

Abstract
DOCK (dedicator of cytokinesis) is an 11-member family of typical guanine nucleotide exchange factors (GEFs) expressed in the brain, spinal cord, and skeletal muscle. Several DOCK proteins have been implicated in maintaining several myogenic processes such as fusion. We previously identified DOCK3 as being strongly upregulated in Duchenne muscular dystrophy (DMD), specifically in the skeletal muscles of DMD patients and dystrophic mice. Dock3 ubiquitous KO mice on the dystrophin-deficient background exacerbated skeletal muscle and cardiac phenotypes. We generated Dock3 conditional skeletal muscle knockout mice (Dock3 mKO) to characterize the role of DOCK3 protein exclusively in the adult muscle lineage. Dock3 mKO mice presented with significant hyperglycemia and increased fat mass, indicating a metabolic role in the maintenance of skeletal muscle health. Dock3 mKO mice had impaired muscle architecture, reduced locomotor activity, impaired myofiber regeneration, and metabolic dysfunction. We identified a novel DOCK3 interaction with SORBS1 through the C-terminal domain of DOCK3 that may account for its metabolic dysregulation. Together, these findings demonstrate an essential role for DOCK3 in skeletal muscle independent of DOCK3 function in neuronal lineages.
Keywords: DOCK3, skeletal muscle, regeneration, GLUT4 processing
Graphical Abstract
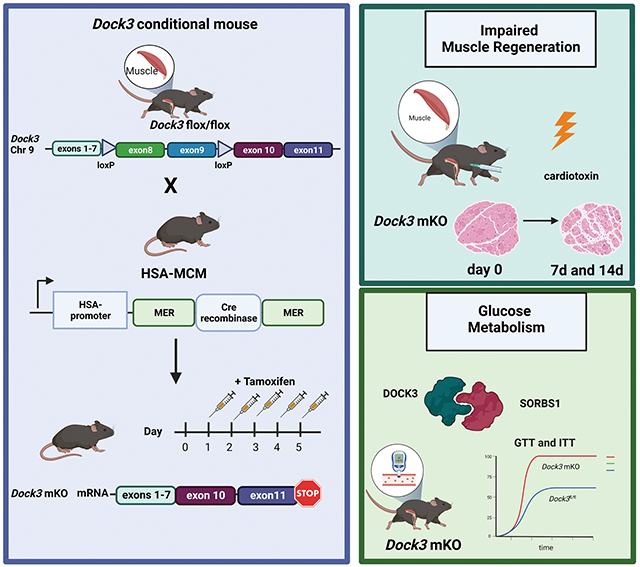
DOCK3 is essential for normal muscle function, muscle regeneration, and regulates metabolism via an interaction with SORBS1. Schematic showing the outline of the conditional Dock3 muscle knockout mouse (Dock3 mKO). Dock3 genetic ablation in the myofiber occurs after Cre-mediated recombination due to the HSA-MerCreMer (HSA-MCM) transgene resulting in the deletion of exons 8 and 9 in the mouse Dock3 flox/flox (Dock3fl/fl) mice. The Dock3 mKO mice have impaired skeletal muscle regeneration as measured via a cardiotoxin-induced TA-muscle injury. The mice also have impaired glucose processing as measured via glucose and insulin tolerance that may be due to the DOCK3 interaction with SORBS1.
Introduction
Skeletal muscle is essential for the body’s locomotive function, maintenance of the skeleton structure, and it retains a trademark capacity for repair and regeneration(1). As an organ, skeletal muscle plays a major role in the processing and utilization of glucose in response to insulin. Through this mechanism, it is responsible for approximately 80% of postprandial glucose uptake from circulation, making it critical to maintaining metabolic homeostasis at the organismal level(2). Many key cell signaling pathways are essential for normal muscle cell regeneration, migration, membrane fusion, repair, and muscle metabolism during growth and development(3). Several Rho GTPases function as molecular switches during cell signaling pathways important to the regulation of the F-actin cytoskeleton(4). Additional downstream Rho signaling effectors, such as RAC1 and CDC42, have been implicated in myogenic processes including myogenic differentiation, fusion, myoblast proliferation, and are known to influence the regenerative capacity within the skeletal muscle(5).
The DOCK gene family is an 11-member class of guanine nucleotide exchange factors capable of influencing multiple pathways involved in cellular fusion, migration, and survival in a myriad of tissue types(6). Many of these DOCK proteins are highly expressed in the brain, spinal cord, and muscle(7). Recent studies have demonstrated that DOCK proteins play essential functional roles in important skeletal muscle processes in health and disease(8). For example, DOCK1 and DOCK5 have been illustrated as crucial players in myoblast fusion(9, 10). Along those same lines, DOCK3 plays a key role in RAC1 activation and WAVE signaling in neurons and skeletal muscle(11-13). Patients with loss-of-function DOCK3 variants present with a variety of developmental disorders such as intellectual disability, developmental delay, ataxia, and muscle hypotonia(14, 15).
Previously, we identified DOCK3 as a dosage-sensitive biomarker of DMD in which disease severity correlated with increased DOCK3 expression in the skeletal muscles of affected patients and dystrophic mdx5cv mice(16). Interestingly, the adult skeletal muscle of the ubiquitous Dock3 KO mice showed a reduction in myofiber diameter and overall structure, reduced muscle mass, and metabolic dysfunction. DOCK3 is expressed in both the central nervous system and in skeletal muscle, thus we sought to understand the role of DOCK3 exclusively in the skeletal muscle by generating a skeletal muscle-specific conditional mouse knockout of Dock3 within the myofiber. We hypothesized that a muscle-specific loss of DOCK3 would disrupt major myogenic processes and protein-protein interactions, subsequently undermining muscle regeneration, metabolism, and overall muscle function. We generated a muscle-specific mouse model (henceforth referred to as Dock3 mKO) to understand the role of DOCK3 in overall muscle health. We evaluated Dock3 mKO mouse models and found mild disruptions in muscle integrity and function using activity tracking, but no evidence of contractile deficits using ex vivo functional assays. We evaluated the role of DOCK3 in muscle repair and showed an impairment in the skeletal muscle’s capacity to repair in the Dock3 mKO mice following a cardiotoxin-induced injury. Finally, we evaluated the impact of DOCK3 on glucose metabolism via its activation of the GLUT4 transporter and identified a novel protein-protein interaction with the insulin adaptor protein Sorbin and SH3 domain containing 1 (SORBS1). We demonstrated that DOCK3 is essential for normal skeletal muscle regeneration and metabolic regulation within the skeletal muscle.
Materials and Methods
Animals
Dock3 conditional knockout mice were generated commercially (Cyagen; Santa Clara, CA). These mice were generated using a CRISPR-Cas9 approach to generate an out-of-frame Dock3 deficient mouse upon Cre-mediated recombination by excision of exons 8 and 9 of the mouse Dock3 transcript (NCBI Reference Sequence: NM_153413). Guide RNAs (gRNAs) targeting the intronic regions flanking mouse Dock3 exons 8 and 9 were used along with a homologous recombination vector were injected into wild type C57BL/6 mouse embryos (Taconic Biosciences; Germantown, NY) to generate Dock3 conditional knockout mice (Dock3fl/fl). The targeting homologous recombination vector contained loxP sites flanking mouse Dock3 exons 8 and 9 and was co-injected with the gRNAs and Cas9 mRNA. F0 founder mice were identified by PCR followed by sequence analysis and were then backcrossed to wild type mice to test germline transmission and F1 animal generation. PCR oligonucleotide primers used to genotype the genomic tail DNA from isolated biopsies from the loxP sites in the Dock3 conditional mice were F: 5’-GAGATGCTGATTTCACTGTCTAGC-3’ and R: 5’-CTCTTATCACTGGCTGAAACTACA-3’. PCR primers for the Cre recombinase transgene used were Forward: 5’-GAACGCACTGATTTCGACCA-3’ Reverse: 5’-GCTAACCAGCGTTTTCGTTC-3. The Dock3 conditional mice Dock3fl/fl were deposited into the Jackson Laboratory (Bar Harbor, ME; stock# 038465). Skeletal myofiber tamoxifen-inducible mice were purchased from Jackson Laboratory and subsequently the Human Skeletal-Actin-MerCreMer (HSA-MCM) (Jackson Labs; Bar Harbor, ME; stock# 025750) and WT (C57BL/6J; stock# 000664) mice were maintained in our animal colony under pathogen-free standard housing conditions. Dock3 ubiquitous KO mice (Jackson Labs; stock# 033736) were originally obtained from the laboratory of Dr. David Shubert (Salk Institute) and have been previously described (10). The mdx5cv (Jackson Labs; stock# 002379) mice were originally obtained from Jackson Labs. All mice were maintained on the C57BL/6J strain background. All mouse strains were maintained under standard housing and feeding conditions with the University of Alabama at Birmingham Animal Resources Facility under pathogen-free, sterile conditions under animal protocol number 21393. Mice were all fed a diet consisting of the Teklad Global Rodent Diets (Envigo; Indianapolis, IN) with ad libitum access to food and water.
GLUT4-receptor assays
WT and Dock3 KO primary mouse muscle cells were harvested from the ubiquitous Dock3 KO mice as previously described(17). Primary mouse myoblasts were grown in Skeletal Muscle Cell Growth Medium (Promocell Cat# C-23060; Heidelberg, Germany) with 20% FBS (ThermoFisher Scientific; Waltham, MA; Cat# 16140071), and incubated at 37°C using a standard primary muscle cell isolation protocol(18). Muscle cells were plated in a 6-well gelatin coated plate at 50,000 cells/well. Muscle cells were then transfected with a pLenti-myc-GLUT4-mCherry (Addgene; Watertown, MA; stock# 64049) for 48 hours and the GLUT4 localization assay was performed as previously described(19). For the myotube differentiation experiment, day 4 WT and Dock3 KO differentiated using serum withdrawal and cultured in 2% horse serum medium (Promocell; Cat# C-23061) myotubes stably infected with the pLenti-myc-GLUT4-mCherry reporter were stimulated with 0.01 mM of 2-deoxyglucose (MilliporeSigma; Cat# D8375) and 100 nM insulin (MilliporeSigma; Cat# 1342106) for 20 minutes before quantifying RFP expression as previously described(19, 20). Immunofluorescent staining was performed using an antibody against DESMIN (MilliporeSigma; Cat# AB907) and VECTASHIELD Antifade mounting medium with DAPI (Vector Laboratories; Cat# H-1200).
Mouse activity tracking
Mouse activity locomotor measurements were performed as previously described(21). Twenty-four hours prior to experiment termination and tissue harvest, mice were analyzed for locomotive activity using the Ethovision XT software platform (Noldus; Leesburg, VA) with isolated individual chambers that recorded motion from mouse head to tail. Mice were acclimated to the room and open-field chambers one day prior to activity and were given a five minute additional adaptation period prior to activity recording. Mouse activity was recorded for six minutes with no external stimulation.
Myofiber diameter calculations
The cross-sectional area (CSA) of the myofibers within the skeletal muscle sections was calculated by quantifying the myofiber areas using a previously described protocol(22). Approximately 600 TA myofibers were counted and CSA (μm2) was measured via several overlapping H&E microscopy images of each section and quantified using Fiji software(23).
DEXA Quantitative Magnetic Resonance (QMR) imaging
Evaluation of body composition comprising of both fat and lean tissue mass in vivo was performed on 4-month-old male Dock3fl/fl and Dock3 mKO mice (10 mice/genotype) using the EchoMRI™ 3-in-1 composition analyzer (software version 2016, Echo Medical; Houston, TX). Individual fat and lean mass measurements were recorded in grams (g) and were analyzed using chi-squared test between Dock3fl/fl and Dock3 mKO mice.
Cardiotoxin-induced skeletal muscle injury
Dock3 mKO and Dock3fl/fl mice were injected in their TA skeletal muscles with 40 μl of cardiotoxin (MilliporeSigma; Cat# 217503) at a 10 μM concentration. The contralateral TA muscle was used as a sham control injection with 1x phosphate-buffered saline (ThermoFisher Scientific; Cat# 10010049). Seven days following injections, mice were euthanized, and their TA skeletal muscles were slow-frozen in Scigen TissuePlus O.C.T. Compound (Fisher Scientific; Hampton, NH Cat# 23-730-571) for histological analysis and snap frozen in liquid nitrogen for molecular analysis.
Immunofluorescence and immunohistochemistry
Mouse skeletal muscles were cryo-frozen in Scigen TissuePlus O.C.T. Compound using an isopentane (FisherScientific; Cat# AC397221000) and liquid nitrogen bath as unfixed tissues. Blocks were later cut on a cryostat into 7-10 μm sections and placed on Fisherbrand Tissue Path Superfrost Plus Gold slides (Fisher Scientific; Cat# FT4981gplus). H&E staining was performed as previously described(24). For immunofluorescent staining, slides were blocked for one hour in 10% goat serum and incubated for one hour at room temperature using a M.O.M kit (Vector Labs Cat# BMK-2202; Newark, CA).
Western blotting
Protein lysates were obtained by homogenizing tissues in M-PER lysis buffer (ThermoFisher; Cat# 78501) with 1x Complete Mini EDTA-free protease inhibitor cocktail tablets (Roche Applied Sciences; Cat# 04693159001; Penzburg Germany). Protein lysates were quantified using a Pierce BCA Protein Assay Kit (ThermoFisher Cat# 23225). Unless stated otherwise, 50 μg of whole protein lysate was used for all immunoblots and resolved on 4-20% Mini-PROTEAN TGX Precast Protein gels (BioRad; Cat# 4561094). Protein samples were transferred to 0.2 μm PDVF membranes (ThermoFisher; Cat# LC2002), blocked in 0.1x TBS-Tween in 5% BSA for one hour, and then gently incubated overnight with primary antibody on a rocker at 4°C. Membranes were washed in 0.1% TBS-tween four times at 10-minute intervals before being incubated with secondary antibodies (either mouse or rabbit IgG) conjugated to HRP for one hour at room temperature with gentle agitation. Following another three washes for 15 minute intervals at room temperature, membranes were then treated with RapidStep ECL Reagent (MilliporeSigma; Cat# 345818-100 ml).
Real-time quantitative PCR
Total RNA was extracted using a miRVana (ThermoFisher, Cat# AM1560) kit while following the manufacturer’s protocol. One microgram of total RNA was reverse transcribed using the Taqman Reverse Transcription kit (Applied Biosystems; Cat# N8080234; Waltham, MA) following the manufacturer’s protocol. TaqMan assay probes were all purchased from ThermoFisher corresponding to each individual transcript. Quantitative PCR (qPCR) TaqMan reactions were performed using TaqMan Universal PCR Master Mix (Applied Biosystems; Cat# 4304437). Relative expression values were calculated using the manufacturer’s software and further confirmed using the 2−ΔΔCt method.
Glucose and insulin tolerance tests
Mice were fasted for eight hours prior to afternoon administration of a bolus of D-glucose (MilliporeSigma; Cat# G8270). Mice were given an intraperitoneal injection at a concentration of 3 mg/gram of mouse bodyweight. Blood glucose was measured on a commercially obtained glucometer (Nipro Diagnostics Inc.; Southampton, UK) using 10 μl of whole serum from tail bleeds. For the insulin tolerance tests, the mice were fasted for five hours prior to afternoon administration of a bolus of human insulin (MilliporeSigma; Cat# 1342106). Mice were given an intraperitoneal injection at a concentration of 3 mg/gram of mouse bodyweight.
Yeast-2-Hybrid
The GAL4-based yeast two hybrid system was used to detect the interaction between recombinant DOCK3 and SORBS1 domains. The bait and prey are expressed as fusion domain constructs to the GAL4 DNA binding domain and GAL4 activation plasmids. Inoculations were then transferred to a 500 mL flask containing 300 mL yeast peptone dextrose (YPD) broth (ThermoFisher Scientific; Cat# A1374501) and incubated at 30°C for 16-18 hours with shaking at 230 rpm. Cultures were incubated at 30°C for 16-18 hours with shaking at 230 rpm in an overnight culture flask containing 300 ml of YPD. Cultures were harvested in 50 ml tubes and centrifuged at 1000 x g for five minutes at room temperature. Cell pellets were resuspended in distilled water and again centrifuged at 1000 x g for five minutes. Pellets were then resuspended in 1.5 ml freshly prepared, sterile 1X TE/1X LiAc solution. Approximately 0.1 μg of plasmid DNA and 0.1 mg of carrier DNA was added to a 1.5 mL tube and mixed. Approximately 0.1 ml of yeast competent cells were then added to each tube and vortexed until well mixed, heat shocked for five minutes in a 42°C water bath, and chilled on ice for 2 minutes. Yeast cultures were then centrifuged for five seconds at 12,000 x g and resuspended in 0.5 μL sterile TE buffer. The cells were plated at 100 μL each on SD/-LEU/-Trp selective transformant agar plates and incubated at 30°C until colonies appeared the next morning.
Co-immunoprecipitation (co-IP)
Protein constructs were expressed in HEK293T cells using Lipofectamine 2000-mediated (Invitrogen, Catalog #11668030; Waltham, MA) plasmid transfection. Expression constructs were subcloned into Vitality hrGFP mammalian expression vectors (Agilent Technologies; Santa Clara, CA; Cat# 240031 and #240032) using standard PCR cloning techniques. HEK293T cells were collected two days post-transfection and lysed in lysis buffer that contained 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and 1:100 Protease/Phosphatase Inhibitor Cocktail (Cell Signaling Technology; Danvers, MA). Cells were then homogenized using an Omni Bead Rupter 12 (Perkin Elmer; Kennesaw, GA). Protein lysates were then incubated on ice for thirty minutes. Lysates were spun down at 10000 x g for ten minutes, and the supernatant was collected for co-IP. Protein levels were quantified using the BCA Kit and normalized (Pierce Protein Biology, Rockford, IL, USA). Approximately 5% of total protein lysate was set aside as the input fraction. Laemmli Buffer plus β-mercaptoethanol was then added to these samples and one mg of total protein lysate was used per co-IP reaction. Approximately 0.5 mg of mouse IgG control (ThermoFisher, Catalog # MA1-213) was used for the control reaction. Co-IP reactions were rotated overnight at 4°C with 100 μl of SureBeads Protein G Magnetic Beads (BioRad; Catalog# 1614013; Hercules, CA). The bead lysates were washed five times in the co-IP buffer using a DynaMag Magnet (ThermoFisher) to pull down the complexes. After this, Laemmli Buffer plus β-mercaptoethanol was added to the beads, which were boiled for five minutes at 100°C. All co-IP reactions were probed using standard western immunoblotting techniques described above. The rabbit DOCK3 (ThermoFisher; Cat# PIPA5100485) and mouse SORBS1 (Sigma-Aldrich; Catalog #SAB4200599; St. Louis, MO) antibodies were used for verifying immunoprecipitation reactions via western immunoblotting. Anti-FLAG M2 magnetic beads (MilliporeSigma; Catalog #M8823) and anti-FLAG M2 monoclonal antibody (MilliporeSigma; Catalog #F1804) were used for co-IP and western immunoblotting reactions. A μMACS HA magnetic bead isolation kit (Miltenyi Biotec; Catalog# 130-091-122) and anti-HA rabbit monoclonal (GenScript; Catalog #A01963) were also used for co-IP and western immunoblotting reactions.
GST pulldown assay
Recombinant SORBS1 protein (Abcam; Cambridge, UK) was incubated with recombinant GST-DOCK3-PXXP or GST alone plasmids (constructs cloned into pGEX-6P-1 plasmid; GE Healthcare; Chicago, IL) in GST reaction buffer (250 mM Tris-HCl at pH 7.4, 500 mM NaCl, 25 mM MgCl2, 5 mM dithiothreitol, 0.5 mM EGTA and 20 mM freshly prepared ATP) for one hour at 4°C on a rotator. Pierce Glutathione Magnetic Agarose Beads (ThermoFisher; Cat# 78602) were then suspended in the GST reaction buffer and added to the reaction mixture for one hour at 4°C with gentle rotation. The beads were then washed four times in reaction buffer using a DynaMag magnet. Laemmli Buffer plus β-mercaptoethanol was added to these samples, which were then boiled for five minutes at 100°C. GST pulldown was verified via immunoblot against the GST epitope (anti-GST; rabbit polyclonal; Abcam; Cat# ab9085).
Proximity Ligation Assay (PLA)
Primary mouse myotubes were generated from 7 day differentiated muscle satellite cells originally isolated from adult (2-4 month old) male C57BL/6J mice using a standard alpha-7-integrin isolation kit (Cat# 130-104-268; Miltenyi Biotec; North Rhine-Westphalia, Germany). Myotubes were cultured on 2-well chamber slides coated with 0.1% rat Collagen I (Cat# 354236; Corning Inc.; Corning, NY). PLA assay was performed using a DuoLink In Situ Probemaker PLUS PLA assay kit (Cat# DUO92009-1KT; MilliporeSigma) following the manufacturer’s instructions.
Muscle physiological function assays
EDL muscles were dissected from anesthetized mice and studied in a phosphate buffer equilibrated with 95% O2, 5% CO2 (35 °C). Contractions were produced using a 150 ms, supramaximal stimulus train (200 μs pulses) with the muscle held at its optimal length (Lo) for tetanic tension. Force was normalized to physiological cross-sectional area as previously described (Huntoon et al. 2018). Each muscle was studied at stimulation frequencies ranging from 30 to 300 Hz (peak force). Fixed-end force values were expressed relative to peak force and fit by a sigmoid curve as previously described(25). Changes in the relationships were evaluated by differences in the inflection point (K, measured in Hz) and slope (H, unitless). Muscles were then subjected to a high active strain protocol consisting of the following sequence: one fix-end trial, 5 lengthening (eccentric) trials, and two fixed-end trials. The fixed-end trials were as described above. The lengthening trials consisted of an initial fixed-end contraction that allowed the muscle to rise to peak force (100 ms duration), followed by a constant velocity stretch at 4 fiber lengths/s (50 mms duration) to a final length of 120% Lo. For the high-strain protocol, force was evaluated at 95 ms of stimulation for both fixed-end and lengthening trials.
Statistical analyses
For most pairwise comparisons, either a chi-squared test for normality or a one-way analysis of variance (ANOVA) with Least Significant Difference (LSD) was performed for all multiple comparisons. GraphPad Prism version 9 software (Graphpad Software; San Diego, CA) was used for all statistical analyses. Statistical significance for the tests are denoted by *p< 0.05, **p<0.01, ***p<0.001, and ****p< 0.0001 and used for all reported data analyses. All graphs were represented as mean +/− SEM.
Results
Generation of a muscle-specific Dock3 knockout mouse
As DOCK3 protein is expressed both within the motor neuron and in the skeletal muscle, we generated a conditional mouse model to differentiate the role of the Dock3 gene exclusively in skeletal muscle. We investigated the specific function of Dock3 in the myofiber by evaluating Dock3-deficient mice in which exons 8 and 9 of the Dock3 gene locus are flanked with loxP sites in the intronic regions (Figure 1A). Upon mating with the mouse model expressing tamoxifen-inducible Cre recombinase driven by the human-skeletal actin promoter (HSA-MerCre-Mer; HSA-MCM), the mice will conditionally ablate Dock3 expression upon tamoxifen administration in the skeletal myofibers (Figure 1A). Genotyping and western blot analyses of Dock3 expression in the tissue extracts of brain and the tibialis anterior (TA) from control and Dock3 ubiquitous KO mice confirmed the ablation of Dock3 from the myofiber. The Dock3fl/fl:HSA-MerCreMer (henceforth referred to as Dock3 mKO) mice showed the deletion of Dock3 expression in tissue extracts from the TA lysates, but not the brain, confirming the tissue-specific deletion of Dock3 from the myofiber (Figures 1B-1E).
Figure 1. Generation and validation of muscle-specific Dock3 conditional knockout mice.

A. Generation of Dock3 mKO schematic. Dock3fl/fl mice containing two loxP sites flanking Dock3 exon 8 and 9 were mated with human-skeletal-actin (HSA)-MerCreMer mouse line to generate the Dock3 mKO mice. When administered tamoxifen (80 mg/kg) over five consecutive days this induces a frameshift mutation resulting in a premature stop codon. B. PCR genotyping agarose gel of Dock3 heterozygous fl/+ alleles to produce homozygous Dock3 flox/flox alleles in skeletal muscle. C. PCR genotyping agarose gel identifying loxP1 site (231 bp) and Cre recombinase (304 bp) in Dock3 flox/flox and WT mice (161 bp) mice in both tibialis anterior (TA) and whole brain lysates (BR). D. Western blot of Dock3 mKO mice indicating a reduction of protein as a result of ablation of Dock3. Low and high exposures of DOCK3 immunoblotting shown as well as a GAPDH loading control. E. Quantification of DOCK3 protein normalized to GAPDH loading control. F. Immunofluorescent staining of adult TA muscles from the Dock3fl/fl and Dock3 mKO mice for LAMININ, DOCK3, and the merged image. Scale bar = 50 μm. G. Quantification of mean fluorescence intensity (RFUs) in the Dock3fl/fl and Dock3 mKO mice. Significance shown as ****p< 0.0001.
Dock3 mKO mice have disrupted skeletal muscle histology and locomotor activity
To evaluate the consequences of Dock3 skeletal muscle ablation, we first analyzed the muscle architecture and morphology of isolated TA muscle fibers of 4-month-old Dock3 mKO mice compared to Dock3fl/fl controls (Figure 2A). We observed a decrease in myofiber cross-sectional area (CSA) and noted smaller myofibers grouped together throughout the Dock3 mKO muscles (Figure 2B). We did not observe a change in centralized myonuclei. However, we did observe muscle fiber atrophy reflected by increased frequency of smaller myofibers in Dock3 mKO mice compared to Dock3fl/fl controls. We sought to characterize how the disruption of Dock3 in the skeletal muscle would impact overall locomotive function by using open field activity tracking to record activity levels in adult mice (Figure 3A). Dock3 mKO mice demonstrated significantly decreased distance traveled and average velocity compared with controls, indicating a reduction in basal locomotor function (Figures 3B-D). These findings are consistent with a decrease in locomotor function previously observed in adult ubiquitous Dock3 KO mice. Interestingly, when we conducted several functional assays on extensor digitorum longus (EDL) muscles isolated from Dock3 mKO and Dock3fl/fl mice we found no significant changes in the muscle’s contractile properties (Figures 3E-3J). This included the relationship between stimulus frequency and force (Figures 3E-G), absolute peak force (Figure 3G), force normalized to the muscle’s physiological cross-sectional area (Figure 3I), and in the muscles resistance to eccentric contractions (Figures 3J and 3K). Therefore, we concluded that loss of Dock3 in the myofiber reduces basal activity independent of undermining the overall contractility and structural integrity of the skeletal muscle.
Figure 2: Muscle-specific loss of Dock3 results in a smaller myofiber sizes.

A. Hematoxylin and eosin (H&E) stainings of TA muscles from Dock3fl/fl vs. Dock3 mKO. Scale bar = 200 μm. B. Quantification of myofiber diameters in Dock3fl/fl vs. Dock3 mKO. Cross-sectional area shown as frequency of fiber sizes over fiber size (μm2). One-way analysis of variance (ANOVA) with a Fisher’s least significant difference (LSD) post hoc test was performed and a * < p-value shown for significance < 0.001 with ns = not significant.
Figure 3. Loss of muscle Dock3 reduces basal activity independent of physiological force.

A. Schematic showing tamoxifen regimen for Dock3fl/fl and Dock3 mKO. Mice were administered an intraperitoneal injection of tamoxifen (80 mg/kg) for five consecutive days followed by a three month washout period before assessing locomotor function. B. Activity tracking traces in Dock3fl/fl vs. Dock3 mKO mice. C. Quantification of total distance traveled (cm) n = 9 mice/cohort, D. Quantification of mouse velocity, n = 9 mice/cohort, E. Force-frequency relationship of EDL muscles from Dock3 mKO vs. Dock3fl/fl EDL mice, n = 5 mice/cohort. F. Inflection point of the force-frequency relationship (K) of EDL muscles from Dock3 mKO vs. Dock3fl/fl mice n = 5 mice/cohort. G. Slope of the force-frequency relationship (H) of EDL muscles from Dock3 mKO vs. Dock3fl/fl mice n = 5 mice/cohort. H. Peak force of EDL muscles from Dock3 mKO vs. Dock3fl/fl mice n = 5 mice/cohort. I. Peak force per physiological cross-sectional area (CSA) of EDL muscles from Dock3 mKO vs. Dock3fl/fl mice, n = 5 mice/cohort. J. Relative isometric force measured during the eccentric contraction protocol for EDL muscles of Dock3 mKO vs. Dock3fl/fl mice, n = 5 mice/cohort. K. Relative force at the conclusion of the eccentric contraction protocol for EDL muscles of Dock3 mKO vs. Dock3fl/fl mice, n = 5 mice/cohort. The following p-values of significance were stated: *p < 0.001, **p < 0.01, and ns = not significant.
Loss of Dock3 at the myofiber inhibits myogenic regeneration after cardiotoxin injury
Previously, we isolated primary myoblasts isolated from Dock3 KO muscle which exhibited impaired regeneration and fusion. We sought to determine if this phenomenon was recapitulated in our Dock3 mKO mice and the degree to which muscle regeneration is impacted by the loss of muscle DOCK3 expression (Figure 4A). We performed an intramuscular injection of cardiotoxin in our Dock3 mKO mice to induce a skeletal muscle injury into the right TA muscle while using the left as a contralateral control receiving a sham injection of phosphate buffered saline (PBS) to evaluate the role of DOCK3 in muscle regeneration. Mice were sacrificed at 7 and 14 days post-injury and evaluated via histological analysis with hematoxylin and eosin (H&E) and Masson’s trichrome to assess myofiber cross-sectional area, myonuclei position, and fibrosis within the muscle (Figures 4B and 4C). We quantified increased levels of centralized myonuclei in the Dock3 mKO mice and observed elevated levels of centralized myonuclei at 7 and 14 days post cardiotoxin injury (Figures 4D and 4E). We also observed that the Dock3 mKO mice had increased fibrosis when compared to the control Dock3fl/fl (Figures 4F and 4G). We compared our the study for 14 days post cardiotoxin TA muscle injury and observed similarly impaired regeneration in the Dock3 mKO mice as previously observed in the Dock3 ubiquitous KO mice(21). These findings were consistent with the high levels of centralized myonuclei and fibrotic areas observed, indicating a delay in regeneration in the skeletal muscle of Dock3 mKO mice and emphasizing the importance of DOCK3 in skeletal muscle.
Figure 4. Dock3 mKO mice show impaired skeletal muscle regeneration following injury.

A. Schematic of cardiotoxin (ctx) induced skeletal muscle TA injury. Two separate cohorts of each Dock3 mKO mice and Dock3fl/fl mice were administered with an intramuscular injection of 10 μM of cardiotoxin at Day 0 and separate cohorts were harvested on days 7 and 14 post-injury. B. Cross-section of injured tibialis anterior stained with immunofluorescent antibody against LAMININ (green), DAPI (blue), and the merged image at days 7 and 14 post ctx injury. Scale bar = 100 μm. C. Masson’s trichrome histochemical analysis of injured TA in Dock3fl/fl vs. Dock3 mKO 7 and 14 days post-injury. D. and E. Quantification of histological images from (C) analyzing % centralized myonuclei per 600 fibers at 7 and 14 days post ctx injury. F. and G. Quantification of histochemical images from (C) with analysis of percent (%) fibrotic area in injured TA of Dock3fl/fl vs. Dock3 mKO 7 and 14 days post-injury. Scale bar = 100 μm (20x images) and 2 mm (1x whole TA images). *p < 0.01, **p < 0.001, ****p< 0.0001, n = 4 mice/cohort.
Adult Dock3 mKO mice have abnormal skeletal muscle mass and metabolism
We previously demonstrated that Dock3 ubiquitous KO mice were glucose intolerant and had decreased weights due to decreased muscle mass. Thus, we sought to understand if the loss of Dock3 in the skeletal muscle would impact whole-body metabolism. Quantitative magnetic resonance (QMR) imagining of adult Dock3 mKO mice revealed increased body weight compared to Dock3fl/fl aged-matched controls (Figure 5A). Conversely, Dock3 mKO mice had significantly increased fat mass compared to Dock3fl/fl aged-matched controls (Figure 5B). No detectable changes in skeletal muscle lean mass were observed in the Dock3 mKO mice (Figure 5C). Being that DOCK3 is known to activate Rho GTPases such as RAC1, a critical regulator of insulin and glucose signaling pathways in skeletal muscle. We measured the ability of the Dock3 mKO mice to respond to a glucose challenge via a glucose tolerance test (GTT). GTT tests revealed no significant changes in glucose processing in the muscle (Figure 5D). However, insulin tolerance tests (ITT) conducted on Dock3 mKO mice revealed whole body hyperglycemia and insulin resistance (Figure 5E). We analyzed the role of DOCK3 in glucose processing within the muscle by isolating primary Dock3 KO myoblasts and infecting with lentiviral GLUT4-RFP(26). Upon insulin stimulation, we observed reduced GLUT4 translocation in the Dock3 KO myoblasts, which supports a defect in glucose uptake and/or processing within the skeletal muscle (Figure 5F). These findings reveal DOCK3 to be a critical regulator of metabolism in the skeletal muscle and that loss of DOCK3 expression in the myofiber undermines important metabolic functioning and insulin processing in the skeletal muscle.
Figure 5. Dock3 mKO mice show increased body mass and whole body hyperglycemia.

A. Quantitative magnetic resonance imaging indicated body weight differences between Dock3fl/fl and Dock3 mKO mice, n = 10 mice/cohort, *p < 0.01. B. Quantitative magnetic resonance imaging indicated differences in fat mass between Dock3fl/fl and Dock3 mKO mice, n = 10 mice/cohort, **p < 0.001. C. Quantitative magnetic resonance imaging indicating differences in lean mass between Dock3fl/fl and Dock3 mKO mice, n = 10 mice/cohort, ns = not significant. D. Glucose Tolerance Test in Dock3fl/fl and Dock3 mKO mice shown. Serum blood glucose level (mg/dl) measured over time (minutes). E. Insulin Tolerance Test in Dock3fl/fl compared to Dock3 mKO mice. n = 8 mice/cohort. Serum blood glucose level (mg/dl) measured over time (minutes). F. WT and Dock3 KO myoblasts transfected with HA-GLUT-RFP. Wheat germ agglutinin stained membranes (green), GLUT4 (RFP), and nuclei are stained with DAPI. Scale bar = 10 μm. One-way ANOVA test with a Fisher’s LSD post hoc test was performed. *p < 0.01 significance values are shown.
DOCK3 interacts with insulin signaling protein, Sorbin and SH3 domain-containing 1 (SORBS1)
Due to the increased fat mass, body weight, and hyperglycemia observed in the Dock3 mKO mice, we explored what potential protein-protein interactions DOCK3 may be involved with regarding glucose uptake. We conducted a yeast two-hybrid neuromuscular cDNA library screen using the C-terminal domain of human DOCK3 protein to identify novel DOCK3 protein interactions (Figure 6A). We identified the insulin adaptor protein, SORBS1 as directly interacting with the C-terminal domain of DOCK3 and confirmed the interaction via secondary yeast amino acid growth selection confirmation (Figure 6B and Figure 6C). SORBS1, also called Cbl-Associated Protein (CAP), is a known insulin adaptor protein whose subcellular localization is essential to downstream insulin signaling events and has been implicated as a secondary signaling pathway critical to insulin-mediated glucose uptake(27). To determine which domains of each protein were critical to their interaction, we conducted a GST-pulldown assay in HEK293T cells overexpressing DOCK3 and SORBS1. The proline rich motif (PXXP) of DOCK3 and the SH3 domains of SORBS1 were identified as the main sites of the DOCK3-SORBS1 protein interaction (Figures 6D-6F). Following these results, we sought to map out which functional domains were critical to the DOCK3-SORBS1 interaction. Overexpression constructs containing full length and deletion of key conserved protein functional domains of DOCK3 and constructs deleting each of the SH3 domains of SORBS1 were generated (Figure 7A). Co-immunoprecipitation confirmed that all three SH3 domains on SORBS1 were essential for the DOCK3-SORBS1 interaction (Figure 7B). This protein-protein interaction between DOCK3 and SORBS1 was further validated in human primary myotubes (Figures 7C-7D). We next probed if endogenous DOCK3 and SORBS1 directed interacted in primary myotubes using a proximity ligation assay (PLA) which uses immunofluorescent labeling of two candidate proteins within 40 nm of each other in a tissue or cell(28). Endogenous DOCK3 and SORBS1 both interacted throughout the primary myotubes indicating that this interaction occurs in muscle and is not an artifact of cell culture overexpression (Figure 7E). We further assessed whether or not GLUT4 processing was directly affected by the loss of DOCK3 in muscle myotubes. Dock3-deficient myotubes showed defective processing of GLUT4 receptor and glucose as upon insulin stimulation compared to wild type myotubes (Supplemental Figures S1). These results identified the DOCK3-SORBS1 interaction as a potential novel source of metabolic regulation that may modulate glucose and insulin signaling in skeletal muscle.
Figure 6. DOCK3 interacts with insulin signaling protein, SH3 domain-containing 1 (SORBS1).

A. DOCK3-SORBS1 yeast-2-hybrid neuromuscular cDNA screening library strategy. B. Selection guide of DOCK3-SORBS1 yeast-2-hybrid amino acid selection. C. Positive interaction of DOCK3-C-terminus and SORBS1 cDNA shown with yeast selective growth. D. DOCK3-SORBS1 co-Immunoprecipitation in HEK293T cells. Immunoprecipitation (IP) performed with DOCK3-GST and immunoblotting (IB) against SORBS-FLAG. E. GST-Pulldown domain constructs shown indicating domains of full-length Dock3 and GST tagged PXXP motif. F. DOCK3-SORBS1 GST-Pulldown immunoblots showing the interaction between recombinant DOCK3 and SORBS1 directly interacting.
Figure 7. DOCK3 interacts with SORBS1 via binding to the SORBS1 SH3 domains.

A. Schematic showing the SORBS1 deletion constructs indicating full length SORBS1, and deletion domains across each SH3 domain. B. The co-IP indicating the interaction and expression of each deletion construct in HEK293T cells. C. The co-IP of DOCK3-SORBS1 in human primary myoblasts. Immunoblot (DOCK3 rabbit polyclonal antibody) with the co-immunoprecipitation (SORBS1-FLAG; FLAG mouse monoclonal antibody) D. The co-IP of SORBS1 deletion constructs containing deletions of each of the SH3 domains in SORBS1 showing the requirements for each in binding to DOCK3. E. Proximity Ligation Assay (PLA) demonstrating endogenous interaction between DOCK3 and SORBS1 in primary mouse myotubes along with a DAPI nuclei stain. Red labeling detects positive interaction between the two proteins. DOCK3 alone (− DOCK3 Ab), SORBS1 alone (− SORBS1 Ab), and no antibody controls shown. Scale bars = 50 and 20 μm (low and high magnification- DOCK3 + SORBS1 only).
Discussion
DOCK3 is a guanine-nucleotide exchange factor whose downstream activation of Rho GTPases impacts a number of pathways that influence cell migration, insulin signaling, and pathways regulating muscle mass(29, 30). Our previous work identified DOCK3 as an important biomarker and dosage-sensitive regulator of Duchenne muscular dystrophy(16). Here, we identified the muscle-specific role of DOCK3 in the myofiber, apart from its role in the motor neuron, and how its expression is critical to normal muscle function, regeneration and glucose processing within the muscle. Furthermore, using a yeast-two-hybrid screen we identified a novel protein-protein interaction with SORBS1, a key glucose and insulin signaling factor that may yield clues into DOCK3’s regulation of skeletal muscle metabolic function via glucose and insulin signaling pathways.
Additional questions remain as DOCK3 has been shown to interact with RAC1, another key regulator of glucose processing in skeletal muscle which may also explain our observed phenotypes in the Dock3 mKO mice(31-33). Our observation of impaired skeletal muscle regeneration following cardiotoxin injury in the Dock3 mKO mice suggests that DOCK3 may play roles in muscle regeneration even though it is not expressed in the muscle satellite or stem cells. DOCK3 has been shown to play key roles in cell migration, actin polymerization, and regulates key signaling pathways such as WAVE which may explain the observed impaired regeneration in the Dock3 mKO mice(12). The observation of Dock3 mKO mice having whole-body hyperglycemia and insulin resistance could be a result of the disruption of the DOCK3-SORBS1 interaction. SORBS1 is part of a small family of adaptor proteins that is known to regulate numerous cellular processes including cell adhesion, cytoskeletal formation, and is required for insulin-stimulated glucose transport(34). A number of studies suggest that genetic variations in SORBS1 could be associated with human disorders such as obesity, diabetes, and insulin resistance(27, 35, 36). Dock3 mKO mice showed significantly increased fat mass and body mass, without any impact on lean mass. The novel interaction between DOCK3 and SORBS1 implies that DOCK3 plays a significant metabolic role in the muscle, specifically involving regulation of insulin-mediated glucose uptake. Admittedly, there are limitations to our studies, as DOCK3 and SORBS1 likely have additional interacting protein partners within the skeletal muscle that likely play critical roles in metabolism and cellular signaling. Moreover, DOCK3’s role in differentiation and cell cycle kinetics may also influence SORBS1 and other downstream interacting proteins in a secondary manner. Further studies are warranted to dissect DOCK3’s additional roles in other lineages using a conditional approach.
Supplementary Material
Acknowledgements
The authors wish to thank members of the Alexander lab including Jeffrey Fairley, Grace Morrison, and Jamie Collins for their assistance with experiments. The authors wish to thank Michael Lopez, Glenn Rowe, and Anna Thalacker-Mercer for their critical evaluation of the manuscript prior to submission. The authors wish to acknowledge Kirk Habegger, Shelly Nason, Jessica Antipenko, and members of the UAB Diabetes Animal Physiology Core for assistance with the metabolic experiments. The authors wish to acknowledge Mary Ballestas and Reid Millican from the UAB Neuroscience NINDS Vector and Virus Core (Funded by NINDS P30 grant number NS047466). Research reported in this publication was supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH, HHS of the National Institutes of Health under award number R01HD095897 awarded to M.S. Alexander. M.S. Alexander is supported by an NIH Office of the Director (OD) U54 grant U54OD030167. A.S. was funded by a NIH NINDS T32 training grant number 5T32NS095775.
Footnotes
Conflict of Interest Statement
The authors declare no conflicts of interest.
Data Availability Statement
The data that support the findings of this study are available in the methods and/or supplementary material of this article.
References
- 1.Chaillou T, and Lanner JT (2016) Regulation of myogenesis and skeletal muscle regeneration: effects of oxygen levels on satellite cell activity. The FASEB Journal 30, 3929–3941 [DOI] [PubMed] [Google Scholar]
- 2.DeFronzo RA, and Tripathy D (2009) Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32 Suppl 2, S157–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sampath SC, Sampath SC, and Millay DP (2018) Myoblast fusion confusion: the resolution begins. Skeletal Muscle 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noviello C, Kobon K, Delivry L, Guilbert T, Julienne F, Maire P, Randrianarison-Huetz V, and Sotiropoulos A (2021) RhoA within myofibers controls satellite cell microenvironment to allow hypertrophic growth. bioRxiv, 2021.2001.2018.426685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samson T, Will C, Knoblauch A, Sharek L, von der Mark K, Burridge K, and Wixler V (2007) Def-6, a guanine nucleotide exchange factor for Rac1, interacts with the skeletal muscle integrin chain alpha7A and influences myoblast differentiation. J Biol Chem 282, 15730–15742 [DOI] [PubMed] [Google Scholar]
- 6.Côté J-F, and Vuori K (2002) Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. Journal of Cell Science 115, 4901–4913 [DOI] [PubMed] [Google Scholar]
- 7.Aguet F, Anand S, Ardlie KG, Gabriel S, Getz GA, Graubert A, Hadley K, Handsaker RE, Huang KH, Kashin S, Li X, MacArthur DG, Meier SR, Nedzel JL, Nguyen DT, Segrè AV, Todres E, Balliu B, Barbeira AN, Battle A, Bonazzola R, Brown A, Brown CD, Castel SE, Conrad DF, Cotter DJ, Cox N, Das S, de Goede OM, Dermitzakis ET, Einson J, Engelhardt BE, Eskin E, Eulalio TY, Ferraro NM, Flynn ED, Fresard L, Gamazon ER, Garrido-Martín D, Gay NR, Gloudemans MJ, Guigó R, Hame AR, He Y, Hoffman PJ, Hormozdiari F, Hou L, Im HK, Jo B, Kasela S, Kellis M, Kim-Hellmuth S, Kwong A, Lappalainen T, Li X, Liang Y, Mangul S, Mohammadi P, Montgomery SB, Muñoz-Aguirre M, Nachun DC, Nobel AB, Oliva M, Park Y, Park Y, Parsana P, Rao AS, Reverter F, Rouhana JM, Sabatti C, Saha A, Stephens M, Stranger BE, Strober BJ, Teran NA, Viñuela A, Wang G, Wen X, Wright F, Wucher V, Zou Y, Ferreira PG, Li G, Melé M, Yeger-Lotem E, Barcus ME, Bradbury D, Krubit T, McLean JA, Qi L, Robinson K, Roche NV, Smith AM, Sobin L, Tabor DE, Undale A, Bridge J, Brigham LE, Foster BA, Gillard BM, Hasz R, Hunter M, Johns C, Johnson M, Karasik E, Kopen G, Leinweber WF, McDonald A, Moser MT, Myer K, Ramsey KD, Roe B, Shad S, Thomas JA, Walters G, Washington M, Wheeler J, Jewell SD, Rohrer DC, Valley DR, Davis DA, Mash DC, Branton PA, Barker LK, Gardiner HM, Mosavel M, Siminoff LA, Flicek P, Haeussler M, Juettemann T, Kent WJ, Lee CM, Powell CC, Rosenbloom KR, Ruffier M, Sheppard D, Taylor K, Trevanion SJ, Zerbino DR, Abell NS, Akey J, Chen L, Demanelis K, Doherty JA, Feinberg AP, Hansen KD, Hickey PF, Jasmine F, Jiang L, Kaul R, Kibriya MG, Li JB, Li Q, Lin S, Linder SE, Pierce BL, Rizzardi LF, Skol AD, Smith KS, Snyder M, Stamatoyannopoulos J, Tang H, Wang M, Carithers LJ, Guan P, Koester SE, Little AR, Moore HM, Nierras CR, Rao AK, Vaught JB, and Volpi S (2020) The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samani A, English KG, Lopez MA, Birch CL, Brown DM, Kaur G, Worthey EA, and Alexander MS (2022) DOCKopathies: A systematic review of the clinical pathologies associated with human DOCK pathogenic variants. Human Mutation n/a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore CA, Parkin CA, Bidet Y, and Ingham PW (2007) A role for the Myoblast city homologues Dock1 and Dock5 and the adaptor proteins Crk and Crk-like in zebrafish myoblast fusion. Development 134, 3145–3153 [DOI] [PubMed] [Google Scholar]
- 10.Laurin M, Fradet N, Blangy A, Hall A, Vuori K, and Côté J-F (2008) The atypical Rac activator Dock180 (Dock1) regulates myoblast fusion in vivo. Proceedings of the National Academy of Sciences 105, 15446–15451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Namekata K, Enokido Y, Iwasawa K, and Kimura H (2004) MOCA Induces Membrane Spreading by Activating Rac1. Journal of Biological Chemistry 279, 14331–14337 [DOI] [PubMed] [Google Scholar]
- 12.Namekata K, Harada C, Taya C, Guo X, Kimura H, Parada LF, and Harada T (2010) Dock3 induces axonal outgrowth by stimulating membrane recruitment of the WAVE complex. Proceedings of the National Academy of Sciences 107, 7586–7591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helbig KL, Mroske C, Moorthy D, Sajan SA, and Velinov M (2017) Biallelic loss-of-function variants in DOCK3 cause muscle hypotonia, ataxia, and intellectual disability. Clin Genet 92, 430–433 [DOI] [PubMed] [Google Scholar]
- 14.Helbig KL, Mroske C, Moorthy D, Sajan SA, and Velinov M (2017) Biallelic loss-of-function variants in DOCK3 cause muscle hypotonia, ataxia, and intellectual disability. Clinical Genetics, n/a-n/a [DOI] [PubMed] [Google Scholar]
- 15.Iwata-Otsubo A, Ritter AL, Weckselbatt B, Ryan NR, Burgess D, Conlin LK, and Izumi K (2018) DOCK3-related neurodevelopmental syndrome: Biallelic intragenic deletion of DOCK3 in a boy with developmental delay and hypotonia. American Journal of Medical Genetics Part A 176, 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reid AL, Wang Y, Samani A, Hightower RM, Lopez MA, Gilbert SR, Ianov L, Crossman DK, Dell’Italia LJ, Millay DP, van Groen T, Halade GV, and Alexander MS (2020) “DOCK3 is a dosage-sensitive regulator of skeletal muscle and Duchenne muscular dystrophy-associated pathologies”. bioRxiv, 2020.2003.2027.010223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Q, Peto CA, Shelton GD, Mizisin A, Sawchenko PE, and Schubert D (2009) Loss of Modifier of Cell Adhesion Reveals a Pathway Leading to Axonal Degeneration. The Journal of Neuroscience 29, 118–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gharaibeh B, Lu A, Tebbets J, Zheng B, Feduska J, Crisan M, Peault B, Cummins J, and Huard J (2008) Isolation of a slowly adhering cell fraction containing stem cells from murine skeletal muscle by the preplate technique. Nat. Protocols 3, 1501–1509 [DOI] [PubMed] [Google Scholar]
- 19.Lim C-Y, Bi X, Wu D, Kim JB, Gunning PW, Hong W, and Han W (2015) Tropomodulin3 is a novel Akt2 effector regulating insulin-stimulated GLUT4 exocytosis through cortical actin remodeling. Nature Communications 6, 5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Somwar R, Kim DY, Sweeney G, Huang C, Niu W, Lador C, Ramlal T, and Klip A (2001) GLUT4 translocation precedes the stimulation of glucose uptake by insulin in muscle cells: potential activation of GLUT4 via p38 mitogen-activated protein kinase. Biochem J 359, 639–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reid AL, Wang Y, Samani A, Hightower RM, Lopez MA, Gilbert SR, Ianov L, Crossman DK, Dell’Italia LJ, Millay DP, van Groen T, Halade GV, and Alexander MS (2020) DOCK3 is a dosage-sensitive regulator of skeletal muscle and Duchenne muscular dystrophy-associated pathologies. Human Molecular Genetics 29, 2855–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mula J, Lee JD, Liu F, Yang L, and Peterson CA (2013) Automated image analysis of skeletal muscle fiber cross-sectional area. Journal of Applied Physiology 114, 148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, and Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nature Methods 9, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beedle AM (2016) Cryosectioning of Contiguous Regions of a Single Mouse Skeletal Muscle for Gene Expression and Histological Analyses. e55058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huntoon V, Widrick JJ, Sanchez C, Rosen SM, Kutchukian C, Cao S, Pierson CR, Liu X, Perrella MA, Beggs AH, Jacquemond V, and Agrawal PB (2018) SPEG-deficient skeletal muscles exhibit abnormal triad and defective calcium handling. Human Molecular Genetics 27, 1608–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Q, Khayat Z, Kishi K, Ebina Y, and Klip A (1998) GLUT4 translocation by insulin in intact muscle cells: detection by a fast and quantitative assay. FEBS Letters 427, 193–197 [DOI] [PubMed] [Google Scholar]
- 27.Baumann CA, Ribon V, Kanzaki M, Thurmond DC, Mora S, Shigematsu S, Bickel PE, Pessin JE, and Saltiel AR (2000) CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature 407, 202–207 [DOI] [PubMed] [Google Scholar]
- 28.Cane G, Leuchowius KJ, Söderberg O, Kamali-Moghaddam M, Jarvius M, Helbing I, Pardali K, Koos B, Ebai T, and Landegren U (2017) Chapter 12 - Protein Diagnostics by Proximity Ligation: Combining Multiple Recognition and DNA Amplification for Improved Protein Analyses. In Molecular Diagnostics (Third Edition) (Patrinos GP, ed) pp. 219–231, Academic Press [Google Scholar]
- 29.Bryan BA, Li D, Wu X, and Liu M (2005) The Rho family of small GTPases: crucial regulators of skeletal myogenesis. Cellular and Molecular Life Sciences CMLS 62, 1547–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiu TT, Jensen TE, Sylow L, Richter EA, and Klip A (2011) Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cell Signal 23, 1546–1554 [DOI] [PubMed] [Google Scholar]
- 31.Sylow L, Jensen TE, Kleinert M, Mouatt JR, Maarbjerg SJ, Jeppesen J, Prats C, Chiu TT, Boguslavsky S, Klip A, Schjerling P, and Richter EA (2013) Rac1 Is a Novel Regulator of Contraction-Stimulated Glucose Uptake in Skeletal Muscle. Diabetes 62, 1139–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raun SH, Ali M, Kjøbsted R, Møller LLV, Federspiel MA, Richter EA, Jensen TE, and Sylow L (2018) Rac1 muscle knockout exacerbates the detrimental effect of high-fat diet on insulin-stimulated muscle glucose uptake independently of Akt. The Journal of Physiology 596, 2283–2299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Mi X, Chen L, Jiang G, Wang N, Zhang Y, Deng W, Wang Z, Chen G, and Wang X (2016) Dock3 Participate in Epileptogenesis Through rac1 Pathway in Animal Models. Molecular Neurobiology 53, 2715–2725 [DOI] [PubMed] [Google Scholar]
- 34.Mandai K, Nakanishi H, Satoh A, Takahashi K, Satoh K, Nishioka H, Mizoguchi A, and Takai Y (1999) Ponsin/SH3P12: an l-afadin- and vinculin-binding protein localized at cell-cell and cell-matrix adherens junctions. J Cell Biol 144, 1001–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lesniewski LA, Hosch SE, Neels JG, de Luca C, Pashmforoush M, Lumeng CN, Chiang SH, Scadeng M, Saltiel AR, and Olefsky JM (2007) Bone marrow-specific Cap gene deletion protects against high-fat diet-induced insulin resistance. Nat Med 13, 455–462 [DOI] [PubMed] [Google Scholar]
- 36.Chang T-J, Wang W-C, Hsiung CA, He C-T, Lin M-W, Sheu WH-H, Chang Y-C, Quertermous T, Chen Y-DI, Rotter JI, Chuang L-M, Hwu C-M, Hung Y-J, Lee W-J, Lee IT, and The, S. S. G. (2018) Genetic variation of SORBS1 gene is associated with glucose homeostasis and age at onset of diabetes: A SAPPHIRe Cohort Study . Scientific Reports 8, 10574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the methods and/or supplementary material of this article.