzed the role of HSF1 in HHV-6A U37-mediated HSE stimulation. KRIBB11 is a well-characterized inhibitor of HSF1 through blocking the HSF1-dependent recruitment of positive transcription elongation factor b (p-TEFb) (44). We confirmed that KRIBB11 had no effect on HEK293T cell proliferation (Fig. 9A), but as shown in Fig. 9B, it impaired HHV-6A U37-dependent stimulation of HSE-luc activity in the presence or absence of heat shock. These observations suggest that HHV-6A U37 specifically stimulates the HSE via the transcription factor HSF1. Taken together, this series of observations suggests that HHV-6A U37 interacts with and activates HSF1 to increase the expression of HSPs.
Fig 9.

KRIBB11 inhibits HSE promoter activation by HHV-6A U37. (A) HEK 293T cells were treated with DMSO or 10 µM KRIBB11. At 18 hours post-treatment, cell viability was measured. Data are shown as the means and standard errors of five independent experiments and are expressed relative to the mean for DMSO-treated cells, which was normalized to 100% (ns, not significant, unpaired Student’s t test). (B) HEK293T cells were co-transfected with HSE-luc together with pRL-CMV as an internal control plasmid, along with either an empty plasmid or plasmid expressing Flag-HHV-6A U37. At 6 hours post-transfection, cells were treated with DMSO or 10 µM KRIBB11, followed by incubation for another 16 hours. For HSE promoter activation, the cells were incubated at 43°C for 30 minutes followed by 5 hours recovering before the luciferase activity was measured. The data are shown as means of five independent experiments and standard error (**P < 0.01, unpaired Student’s t test).
The role of heat shock signaling in HHV-6A replication
Next, we focused on whether heat shock signaling also takes place in HHV-6A-infected cells. The HHV-6A permissive CD4+ T cell line, JJhan, was either mock-infected or infected with HHV-6A U1102, and HSP expression was then analyzed. First, we investigated the accumulation of HSE-regulated proteins including Hsp90 in mock- and HHV-6A-infected cells but failed to detect differences between samples by immunoblotting. When JJhan cells were infected with HHV-6A, some of them produced viral proteins, while others did not express viral proteins. Thus, we analyzed Hsp90 accumulation in the cells expressing HHV-6A protein by immunofluorescence to distinguish those expressing viral protein. JJhan cells were mock-infected or infected with HHV-6A U1102 for 72 hours and then analyzed. Viral tegument protein U14 was detected in about 40% of the infected cells but not in mock-infected cells (Fig. 10A). Accumulation of Hsp90 was significantly enhanced in the U14-expressing cells compared to the uninfected cells, whereas tubulin was not (Fig. 10A and B). Of note, accumulation of Hsp90 was not enhanced in U14-negative HHV-6A infected cells, indicating that the presence of cell debris in viral stock was unlikely to be causing this enhancement. These results suggest that HHV-6A infection results in the accumulation of HSPs, similar to what is reported for other herpesviruses (45 – 47).
Fig 10.

HHV-6A infection results in accumulation of Hsp90. (A) JJhan cells were mock-infected or infected with HHV-6A U1102. At 72 hours post-infection, the cells were fixed and observed by confocal microscopy. Bars, 20 µm. (B) The mean fluorescence intensity (MFI) of Hsp90 or α-tubulin (>6 cells per sample) was quantified. Data are shown as means of three independent experiments ± standard error (*P < 0.05, unpaired Student’s t test). The mean value in the mock-infected cells was normalized to 100% relative MFI.
To investigate the role of HHV-6 U37 in infected cells, we first attempted to construct a U37-deletion mutant using bacterial artificial chromosome (BAC) mutagenesis. We generated HHV-6A-ΔU37 BAC harboring the deletion in U37 and HHV-6A-ΔU37-repair BAC, in which the U37 deletion in HHV-6A-ΔU37 BAC was repaired (Fig. 11A). JJhan cells were transfected with these BAC DNAs and incubated for a few days after which the transfected cells were co-cultured with CBMCs for 3–5 days. At that time, an increase in the number of cells expressing green fluorescent protein (GFP) and cytopathic effects (CPEs) were observed in cells transfected with wild-type HHV-6A BAC or HHV-6A-ΔU37-repair BAC but not with HHV-6A-ΔU37 BAC (Fig. 11B). These results suggest that HHV-6A U37 is essential for viral replication, similar to HCMV nuclear matrix protein UL53 (48, 49). Thus, we could not further analyze the role of U37 for HSP accumulation in infected cells.
Fig 11.

HHV-6A-ΔU37 could not be reconstituted from BAC DNA. (A) Schematic diagrams of the genome structure of wild-type HHV-6A and the relevant domains of the recombinant HHV-6A BAC used in this study. Line 1, HHV-6A BAC (wild-type) genome; line 2, domain of the U36 to the U38 gene; line 3, domains of the U37 gene; line 4 and 5, recombinant HHV-6A BAC with mutations in U37. (B) JJhan cells were transfected with BAC DNA of HHV-6A, HHV-6A-ΔU37 or HHV-6A-ΔU37-repair. The transfected cells were co-cultured with CBMCs 4–6 days post-transfection, and viruses were propagated. GFP fluorescence images and light microscopy images of each type of infected cell are shown. Representative images of three independent experiments are shown. Scale bar indicates 100 µm.
Finally, because HSPs accumulated in HHV-6A-infected cells, we investigated whether HSE activation and HSP accumulation enhanced viral gene expression and replication. JJhan cells were mock-infected or infected with HHV-6A U1102 for 24 hours and then left untreated, or treated with the HSF1 inhibitor KRIBB11, Hsp90 inhibitor 17-AAG (50) or Hsp70 inhibitor VER-155008 (51). The cells were then incubated for a further 48 hours to allow HHV-6A propagation before analysis. As shown in Fig. 12A, KRIBB11, 17-AAG or VER-155008 treatment reduced the accumulation of viral proteins U14 and gQ1, and also viral yields (Fig. 12A and B ). KRIBB11 or 17-AAG treatment had minor effects on the proliferation of JJhan cells, whereas VER-155008 treatment slightly reduced it (Fig. 12C). Taken together, these results suggest that heat shock signaling is important for viral gene expression and viral growth in JJhan cells.
Fig 12.
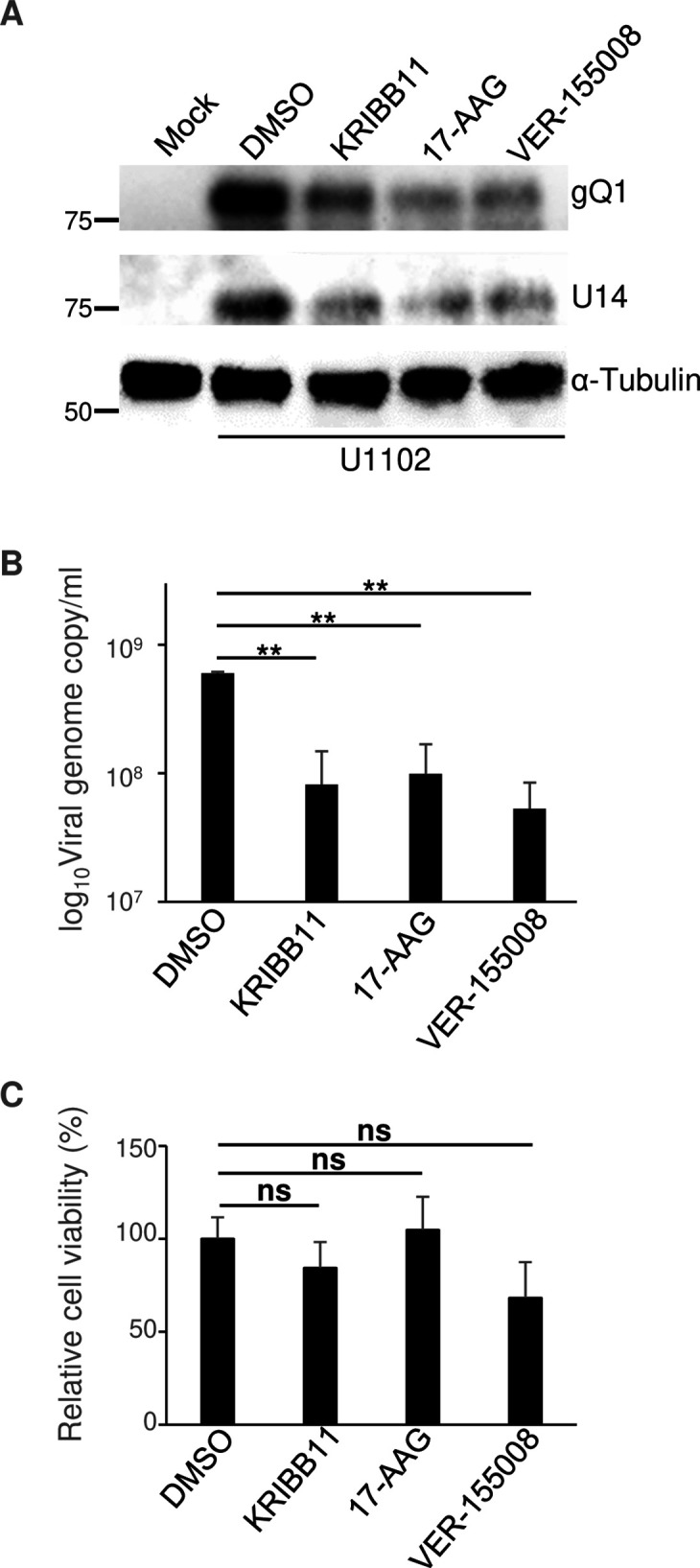
Inhibitors of the heat shock response impair HHV-6A replication. (A and B) JJhan cells were mock-infected or infected with HHV-6A U1102 and were then treated with DMSO, 10 µM KRIBB11, 10 µM 17-AAG, or 10 µM VER-155008 6 hours after infection. At 72 hours post-infection, cells were analyzed using immunoblotting (A) with the indicated antibodies, while genome copy numbers were measured using qPCR (B) following DNA extraction from the supernatant of the infected cells. The data are shown as means of three independent experiments and standard error (**P < 0.01, Tukey’s test). (C) JJhan cells were treated with DMSO, 10 µM KRIBB11, 10 µM 17-AAG, or 10 µM VER-155008. After 72 hours post-treatment, cell viability was measured. Data are shown as the means and standard errors of three independent experiments and are expressed relative to the mean for DMSO treated cells, which was normalized to 100% (ns, not significant, Tukey’s test).
DISCUSSION
The heat shock response is considered to be a cellular defense mechanism against the deleterious effects of physiological stress due to protein misfolding, degradation, and the accumulation of insoluble aggregates (41). These stress signals result in the activation of HSF1 and synthesis of HSPs, many of which function as molecular chaperones (41). Numerous reports have shown that up-regulation of the heat shock response occurs during herpesviral infection via viral protein expression (45 – 47). In the present study, we identified novel interactions between herpes viral proteins and the heat shock response. We demonstrated that HHV-6A nuclear matrix protein U37 activated the HSE through its interaction with HSF1. In the presence of HHV-6A U37, Hsp90 accumulated. Furthermore, we demonstrated that HSF1 and HSPs were important for HHV-6A gene expression and viral replication.
Our results emphasize the importance of heat shock signals in the viral life cycle. Accumulation of HSPs in the nucleus was observed in cells infected with HSV-1 or HCMV (46, 47, 52) and HSP inhibitors impaired viral replication (47, 53 – 58). Because viral DNA polymerase is a client of Hsp90 (47), the accumulation of HSPs is considered to support viral genome replication. In agreement with these observations, we showed that inhibitors of HSF1, Hsp70, or Hsp90 impaired the replication of HHV-6A. Thus, the importance of the heat shock response is highly conserved in viruses belong to Herpesviridae. However, enhancement of the HSE promoter by HCMV UL53 was not observed, even though the role of NEC in nucleocytoplasmic transport of viral capsids is conserved in all subfamilies of the Herpesviridae. Similarly, HHV-6A U37 did not impair the IFN-β cascade, which is blocked by HSV-1 UL31. The region in HHV-6A U37 responsible for HSE activation mapped to its N-terminus and because this region in U37 homologs is less conserved, this might be responsible for these differences. In contrast, the amino acid sequences of HHV-6A U37 and HHV-6B U37 are very similar, with a sequence identity and similarity score of 97% and 100%, respectively. Based on this, we believe that HHV-6A U37 and HHV-6B U37 would both have similar properties. The unexpected diversity of NEC functions is an interesting issue for understanding the differences between the nine human herpesviruses.
Enhancement of the HSE in HHV-6A U37-expressing cells could be mediated by abnormal aggregation of U37 because HSV-1 UL31 or UL34 are unstable without each other (14). However, deletion of the N-terminus of HHV-6A U37 abolished this enhancement, suggesting a specific role of HHV-6A U37 in the heat shock response. Furthermore, interactions between HHV-6A U37 and HSF1 were detected by NanoBiT assays, and we therefore concluded that HHV-6A U37 specifically activates HSF1 to enhance the HSE in HSP promoters. In the present study, we could not show the importance of U37 in HHV-6A-infected cells, as U37 homologs in betaherpesviruses are concluded to be essential for viral replication (48, 49).
In addition to the role of HHV-6A U37, other viral proteins possibly contribute to this activation in infected cells. Previous reports revealed that IE proteins mediate heat shock responses in HSV-1 and HCMV (46, 59). Because IE proteins are expressed immediately after infection and before the expression of viral DNA polymerase, HHV-6A IE proteins might activate the heat shock response. Interestingly, the HSE was not activated by HHV-6A U37 in the presence of HHV-6A U34. Thus, HHV-6A U37 possibly activates the heat shock response at an early time after infection but loses this ability after accumulation of HHV-6A U34 at the INM. This negative feedback might be important for evasion of host antiviral machinery mediated by heat shock response (60). However, both HHV-6A U34 and HHV-6A U37 are characterized as late proteins and are presumably expressed at the same time (61). Further study is required to determine how and when HHV-6A U37 activates the HSE in infected cells. Alternatively, proteins other than U37, expressed later after infection, possibly activate the heat shock response. In any case, heat shock signaling is regulated in infected cells, but the details have not been elucidated yet.
In addition to their role in regulating viral DNA polymerase, HSPs support various viral and cellular proteins which mediate viral entry, capsid transport or viral assembly (47, 53 – 58). Furthermore, the heat shock response is important for protecting cells from the stress caused by viral infection. Thus, the heat shock response is expected to be a therapeutic target for herpesvirus infection. Our findings provide important insights into novel interactions between herpesviruses and the heat shock response, and might suggest new targets for antiviral therapy for HHV-6A.
MATERIALS AND METHODS
Cells and viruses
HEK293T cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 8% fetal bovine serum (FBS). The human T-lymphoblastoid cell line, JJhan, was cultured in RPMI 1640 medium containing 8% FBS. Umbilical cord blood mononuclear cells (CBMCs) were cultured as described previously (62, 63). CBMCs were purchased from the Cell Bank of the RIKEN BioResource Center, Tsukuba, Japan and used to make viral stocks. The use of CBMCs in this study was approved by the Ethics Committee of Kobe University Graduate School of Medicine (approval No. 1209). The HHV-6A strain U1102 was used for this study, and HHV-6 cell-free virus was prepared from CBMCs as described previously (62, 63). Briefly, CBMCs infected with HHV-6A U1102 were lysed by freezing and thawing once at −80°C. Cell debris was removed by centrifugation at 1,500 g for 5 minutes and the supernatants were then used for virus infections. For infecting the target cell line JJhan with HHV-6A virus stock, 1 × 106 cells were collected by centrifugation and resuspended in 1.5 mL of RPMI medium with 200 µL of virus stock containing 1.6 × 108 genome copies of the virus or medium only as a mock control. The cells were then centrifuged at 35 °C for 30 minutes at 1,700 g in 24-well plates, followed by culturing in RPMI supplemented with 2% FBS. Transfection experiments were performed using Lipofectamine 3000 (Thermo Fisher Scientific) as previously described (64, 65). To inhibit proteasomal degradation, the cells were treated with 100 nM MG132 for 16 hours before collection.
Cell viability determination
Five thousand cells/well were cultured in 96-well plates with 100 µL of medium/well. The cells were treated with DMSO, 10 µM KRIBB11, 10 µM 17-AAG, or 10 µM VER-155008. Three hours prior to determining cell viability, 10 µL of Cell Counting Kit-8 solution (Dojindo) was added to each well. The amount of formazan dye generated by the dehydrogenase in living cells was determined by measuring the absorbance at 450 nm using a microplate reader, following the manufacturer’s instructions.
Plasmids
The coding sequences of HHV-6A U37 were codon-optimized, fused with Flag at the N-terminus, and cloned into pcDNA3.1+ (Thermo Fisher Scientific) to express Flag-HHV-6A U37. The coding sequences of HHV-6A U34 were codon-optimized, fused with Strep-tag at the C-terminus, and cloned into pcDNA3.1+ to express HHV-6A U34-strep. To construct the HHV-6A U34-Strep/Flag-HHV-6A U37 co-expression plasmid, the stop codon of HHV-6A U34-Strep was removed, fused to P2A sequence, and cloned into the N-terminus of the Flag-HHV-6A U37 coding sequence in its expression plasmid. The HHV-6A U34-Strep/Flag-HHV-6A U37 co-expression plasmid encodes the bipartite fusion protein of HHV-6A U34-Strep, P2A self-cleaving peptides, and HHV-6A Flag-U37 to produce U34-Strep-P2A and Flag-U37 separately. To express HCMV UL50 and HCMV UL53, their coding sequences were amplified by PCR from the genome of the HCMV Merlin strain and cloned into pcDNA3 (Thermo Fisher Scientific) similar to HHV-6A U34 and U37. Plasmid FLAG-HSF1 was a gift from Stuart Calderwood (Addgene plasmid # 32537) (66) and was used to express HSF1. For NanoBiT assays, the coding sequences of HHV-6A U34 or HSF1 were amplified by PCR from the plasmids described above and cloned into pTK-C-SmBiT (Promega). They are designated pTK-HHV-6A U34-SmBit or pTK-HSF1-SmBit, respectively. The coding sequences of HHV-6A U37 or cDNA of HSP90AA1 from CBMCs were cloned into pTK-C-LgBiT (Promega) and designated pTK-HHV-6A U37-LgBit or pTK-Hsp90-LgBit, respectively. The plasmid pBS-U3791-590-Kan was constructed as follows: part of the coding sequence of HHV-6A U37 (nucleotide position 91–590) was amplified by PCR and cloned into pBlueScript KS (+), and designated pBS-HHV-6A-U3791-590. The kanamycin resistance cassette, amplified from pEP-KanS (67) using the primers 5′-AACACACCTGCCTAGAATTCTCCCCCTACGCTAACGAACAAATCTCCAAAAGCGCCTGCCTAGGGATAACAGGGTAATCGATTT-3′ and 5′- GCGAATTCGCATATGCTAGCCAGTGTTACAACCAATTAACC-3′ was then cloned into the internal EcoRI site in pBS-HHV-6A U3791-590 and designated pBS-HHV-6A U3791-590-kan.
Antibodies
For immunoblotting and immunofluorescence analysis, we used mouse monoclonal antibodies against α-tubulin (DM1A; Sigma), Hsp90 (AC88, Abcam), FLAG (M2; Sigma), and Strep-tag II (4F1; MBL), and a rabbit monoclonal antibody against HSF1 pS326 (EP1713Y; Abcam). Monoclonal antibodies to HHV-6A U14 (AU14) and gQ1 (AgQ1-119) were produced and used as previously described (68, 69).
Luciferase assay
HEK293T cells were co-transfected with either a pGL4.32 (NF-κB-luc; Promega), p125-Luc [IFN-β-luc (70)], pGL4.41 (HSE-luc; Promega), or pGL4.29 (CRE-luc; Promega) reporter plasmid along with an empty plasmid or plasmid carrying Flag-HHV-6A U37. As an internal control, pRL-CMV (Promega) containing the Renilla luciferase gene driven by the cytomegalovirus (CMV) promoter was co-transfected into the target cells. At 24 hours post-transfection, firefly and Renilla luciferase activities were independently assayed using the dual-luciferase reporter assay system (Promega). The luciferase activity (Fluc/Rluc) was calculated as (firefly luciferase activity)/(Renilla luciferase activity) (62, 65). To analyze the effect of different stimuli, transfected cells were untreated or treated with 10 ng TNF-α or 10 µM colforsin as an inducer of NF-κB and CRE, respectively, 6 hours after transfection, followed by incubation for a further 18 hours. For IFN-β-luc, the MAVS expression plasmid was co-transfected with the reporter plasmids. For HSE-luc, the transfected cells were incubated at 43°C for 30 minutes 18 hours post-transfection, followed by incubation at 37°C for 5 hours before measuring the luciferase activity. To inhibit HSF1, the transfected cells were treated with 10 µM of KRIBB11 or mock-treated 6 hours post-transfection and further incubated for 18 hours before luciferase analysis.
Immunoblotting and immunofluorescence
Immunoblotting and immunofluorescence was performed as described previously (19, 22, 63, 71). The band intensities of HSF1 pS326 were evaluated using Image J software and normalized to HSF1. For immunofluorescence, the cells were fixed with 4% paraformaldehyde (for transfected cells) or methanol/acetone (for infected cells) and stained with the indicated antibodies. Nuclear DNA was stained with Hoechst 33342 and specific signals were detected using a confocal laser-scanning microscope (LSM800 microscope; Zeiss). For quantification of fluorescence, images of each cell acquired by the LSM800 microscope were analyzed using the Histo function in ZEN3.1 software (Zeiss) as previously reported (22, 72).
NanoBiT assay
HEK293T cells were transfected with pTK-HHV-6A U34-SmBit or pTK-HSF1-SmBit together with pTK-HHV-6A U37-LgBit or pTK-Hsp90-LgBit. After 24 hours, Nano-Glo Live Cell Reagent (Promega) was added and luminescence was detected using a microplate reader. The combination of pTK-HHV-6A U37-LgBit and the HaloTag-SmBiT vector was used as a negative control.
Construction of recombinant HHV-6A BAC
To construct HHV-6A-ΔU37 BAC, harboring a deletion in U37 (nucleotide position 201–500) (Fig. 11), a two-step Red-mediated recombination system in Escherichia coli GS1783 harboring the HHV-6A BAC genome (73) was employed using the primers 5′- TACGAACAGAAATTTCTAGCGATCATAAAACTGCCAATTACCGGTAAAGAATGTTCGAAAGAAAACAAAAAGGATGACGACGATAAGTAGGG-3′ and 5′- CGAGACTGGAAAACTATGTGCATTCCGAGCTTTTGTTTTCTTTCGAACATTCTTTACCGGTAATTGGCAGCAACCAATTAACCAATTCTGATTAG-3′. The HHV-6A-ΔU37-repair BAC, in which the U37 deletion in HHV-6A-ΔU37 BAC was repaired (Fig. 11), was generated as described above, except the primers 5′-AAGCTTTACTTACAACTGAA-3′ and 5′-ACTATTTGTTTAATGCATTC-3′ were used with pBS-HHV-6A U3791-590-Kan as the template. BAC DNA of HHV-6A (wild type), HHV-6A-ΔU37, or HHV-6A-ΔU37-repair was extracted from E. coli using NucleoBond BAC100 (Macherey-Nagel) and then resuspended in pH 8.0 Tris-EDTA (10 mM Tris-HCl, 1 mM EDTA). Two million JJhan cells were transfected with 3 µg of BAC DNA using the NEPA21 Super Electroporator (NEPAGENE) following the manufacturer’s instructions. The cells were incubated for a few days after electroporation and then co-cultured with 2–3 × 106 CBMCs. An increase in the number of cells expressing GFP showing CPE was observed after co-culture with CBMCs for 3–5 days.
Calculation of virus genome copy numbers
1 × 106 cells (JJhan) were infected with U1102 (1.6 × 108 genome copies). At 6 hours post-infection, infected cells were divided into four aliquots. Each aliquot was treated with either 10 µM KRIBB11, 10 µM 17-AAG (Hsp90 inhibitor), or 10 µM VER-155008 (Hsp70 inhibitor), or left untreated. At 72 hours after infection, the infected cells were freeze-thaw lysed and DNA was extracted from the supernatant of the cells treated under each condition using the DNeasy Blood & Tissue Kit (QIAGEN). The genome copy number per milliliter of infected cells was quantified by qPCR using SYBR Select Master Mix (Thermo Fisher Scientific). The sequences of primers used for this purpose were 5ʹ-CGCTAGGTTGAGAATGATCGA-3ʹ (forward) and 5ʹ-CAAAGCCAAATTATCCAGAGCG-3ʹ (reverse) as described previously (63).
Statistical analysis
For comparisons of two groups, statistical analysis was performed using the unpaired Student’s t test. Tukey’s test was used for multiple comparisons. A P-value > 0.05 was considered not significant (ns).
ACKNOWLEDGMENTS
We thank Yasuyo Ueda for their excellent technical assistance, S. Calderwood for kindly providing reagents, and lab members for helpful comments.
This study was supported by Grants for Scientific Research from the Japan Society for the Promotion of Science (JSPS), the Ministry of Education, Culture, Science, Sports and Technology of Japan (MEXT) Leading Initiative for Excellent Young Researchers Grant, contract research funds from the Japan Program for Infectious Diseases Research and Infrastructure (20wm0325005h), and Precursory Research for Innovative Medical Care (PRIME, 22gm6410022h) from the Japan Agency for Medical Research and Development (AMED) and grants from the Takeda Science Foundation and MSD Life Science Foundation, Public Interest Incorporated Foundation.
Contributor Information
Jun Arii, Email: jarii@med.kobe-u.ac.jp.
Felicia Goodrum, The University of Arizona, Tucson, Arizona, USA .
REFERENCES
- 1. Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559 [DOI] [PubMed] [Google Scholar]
- 2. Arii J. 2021. Host and viral factors involved in nuclear egress of herpes simplex virus 1. Viruses 13:754. doi: 10.3390/v13050754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Häge S, Marschall M. 2022. ’'Come together'-the regulatory interaction of herpesviral nuclear egress proteins comprises both essential and accessory functions. Cells 11:1837. doi: 10.3390/cells11111837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol 75:8803–8817. doi: 10.1128/jvi.75.18.8803-8817.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuan MI, O’Dowd JM, Chughtai K, Hayman I, Brown CJ, Fortunato EA. 2016. Human cytomegalovirus nuclear egress and secondary envelopment are negatively affected in the absence of cellular p53. Virology 497:279–293. doi: 10.1016/j.virol.2016.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bigalke JM, Heldwein EE. 2015. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J 34:2921–2936. doi: 10.15252/embj.201592359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lye MF, Sharma M, El Omari K, Filman DJ, Schuermann JP, Hogle JM, Coen DM. 2015. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J 34:2937–2952. doi: 10.15252/embj.201592651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leigh KE, Sharma M, Mansueto MS, Boeszoermenyi A, Filman DJ, Hogle JM, Wagner G, Coen DM, Arthanari H. 2015. Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc Natl Acad Sci U S A 112:9010–9015. doi: 10.1073/pnas.1511140112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zeev-Ben-Mordehai T, Weberruß M, Lorenz M, Cheleski J, Hellberg T, Whittle C, El Omari K, Vasishtan D, Dent KC, Harlos K, Franzke K, Hagen C, Klupp BG, Antonin W, Mettenleiter TC, Grünewald K. 2015. Crystal structure of the herpesvirus nuclear egress complex provides insights into inner nuclear membrane remodeling. Cell Rep 13:2645–2652. doi: 10.1016/j.celrep.2015.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Muller YA, Häge S, Alkhashrom S, Höllriegl T, Weigert S, Dolles S, Hof K, Walzer SA, Egerer-Sieber C, Conrad M, Holst S, Lösing J, Sonntag E, Sticht H, Eichler J, Marschall M. 2020. High-resolution crystal structures of two prototypical beta- and gamma-herpesviral nuclear egress complexes unravel the determinants of subfamily specificity. J Biol Chem 295:3189–3201. doi: 10.1074/jbc.RA119.011546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schweininger J, Kriegel M, Häge S, Conrad M, Alkhashrom S, Lösing J, Weiler S, Tillmanns J, Egerer-Sieber C, Decker A, Lenac Roviš T, Eichler J, Sticht H, Marschall M, Muller YA. 2022. The crystal structure of the varicella-zoster Orf24-Orf27 nuclear egress complex spotlights multiple determinants of herpesvirus subfamily specificity. J Biol Chem 298:101625. doi: 10.1016/j.jbc.2022.101625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thorsen MK, Draganova EB, Heldwein EE. 2022. The nuclear egress complex of epstein-barr virus buds membranes through an oligomerization-driven mechanism. PLoS Pathog 18:e1010623. doi: 10.1371/journal.ppat.1010623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walzer SA, Egerer-Sieber C, Sticht H, Sevvana M, Hohl K, Milbradt J, Muller YA, Marschall M. 2015. Crystal structure of the human cytomegalovirus pUL50-pUL53 core nuclear egress complex provides insight into a unique assembly scaffold for virus-host protein interactions. J Biol Chem 290:27452–27458. doi: 10.1074/jbc.C115.686527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bigalke JM, Heuser T, Nicastro D, Heldwein EE. 2014. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun 5:4131. doi: 10.1038/ncomms5131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hagen C, Dent KC, Zeev-Ben-Mordehai T, Grange M, Bosse JB, Whittle C, Klupp BG, Siebert CA, Vasishtan D, Bäuerlein FJB, Cheleski J, Werner S, Guttmann P, Rehbein S, Henzler K, Demmerle J, Adler B, Koszinowski U, Schermelleh L, Schneider G, Enquist LW, Plitzko JM, Mettenleiter TC, Grünewald K. 2015. Structural basis of vesicle formation at the inner nuclear membrane. Cell 163:1692–1701. doi: 10.1016/j.cell.2015.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang K, Baines JD. 2011. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc Natl Acad Sci U S A 108:14276–14281. doi: 10.1073/pnas.1108564108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang K, Wills E, Lim HY, Zhou ZH, Baines JD. 2014. Association of herpes simplex virus pUL31 with capsid vertices and components of the capsid vertex-specific complex. J Virol 88:3815–3825. doi: 10.1128/JVI.03175-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rönfeldt S, Klupp BG, Franzke K, Mettenleiter TC. 2017. Lysine 242 within helix 10 of the pseudorabies virus nuclear egress complex pUL31 component is critical for primary envelopment of nucleocapsids. J Virol 91:e01182-17. doi: 10.1128/JVI.01182-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takeshima K, Arii J, Maruzuru Y, Koyanagi N, Kato A, Kawaguchi Y. 2019. Identification of the capsid binding site in the herpes simplex virus 1 nuclear egress complex and its role in viral primary envelopment and replication. J Virol 93:e01290-19. doi: 10.1128/JVI.01290-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee C-P, Liu P-T, Kung H-N, Su M-T, Chua H-H, Chang Y-H, Chang C-W, Tsai C-H, Liu F-T, Chen M-R, Sun R. 2012. The ESCRT machinery is recruited by the viral BFRF1 protein to the nucleus-associated membrane for the maturation of epstein-barr virus. PLoS Pathog 8:e1002904. doi: 10.1371/journal.ppat.1002904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee C-P, Liu G-T, Kung H-N, Liu P-T, Liao Y-T, Chow L-P, Chang L-S, Chang Y-H, Chang C-W, Shu W-C, Angers A, Farina A, Lin S-F, Tsai C-H, Bouamr F, Chen M-R, Longnecker RM. 2016. The ubiquitin ligase Itch and ubiquitination regulate BFRF1-mediated nuclear envelope modification for Epstein-Barr virus maturation. J Virol 90:8994–9007. doi: 10.1128/JVI.01235-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arii J, Watanabe M, Maeda F, Tokai-Nishizumi N, Chihara T, Miura M, Maruzuru Y, Koyanagi N, Kato A, Kawaguchi Y. 2018. ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat Commun 9:3379. doi: 10.1038/s41467-018-05889-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arii J, Takeshima K, Maruzuru Y, Koyanagi N, Nakayama Y, Kato A, Mori Y, Kawaguchi Y, Frappier L. 2022. Role of the arginine cluster in the disordered domain of herpes simplex virus 1 Ul34 for the recruitment of ESCRT-III for viral primary envelopment. J Virol 96:e0170421. doi: 10.1128/JVI.01704-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roberts KL, Baines JD. 2011. UL31 of herpes simplex virus 1 is necessary for optimal NF-kappaB activation and expression of viral gene products. J Virol 85:4947–4953. doi: 10.1128/JVI.00068-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sherry MR, Hay TJM, Gulak MA, Nassiri A, Finnen RL, Banfield BW. 2017. The herpesvirus nuclear egress complex component, UL31, can be recruited to sites of DNA damage through poly-ADP ribose binding. Sci Rep 7:1882. doi: 10.1038/s41598-017-02109-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gong L, Ou X, Hu L, Zhong J, Li J, Deng S, Li B, Pan L, Wang L, Hong X, Luo W, Zeng Q, Zan J, Peng T, Cai M, Li M. 2022. The molecular mechanism of herpes simplex virus 1 UL31 in antagonizing the activity of IFN-beta. Microbiol Spectr 10:e0188321. doi: 10.1128/spectrum.01883-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aubin JT, Collandre H, Candotti D, Ingrand D, Rouzioux C, Burgard M, Richard S, Huraux JM, Agut H. 1991. Several groups among human herpesvirus-6 strains can be distinguished by Southern blotting and polymerase chain-reaction. J Clin Microbiol 29:367–372. doi: 10.1128/jcm.29.2.367-372.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Campadelli-Fiume G, Guerrini S, Liu X, Foà-Tomasi L. 1993. Monoclonal antibodies to glycoprotein B differentiate human herpesvirus 6 into two clusters, variants A and B. J Gen Virol 74:2257–2262. doi: 10.1099/0022-1317-74-10-2257 [DOI] [PubMed] [Google Scholar]
- 29. Wyatt LS, Balachandran N, Frenkel N. 1990. Variations in the replication and antigenic properties of human herpesvirus 6 strains. J Infect Dis 162:852–857. doi: 10.1093/infdis/162.4.852 [DOI] [PubMed] [Google Scholar]
- 30. Ablashi D, Agut H, Alvarez-Lafuente R, Clark DA, Dewhurst S, DiLuca D, Flamand L, Frenkel N, Gallo R, Gompels UA, Höllsberg P, Jacobson S, Luppi M, Lusso P, Malnati M, Medveczky P, Mori Y, Pellett PE, Pritchett JC, Yamanishi K, Yoshikawa T. 2014. Classification of HHV-6A and HHV-6B as distinct viruses. Arch Virol 159:863–870. doi: 10.1007/s00705-013-1902-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamanishi K, Mori Y, Pellet PE. 2013. Human Herpesviruses 6 and 7, p 2058–2079. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields Virology, 6th ed. Lippincott-Williams &Wilkins, Philadelphia, PA. [Google Scholar]
- 32. Alvarez-Lafuente R, García-Montojo M, De las Heras V, Bartolomé M, Arroyo R. 2006. Clinical parameters and HHV-6 active replication in relapsing-remitting multiple sclerosis patients. J Clin Virol 37:S24–S26. doi: 10.1016/S1386-6532(06)70007-5 [DOI] [PubMed] [Google Scholar]
- 33. Caselli E, Zatelli MC, Rizzo R, Benedetti S, Martorelli D, Trasforini G, Cassai E, degli Uberti EC, Di Luca D, Dolcetti R. 2012. Virologic and immunologic evidence supporting an association between HHV-6 and hashimoto's thyroiditis. PLoS Pathog 8:e1002951. doi: 10.1371/journal.ppat.1002951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morimoto RI, Santoro MG. 1998. Stress-inducible responses and heat shock proteins: new pharmacologic targets for cytoprotection. Nat Biotechnol 16:833–838. doi: 10.1038/nbt0998-833 [DOI] [PubMed] [Google Scholar]
- 35. Pirkkala L, Nykänen P, Sistonen L. 2001. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J 15:1118–1131. doi: 10.1096/fj00-0294rev [DOI] [PubMed] [Google Scholar]
- 36. Bolhassani A, Agi E. 2019. Heat shock proteins in infection. Clin Chim Acta 498:90–100. doi: 10.1016/j.cca.2019.08.015 [DOI] [PubMed] [Google Scholar]
- 37. Funk C, Ott M, Raschbichler V, Nagel C-H, Binz A, Sodeik B, Bauerfeind R, Bailer SM, Everett RD. 2015. The herpes simplex virus protein pUL31 escorts nucleocapsids to sites of nuclear egress, a process coordinated by its N-terminal domain. PLoS Pathog 11:e1004957. doi: 10.1371/journal.ppat.1004957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wilkie AR, Sharma M, Coughlin M, Pesola JM, Ericsson M, Lawler JL, Fernandez R, Coen DM. 2022. Human cytomegalovirus nuclear egress complex subunit, UL53, associates with capsids and myosin VA, but is not important for capsid localization towards the nuclear periphery. Viruses 14:479. doi: 10.3390/v14030479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Camozzi D, Pignatelli S, Valvo C, Lattanzi G, Capanni C, Dal Monte P, Landini MP. 2008. Remodelling of the nuclear lamina during human cytomegalovirus infection: role of the viral proteins pUL50 and pUL53. J Gen Virol 89:731–740. doi: 10.1099/vir.0.83377-0 [DOI] [PubMed] [Google Scholar]
- 40. Schmeiser C, Borst E, Sticht H, Marschall M, Milbradt J. 2013. The cytomegalovirus egress proteins pUL50 and pUL53 are translocated to the nuclear envelope through two distinct modes of nuclear import. J Gen Virol 94:2056–2069. doi: 10.1099/vir.0.052571-0 [DOI] [PubMed] [Google Scholar]
- 41. Akerfelt M, Morimoto RI, Sistonen L. 2010. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol 11:545–555. doi: 10.1038/nrm2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ali A, Bharadwaj S, O’Carroll R, Ovsenek N. 1998. HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol Cell Biol 18:4949–4960. doi: 10.1128/MCB.18.9.4949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. 1998. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94:471–480. doi: 10.1016/s0092-8674(00)81588-3 [DOI] [PubMed] [Google Scholar]
- 44. Yoon YJ, Kim JA, Shin KD, Shin DS, Han YM, Lee YJ, Lee JS, Kwon BM, Han DC. 2011. KRIBB11 inhibits HSP70 synthesis through inhibition of heat shock factor 1 function by impairing the recruitment of positive transcription elongation factor b to the HSP70 promoter. J Biol Chem 286:1737–1747. doi: 10.1074/jbc.M110.179440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Santomenna LD, Colberg-Poley AM. 1990. Induction of cellular HSP70 expression by human cytomegalovirus. J Virol 64:2033–2040. doi: 10.1128/JVI.64.5.2033-2040.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Burch AD, Weller SK. 2004. Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J Virol 78:7175–7185. doi: 10.1128/JVI.78.13.7175-7185.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Burch AD, Weller SK. 2005. Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone HSP90 for proper localization to the nucleus. J Virol 79:10740–10749. doi: 10.1128/JVI.79.16.10740-10749.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A 100:12396–12401. doi: 10.1073/pnas.1635160100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sharma M, Kamil JP, Coughlin M, Reim NI, Coen DM. 2014. Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J Virol 88:249–262. doi: 10.1128/JVI.02358-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Solit DB, Zheng FF, Drobnjak M, Münster PN, Higgins B, Verbel D, Heller G, Tong W, Cordon-Cardo C, Agus DB, Scher HI, Rosen N. 2002. 17-allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res 8:986–993. [PubMed] [Google Scholar]
- 51. Massey AJ, Williamson DS, Browne H, Murray JB, Dokurno P, Shaw T, Macias AT, Daniels Z, Geoffroy S, Dopson M, Lavan P, Matassova N, Francis GL, Graham CJ, Parsons R, Wang Y, Padfield A, Comer M, Drysdale MJ, Wood M. 2010. A novel, small molecule inhibitor of HSC70/HSP70 potentiates HSP90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother Pharmacol 66:535–545. doi: 10.1007/s00280-009-1194-3 [DOI] [PubMed] [Google Scholar]
- 52. Ohgitani E, Kobayashi K, Takeshita K, Imanishi J. 1999. Biphasic translocation of a 70 kDa heat shock protein in human cytomegalovirus-infected cells. J Gen Virol 80:63–68. doi: 10.1099/0022-1317-80-1-63 [DOI] [PubMed] [Google Scholar]
- 53. Basha W, Kitagawa R, Uhara M, Imazu H, Uechi K, Tanaka J. 2005. Geldanamycin, a potent and specific inhibitor of HSP90, inhibits gene expression and replication of human cytomegalovirus. Antivir Chem Chemother 16:135–146. doi: 10.1177/095632020501600206 [DOI] [PubMed] [Google Scholar]
- 54. Sun X, Bristol JA, Iwahori S, Hagemeier SR, Meng Q, Barlow EA, Fingeroth JD, Tarakanova VL, Kalejta RF, Kenney SC. 2013. HSP90 inhibitor 17-DMAG decreases expression of conserved herpesvirus protein kinases and reduces virus production in epstein-barr virus-infected cells. J Virol 87:10126–10138. doi: 10.1128/JVI.01671-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Song X, Wang Y, Li F, Cao W, Zeng Q, Qin S, Wang Z, Jia J, Xiao J, Hu X, Liu K, Wang Y, Ren Z. 2021. HSP90 inhibitors inhibit the entry of herpes simplex virus 1 into neuron cells by regulating cofilin-mediated F-actin reorganization. Front Microbiol 12:799890. doi: 10.3389/fmicb.2021.799890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhong M, Zheng K, Chen M, Xiang Y, Jin F, Ma K, Qiu X, Wang Q, Peng T, Kitazato K, Wang Y. 2014. Heat-shock protein 90 promotes nuclear transport of herpes simplex virus 1 capsid protein by interacting with acetylated tubulin. PLoS One 9:e99425. doi: 10.1371/journal.pone.0099425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li F, Jin F, Wang Y, Zheng D, Liu J, Zhang Z, Wang R, Dong D, Zheng K, Wang Y. 2018. HSP90 inhibitor AT-533 blocks HSV-1 nuclear egress and assembly. J Biochem 164:397–406. doi: 10.1093/jb/mvy066 [DOI] [PubMed] [Google Scholar]
- 58. Qin S, Hu X, Lin S, Xiao J, Wang Z, Jia J, Song X, Liu K, Ren Z, Wang Y. 2021. HSP90 inhibitors prevent HSV-1 replication by directly targeting UL42-HSP90 complex. Front Microbiol 12:797279. doi: 10.3389/fmicb.2021.797279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Caswell R, Hagemeier C, Chiou CJ, Hayward G, Kouzarides T, Sinclair J. 1993. The human cytomegalovirus 86K immediate early (IE) 2 protein requires the basic region of the TATA-box binding protein (TBP) for binding, and interacts with TBP and transcription factor TFIIB via regions of IE2 required for transcriptional regulation. J Gen Virol 74:2691–2698. doi: 10.1099/0022-1317-74-12-2691 [DOI] [PubMed] [Google Scholar]
- 60. Hasday JD, Singh IS. 2000. Fever and the heat shock response: distinct, partially overlapping processes. Cell Stress Chaperones 5:471–480. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yao K, Mandel M, Akyani N, Maynard K, Sengamalay N, Fotheringham J, Ghedin E, Kashanchi F, Jacobson S. 2006. Differential HHV-6A gene expression in T cells and primary human astrocytes based on multi-virus array analysis. Glia 53:789–798. doi: 10.1002/glia.20333 [DOI] [PubMed] [Google Scholar]
- 62. Aktar S, Arii J, Tjan LH, Nishimura M, Mori Y, Goodrum F. 2021. Human herpesvirus 6A tegument protein U14 induces NF-kappaB signaling by interacting with P65. J Virol 95:e0126921. doi: 10.1128/JVI.01269-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Aktar S, Arii J, Nguyen TTH, Huang JR, Nishimura M, Mori Y, Goodrum F. 2022. ATF1 restricts human herpesvirus 6A replication via beta interferon induction. J Virol 96:e0126422. doi: 10.1128/jvi.01264-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Arii J, Maeda F, Maruzuru Y, Koyanagi N, Kato A, Mori Y, Kawaguchi Y. 2020. ESCRT-III controls nuclear envelope deformation induced by progerin. Sci Rep 10:18877. doi: 10.1038/s41598-020-75852-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. doi: 10.1038/nature09420 [DOI] [PubMed] [Google Scholar]
- 66. Wang X, Grammatikakis N, Siganou A, Calderwood SK. 2003. Regulation of molecular chaperone gene transcription involves the serine phosphorylation, 14-3-3 epsilon binding, and cytoplasmic sequestration of heat shock factor 1. Mol Cell Biol 23:6013–6026. doi: 10.1128/MCB.23.17.6013-6026.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096 [DOI] [PubMed] [Google Scholar]
- 68. Mori Y, Akkapaiboon P, Yang X, Yamanishi K. 2003. The human herpesvirus 6 U100 gene product is the third component of the gH-gL glycoprotein complex on the viral envelope. J Virol 77:2452–2458. doi: 10.1128/jvi.77.4.2452-2458.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Takemoto M, Koike M, Mori Y, Yonemoto S, Sasamoto Y, Kondo K, Uchiyama Y, Yamanishi K. 2005. Human herpesvirus 6 open reading frame U14 protein and cellular P53 interact with each other and are contained in the virion. J Virol 79:13037–13046. doi: 10.1128/JVI.79.20.13037-13046.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. 1998. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/P300. EMBO J 17:1087–1095. doi: 10.1093/emboj/17.4.1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Arii J, Takeshima K, Maruzuru Y, Koyanagi N, Kato A, Kawaguchi Y. 2019. Roles of the interhexamer contact site for hexagonal lattice formation of the herpes simplex virus 1 nuclear egress complex in viral primary envelopment and replication. J Virol 93:e00498-19. doi: 10.1128/JVI.00498-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Maeda F, Arii J, Hirohata Y, Maruzuru Y, Koyanagi N, Kato A, Kawaguchi Y. 2017. Herpes simplex virus 1 UL34 protein regulates the global architecture of the endoplasmic reticulum in infected cells. J Virol 91:e00271-17. doi: 10.1128/JVI.00271-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tang H, Kawabata A, Yoshida M, Oyaizu H, Maeki T, Yamanishi K, Mori Y. 2010. Human herpesvirus 6 encoded glycoprotein Q1 gene is essential for virus growth. Virology 407:360–367. doi: 10.1016/j.virol.2010.08.018 [DOI] [PubMed] [Google Scholar]