

Abstract
Cardiac fibrosis is a pathological condition characterized by excessive accumulation of extracellular matrix components within the myocardium, which can lead to impaired cardiac function and heart failure. Studies have shown that lymphocytes including B and T cells play important roles in the development and progression of cardiac fibrosis after a myocardial infarction. In this review, we focus on the regulation of cardiac fibrosis by lymphocyte subsets, with a particular emphasis on CD4+ and CD8+ T cells and their effects on fibroblasts and cardiac remodeling. We also highlight areas for further exploration of the interactions between T cells and fibroblasts necessary for understanding and treating cardiac fibrosis and heart failure.
Keywords: B cells, fibroblasts, fibrosis, myocardial infarction, T cells
INTRODUCTION
Because of ischemic injury, the loss of healthy myocytes postmyocardial infarction (MI) initiates left ventricular remodeling resulting in dilation of the left ventricle (LV), increased loading volume, and an increase in the mechanical stress applied to the myocardium. Fibroblasts are primary mesenchymal cells that maintain the cellular structural frameworks by producing extracellular matrix (ECM) proteins. In myocardial tissue, ECM production is tightly regulated based on the deposition of new collagen and the degradation of old collagen (1). Fibroblast function is dependent on both mechanical and chemical stimuli, which change in the post-MI environment (2). Activated fibroblasts can remodel and support the infarcted myocardium by contributing to physiological and pathophysiological signaling processes, including initiating the production of the collagenous scar.
Myocardial fibrosis is categorized into two distinct types: reactive and replacement fibrosis. Reactive fibrosis occurs because of alterations in collagen turnover and usually progresses without the loss of cardiomyocytes (3). Replacement fibrosis occurs after the loss of cardiomyocytes whereby the injury site undergoes repair resulting in a fibrotic scar. If left unchecked, the overproduction of collagen can lead to pathological fibrosis and impaired cardiac function. In contrast, excessive degradation of the ECM after MI can result in possible rupture of the left ventricle or other areas of the vasculature (4). This balance in regulating ECM turnover is key when discussing cardiac fibrosis post-MI.
One facet of the cellular response that facilitates remodeling and fibroblast production of the ECM is the adaptive immune system. The adaptive immune system consists of cells termed lymphocytes, which are longer lived compared with innate cells due to their role in immunological memory (5). Lymphocytes such as T and B cells infiltrate the injury site and enhance the inflammatory stimuli within the post-MI microenvironment, facilitating in the fibroblast phenotypic switch and scar formation (6–8). The focus of this review is to examine the role of T and B cells on fibroblast activation and ECM scar formation over the divergent phases of post-MI healing (Fig. 1).
Figure 1.

Lymphocytes are a diverse set of immune cells within adaptive immunity. Lymphocytes are characterized by specific receptor markers (illustrated in cellular representative images) needed for activation and can regulate cardiac fibrosis that occurs through varying mechanisms. Although some subsets have demonstrated direct actions on cardiac fibroblast through secretion of growth factors, other lymphocytes have demonstrated more of an indirect role by regulating matrix metalloproteases and innate immune cells including macrophages. In turn, fibroblasts can modulate lymphocyte recruitment and phenotypes. ECM, extracellular matrix; MMP, matrix metalloproteinase; TGF-β, tissue growth factor-β.
Fibroblast Regulation of Lymphocytes
Adverse post-MI remodeling is usually characterized by inflammatory cell accumulation in the extravascular connective tissue (9, 10). As discussed earlier after infiltration into the injured zone, lymphocytes can interact with fibroblasts facilitating the generation or breakdown of the infarct scar. This cellular crosstalk is not one-sided as research has shown fibroblasts can facilitate in activation of the adaptive immune system (8, 11, 12). During the first initial days of post-MI healing (i.e., inflammatory phase), fibroblasts are prompted by the massive cardiomyocyte death in the infarcted area to secrete proinflammatory cytokines including interleukin (IL)-1β and IL-6, which facilitate the recruitment of immune cells to the injury site (8). Cardiac fibroblasts in the injured and noninjured zone are also responsible for releasing inflammatory proteins that degrade cardiac ECM allowing T-cell migration into the infarcted region (1). This results in an initial increase in ECM degradation and a decrease in ECM synthesis in the infarcted area (1).
In the proliferative phase, fibroblasts begin to migrate and proliferate throughout the infarcted region of the tissue, whereas the inflammatory phase begins to resolve. Migrating fibroblasts undergo phenotypic changes and lay down matrix proteins, like collagen to stimulate wound contraction, establishing the fibrotic scar (1). This initial production of ECM facilitates not only cell migration and wound contraction but also microenvironmental changes that stimulate immune cell efferocytosis and lymphatic drainage (6, 13). Tumor-derived fibroblasts induce sprouting, altered vasculature permeability and increased the expression of prolymphangiogenic genes (14). Similarly, in a rat permanent occlusion model of MI, fibroblasts expressing VCAM efficiently mobilized the lymphatic endothelial cells into the infarcted area thus promoting lymphangiogenesis and improving cardiac function (12).
Ngwenyama et al. (11) demonstrated that cardiac fibroblasts can regulate T-cell activation through contact-dependent mechanisms by expressing major histocompatibility complex (MHC)-II in a mouse model of pressure overload. If activation of circulating or resident lymphocytes becomes self-perpetuating, this can lead to chronic tissue destruction (15). Conditional deletion of MHC-II in cardiac fibroblasts was found to attenuate pressure-overload cardiac remodeling and dysfunction further illustrating the importance of fibroblast-mediated lymphocyte activation (11). Whether fibroblast-mediated antigen presentation is also key in post-MI remodeling has not been evaluated. These studies point to a more direct role of fibroblasts in the regulation of lymphocytes post-MI. Regulation of activation and phenotype between lymphocytes and fibroblast adds to the balance in ECM degradation and deposition, thereby playing a critical role in cardiac remodeling post-MI (16).
T Cells
T-cell infiltration into the myocardium post-MI is both necessary and sufficient to stimulate fibrosis and LV dysfunction indicating a functional role for T cells in adverse cardiac remodeling (17). Studies evaluating the role of T cells during development of ischemic heart failure have demonstrated the complexity of cross talk between T cells and resident fibroblasts (Fig. 1). There is a growing consensus that T cells are involved in the induction of cardiac fibrosis through both direct adhesion and actions on cardiac fibroblasts in addition to secretion of a variety of inflammatory molecules including tumor necrosis factor (TNF)α and IL-1β (17). Secretion of these factors likely has an indirect role in the regulation of fibroblast ECM deposition and proliferation through polarization of macrophages as well as fibroblasts (18, 19).
T cells originate in the bone marrow as hematopoietic stem cells before circulating to the thymus where they engraft and develop into naive T cells (20). Although all T cells are characterized by the CD3 receptor, in the thymus, T cells undergo differentiation to become either CD4+ or CD8+ T cells (21). T-cell activation is a multistep process that initiates in the lymph nodes. Because of ischemic injury, the myocardium releases damage-associated molecular patterns (DAMPs), which serve as antigens that initiate the immune response. Antigen-presenting cells (e.g., dendritic cells) internalize these DAMPs and upregulate major histocompatibility complexes (MHCs) class I and II to display the antigen-derived peptides for T-cell interaction and activation (22). Surface expression of costimulatory signals (e.g., CD28, CD80/86) and production of cytokines [e.g., interferon (IFN)-γ] are also necessary for effective T-cell activation and polarization (22–24).
On stimulation, T cells can differentiate into CD4+ helper T cells (Th1, Th2, and Th17), CD4+ Tregs, and CD8+ (cytotoxic) T cells (Fig. 2). These different T-cell subsets have diverse expression patterns facilitating immune response and driving the various roles of T cells in post-MI remodeling. Th1 cells are defined by the expression of lineage cytokine IFN-γ and the master transcription factor transforming growth factor (TGF)-β, whereas Th2 cells are defined by IL-4, IL-5, IL-13, and transcription factor GAΤA3 (26–28). In contrast, Th17 cells are defined by the lineage cytokine IL17 and IL23 along with the master transcription factor RORγt (29). The stimulus necessary for T-cell polarization is also variable between the T-cell subtypes. Th1 cells become activated with IL-12 or IFN-γ stimulation, whereas Th2 cells are activated via IL-4 and/or IL-23 stimulation in addition to antigen presentation (26, 27). Th17 cells demonstrate increased plasticity, transitioning to a more IFN-γ-secreting Th1-like cell when in the absence of TGF-β (30, 31). Without TGF-β, both IL-23 and IL-12 suppress IL-17 production and enhance IFN-γ production in a STAT4- and Tβ-dependent manner (31). Because of the complex nature of T-cell subsets and context-dependent activity, generating a concise summary of T-cell function in the heart continues to be a difficult task. Below we will summarize the findings in the literature on T cells and their roles post-MI.
Figure 2.
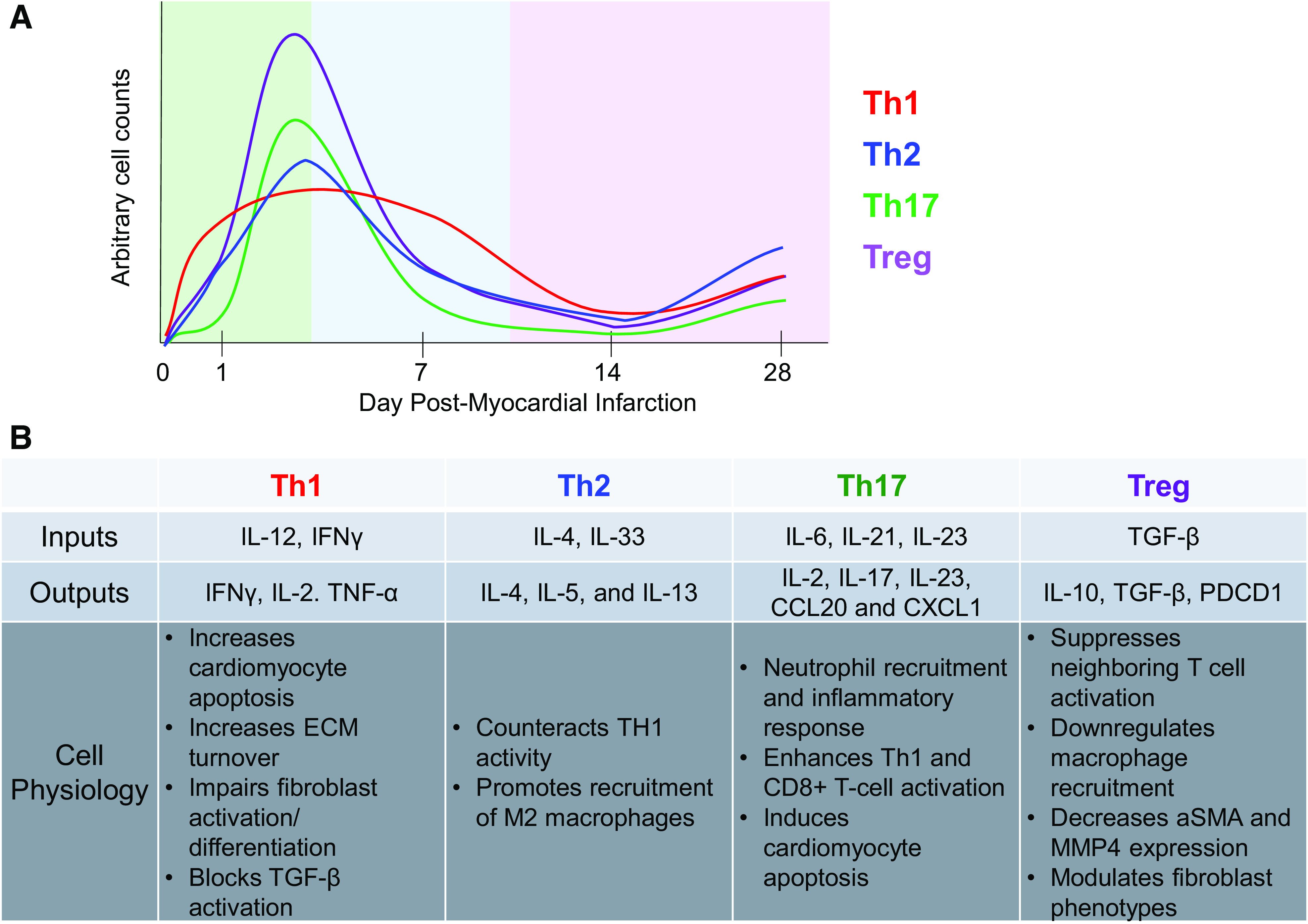
A: temporal phenotypic changes in CD4+ T cells during the inflammation (green), proliferation (blue), and maturation (purple) phases of cardiac remodeling post-MI. Arbitrary cell counts mapped are based on findings presented in Kumar et al. (25). B: summary of the roles of CD4+ T cells postmyocardial infarction in the permanent occlusion model. α-SMA, α-smooth muscle actin; ECM, extracellular matrix; IFN, interferon; IL, interleukin; MMP, matrix metalloproteinase; PDCD, programmed cell death protein; TNFα, tumor necrosis factor α; TGF-β, tissue growth factor-β.
CD4+ T Cells
Although there are resident CD4+ T cells in the heart, there is a noticeable infiltration seen within the first week after ischemia-reperfusion (I/R) and permanent occlusion (25, 32). Kumar et al. (25) demonstrated that cardiac CD4+ T cells exhibit two distinct patterns of transmigration. During the acute phase of healing, there is a rapid CD4+ T-cell response peaking at post-MI day 3 and returning to baseline by 14 days. During the chronic phase of cardiac healing, CD4+ T cells undergo a second phase of activation with a 20-fold increase in the number of cells in the LV compared with sham-operated mice. These biphasic kinetics were observed in all major CD4+ T-cell subsets including Th1, Th2, Th17, and regulatory T cells, suggesting a global change (25).
CD4+ T cells and, importantly their subsets, have a complex mechanism that coordinates necrotic tissue and debris clearance while simultaneously regulating scar formation and immune resolution post injury (23). In multiple studies conducted, there has been evidence that in mice lacking CD4+ T cells (CD4−/−), there was insufficient cardiac healing that ultimately affected cardiac function (23, 33). In contrast, depletion of CD4+ T cells using a Cre-inducible knockout during the chronic phase of healing (weeks 4–8 post-MI using permanent occlusion) demonstrated that CD4+ T cells promote adverse remodeling of the heart through excessive fibrosis and progressive cardiac dysfunction (25). These studies suggest that CD4+ T-cell phenotypes are different during the distinct phases of healing. Temporal immunomodulation of CD4+ T cells may be a viable translatable modality for targeting ischemic heart failure.
Th1.
Th1 cells are associated with an increase in cardiomyocyte apoptosis, dysregulation of ECM turnover, and impaired myofibroblast differentiation leading to cardiac rupture. Th1 cells have been connected to an imbalance in ECM turnover and in fibroblast activation (34). Th1 cells produce cytokines such as IFN-γ, TNF-α, and IL-2, which activate macrophages and cytotoxic (CD8+) T cells. Th1 cells also block activation of fibroblasts in part through IFN-γ inhibition of TGF-β ultimately affecting the production of collagen in a profibrotic environment (35, 36).
Yang et al. (37) examined RAG1-knockout mice, which are deficient in both B and T cells, and showed a significant decrease in infarct size, by over 30%, compared with control. After adoptive transfer of CD4+ T cells, the infarct sizes increased significantly and nearly returned to levels comparable with control mice (37). Interestingly, this effect was not achieved when CD4+ T cells from IFN-γ-deleted mice were used indicating IFN-γ signaling was partially responsible for effects on the infarct. In a separate study, Yan et al. (34) established that IFN-γ reversed the TGF-β induction of α-SMA expression and collagen I and II in isolated cardiac fibroblasts, further demonstrating the role of Th1 cells in regulating cardiac fibrosis through IFN-γ signaling.
IL-2 is also secreted mainly by conventional Th1 cells and has been shown to stimulate fibroblast activation (38). Fibroblasts express receptors for IL-2, specifically the high-affinity receptor IL-2Rα/β/γ complex (38). When IL-2 binds to these receptors on fibroblasts, it can activate several signaling pathways, including the JAK-STAT, MAPK, and PI3K pathways (39). These signaling pathways induce the production of various cytokines, such as IL-6, IL-8, and TGF-β, which promote fibroblast activation and proliferation (40). Interestingly, using the autophagic inhibitor 3MA, Kang et al. (41) demonstrated that IL-2 stimulation initiated apoptosis in lung fibroblast, suggesting that autophagy promotes cell survival in response to IL-2 and is required for IL-2-induced cell proliferation. After an ischemic event, autophagy is activated to protect cardiomyocytes against injury and suppress the inflammatory response (42, 43). Whether myocardial autophagy after an MI modulates cardiac fibroblasts’ response to Th1 production of IL-2, thus facilitating scar formation and cardiac healing, is not clear. Nonetheless, Th1 cells in the post-MI heart are one of the key immune populations to regulate fibroblast activation and cardiac fibrosis through both direct and indirect mechanisms.
Th2.
Although there have been multiple studies evaluating the role of Th1 cells on post-MI healing, less is known about the anti-inflammatory Th2 cells. In mouse permanent occlusion model of MI, markers of Th2 cells are not observed in great numbers during the acute phase of healing (first 7 days); however, by 8 wk post-MI, Th2 cells become the predominant T-cell phenotype in the LV (32, 44). Th2 cells counteract Th1 actions by secreting immune modulators including IL-4, IL-10, and IL-13. IL-4 stimulates GATA-3 expression in a STAT-6-dependent mechanism, further increasing IL-4 and inhibiting the production of IFN-γ (45). IL-4 and IL-13 have also been shown to promote recruitment of monocyte-derived M2 macrophages facilitating cardiac fibroblast phenotypic changes through a more indirect mechanism (46, 47). Based on these studies, Th2 cells are both anti-inflammatory and profibrotic; nevertheless, how to stimulate Th2-cell upregulation in a controlled manner to improve post-MI healing requires further evaluation.
Th17.
Th17 cells increase early peaking at around day 3 post-MI in the permanent occlusion MI model and become one of the predominant T-cell phenotypes around 8 wk post-MI implying they may have a major role in the chronic cardiac remodeling process (25, 44). Nevertheless, the role of Th17 cells post-MI is an understudied area. Many of the cytokines secreted by Th17 cells such as Cxcl1, IL-2, and Ccl20 modulate the immune system in models of multiple sclerosis, cancer, and hypertension (48–51). For example, Cxcl1 is a known regulator of neutrophil recruitment, whereas IL-2 and Ccl20 have been shown to enhance Th1 and CD8+ T-cell function (49–51). Deletion of IL-17, a primary Th17 marker, led to reduced cardiomyocyte apoptosis and neutrophil infiltration, which ultimately improved survival and attenuated LV dilation at post-MI day 28 using the I/R model (52). This study, however, did not directly evaluate Th17 cells as there are other cellular sources of IL-17 in the post-MI setting (7, 52).
Tregs.
Tregs increase gradually over the first week in the permanent occlusion model of MI and are considered beneficial for cardiac healing by maintaining immune homeostasis and suppressing excessive or aberrant immune responses. By combining transgenic T-cell receptor models, genetic barcoding of endogenous heart-specific T cells, and single-cell transcriptomics, Delgobo et al. (53) found that after permanent occlusion of the coronary artery the myocardium is enriched for Tregs exhibiting a genetic signature including Izumo1r, Tgfb1, and Pdcd1 expression. These Tregs were found to target a myocyte-specific antigen (i.e., myosin heavy α-chain-derived peptide) and acquired FOXP3 expression in the infarcted myocardium (53). The myosin-specific Tregs protected the myocardium by dampening post-MI inflammation and suppressing neighboring T-cell activation (53). Similarly in a model of dermal injury, Tregs limited the antifibrotic role of Th17 cells by suppressing production of mediators for fibroblast migration, IL-22 and CXCL8 (54). Depletion of Tregs has also been shown to effect the innate immune response leading to increased proinflammatory M1 macrophages and impaired M2-like differentiation (24). Saxena et al. (55) demonstrated Treg depletion accelerated ventricular dilation and accentuated apical remodeling indirectly because of an increase in Ccl2 and macrophage recruitment.
In addition to regulation of the immune system, coculture of Tregs with cardiac fibroblasts has been shown to decrease αSMA and matrix metalloproteinase (MMP)-3 expression and attenuate fibroblast-mediated contraction of collagen pads suggesting a direct antifibrotic role (55). Furthermore, expansion of Tregs by CD28 antibodies significantly decreased MMP-mediated degradation of collagen leading to improved survival and fewer cardiac ruptures during the first 7 days after permanent occlusion (24). In a model of hypertensive hearts, adoptive transfer of Tregs attenuated cardiac fibrosis through a reduction of the TGF-β1 system (56). In contrast, some studies have indicated that Tregs release the profibrotic molecule TGF-β, which would be critical for conversion of fibroblasts to an activated, collagen-producing phenotype (40, 55). Bansal et al. (57) revealed Tregs can begin to lose the immunomodulator function becoming an inflammatory and antiangiogenic phenotype. These dysfunctional Tregs play an essential pathogenetic role in chronic ischemic heart failure by promoting immune activation and pathological LV remodeling. This study demonstrates that the restoration of normal Treg function may be a viable approach to therapeutic immunomodulation as this may facilitate restoration of tissue architecture and function without causing excessive scarring or fibrosis. Despite our improved progress in beginning to understand the dual role for Tregs in regulation of fibrosis and fibroblast activation, additional studies are needed before we can consider Tregs as a feasible cellular therapy. Future studies must focus on understanding the exact mechanisms and factors within the microenvironment that stimulate Tregs toward either antifibrotic or profibrotic phenotype.
CD8+ T Cells
CD8+ T cells were not considered to play a significant role in the wound healing response post-MI. The best-known role of CD8+ T cells is to mediate viral clearance by using a variety of cytotoxic mechanisms (58, 59). Similar to CD4+ T cells, CD8+ T cells also exhibit a biphasic temporal response after an MI. CD8+ T cells are observed in the myocardium of the permanent occlusion MI model as early as 24 h within the heart and peak at day 7 (32, 60). At post-MI day 14, the second wave of infiltration occurs with CD8+ T cells remaining in the infarct as long as 8 wk post-MI (44).
Over the past decade, there has been an increased interest in understanding the role of CD8+ T cells post-MI with the majority focusing on cellular cytotoxicity. Excessive or prolonged activation of CD8+ T cells contributes to tissue damage due to high levels of cytotoxic molecules such as granzymes and perforin, which induce cardiac apoptosis and likely contribute to infarct expansion (60). Plasma levels of Granzyme B, a CD8+ T cell-secreted protein, correlated with increased mortality in patients with MI, suggesting a negative role for CD8+ T cells in mediating post-MI remodeling (60). In support of this finding, Ilatovskaya et al. (61) demonstrated that genetic depletion of CD8+ T cells resulted in an accelerated healing response after induction of MI using the permanent occlusion model ultimately leading to an attenuation of LV dilation and improved cardiac function. Similarly, Santos-Zas et al. (60) demonstrated that CD8+ T cells amplified myocardial injury and exaggerated cardiac dysfunction through Granzyme B-mediated mechanisms.
Despite the majority of evidence suggesting a negative role for CD8+ T cells, there is speculation on whether CD8+ T cells strictly attenuate fibrotic deposition, or if there are subsets that promote cardiac fibrosis as well. This idea that there is a cardioprotective CD8+ T-cell subset is supported by clinical data, whereby a drop in circulating CD8+ T cells within an hour following percutaneous coronary intervention was associated with poor prognosis (62). This is in contrast to previously mentioned clinical studies where CD8+ T-cell markers were measured at the time of admission (60) suggesting effects from CD8+ T cells are likely temporally dependent. In a rat model of permanent occlusion, Curato et al. (63) identified a cardioprotective CD8+ T-cell population that expressed the angiotensin receptor AT2R. Transplantation of this CD8+ T-cell subtype resulted in a decrease in cardiac injury.
CD8+ T cells also help to balance the inflammatory microenvironment by responding to the injury sites and facilitating in the influx of other immune cells including macrophages (61). Once recruited to the injury site, CD8+ T cells produce various cytokines, such as IFN-γ and TNF-α. Although CD8+ T cells produce an abundance of IFN-γ, recruited inflammatory cells represent the primary source of IFN-γ in a post-MI setting indirectly affecting fibrosis (64). The idea that CD8+ T cells initiate signaling pathways in post-MI cardiac fibrosis is also seen in postunilateral ureter obstruction renal fibrosis whereby CD8+ T cells secreted IFN-γ and TNF-α to increase proinflammatory macrophages (65). This increase in proinflammatory macrophages further elevates CD8+ T-cell recruitment and cytokine secretion forming a positive feedback mechanism for CD8+ T cells in post-MI cardiac environments. There is limited research in the direct effects of IFN-γ but Hellkvist et al. (64) showed that IFN-γ induced proliferation of rat cardiac fibroblasts during posttransplantation rejection through increased hyaluronan production. As we learn more about CD8+ T cells, evidence demonstrates that CD8+ T-cell populations are heterogeneous in terms of phenotype, function, and distribution.
B Cells
Although the literature is limited, it is believed that B cells have a small role in post-MI remodeling and fibrotic scar deposition. Mo et al. (66) discovered that B-cell levels in the myocardium, spleen, and blood were highest at day 5 after permanent occlusion of the coronary artery compared with sham mice. Similarly in the I/R model, B cells were found to peak in the heart at post-MI day 3 before returning to physiological levels by day 5 (32). Like T cells, B cells produce inflammatory mediators, which facilitate in post-MI healing; however, unlike T cells, B cells require additional stimuli and further differentiation to begin cytokine production (67). B cells in a state of homeostasis function as a surveillance system that only becomes activated and expands with infection or injury. After the infection or injury is resolved, B cells return to their inactivated state (68).
Mice lacking B cells were found to have decreased Masson Trichrome staining at 14 days post-MI indicating decreased fibrosis after permanent occlusion (66). Interestingly, the potent fibroblast activator, TGF-β, was also found to be decreased in the myocardium at post-MI day 5 suggesting decreased signaling for fibroblast activation may be responsible for the reduced fibrosis in the B cell-depleted model. Whether or not this is directly through B-cell regulation, or a more indirect mechanism, was not evaluated.
In contrast, Goodchild et al. (69) showed that B-cell injections into the hearts of Sprague–Dawley rats at time of coronary artery permanent occlusion had better functional outcomes compared with saline controls. Their study demonstrated that B-cell supplementation decreased myocardial apoptosis compared with rats receiving saline injections post-MI (69). These data seem to suggest that B cells would be beneficial potentially by modulating myocyte viability.
Mice lacking B cells were also shown to have reduced MMP-9 expression indicating a decrease in ECM turnover (66). In line with these studies, Storch et al. (70) demonstrated that B cells were able to stimulate secretion of inflammatory proteins including MMP-3 in human synovial fibroblasts after coculture. Although neither of these studies directly investigated the molecular signature responsible for B-cell and fibroblast communication, it can be hypothesized that B cells are able to release cytokines that activate fibroblasts and promote fibrosis through increased ECM turnover and deposition. Further research is needed on the roles B cells have on cardiac fibrosis and potential influences on fibroblast physiology post-MI.
Future Directions
Multiple studies have demonstrated B and T cells regulate cardiac fibrosis through signaling pathways that regulate fibroblast phenotype and activation. The direct binding of T cells to cardiac fibroblasts and the release of cytokines both play major roles in the production and breakdown of cardiac ECM during the remodeling phase post-MI. Despite considerable progress in the research of T cells and cardiac remodeling, the exact mechanisms of T-cell regulation of fibroblasts and effects on the fibrotic environment are not fully understood. Since T cells are heterogeneous and subject to temporal differentiation and changes in activity, it is hard to exclusively categorize the effects of T-cell involvement post-MI. The focus of future research should be on determining mechanistic pathways and trends that affect cardiac healing and scar formation to reveal T cell-specific contributions to wound healing over time.
In the past decade, studies using chimeric antigen receptor (CAR) T cells have shown potential for treatment of pressure-overload-induced heart failure (71, 72). Aghajanian et al. (71) identified fibroblast activation protein (FAP) as an endogenous cardiac fibroblast target. Treating angiotensin II/phenylephrine-exposed mice with engineered CAR T cells against FAP resulted in a significant reduction of cardiac fibrosis and restoration of function (71). As CAR T-cell use for cancer therapy has been shown to have potential off-target effects (73), Rurik et al. (72) engineered transient CAR T cells using a CD5-targeted lipid nanoparticle and demonstrated similar protective effects in the angiotensin/phenylephrine mouse model. Although these studies demonstrate proof of concept for the use of CAR T cells in a model of nonischemic heart failure, there is some concern regarding how these engineered cells would affect the post-MI remodeling process in which fibrosis is necessary for replacing the necrotic, injured tissue. The indefinite permanence of engineered T cells may be detrimental for post-MI remodeling, especially during the initial stages of healing. Future studies using an off switch would need to be performed if the use of CAR T cells is to be considered for post-MI treatment.
Biological risk factors including sex and age also contribute to the regulation of cardiac fibrosis and inflammatory responses. A study examining sex differences in regulatory T cells found that middle-aged female mice exhibited increased levels of proinflammatory been shown to have T cells and decreased levels of anti-inflammatory Tregs in adipose tissue compared with age-matched males likely contributing to increased cardiovascular disease burden in aging women (74). Estrogen directly benefits MI remodeling by increasing endothelial nitric oxide synthase (eNOS) activity and decreasing leukocyte accumulation after I/R injury (75, 76). Estrogen receptor (ER)β has also been shown to be protective through TGF-β stimulation of cardiac fibroblasts resulting in elevated ECM production and a more stable scar after MI using the permanent occlusion model (77). Similarly, using a novel ERβ agonist post-MI, Rosenzweig et al. (78) demonstrated estrogen receptor signaling blunted CD4+ T-cell numbers in the permanent occlusion model leading to improvements in left ventricular remodeling in both sexes.
However, not all studies have shown estrogen to be protective. At physiological levels, estrogen had no significant effect on mortality but improved cardiac physiology, reduced fibrosis, and increased capillary density (79). Doses that raised plasma estrogen far beyond the physiological level exacerbated cardiac fibrosis, hypertrophy, and LV dysfunction and dilatation. This study highlights the complexity of estrogen in the regulation of the immune response and cardiovascular disease.
Similarly, with age there is a significant transition with a loss of naïve T cells and an increase in effector and memory phenotypes (9, 80). The most fundamental determinant of biological sex is the presence of the sex chromosomes (X or Y), which are present in every cell of the body. One possible mechanism of age-associated immune senescence is changes in transcriptional regulation (81, 82). In mammalian female T cells, the inactive X chromosome is predisposed to reactivate, resulting in the overexpression of immune-related genes including CD40LG, Cxcr3, and Foxp3 (83, 84). Ye et al. (85) demonstrated that with increased age, YY1 transcripts, which are critical for maintenance of X chromosome gene expression, are decreased in T cells. Whether this decrease in YY1 with age facilitates a loss of XCI in females is unknown. The relationships between age, sex, and inflammatory response and subsequent fibrosis deserve greater examination.
GRANTS
This work was supported by National Institutes of Health Grants UL1TR001450, R25GM113278, and T32GM132055 and Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development Grant BX003922.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.L. conceived and designed research; M.L., A.C., S.D., and K.Y.D.-P. prepared figures; M.L., A.C., S.D., and K.Y.D.-P. drafted manuscript; M.L., A.C., S.D., and K.Y.D.-P. edited and revised manuscript; M.L., A.C., S.D., and K.Y.D.-P. approved final version of manuscript.
REFERENCES
- 1. Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res 365: 563–581, 2016. doi: 10.1007/s00441-016-2431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Khalil NN, McCain ML. Engineering the cellular microenvironment of Ppst-infarct myocardium on a chip. Front Cardiovasc Med 8: 709871, 2021. doi: 10.3389/fcvm.2021.709871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Díez J, González A, Kovacic JC. Myocardial interstitial fibrosis in nonischemic heart disease, part 3/4: JACC focus seminar. J Am Coll Cardiol 75: 2204–2218, 2020. doi: 10.1016/j.jacc.2020.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frangogiannis NG. The extracellular matrix in myocardial injury, repair, and remodeling. J Clin Invest 127: 1600–1612, 2017. doi: 10.1172/JCI87491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ratajczak W, Niedźwiedzka-Rystwej P, Tokarz-Deptuła B, Deptuła W. Immunological memory cells. Cent Eur J Immunol 43: 194–203, 2018. doi: 10.5114/ceji.2018.77390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ceccato TL, Starbuck RB, Hall JK, Walker CJ, Brown TE, Killgore JP, Anseth KS, Leinwand LA. Defining the cardiac fibroblast secretome in a fibrotic microenvironment. J Am Heart Assoc 9: e017025, 2020. doi: 10.1161/JAHA.120.017025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan X, Shichita T, Katsumata Y, Matsuhashi T, Ito H, Ito K, Anzai A, Endo J, Tamura Y, Kimura K, Fujita J, Shinmura K, Shen W, Yoshimura A, Fukuda K, Sano M. Deleterious effect of the IL-23/IL-17A axis and γδT cells on left ventricular remodeling after myocardial infarction. J Am Heart Assoc 1: e004408, 2012. doi: 10.1161/JAHA.112.004408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol 70: 74–82, 2014. doi: 10.1016/j.yjmcc.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ponnappan S, Ponnappan U. Aging and immune function: molecular mechanisms to interventions. Antioxid Redox Signal 14: 1551–1585, 2011. doi: 10.1089/ars.2010.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zaidi Y, Aguilar EG, Troncoso M, Ilatovskaya DV, DeLeon-Pennell KY. Immune regulation of cardiac fibrosis post myocardial infarction. Cell Signal 77: 109837, 2021. doi: 10.1016/j.cellsig.2020.109837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ngwenyama N, Kaur K, Bugg D, Theall B, Aronovitz M, Berland R, Panagiotidou S, Genco C, Perrin MA, Davis J, Alcaide P. Antigen presentation by cardiac fibroblasts promotes cardiac dysfunction. Nat Cardiovasc Res 1: 761–774, 2022. doi: 10.1038/s44161-022-00116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iwamiya T, Segard BD, Matsuoka Y, Imamura T. Human cardiac fibroblasts expressing VCAM1 improve heart function in postinfarct heart failure rat models by stimulating lymphangiogenesis. PLoS One 15: e0237810, 2020. doi: 10.1371/journal.pone.0237810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brown FD, Turley SJ. Fibroblastic reticular cells: organization and regulation of the T lymphocyte life cycle. J Immunol 194: 1389–1394, 2015. doi: 10.4049/jimmunol.1402520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lugo-Cintrón KM, Ayuso JM, Humayun M, Gong MM, Kerr SC, Ponik SM, Harari PM, Virumbrales-Muñoz M, Beebe DJ. Primary head and neck tumour-derived fibroblasts promote lymphangiogenesis in a lymphatic organotypic co-culture model. EBioMedicine 73: 103634, 2021. doi: 10.1016/j.ebiom.2021.103634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zaidi Y, Corker A, Vasileva VY, Oviedo K, Graham C, Wilson K, Martino J, Troncoso M, Broughton P, Ilatovskaya DV, Lindsey ML, DeLeon-Pennell KY. Chronic Porphyromonas gingivalis lipopolysaccharide induces adverse myocardial infarction wound healing through activation of CD8+ T cells. Am J Physiol Heart Circ Physiol 321: H948–H962, 2021. doi: 10.1152/ajpheart.00082.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Venugopal H, Hanna A, Humeres C, Frangogiannis NG. Properties and functions of fibroblasts and myofibroblasts in myocardial infarction. Cells 11: 1386, 2022. doi: 10.3390/cells11091386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blanton RM, Carrillo-Salinas FJ, Alcaide P. T-cell recruitment to the heart: friendly guests or unwelcome visitors? Am J Physiol Heart Circ Physiol 317: H124–H140, 2019. doi: 10.1152/ajpheart.00028.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barbul A, Breslin RJ, Woodyard JP, Wasserkrug HL, Efron G. The effect of in vivo T helper and T suppressor lymphocyte depletion on wound healing. Ann Surg 209: 479–483, 1989. doi: 10.1097/00000658-198904000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Diaz A, Munoz E, Johnston R, Korn JH, Jimenez SA. Regulation of human lung fibroblast alpha 1(I) procollagen gene expression by tumor necrosis factor alpha, interleukin-1 beta, and prostaglandin E2. J Biol Chem 268: 10364–10371, 1993. [PubMed] [Google Scholar]
- 20. Zlotoff DA, Bhandoola A. Hematopoietic progenitor migration to the adult thymus. Ann NY Acad Sci 1217: 122–138, 2011. doi: 10.1111/j.1749-6632.2010.05881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bradshaw AD, DeLeon-Pennell KY. T-cell regulation of fibroblasts and cardiac fibrosis. Matrix Biol 91-92: 167–175, 2020. doi: 10.1016/j.matbio.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Van der Borght K, Scott CL, Nindl V, Bouché A, Martens L, Sichien D, Van Moorleghem J, Vanheerswynghels M, De Prijck S, Saeys Y, Ludewig B, Gillebert T, Guilliams M, Carmeliet P, Lambrecht BN. Myocardial infarction primes autoreactive T cells through activation of dendritic cells. Cell Rep 18: 3005–3017, 2017. doi: 10.1016/j.celrep.2017.02.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, Kerkau T, Frantz S. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 125: 1652–1663, 2012. doi: 10.1161/CIRCULATIONAHA.111.044164. [DOI] [PubMed] [Google Scholar]
- 24. Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, Ertl G, Kerkau T, Frantz S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res 115: 55–67, 2014. doi: 10.1161/CIRCRESAHA.115.303895. [DOI] [PubMed] [Google Scholar]
- 25. Kumar V, Prabhu SD, Bansal SS. CD4+ T-lymphocytes exhibit biphasic kinetics post-myocardial infarction. Front Cardiovasc Med 9: 992653, 2022. doi: 10.3389/fcvm.2022.992653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol 10: 225–235, 2010. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, Guo L, Yagi R, Yamane H, Punkosdy G, Feigenbaum L, Zhao K, Paul WE. The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity 37: 660–673, 2012. doi: 10.1016/j.immuni.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q, Killeen N, Urban JF Jr, Guo L, Paul WE. Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat Immunol 5: 1157–1165, 2004. doi: 10.1038/ni1128. [DOI] [PubMed] [Google Scholar]
- 29. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30: 576–587, 2009. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci USA 112: 7061–7066, 2015. doi: 10.1073/pnas.1415675112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT. Late developmental plasticity in the T helper 17 lineage. Immunity 30: 92–107, 2009. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K, Sano M. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol 62: 24–35, 2013. doi: 10.1016/j.yjmcc.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 33. Hardeman SW, Soloway MS. Urethral recurrence following radical cystectomy. J Urol 144: 666–669, 1990. doi: 10.1016/s0022-5347(17)39549-6. [DOI] [PubMed] [Google Scholar]
- 34. Yan X, Zhang H, Fan Q, Hu J, Tao R, Chen Q, Iwakura Y, Shen W, Lu L, Zhang Q, Zhang R. Dectin-2 deficiency modulates Th1 differentiation and improves wound healing after myocardial infarction. Circ Res 120: 1116–1129, 2017. doi: 10.1161/CIRCRESAHA.116.310260. [DOI] [PubMed] [Google Scholar]
- 35. Ulloa L, Doody J, Massagué J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature 397: 710–713, 1999. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]
- 36. Lee JW, Oh JE, Rhee KJ, Yoo BS, Eom YW, Park SW, Lee JH, Son JW, Youn YJ, Ahn MS, Ahn SG, Kim JY, Lee SH, Yoon J. Co-treatment with interferon-γ and 1-methyl tryptophan ameliorates cardiac fibrosis through cardiac myofibroblasts apoptosis. Mol Cell Biochem 458: 197–205, 2019. doi: 10.1007/s11010-019-03542-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA, French BA, Linden J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation 114: 2056–2064, 2006. doi: 10.1161/CIRCULATIONAHA.106.649244. [DOI] [PubMed] [Google Scholar]
- 38. Kino T, Khan M, Mohsin S. The regulatory role of T cell responses in cardiac remodeling following myocardial infarction. Int J Mol Sci 21: 5013, 2020. doi: 10.3390/ijms21145013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou X, Xing J, Tang X, Sheng X, Chi H, Zhan W. Interleukin-2 (IL-2) interacts with IL-2 receptor beta (IL-2Rβ): its potential to enhance the proliferation of CD4+ T lymphocytes in flounder (Paralichthys olivaceus). Front Immunol 11: 531785, 2020. doi: 10.3389/fimmu.2020.531785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frangogiannis N. Transforming growth factor-β in tissue fibrosis. J Exp Med 217: e20190103, 2020. doi: 10.1084/jem.20190103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kang R, Tang D, Lotze MT, Zeh Iii HJ. Autophagy is required for IL-2-mediated fibroblast growth. Exp Cell Res 319: 556–565, 2013. doi: 10.1016/j.yexcr.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sciarretta S, Yee D, Shenoy V, Nagarajan N, Sadoshima J. The importance of autophagy in cardioprotection. High Blood Press Cardiovasc Prev 21: 21–28, 2014. doi: 10.1007/s40292-013-0029-9. [DOI] [PubMed] [Google Scholar]
- 43. Wang L, Li Y, Ning N, Wang J, Yan Z, Zhang S, Jiao X, Wang X, Liu H. Decreased autophagy induced by β1-adrenoceptor autoantibodies contributes to cardiomyocyte apoptosis. Cell Death Dis 9: 406, 2018. doi: 10.1038/s41419-018-0445-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, Prabhu SD. Activated T lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circ Heart Fail 10: e003688, 2017. doi: 10.1161/CIRCHEARTFAILURE.116.003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maier E, Duschl A, Horejs-Hoeck J. STAT6-dependent and -independent mechanisms in Th2 polarization. Eur J Immunol 42: 2827–2833, 2012. doi: 10.1002/eji.201242433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shintani Y, Ito T, Fields L, Shiraishi M, Ichihara Y, Sato N, Podaru M, Kainuma S, Tanaka H, Suzuki K. IL-4 as a repurposed biological drug for myocardial infarction through augmentation of reparative cardiac macrophages: proof-of-concept data in mice. Sci Rep 7: 6877, 2017. doi: 10.1038/s41598-017-07328-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hofmann U, Knorr S, Vogel B, Weirather J, Frey A, Ertl G, Frantz S. Interleukin-13 deficiency aggravates healing and remodeling in male mice after experimental myocardial infarction. Circ Heart Fail 7: 822–830, 2014. doi: 10.1161/CIRCHEARTFAILURE.113.001020. [DOI] [PubMed] [Google Scholar]
- 48. Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, Delpire E, Harrison DG, McDonough AA. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-γ-/- and interleukin-17A-/- mice. Hypertension 65: 569–576, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 31: 787–798, 2009. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ankathatti Munegowda M, Deng Y, Mulligan SJ, Xiang J. Th17 and Th17-stimulated CD8+ T cells play a distinct role in Th17-induced preventive and therapeutic antitumor immunity. Cancer Immunol Immunother 60: 1473–1484, 2011. doi: 10.1007/s00262-011-1054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wojkowska DW, Szpakowski P, Glabinski A. Interleukin 17A promotes lymphocytes adhesion and induces CCL2 and CXCL1 release from brain endothelial cells. Int J Mol Sci 18: 1000, 2017. doi: 10.3390/ijms18051000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liao YH, Xia N, Zhou SF, Tang TT, Yan XX, Lv BJ, Nie SF, Wang J, Iwakura Y, Xiao H, Yuan J, Jevallee H, Wei F, Shi GP, Cheng X. Interleukin-17A contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and neutrophil infiltration. J Am Coll Cardiol 59: 420–429, 2012. doi: 10.1016/j.jacc.2011.10.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Delgobo M, Weiß E, Ashour D, Richter L, Popiolkowski L, Arampatzi P, Stangl V, Arias-Loza P, Mariotti-Ferrandiz E, Rainer PP, Saliba AE, Ludewig B, Hofmann U, Frantz S, Campos Ramos G. Myocardial milieu favors local differentiation of regulatory T cells. Circ Res 132: 565–582, 2023. doi: 10.1161/CIRCRESAHA.122.322183. [DOI] [PubMed] [Google Scholar]
- 54. Crome SQ, Clive B, Wang AY, Kang CY, Chow V, Yu J, Lai A, Ghahary A, Broady R, Levings MK. Inflammatory effects of ex vivo human Th17 cells are suppressed by regulatory T cells. J Immunol 185: 3199–3208, 2010. doi: 10.4049/jimmunol.1000557. [DOI] [PubMed] [Google Scholar]
- 55. Saxena A, Dobaczewski M, Rai V, Haque Z, Chen W, Li N, Frangogiannis NG. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am J Physiol Heart Circ Physiol 307: H1233–H1242, 2014. doi: 10.1152/ajpheart.00328.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kanellakis P, Dinh TN, Agrotis A, Bobik A. CD4+CD25+Foxp3+ regulatory T cells suppress cardiac fibrosis in the hypertensive heart. J Hypertens 29: 1820–1828, 2011. doi: 10.1097/HJH.0b013e328349c62d. [DOI] [PubMed] [Google Scholar]
- 57. Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, Prabhu SD. Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation 139: 206–221, 2019. doi: 10.1161/CIRCULATIONAHA.118.036065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, Chisari FV. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol 77: 68–76, 2003. doi: 10.1128/jvi.77.1.68-76.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schmidt ME, Varga SM. The CD8 T cell response to respiratory virus infections. Front Immunol 9: 678, 2018. doi: 10.3389/fimmu.2018.00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Santos-Zas I, Lemarie J, Zlatanova I, Cachanado M, Seghezzi JC, Benamer H, Goube P, Vandestienne M, Cohen R, Ezzo M, Duval V, Zhang Y, Su JB, Bizé A, Sambin L, Bonnin P, Branchereau M, Heymes C, Tanchot C, Vilar J, Delacroix C, Hulot JS, Cochain C, Bruneval P, Danchin N, Tedgui A, Mallat Z, Simon T, Ghaleh B, Silvestre JS, Ait-Oufella H. Cytotoxic CD8+ T cells promote granzyme B-dependent adverse post-ischemic cardiac remodeling. Nat Commun 12: 1483, 2021. doi: 10.1038/s41467-021-21737-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ilatovskaya DV, Pitts C, Clayton J, Domondon M, Troncoso M, Pippin S, DeLeon-Pennell KY. CD8+ T-cells negatively regulate inflammation post-myocardial infarction. Am J Physiol Heart Circ Physiol 317: H581–H596, 2019. doi: 10.1152/ajpheart.00112.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Boag SE, Das R, Shmeleva EV, Bagnall A, Egred M, Howard N, Bennaceur K, Zaman A, Keavney B, Spyridopoulos I. T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients. J Clin Invest 125: 3063–3076, 2015. doi: 10.1172/JCI80055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Curato C, Slavic S, Dong J, Skorska A, Altarche-Xifró W, Miteva K, Kaschina E, Thiel A, Imboden H, Wang J, Steckelings U, Steinhoff G, Unger T, Li J. Identification of noncytotoxic and IL-10-producing CD8+AT2R+ T cell population in response to ischemic heart injury. J Immunol 185: 6286–6293, 2010. doi: 10.4049/jimmunol.0903681. [DOI] [PubMed] [Google Scholar]
- 64. Hellkvist J, Tufveson G, Gerdin B, Johnsson C. Characterization of fibroblasts from rejecting tissue: the hyaluronan production is increased. Transplantation 74: 1672–1677, 2002. doi: 10.1097/00007890-200212270-00004. [DOI] [PubMed] [Google Scholar]
- 65. Gao M, Wang J, Zang J, An Y, Dong Y. The mechanism of CD8+ T cells for reducing myofibroblasts accumulation during renal fibrosis. Biomolecules 11: 990, 2021. doi: 10.3390/biom11070990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mo F, Luo Y, Yan Y, Li J, Lai S, Wu W. Are activated B cells involved in the process of myocardial fibrosis after acute myocardial infarction? An in vivo experiment. BMC Cardiovasc Disord 21: 5, 2021. doi: 10.1186/s12872-020-01775-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vazquez MI, Catalan-Dibene J, Zlotnik A. B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine 74: 318–326, 2015. doi: 10.1016/j.cyto.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Garcillán B, Figgett WA, Infantino S, Lim EX, Mackay F. Molecular control of B-cell homeostasis in health and malignancy. Immunol Cell Biol 96: 453–462, 2018. doi: 10.1111/imcb.12030. [DOI] [PubMed] [Google Scholar]
- 69. Goodchild TT, Robinson KA, Pang W, Tondato F, Cui J, Arrington J, Godwin L, Ungs M, Carlesso N, Weich N, Poznansky MC, Chronos NA. Bone marrow-derived B cells preserve ventricular function after acute myocardial infarction. JACC Cardiovasc Interv 2: 1005–1016, 2009. doi: 10.1016/j.jcin.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 70. Störch H, Zimmermann B, Resch B, Tykocinski LO, Moradi B, Horn P, Kaya Z, Blank N, Rehart S, Thomsen M, Lorenz HM, Neumann E, Tretter T. Activated human B cells induce inflammatory fibroblasts with cartilage-destructive properties and become functionally suppressed in return. Ann Rheum Dis 75: 924–932, 2016. doi: 10.1136/annrheumdis-2014-206965. [DOI] [PubMed] [Google Scholar]
- 71. Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, Wang T, Bedi K, Morley MP, Linares Saldana RA, Bolar NA, McDaid K, Assenmacher CA, Smith CL, Wirth D, June CH, Margulies KB, Jain R, Puré E, Albelda SM, Epstein JA. Targeting cardiac fibrosis with engineered T cells. Nature 573: 430–433, 2019. doi: 10.1038/s41586-019-1546-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rurik JG, Tombácz I, Yadegari A, Méndez Fernández PO, Shewale SV, Li L, Kimura T, Soliman OY, Papp TE, Tam YK, Mui BL, Albelda SM, Puré E, June CH, Aghajanian H, Weissman D, Parhiz H, Epstein JA. CAR T cells produced in vivo to treat cardiac injury. Science 375: 91–96, 2022. doi: 10.1126/science.abm0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hu JR, Patel A, Huang S, Su YR, Dahlman KB, Tomasek K, Zhang Y, O'Neil RT, O'Neal JF, Turker I, Johnson DB, Salem JE, Moslehi JJ, Oluwole O. High sensitivity troponin T and NT-proBNP in patients receiving chimeric antigen receptor (CAR) T-cell therapy. Clin Hematol Int 3: 96–102, 2021. doi: 10.2991/chi.k.210718.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ahnstedt H, Roy-O'Reilly M, Spychala MS, Mobley AS, Bravo-Alegria J, Chauhan A, Aronowski J, Marrelli SP, McCullough LD. Sex differences in adipose tissue CD8+ T cells and regulatory T cells in middle-aged mice. Front Immunol 9: 659, 2018. doi: 10.3389/fimmu.2018.00659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 407: 538–541, 2000. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yang S, Bae L, Zhang L. Estrogen increases eNOS and NOx release in human coronary artery endothelium. J Cardiovasc Pharmacol 36: 242–247, 2000. doi: 10.1097/00005344-200008000-00015. [DOI] [PubMed] [Google Scholar]
- 77. Zhang JB, Guo CL. Protective effect and mechanism of estrogen receptor β on myocardial infarction in mice. Exp Ther Med 14: 1315–1320, 2017. doi: 10.3892/etm.2017.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rosenzweig R, Kumar V, Gupta S, Bermeo-Blanco O, Stratton MS, Gumina RJ, Bansal SS. Estrogen receptor-β agonists modulate T-lymphocyte activation and ameliorate left ventricular remodeling during chronic heart failure. Circ Heart Fail 15: e008997, 2022. doi: 10.1161/CIRCHEARTFAILURE.121.008997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhan E, Keimig T, Xu J, Peterson E, Ding J, Wang F, Yang XP. Dose-dependent cardiac effect of oestrogen replacement in mice post-myocardial infarction. Exp Physiol 93: 982–993, 2008. doi: 10.1113/expphysiol.2008.042788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Naylor K, Li G, Vallejo AN, Lee WW, Koetz K, Bryl E, Witkowski J, Fulbright J, Weyand CM, Goronzy JJ. The influence of age on T cell generation and TCR diversity. J Immunol 174: 7446–7452, 2005. doi: 10.4049/jimmunol.174.11.7446. [DOI] [PubMed] [Google Scholar]
- 81. Park J, Belden WJ. Long non-coding RNAs have age-dependent diurnal expression that coincides with age-related changes in genome-wide facultative heterochromatin. BMC Genomics 19: 777, 2018. doi: 10.1186/s12864-018-5170-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Casillas MA Jr, Lopatina N, Andrews LG, Tollefsbol TO. Transcriptional control of the DNA methyltransferases is altered in aging and neoplastically-transformed human fibroblasts. Mol Cell Biochem 252: 33–43, 2003. doi: 10.1023/a:1025548623524. [DOI] [PubMed] [Google Scholar]
- 83. Ritzel RM, Crapser J, Patel AR, Verma R, Grenier JM, Chauhan A, Jellison ER, McCullough LD. Age-associated resident memory CD8 T cells in the central nervous system are primed to potentiate inflammation after ischemic brain injury. J Immunol 196: 3318–3330, 2016. doi: 10.4049/jimmunol.1502021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang J, Syrett CM, Kramer MC, Basu A, Atchison ML, Anguera MC. Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X. Proc Natl Acad Sci USA 113: E2029–E2038, 2016. doi: 10.1073/pnas.1520113113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ye Z, Li G, Kim C, Hu B, Jadhav RR, Weyand CM, Goronzy JJ. Regulation of miR-181a expression in T cell aging. Nat Commun 9: 3060, 2018. doi: 10.1038/s41467-018-05552-3. [DOI] [PMC free article] [PubMed] [Google Scholar]