


Abstract
DNA damage formation and repair are tightly linked to protein–DNA interactions in chromatin. We have used minichromosomes in yeast as chromatin substrates in vivo to investigate how nucleotide excision repair (NER) and repair by DNA-photolyase (photoreactivation) remove pyrimidine dimers from an origin of replication (ARS1). The ARS1 region is nuclease sensitive and flanked by nucleosomes on both sides. Photoreactivation was generally faster than NER at all sites. Site-specific heterogeneity of repair was observed for both pathways. This heterogeneity was different for NER and photoreactivation and it was altered in a minichromosome where ARS1 was transcribed. The results indicate distinct interactions of the repair systems with protein complexes bound in the ARS region (ORC, Abf1) and a predominant role of photolyase in CPD repair of an origin of replication.
INTRODUCTION
Nucleotide excision repair (NER) and DNA repair by photolyase in the presence of light (photoreactivation, PR) are the major pathways to remove UV-induced DNA lesions from the genome thereby preventing mutagenesis and cell death (1–5). Recent years have shown that both repair pathways are intimately modulated by protein–DNA interactions in chromatin (6). In particular, folding of DNA in nucleosomes restricts accessibility of DNA and slows down NER (7,8) as well as PR (9), whereas DNA lesions exposed in linker DNA are more rapidly repaired. Moreover, heterogeneity of NER observed in promoter regions (10–12) indicated that sequence-specific proteins remain bound to damaged DNA and modulate damage recognition and processing (13–15). On the other hand, DNA lesions can prevent binding of sequence-specific proteins in vitro (16,17). DNA lesions can maintain or disrupt protein–DNA complexes in vivo as suggested by the differential accessibility of the SNR6- and GAL10 TATA-boxes to photolyase (12).
Origins of replication are functional elements of chromosomes which regulate the site and timing of DNA duplication. In contrast with higher eukaryotes, yeast Saccharomyces cerevisiae has well-defined sequence elements (autonomously replicating sequences, ARSs), which serve as origins of replications. They confer extrachromosomal maintenance to plasmids and many act as origins of replication in their chromosomal context (18). The ARS sequences contain an A element with an 11-bp ARS consensus sequence (ACS) which is essential for origin function and a flanking B domain with three elements B1, B2 and B3 (19). Chromatin analysis of ARS1 in different minichromosomes revealed that the B domain is sensitive to nuclease digestion and by this criterium free of nucleosomes, whereas the A element is located in the edge of a positioned nucleosome (20–22).
The A element binds the origin of replication complex (ORC) (23). Interaction of ORC was also shown with the neighbouring B1 element (24). The B3 element functions as a binding site for the ARS-binding factor 1 (Abf1) (25). The ORC and Abf1 are bound to ARS1 during the whole cell cycle, but the composition alternates between the postreplicative complex (post-RC) during S, G2 and M phase and the prereplicative complex (pre-RC) during G1 phase (26,27).
Studies on repair of UV-induced DNA lesions in yeast minichromosomes showed that both NER and PR are affected by local chromatin structures (7,9,28). Repair by photolyase was more efficient than NER in active open promoters, which suggests an important role of PR in the restoration of these regulatory regions (9). Despite the existence of an ‘open’ chromatin structure of ARS1 (nuclease-sensitive region), NER was surprisingly slow (28), and PR was heterogeneous with both fast and slowly repaired sites (9). These results suggested a possible interference with ARS1 binding protein complexes.
Here, we have addressed this topic in detail using high resolution DNA repair analysis. We found that PR is the predominant mechanism to remove cyclobutane pyrimidine dimers (CPDs) from the ARS1 region. Moreover, we noticed distinct repair heterogeneity by PR and NER, indicating that PR and NER differentially interact with chromatin structure of the ARS1 region.
MATERIALS AND METHODS
Yeast strains
JMY1 [MATa, his3-Δ1 trp1-289 rad1-Δ ura3-52 YRpTRURAP (URA3 ARS1)]; FTY117 [MATa his3-Δ1 trp1-289 rad1-Δ ura3-52 YRpCS1 (HIS3 TRP1 ARS1)] was generated from JMY1 by selecting for plasmid loss and subsequent transformation with YRpCS1 (9); FTY23 [MATα his3-1 ura3-52 trp1 gal2 gal10 cir0 YRpTRURAP (URA3 ARS1)] (7).
UV irradiation and repair in vivo
The NER and PR experiments were done as described (7,9,29). Briefly, yeast cells were grown at 30°C in SD minimal medium (2% dextrose, 0.67% Yeast Nitrogen Base without amino acids) supplemented with the required amino acids and uracil to a final density of ~3 × 107 cells/ml. Cells were harvested and resuspended in SD minimal medium to yield ∼1.5 × 107 to 4 × 107 cells/ml. Aliquots of 250 ml were irradiated with UV light (predominantly 254 nm using Sylvania G15T8 germicidal lamps). After irradiation, the medium was supplemented with the appropriate amino acids or uracil. For PR, the cell suspensions were exposed to photoreactivating light (Sylvania Type F15 T8/BLB bulbs, peak emission at 375 nm) at 1.5 mW/cm2 for up to 120 min and at temperatures between 22 and 26°C. Aliquots were collected and immediately chilled on ice. For NER, cells were incubated at 30°C in the dark. All manipulations were carried out in rooms equipped with safety light (Sylvania GE ‘Gold’, fluorescent light). DNA purification was done by phenol extractions or by Qiagen Genomic tips 500/G (Qiagen, Hilden, Germany). Purified DNA was cut to completion with EcoRI and purified using Elutip-D columns (Schleicher and Schuell, Dassel, Germany) or by the QIAEX buffer desalting and concentration protocol (Qiagen) and finally dissolved in 10 mM Tris–HCl, 1 mM EDTA, pH 8.0.
UV irradiation and photoreactivation of DNA
For damage induction in vitro, DNA from non-irradiated cells (50 µl, 10 mM Tris–HCl, 1 mM EDTA, pH 8.0, 100–200 ng/µl) was exposed to UV. For PR in vitro, DNA was mixed with Escherichia coli photolyase to a final concentration of 0.1–0.2 µg photolyase per µg irradiated DNA in 50 mM Tris pH 7.4, 50 mM NaCl, 1 mM EDTA, 10 mM DTT, 5% glycerol and 50 µg/ml BSA, and exposed to photoreactivating light (Sylvania Type F15 T8/BLB bulbs at 2 mW/cm2) for 30–40 min. DNA was purified and dissolved in 10 mM Tris–HCl, 1 mM EDTA pH 8.0.
Primer extension
Primers were purified by PAGE. Top strand primer was: 5′-CAACCCCCTGCGATGTATATTTTCC-3′ (corresponding to nucleotides 2115–2091 of YRpTRURAP and nucleotides 954–930 of YRpCS1). Bottom strand primer was: 5′-GGTGATGCGCTTAGATTAAATGGCG-3′ (nucleotides 1822–1847 of YRpTRURAP and 661–685 of YRpCS1). Primer (10 pmol) in 70 mM Tris–HCl pH 7.6, 10 mM MgCl2, 5 mM DTT was labelled at the 5′-end using 10 U T4-polynucleotide kinase (New England BioLabs, Beverly, MA) and 10–15 pmol [γ-32P]ATP (5000 Ci/mmol, 10 mCi/ml, Amersham-Pharmacia, Uppsala, Sweden) in a volume of 30 µl, at 37°C for 30–60 min and stopped with 70 µl 10 mM Tris–HCl pH 8.0, 1 mM EDTA. Non-incorporated nucleotides were removed using the Quick Spin™ columns for radiolabelled DNA purification (Boehringer Mannheim). For the analysis of NER, primer extension was done as described (7). For PR, primer extension was slightly modified: 1–10 µl DNA (5 ng) was mixed with 20 µl endlabelled primer (0.6–0.8 pmol), 4 µl 10× Qiagen Taq buffer [100 mM Tris–HCl, pH 8.7, 500 mM KCl, (NH4)2SO4, 15 mM MgCl2], 1.2 µl of nucleotide mixture (5 mM of each dATP, dCTP, dGTP, dTTP; Pharmacia Ultrapure dNTPs) adjusted to a final volume of 40 µl and overlaid with 25 µl paraffin. Samples were heated at 95°C for 5 min, and then chilled in ice. Taq polymerase (Qiagen Taq DNA polymerase; 1 U in 5 µl 1× Taq buffer) was added and the samples were subjected to 30 primer extension cycles using the Perkin Elmer PCR thermocycler (denaturation at 94°C for 45 s, annealing at 58°C for 4.5 min and extension at 72°C for 2 min). DNA was ethanol precipitated and analysed in 5 or 8% (w/v) sequencing gels (30). The gels were dried on Whatman DE 81 papers and exposed on X-ray films (Fuji RX, Tokyo, Japan) or quantified using a PhosphorImager (Molecular Dynamics). Sequencing reactions were done in parallel by the chain termination method using Taq polymerase and non-irradiated DNA.
Quantitation
To obtain the fraction of molecules that contain a dimer at a defined site, the signal of individual bands or clusters of bands was measured and divided by the signal of the whole lane. The same procedure was done for the lanes containing non-irradiated DNA or DNA treated by photolyase to remove CPDs (‘photoreactivated DNA’). The value from non-irradiated DNA was subtracted as non-specific background. The fraction of CPDs was calculated by subtraction of ‘photoreactivated DNA’. To generate repair curves, the damage at each repair time was normalised with respect to the initial damage (no repair, 100% damage). The data points were fitted in an exponential curve using Kaleidagraph 3.0.1. (Abelbeck Software, Reading, PA) and the time was calculated, where 50% of the CPDs were removed (T50% value).
RESULTS
Photoreactivation in ARS1 of YRpTRURAP
Figure 1 illustrates DNA repair by photolyase in the ARS1 region of minichromosome YRpTRURAP in strain JMY1 (rad1ΔYRpTRURAP) which is defective in NER. The chromatin structure schematically illustrates the location of the ARS1 elements (A, B1, B2, B3) and the position of the nucleosomes which flank the nuclease-sensitive ARS1 region (20,21).
Figure 1.

CPD repair by photolyase in ARS1 of minichromosome YRpTRURAP. (A and B) Primer extension products of the bottom strand (short and long gel run, respectively). (C) Primer extension products of the top strand. Chromatin structure is illustrated according to F.Thoma (21), S.Tanaka and F.Thoma, unpublished results: positioned nucleosomes (ovals), presumed nucleosome position (dashed oval); ARS1 elements (boxes, according to Diffley and Cocker, 32): B3 (position 1912–1929), B2 (position 1959–1969), B1 (position 1996–2009), A (position 2018–2028). Indicated are: pyrimidine sites used for repair calculation (filled boxes, numbers refer to the 5′ nucleotide in the YRpTRURAP sequence), sites not quantified due to low signal intensity (open boxes). The lanes represent: dideoxy-sequencing reactions A, G, C and T (lanes 1–4); DNA damaged in vitro with 40 J/m2 (lane 5); DNA of non-irradiated cells (lane 6); DNA of cells irradiated with 100 J/m2 (chromatin, lanes 7–15), photoreactivated for 3–120 min (lanes 9–14) or incubated in the dark for 120 min (lane 15); damaged DNA (as in lane 8), but treated with E.coli photolyase to remove CPDs and to display 6-4PPs and other non-CPD lesions (lane 7).
UV lesions are assigned to dipyrimidine sites and polypyrimidine clusters by comparing the primer extension products of irradiated DNA with sequencing lanes (Fig. 1, lanes 1–4). Taq polymerase stops frequently 1 nt 3′ of a damaged dipyrimidine which is consistent with previous observations (31). DNA from non-irradiated cells reveals the background of non-specific Taq stops (Fig. 1, lanes 6). DNA from irradiated cells (repair time 0 min) displays the initial UV damage generated in vivo (Fig. 1, lanes 8). The intensities of bands reflect the frequency of lesions at individual sites. Taq polymerase stops at CPDs and 6-4 photoproducts (6-4PPs) (31). To identify the fraction of non-CPD photoproducts (predominantly 6-4PPs), the CPDs were removed from damaged DNA (time 0 min) by PR in vitro using E.coli photolyase (Fig. 1, lanes 7).
Calculation of the initial damage shows that 5–8% of the minichromosomes have one CPD in the ~200 bp long ARS1. The yield of damage generated by 40 J/m2 in naked DNA was similar to that generated in cells by 100 J/m2 consistent with previous observations (31) (compare lanes 5 and 8). The damage patterns in DNA and chromatin are similar and quantitative analysis revealed only differences below a factor of two (data not shown). Thus, under those conditions, there is no dramatic photofootprint generated by proteins bound in the ARS1 region.
The intensity of bands at pyrimidine sites decreased with increasing PR time (Fig. 1, lanes 8–14) but no change was observed when cells were kept in the dark (Fig. 1, lane 15). This demonstrates efficient DNA repair by photolyase and inactivation of NER by deletion of the RAD1 gene (rad1Δ). From visual inspection, it is obvious that there is substantial repair heterogeneity with fast and slowly repaired sites around the A and B1 elements in the nucleosome (Fig. 1B) as well as in the B2 and B3 region (Fig. 1A and C).
To obtain a quantitative picture of CPD repair by photolyase, the fraction of molecules damaged at a particular site was calculated for all sites and repair times and the fraction of non-CPD photoproducts (Fig. 1, lanes 7) was subtracted. Repair curves were calculated only if the initial yield of CPDs was at least 0.1%. In our experiments, the damage was sufficiently high at most pyrimidine clusters containing three or more pyrimidines (dark boxes). C-rich pyrimidine clusters and TC, CT, CC and most TT dipyrimidine sites were excluded from the quantitative analysis, since CPD yields were too low or the level of 6-4PPs was too high (open boxes). However, signals of lesions which are flanked by adenines were sufficiently high, e.g. TT 1943 (Fig. 1C) and TT 2056 (Fig. 1B).
Heterogeneity of photoreactivation in ARS1
The repair curves for CPD repair by photolyase are shown in Figure 2. They quantitatively confirm the repair heterogeneity. Some sites are rapidly repaired; other lesions are rather slowly removed. Some data points are best fitted with an exponential curve. At other sites, the repair process appears to be biphasic with a rapid and a slow component. The curves were used to calculate half-lives of DNA lesions (T50%). The T50% values were graphically summarised (see Fig. 5A). The average half-lives were calculated for the ARS1 region without the flanking nucleosome. The average of 11 sites in the top strand (Figs 1C and 2B, all sites) was 12.1 ± 8.4 min. The average of 12 sites in the bottom strand (Fig. 1A, sites TCTTTT 1895–TTT 2028, Fig. 2A) was 10.1 ± 6.7 min. These results show that PR is very efficient in the nucleosome-free region of ARS1. The average values of top and bottom strand are similar; thus, there is no general strand bias for PR in ARS1.
Figure 2.

Repair curves for PR in the bottom (A) and top strand (B). Initial DNA damage for each site was set to 100% (Fig. 1, lanes 8); initial damage without CPDs (Fig. 1, lanes 7) was set to 0%. Graphs show DNA damage (%) versus repair time (minutes), data points used to fit with exponential curves (circles, thick lines), data points not used for exponential curves (squares), biphasic curves with fast exponential and slow linear curves (CTTTTC 2002, CTT 2006, TCTTTT 2015, TT 1968, TTT 1971, CTT 1979, CCTTTTT 1991). Data points to calculate positions 1965–2053 and 1896–1991 were averaged from two gels. (There is no significant difference between results from different gels.) All other values were obtained from one gel.
Figure 5.

Comparison of DNA repair in ARS1 of YRpTRURAP with chromatin structure. (A) PR; (B) NER. Indicated are a nucleosome positions (ovals), presumed nucleosome positions (dashed ovals) and functional elements of ARS1 (boxes B3, B2, B1 and A). Bars show the time (min) used to remove 50% of the lesions (T50%). T50% values were calculated from repair curves (Figs 2 and 4). Asterisks indicate data points which could be quantified in one panel only (A or B).
The graphic display of repair curves illustrates that the PR in vivo is not determined by the DNA sequence, since the same sequences at different positions of the ARS1 region generate clearly different repair curves (e.g. Fig. 2A, compare TCTTTT 1895 and TCTTTT 2015). Thus, the heterogeneity of repair curves is likely to reflect an interaction of photolyase with proteins bound in the nuclease-sensitive region and the flanking nucleosome.
The slowest repair was observed in TTTTCCT 2096 within the nucleosome flanking the A element (T50% = 76 min), whereas repair at the edges of the nucleosome was much faster (e.g. TCTT 2045, T50% = 4 min). This result is consistent with repair in other nucleosomes (B.Suter and F.Thoma, unpublished results) and suggests a modulation of PR by nucleosomes. In addition, a distinct modulation of PR was found within the B3 element. Repair in TTTC 1914 (T50% = 21 min; Fig. 2B) and in TTT 1913 (T50% = 18 min; Fig. 2A) was about three to four times slower than in TTTTT 1925 (T50% = 6 min; Fig. 2A). Since the B3 element is the binding site for Abf1, Abf1 binding is a likely explanation for the observed heterogeneity of PR in B3. The result implies that Abf1 remains bound despite the presence of the UV lesion. In the A element, which is the binding site for the ORC (32,33), repair was surprisingly fast (TTTT 2028, T50% = 14 min; TTT 2021, T50% = 5 min; Fig. 2A). Thus ORC did not strongly inhibit photolyase or the ORC was displaced by the DNA lesion.
For slowly repaired lesions in B1 (bottom strand; CTTTTC 2002, CTT 2006, TCTTTT 2015) and between B3 and B1 (top strand; TT 1968, TTT 1971, CTT 1979, CCTTTT 1991), the calculated data are best described by biphasic curves (Fig. 2A and B). These curves consist of a fast early component, which fits exponentially, and a slow late component, which fits a linear function. This result might reflect two different populations of minichromosomes, one containing proteins that inhibit repair.
In summary, the modulation of photorepair strongly suggests that photolyase is affected by nucleosomes and by other proteins in the ARS1 region. It indicates that those proteins remain bound despite of the DNA lesion.
Nucleotide excision repair in ARS1: a comparison with photoreactivation
NER in ARS1 of YRpTRURAP was analysed in strain FTY23 (Figs 3, 4 and 5B). NER does not discriminate between CPDs and 6-4PPs. Therefore non-irradiated DNA was used for background subtraction. Since 6-4PPs are generated at 3–10 times lower yields than CPDs, the repair results preferentially relate to CPD repair and are comparable to the PR data. The repair data were fitted to exponential curves (Fig. 4) and the T50% values were calculated and compared with PR (Fig. 5).
Figure 3.

Primer extension analysis of NER (dark repair) in ARS1 of minichromosome YRpTRURAP. UV photoproducts in the bottom (A) and top strand (B). Irradiation conditions, repair times, chromatin structure, ARS1 elements and pyrimidine sites are illustrated as described in Figure 1. Dideoxy-sequencing reactions A, G, C and T (lanes 1–4); DNA irradiated in vitro (40 J/m2, lane 5); DNA of non-irradiated cells (0 J/m2, lane 6); DNA of irradiated cells (chromatin; 100 J/m2, lanes 7–13). For NER, cells were incubated in the dark for 20–150 min (lanes 8–13).
Figure 4.

Repair curves for NER in the bottom (A) and top strand (B). Initial DNA damage for each site was set to 100% (Fig. 3, lanes 7). Background from non-irradiated DNA (Fig. 3, lanes 6) was set to 0%. Plotted is DNA damage (%) versus repair time (minutes). Repair curves represent exponential fits. Data points are averages of two (top strand) and three (bottom strand) independent experiments (material of the same experiments as described by R.-E.Wellinger and F.Thoma, 7).
In contrast with PR, NER follows similar kinetics at all lesions (Fig. 4A and B). NER in the nucleosome-free ARS1 is characterised by an average of T50% values of 80.3 ± 22.4 min in the top strand (11 lesions, CTCT 1895–TCT 2016). The average of 12 lesions in the bottom strand (TCTTTT 1895–TTT 2028) is 73.1 ± 20.3 min and not significantly different from the top stand. Thus, averaged T50% values indicate the absence of strand specificity for NER in ARS1. Compared with PR, repair by NER is roughly six to seven times slower in the entire ARS1 region and no site was found which is faster repaired by NER than by PR (note the different scales for NER and PR in Fig. 5). Thus, under our conditions, PR is the predominant mechanism for CPD repair in ARS1. Surprisingly, NER in ARS1 was slower than NER in the nucleosome-free URA3 promoter of the same minichromosome (the same repair experiment; T50% ∼40 min) (7) indicating differential modulation of NER in nucleosome-free regions.
T50% values of NER vary <4-fold (Fig. 5B). The slowest repair was in TTTTCCT 2096 (T50% = 167 min) of the flanking nucleosome. Fast repair was found around B3 (e.g. CTCT 1896, T50% = 44 min, TTTCTT 1935, T50% = 55 min) and slow repair was detected in B1 and A elements (e.g. TCT 2016, T50% = 94 min, TTTT 2028, T50% = 107 min). In the B3 region, NER was slow in TTTC 1914 (T50% = 130 min) and TTT 1913 (T50% = 93 min). These sites were also slowly repaired by photolyase (Fig. 5). Hence, NER and PR were modulated similarly in the ARS1 flanking nucleosome and in the B3 region. However, in contrast with the strong heterogeneity in PR in B2 and A, NER is only moderately modulated.
Altered modulation of photoreactivation in ARS1 of minichromosome YRpCS1
To further test whether the repair heterogeneity depends on the DNA sequence or on proteins interacting with the ARS1 region, PR was analysed in the ARS1 region of minichromosome YRpCS1 (Fig. 6). In this minichromosome, the ARS1 is rarely transcribed from an upstream promoter, which partially impairs replication. Transcription stops at the ABF1 site (34). However, the chromatin structure of the ARS1 regions (determined by nuclease digestion), is similar to that of YRpTRURAP (21).
Figure 6.
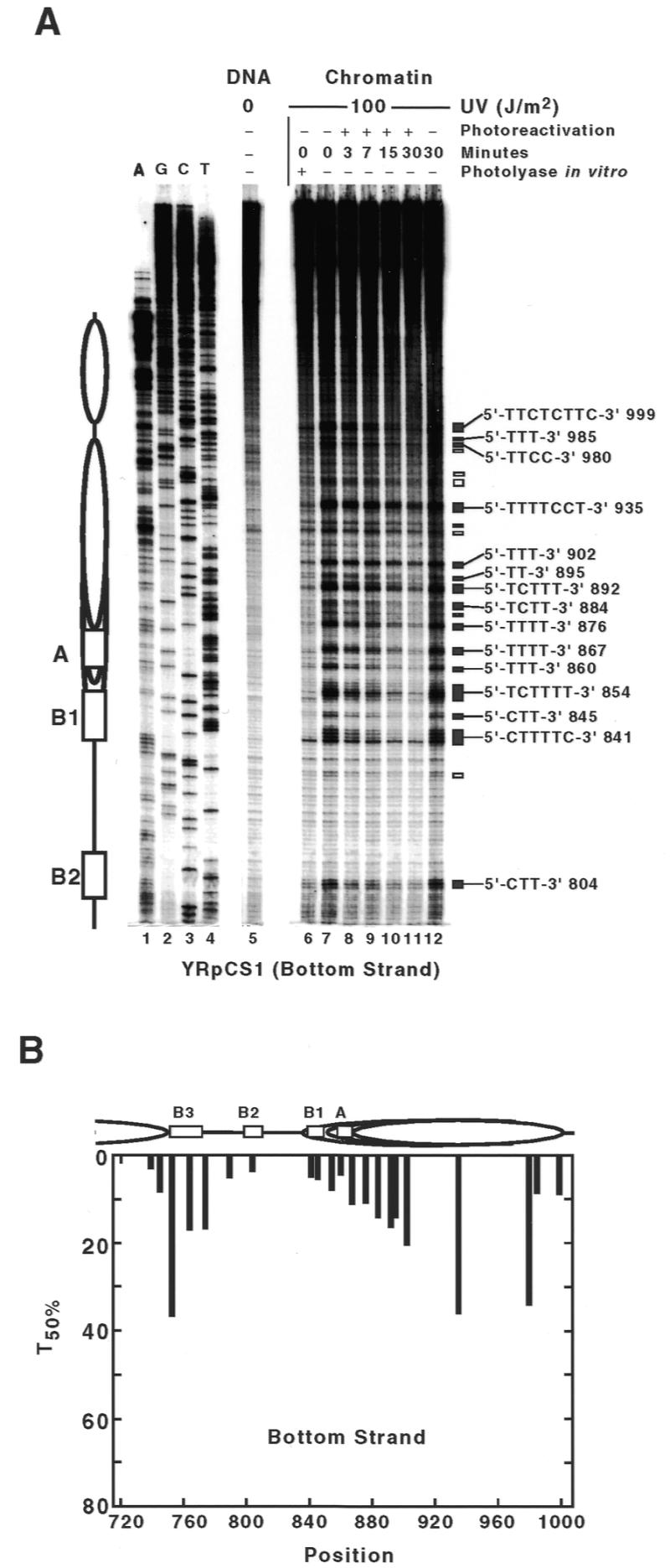
CPD repair by photolyase in the bottom strand of ARS1 in minichromosome YRpCS1. (A) Primer extension; (B) comparison of DNA repair with chromatin structure. Conditions and structural interpretation are indicated as in Figures 1 and 5. Note that the nucleotide positions in YRpCS1 and YRpTRURAP differ by 1161 bp. The position of ARS1 elements are B3 (751–767), B2 (798–808), B1 (835–848) and A (858–868).
The modulation of PR was different at some but not all sites compared with YRpTRURAP (compare Fig. 6B with Fig. 5A). Different repair was found in the B1 and B2 regions, where lesions are ~4-fold faster repaired in YRpCS1 than in YRpTRURAP (see TCTTTT 854, CTT 845, CTTTTC 841; Fig. 6A). Fast repair of this region in YRpCS1 suggests an improved accessibility of photolyase to the DNA. Less heterogeneous repair of this region demonstrates that the observed modulation of CPD repair by photolyase in YRpTRURAP is not a sequence-related effect. In the A element, repair in YRpCS1 is similar to YRpTRURAP (compare lesion TTTT 867, Fig. 6A, with TTTT 2028, Fig. 1B). This indicates that the DNA is well accessible to photolyase in the A element of both minichromosomes. Altered modulation in YRpCS1 was also found in the nucleosome position flanking ARS1 (Fig. 6B). Lesions in TTTTCCT 935 were repaired faster in YRpCS1 than the corresponding lesions in YRpTRURAP, whereas lesions at the edge of the nucleosome were repaired slower (e.g. TTT 902). Altered repair in the ARS1 flanking nucleosome might reflect a slightly altered nucleosome position.
DISCUSSION
DNA damage formation and repair is tightly linked to protein–DNA interactions in chromatin. This has been inferred from promoter regions interacting with regulatory proteins as well as for inhibition of repair in nucleosomes (6,13,14). Here, we have investigated the individual contributions of NER and PR to remove UV-induced DNA lesions from active origins of replication ARS1 in yeast S.cerevisiae using minichromosomes as model substrates.
In a previous low resolution study, we noticed that NER was relatively slow in ARS1 (28). This was surprising, since the ARS1 region was known to be nuclease sensitive (20,21), thus presumably free of nucleosomes. One hypothesis is that proteins of the replication complex inhibit damage accessibility and processing or that chromatin remodels after damage formation, e.g. by folding of ARS1 in a nucleosome. The results shown here demonstrate very efficient repair by photolyase within the nuclease-sensitive region for both strands and a much slower repair by NER. First, this demonstrates that PR is the predominant mechanism for repair of the major UV photoproducts in ARS1. NER is slower, but responsible for removal of the non-CPD lesions. The low amount of 6-4PPs does not account for the slow NER. Second, since PR is slow in nucleosomes, but fast in non-nucleosomal regions and linker DNA (9), photolyase can be used in vivo to assess whether a damage is incorporated in a nucleosome or not. Fast repair by photolyase in the ARS1 region shows that the CPDs are accessible and not packaged in nucleosomes. Therefore, we can conclude that no remodelling or rearrangement of nucleosomes occurs in the ARS1 region as a consequence of DNA damage induction.
Slow removal of DNA lesions by NER can be explained in two ways. First, photolyase might be more abundant than the NER proteins and therefore hits the lesions preferentially. Second, in contrast to PR, NER is a complex multistep pathway, which includes damage recognition, excision of the lesion and repair synthesis. It might, therefore, take more time to assemble the NER machinery and to execute the repair process. This could also be affected by limiting amounts of one of the NER components.
Interference of NER with regulatory proteins has been used as an argument to explain inefficient repair at binding sites of transcription factors in promoter regions (10,11,35). Moreover, TATA-binding protein was shown to inhibit repair by photolyase in vitro and in the initiation complex of the SNR6 gene in living yeast cells (12). Here, we observed a modulation of NER and PR in ARS1. Since that region is nucleosome free, the observed heterogeneity most likely relates to interference with proteins that are associated with ARS1 (26). The altered heterogeneity pattern which was observed in YRpCS1 supports this conclusion. In that particular minichromosome, rare transcription elongates through ARS1 and stops at the Abf1 binding site (B3) (34). Transcription possibly disturbs protein–DNA interactions in the ARS1 region, which leads to an altered repair heterogeneity.
Generally, the repair heterogeneity by NER appears less pronounced than by PR reflecting different modes of damage recognition and processing. Saccharomyces cerevisae photolyase is a moderately sized protein of 565 amino acids (36), which apparently works as a single protein and does not require a lot of space. In contrast, processing of a lesion by NER requires building up large complexes (37). Several proteins are required for damage recognition and many more for the excision of the lesion and DNA-repair synthesis (2,6). The excision complex generates a footprint of ∼30 bp (38) and requires ∼100 bp substrate size for excision (39). Since the spatial requirement for NER is considerably higher than that of photolyase, assembly of NER factors could be affected by bound factors further apart from the lesion site. Thus, function of NER proteins may not only be affected at the level of DNA damage recognition, but also by impaired assembly of the repair machinery.
Recently, Abf1 was identified as a component of the Rad7/Rad16 NER subcomplex suggesting a role for Abf1 protein during NER in yeast (40). In this context, it is interesting to note that the Abf1 binding site was slowly repaired by NER and PR indicating that Abf1 itself remains bound after damage formation. If Abf1 would cooperate with Rad7/Rad16 in damage recognition, using ARS1 as a starting point for DNA repair, we intuitively would expect faster repair at this site. The averaged T50% values for NER in ARS1 (80 min for the top strand and 73 min for the bottom strand) are, however, even higher than the average T50% values of the URA3 promoter (∼40 min) (7), which does not contain an Abf1 site and is also nuclease sensitive. Thus, in the case of the Abf1 binding site of ARS1, Abf1 does not seem to have a stimulatory role for NER, but it will be interesting to investigate whether Abf1 has an effect on repair of the other lesions and other chromatin regions.
At several sites, we observed biphasic repair curves for NER. Since the experiments were done with growing cultures, the most convenient explanation is that the different repair rates reflect different subpopulations of minichromosomes. The slow component of the repair course may reflect repair of minichromosomes in G1 cells, where B1 and B2 elements are bound by components of the pre-RC that renders lesions inaccessible (26). In the other phases of the cell cycle, lesions in B1 and B2 elements would be accessible and fast repaired. This interpretation is consistent with the ORC and cell cycle-dependent modulation of UV photofootprints in the ARS region reported recently (41).
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr A. Sancar for photolyase and Dr U. Suter for continuous support. This work was supported by grants from the Swiss National Science Foundation and by the Swiss Federal Institute of Technology (ETH, grants to F.T.)
REFERENCES
- 1.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington DC.
- 2.de Laat W.L., Jaspers,N.G. and Hoeijmakers,J.H. (1999) Genes Dev., 13, 768–785. [DOI] [PubMed] [Google Scholar]
- 3.Wood R.D. (1999) Biochimie, 81, 39–44. [DOI] [PubMed] [Google Scholar]
- 4.Yasui A., Eker,A.P., Yasuhira,S., Yajima,H., Kobayashi,T., Takao,M. and Oikawa,A. (1994) EMBO J., 13, 6143–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sancar A. (1996) Science, 272, 48–49. [DOI] [PubMed] [Google Scholar]
- 6.Thoma F. (1999) EMBO J., 18, 6585–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wellinger R.E. and Thoma,F. (1997) EMBO J., 16, 5046–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tijsterman M., de Pril,R., Tasseron-de Jong,J.G. and Brouwer,J. (1999) Mol. Cell. Biol., 19, 934–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suter B., Livingstone-Zatchej,M. and Thoma,F. (1997) EMBO J., 16, 2150–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao S.W., Drouin,R. and Holmquist,G.P. (1994) Science, 263, 1438–1440. [DOI] [PubMed] [Google Scholar]
- 11.Tu Y.Q., Tornaletti,S. and Pfeifer,G.P. (1996) EMBO J., 15, 675–683. [PMC free article] [PubMed] [Google Scholar]
- 12.Aboussekhra A. and Thoma,F. (1999) EMBO J., 18, 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tornaletti S. and Pfeifer,G.P. (1996) Bioessays, 18, 221–228. [DOI] [PubMed] [Google Scholar]
- 14.Smerdon M.J. and Conconi,A. (1999) Prog. Nucleic Acids Res. Mol. Biol., 62, 227–255. [DOI] [PubMed] [Google Scholar]
- 15.Smerdon M. and Thoma,F. (1998) In Nickoloff,J.A. and Hoekstra,M.F. (eds), DNA Damage and Repair, Vol. 2: DNA Repair in Higher Eukaryotes. Humana Press, Totowa, NJ, pp. 199–222.
- 16.Tommasi S., Swiderski,P.M., Tu,Y., Kaplan,B.E. and Pfeifer,G.P. (1996) Biochemistry, 35, 15693–15703. [DOI] [PubMed] [Google Scholar]
- 17.Liu X., Conconi,A. and Smerdon,M.J. (1997) Biochemistry, 36, 13710–13717. [DOI] [PubMed] [Google Scholar]
- 18.Fangman W.L. and Brewer,B.J. (1991) Annu. Rev. Cell. Biol., 7, 375–402. [DOI] [PubMed] [Google Scholar]
- 19.Marahrens Y. and Stillman,B. (1992) Science, 255, 817–823. [DOI] [PubMed] [Google Scholar]
- 20.Thoma F., Bergman,L.W. and Simpson,R.T. (1984) J. Mol. Biol., 177, 715–733. [DOI] [PubMed] [Google Scholar]
- 21.Thoma F. (1986) J. Mol. Biol., 190, 177–190. [DOI] [PubMed] [Google Scholar]
- 22.Losa R., Omari,S. and Thoma,F. (1990) Nucleic Acids Res., 18, 3495–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bell S.P. and Stillman,B. (1992) Nature, 357, 128–134. [DOI] [PubMed] [Google Scholar]
- 24.Rao H. and Stillman,B. (1995) Proc. Natl Acad. Sci. USA, 92, 2224–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diffley J.F. and Stillman,B. (1988) Proc. Natl Acad. Sci. USA, 85, 2120–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diffley J.F., Cocker,J.H., Dowell,S.J. and Rowley,A. (1994) Cell, 78, 303–316. [DOI] [PubMed] [Google Scholar]
- 27.Santocanale C. and Diffley,J.F. (1996) EMBO J., 15, 6671–6679. [PMC free article] [PubMed] [Google Scholar]
- 28.Smerdon M.J. and Thoma,F. (1990) Cell, 61, 675–684. [DOI] [PubMed] [Google Scholar]
- 29.Suter B., Livingstone-Zatchej,M. and Thoma,F. (1999) Methods Enzymol., 304, 447–461. [DOI] [PubMed] [Google Scholar]
- 30.Maniatis T., Fritsch,E. and Sambrook,J. (1982) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, NY.
- 31.Wellinger R.E. and Thoma,F. (1996) Nucleic Acids Res., 24, 1578–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diffley J.F. and Cocker,J.H. (1992) Nature, 357, 169–172. [DOI] [PubMed] [Google Scholar]
- 33.Bell S.P. and Stillman,B. (1992) Nature, 357, 128–134. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka S., Halter,D., Livingstone-Zatchej,M., Reszel,B. and Thoma,F. (1994) Nucleic Acids Res., 22, 3904–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tu Y., Bates,S. and Pfeifer,G.P. (1997) J. Biol. Chem., 272, 20747–20755. [DOI] [PubMed] [Google Scholar]
- 36.Sancar G.B. (1985) Nucleic Acids Res., 13, 8231–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Svejstrup J.Q., Wang,Z.G., Feaver,W.J., Wu,X.H., Bushnell,D.A., Donahue,T.F., Friedberg,E.C. and Kornberg,R.D. (1995) Cell, 80, 21–28. [DOI] [PubMed] [Google Scholar]
- 38.Wakasugi M. and Sancar,A. (1998) Proc. Natl Acad. Sci. USA, 95, 6669–6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang J.C. and Sancar,A. (1994) J. Biol. Chem., 269, 19034–19040. [PubMed] [Google Scholar]
- 40.Reed S.H., Akiyama,M., Stillman,B. and Friedberg,E.C. (1999) Genes Dev., 13, 3052–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujita M., Hori,Y., Shirahige,K., Tsurimoto,T., Yoshikawa,H. and Obuse,C. (1998) Genes Cells, 3, 737–749. [DOI] [PubMed] [Google Scholar]