


Abstract
DNA·RNA hybrid duplexes are substrates of RNase H and reverse transcriptase. The crystal structure of a hybrid duplex, d(5′-CTCTTCTTC-3′)·r(5′-gaagaagag-3′) (the uppercase letters indicate DNA and lowercase letters RNA), with a polypurine RNA strand and a complementary DNA strand has been determined at 1.8 Å resolution. The structure was refined first at 1.9 Å by XPLOR and subsequently by CNS at 1.8 Å. The hybrid is found in a standard A-form conformation with all the sugars in the C3′-endo puckering. The 5′-terminal base dC of the DNA strand was clearly visible in the electron density map of the present structure, in contrast to the previously reported structure d(TTCTTBr5CTTC)·r(gaagaagaa) where the 5′-terminal base dT was not visible, leaving the terminal rA unpaired. Thus, the comparison of the terminal base pairs, C·g versus T·a, in the two hybrid crystal structures provides information on the stability of these base pairs in hydrogen bonding (three versus two) and base stacking interactions. The differences in the terminal base pairs produce different kinks in the two structures. Minor groove widening is observed in the present structure at a distinctive kink in the lower half of the duplex, in contrast to the small widening of the minor groove and a very slight bend in the upper half of the T·a structure.
INTRODUCTION
Hybrid duplexes formed by one DNA strand and the other RNA strand are of great importance in biological functions as in gene therapy and antisense technology. (For simplicity, we refer to hybrid duplexes when we have one strand of DNA and the other strand RNA, while we refer to chimeric duplexes as containing a mixture of DNA and RNA in the same strand.) The enzymes RNase H and reverse transcriptase, which has a RNase H domain, catalyze the hydrolysis of the RNA strand in the hybrid duplex. However, the polypurine tract (PPT), r(aaaagaaaagggggga), located at the 3′-end of the U3 region of the human immunodeficiency virus (HIV-1) RNA genome is not digested by the reverse transcriptase. This PPT serves as the second primer for the plus-strand DNA synthesis (1). The Los Alamos HIV sequence compendium (2) reveals that many other polypurine stretches in the virus genome with comparable lengths to the PPT (10–20 purines) are digested. However, mutation studies changing the specific sequence of the PPT still largely preserve its resistance to the digestion by RNase H (3). Studying the structure of hybrid duplexes with polypurine RNA strands is therefore relevant and might provide preliminary information about the enzyme–substrate complex.
In the previous hybrid structure with a polypurine RNA strand and a polypyrimidine DNA strand, d(TTCTTBr5CT-TC)·r(gaagaagaa) (referred to as TTC), the 5′ terminal base T of the DNA strand was disordered and not visible (4). Here we have determined the hybrid structure, d(CTCTTCTTC)·r(gaa-gaagag) (referred to as CTC), where the first base pair C1·g18 replaces the previous T1·a18 base pair and the cytosine 6 is not brominated (Fig. 1). We hoped that the three-hydrogen bonded C·g Watson–Crick base pair would be visible and more stable than the two-hydrogen bonded T·a base pair.
Figure 1.

The numbering schemes for (a) this structure and (b) the previously determined TTC hybrid duplex (4).
MATERIALS AND METHODS
Sample preparation and data collection
The nonamer DNA (CTCTTCTTC) and RNA (gaagaagag) fragments were synthesized in house using phosphoramidite chemistry. Purification and annealing of the two strands were conducted as before (4). Crystals were grown by the hanging-drop vapor diffusion method using 1 mM hybrid duplex, 400 mM MgCl2, 2 mM spermine tetrachloride, 100 mM sodium cacodylate buffer (pH 6.0) and 5% (v/v) methyl-2,4-pentanediol (MPD), against 40% MPD in the reservoir at room temperature. Data were collected to 1.8 Å resolution at –10°C using an in-house Raxis IIc imaging-plate system equipped with a Rigaku rotating anode generator and graphite monochromated CuKα radiation (1.5418 Å). The crystal measuring 0.3 × 0.1 × 0.1 mm3, was indexed in the hexagonal space group P61 with unit cell dimensions a = b = 49.15 Å and c = 46.13 Å. The data were processed using the program DENZO and SCALEPACK (5). Data collection statistics are summarized in Table 1.
Table 1. Crystal data and refinement statistics for d(CTCTTCTTC)·r(gaagaagag).
| Crystal system | Hexagonal |
| Space group | P61 |
| Cell parameters | |
| a (Å) | 49.15 |
| b (Å) | 49.15 |
| c (Å) | 46.13 |
| γ (°) | 120 |
| Volume/base pair (Å3) | 1590 |
| Resolution (Å) | 1.8 |
| No. of unique reflections [F ≥ 2σ(F)] | 5382 |
| Data completeness (%) | 91.0 |
| Rsym (%) on intensity | 6.7 |
| R-work | 17.9 |
| R-free | 20.5 |
| RMSD from ideal geometry | |
| Parameter file | dna-rna.param |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.26 |
| Torsion angles (°)a | 8.08 |
aC3′-endo DNA dihedrals are used in calculating the RMSD although no dihedral restraints were used for the sugars during the refinement.
Structure solution and refinement
Although the space group and cell dimensions were similar between the present (CTC) and the previous (TTC) structures, we could not solve the present structure by placing the previous coordinate directly. Therefore, a molecular replacement search was performed using the program AMoRe (6) with a model of the TTC structure with the terminal base pair changed to C·g. One outstanding peak in the rotational and translational searches made the structure solution trivial. The structure was then refined using XPLOR (7) with the improved DNA–RNA parameter file (8). A subset of reflections (10%) was kept for the R-free calculation (9) and was not included in the refinement. A rigid body refinement followed by simulated annealing dropped the R-work and R-free values to 27.8 and 31.2%, respectively, for 4793 reflections between 10 and 1.9 Å. After several cycles of conjugate gradient energy minimization and restrained individual B-factor refinements, water molecules were added. The water densities corresponding to 3σ in the Fo-Fc difference Fourier map and simultaneously satisfying 1σ in the 3Fo-2Fc map were used. Thirty-seven water molecules were located and included in the iterative cycles of positional and B-factor refinements with the R-work/R-free converged at 19.9/26.2%. The structure was then refined with the program package CNS (10) with the data resolution extended to 1.8 Å. The overall anisotropic B-factor and bulk solvent corrections were applied to the reflection data. The final structure contains six additional water molecules. Inclusion of all 43 water molecules lowered the R-work/R-free to 17.9/20.5% for the 5382 unique reflections (F > 2σF) in the resolution range 10–1.8 Å. The CNS refinement statistics are summarized in Table 1. The coordinates and structure factors have been deposited in the Nucleic Acid Database (11), access code ah005.
RESULTS AND DISCUSSION
The hybrid duplex adopts an A-form conformation with a stable terminal C1·g18 base pair
The hybrid duplex is found in the A-form conformation (Fig. 2a) and all of the helical parameters conform to the A family, which is also observed in the previous TTC structure. The helical parameters were calculated by the program of Lavery and Sklenar (12). The present structure is characterized by an average global twist angle of 32.3° and an average global base pair inclination angle of 7.9°. The base pairs are displaced (dx) by an average of –4.0 Å with an average rise of 3.0 Å. All of the bases are anti and sugars are in the C3′-endo puckering. Superposition of the eight common base pairs of the current structure and the TTC hybrid duplex gives a root mean square deviation (RMSD) of 1.01 Å (Fig. 2b). In contrast to the TTC hybrid where the 5′ terminal T1 is disordered and not visible, the C1 residue in this structure could be clearly located, confirming that the three-hydrogen bonded C1·g18 base pair of the present structure is more stable than the two-hydrogen bonded T1·a18 base pair of the previous structure. The additional hydrogen bond in the C·g base pair seemingly provides the stabilizing force to lock the C1 base in place. Figure 3 shows that there is a slight improvement in the base stacking of the terminal C1 residue when compared to that of the deduced T1 residue of the previous structure (4). In Figure 3a, the C1·g18 base pair moves inwards, while in Figure 3b the T1·a18 moves outwards. Thus, both the C·g hydrogen bonding and base stacking contribute to the stability of the hybrid structure. It is known that duplex structures with more G·C base pairs are more stable than those with more A·T base pairs (13). In fact, a survey of all the oligonucleotide structures deposited in the Nucleic Acid Database shows that only a few oligonucleotides with A·T base pairs at the terminal position have been crystallized, evidencing that the two-hydrogen bonded A·T base pair is intrinsically less stable than the three-hydrogen bonded G·C base pair.
Figure 2.

Stereo views of (a) the hybrid duplex with the σ weighted 2Fo-Fc density map contoured at 1σ level, and (b) superposition of the eight common base pairs of the present hybrid duplex (thick lines) with the previous TTC structure (thin lines).
Figure 3.

The intramolecular stacking of the terminal base pairs in (a) the present structure, C1·g18 and (b) the previous TTC structure. The missing T1 is indicated by broken lines. There is a slight improvement in stacking for the C1 residue (a) as compared to the T1 (b).
Conformational variations of the hybrid duplexes
Although both the present structure and the previous TTC hybrid exhibit standard A-form conformation, they display some structural differences. Despite that the two hybrid duplexes have a small RMSD of 1.01 Å, the present structure has a distinctive 25° bend towards the major groove at the lower half (residue 6), in contrast to the slight bend in the upper half of the TTC hybrid (Fig. 4). As the duplex bends toward the major groove, the minor groove is slightly opened up around the bending site. Figure 5 shows that the current structure has a relatively wide minor groove at the bending site, while the earlier hybrid has only a slight widening in the minor groove in the upper half of the molecule. These conformational variations may be induced by different packing environments of the hybrid duplexes. In both crystals the abutting interactions dominate the packing (Fig. 6a). In the present structure the C1·g18 pair lies close to the minor groove of a symmetry-related molecule with different abutting interactions to the TTC structure. These abutting interactions involve extensive van der Waals contacts between the terminal C1·g18 base pair surface of the approaching hybrid duplex and the DNA sugar rings of the symmetry-related molecule, and hydrogen bonding interactions between the base atoms and the phosphate oxygens in the RNA (Fig. 6b). In particular, the 2′-OH group of g18 forms bifurcated hydrogen bonds with the 2′-OH group of a12* (a single ‘ribose zipper’) and O4′ of a13* of the symmetry-related RNA strand (*). In contrast, the a18 base of the TTC hybrid moves 4–5 Å toward the DNA strand, thus avoiding the above interactions with the RNA strand while forming a different ‘ribose zipper’ between 2′-hydroxyls of a18 and a13* (Fig. 6c). This difference in the RNA strand is manifested in the RMSD of the individual strands of two structures. When individual DNA or RNA strands are superimposed between the present and the previous structures, it shows that the two RNA strands have a larger deviation (RMSD 1.06 Å) than the DNA strands (RMSD 0.78 Å).
Figure 4.
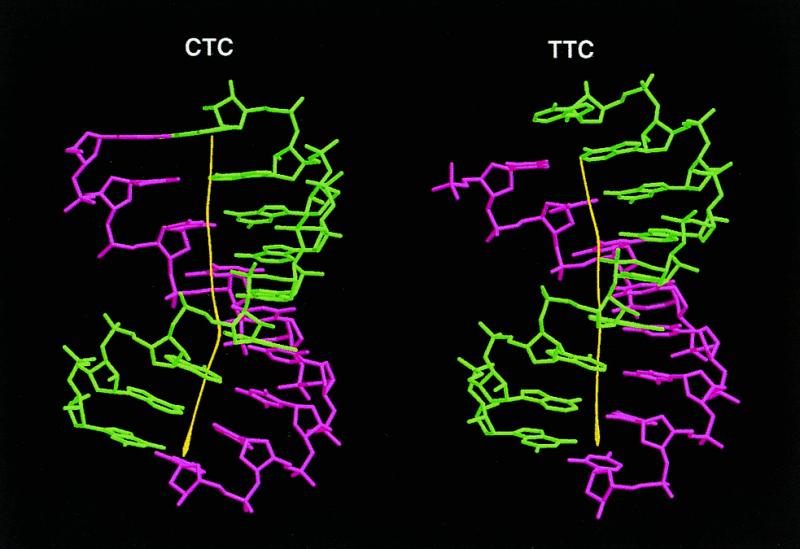
Marked kinking at the lower half of the helix observed in the present structure (left), with the bending angle >25° toward the major groove, compared to the slight kinking in the upper half of the previous TTC duplex (right). DNA strands are shown in pink and RNA strands in green.
Figure 5.

The minor groove width in the present structure (solid lines) is larger than that of the previous TTC structure (dashed lines) at the bending site.
Figure 6.
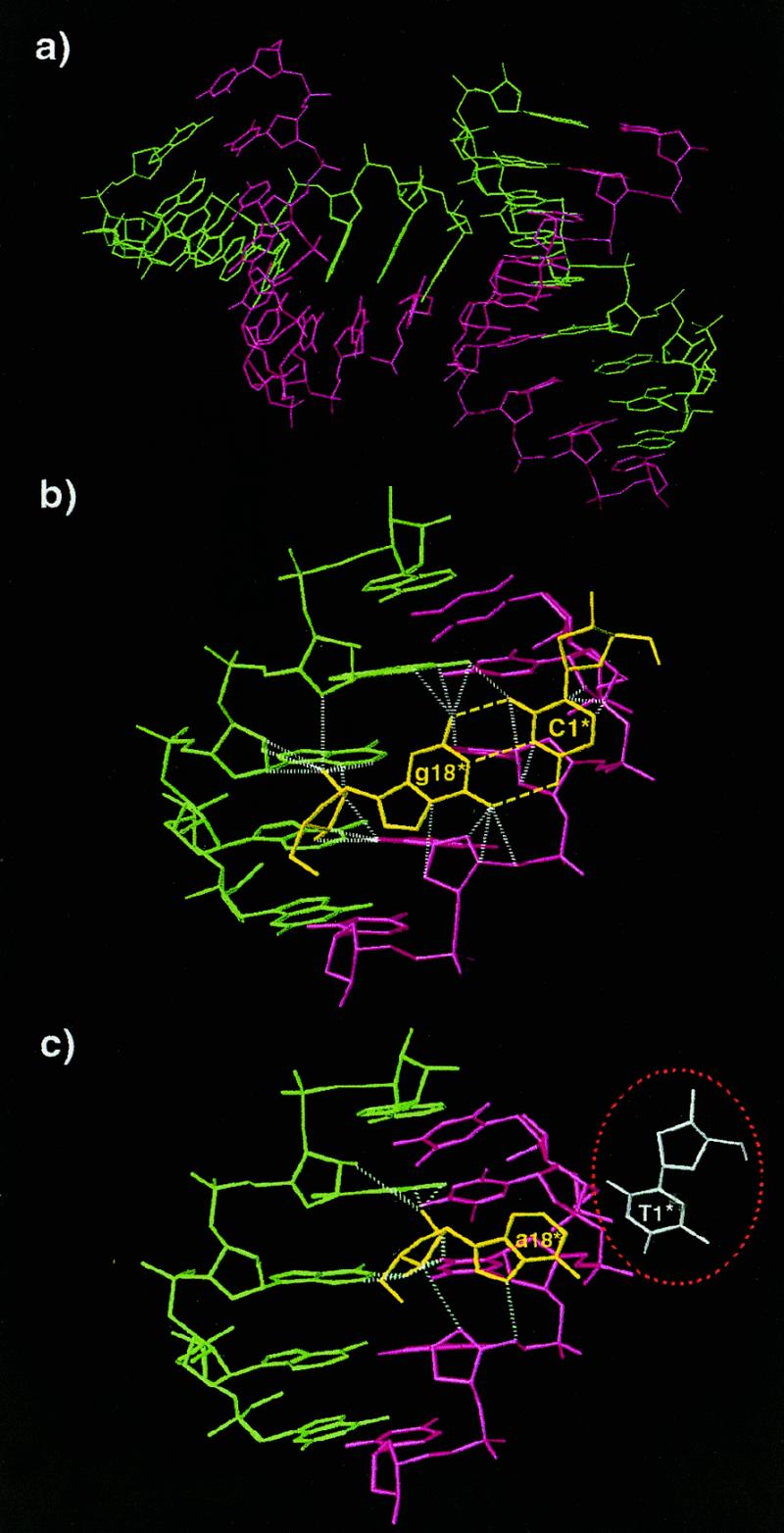
(a) Symmetry-related molecules showing the abutting interaction involving the A-DNA strand (pink). The RNA strand is shown in green. (b) Interactions involving the terminal C1·g18 base pair (yellow). Broken yellow lines indicate the base pair hydrogen bonds. Dashed white lines indicate close contacts and hydrogen bonding interactions. (c) Interactions involving the terminal T1·a18 base pair in the previous TTC structure. Color coding is the same as in (b). The missing T1 base is circled in red. Note that T1 has hardly any stacking interactions.
Crystal packing and hydration
The crystal packing seems to be dominated by the DNA strand (A-DNA), where the terminal base pair abuts into the minor groove of symmetry-related DNA sugar–phosphate backbone (Fig. 6a). Compared to the C2′-endo B-DNA with pseudo-continuous helical packing, the C3′-endo A-DNA has abutting interactions which form a more compact packing network in the hybrid molecules minimizing the hydrophobic surface areas (14). In fact the abutting interactions are also seen in crystal structures of other hybrid duplexes (15–17) and chimeric duplexes (18–22) (Table 2).
Table 2. Single crystal structures for DNA·RNA hybrid/chimer duplexes.
| Sequencea | Structureb | Crystal packing | Reference |
|---|---|---|---|
| Hybrid | |||
| (gaagaagag)·(CTCTTCTTC) | A-form | Abutting | This work |
| (gaagaagaa)·(TTCTTBr5CTTC) | A-form | Abutting | (4) |
| (gaagagaagc)·(GCTTCTCTTC) | A-form | Abutting | (15) |
| Hammerhead ribozyme with a DNA substrate | A-form | Abutting and end-to-end stacking | (16) |
| (uucgggcgcc)·(GGCGCCCGAA) | RNA: C3′-endo | Abutting and end-to-end stacking | (17) |
| DNA: C2′-endo for AA | |||
| C3′-endo for others | |||
| Chimer | |||
| (gcgTATACGC)2 | A-form | Abutting | (18) |
| (GCGTaTACGC)2 | A-form | Abutting | (19) |
| (gCGTATACGC)2 | A-form | Abutting | (19) |
| (CCGGCgCCGG)2 | A-form | Abutting | (20) |
| (gcgTATACCC)·(GGGTATACGC) | A-form | Abutting | (21) |
| (gcaguggc)·(gccaCTGC) | A-form | Abutting and end-to-end stacking | (22) |
aRNA is in lowercase letters and underlined.
bDiscussions are restricted to the hybrid portion of the duplex.
A total of 43 water molecules are found hydrating the present hybrid duplex. Of these, 5 are located in the minor groove, 20 in the major groove and 18 are associated with the sugar–phosphate backbone. Eight of the sugar–phosphate backbone water molecules are also involved in bridging the O2′-hydroxyl groups with base N3 atoms in the minor groove or bridging the phosphate oxygen atoms with the base N7 atoms in the major groove. Compared to the 55 waters in the previous TTC structure, there is a substantial reduction in the number of waters found in this structure. The difference might be caused by the variation in the crystallization conditions. In the present study, a MgCl2 concentration of 400 mM was used in the hanging drop, instead of the 0.5 mM cobalt hexamine chloride in the earlier study. Thus, the high salt concentration used might be responsible for the fewer water molecules observed in this structure.
CONCLUSION
In this study we observe the hybrid duplex with polypurine RNA strand adopting A-form conformation similar to our previous structure (4). Most of the minor grove widening and helix bending probably occur because of packing interactions. In summary, the terminal base pair C1·g18 of a symmetry-related molecule abuts into the minor groove of another hybrid duplex thus creating a bend in the molecule. It is believed that the RNase H attacks the hybrid through the minor groove and a model of the enzyme binding to a hybrid duplex with intermediate minor groove width was proposed (23,24). Studying the dimensional change of the minor groove is therefore particularly relevant for the understanding of the enzyme–substrate recognition process. However, the wide A-form minor groove we observed here is not compatible with the intermediate between A- and B-forms suggested for RNase H binding (23). Nevertheless, the width change in the minor groove of the hybrid duplex could shed light on the scenario when it binds to RNase H. Conformational flexibility has been observed in other hybrid/chimer duplex and is believed to be important in facilitating the RNase H–hybrid duplex interactions (22). As we noticed in our previous study, it seems that hybrids with high purine content in the RNA strand have the tendency to adopt the A-form conformation, indicating that an A-form geometry of the HIV-1 PPT might contribute to protect this biological second primer from RNase H digestion (4). However, this alone cannot explain why other hybrid duplexes with polypurine RNA stretch, as found in the Los Alamos HIV sequence compendium (2), are digested. In this respect, it certainly deserves further investigation of the PPT structure with the actual sequence and also an enzyme–substrate complex.
Acknowledgments
ACKNOWLEDGEMENTS
We acknowledge the support of the National Institutes of Health grant GM-17378 and the Board of Regents of Ohio for an Ohio Eminent Scholar Chair and Endowment to M.S. We also acknowledge the Hays Consortium Investment Fund by the Regions of Ohio for partial support for purchasing the R-axis IIc imaging plate.
NDB/PDB accession no. ah005
REFERENCES
- 1.Coffin J.M. (1996). In Fields,B.N., Knipe,D.M., Howly,P.M., Chanock,R.M., Melnick,J.L., Monath,T.P., Roizeman,B. and Straus,S.E., (eds), Virology, 3rd Edn. Lippincott-Raven Publishers, New York, NY, pp. 1767–1847. [Google Scholar]
- 2.Korber B., Kuiken,C.L., Foley,B., Hahn,B., McCutchan,F., Mellors,J.W. and Sodroski,J. (eds), Human Retroviruses and AIDS 1998: A Compilation and Analysis of Nucleic Acid and Amino Acid Sequences. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM.
- 3.Rattery A.J. and Champoux,J.J. (1989) J. Mol. Biol., 288, 455–466. [Google Scholar]
- 4.Xiong Y. and Sundaralingam,M. (1998) Structure, 6, 1493–1501. [DOI] [PubMed] [Google Scholar]
- 5.Otwinowski Z. and Minor,W. (1997) In Carter,C.W.,Jr and Sweet,R.M. (eds), Methods in Enzymology, Volume 276: Macromolecular Crystallography, Part A. Academic Press, New York, NY, pp. 307–326.
- 6.Navaza J. (1994) Acta Crystallogr., A50, 157–163. [Google Scholar]
- 7.Brunger A.T. (1992) Nature, 355, 472–474. [DOI] [PubMed] [Google Scholar]
- 8.Parkinson G., Vojtechovsky,J., Clowney,L., Brunger,A.T. and Berman,H.M. (1996) Acta Crystallogr., D52, 57–64. [DOI] [PubMed] [Google Scholar]
- 9.Brunger A.T. (1994) XPLOR Manual, Version 3.1. Yale University, New Haven, CT.
- 10.Brunger A.T., Adams,P.D., Clore,G.M., DeLano,W.L., Gros,P., Grosse-Kuntsleve,R.W., Jiang,J.S., Kuszewski,J., Nilges,M., Pannu,N.S., Read,R.J. Rice,L.M., Simonson,T. and Warren,G.L. (1998) Acta Crystallogr., D54, 905–921. [DOI] [PubMed] [Google Scholar]
- 11.Berman H.M., Olson,W.K., Beveridge,D.L., Westbrook,J., Gelbin,A., Demeny,T., Hsieh,S.H., Srinivascan,A.R. and Schneider,B. (1992). Biophys. J., 63, 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavery R. and Sklenar,H. (1988) J. Biomol. Struct. Dyn., 6, 63–91. [DOI] [PubMed] [Google Scholar]
- 13.Marmur J. and Doty,P. (1962) J. Mol. Biol., 5, 109–118. [DOI] [PubMed] [Google Scholar]
- 14.Sundaralingam M. and Biswas,R. (1997) J. Biomol. Struct. Dyn., 15, 173–176. [DOI] [PubMed] [Google Scholar]
- 15.Conn G.L., Brown,T. and Leonard,G.A. (1999) Nucleic Acids Res., 27, 555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Play H.W., Flaherty,K.M. and McKay,D.B. (1994) Nature, 372, 68–74. [DOI] [PubMed] [Google Scholar]
- 17.Horton N.C. and Finzel,B.C. (1996) J. Mol. Biol., 264, 521–533. [DOI] [PubMed] [Google Scholar]
- 18.Wang A.H.-J., Fujii,S., Van Boom,J.H., van der Marel,G.A., van Boekel,S.A.A. and Rich,A. (1982) Nature, 299, 601–604. [DOI] [PubMed] [Google Scholar]
- 19.Egli M., Usman,N., Zhang,S. and Rich,A. (1992) Proc. Natl Acad. Sci. USA, 89, 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ban C., Ramakrishnan,B. and Sundaralingam,M. (1994) J. Mol. Biol., 236, 275–285. [DOI] [PubMed] [Google Scholar]
- 21.Egli M., Usman,N. and Rich,A. (1993) Biochemistry, 32, 3221–3237. [PubMed] [Google Scholar]
- 22.Mueller U., Maier,G., Onori,A.M., Cellai,L., Heumann,H. and Heinemann,U. (1998) Biochemistry, 37, 12005–12011. [DOI] [PubMed] [Google Scholar]
- 23.Fedoroff O.Y., Salazar,M. and Reid,B.R. (1993) J. Mol. Biol., 233, 509–523. [DOI] [PubMed] [Google Scholar]
- 24.Salazar M., Fedoroff,O.Y., Miller,J.M., Ribeiro,N.S. and Reid,B.R. (1993) Biochemistry, 32, 4207–4215. [DOI] [PubMed] [Google Scholar]