


Abstract
Post-transcriptional regulation is an important mechanism in cellular response to stimuli, allowing for the rapid and discrete expression of relevant proteins. Genes regulated by this mechanism have specific cis-acting elements, frequently in their 3′ untranslated regions (UTRs), that have been shown to serve as recognition sites for trans-acting RNA-binding proteins. Unfortunately, the identification of specific mRNA ligands for different RNA binding proteins in vivo has been limited by a lack of adequate methodology. We have developed a novel technique that addresses this shortcoming, using immunoprecipitation of RNA binding proteins from polysomes followed by RT–PCR and library screening to identify the in vivo mRNA ligands of RNA binding proteins. Utilizing this approach, we have identified 32 known and 16 novel mRNAs specifically bound by the heterogeneous nuclear ribonucleoprotein (hnRNP) A2. Of the clones identified, 74% contained AU-rich elements and/or poly-uridine tracts in their 3′ UTRs, cis-acting elements that have been established as impacting mRNA stability. The high percentage of clones containing these uridine-rich sequences compares favorably with the high affinity binding of poly-uridine RNA by hnRNP A2 in vitro. These data thus support the representative nature of the technique.
INTRODUCTION
Cellular responses to stimuli, such as mitogenic activation or hypoxia, are associated with diverse and widespread changes in gene expression at both the transcriptional and post-transcriptional levels (1–3). Post-transcriptional regulation, in particular, provides the means by which a cell can rapidly alter gene expression (4). A critical component of post-transcriptional regulation involves modulation of the turnover or translation rates of labile mRNAs, such as cytokines and proto-oncogenes, which transduce rapid and discrete cellular responses (5). This complex regulatory mechanism has been shown to utilize cis-acting elements located in mRNA sequences, frequently, but not exclusively, in the 3′ untranslated region (3′ UTR) (5–8). These cis-acting elements are thought to serve as recognition sites for the binding of specific trans-acting factors. The interaction of these trans-acting factors with specific cis-acting elements is thought to modulate mRNA translation and stability, although the exact mechanism(s) by which this occurs is unclear (8,9).
Cytokine and proto-oncogene mRNA are typically labile, displaying half-lives of <30 min; whereas in response to some stimuli, such as mitogen activation, these mRNAs are stabilized, often by >10-fold (1–4,8). Sequence analysis of the 3′ UTR of many labile mRNAs frequently reveals adenosine–uridine-rich elements (AURE). These AU-rich regions often contain repeats of the pentamer AUUUA, which has been established as a cis-acting element impacting mRNA stability (5,10,11). Additionally, recent work suggests that GUUUG repeats or poly-uridine stretches with interspersed purines also impact the stability of labile mRNAs (12–18). Together, these data suggest that multiple, uridine-rich elements in the 3′ UTR modulate the turnover of mRNA.
Several lines of evidence suggest that trans-acting AURE-specific mRNA binding proteins are responsible for regulating mRNA decay (18–24). An AURE in the 3′ UTR does not necessarily confer increased mRNA turnover (23). AURE-dependent mRNA turnover and translation can be modulated in vivo by various stimuli and signals associated with cellular activation, differentiation, or stress (4,25–27). Finally, differential mRNA turnover and translation of two mRNAs with similar AURE can occur in the same cell (27–30). While the specific molecular mechanisms for AURE-mediated mRNA decay remain unclear, recent work suggests the involvement of AUF-1/heterogeneous nuclear ribonucleoprotein (hnRNP) D and the heat shock–ubiquitin–proteasome pathway (31). Additional studies have implicated specific RNA-binding proteins as important in AURE-dependent turnover (32–35), such as HuR (ELAV), which binds to and stabilizes VEGF mRNA (32,33).
Despite the progress made in understanding the role of specific proteins in regulating mRNA turnover, numerous questions remain. Is the binding of these trans-acting factors restricted to AU-rich elements containing mRNA? Does the binding of these proteins always effect changes in mRNA turnover? Do stimuli that alter AURE-mediated turnover alter binding of these trans-acting factors?
To address these issues, it would be useful to be able to identify the mRNA ligands of a specific RNA binding protein. Few techniques exist to provide information about the mRNA ligands of a specific trans-acting factor in vivo. Typically, in vitro binding approaches are used to identify the ‘ideal’ RNA ligands of specific trans-acting factors. These assays examine the binding specificity of a recombinant RNA binding protein to synthetic RNA oligonucleotides (SELEX) or a pool of tissue-specific mRNA (36,37), with the ‘winner’ sequence used to identify possible mRNA ligands. This approach has been variable in successfully predicting mRNA ligands (33,38,39). There are several potential shortcomings for this approach including the use of recombinant proteins that lack appropriate post-translational modifications or the effects of either heteromeric or homomeric protein–protein interactions. Moreover, the use of synthetic oligoribonucleotides does not allow identification of RNA tertiary structures important for protein recognition and binding (40,41). Perhaps most importantly, these studies are limited by their relevance, since it is never clear that the RNA binding protein–mRNA interaction identified in vitro ever occurs in vivo.
To redress the lack of in vivo data, we have developed a reproducible technique for isolating and identifying the mRNA ligands for known RNA binding proteins associated with polysomes. For this study, we examined the mRNA ligands of hnRNP A2, a member of the hnRNP family of RNA binding proteins. We selected hnRNP A2 because of the association between its cytoplasmic overexpression and non-small cell lung cancer (42–48). Additional data implicate hnRNP A2 as an overexpressed cytosolic tumor-specific antigen in various forms of cancer (34, B.J.Hamilton and W.F.C.Rigby, unpublished observation), further suggesting a potential role in neoplastic transformation. Utilizing specific antisera against hnRNP A2, we demonstrated its association with polysomes and poly(A) RNA in vivo and identified 32 known and 16 novel mRNA ligands specifically bound by hnRNP A2.
MATERIALS AND METHODS
Materials
Pepstatin A, leupeptin and Pefabloc were purchased from Boehringer-Mannheim (Indianapolis, IN). [α-32P]dCTP (3000 Ci/mmol)was purchased from New England Nuclear (Boston, MA). The phorbol 12-myristate 13-acetate (PMA) activated (200 nM, for 4 days) THP-1 Lambda ZAP II cDNA library was a generous gift from T. Y. Chang (49).
Isolation of polysomes
Polysomes were isolated from the THP-1 myelomonocytic cell line as well as the leukocytes of a myeloid leukemic patient with a high circulating blast count (34). Cells (2 × 108) were washed three times with 1× phosphate buffered saline and resuspended in 3.5 ml of buffer A (10 mM Tris–HCl pH 7.6, 1 mM KAc, 1.5 mM MgAc, 2 mM DTT, 1 µg/ml pepstatin A, 1 µg/ml leupeptin, 1 mM Pefabloc). Cells were lysed by 20 strokes with a Teflon pestle homogenizer at 1500 r.p.m. and centrifuged at 12 000 g for 10 min to pellet the nuclei. The supernatant was layered over 7.5 ml of buffer B (buffer A plus 30% sucrose w/v) in a Beckman ultra-centrifuge tube and spun for 5 h at 36 000 r.p.m. in a SW41 rotor. The S-130 supernatant fraction was removed and saved, and the pellet was washed twice with 0.5 ml of buffer A. The pellet was then resuspended in 0.5 ml of buffer A, an OD A260 reading was taken and the sample was stored as aliquots at –80°C.
Immunoprecipitation
For polysomal immunoprecipitations, 500 µg of either the isotype control IgG1 antibody, P3, the hnRNP A2 peptide specific monoclonal IgG1 antibody, EF-67, or the hnRNP A1 peptide specific polyclonal antisera ACT-1 (50), were bound to 3 mg swollen protein A Sepharose beads by incubating overnight at 4°C with continuous rotation in IP buffer (10 mM Tris–HCl pH 7.6, 1.5 mM MgCl2, 100 mM NaCl, 0.5% Triton X-100, 1 mM Pefabloc and 1 µg/ml each leupeptin and pepstatin A). The beads were then washed three times with 500 µl of IP buffer. The EF-67 anti-hnRNP A2 monoclonal antibody, which is specific for the C-terminus of hnRNP A2, shows no cross-reactivity with hnRNP A1 (34).
Polysomes (2.0 A260) were pre-cleared for 2 h at 4°C with the P3 beads in IP buffer with continuous rotation. The beads were pelleted and the supernatant was then incubated with either the EF-67 or ACT-1 loaded beads and the immunoprecipitation repeated as above. Following immunoprecipitation, the beads were pelleted, washed six times with IP buffer and then used for RNA isolation and RT–PCR.
Immunoprecipitation for immunoblotting was performed in parallel using the P3, EF-67, ACT-1 and protein A Sepharose beads (no antibody), using A260 2.0 polysomes used for each immunoprecipitation. Following immunoprecipitation and washing as above, 50 µl of 2× western loading buffer [100 mM Tris–Cl (pH 6.8), 4% SDS, 0.2% bromophenol blue, 20% glycerol, 200 mM DTT] were added to the beads. The samples were boiled in loading buffer, resolved by 12% SDS–PAGE, then electro-transferred to nitrocellulose.
Isolation of RNA from immunoprecipitation and RT–PCR
Each immunoprecipitate was digested with RNase free Proteinase K (50 ng/ml) (10 mM Tris–HCl pH 7.6, 5 mM EDTA) in 100 µl for 30 min at 37°C, extracted twice with an equal volume of 25:24:1 phenol/chloroform/isoamylalcohol, once with an equal volume of 49:1 chloroform/isoamyl alcohol and ethanol precipitated.
Immunoprecipitable RNA was reverse transcribed with an oligo(dT) 20mer, using Superscript II reverse transcriptase (Life Technologies, Gaithersburg, MD). The RNA/cDNA duplex was heated to 95°C for 10 min, and purified by column chromatography (Chroma-Spin-30 column; Clontech, Palo Alto, CA) to remove unincorporated primer, RNA fragments and salts. A homopolymer poly d(A) tail was generated at the 3′ end of the single-stranded cDNA with dATP and terminal transferase (Stratagene, La Jolla, CA) thereby allowing amplification by PCR with a single 28mer oligonucleotide primer (5′-GGAATTCCTTTTTTTTTTTTTTTTTTTT-3′) containing an EcoRI restriction endonuclease site at the 5′ end. Following column chromatography, PCR was performed using standard conditions (PCR Core Kit; Boehringer-Mannheim) and 30 cycles at the following temperatures: 94°C for 1 min, 48°C for 1 min, 72°C for 2 min. Amplification was confirmed by agarose gel electrophoresis.
Library screening and sequencing
The amplified cDNAs from the immunoprecipitations were purified by column chromatography and used to generate [32P]dCTP-labeled random primed probes (Rad-Prime DNA Labeling, Life Technologies). Each of the 32P-labeled probes was used to screen lifts from a THP-1 Lambda ZAP cDNA library. Immunoprecipitation 1 utilized the PMA activated THP-1 cell ZAP II library, while immunoprecipitation 2 utilized an LPS activated THP-1 cell ZAP Express Library constructed in our laboratory (Stratagene). Following library plating on 150 mm plates, at 50 000 p.f.u./plate, replica nylon lifts were made. The lifts were denatured in 0.5 M NaOH/1.5 M NaCl for 2 min, neutralized in 0.5 M Tris–HCl (pH 7.5)/1.5 M NaCl for 5 min and rinsed in 0.2 M Tris–HCl (pH 7.5)/2× SSC for 30 s. The library lifts were prehybridized with ExpressHyb (Clontech) for 30 min at 68°C, and then hybridized for 1 h at 68°C with 2 × 106 c.p.m./ml of either P3 or EF-67 random-primed DNA probe generated from the RT–PCR. Lifts were washed in 2× SSC/0.05% SDS at 25°C for 40 min with two changes of wash buffer, then in 0.1× SSC/0.1% SDS at 50°C for 40 min with one change of wash buffer, and used for autoradiography. Replica lifts were compared for overlapping binding. EF-67 positive plaques were picked and used for secondary screening and positive plaques from the secondary screening were picked and the phagemid excised according to the manufacturer’s protocol (Stratagene). Excised phagemids were examined by agarose gel electrophoresis following digestion with restriction endonucleases, sequenced using the ABI Prism Dye Terminator Cycle Sequencing kit (Perkin Elmer Corp., Wellesley, MA), and searched on the NCBI database using the BLAST search program (51).
Slot blotting
Slot blotting was performed using 100 ng of clone insert cDNA per slot. The cDNA was denatured in 0.3 M NaOH, boiled for 10 min and placed on ice. An equal volume of 4 M NH4OAc was added, to neutralize the NaOH, followed by sufficient 20× SSC for a final concentration of 10× SSC. Samples were then slotted onto a nylon membrane, allowed to stand for 15 min followed by administration of a vacuum for 15 min. Following crosslinking in a Stratalinker, the membranes were probed as described above.
RESULTS
hnRNP A2 is on THP-1 polysomes and is immunoprecipitated by EF-67
Prior studies have demonstrated that hnRNP A2 is on the polysomes of normal tissues such as cortical neurons, lymphocytes and cell lines (34, B.J.Hamilton and W.F.C.Rigby, unpublished observation). As expected, immunoblotting of polysomes from THP-1 cells showed two hnRNP A2 reactive bands corresponding to hnRNP A2 (36 kDa) and its alternately spliced form hnRNP B1 (38 kDa). We then demonstrated the ability to specifically immunoprecipitate hnRNP A2 from THP-1 polysomes with EF-67 (Fig. 1). No hnRNP A2 was immunoprecipitable with an irrelevant isotype control, P3, with the anti-hnRNP A1 polyclonal antisera ACT-1, or with protein A beads alone. In other studies, we have demonstrated that immunoprecipitation of hnRNP A2 from polysomal, cytoplasmic and nuclear fractions does not co-immunoprecipitate hnRNP A1 (34). From these studies, we concluded that we could specifically immunoprecipitate hnRNP A2 complexed to RNA from polysomes.
Figure 1.
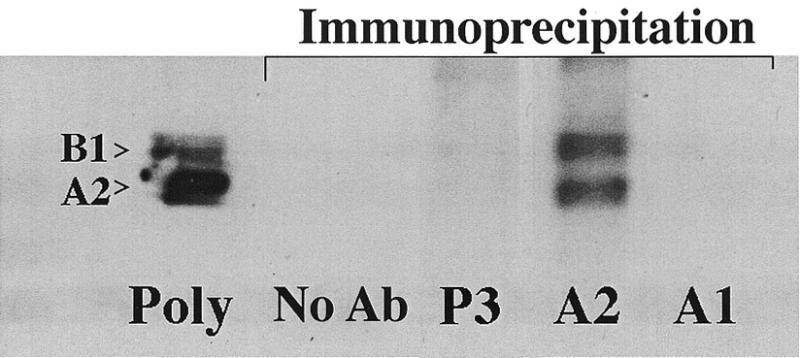
hnRNP A2 can be selectively immunoprecipitated from THP-1 polysomes. Two absorbance units at OD A260 of THP-1 polysomes were individually immunoprecipitated with P3, anti-hnRNP A2 (EF-67), anti-hnRNP A1 (ACT-1) or no antibody (protein A Sepharose beads alone), and resolved by SDS–PAGE and immunoblotting. Two bands, representing hnRNP A2 (36 kDa) and B1 (38 kDa) are present in the polysome (also 2 OD A260) and EF-67 immunoprecipitate lanes only, following blotting with EF-67.
Isolation of RNA from polysome immunoprecipitation and RT–PCR
To determine the in vivo mRNA ligands of hnRNP A2, we sought a simple method to isolate the RNA from immunoprecipitated polysomes (Fig. 2). Serial immunoprecipitations with P3 then EF-67 were performed. Immunoprecipitates were treated with RNase-free proteinase K, phenol/chloroform extracted and ethanol precipitation and reverse transcribed using oligo(dT). The single-stranded cDNA was tailed with dATP and terminal transferase and amplified by PCR using a single oligonucleotide primer and 30 cycles as outlined in the Materials and Methods.
Figure 2.

Schematic outline of the isolation of in vivo mRNA ligands of hnRNP A2.
Initially, we utilized polysomes extracted from a patient with acute myeloid leukemia and a high circulating blast count. The RT–PCR results from immunoprecipitable RNA are shown in Figure 3. RNA that was non-specifically associated with the isotype control, P3, yielded a low molecular weight smear ranging from approximately 100 to 500 bases. A higher molecular weight smear is evident in the hnRNP A2 lane ranging from approximately 100 to 2000 bases. These results demonstrate that RNA can be isolated from immunoprecipitated polysomes and amplified using a single primer by RT–PCR. Importantly, the longer cDNA length obtained with the EF-67 immunoprecipitation provides support for the specificity of the reverse transcription and amplification. The short cDNAs generated from P3 immunoprecipitation probably result from a non-specific interaction with partially degraded mRNA, which still retained at least a portion of its poly(A) tail.
Figure 3.
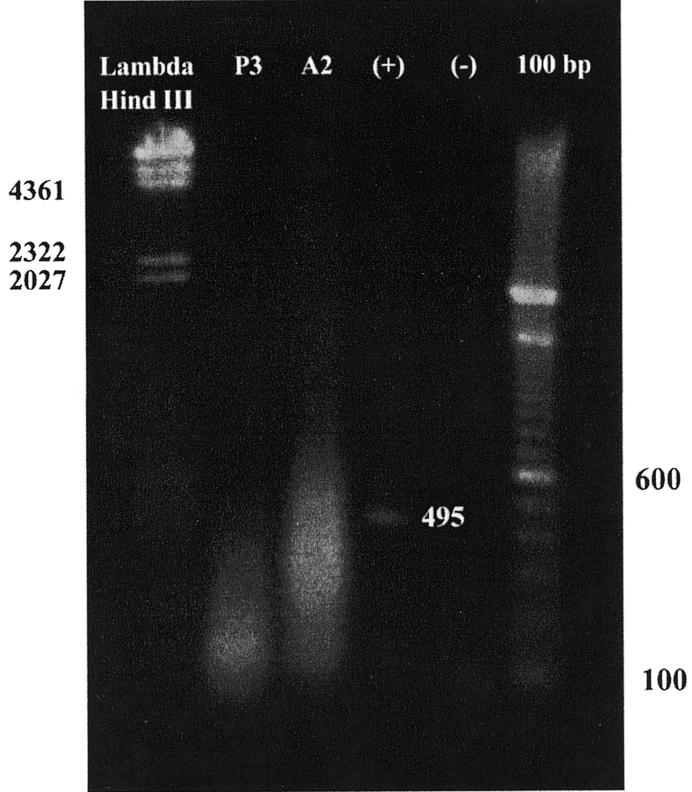
PCR amplification of hnRNP A2 associated polysomal RNA. PCR amplification of the RNA isolated by immunoprecipitation of leukemic polysomes was analyzed by agarose gel electrophoresis and ethidium bromide staining. From left to right, the lanes are as follows: Lambda HindIII markers; amplified cDNA from the P3 immunoprecipitation; amplified cDNA from the EF-67 immunoprecipitation (hnRNP A2); positive control using full-length GM-CSF giving a single 495 bp band, the expected size for the primers used; negative control containing no DNA giving no bands, but a faint smear of primer; 100 bp marker ladder.
Library screen of a PMA activated THP-1 Lambda Zap II cDNA library
Having established a method to isolate and amplify immunoprecipitated RNA–protein complexes from polysomes, we used library screening to identify the ligands and confirm that we had specifically isolated mRNA. PCR amplified cDNA from the leukemic polysome immunoprecipitates was used to generate a random primed radiolabeled probe, which was used to screen a PMA activated THP-1 Lambda Zap II library. Duplicate nylon lifts were made from each plate and probed with either the P3 or EF-67 random primed probes. No overlap of plaques bound by P3 and by EF-67 was observed on the duplicate lifts, indicating that the EF-67 immunoprecipitation of hnRNP A2 from polysomes was specific. A total of 34 plaques containing 29 unique clones were isolated from the EF-67 screening (Table 1). Among the 29 individual clones, five were novel sequences not found in the NCBI database by BLAST search (51). Of the remaining 24 clones, four matched human genomic clones and the other 20 matched known mRNAs in the database. As the library used for screening was generated from mRNA, all of these clones represent expressed messages. Also shown in Table 1 are the presence of AURE and poly-uridine tracts, which will be discussed later.
Table 1. Clones from the immunoprecipitation of leukemic polysomes.
| Blot position | Clone name | Nonamer (UUAUUUAU/AU/A) | Pentamer (AUUUA) | Poly(U) tract(s) |
|---|---|---|---|---|
| A-1 | EPA glycoprotein | – | – | – |
| A-2 | Vimentin | – | + (1) | + |
| A-3 | Chrom 6p22.3–24.3 | |||
| A-4 | Novel sequence | |||
| A-6 | Heparin sulfate proglycan core | – | + (6) | + |
| A-7 | Ribosomal protein L44 | – | – | – |
| A-8 | Novel sequence | |||
| A-9 | Ribosomal protein L18a | – | – | – |
| A-11 | KM 102-derived reductase-like factor | + (1) | + (2) | + |
| A-12 | Novel sequence | |||
| B-1 | Chromo Xp11.23–11.3 | |||
| B-2 | Chromo Xq25 | |||
| B-3 | Cyclooxygenase I | – | + (1) | + |
| B-4 | sec61 homolog | – | – | + |
| B-6 | SOM172 (phospholipase C B3) | – | – | + |
| B-7 | PEA-15 astrocytic phosphoprotein | – | – | + |
| B-8 | Cathepsin B | – | – | + |
| B-9 | Inducible poly(A)-binding protein | – | + (1) | + |
| B-11 | Chromo 22q12 matches EST | |||
| B-12 | Adenylyl cyclase associated protein | – | – | + |
| C-1 | Novel sequence | |||
| C-2 | GTF2I (transcription factor) | – | + (1) | + |
| C-3 | β-actin | – | – | + |
| C-4 | Ribosomal protein L30 | – | – | – |
| C-6 | Novel sequence | |||
| C-7 | Heme oxygenase (decycling) 1 | – | + | |
| C-8 | Novel sequence | |||
| C-9 | Cytochrome oxidase subunit II | – | – | – |
| C-11 | TAGLN2 | – | + (3) | – |
Clones identified from the first immunoprecipitation. List of the 29 clones from the first immunoprecipitate examined by slot blotting, along with the presence or absence of the indicated 3′ UTR cis-elements, which are defined as follows: 3′ UTR, the region of the message from the stop codon to the start of the poly(A) tail; pentamer, the sequence AUUUA (ATTTA for cDNA); nonamer, the sequence UUAUUUAU/AU/A (TTATTTAT/AT/A in cDNA); and poly(U) tract, UUUU occurring four times or UUUUU occurring twice. The number in parentheses in the pentamer column indicates the number of pentamers present in the 3′ UTR. The blot position corresponds to the location of the clone on the slot blot in Figure 4.
Confirmation of specificity using slot blotting
The reproducibility and specificity of the technique was then assessed using slots blots of the 29 mRNA ligands identified from the library screen. New immunoprecipitations of leukemic polysomes with P3 and EF-67 were performed and the immunoprecipitable RNA subjected to RT–PCR. The RT–PCR from each IP was then labeled with 32P by random priming and hybridized with the slot blots containing the 29 clones identified above (Fig. 4).
Figure 4.
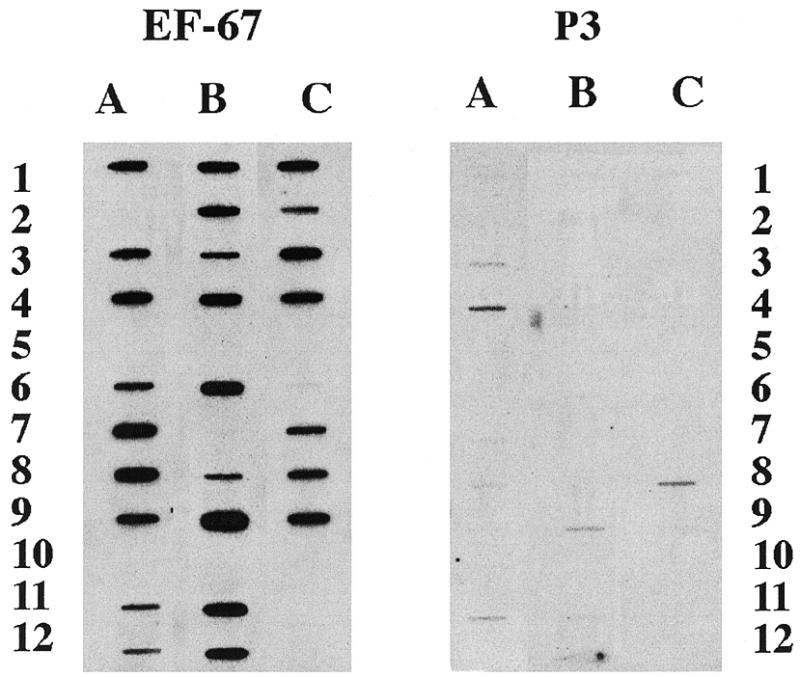
Slot blots of clones from the first immunoprecipitation. Slot blots of 100 ng of insert cDNA from each unique clone isolated by library screening, PMA activated THP-1 library, with RT–PCR of hnRNP A2-associated polysomal RNA was probed at a concentration of 2 × 106 c.p.m./ml and used for autoradiography on the same piece of film with identical exposures shown. The probe was generated by 32P random prime-labeling the RT–PCR of a second immunoprecipitation with the indicated antisera from leukemic polysomes. The identity of each clone is listed in Table 1.
Autoradiography demonstrated specific hybridization with 26 of the 29 clones. Three of the clones initially isolated (position A-2, B-7 and C-11) did not bind either P3 of EF-67 probe with this screen, while a novel sequence (C-6) bound the EF-67 probe weakly. These four clones were classified as false positives, yielding a positive identification rate of >86% in repeat immunoprecipitations. These data indicate both the reproducibility and specificity of this approach. It should be noted that this strategy of analysis (probing with RT–PCR of a second immunoprecipitation) was not optimized to detect quantitative differences. Despite the non-linear nature of the PCR conditions used in this experiment, the observed reproducibility between immunoprecipitations suggests the robust nature of the technique.
Replication of the technique using THP-1 polysomes
In order to refine and confirm our technique, as well as to continue to catalog the in vivo mRNA ligands of hnRNP A2, we extended our study to the THP-1 myelomonocytic cell line. Polysomes from LPS activated (1 µg/ml, 1 h) THP-1 cells were isolated, immunoprecipitated as above and the isolated RNA was used to generate a cDNA probe and screen an LPS activated (1 µg/ml, 1 h) THP-1 ZAP Express library.
This screen identified 38 candidate clones of which 36 were identified as unique by sequencing (Table 2). As before, the reproducibility was determined by slot blotting using 32P labeling of RT–PCR from a second set of P3/EF-67 immunoprecipitations as well as a P3/ACT-1 immunoprecipitation (Fig. 5). No significant hybridization with the hnRNP A2-associated cDNA clones was seen with the RT–PCR of immunoprecipitates performed with anti-hnRNP A1 antibodies. Specifically, P3 and A1 were equivalent in their lack of overlap with A2. These data provide additional support for the specificity of our technique which is of considerable importance as hnRNP A1 and A2 have a high level of homology and bind poly(U) with high affinity (52,53). Despite high affinity binding for poly(U), the lack of overlap between hnRNP A2 and A1 in their in vivo mRNA association is consistent with previously observed differences in their mRNA binding specificity (34).
Table 2. Clones from the of immunoprecipitation of THP-1 polysomes.
| Blot position | Clone name | Nonamer (UUAUUUAU/AU/A) | Pentamer (AUUUA) | Poly U tract(s) |
|---|---|---|---|---|
| A-1 | mRNA for unknown product | – | + (1) | – |
| A-2 | Ribosomal protein L-27 | – | – | – |
| A-3 | Novel sequence | |||
| A-4 | γ1 actin | – | – | + |
| A-5 | transcription factor CBFB | – | + (5) | + |
| A-6 | co-β-glucosidase | – | – | + |
| A-7 | Chromosome Xp22 | |||
| A-8 | Granulin/epithelin 1 and 2 | – | – | – |
| A-9 | Novel sequence | |||
| A-10 | Calnexin | – | + (1) | + |
| A-11 | Origin recognition complex subunit 5 | – | + (1) | + |
| A-12 | Y box binding protein-1 | – | – | + |
| B-1 | c-myc | – | + (4) | + |
| B-2 | Cytochrome oxidase subunit II | – | – | – |
| B-3 | aac11 | – | + (3) | + |
| B-4 | Chromosome 22 | |||
| B-5 | Novel sequence | |||
| B-6 | Ribosomal protein S18 | – | – | – |
| B-7 | TI-227H (novel sequence) | |||
| B-8 | Novel sequence | |||
| B-9 | MHC HLA-E | – | – | – |
| B-10 | Ribosomal protein S-7 | – | – | – |
| B-11 | PCTAIRE-1 | – | – | – |
| B-12 | Human clone 25077 | – | + (1) | + |
| C-1 | CTP-synthase (CTPS) | – | – | + |
| C-2 | Chromosome 4p16.3-Huntington’s region | |||
| C-3 | Chromo 17 | |||
| C-4 | RSU-1/RSP-1 | – | + (2) | + |
| C-5 | KIAA0102 | – | + (1) | + |
| C-6 | HOXA13 | – | – | – |
| C-7 | Human clone for unknown mRNA | + (1) | + (2) | + |
| C-8 | Protein disulfide isomerase | – | – | + |
| C-9 | α-enolase | – | – | – |
| C-10 | Cytochrome oxidase subunit I | – | – | – |
| C-11 | Initiation factor 4B | – | + (2) | + |
| C-12 | Phosphatidylinositol synthase (PIS) | + (1) | + (1) | + |
Clones identified from the second immunoprecipitation. List of the 36 clones from the second immunoprecipitate examined by slot blotting, along with the presence or absence of the indicated 3′ UTR cis-elements, which are defined as follows: 3′ UTR, the region of the message from the stop codon to the start of the poly(A) tail; pentamer, the sequence AUUUA (ATTTA for cDNA); nonamer, the sequence UUAUUUAU/AU/A (TTATTTAT/AT/A in cDNA); and poly(U) tract, UUUU occurring four times or UUUUU occurring twice. The number in parentheses in the pentamer column indicates the number of pentamers present in the 3′ UTR. The blot position corresponds to the location of the clone on the slot blot in Figure 5.
Figure 5.
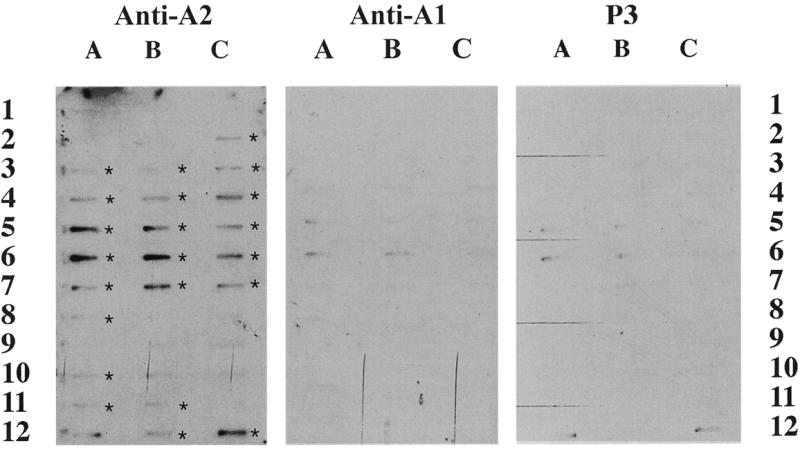
Slot blots of clones from the second immunoprecipitation. Slot blots of 100 ng of insert cDNA from each unique clone isolated by library screening, LPS activated THP-1 library, with RT–PCR of hnRNP A2-associated polysomal RNA were probed at a concentration of 2 × 106 c.p.m./ml and used for autoradiography on the same piece of film. The probe was generated by 32P random primer labeling of the RT–PCR of a set of second immunoprecipitation with the indicated antisera from LPS + THP-1 polysomes. Asterisks indicate positive clones as determined by densitometery. The identity of each clone is listed in Table 2.
In this screen, 23 clones hybridized with the EF-67 probe, of which one (A-12) was not clearly different from that hybridized with the isotype control (P3) immunoprecipitation and RT–PCR. The finding that 22 out of 36 (61%) clones were found to be hnRNP A2-associated on a repeat immunoprecipitation provides a further index of the reproducibility of this technique. The 22 positive clones are represented by five novel or unknown mRNAs, four matches to chromosomal regions and 13 matches to sequences in the NCBI database (Table 2).
Thirteen of the clones failed to hybridize the EF-67 probe generated from the second immunoprecipitation/RT–PCR. This resulted in a false positive rate of 39%; one possible factor in the higher false positive rate observed with the THP-1 cells repeat IP/RT–PCR was our use of an entirely different polysome preparation, performed several months later. This is in contrast to the previous experiment with a 12% false positive, where the repeat IP/RT–PCR was performed using the same polysome aliquot as the initial IP. Nevertheless, false positives were easily discriminated by slot blotting.
Analysis of the 3′ UTRs of the hnRNP A2 mRNA ligands
The isolation of polysomal hnRNP A2 from human leukemic and myelomonocytic cell line THP-1 polysomal preparations resulted in the identification of 48 specific mRNA ligands that were confirmed by repeat immunoprecipitations and slot blotting. Table 3 presents an analysis of the 31 (of the 48) clones present in the NCBI database as identified genes. Of these 31 clones, 14 (44%) contain the pentamer (AUUUA) in their 3′ UTR, which has been shown to have a destabilizing effect on mRNA (54,55). All of these 14 AURE-containing clones, as well as nine additional clones, contained poly(U) tracts, defined by at least two tracts with a minimum of five uridines (56) or at least four tracts of four uridines (57). Thus 23 out of 31 (74%) contained poly(U) tracts. These data add further support to the specificity of our technique, as 74% of the identified mRNA ligands contain cis-acting elements in their 3′ UTR shown to be bound by hnRNP A2 with high affinity (34,53).
Table 3. Organization of all confirmed and identified mRNA ligands of hnRNP A2 based on 3′ UTR features.
| Pentamer and poly(U) tracts | Poly(U) tracts |
| 1. Heparin sulfate proteoglycan core protein | 1. SEC61 homolog |
| 2. KM 102-derived reductase-like factor | 2. Phospholipase c B3 |
| 3. Cyclooxygenase I | 3. Cathepsin B |
| 4. Inducible poly(A)-binding protein | 4. Adenylyl cyclase associated protein |
| 5. GTF2I (transcription factor) | 5. β-actin |
| 6. CBFB (transcription factor) | 6. Heme oxygenase (decyclin) 1 |
| 7. Calnexin | 7. γ-actin |
| 8. Origin recognition complex subunit 5 | 8. Co-β-glucosidase |
| 9. AAC-11 | 9. Y Box binding protein 1 |
| 10. Human clone 25077 | |
| 11. RSU-1/RSP-1 | Ribosomal proteins |
| 12. KIA0102 | 1. L18a |
| 13. Unknown clone (accession no. AF009205) | 2. L30 |
| 14. Phosphatidylinositol synthase | 3. L44 |
| 4. S18 | |
| Nonamer | |
| 1. KM 102-derived reductase-like factor | Other genes |
| 2. Unknown clone (accession no. AF009205) | 1. Epithelin 1 and 2 |
| 2. Cytochrome oxidase subunit II | |
| 3. Pctaire-1 | |
| 4. HOXA13 |
Summary of the all of the clones from the two immunoprecipitations that demonstrated EF-67 specific binding. Clones are organized according to the presence or absence or the listed 3′ UTR cis-elements. All of the pentamer containing clones also contain poly(U) tracts, which are defined as a least two tracts with a minimum of five uridines or at least four tracts with a minimum of four uridines.
Eight of the 31 (26%) clones contained neither pentamers nor poly(U) tracts. Of these eight, four encoded ribosomal proteins. Their presence in each immunoprecipitation is of some interest because they all have fairly short (average length 42 nt) 3′ UTRs. Nevertheless, the L30 ribosomal protein mRNA contains a pentamer-like sequence, AUUUUUA, in its 3′ UTR. It is possible that these messages are up-regulated in response to proliferation, as observed with the up-regulation of S18 in a number of tumors (58).
DISCUSSION
In this report, we describe a technique for identifying RNA ligands of specific RNA binding proteins in vivo. This approach utilizes immunoprecipitation of RNA–protein complexes, RT–PCR and library screening, ensuring the validity of the interaction between mRNA and RNA-binding protein. The reproducible nature of the mRNA identified by this technique was demonstrated by a high level of fidelity between repeat immunoprecipitations in two different cell lines. The specificity of this technique was confirmed by the lack of hybridization of the cDNA from an isotype control immunoprecipitation as well as an anti-hnRNP A1 immunoprecipitation, an RNA binding protein with a high level of homology to hnRNP A2. Additionally, the absence of highly abundant messages isolated in the library screen argues against the notion that hnRNP A2 interacts with most or all polysome associated proteins. Moreover, of the various RNA species identified, 74% contained poly(U) tracts, which are bound by hnRNP A2 with high affinity (53).
In contrast to the in vitro approaches that have been used to identify the mRNA ligands of RNA binding proteins (36,37), this technique offers a number of advantages. First and foremost, our technique not only guarantees the specificity of the mRNA–RNA binding protein interaction, but also the existence of this interaction in vivo. Additionally, this approach utilizes native protein rather than recombinant protein, guaranteeing the appropriate effects of post-translational modifications as well as the possible effects of protein–protein interactions in influencing mRNA binding activity. This latter effect is important as several studies indicate that RNA binding proteins are phosphoproteins (59,60) as well as multimeric (61,62). The most significant drawback of our technique, relative to in vitro techniques, is the inability to ascertain if the RNA binding protein is directly interacting with the message or whether the interaction is through another protein. This may or may not be a factor in this particular study as we have shown that hnRNP A2 and L can directly interact on polysomes, yet can independently bind GLUT-1 mRNA 3′ UTR (34)
Furthermore, our technique utilizes RT–PCR of immunoprecipitable mRNA for library screening to maximize identification of the ligands of the RNA binding protein, in contrast to other approaches (37,63). This strategy also provides a reliable and efficient means of confirming in vivo associated, positive mRNA ligands by slot blotting. This is in contrast to the technique presented by Bhattacharya et al. (37), which utilizes recombinant AUF-1/hnRNP D affinity chromatography to select mRNA ligands. With this technique, ‘true’ positives are identified following reiterated column washes, a more ambiguous and arbitrary methodology, as noted by the authors. Additionally, these studies differ in that we employed a more specific 3′ UTR classification, requiring the presence of an AUUUA pentamer or poly(U) tracts (four uridines four times or five uridines twice) in the 3′ UTR of the message, all of which have been shown to be binding sites RNA binding proteins (54–57). If we employ the less stringent definition described by Bhattacharya et al. (37) simply requiring the presence of AU-rich sequences, then >90% of the cDNA specifically associated with hnRNP A2 contained 3′ UTR AUREs.
Despite these differences, it is interesting to note that several of the mRNAs interacting with hnRNP A2 are similar to mRNA ligands identified as binding to AUF-1 in vitro (37). These include Cathepsin B, proteoglycan core protein (heparin sulfate versus hematopoietic), and poly(A) binding protein (inducible versus non-inducible). Additionally, both sets of identified ligands contain several transcription factors. A number of ribosomal mRNAs were also identified as bound by AUF1/hnRNP D, including one, L44, which we also isolated.
Interestingly, our approach also identified β-actin (position C-3, Fig. 4) and γ-actin (position A4, Fig. 5). Both of these genes contain poly(U) tracts in their 3′ UTRs, which could indicate cis-element binding sites for trans-acting factors, and both are specific to the EF-67 probing. These results would argue against the notion that the mRNAs isolated by the immunoprecipitation were present solely due to non-specific interactions as a consequence of their abundant representation in the cell, as the isotype control should also have contained these clones. Additionally, we did not isolate abundant messages present in THP-1 cells such as class I major histocompatibility complex, β-2-microglobulin, or glyceraldehyde-3-phosphate dehydrogenase.
It should be noted that the two screens we performed did not provide any congruence in the hnRNP A2 ligands identified. This was observed despite high levels of reproducibility and specificity of the isolated mRNA ligands within each cell type. There are several potential reasons for this observation. First, the messages that were identified in the two screens are from very different sources of cells, leukemic blasts versus the human myelomonocytic cell line THP-1. Additionally, the libraries that were screened, while both from THP-1 cells, were activated in different ways, the first with PMA (200 nM) for 4 days, the second with LPS (1 µg/ml) for 1 h. Finally, the lack of congruence strongly suggests that the ligands of any RNA binding protein may vary considerably between cell types.
These findings provide a basis for additional studies examining not only hnRNP A2 in other cell lines and under differing conditions of activation, but also examination of other RNA-binding proteins. By using polysomes as a source for the RNA–protein interaction, an in vivo assessment of the specific ligands bound by an RNA-binding protein can be determined, thus avoiding the problems of non-specific interaction or validity of the interaction associated with in vitro assays.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by a Merit Review Award from the Department of Veterans Affairs and the NIH (RO1AI34928, T32 AR07576) and the Grimshaw-Gudewicz Foundation.
REFERENCES
- 1.Shih S.C. and Claffey,K.P. (1998) Int. J. Exp. Pathol., 79, 347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Day D.A. and Tuite,M.F. (1998) J. Endocrinol., 157, 361–371. [DOI] [PubMed] [Google Scholar]
- 3.Ladomery M. (1997) Bioessays, 19, 903–909. [DOI] [PubMed] [Google Scholar]
- 4.Siomi H. and Dreyfuss,G. (1997) Curr. Opin. Genet. Dev., 7, 345–353. [DOI] [PubMed] [Google Scholar]
- 5.Shaw G. and Kamen,R. (1986) Cell, 46, 659–669. [DOI] [PubMed] [Google Scholar]
- 6.Reeves R., Elton,T., Nissen,M.S., Lehn,D. and Johnson,K.R. (1987) Proc. Natl Acad. Sci. USA, 84, 6531–6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson T. and Treisman,R. (1988) Nature, 366, 396–399. [DOI] [PubMed] [Google Scholar]
- 8.Ross J. (1995) Microbiol. Rev., 59, 423–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klausner R.D. and Harford,J.B. (1989) Science, 246, 870–872. [DOI] [PubMed] [Google Scholar]
- 10.Chen C.Y. and Shyu,A.B. (1995) Trends Biochem. Sci., 20, 465–470. [DOI] [PubMed] [Google Scholar]
- 11.Gillis P. and Malter,J.S. (1991) J. Biol. Chem., 266, 3172–3177. [PubMed] [Google Scholar]
- 12.Shimizu N., Ohta,M., Fujiwara,C., Sagara,J., Mochizuki,N., Oda,T. and Utiyama,H. (1991) J. Biol. Chem., 266, 12157–12161. [PubMed] [Google Scholar]
- 13.Chen C.Y. and Shyu,A.B. (1994) Mol. Cell. Biol., 14, 8471–8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine T.D., Gao,F., King,P.H., Andrews,L.G. and Keene,J.D. (1993) Mol. Cell. Biol., 13, 3494–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng H. and Lever,J.E. (1993) J. Biol. Chem., 270, 23996–24003. [DOI] [PubMed] [Google Scholar]
- 16.Chung S., Jiang,L., Cheng,S. and Furneaux,H. (1996) J. Biol. Chem., 271, 11518–11524. [DOI] [PubMed] [Google Scholar]
- 17.Lagnado C.A., Brown,C.Y. and Goodall,G.J. (1994) Mol. Cell. Biol., 14, 7984–7995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.You Y., Chen,C.Y. and Shyu,A.B. (1992) Mol. Cell. Biol., 12, 2931–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amara F.M., Entwistle,J., Kuschak,T.I., Turley,E.A. and Wright,J.A. (1996) J. Biol. Chem., 271, 15279–15284. [DOI] [PubMed] [Google Scholar]
- 20.Kiledjian M., DeMaria,C.T., Brewer,G. and Novick,K. (1997) Mol. Cell. Biol., 17, 4870–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vakalopoulou E., Schaack,J. and Shenk,T. (1991) Mol. Cell. Biol., 11, 3355–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shetty S., Kumar,A. and Idell,S. (1997) Mol. Cell. Biol., 17, 1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sirenko O.I., Lofquist,A.K., DeMaria,C.T., Morris,J.S., Brewer,G. and Haskill,J.S. (1997) Mol. Cell. Biol., 17, 3898–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aiello L.P., Robinson,G.S., Lin,Y.W., Nishio,Y. and King,G.L. (1994) Proc. Natl Acad. Sci. USA, 91, 6231–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindsten T., June,C.H., Ledbetter,J.A., Stella,G. and Thompson,C.B. (1989) Science, 244, 339–343. [DOI] [PubMed] [Google Scholar]
- 26.Shyu A.-B., Greenberg,M.E. and Belasco,J.G. (1989) Genes Dev., 3, 60–72. [DOI] [PubMed] [Google Scholar]
- 27.Nagy E., Buhlmann,J.E., Henics,T., Waugh,M. and Rigby,W.F.C. (1994) Cell. Immunol., 159, 140–151. [DOI] [PubMed] [Google Scholar]
- 28.Kruys V., Marinx,O., Shaw,G., Deschamps,J. and Huez,G. (1989) Science, 245, 852–855. [DOI] [PubMed] [Google Scholar]
- 29.Moreira A.L., Sampaio,E.P., Zmuidzinas,A., Frindt,P., Smith,K.A. and Kaplan,G. (1993) J. Exp. Med., 177, 1675–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee J.C., Laydon,J.T., McDonnell,P.C., Gallagher,T.F., Kumar,S., Green,D., McNulty,D., Blumenthal,M.J., Heys,J.R., Landvatter,S.W. et al. (1994) Nature, 372, 739–746. [DOI] [PubMed] [Google Scholar]
- 31.Laroia G., Cuesta,R., Brewer,G. and Schneider,R.J. (1999) Science, 284, 499–502. [DOI] [PubMed] [Google Scholar]
- 32.Levy N.S., Chung,S., Furneaux,H. and Levy,A.P. (1998) J. Biol. Chem., 273, 6417–6423. [DOI] [PubMed] [Google Scholar]
- 33.Jain R.G., Andrews,L.G., McGowan,K.M., Pekala,P.H. and Keene,J.D. (1997) Mol. Cell. Biol., 17, 954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamilton B.J., Nichols,R.C., Tsukamoto,H., Boado,R.J., Pardridge,W.M. and Rigby,W.F.C. (1999) Biochem. Biophys. Res. Commun., 261, 646–651. [DOI] [PubMed] [Google Scholar]
- 35.Hamilton B.J., Nagy,E., Malter,J.S., Arrick,B.A. and Rigby,W.F.C. (1993) J. Biol. Chem., 268, 8881–8887. [PubMed] [Google Scholar]
- 36.Gao F.-B., Carson,C.C., Levine,T. and Keene,J.D. (1994) Proc. Natl Acad. Sci. USA, 91, 11207–11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhattacharya S., Giordano,T., Brewer,G. and Malter,J.S. (1999) Nucleic Acids Res., 27, 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dreyfuss G., Matunis,M.J., Pinol-Roma,S. and Burd,C.G. (1993) Annu. Rev. Biochem., 62, 289–321. [DOI] [PubMed] [Google Scholar]
- 39.Dember L.M., Kim,N.D., Liu,K.Q. and Anderson,P. (1996) J. Biol. Chem., 271, 2783–2788. [DOI] [PubMed] [Google Scholar]
- 40.Casey J.L., Hentze,M.W., Koeller,D.M., Caughman,S.W., Rouault,T.A., Klausner,R.D. and Harford,J.B. (1988) Science, 240, 924–928. [DOI] [PubMed] [Google Scholar]
- 41.Mullner E.W. and Kuhn,L.C. (1988) Cell, 53, 815–825. [DOI] [PubMed] [Google Scholar]
- 42.Sueoka E., Goto,Y., Sueoka,N., Kai,Y., Kozu,T. and Fujiki,H. (1999) Cancer Res., 59, 1404–1407. [PubMed] [Google Scholar]
- 43.Montuenga L.M., Zhou,J., Avis,I., Vos,M., Martinez,A., Cuttitta,F., Treston,A.M., Sunday,M. and Mulshine,J.L. (1998) Am. J. Respir. Cell. Mol. Biol., 19, 554–562. [DOI] [PubMed] [Google Scholar]
- 44.Zhou J., Mulshine,J.L., Ro,J.Y., Avis,I., Yu,R., Lee,J.J., Morice,R., Lippman,S.M. and Lee,J.S. (1998) Clin. Cancer Res., 4, 1631–1640. [PubMed] [Google Scholar]
- 45.Tockman M.S., Mulshine,J.L., Piantadosi,S., Erozan,Y.S., Gupta P.K., Ruckdeschel,J.C., Taylor,P.R., Zhukov,T., Zhou,W.H., Qiao,Y.L. and Yao,S.X. (1997) Clin. Cancer Res., 3, 2237–2246. [PubMed] [Google Scholar]
- 46.Qiao Y.L., Tockman,M.S., Li,L., Erozan,Y.S., Yao,S.X,. Barrett,M.J., Zhou,W.H., Giffen,C.A., Luo,X.C. and Taylor,P.R. (1997) Cancer Epidemiol. Biomarkers. Prev., 6, 893–900. [PubMed] [Google Scholar]
- 47.Tockman M.S. (1996) J. Cell. Biochem. Suppl., 25, 177–184. [PubMed] [Google Scholar]
- 48.Zhou J., Mulshine,J.L., Unsworth,E.J,. Scott,F.M., Avis,I.M., Vos,M.D. and Treston,A.M. (1996) J. Biol. Chem., 271, 10760–10766. [DOI] [PubMed] [Google Scholar]
- 49.Matsumoto A., Nito,M., Itakura,H., Ikemoto,S., Asaoka,H., Hayakawa,I., Kanamori,H., Aburatani,H, Takaku,F., Suzuki,H., Kobari,Y., Miyai,T., Takahashi,K., Cohen,E.H., Wydro,R., Housman,D.E. and Kodama,T. (1990) Proc. Natl Acad. Sci. USA, 87, 9133–9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hamilton B.J., Burns,C.M., Nichols,R.C. and Rigby,W.F.C. (1997) J. Biol. Chem., 272, 28732–28741. [DOI] [PubMed] [Google Scholar]
- 51.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 52.Burd C.G. and Dreyfuss,G. (1994) EMBO J., 13, 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Swanson M.S. and Dreyfuss,G. (1988) Mol. Cell. Biol., 8, 2237–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang W., Furneaux,H., Cheng,H., Caldwell,M.C., Hutter,D., Liu,Y., Holbrook,N. and Gorospe,M. (2000) Mol. Cell. Biol., 20, 760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gay E. and Babajko,S. (2000) Biochem. Biophys. Res. Commun., 267, 509–515. [DOI] [PubMed] [Google Scholar]
- 56.Gorlach M., Burd,C.G. and Dreyfuss,G. (1994) J. Biol. Chem., 269, 23074–23078 [PubMed] [Google Scholar]
- 57.Temsamani J. and Pederson,T. (1996) J. Biol. Chem., 271, 24922–24926. [DOI] [PubMed] [Google Scholar]
- 58.Chassin D., Benifla,J.L., Delattre,C., Fernandez,H., Ginisty,D., Janneau,J.L., Prade,M., Contesso,G., Caillou,B., Tournaire,M. et al. (1994) Cancer Res., 54, 5217–5223. [PubMed] [Google Scholar]
- 59.Pinol-Roma S. and Dreyfuss,G. (1993) Mol. Cell. Biol., 13, 5762–5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taylor G.A., Thompson,M.J., Lai,W.S. and Blackshear,P.J. (1995) J. Biol. Chem., 270, 13341–13347. [DOI] [PubMed] [Google Scholar]
- 61.Barnett S.F., Theiry,T.A. and LeStourgeon,W.M. (1991) Mol. Cell. Biol., 11, 864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kiledjian M., DeMaria,C.T., Brewer,G. and Novick,K. (1997) Mol. Cell. Biol., 17, 4870–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chu E., Copur,S.M., Ju,J., Chen,T.M., Khleif,S., Voeller,D.M., Mizunuma,N., Patel,M., Maley,G.F., Maley,F. and Allegra,C.J. (1999) Mol. Cell. Biol., 19, 1582–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]