

Abstract
Purpose:
This study focused on the genetic screening of Myocilin (MYOC), Cytochrome P450 family 1 subfamily B member 1 (CYP1B1), Optineurin (OPTN), and SIX homeobox 6 (SIX6) genes in a family with coexistence of primary congenital glaucoma (PCG) and juvenile open-angle glaucoma (JOAG).
Methods:
Sanger sequencing was used to examine the coding region of all four genes. Six different online available algorithms were used for the pathogenicity prediction of missense variant. Structural analysis was done using Garnier–Osguthorpe–Robson (GOR), PyMol, ChimeraX, and Molecular Dynamic (MD) Simulations (using Graphics Processing Unit (GPU)-enabled Desmond module of Schrödinger).
Results:
There were a total of three sequence variants within the family. All seven algorithms determined that a single mutation, G538E, in the OPTN gene is pathogenic. The loops connecting the strands became more flexible, as predicted structurally and functionally by pathogenic mutations. Mutations create perturbations and conformational rearrangements in proteins, hence impairing their functioning.
Conclusion:
In this study, we describe a North Indian family in which members were having JOAG and PCG due to a rare homozygous/heterozygous mutation in OPTN. The coexistence of two types of glaucoma within a single pedigree suggests that certain OPTN mutations may be responsible for the onset of different glaucoma phenotypes.
Keywords: Cytochrome P450, juvenile open-angle glaucoma, myocilin, optineurin, primary congenital glaucoma
Glaucoma is a group of related optic ailments that can cause progressive vision loss and complete blindness by acquired loss of retinal ganglion cells (RGCs) damaging a nerve in the back of our eye called the optic nerve, which causes visual impairment and leads to absolute blindness if left untreated.[1,2,3,4] The distinguishing characteristic of glaucoma is its specific pattern of peripheral loss of vision.[5,6] Glaucoma is broadly classified into two categories: primary glaucoma and secondary glaucoma, which are further divided into open-angle and angle-closure on the basis of anterior chamber anatomy.[3]
On the basis of disease onset-age, the primary open-angle glaucoma (POAG) can be classified into three subtypes: primary congenital glaucoma (PCG) with onset-age 0–3 years of age, juvenile open-angle glaucoma (JOAG) with onset-age 3–40 years and adult-onset POAG with onset-age above 40 years.
A primary congenital glaucoma is a severe form of the disease characterized by abnormal development of trabecular meshwork and elevated intraocular pressure (IOP) at birth or within the first year of life, which results in photophobia, extreme tearing, and buphthalmos (enlargement of the eye globe).[7] PCG is bilateral in almost 80% of the cases.[8]
Juvenile open-angle glaucoma (JOAG) is a rare subset of POAG having an onset-age of 3–40 years.[9] An International Consensus Classification proposed by the Childhood Glaucoma Research Network states that the earliest onset-age for JOAG is considered to be 4 years, and the upper limit ranges up to 30 or 40 years, and after this, it will be considered as adult-onset POAG.[10] Important diagnostic characteristics include an IOP of more than 21 mmHg, open iridocorneal angles, and glaucomatous optic neuropathy (with or without any visual field abnormalities). JOAG is typically accompanied by a more severe and rapidly developing disease than POAG in adults. Individuals with this condition frequently develop high intraocular pressures (IOP >30 mmHg) that are resistant to treatment.[11] Although topical medications may temporarily reduce IOP, surgical treatment is often required for optimal IOP control. The incidence rate is higher in males.[12] Although JOAG is typically found in both eyes, up to 25% of individuals have been observed to have only one affected eye. Among different groups, JOAG is more or less likely to run in the family; among Koreans, it is 28%, among Indians, it is 30%, among Americans it is 43%, and among Africans, it can reach as high as 88%.[12]
Recent data indicate that the JOAG is genetically diverse. In addition to autosomal dominant inheritance with diverse penetrance and expressivity, autosomal recessive inheritance and sporadic occurrence are common in specific ethnic groups. Genetic studies on various ethnic studies have found the gene involved in the pathogenesis of JOAG. Myocilin (MYOC) was the first gene linked to the pathogenesis of JOAG.[9,12,13] Further research have revealed various other gene linked to JOAG including Cytochrome P450 family 1 subfamily B member 1 (CYP1B1),[14,15,16,17] Optineurin (OPTN),[15,18] latent transforming growth factor beta-binding protein 2 (LTBP2),[19] TANK-binding kinase 1 (TBK1), C3- and PZP-like alpha-2-macroglobulin domain containing 8 (CPAMD8),[20] and EGF-containing fibulin-like extracellular matrix protein 1 (EFEMP1).[21] While the genes associated with PCG includes CYP1B1,[22] MYOC,[22] LTBP2,[23] forkhead box C1 (FOXC1),[24] and collagen type I alpha 1 chain (COL1A1).[25]
OPTN gene is located on chromosome 10. It encodes an adapter protein that mediates multiple physiological functions, including signaling, vesicle trafficking, and autophagy. It comprises 16 exons out of which 3 are noncoding exons in the 5’ UTR and the rest 13 are coding exons that code for Optn protein formed of amino acids at position 577.[26] Optn protein is organized into various distinct domains like LC-3 interacting region (LIR), Ubiquitin-binding domain (UBD), zinc finger (ZF), and leucine zipper (LZ).[27] Mutations in OPTN are associated with POAG, JOAG,[15] and amyotrophic lateral sclerosis.[28] The OPTN mutations associated with glaucoma are mostly missense mutations. Optn interacts with various proteins including Rab8, myosin VI, Huntingtin (htt), TBC1 domain family member 17 (TBC1D17), and transferrin receptor to mediate various membrane vesicle trafficking pathways. It is an autophagy receptor that mediates cargo-selective as well as nonselective autophagy.[26] Glaucoma-associated mutants of OPTN, E50K, and M98K lead to the death of RGCs by causing defects in vesicle trafficking, autophagy, and signaling.[29] In the present study, we characterized the clinical manifestations of a North Indian family with glaucoma and investigated its molecular basis in order to expand the spectrum of MYOC, CYP1B1, OPTN, and SIX6 gene mutations in the Indian population.
Methods
Clinical observations and diagnosis
A large North Indian family having 16 members spanning three generations was enrolled in this study. The proband was a 3-year-old boy presenting in the glaucoma clinic. The proband was a hereditary case of glaucoma with two other family members (his sister and mother) were also suffering from the disease. This study was approved by the IHEC (Institution Human Ethical Committee) and the BREC (Biomedical Research Ethics Committee).
After obtaining written informed consent from all participants or their legal guardians, where required, their medical history was collected. Comprehensive ophthalmologic examinations were done for all the participants of the family. The examination includes the best-corrected visual acuity according to Snellen’s chart, IOP measurement, anterior chamber angle evaluation, and vertical optic cup-to-disc ratio (C/D ratio) assessment.
Approximately 5 ml of blood was drawn from all participating family members, and the samples were stored in K2EDTA vacutainers. Genomic DNA was extracted as previously described. The medical records including IOP, visual acuity, and Vertical cup-to-disc ratio (VCDR) were collected to analyze the genotype–phenotype correlation of the patients.
Mutation screening using Sanger sequencing
Four genes including CYP1B1, MYOC, OPTN, and SIX6 were analyzed for any mutation in the proband and its family. Polymerase Chain Reaction (PCR) was used to amplify all the coding exons and intron-exon junctions of the genes using specific primers. The primers for CYP1B1 were designed using Primer3Plus, while already published primers were used for the amplification of MYOC,[30] OPTN,[31] and SIX6[32] gene. PCR reactions were performed in 50 μl containing 110–150 ng of genomic DNA with 5 μl of taq buffer, 2.5 μl of 25 mM MgCl2, 2.5 μl of dNTPs, 2 μl of each primer, and 1.5 μl of Taq DNA Polymerase (Genei Labs). The temperature profiles included initial denaturation at 94ºC for 5 minutes followed by 35 cycles consisting of 60-second denaturation at 94ºC, annealing at 54–60ºC for 45 to 60 seconds, and extension at 72ºC for 60 seconds. The PCR products obtained were visualized on 1.8% agarose gel. The amplified PCR products were prepared for Sanger Sequencing using the PCR Product Purification Kit (Favorgen, Biotech Corp.). DNA Sanger sequencing was performed on purified PCR products by Barcode Biosciences Pvt Ltd.
Pathogenicity prediction analysis
MYOC, CYP1B1, OPTN, and SIX6 DNA sequences were compared with ensemble gene reference sequence ENSG00000034971, ENSG00000138061, ENSG00000123240, and ENSG00000184302, respectively, using the CLUSTALW[33] program.
ClinVar and gnomAd were used to look for the identified variation in all the genes., PROVEAN (Protein Variation Effect Analyzer),[34] PolyPhen2,[35,36,37] Mutation Taster, SNAP2, I Mutant2.0, and MutaPred2 algorithms were used to predict the pathogenicity of detected missense sequence variants.[38]
Homology analysis of protein
The Human Optn protein sequence (ensemble gene reference sequence ENSG00000184302) was compared to the Optn protein sequence of Chimpanzee (ensemble gene reference sequence ENSPTRG00000002298), Monkey (ensemble gene reference sequence ENSMMUG00000005601), Cat (ensemble gene reference sequence ENSFCAG00000003917), Mouse (ensemble gene reference sequence ENSMUSG00000026672), and Chicken (ensemble gene reference sequence ENSGALG00010005383) using the CLUSTALW program. The sequence logo was prepared using WebLogo[39] to show the important regions of the protein from an evolutionary point of view.
Structural and functional analysis
Utilizing Garnier–Osguthorpe–Robson (GOR) software,[40] the effect of mutation G538E on the secondary structure of Optn was predicted. The GOR software computes the probability of the four structural classes (helix, sheet, turn, and loop) from the generated matrices based on the central residue and its neighbors to infer the secondary structure of the sequence.
For 3D structural analysis human OPTN 3D structure was obtained from the Alpha fold database (https://alphafold.ebi.ac.uk/). Wild-type OPTN structure was employed to create disease mutant p.G538E structure by substituting wild residue glycine with disease-associated residue glutamic acid to elucidate the possible structural effects of the mutation. The structural changes in the mutant protein were analyzed using Pymol[41] and ChimeraX.[42] The change in H-bonds and electrostatic potential were analyzed to predict the structural changes.
Functional analysis was done by using Molecular Dynamic (MD) Simulation. For the wild-type (WD) and G538E mutant, all atom MD simulations were run for 10 ns under explicit water solvent conditions. MD simulations were done at 300 K at the molecular mechanics level using the GROMOS96 43a1 force field in the GROMACS 5.1.2 software package. All systems were immersed in a 10 Å cubic box of water molecules. The systems were then immersed in a box with a simple point charge (SPC16) water model. Na+ and Cl− ions were further aided in the systems for neutralizing and maintaining physiological concentrations (0.15 M). All of the systems were minimized using the steepest descent of 5,000 steps. The final simulations were run for 10 ns for each system, using a leap-frog integrator to track the time evolution of trajectories. Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF) were calculated to depict the functional changes in the protein molecules that occurred due to the mutation.[43]
Results
Clinical examination
The pedigree of the family included in the present study is shown in Fig. 1 The III2 proband experienced buphthalmos, symptoms of vision loss, itching, and headache for the first time at 2 years of age. The proband was diagnosed with PCG by the clinician. The IOP at the presentation were 26.3mmHg and 34.5 mmHg and the cup-to-disc ratio of 0.9:1/0.9:1 for the right and left eye, respectively. The patient exhibited severe visual field damage and low vision (Right: light perception +ve; Left: light perception +ve).
Figure 1.
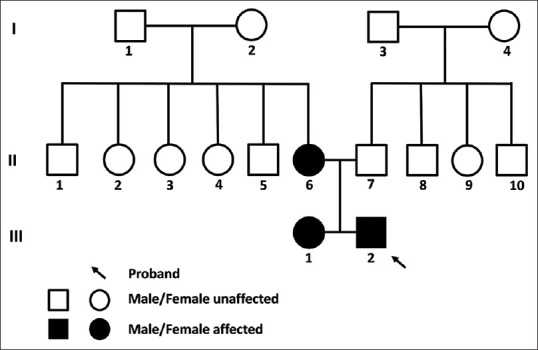
Pedigree of the family affected with JOAG
Of the remaining 15 members, the mother of the proband was suffering from JOAG whereas the sister of the proband at 4 years of age was suffering from PCG. The rest of the unaffected family members have normal IOP levels, C/D ratio, and visual acuity. All patients showed symptoms of headache and vision loss before diagnosis (also buphthalmos in the case of PCG patients). The mean IOP values were 32.4 mmHg (range: 26.3–42 mmHg) and 33 mmHg (range: 32–34.5 mmHg) for the right and left eye, respectively. The C/D ratios of the patients were OA/0.9:1, 0.7:1/0.75:1, and 0.9:1/0.9:1 for II6, III1, and III2, respectively. Due to the severity, the visual acuity is affected in all the patients i.e., Pl-ve : HM +ve, 6/60 : Pl+ve, Pl+ve : Pl+ve for II6, III1, III2 respectively (patient cards are attached as supplementary material). While the unaffected members have a C/D ratio and visual acuity in the normal range. The clinical details of the family members if included in Table 1.
Table 1.
Clinical data of the affected and unaffected members of the family
| Pedigree number | Phenotype | Sex | Age | IOP OD/OS | Age at diagnosis | Buphthalmos | Corneal diameter (in mm) | Corneal striae | C/D ratio OD/OS | BCVA OD/OS | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||||||
| Vertical | Horizontal | OD | OS | |||||||||||
|
|
|
|||||||||||||
| OD | OS | OD | OS | |||||||||||
| Affected family members | ||||||||||||||
| II6 | JOAG | F | 26 | 42/32 mmHg | 18 | Absent | – | – | – | – | – | –– | OA/0.9:1 | Pl-ve: HM+ve |
| III1 | PCG | F | 4 | 26.3/32.6 mmHg | 2.5 | Present | 13 | 14 | 14 | 14.5 | - | – | 0.7:1/0.75:1 | 6/60 : Pl+ve |
| III2 | PCG | M | 3 | 29/34.5 mmHg | 2 | Present | 16.5 | 16.5 | 17.0 | 17.0 | At 3 and 5:30 o’clock position | At 6 o’clock position | 0.9:1/0.9:1 | Pl+ve : Pl+ve |
| Unaffected family members | ||||||||||||||
| II5 | Normal | M | 29 | 14/17 mmHg | – | Absent | – | – | – | – | – | – | 0.3:1/0.4:1 | 6/6 : 6/6 |
| II7 | Normal | M | 30 | 13/15 mmHg | – | Absent | – | – | – | – | – | – | 0.3:1/0.3:1 | 6/12 : 6/6 |
| II9 | Normal | F | 34 | 15/16 mmHg | – | Absent | – | – | – | – | – | – | 0.3:1/0.35:1 | 6/6 : 6/18 |
BCVA, best corrected visual acuity; OD/OS, oculus dexter/oculus sinister
Sequencing and comparative analysis
Sanger sequencing was done to discover any missense changes in MYOC, CYP1B1, OPTN, and SIX6 in the family members of the proband. The coding region of these genes was amplified using PCR and the purified products were sent for Sanger sequencing. The sequencing results were compared with the reference sequences from the ensemble database. A total of three mutations were found in MYOC (g.171652247G>C), CYP1B1 (g.38071007T>C), and OPTN gene (g.13136745 G>A). No variants were found in the SIX6 gene. After comparing with the reference sequence, only the OPTN missense mutation (g.13136745 G>A, c.1613G>A: p.G538E) was found to be pathogenic by protein function prediction software PROVEAN, PolyPhen2, Mutation Taster, MutPred2, SNAP2 and IMutant2.0 [Table 2]. The mutation (G>A) was located in the coding region of exon 16 at base 1916, leading to the mutation of amino acid at position 538 of the encoded protein from glycine to glutamic acid. Multiple sequence alignment analyses indicated that this gene shows a high degree of conservation among different species. The detected mutation was not present in ExAC, 1000 genomes, and gnomAD database, but a single entry with insufficient information was present on ClinVar. Three family members (two heterozygous and one homozygous) of the family were harboring this mutation, and all of them are suffering from advanced stages of glaucoma (2 PCG and 1 with JOAG) with a high IOP value and particular damage to the optic nerve. The mutation was not present in the unaffected members of the family. The Sanger sequencing results and the protein homology analysis are depicted in Fig. 2. The pathogenic mutation in the OPTN gene was further analyzed to predict the structural and functional effects of the mutation.
Table 2.
Pathogenicity prediction of the sequence variation using six different algorithms
| Gene Name | Genomic Position and Base Change | cDNA Position | Codon change | Amino acid change | No. of Patients with the change (H: homozygous, h: heterozygous) | Pathogenicity prediction | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| PROVEAN | Poly Phen2 | Mutation Taster | IMutant2.0 | SNAP2 | Mut Pred2 | ||||||||
|
|
|
||||||||||||
| ΔΔG (kcal/mol) DDG value | Stability Prediction SVM2 | Score | Prediction | ||||||||||
| MYOC | g.171652247G>C | c.365 | GGC>GCC | p.Gly122Ala | 1 (h) | Neutral | Benign | Polymorphism | -72 | Neutral | 0.52 | Decrease | 0.061 |
| CYP1B1 | g.38071007T>C | c.1347 | GAT>GAC | p.Asp449= | 1 (h) | Not applicable | |||||||
| OPTN | g.13136745 G>A | c.1613 | GGA>GAA | p.G538E | 3 (1H, 2h) | Deleterious | Probably damaging | Disease Causing | 0.13 | Increase | 34 | effect | 0.373 |
Figure 2.

Sequencing and comparative analysis. (a) Results of Sanger sequencing showing normal sequence (II7), heterozygous mutation (II6, III2), and homozygous mutation (III1); (b) Homology analysis of the protein between humans and other chordate species; and (c) Word Logo showing high degree of conservation among different species at position 538 of amino acid
Changes in protein secondary structure and 3D structure
The change in amino acid at position 538 from glycine to glutamic acid resulted in structural changes in the protein. Glycine (NH2-CH2-COOH) is the simplest amino acid having no charge, whereas glutamic acid (C5H9NO4) is negatively charged and polar in nature. The secondary conformations of amino acid from position 500 to 570 are depicted in Fig. 3. Due to p.G538E, there is a change in the secondary structure of amino acid at positions 533–539. In the WD protein, the amino acids at positions 533–536 were in a sheet conformation, 537 was in a turn conformation, and 538–539 were coil conformation. Due to the change at position 538, the amino acids at positions 533–539 were changed to the helical conformation.
Figure 3.

The secondary conformations of amino acid from 500 to 570 in (a) WT and (b) Mutated protein using GOR software
The amino acid glutamic acid at position 538 clashes with the amino acid glutamic acid at position 540 causing a structural deviation in the protein. The clashing between the particular atoms of amino acids at positions 538 and 540 were shown as yellow lines. Due to these clashes, there might be a structural change in the protein to avoid these clashes. Due to the differences in the net charge of the two amino acids, there is an increase in the net negative charge in the area of mutation. The functional effect of this change is further confirmed by doing MD simulations. The change in the structure and electrostatic potential has been shown in Fig. 4.
Figure 4.
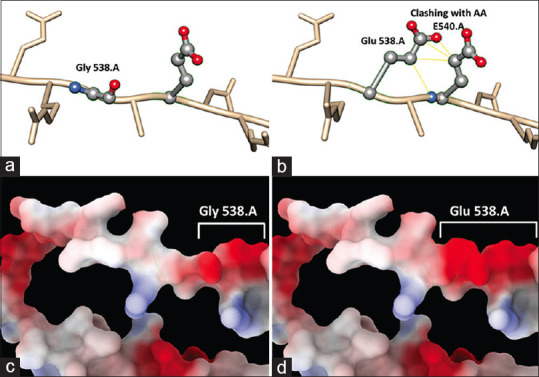
Changes in the 3D structure of the protein. (a) 3D structure showing WT protein with glycine at position 538 and (b) showing mutated protein with glutamic acid at position 538 clashing with amino acid at position 540. (c and d) showing the changes in electrostatic charge between WT and mutated protein, respectively
Molecular dynamic simulation
MD simulation is helpful in assessing the movement of atoms in a high-complexity macromolecule. The RMSD and the RMSF are the two most commonly used entities to measure the fluctuations in the structure of the macromolecule-like proteins. The RMSD data for the WD and mutated protein are displayed in the graphical form in Fig. 5a, where we can easily see a measurable change in the values between both proteins. The average RMSD value for WD and the mutant is 0.75 and 1.75 nm, respectively. There is a difference of 1 nm in the RMSD values, which show that there is a decrease in the stability of the protein due to the mutation G538E. Differences in the RMSD values for WT and mutant protein show that there are conformational changes in the protein due to the mutation G538E. The RMSF allows us to analyze the fluctuation of residual variation. The RMSF values of the WD and the mutant in Fig. 5b show differences at residue 180–200 (RMSF 12.3 Å for WD and 6 Å for G538E mutant) and 450–550 (average RMSF of 3 and 2 Å for WD and G538E mutant, respectively, from residue 450 to 520, for residue 520–550 the average RMSF was 6 Å and 4 Å for WT and G538E mutant, respectively) where the values for the mutant are lower than the WT suggesting a decrease in the flexibility of the protein due to the mutation. The mutant G538E protein becomes more compact leading to the decrease in the flexibility of the protein. The change in RMSD and the RMSF values indicates the potential cause behind the pathogenic behavior of the mutation as suggested by the protein function prediction software.
Figure 5.

MD simulation results of the WT and mutated protein. (a and b) Graphical representation of the RMSD and RMSF values, respectively, showing differences between WT and mutated protein
Discussion
Optineurin mutations were primarily linked to Normal Tension Glaucoma (NTG, a rare subset of glaucoma having IOP in the normal range, i.e., 11–21 mmHg) and amyotrophic lateral sclerosis (ALS). But population-based studies have revealed the role of optineurin in the pathogenesis of POAG and JOAG also.[15] But there are no studies indicating the role of pathogenic OPTN mutations in the progression of PCG. Optineurin is involved in various cellular mechanisms including signaling, vesicle trafficking, and autophagy through interaction with protein partners like Rab8, myosin VI, htt, TBC1D17 in various types of cells including trabecular meshwork (TM) in the eyes.[44]
Optineurin colocalizes with htt in the Golgi apparatus, where it contributes to post-Golgi trafficking and Golgi organization. Optineurin also acts as a linking unit between Myosin VI, Rab8, and htt,[45] which is also affected by the mutation G538E. The C-terminal of optineurin binds with huntingtin, which has been shown to bind with Huntingtin-associated protein 1 (HAP1). HAP1 interacts with the plus (+)-end-directed microtubule motor protein dynein as well as dynein activator dynactin. With the help of huntingtin, optineurin helps in maintaining the structure of the Golgi complex intact and in the vesicular transport of various biomolecules including proteins.[46] But due to G538E mutation, the optineurin fails to interact with huntingtin (as suggested by the Protein function prediction software Mutation Taster) which in turn causes the fragmentation of the Golgi complex and interrupts the vesicular transport of proteins across the cells as shown in Fig. 6. This can lead to cell misfunctioning and ultimately autophagy of TM cells. Abnormal development of TM results in an increase in IOP levels, which is the main driving force behind the development of glaucoma.[47]
Figure 6.

Diagrammatic illustration of the possible interaction of optineurin with motor protein complexes at the Golgi complex. (a) Interaction involving WT optineurin and (b) showing the Golgi complex fragmentation due to the mutation G538E in Optn protein
Conclusion
In this study, we describe a North Indian family in which members were having JOAG and PCG due to a rare homozygous/heterozygous mutation in OPTN. This type of familial inheritance of JOAG and PCG has previously been documented in a Pakistani study with a homozygous mutation in the CYP1B1 gene.[48] In addition, the p.G538E mutation in OPTN would be the first reported mutation in Indian JOAG and PCG patients. The coexistence of two types of glaucoma within a single pedigree suggests that certain OPTN mutations may be responsible for the onset of different glaucoma phenotypes. With this new mutation associated with glaucoma, specific genetic testing kits may be developed to identify children in a family who may be at risk for the disease and to initiate disease surveillance and conventional treatments sooner in order to prevent any chances of vision loss. The results of this study may lead to the development of treatments designed to prevent vision loss in children with the OPTN mutation. Further functional analysis can reveal the exact mechanism lying behind the pathogenesis of this particular optineurin mutant.
Financial support and sponsorship
Council of Scientific and Industrial Research, New Delhi, India.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Huang X, Li M, Guo X, Li S, Xiao X, Jia X, et al. Mutation analysis of seven known glaucoma-associated genes in Chinese patients with glaucoma. Invest Ophthalmol Vis Sci. 2014;55:3594–602. doi: 10.1167/iovs.14-13927. [DOI] [PubMed] [Google Scholar]
- 2.Wiggs JL, Pasquale LR. Genetics of glaucoma. Hum Mol Genet. 2017;26:R21–7. doi: 10.1093/hmg/ddx184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zukerman R, Harris A, Oddone F, Siesky B, Verticchio Vercellin A, Ciulla TA. Glaucoma heritability:Molecular mechanisms of disease. Genes. 2021;12:1135. doi: 10.3390/genes12081135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yadav M, Tanwar M. Impact of COVID-19 on glaucoma management: A review. [[Last accessed on 2022 Dec 28]];Front Ophthalmol. 2022 2 doi: 10.3389/fopht.2022.1003653. Available from: https://www.frontiersin.org/articles/10.3389/fopht. 2022.1003653 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonas JB, Aung T, Bourne RR, Bron AM, Ritch R, Panda-Jonas S. Glaucoma. Lancet. 2017;390:2183–93. doi: 10.1016/S0140-6736(17)31469-1. [DOI] [PubMed] [Google Scholar]
- 6.Kastner A, King AJ. Advanced glaucoma at diagnosis:Current perspectives. Eye. 2020;34:116–28. doi: 10.1038/s41433-019-0637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melki R, Colomb E, Lefort N, Brézin AP, Garchon HJ. CYP1B1 mutations in French patients with early-onset primary open-angle glaucoma. J Med Genet. 2004;41:647–51. doi: 10.1136/jmg.2004.020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanwar M, Dada T, Sihota R, Dada R. Mitochondrial DNA analysis in primary congenital glaucoma. Mol Vis. 2010;16:518–33. [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta V, Somarajan BI, Gupta S, Walia GK, Singh A, Sofi R, et al. The mutational spectrum of Myocilin gene among familial versus sporadic cases of Juvenile onset open angle glaucoma. Eye. 2021;35:400–8. doi: 10.1038/s41433-020-0850-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thau A, Lloyd M, Freedman S, Beck A, Grajewski A, Levin AV. New classification system for pediatric glaucoma:Implications for clinical care and a research registry. Curr Opin Ophthalmol. 2018;29:385–94. doi: 10.1097/ICU.0000000000000516. [DOI] [PubMed] [Google Scholar]
- 11.Turalba AV, Chen TC. Clinical and genetic characteristics of primary juvenile-onset open-angle glaucoma (JOAG) Semin Ophthalmol. 2008;23:19–25. doi: 10.1080/08820530701745199. [DOI] [PubMed] [Google Scholar]
- 12.Selvan H, Gupta S, Wiggs JL, Gupta V. Juvenile-onset open-angle glaucoma –A clinical and genetic update. Surv Ophthalmol. 2022;67:1099–117. doi: 10.1016/j.survophthal.2021.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan AO. Genetics of primary glaucoma. Curr Opin Ophthalmol. 2011;22:347–55. doi: 10.1097/ICU.0b013e32834922d2. [DOI] [PubMed] [Google Scholar]
- 14.Bayat B, Yazdani S, Alavi A, Chiani M, Chitsazian F, Tusi BK, et al. Contributions of MYOC and CYP1B1 mutations to JOAG. Mol Vis. 2008;14:508–17. [PMC free article] [PubMed] [Google Scholar]
- 15.Huang C, Xie L, Wu Z, Cao Y, Zheng Y, Pang CP, et al. Detection of mutations in MYOC, OPTN, NTF4, WDR36 and CYP1B1 in Chinese juvenile onset open-angle glaucoma using exome sequencing. Sci Rep. 2018;8:4498. doi: 10.1038/s41598-018-22337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Souzeau E, Hayes M, Zhou T, Siggs OM, Ridge B, Awadalla MS, et al. Occurrence of CYP1B1 Mutations in Juvenile Open-Angle Glaucoma With Advanced Visual Field Loss. JAMA Ophthalmol. 2015;133:826–33. doi: 10.1001/jamaophthalmol.2015.0980. [DOI] [PubMed] [Google Scholar]
- 17.Su CC, Liu YF, Li SY, Yang JJ, Yen YC. Mutations in the CYP1B1 gene may contribute to juvenile-onset open-angle glaucoma. Eye Lond Engl. 2012;26:1369–77. doi: 10.1038/eye.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang M, Chen JH, Wu P, Huang C, HUANG Y, Chen H, et al. Mutation screen of MYOC, OPTN, WDR36 and NTF4 in juvenile-onset open-angle glaucoma. Invest Ophthalmol Vis Sci. 2015;56:2546. [Google Scholar]
- 19.Saeedi O, Yousaf S, Tsai J, Palmer K, Riazuddin S, Ahmed ZM. Delineation of novel compound heterozygous variants in LTBP2 associated with juvenile open angle glaucoma. Genes. 2018;9:527. doi: 10.3390/genes9110527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siggs OM, Souzeau E, Taranath DA, Dubowsky A, Chappell A, Zhou T, et al. Biallelic CPAMD8 variants are a frequent cause of childhood and juvenile open-angle glaucoma. Ophthalmology. 2020;127:758–66. doi: 10.1016/j.ophtha.2019.12.024. [DOI] [PubMed] [Google Scholar]
- 21.Gupta V, Somarajan BI, Gupta S, Mahalingam K, Kumar M, Singh A. Association of EFEMP1 with juvenile-onset open angle glaucoma in a patient with concomitant COL11A1-related Stickler syndrome. Ophthalmic genetics. 2023;44:281–5. doi: 10.1080/13816810.2022.2109682. [DOI] [PubMed] [Google Scholar]
- 22.Kim HJ, Suh W, Park SC, Kim CY, Park KH, Kook MS, et al. Mutation spectrum of CYP1B1 and MYOC genes in Korean patients with primary congenital glaucoma. Mol Vis. 2011;17:2093–101. [PMC free article] [PubMed] [Google Scholar]
- 23.Narooie-Nejad M, Paylakhi SH, Shojaee S, Fazlali Z, Rezaei Kanavi M, Nilforushan N, et al. Loss of function mutations in the gene encoding latent transforming growth factor beta binding protein 2, LTBP2, cause primary congenital glaucoma. Hum Mol Genet. 2009;18:3969–77. doi: 10.1093/hmg/ddp338. [DOI] [PubMed] [Google Scholar]
- 24.Siggs OM, Souzeau E, Pasutto F, Dubowsky A, Smith JEH, Taranath D, et al. Prevalence of FOXC1 Variants in Individuals With a Suspected Diagnosis of Primary Congenital Glaucoma. JAMA Ophthalmol. 2019;137:348–55. doi: 10.1001/jamaophthalmol.2018.5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mauri L, Uebe S, Sticht H, Vossmerbaeumer U, Weisschuh N, Manfredini E, et al. Expanding the clinical spectrum of COL1A1 mutations in different forms of glaucoma. Orphanet J Rare Dis. 2016;11:108. doi: 10.1186/s13023-016-0495-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ryan TA, Tumbarello DA. Optineurin:A coordinator of membrane-associated cargo trafficking and autophagy. Front Immunol. 2018;9:1024. doi: 10.3389/fimmu.2018.01024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kachaner D, Génin P, Laplantine E, Weil R. Toward an integrative view of Optineurin functions. Cell Cycle. 2012;11:2808–18. doi: 10.4161/cc.20946. [DOI] [PubMed] [Google Scholar]
- 28.Toth RP, Atkin JD. Dysfunction of optineurin in amyotrophic lateral sclerosis and glaucoma. Front Immunol. 2018;9:1017. doi: 10.3389/fimmu.2018.01017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swarup G, Sayyad Z. Altered functions and interactions of glaucoma-associated mutants of optineurin. Front Immunol. 2018;9:1287. doi: 10.3389/fimmu.2018.01287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhuo YH, Wang M, Wei YT, Huang YL, Ge J. Analysis of MYOCgene mutation in a Chinese glaucoma family with primary open-angle glaucoma and primary congenital glaucoma. Chin Med J (Engl) 2006;119:1210–4. [PubMed] [Google Scholar]
- 31.Ayala-Lugo RM, Pawar H, Reed DM, Lichter PR, Moroi SE, Page M, et al. Variation in optineurin (OPTN) allele frequencies between and within populations. Mol Vis. 2007;13:151–63. [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X, Sun NN, Zhao ZN, He SX, Zhang M, Zhang DD, et al. Coinheritance of OLFM2 and SIX6 variants in a Chinese family with juvenile-onset primary open-angle glaucoma:A case report. World J Clin Cases. 2021;9:697–706. doi: 10.12998/wjcc.v9.i3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W:Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi Y. Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine - BCB '12. Orlando, Florida: ACM Press; 2012. [[Last accessed on 2022 Jun 20]]. A fast computation of pairwise sequence alignment scores between a protein and a set of single-locus variants of another protein; pp. 414–7. Available from: http://dl.acm.org/citation.cfm?doid=2382936.2382989 . [Google Scholar]
- 35.Tanwar M, Dada T, Sihota R, Das TK, Yadav U, Dada R. Mutation spectrum of CYP1B1 in North Indian congenital glaucoma patients. Mol Vis. 2009;15:1200–9. [PMC free article] [PubMed] [Google Scholar]
- 36.Tanwar M, Dada T, Sihota R, Dada R. Identification of four novel cytochrome P4501B1 mutations (p. I94X, p. H279D, p. Q340H, and p. K433K) in primary congenital glaucoma patients. Mol Vis. 2009;15:2926–37. [PMC free article] [PubMed] [Google Scholar]
- 37.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2:Mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 39.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo:A sequence logo generator. Genome Res. 2004;14:1188–90. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garnier J, Osguthorpe DJ, Robson B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J Mol Biol. 1978;120:97–120. doi: 10.1016/0022-2836(78)90297-8. [DOI] [PubMed] [Google Scholar]
- 41.PyMOL | pymol.org. [[Last accessed on 2022 Oct 18]]. Available from: https://pymol.org/2/
- 42.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 43.Amir M, Mohammad T, Kumar V, Alajmi MF, Rehman MT, Hussain A, et al. Structural analysis and conformational dynamics of STN1 gene mutations involved in coat plus syndrome. Front Mol Biosci. 2019;6:41. doi: 10.3389/fmolb.2019.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ying H, Yue BYJT. Cellular and molecular biology of optineurin. Int Rev Cell Mol Biol. 2012;294:223–58. doi: 10.1016/B978-0-12-394305-7.00005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sahlender DA, Roberts RC, Arden SD, Spudich G, Taylor MJ, Luzio JP, et al. Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J Cell Biol. 2005;169:285–95. doi: 10.1083/jcb.200501162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harjes P, Wanker EE. The hunt for huntingtin function:Interaction partners tell many different stories. Trends Biochem Sci. 2003;28:425–33. doi: 10.1016/S0968-0004(03)00168-3. [DOI] [PubMed] [Google Scholar]
- 47.Agarwal R, Gupta SK, Agarwal P, Saxena R, Agrawal SS. Current concepts in the pathophysiology of glaucoma. Indian J Ophthalmol. 2009;57:257–66. doi: 10.4103/0301-4738.53049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bashir R, Tahir H, Yousaf K, Naz S, Naz S. Homozygous p. G61E mutation in a consanguineous Pakistani family with co-existence of juvenile-onset open angle glaucoma and primary congenital glaucoma. Gene. 2015;570:295–8. doi: 10.1016/j.gene.2015.07.014. [DOI] [PubMed] [Google Scholar]