

Abstract
Background:
Severe hypercholesterolemia, defined as low-density lipoprotein cholesterol (LDL-C) measurement ≥ 190 mg/dl, is associated with increased risk for coronary artery disease (CAD). Causes of severe hypercholesterolemia include: monogenic familial hypercholesterolemia (FH), polygenic hypercholesterolemia, elevated lipoprotein(a) [Lp(a)] hypercholesteremia, polygenic hypercholesterolemia with elevated Lp(a) (two-hit), or non-genetic hypercholesterolemia. The added value of using a genetics approach to stratifying risk of incident CAD among those with severe hypercholesterolemia versus using LDL-C levels alone for risk stratification is not known.
Methods:
To determine whether risk stratification by genetic cause provided better 10-year incident CAD risk stratification than LDL-C level, a retrospective cohort study comparing incident CAD risk among severe hypercholesterolemia subtypes (genetic and non-genetic causes) was performed among 134,185 UK Biobank participants. Analyses were limited to unrelated, White British or Irish participants with available exome sequencing data. Participants with cardiovascular disease at baseline were excluded from analyses of incident CAD.
Results:
Of 134,185 individuals, 70,637 (52.6%) were female, and the mean (SD) age was 56.7 (8.0) years. 9.0% of the cohort met severe hypercholesterolemia criteria. Participants with LDL-C between 210-229 mg/dL and LDL-C ≥230 mg/dL showed modest increases in incident CAD risk relative to those with LDL-C between 190-209 mg/dL (hazard ratio [HR] (95% confidence interval [CI]) 210-229 mg/dL: 1.3 (1.1-1.6); ≥230 mg/dL: 1.3 (1.0-1.7). In contrast, when risk was stratified by genetic subtype, monogenic FH, elevated Lp(a), and two-hit hypercholesterolemia subtypes had increased rates of incident CAD relative to the non-genetic hypercholesterolemia subtype (HR (95% CI): monogenic FH: 2.3 (1.4-4.0); elevated Lp(a): 1.6 (1.2-2.0); two-hit: 1.9 (1.4-2.6)), while polygenic hypercholesterolemia did not.
Conclusions:
Genetics-based subtyping for monogenic FH and Lp(a) in those with severe hypercholesterolemia provided better stratification of 10-year incident CAD risk than LDL-C-based stratification.
Keywords: CAD, ASCVD, FH, hypercholesterolemia, Genetics, Polygenic, lipoprotein(a), LDL
Graphical Abstract
Introduction
Severe hypercholesterolemia is associated with increased risk for coronary artery disease (CAD)1,2. Current guidelines define severe hypercholesterolemia as the presence of a low-density lipoprotein cholesterol (LDL-C) measurement ≥ 190 mg/dl. Several distinct genetic determinants are known to cause the disorder including the presence of a monogenic familial hypercholesterolemia (FH) causing variant3,4, an elevated serum concentration of lipoprotein (a) (Lp(a)) which is primarily controlled by genetic variation in and near the LPA gene5, and polygenic hypercholesterolemia caused by the inheritance of a large burden of common alleles that are each associated with incremental increases of LDL-C6,7.
There is controversy in the field regarding the clinical value of measuring genetic determinants of severe hypercholesterolemia8. The controversy stems from the hypothesis that LDL-C levels alone carry sufficient information to guide therapy without measuring genetic markers of chronic, lifelong exposure to elevated LDL-C. Supporting the argument for identifying genetic determinants is the observation that patients with a monogenic FH variant have three times higher CAD risk than those without a variant at any given level of LDL-C9 and the knowledge that Lp(a) is a predictor of CAD risk independent of other risk factors5. Polygenic hypercholesterolemia is also thought to increase CAD risk, but its independence from LDL-C is less well established6.
In this study, we examined four genetic determinants of severe hypercholesterolemia in the UK Biobank. For each determinant, we established its contribution to disease incidence and the proportion of risk among severe hypercholesterolemia cases directly attributable to exposure. Next, we created a severe hypercholesterolemia cohort and classified participants into five mutually exclusive subtypes defined by the four genetic determinants: 1) monogenic FH caused by a pathogenic or likely pathogenic (P/LP) variant in a canonical FH gene 2) polygenic hypercholesterolemia caused by an LDL-C polygenic score in the top decile of the population10, 3) elevated Lp(a) hypercholesterolemia defined as a serum Lp(a) concentration ≥125 nmol/L (50mg/dL)11,12 4) two-hit hypercholesterolemia defined as coexisting elevated Lp(a) and polygenic hypercholesterolemia, and 5) non-genetic, defined as the absence of a genetic or molecular determinant13. We also investigated the impact of lipid lower medication (LLM) on outcomes by genetic subtype. Time-to-event analyses were used to test if subtyping provides risk stratification of 10-year incident CAD beyond stratification by LDL-C levels.
Methods
Availability of Data
All anonymized electronic health record (EHR), self-report, and genetic data used here can be obtained directly from the UK Biobank. Analysis scripts are available upon reasonable request to the authors.
Participants
This study was approved by the Geisinger Institutional Review Board. The UK Biobank is a large epidemiological cohort with genetic data, self-reported health data, and linkage to hospital inpatient records14. UK Biobank participants were 40-69 years old when recruited. This study was conducted from January 2021 to January 2023, under UK Biobank project number 49945. The UK Biobank has ethical approval from the North West Multi-Centre Ethics Committee. All participants provided informed consent to participate in UK Biobank projects.
Several exclusion criteria were applied to the 200,602 participants with exome sequences available for analysis (Figure 1). To reduce confounding by genetic ancestry and relatedness between participants, the cohort was filtered to only include individuals of white British or Irish ancestry. First-degree relatives were removed using the UK Biobank tools package in R15. Those with missing LDL-C, high-density lipoprotein cholesterol (HDL), systolic blood pressure measurements, or smoking status were also excluded. Exome sequences of the remaining 149,326 UK Biobank participants were screened for FH-causing variants in canonical FH genes (Table S1; Supplemental Methods)16. 65 unique FH-causing variants were identified in 357 individuals, 204 of whom had LDL-C ≥ 190mg/dL. Individuals who did not have a monogenic FH variant but had a secondary cause of hypercholesterolemia (Table S2) or were missing imputed genotype data or Lp(a) measurements were also excluded.
Figure 1.

Flow chart of study design. 130,091 UK Biobank participants with exome sequences available were classified by molecular and genetic determinants of severe hypercholesterolemia. 11,738 participants with severe hypercholesterolemia were classified into subtypes based on the presence of corresponding determinants. Those without a determinant were classified as the non-genetic subtype. LDL-C = low-density lipoprotein cholesterol; HDL = high-density lipoprotein cholesterol; BP = blood pressure; FH = familial hypercholesterolemia; PRS = polygenic risk score; Lp(a) = lipoprotein (a); CAD = coronary artery disease.
Genetic instruments for prediction of polygenic hypercholesterolemia and elevated Lp(a) were calculated from imputed genotype array data. This study used genotypes imputed from the Haplotype Reference Consortium reference panel17. Polygenic risk for hypercholesterolemia was estimated in the filtered cohort using the PRS-CS software. For variant weights, we used summary statistics from a genome-wide association study of quantitative LDL-C levels performed on a European ancestry sample reported by the Global Lipids Genetics Consortium7. Genetically-predicted Lp(a) was calculated using the same genetic instrument for predicted Lp(a) levels created by Burgess et al.18 and reported in the UK Biobank by Trinder et al.5 This Lp(a) score includes 43 variants in the LPA gene region (Supplemental Methods).
Classification of Severe Hypercholesterolemia Determinants and Subtypes
Those with lipid-lowering medication (LLM) use at baseline (defined by self-reported data collected during an in-person interview and questionnaire) (Table S3) had LDL-C values adjusted by dividing direct LDL-C by 0.7 to approximate the untreated state (Supplemental Methods)9. Those with LDL-C ≥ 190 mg/dL after adjustment were considered to have severe hypercholesterolemia. This approach was supported by sensitivity analyses (Table S4, Table S5).
The severe hypercholesterolemia cohort, containing only individuals with LDL-C ≥ 190 mg/dL, was stratified using two approaches: 1) LDL-C-based and 2) subtyping-by genetic determinant. In the LDL-C-based stratification, the cohort was divided into three groups: LDL-C ≥ 190 and < 210 mg/dL, LDL-C ≥ 210 and < 230 mg/dL, and ≥ 230 mg/dL. For the genetic stratification, each participant was classified into one of five mutually exclusive subtypes based on genetic determinants of severe hypercholesterolemia: 1) monogenic FH caused by a P/LP variant in a canonical FH gene 2) polygenic hypercholesterolemia defined by an LDL-C polygenic score in the top decile of the population, a commonly used threshold for designating high genetic risk in analyses of polygenic risk scores, including hypercholesterolemia10,19 and atherosclerotic cardiovascular disease (ASCVD)6 3) elevated Lp(a) hypercholesterolemia defined as a serum Lp(a) concentration ≥125 nmol/L (50 mg/dL) based on the 2018 American Heart Association guidelines for high risk12 4) two-hit hypercholesterolemia defined as coexisting elevated Lp(a) and polygenic hypercholesterolemia, and 5) non-genetic, defined as the absence of a genetic or molecular determinant13 (Table S6).
Baseline Clinical Characteristics and Outcomes
CAD and cardiovascular disease (CVD) phenotypes were defined by self-reported disease and inpatient diagnoses as described in Elliot et al.20. CAD was defined as heart attack or myocardial infarction and related sequelae, identified using ICD-9 and ICD-10 codes and self-report; and coronary angioplasty or coronary artery bypass grafts, identified using OPCS-4 codes and self-report (Table S7). The CVD definition includes CAD in addition to angina, transient ischemic attack, and ischemic stroke. Peripheral artery disease (PAD) and aortic valve stenosis were defined following Bjornsson et al.21. The American Heart Association’s (AHA) Lifestyle Score (poor, intermediate, or ideal) was determined for UK Biobank participants following Said et al.22 (Supplemental Methods).
Statistical Analyses
To calculate the likelihood of having severe hypercholesterolemia at baseline for each determinant relative to the remainder of the cohort, generalized linear models stratified by age binned in 5 year increments, sex, and controlling for the first five principal components of ancestry were performed in R version 4.1.123. Population attributable fraction (PAF) is the proportional reduction in disease that would occur if exposure to a risk factor were removed from the population. Attributable risk proportion (ARP) is an estimate of the proportion of risk among the exposed that can be directly attributed to the exposure of interest. PAF and ARP were calculated using the twoxtwo library in R24.
Linear and logistic regression were used to determine whether each genetically-defined subtype differed from the non-genetic subtype in baseline CAD risk factors, comorbidities, and self-reported family history of heart disease. All regression analyses controlled for age, sex, and the first five principal components of ancestry. Forest plots were visualized using the ggplot2 package in R25. P-values were adjusted for multiple tests using Benjamini-Hochberg correction and considered significant if adjusted P<0.0526.
Due to the increased risk of myocardial infarction (defined here as CAD) in patients with a history of myocardial infarction, angina, transient ischemic attack, or ischemic stroke (defined here as CVD), individuals with pre-existing CVD were removed prior to the analysis of incident CAD27. Incident CAD was defined as a first-time CAD event occurring over the 10 years immediately following assessment at the UK Biobank assessment center. Cox proportional hazard regression models were fitted using the survival package in R28, stratifying by age, sex, baseline LLM use, smoking status, diabetes status, and controlling for systolic blood pressure (BP), HDL, LDL-C and the first five principal components of ancestry, where appropriate. A sensitivity analysis additionally controlling for body mass index (BMI) and C-reactive protein was also performed. The model was stratified by covariates when necessary to meet proportionality assumptions. The proportionality assumption of every Cox model was tested with the cox.zph function, and all results were non-significant. Kaplan-Meier plots were generated using the survminer package in R29. Absolute risk of 10-year incident CAD was calculated following the method described by Austin (2010)30.
RESULTS
Prevalence of Determinants of Severe Hypercholesterolemia
The UK Biobank cohort comprised 130,091 individuals with exome sequence and genotype array data that met study inclusion criteria at the time of study (Table 1; Figure 1). 357 individuals (1 in 418; 0.24%) had a P/LP variant in an FH gene (Table S8); 3,325 (1 in 39; 2.6%) had a coexisting high LDL-C polygenic risk score (PRSLDL-C) and serum concentration of Lp(a) ≥ 125 nmol/L; 9,657 (1 in 13; 7.4%) had a high PRSLDL-C alone; and 17,940 (1 in 7; 13.8%) had a serum concentration of Lp(a) ≥ 125 nmol/L alone. Of these, 11,738 individuals (1 in 11, 9.0%) met criteria for severe hypercholesterolemia (LDL-C ≥ 190 mg/dL). The proportion of the five subtypes in the severe hypercholesterolemia cohort was 1.7% monogenic FH (n=204), 7.8% two-hit hypercholesterolemia (n=913), 17.6% polygenic hypercholesterolemia (n=2,070), 17.2% elevated Lp(a) (n=2,023), and 55.6% non-genetic (n=6,528) (Table 2).
Table 1.
Demographics
| Sample Size (N) | 130,091 |
| Age (Mean (SD)) | 56.68 (8.01) |
| Sex (% Female) | 52.6 |
| LDL-C (mg/dL) ((Mean (SD)) | 137.91 (33.27) |
| LDL-C (est. untreated) (mg/dL) ((Mean (SD)) | 145.73 (33.01) |
| BMI (Mean (SD)) | 27.22 (4.62) |
| Systolic BP (mmHg) (Mean (SD)) | 137.72 (18.49) |
| HDL-C (mg/dL) (Mean (SD)) | 56.71 (14.73) |
| Smoking (%) | 9.3 |
| Cholesterol-lowering medication (%) | 16.8 |
| Years follow-up ((Mean (IQR)) | 11.31 (1.63) |
Table 2.
Clinical characteristics and comorbidities of subtypes in the severe hypercholesterolemia cohort (n = 11,738).
| Non-genetic | Monogenic | Two-hit | Polygenic | Elevated Lp(a) | |
|---|---|---|---|---|---|
| Sample Size (N) | 6528 | 204 | 913 | 2070 | 2023 |
| Prevalence | 1 in 2 | 1 in 58 | 1 in 13 | 1 in 6 | 1 in 6 |
| Age (Mean (SD)) | 58.88 (6.99) | 56.9 (7.23) | 58.1 (7.31) | 58.02 (7.21) | 59.07 (6.67) |
| Sex (% Female) | 55.5 | 64.7 | 57.6 | 54.9 | 61.2 |
| LDL-C (est. untreated) (mg/dL) ((Mean (SD)) | 210.19 (21.46) | 236.1 (40.46) | 214.74 (24.74) | 212.68 (23.37) | 211.45 (22.58) |
| BMI (Mean (SD)) | 28.13 (4.24) | 27.57 (4.92) | 27.68 (4.5) | 28.03 (4.3) | 27.99 (4.43) |
| Systolic BP (mmHg) (Mean (SD)) | 143.04 (18.42) | 137.98 (18.82) | 141.21 (18.71) | 141.47 (18.52) | 143.02 (18.7) |
| HDL-C (mg/dL) (Mean (SD)) | 57.14 (13.11) | 55.67 (11.78) | 59.47 (14.45) | 57.33 (12.75) | 59.18 (13.59) |
| Triglycerides (mg/dL) (Mean (SD)) | 194.44 (74.71) | 146.53 (71.81) | 173.15 (72.44) | 187.83 (74.6) | 182.92 (72.35) |
| BP meds (%) | 23.7 | 20.1 | 20.4 | 19.6 | 26.1 |
| Smoking (%) | 11.3 | 8.3 | 10.5 | 9.6 | 9.7 |
| Cholesterol-lowering medication (%) | 29.8 | 56.4 | 32 | 28.1 | 32.5 |
| AHA Lifestyle Score (Mean (SD)) 1 | 0.99 (0.44) | 1.06 (0.49) | 1.01 (0.45) | 1.02 (0.43) | 1.03 (0.44) |
| Townsend Deprivation Index (Mean (SD)) 2 | −1.58 (2.9) | −1.47 (3.09) | −1.61 (2.94) | −1.6 (2.89) | −1.55 (2.82) |
| Prevalent T1D (%) | 14/6528 (0.2) | 2/204 (1) | 5/913 (0.5) | 2/2070 (0.1) | 4/2023 (0.2) |
| Prevalent T2D (%) | 18/6514 (0.3) | 0/202 (0) | 3/908 (0.3) | 3/2068 (0.1) | 4/2019 (0.2) |
| Prevalent CAD (%) | 2 | 3.9 | 1.4 | 1.7 | 2.9 |
| Prevalent CVD (%) | 6.7 | 8.3 | 4.9 | 4.7 | 6.6 |
| Prevalent PAD (%) | 0.4 | 1 | 0.5 | 0.1 | 0.5 |
| Prevalent AVS (%) | 0.2 | 0.5 | 0.1 | 0.1 | 0.2 |
AHA Lifestyle Score: A higher score indicates a more ideal lifestyle (0 = poor, 1 = intermediate, 2 = ideal).
Townsend Deprivation Index: A higher (less negative) score corresponds to a lower socioeconomic status.
Attributable Risk of Determinants to Severe Hypercholesterolemia
Individuals with an FH-causing variant were at the highest risk of severe hypercholesterolemia relative to the remainder of the cohort (adjusted odds ratio [OR], 13.9; 95% confidence interval [CI], 11.2-17.2) (Figure 2). Individuals with two determinants: both a PRSLDL-C in the top decile and a serum concentration of Lp(a) ≥ 125 nmol/L (OR, 4.1; 95% CI, 3.7-4.4), a PRSLDL-C in the top decile alone (OR, 3.2; 95% CI, 3.0-3.4), and serum concentration of Lp(a) ≥ 125 nmol/L alone (OR, 1.3; 95% CI, 1.2-1.4) were also at increased risk of having severe hypercholesterolemia. The PAF of FH-causing variants, two determinants, PRSLDL-C in the top decile alone, and a serum concentration Lp(a) ≥ 125 nmol/L alone were 1.5%, 5.4%, 11.0%, and 4.0% respectively; and the ARP were 84.4%, 68.9%, 62.5%, and 23.2% respectively (Table S9).
Figure 2.

Genetic determinants of severe hypercholesterolemia. A fan plot shows prevalence, odds ratio, and population attributable fraction for each determinant of severe hypercholesterolemia. The x and y coordinates of each point indicate the prevalence and odds ratio (OR), respectively, of each determinant. The population attributable fraction is represented by the y-axis intercept of the blue line connected to each point. The dashed horizontal black line demarcates an odds ratio of 1.
Clinical Characteristics of Severe Hypercholesterolemia Subtypes
The monogenic FH subtype was associated with the most striking differences in clinical characteristics relative to the non-genetic subtype including younger age, more likely to be female, higher LDL-C, lower HDL, lower triglycerides, higher rates of lipid-lowering medication at baseline, and use of a stronger statin compound (atorvastatin or rosuvastatin) (Figure S1A, Figure S2). The non-genetic subtype was associated with a poor AHA lifestyle score compared to polygenic and elevated Lp(a) hypercholesterolemia subtypes. The prevalence of comorbidities at baseline were similar among the subtypes with these exceptions: the monogenic FH and elevated Lp(a) hypercholesterolemia subtypes were associated with a higher prevalence of CAD at baseline (monogenic OR, 3.0; 95% CI, 1.3-6.0; elevated Lp(a) OR, 1.6; 95% CI, 1.2-2.2), and polygenic hypercholesterolemia was associated with a lower prevalence of type 2 diabetes (OR, 0.6; 95% CI, 0.5-0.8) and CVD (OR, 0.7; 95% CI, 0.6-0.9) (Figure S1B). The monogenic FH and elevated Lp(a) hypercholesterolemia subtypes were associated with the highest odds of reporting a first-degree relative with heart disease (monogenic OR, 2.0; 95% CI, 1.5-2.7; elevated Lp(a) OR, 1.3; 95% CI, 1.2-1.4) (Figure S3).
LDL-C Level Versus Subtyping for Risk Stratification of 10-Year Incident CAD
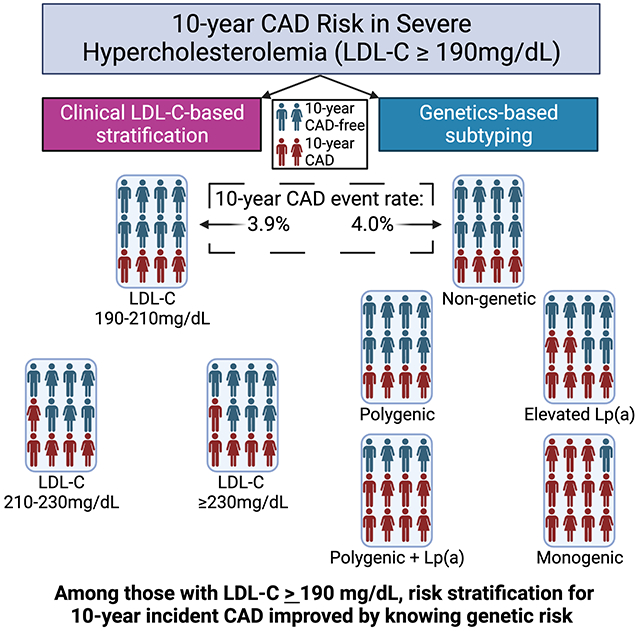
We observed 485 incident CAD events after removing 729 participants with prevalent CVD at baseline, corresponding to an event rate of 0.46% per person-year. Using the LDL-C-based stratification of the cohort, the LDL-C 210-229 mg/dL group (n=2,588) and the LDL-C ≥ 230 mg/dL group (n=1,593) were both associated with increased risk of incident CAD relative to the LDL-C 190-209 mg/dL group (n=6,828) (210-229 HR, 1.3; 95% CI, 1.1-1.7; ≥ 230 HR, 1.3; 95% CI, 1.0-1.7) (Figure 3A).
Figure 3.

Severe hypercholesterolemia stratified by LDL-C and genetic subtype. Kaplan-Meier plots show 10-year incident CAD risk among UK Biobank participants A) binned into three groups of increasing LLM-adjusted LDL-C: 190-210 mg/dL (reference), 210-230 mg/dL and >230 mg/dL and B) binned by non-genetic (reference) and genetic subtypes. Cox proportional-hazard ratios and 95% confidence intervals adjusted for CAD risk factors and principal components of ancestry are shown.
In the genetic determinant subtype analysis, the monogenic FH (n=187), two-hit (n=868), and elevated Lp(a) subtypes (n=1,889) were associated with increased risk of incident CAD relative to the non-genetic subtype (monogenic FH HR, 2.3; 95% CI, 1.4-4.0; two-hit HR, 1.9; 95% CI, 1.4-2.6; elevated Lp(a) HR, 1.5; 95% CI, 1.2-2.0) (Figure 3B). Within the monogenic FH subtype, those with an LDLR loss-of-function variant (n=17) had the highest incident CAD risk (HR, 8.9; 95% CI, 2.4-32.7), however the clinical significance cannot be determined due to the small sample size (Figure S4). The polygenic hypercholesterolemia subtype (n=1,972) was not associated with increased incident CAD. Having an LDL-C ≥ 230 mg/dL was associated with a 1.2 absolute 10-year incident CAD risk difference relative to having an LDL-C between 190-209 mg/dL (Table S10). In contrast, having the monogenic subtype was associated with a 4.8% absolute 10-year incident CAD risk difference relative to having the non-genetic subtype. The results were not sensitive to whether the polygenic and two-hit subtypes were defined by the top decile or top quartile of LDL-C PRS, another threshold that has been used for designating high genetic risk of hypercholesterolemia31 (Table S11). Using genetically predicted Lp(a) instead of measured Lp(a) did not meaningfully affect the results (Figure S5).
The previous analyses were repeated separately in males and females. In males, LDL-C-based stratification did not result in a significantly increased risk of incident CAD in either the LDL-C 210-229 mg/dL group or the LDL-C ≥ 230 mg/dL group relative to the LDL-C 190-209 mg/dL group (Figure S6A). When using genetic subtyping of males, the monogenic FH (HR, 2.3; 95% CI, 1.1-4.5) and two-hit (HR, 1.8; 95% CI, 1.3-2.6) subtypes were associated with increased risk of incident CAD relative to non-genetic (Figure S6B). In females, LDL-C-based stratification was incoherent, with a significantly increased CAD risk observed in the LDL-C 210-229 mg/dL group relative to the LDL-C 190-209 mg/dL group (HR, 1.8; 95% CI, 1.3-2.7), but no significant association between the LDL-C ≥ 230 mg/dL group relative to the LDL-C 190-209 mg/dL group (Figure S6C). When using genetic subtyping of females, the monogenic FH (HR, 3.2; 95% CI, 1.2-8.1), two-hit (HR, 2.3; 95% CI, 1.3-4.0), and elevated Lp(a) (HR, 2.2; 95% CI, 1.5-3.3) subtypes were associated with increased risk of incident CAD relative to non-genetic (Figure S6D).
LLM Sensitivity Analyses
Relative to those in the severe hypercholesterolemia cohort who were untreated with LLM at baseline, the LLM treated subgroup had an overall higher burden of CAD risk factors (Table S12), a higher prevalence of more severe LDLR loss-of-function variants (Figure S7), a higher incidence of CAD when not accounting for clinical risk factors, and a lower risk of incident CAD (HR, 0.7; 95% CI, 0.6-0.9) when controlling for clinical risk factors (Table S13). To address these complexities, the analysis of incident CAD risk by severe hypercholesterolemia subtype was repeated separating the LLM-treated and untreated subgroups. In the LLM-treated subgroup, the monogenic FH and the two-hit subtypes were associated with increased risk of incident CAD relative to the non-genetic subtype (monogenic HR, 3.1; 95% CI, 1.6-5.9; two-hit HR, 1.8; 95% CI, 1.1-3.0) (Figure 4). The remaining subtypes were not significantly different in risk from the non-genetic subtype. In the LLM-untreated subgroup, the two-hit subtype and the elevated Lp(a) subtype were associated with increased risk of CAD (two-hit HR, 1.9; 95% CI, 1.3-2.8; elevated Lp(a) HR, 1.7; 95% CI, 1.3-2.3). Too few individuals with the monogenic FH subtype were untreated at baseline (n=88), therefore while higher rates of CAD were observed relative to the non-genetic subtype, the difference did not achieve statistical significance (HR=1.5; 95% CI, 0.5-4.2). Polygenic hypercholesterolemia was not different from the non-genetic subtype with respect to CAD risk in either subgroup. Controlling for BMI and C-reactive protein did not affect the results (Table S14).
Figure 4.

10-year incident CAD risk stratified by LLM use. Forest plot shows 10-year incident CAD risk among LLM-treated (blue) and LLM-untreated (red) UK Biobank participants with a severe hypercholesterolemia subtype compared to non-genetic severe hypercholesterolemia. Cox proportional-hazard ratios with 95% confidence intervals are shown.
Discussion
Our results show that subtyping severe hypercholesterolemia by genetic determinants adds substantial information for incident CAD risk stratification compared to observable LDL-C levels. Furthermore, observable LDL-C levels are a poor proxy for the excess CAD risk conferred by genetic determinants as the risk gradient for 20 mg/dL incremental increases in LDL-C is much less than that observed by the presence of a monogenic FH variant or the combination of elevated Lp(a) and polygenic hypercholesterolemia. Our results show that polygenic hypercholesterolemia by itself does not confer substantial excess risk compared to LDL-C measurement and isolated Lp(a) is intermediate between these risk groups.
The investigation of polygenic and monogenic contributions to severe hypercholesterolemia and incident CAD reveals important distinctions in their prevalence and risk profile. Among the genetic determinants examined, the largest fraction (PAF = 11.0%) of severe hypercholesterolemia cases was attributable to polygenic risk in the top decile of the population, consistent with high heritability (h2 = 40-50%) estimates of LDL-C32. However, the rate of incident CAD among those with polygenic hypercholesterolemia was not different from that in individuals with non-genetic hypercholesterolemia, indicating that polygenic risk does not provide additional risk stratification information. In contrast, the monogenic FH and the two-hit subtypes were associated with similar increases in incident CAD risk (HR=2.3 vs. 1.9). This equivalence in risk is supported by data from the Copenhagen General Population Study which identified levels of Lp(a) that were associated with a similar future risk of CAD as those with FH33.
Genetic determinants are a major cause (ARP > 20%) of severe hypercholesterolemia among individuals with a genetic subtype, and the primary cause (ARP > 60%) in all severe hypercholesterolemia subtypes except elevated Lp(a). Comorbidities, CAD risk factors, medication use, and family history were different between subtypes of severe hypercholesterolemia. The monogenic FH subtype was associated with the highest prevalence of CAD at baseline, consistent with lifelong exposure to elevated LDL-C. Individuals with the monogenic FH subtype had fewer CAD risk factors relative to other subtypes, including a higher prevalence of female sex. The bias towards female sex may reflect premature mortality in men with an FH variant. This study confirms an association between low triglyceride levels and monogenic FH among severe hypercholesterolemia cases34. This finding supports the use of low triglyceride levels for phenotype-based identification of FH in EHR data35. Similarly, the higher family history of heart disease among those with the monogenic FH subtype demonstrates the importance of family history as a component of the FH clinical definition. An association with the elevated Lp(a) subtype and a family history of heart disease was also observed. A high serum concentration of Lp(a) is known to mimic the FH clinical phenotype through increased risk of hypercholesterolemia, CAD, and family history. This reveals a limitation in the specificity of using phenotypic definitions and diagnostic scores to identify FH in population-cohorts36.
The sensitivity analysis by LLM use revealed differences between subtypes that may inform treatment strategies. In the LLM treated group, the most excess CAD risk was observed among those with monogenic FH, further evidence of pervasive undertreatment of this disorder37. Despite the highest proportion of individuals reporting LLM use among genetic subtypes, this result may indicate that monogenic FH patients have treatment initiated too late in life38,39. Those with monogenic FH have been exposed to lifelong high levels of cholesterol, contributing to higher rates of incident CAD despite statin treatment; those with polygenic inheritance may have less chronic exposure40. The two-hit hypercholesterolemia subtype was associated with a two-fold increase of CAD risk in both the LLM-treated and untreated subgroups. Similar to monogenic FH, the excess risk associated with two-hit hypercholesterolemia in the LLM-treated group underscores the importance of early measurement of Lp(a) for risk stratification.
The elevated Lp(a) genetic subtype was defined using a widely accepted threshold of high Lp(a) serum concentration instead of a genetic instrument based on LPA genotypes. A previous study of the UK Biobank demonstrated that elevated Lp(a) defined with LPA genetic scores or serum concentration of Lp(a) have equivalent CAD risk5, a finding that was validated in a sensitivity analysis here.
Limitations
LLM use reported in the UK Biobank includes incomplete data on strength, frequency, and timeframe of use, as well as adherence to prescribed therapy, making it difficult to determine the precise therapy regimen the patient was taking when the LDL-C was measured. Adjusted LDL-C estimates are based on a single LDL-C measurement taken at baseline. The cumulative lifetime exposure to severe hypercholesterolemia was not accounted for. Estimates of FH variant prevalence and incident CAD risk in the monogenic FH subtype were likely underestimated here due to 1) a high rate of premature death in patients with an FH variant27 2) the high prevalent CAD event rate in the monogenic FH subtype, 3) the omission of structural variations in LDLR including loss-of-function copy number variants (CNV) from analyses, and 4) the healthy volunteer selection bias in the UK Biobank41. The monogenic FH subtype was not stratified by polygenic score or Lp(a) as reported in other studies due to sample size constraints42-44. Further, we did not analyze the impact on outcomes related to the presence of an FH variant in those with an LDL-C < 190 mg/dl. Due to low sample sizes of non-European populations, analyses were performed exclusively on the European subset of participants. These results will need to be confirmed in populations of non-European ancestry.
Conclusions
Incorporating genetic causes of severe hypercholesterolemia, particularly monogenic FH and Lp(a), into CAD risk assessment can be used to identify those severe hypercholesterolemia patients at highest risk for an imminent coronary artery disease event.
Supplementary Material
Highlights.
A large proportion of severe hypercholesterolemia cases were attributable to genetic determinants.
Among severe hypercholesterolemic participants, genetic subtyping stratified incident CAD risk more than LDL-C-based subtyping.
Individuals with an FH-causing variant had high incidence of CAD even when treated and would benefit from earlier diagnosis and treatment.
Those with elevated Lp(a) and two-hit hypercholesterolemia had high incidence of CAD if untreated and would benefit from earlier diagnosis and targeted treatment.
Acknowledgements
The authors thank Dr. H. Lester Kirchner, PhD for guidance in statistical analyses, and Dr. Scott M. Myers, MD, and Brenda M. Finucane, MS, for providing feedback on the manuscript.
Funding
This study was supported by NHLBI, Bethesda, MD (grant number: 1R01HL159182-01A1)
Abbreviations
- AHA
American Heart Association
- ARP
attributable risk proportion
- ASCVD
atherosclerotic cardiovascular disease
- BMI
body mass index
- BP
blood pressure
- CAD
coronary artery disease
- CI
confidence interval
- CVD
cardiovascular disease
- EHR
electronic health record
- FH
familial hypercholesterolemia
- HDL
high-density lipoprotein cholesterol
- HR
hazard ratio
- LDL-C
low-density lipoprotein cholesterol
- LLM
lipid lower medication
- Lp(a)
lipoprotein(a)
- OR
odds ratio
- P/LP
pathogenic or likely pathogenic
- PAD
peripheral artery disease
- PAF
population attributable fraction
- PRS
polygenic risk score
- SD
standard deviation
Footnotes
Conflicts of interest
ASFB: None
SSG: Consultant, Esperion
LKJ: Consultant, Novartis
EJS: Received a research grant from Amgen.
MTO: None
Data availability statement
The data underlying this article were accessed from the UK Biobank (biobank.ndph.ox.ac.uk). The derived datasets can be generated with permission from the UK Biobank.
References
- 1.Gidding SS, Champagne MA, de Ferranti SD, et al. The Agenda for Familial Hypercholesterolemia: A Scientific Statement From the American Heart Association. Circulation. 2015;132:2167–2192. [DOI] [PubMed] [Google Scholar]
- 2.Watts GF, Gidding SS, Mata P, et al. Familial hypercholesterolaemia: evolving knowledge for designing adaptive models of care. Nat Rev Cardiol. 2020;17:360–377. [DOI] [PubMed] [Google Scholar]
- 3.Hu P, Dharmayat KI, Stevens CAT, et al. Prevalence of Familial Hypercholesterolemia Among the General Population and Patients With Atherosclerotic Cardiovascular Disease. Circulation. 2020;141:1742–1759. [DOI] [PubMed] [Google Scholar]
- 4.Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J Am Coll Cardiol. 2020;75:2553–2566. [DOI] [PubMed] [Google Scholar]
- 5.Trinder M, Uddin MM, Finneran P, Aragam KG, Natarajan P. Clinical Utility of Lipoprotein(a) and LPA Genetic Risk Score in Risk Prediction of Incident Atherosclerotic Cardiovascular Disease. JAMA Cardiology. Published online 2020. doi: 10.1001/jamacardio.2020.5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trinder M, Francis GA, Brunham LR. Association of Monogenic vs Polygenic Hypercholesterolemia With Risk of Atherosclerotic Cardiovascular Disease. JAMA Cardiol. 2020;5:390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graham SE, Clarke SL, Wu KHH, et al. The power of genetic diversity in genome-wide association studies of lipids. Nature. 2021;600:675–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sniderman AD, Glavinovic T, Thanassoulis G. Key Questions About Familial Hypercholesterolemia: JACC Review Topic of the Week. J Am Coll Cardiol. 2022;79:1023–1031. [DOI] [PubMed] [Google Scholar]
- 9.Khera AV, Won HH, Peloso GM, et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J Am Coll Cardiol. 2016;67:2578–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Talmud PJ, Shah S, Whittall R, et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet. 2013;381:1293–1301. [DOI] [PubMed] [Google Scholar]
- 11.Trinder M, DeCastro ML, Azizi H, et al. Ascertainment Bias in the Association Between Elevated Lipoprotein(a) and Familial Hypercholesterolemia. J Am Coll Cardiol. 2020;75:2682–2693. [DOI] [PubMed] [Google Scholar]
- 12.Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139:e1082–e1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fahed AC, Wang M, Patel AP, et al. Association of the Interaction Between Familial Hypercholesterolemia Variants and Adherence to a Healthy Lifestyle With Risk of Coronary Artery Disease. JAMA Netw Open. 2022;5:e222687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanscombe KB, Coleman JRI, Traylor M, Lewis CM. ukbtools: An R package to manage and query UK Biobank data. PLoS One. 2019;14:e0214311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Hout CV, Tachmazidou I, Backman JD, et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature. 2020;586:749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loh PR, Danecek P, Palamara PF, et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet. 2016;48:1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burgess S, Ference BA, Staley JR, et al. Association of LPA Variants With Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol. 2018;3:619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saadatagah S, Jose M, Dikilitas O, et al. Genetic basis of hypercholesterolemia in adults. NPJ Genom Med. 2021;6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elliott J, Bodinier B, Bond TA, et al. Predictive Accuracy of a Polygenic Risk Score-Enhanced Prediction Model vs a Clinical Risk Score for Coronary Artery Disease. JAMA. 2020;323:636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Björnsson E, Thorgeirsson G, Helgadóttir A, et al. Large-Scale Screening for Monogenic and Clinically Defined Familial Hypercholesterolemia in Iceland. Arterioscler Thromb Vasc Biol. 2021;41:2616–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Said MA, Verweij N, van der Harst P. Associations of Combined Genetic and Lifestyle Risks With Incident Cardiovascular Disease and Diabetes in the UK Biobank Study. JAMA Cardiol. 2018;3:693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.R Core Team. R: A Language and Environment for Statistical Computing. Published online 2020. Accessed November 1, 2021. https://www.R-project.org/
- 24.Nagraj VP. twoxtwo: Work with Two-by-Two Tables. Published online 2021. https://CRAN.R-project.org/package=twoxtwo [Google Scholar]
- 25.Wickham H. ggplot2: Elegant Graphics for Data Analysis. Published online 2016. http://ggplot2.org [Google Scholar]
- 26.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300. [Google Scholar]
- 27.Duell PB, Gidding SS, Andersen RL, et al. Longitudinal low density lipoprotein cholesterol goal achievement and cardiovascular outcomes among adult patients with familial hypercholesterolemia: The CASCADE FH registry. Atherosclerosis. 2019;289:85–93. [DOI] [PubMed] [Google Scholar]
- 28.Therneau TM. A Package for Survival Analysis in R.; 2021. Accessed November 2, 2021. https://CRAN.R-project.org/package=survival [Google Scholar]
- 29.Kassambara A, Kosinski M, Biecek P. survminer: Drawing Survival Curves using “ggplot2.” Published online 2021. Accessed November 2, 2021. https://CRAN.R-project.org/package=survminer [Google Scholar]
- 30.Austin PC. Absolute risk reductions and numbers needed to treat can be obtained from adjusted survival models for time-to-event outcomes. J Clin Epidemiol. 2010;63:46–55. [DOI] [PubMed] [Google Scholar]
- 31.Mariano C, Alves AC, Medeiros AM, et al. The familial hypercholesterolaemia phenotype: Monogenic familial hypercholesterolaemia, polygenic hypercholesterolaemia and other causes. Clin Genet. 2020;97:457–466. [DOI] [PubMed] [Google Scholar]
- 32.Pilia G, Chen WM, Scuteri A, et al. Heritability of cardiovascular and personality traits in 6,148 Sardinians. PLoS Genet. 2006;2:e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hedegaard Berit Storgaard, Bork Christian Sørensen, Kaltoft Morten, et al. Equivalent Impact of Elevated Lipoprotein(a) and Familial Hypercholesterolemia in Patients With Atherosclerotic Cardiovascular Disease. J Am Coll Cardiol. 2022;80:1998–2010. [DOI] [PubMed] [Google Scholar]
- 34.Futema M, Whittall RA, Kiley A, et al. Analysis of the frequency and spectrum of mutations recognised to cause familial hypercholesterolaemia in routine clinical practice in a UK specialist hospital lipid clinic. Atherosclerosis. 2013;229:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weng S, Kai J, Akyea R, Qureshi N. Detection of familial hypercholesterolaemia: external validation of the FAMCAT clinical case-finding algorithm to identify patients in primary care. Lancet Public Health. 2019;4:e256–e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birnbaum RA, Horton BH, Gidding SS, Brenman LM, Macapinlac BA, Avins AL. Closing the gap: Identification and management of familial hypercholesterolemia in an integrated healthcare delivery system. J Clin Lipidol. Published online February 2, 2021. doi: 10.1016/j.jacl.2021.01.008 [DOI] [PubMed] [Google Scholar]
- 37.Abul-Husn NS, Manickam K, Jones LK, et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science. 2016;354. doi: 10.1126/science.aaf7000 [DOI] [PubMed] [Google Scholar]
- 38.Mickiewicz A, Futema M, Ćwiklinska A, et al. Higher Responsiveness to Rosuvastatin in Polygenic versus Monogenic Hypercholesterolaemia: A Propensity Score Analysis. Life. 2020;10. doi: 10.3390/life10050073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D’Erasmo L, Minicocci I, Di Costanzo A, et al. Clinical Implications of Monogenic Versus Polygenic Hypercholesterolemia: Long‐Term Response to Treatment, Coronary Atherosclerosis Burden, and Cardiovascular Events. J Am Heart Assoc. 2021;10:e018932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60:2631–2639. [DOI] [PubMed] [Google Scholar]
- 41.Batty GD, Gale CR, Kivimäki M, Deary IJ, Bell S. Comparison of risk factor associations in UK Biobank against representative, general population based studies with conventional response rates: prospective cohort study and individual participant meta-analysis. BMJ. 2020;368:m131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trinder M, Paquette M, Cermakova L, et al. Polygenic Contribution to Low-Density Lipoprotein Cholesterol Levels and Cardiovascular Risk in Monogenic Familial Hypercholesterolemia. Circulation: Genomic and Precision Medicine. 2020;13:515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paquette M, Bernard S, Thanassoulis G, Baass A. LPA genotype is associated with premature cardiovascular disease in familial hypercholesterolemia. J Clin Lipidol. 2019;13:627–633.e1. [DOI] [PubMed] [Google Scholar]
- 44.Oetjens MT, Kelly MA, Sturm AC, Martin CL, Ledbetter DH. Quantifying the polygenic contribution to variable expressivity in eleven rare genetic disorders. Nat Commun. 2019;10:4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fahed AC, Wang M, Homburger JR, et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat Commun. 2020;11:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ge T, Chen CY, Ni Y, Feng YCA, Smoller JW. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun. 2019;10:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lloyd-Jones DM, Hong Y, Labarthe D, et al. Defining and setting national goals for cardiovascular health promotion and disease reduction: the American Heart Association’s strategic Impact Goal through 2020 and beyond. Circulation. 2010;121:586–613. [DOI] [PubMed] [Google Scholar]
- 51.Mozaffarian D. Dietary and Policy Priorities for Cardiovascular Disease, Diabetes, and Obesity: A Comprehensive Review. Circulation. 2016;133:187–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All anonymized electronic health record (EHR), self-report, and genetic data used here can be obtained directly from the UK Biobank. Analysis scripts are available upon reasonable request to the authors.
The data underlying this article were accessed from the UK Biobank (biobank.ndph.ox.ac.uk). The derived datasets can be generated with permission from the UK Biobank.