

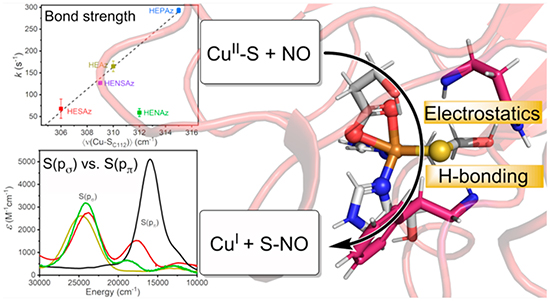
Abstract
Much progress has been made in understanding the roles of the secondary coordination sphere (SCS) in tuning redox potentials of metalloproteins. In contrast, the impact of SCS on reactivity is much less understood. A primary example is how copper proteins can promote S-nitrosylation (SNO), which is one of the most important dynamic post-translational modifications, and is crucial in regulating nitric oxide storage and transportation. Specifically, the factors that instill CuII with S-nitrosylating capabilities and modulate activity are not well understood. To address this issue, we investigated the influence of the primary and secondary coordination sphere on CuII-catalyzed S-nitrosylation by developing a series of azurin variants with varying catalytic capabilities. We have employed a multidimensional approach involving electronic absorption, S and Cu K-edge XAS, EPR, and resonance Raman spectroscopies together with QM/MM computational analysis to examine the relationships between structure and molecular mechanism in this reaction. Our findings have revealed that kinetic competency is correlated with three balancing factors, namely Cu–S bond strength, Cu spin localization, and relative S(ps) vs S(pp) contributions to the ground state. Together, these results support a reaction pathway that proceeds through the attack of the Cu–S bond rather than electrophilic addition to CuII or radical attack of SCys. The insights gained from this work provide not only a deeper understanding of SNO in biology but also a basis for designing artificial and tunable SNO enzymes to regulate NO and prevent diseases due to SNO dysregulation.
Graphical Abstract
1. INTRODUCTION
Metalloenzymes perform a stunning range of chemistry under limited environmental conditions conducive to life with a relatively small range of biologically available ligands and metallocofactors. The properties of these metalloenzymes are largely governed not only by the primary metal-coordinating ligand sphere (PCS), but often complex secondary coordination sphere (SCS) environments that provide weak non-covalent effects, such as electrostatic, hydrophobic, steric, and hydrogen bonding interactions. Artificial metalloenzymes, designed using both de novo and re-engineering approaches, have provided an excellent basis for systematically understanding how the SCS modulates the properties of metal centers.1–11 In particular, extensive investigations of the influence of the SCS on metal redox potentials have resulted in the ability to rationally and precisely tune redox potentials over wide ranges.12–17 However, understanding the impact of these effects on tuning the reactivity of metal centers still remains at the frontier of metalloenzyme design.
S-Nitrosylation (SNO) provides an ideal reaction for disentangling the correlations between reactivity and metal coordination sphere due to both the relative simplicity of the reaction itself and the consequences of its function (Scheme 1). S-Nitrosylation is a reversible post-translational protein modification that serves as a significant route for cellular NO signaling,18 controlling functions such as apoptosis,19 muscle contraction,20 and vasodilation.21 As a result, dysregulation of S-nitrosylation has been correlated with a plethora of diseases,22 including cancer initiation,23–29 Alzheimer’s disease,30–33 Parkinson’s syndrome,34–38 and cardiovascular disease.39–41 There is significant evidence that this process relies on metalloproteins to catalyze the S-nitrosylation of protein cysteine thiols, followed by transfer of NO by trans-S-nitrosylation to other acceptor proteins or low molecular weight thiols such as glutathione or coenzyme A.18,42 While S-nitrosylation has been shown to be catalyzed by CuII- and FeII-containing enzymes, such as ceruloplasmin and hemoglobin,18,43–45 thousands of potential S-nitrosylated proteins have been identified using proteomics methods,22,46–50 highlighting the potential role of less well-characterized metalloenzymes in the regulation or dysregulation of NO.
Scheme 1.
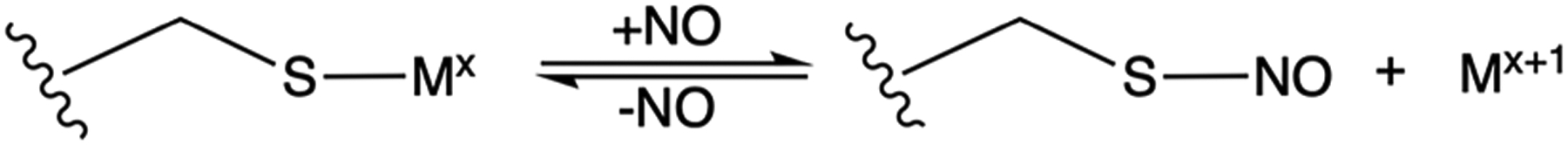
Metal Mediated S-Nitrosylation
Despite the importance of the function and the large number of metalloproteins involved in the function, little is known regarding the electronic and geometric factors that define the S-nitrosylating activity in metalloenzymes. To address this issue, we and others reported the generation of reversible CuII-catalyzed S-nitrosylation in an engineered azurin (Az).51 Wild-type azurin (WTAz) contains a blue copper site involving CuII coordinated to Cys112, His117, and His46 in a trigonal plane, with additional weak axial interactions with Met121 and the carbonyl of Gly45 at 3.3 and 3.0 Å, respectively (Figure 1). Although WTAz has been shown to interact with NO to form a photolabile {CuNO}10 species,52,53 S-nitrosylation has not been observed. However, mutations of His46 and Met121 to Glu and His, respectively, generated a red copper center, transforming the trigonal planar geometry of WTAz to a tetragonally distorted square planar geometry that closely modeled that in nitrosocyanin and catalyzed rapid formation of the CuI–SC112NO product. Further inclusion of an F114P mutation to M121H/H46E azurin (HEAz), which lowered the reduction potential of by ~107 mV at pH 8, resulted in a further increase in the reaction rate.51 Importantly, these studies demonstrated that a broader array of CuII enzymes outside of ceruloplasmin have the capability of catalyzing intramolecular S-nitrosylation.
Figure 1.
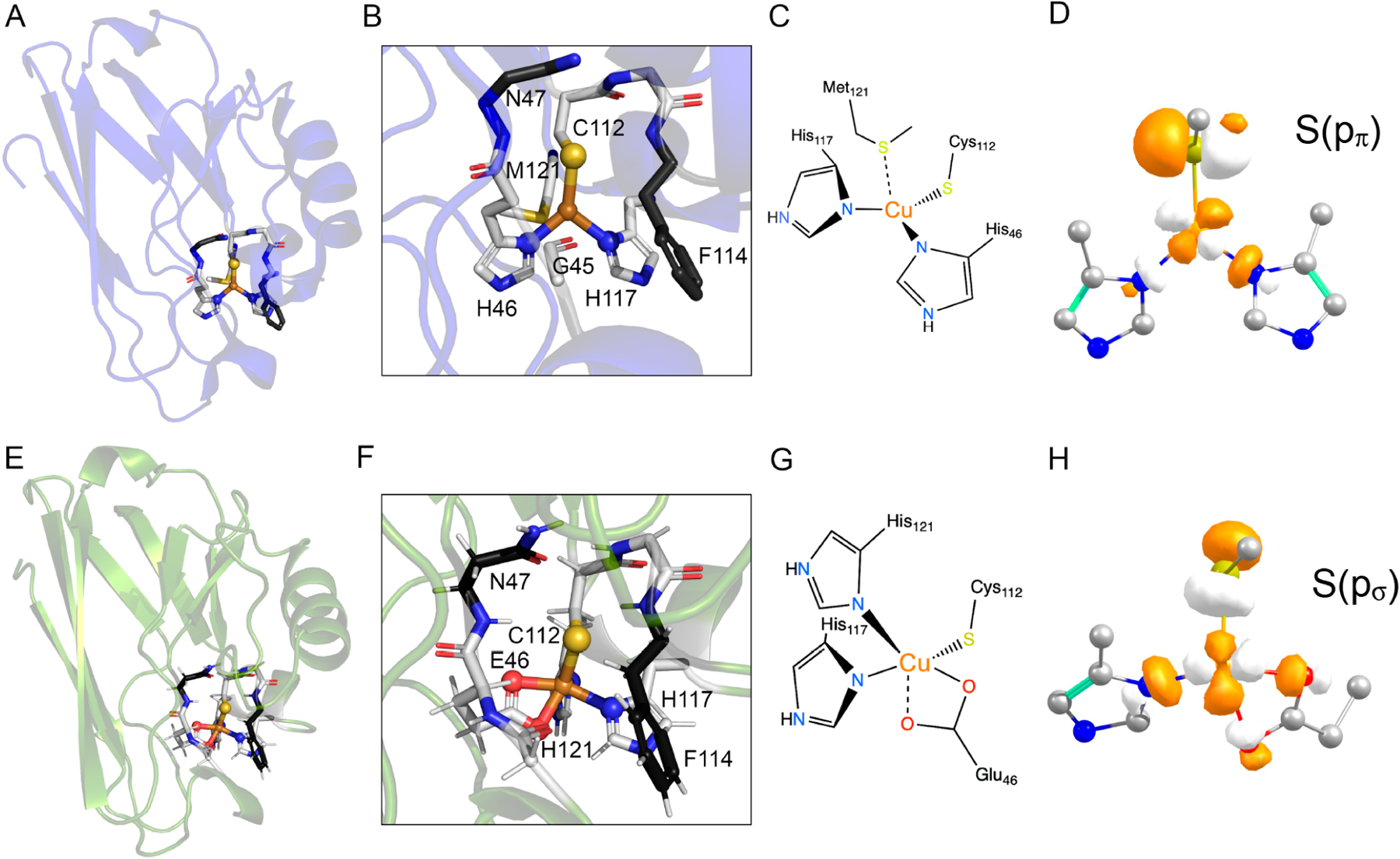
(A) The Crystal structure of reduced wild-type azurin (WTAz) (PDB ID: 1E5Y54). (B) Highlight of the primary coordination sphere of WTAz. (C) Schematic of the primary coordination environment of WT Az. (D) Isosurface of the β-LUMO (lowest unoccupied molecular orbital) of WTAz. (E) QM/MM optimized structure of M121H/H46E azurin (HEAz) based on the X-ray diffraction structure (PDB ID: 4WKX51). (F) Highlight of the primary coordination sphere generated by the 1Cys, 2His, and 1Glu binding motif in HEAz, and the secondary coordination sphere residues targeted in the present study. (G) Schematic of the primary coordination environment in HEAz. (H) Isosurface of the α-HOMO (highest occupied molecular orbital) of HEAz. In B and E, atoms are colored as follows: Cu (orange), S (yellow), N (blue), and O (red). Primary coordination sphere C atoms are shown in light gray, while targeted secondary coordination sphere C atoms are shown in dark gray. Calculations in D and H were calculated at the TPSSh/def2-TZVP level and plotted with isovalues of 0.05.
Despite successes in conferring SNO reactivity in re-engineered azurin, the factors that instill copper with S-nitrosylating capabilities and further modulate activity are not well understood. A detailed understanding of these factors will provide not only deeper insight into native SNO enzymatic function but also the further design of novel SNO enzymes to prevent SNO dysregulation underlying many diseases. Toward this goal, we present an investigation of the influence of the PCS and SCS on the CuII-promoted S-nitrosylation of cysteine in Az. We begin by looking at the influence of the relative positioning of Glu and His in the PCS of the CuII(His)2(Glu)-(Cys) binding motif, and further explore the influence of H-bonding and electrostatics in the SCS by varying residues at positions 47 and 114 for the most active PCS variant, HEAz (Figure 1F).1,14,51 To understand the underlying factors that modulate S-nitrosylating reactivity, we employ a multifaceted spectroscopic approach combined with DFT and ab initio-based quantum mechanics/molecular mechanics (QM/MM) calculations to characterize the electronic and geometric structures of HEAz and corresponding SCS variants. Together, these results highlight three factors, namely, S(ps)/S(pp) mixing, Cu–S bond strength, and CuII spin density, that interplay to modulate S-nitrosylating activity. Our findings further support that S-nitrosylation is promoted through the attack of the Cu–S bond itself, rather than electrophilic attack of CuII or radical attack of S. These studies have led to a conclusion that the apparent requirement of red copper for S-nitrosylation in the Az largely arises from steric effects, where attack of the Cu–S bond in blue copper is likely hindered due to the in-plane orientation of the Cu–S antibonding interaction.51,54
2. EXPERIMENTAL METHODS
2.1. Site Directed Mutagenesis, Protein Expression.
Plasmids containing the M121E (M121EAz), M121H/H46E (HEAz), M121H/H46E/N47S (HESAz), M121H/H46E/F114N (HENAz), M121H/H46E/F114N/N47S (HENSAz), and M121H/H46E/F114P (HEPAz) species were constructed by site-directed mutagenesis using wild-type azurin (pET9a) as a template for the quick-change polymerase chain reaction procedure. The mutations were confirmed by sequencing. Proteins were expressed in BL21(DE3) E. coli (Novagen, Madison, WI). Cultures were grown in LB media for 8 h at 37 °C and used to inoculate flasks containing 1.5 L of 2x YT media containing 50 mg/L Kanamycin (held in a 4 L flask), which were further grown at 25 °C while shaking at 210 rpm. Further details regarding cell harvest and protein purification are provided in the SI.
2.2. Kinetic Assessment.
Experiments were performed as previously described for HEPAz.51 Measurements were performed using an Applied Photophysics Ltd. (Leatherhead, U.K.) SX18.MV stopped-flow spectrometer equipped with a 256-element photodiode array detector. The instrument was prepared for anaerobic measurements by rinsing the lines several times with a degassed buffer solution.
50 mM MES buffer (pH 6.0) was used for all experiments. All solutions of CuII-Az (M121EAz, HEAz, HESAz, HENAz, HENSAz, HEPAz) were degassed and equilibrated with Ar using a Schlenk line. Two-syringe mixing was employed to mix equal volumes of 50 μM CuII-Az and NO saturated (2 mM) buffer solutions to obtain the pseudo first-order rate kinetics.
2.3. UV/vis Spectroscopy.
UV/vis spectroscopy was performed by using a Cary 60 spectrometer. Samples were prepared at approximately 100 μM [Az] based on ε280 = 8000 M−1 cm−1, and metalated by the slow addition of subsequent 0.1 equiv of CuIISO4 until 1 equiv was reached. Samples were passed through a PD10 column and reconcentrated to approximately 100 μM [CuII-Az]. Quantification of CuII in UV/vis samples was performed by spin integration via X-band EPR at 85 K. These concentrations were further used to determine the spectral molar extinction coefficients.
2.4. Resonance Raman Spectroscopy.
Resonance Raman samples were excited at 379.5 nm using continuous wave excitation from an Ar+ ion (Innova Sabre 25/7) laser. The samples were immersed in a liquid nitrogen cooled (77 K) EPR finger dewar (Wilmad). Data were recorded for 10 min with an incident power of ~5 mW at the sample. To minimize photodecomposition, samples were manually rotated (~10 rotations/min) during the measurement. The spectra were recorded using a Spex 1877 CP triple monochromator with 1200, 1800, and 2400 grooves/mm holographic spectrograph gratings and detected by an Andor Newton CCD cooled to −80 °C. Resonance Raman data were processed using SpectraGryph software, version 1.2 (Dr. Friedrich Menges SoftwareEntwicklung, Oberstdorf, Germany).
2.5. Electron Paramagnetic Resonance Spectroscopy.
Continuous wave (CW) electron paramagnetic resonance (EPR) measurements were performed at X-band frequency (~9.65 GHz) using a Bruker EMXplus spectrometer equipped with a Bruker high-sensitivity cavity (ER4119HS) and a nitrogen-flow cryostat (ESR 900). A high-sensitivity Bruker Premium-X bridge with an integrated microwave frequency counter was employed along with a magnetic field controller (ER083CS) that was calibrated using a Bruker NMR field probe (ER035M). All measurements utilized a 4 G modulation amplitude with a 100 kHz modulation frequency, an 80.92 ms time constant, and a 1 mW microwave power. All spectra were recorded on frozen solutions held at 85 K. All spectra were processed and analyzed using the software package EasySpin (version 6.0.0),55 as implemented in Matlab (version R2022b). Spin quantification of samples presented for UV/vis measurements was performed using a 1 mM CuSO4 standard.
2.6. X-ray Absorption Spectroscopy.
S K-edge X-ray absorption spectroscopy (XAS) measurements were acquired at wiggler side-state beamline 4–3 of the Stanford Synchrotron Radiation Lightsource (SSRL). The SPEAR storage ring operated at 3 GeV in top-off mode with an ~500 mA ring current. A liquid N2-cooled double-crystal monochromator with Si(111) crystals at ϕ = 0° was used to select the incoming X-ray energy with an intrinsic resolution (ΔE/E) of ~1.4 × 10−4. The X-ray beam size was 1 × 1.8 mm2 (v × h) at the sample position. Samples were maintained at cryogenic temperatures using a liquid He flow cryostream at the SSRL to minimize radiation damage and maintain an inert sample environment. Fluorescence measurements were recorded using a 7-element solid-state silicon drift detector. Prior to measurements, each sample was checked for signs of radiation damage by performing subsequent 1.5 min scans over the same sample spot. These tests showed significant sensitivity to irradiation at the S K-edge (~2472 eV), and required detuning of the monochromator to ~2% of the initial flux in order to enable X-ray irradiation times of up to 5 min. This rate of damage was consistent across all measured protein samples in the present study. All S K-edge XAS scans were collected by scanning the incident energy from 2400 to 2761 eV using regional scanning: 2400–2460 eV (10 eV step, 0.5 s/pt), 2460–2465 eV (1 eV step, 0.5 s/pt), 2465–2475 eV (0.1 eV step, 1 s/pt), 2475–2761 eV (k = 0.05 Å−1 step, 0.5 s/pt). Energy calibration was performed by intermittent measurement of a Na2S2O3·5H2O standard with the first inflection point set to 2472 eV.
Cu K-edge XAS measurements were acquired at beamline 7–3 of SSRL. A liquid N2-cooled double-crystal monochromator with Si(220) crystals at ϕ = 0° was used to select the incoming X-ray energy with an intrinsic resolution (ΔE/E) of ~0.6 × 10−4. The X-ray beam size was 1 × 6 mm2 (v × h) at the sample position. Samples were maintained at cryogenic temperatures using a liquid He flow cryostat at SSRL (~20 K) to minimize radiation damage and maintain an inert sample environment. Fluorescence measurements were recorded using a Canberra 30-element Ge monolith solid-state detector. Prior to measurements, each sample was checked for signs of radiation damage by performing subsequent 1.5 min scans over the same sample spot. Similar to S K-edge measurements and other measurements of CuII, the pre-edge feature of measured samples showed very high sensitivity to photoreduction. To enable beam stability for up to 13 min (required for extended X-ray absorption fine structure [EXAFS] scans), beam flux was reduced by 99.5% by a combination of monochromator detuning and Al filters. Cu XAS scans focused on the X-ray absorption near edge spectral region (XANES) were collected by scanning the incident energy from 8800 to 9543 eV, while scans focused on the EXAFS from 8800 to 9904 eV and calibrated by simultaneous measurement of a Cu foil, with the first inflection point set to 8989 eV.
For both S K-edge and Cu K-edge measurements, individual scans were normalized to the incident photon flux, energy calibrated, and merged using the program Larch.56 For S K-edge spectra, background subtraction and normalization were performed using a linear regression for the pre-edge region from 2460 to 2466 eV, and a quadratic polynomial regression for the postedge region from 2482 to 2572 eV. For Cu K-edge spectra, background subtraction and normalization were performed using a linear regression for the postedge region from 8840 to 8960 eV, and a quadratic polynomial regression for the postedge region from 9040 to 9904 eV. Cu K-edge XAS spectra were splined from k = 0–15 Å−1 using an R-background of 1.0 and k-weight of 2. EXAFS fitting was performed using FEFF 8.057–59 via the software package Larch.56 Further details regarding EXAFS data analysis are provided in SI.
2.7. QM/MM Calculations.
QM/MM models of each red copper azurin protein were performed based on the available crystal structure of M121H/H46E Az (PDB ID: 4KWX).51 All QM/MM calculations were performed using ASH,60 which provides an interface for the QM program ORCA,61,62 version 5.0.3, with the MM program OpenMM.63 Details regarding model setup, choice of QM space, computational methodology, and additional ab initio calculations are provided in SI.
3. RESULTS
3.1. UV/vis Spectroscopy and Measuring NO Reactivity.
The absorption spectrum of WTAz is dominated by a feature at ~16 000 cm−1 that has previously been assigned to the ligand-to-metal charge-transfer (LMCT) transition (Figure 2A).64 The intensity of this transition increases with increasing antibonding S(pπ) character (Figure 1D) in the singly occupied molecular orbital (SOMO), . The replacement of Met121 at the axial position with Glu (M121EAz) provides an additional PCS ligating residue65 and results in a decrease in intensity and increase in energy of the transition to 17700 cm−1, as well as the appearance of a second feature arising from the LMCT at ~24 000 cm−1, consistent with previous observations.66 The interaction is visualized in Figure 1H. Similar to the LMCT, the LMCT gains intensity through increased S(pσ) character in the SOMO.67 By further effectively “rotating” the position of Glu relative to the Cu–S bond by M121H/H46E substitutions (Figure 2A), we observe both an increase in energy of the to 24 600 cm−1, and near complete elimination of the .
Figure 2.
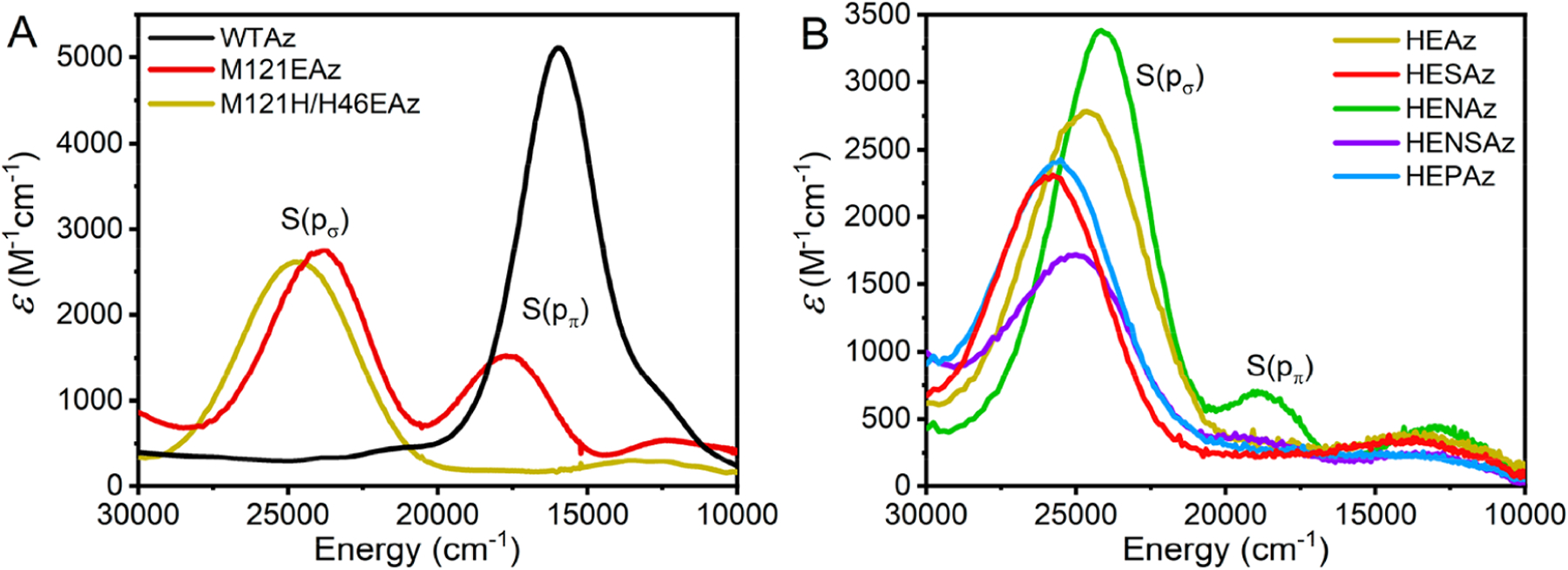
(A) Comparison of the UV–vis spectra of WTAz with those of Glu-substituted M121EAz and M121H/H46EAz (HEAz). (B) Comparison of UV/vis spectra of HEAz and corresponding secondary coordination sphere mutations. Extinction coefficients were determined by X-band EPR-based spin quantification. All variants are CuII-coordinated.
Reaction of either M121EAz or M121H/H46EAz (HEAz) with a stoichiometric excess of NO leads to a significant decrease in the intensities of both and features, indicative of the reduction of CuII → CuI. Simultaneously, a new feature arises at 29 700 cm−1, which has previously been assigned to the formation of a S-nitrosyl species (Figure S2).51,68 The assignment of this feature to S-nitrosyl formation is further supported by mass-spectrometric measurements, which display a clear +29 Da feature above the anticipated protein mass (Figure S3), from the formation of S-nitrosyl species. In the presence of 3 equiv of NO (provided by 2 equiv of DEA-NONOate), S-nitrosylation by M121EAz reaches 40%, while HEAz proceeds by 95%. Additionally, M121EAz reacts 6 orders of magnitude slower than HEAz under pseudo first-order rate conditions (Figure S4, Table 1).
Table 1.
Reaction Rates and Corresponding Estimates of ΔG‡ for M121EAz and HEAz Variants at 20 °C
| k (s−1) | ΔG‡ (kcal/mol) | |
|---|---|---|
| M121EAz | 10 ± 3 × 10−4 | 17.6 |
| HEAz (M121H/H46E) | 165 ± 12 | 10.0 |
| HESAz (M121H/H46E/N47S) | 68 ± 22 | 10.5 |
| HENAz (M121H/H46E/F114N) | 59 ± 9 | 10.6 |
| HENSAz (M121H/H46E/F114N/N47S) | 127 ± 2 | 10.2 |
| HEPAz (M121H/H46E/F114P) | 293 ± 5 | 9.7 |
Due to the much higher reactivity toward S-nitrosylation observed for HEAz, we further explored the influence of the SCS on the S-nitrosylating capabilities of this variant. UV/vis spectra of variants containing additional N47S (HESAz), F114N (HENAz), F114N/N47S (HENSAz), and F114P (HEPAz) mutations are presented in Figure 2B, with energetic positions and extinction coefficients provided in Table S1. For all of these HEAz-based variants, the LMCT feature remains dominant; however, a significant feature is observed at ~18 900 cm−1 for HENAz. This feature also appears in HENSAz ~19 200 cm−1, although to a much lesser extent.
To better understand how the PCS and SCS modulate Cu-catalyzed S-nitrosylation, the pseudo first-order reaction rates were determined for the HEAz, HESAz, HENAz, HENSAz, and HEPAz variants by measuring A415 (24 100 cm–1) changes over time using stopped-flow UV/vis spectroscopy (Figure S4). The pseudo first-order reaction rate of HEPAz (293 ± 5 s−1) is consistent with our previous observations.51 HEAz appears somewhat slower, with a rate of 165 ± 12 s−1, followed by HENSAz (127 ± 2 s−1), HESAz (68 ± 22 s−1), and HENAz (59 ± 9 s−1). Using the Arrhenius equation, a variation of ~1 kcal mol−1 in ΔG‡ between the slowest (HENAz) and fastest (HEPAz) variants was found. Assuming an invariant preexponential factor A, ΔG‡ was estimated for each variant based on the previously determined ΔG‡ of HEPAz of 9.7 kcal/mol (Table 1).51
3.2. Investigating the Cu–S Covalent Interaction.
To better understand the underlying factors contributing to the S-nitrosylating activity observed between PCS and SCS Az variants containing the CuII-(His)2(Cys)(Glu) binding motif, we first looked to characterize the S and Cu character of the SOMO. To do so, two complementing approaches were taken. Sulfur K-edge XAS provides one of the most direct experimental probes of the covalency of the Cu–S interaction.69 A pre-edge feature of ~2469 eV is commonly observed in S-coordinated metals with partially occupied valence shells. The pre-edge intensity reflects the S(3p) character of the ground state wave function, providing a direct probe of the covalency of the metal–sulfur bond. Meanwhile, CW EPR can provide insight into metal–ligand covalency, where the CuII character in the SOMO can be derived from the parallel hyperfine interaction (A||) of the single unpaired electron with the I = 3/2 Cu nucleus.
3.2.1. S K-Edge XAS.
The S K-edge XAS of M121EAz and HEAz variants is displayed in Figure 3 (pre-edge region) and Figure S5 (full scale edge). The pre-edge feature observed between 2469.5 and 2570.0 eV arises from the S(1s) → ψ*(SOMO) transition, where the predominant , character of the SOMO makes this a formal LMCT-type transition. The energy of this feature depends on the relative energy of the SOMO to the S(1s), and the intensity is directly dependent on the degree of Sp mixing in the ground state wave function. M121EAz, HEAz, and HENAz appear nearly identical in pre-edge intensity and energetic position. Meanwhile, HESAz, HENSAz, and HEPAz all display a significant decrease in pre-edge intensity accompanied by an increase in energy to ~2469.7 eV, again consistent with the UV/vis spectroscopic observations shown in Figure 2.
Figure 3.
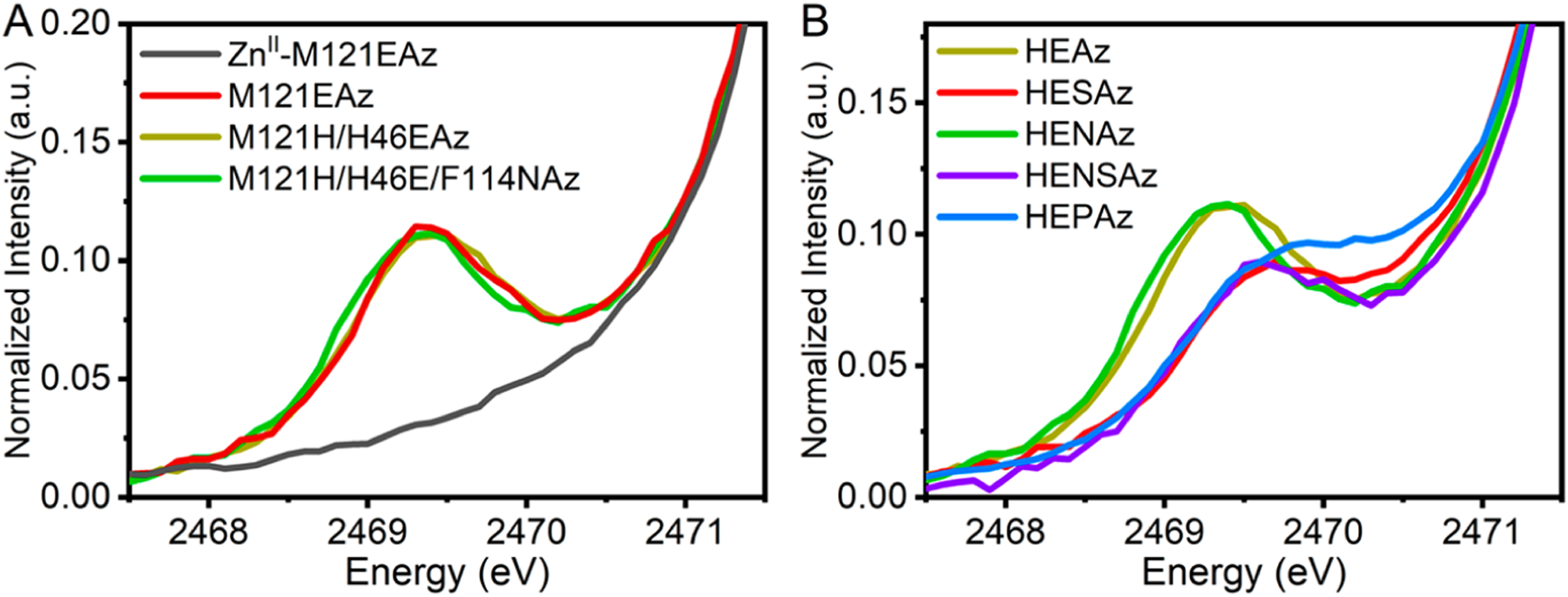
Comparison of the S K-edge pre-edge features observed among the (A) CuII-coordinated M121EAz, HEAz, and HENAz variants (ZnII-coordinated M121EAz which lacks the pre-edge feature is included as a background) and (B) the HEAz SCS variants.
To quantify the relative covalency of the Cu–S interaction, the pre-edge spectra were simulated by using a single Gaussian band combined with a background spline to compensate for the rising S(1s) → S(4p) edge. The S(p) contribution was determined by integrated intensity, using WTAz as a reference with 45 ± 3% Sp character.14 The Sp character for each variant is summarized in Table 2 based on the integrated intensity of the respective pre-edge features. Significantly lower Sp character is observed for all variants relative to reports for WTAz (~45%14), consistent with previous observations for the red copper proteins nitrosocyanin and BSco.70,71 The Sp character varies considerably among the HEAz variants, ranging from as low as 8% in HESAz to as high as 19% for HEAz and HENAz. We note that despite large differences in S-nitrosylating activity and S(pσ)/S(pπ) contributions in the UV–vis spectra, HEAz, HENAz, and M121EAz appear very similar in terms of Cu–S covalency.
Table 2.
Summary of S K-edge XAS Pre-edge Fitting
| Variant | Pre-edge Energy (eV) | Covalency (% Sp) |
|---|---|---|
| M121EAz | 2469.3 | 20 ± 1 |
| HEAz | 2469.4 | 19 ± 1 |
| HESAz | 2469.7 | 8 ± 1 |
| HENAz | 2469.4 | 19 ± 1 |
| HENSAz | 2469.6 | 15 ± 1 |
| HEPAz | 2469.8 | 12 ± 1 |
3.2.2. EPR Spectroscopy.
While S K-edge provides a direct measure of Sp contributions to the ground state wave function, determination of the degree of spin localization at CuII and other ligands requires an alternative approach. To further probe the electronic ground state, CW X-band EPR measurements were performed, as shown in Figure 4. A summary of the fit spin Hamiltonian parameters is provided in Table 3. These data reveal similar axial (g|| > g⊥ > ge) signals for M121EAz and the HEAz variants, consistent with a SOMO in the ground state. Values of g|| ≈ 2.24 and a relatively large A|| are indicative of a distorted-square-planar geometry at CuII in the M121EAz and HEAz variants. The larger values of g|| for HESAz, HEPAz, and HENSAz are also consistent with the reduced %Sp contribution to the SOMO.
Figure 4.
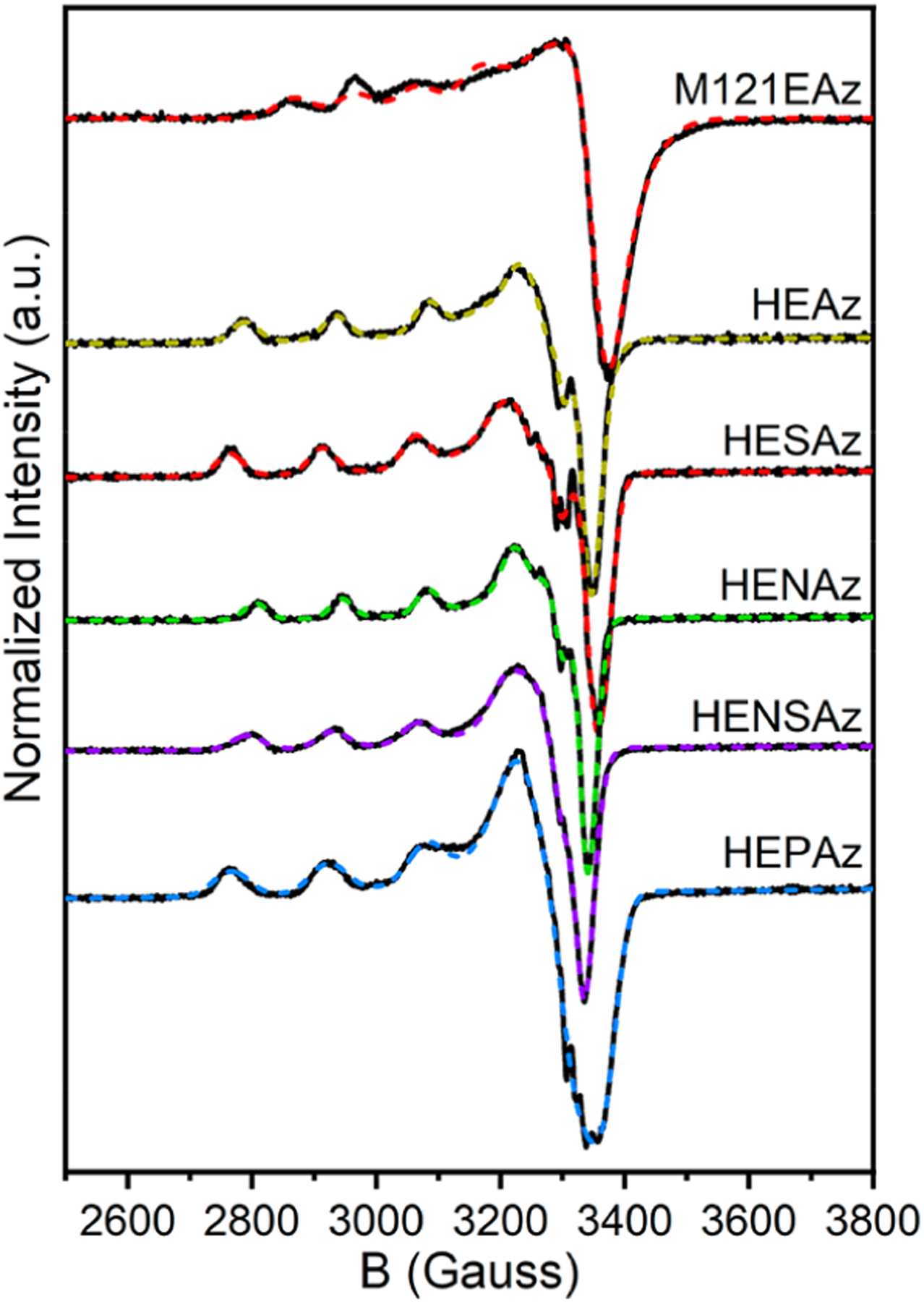
CW X-band EPR spectra of M121EAz and HEAz SCS variants. Experimental spectra are provided as solid black lines, and simulated spectra using the spin-Hamiltonian parameters summarized in Table 3 are displayed as dashed, colored lines. Spectra were collected at 85 K using 1 mW power.
Table 3.
Summary of Fitting Parameters for X-Band EPR Spectra of HEAz Variantsa
| Variant | g | A (MHz) | Aiso(MHz) | α 2 |
|---|---|---|---|---|
| WTAz | [2.262, 2.056, 2.042] | [−177, 33, 33] | −37 | 0.49 |
| M121EAz | [2.285, 2.061, 2.059] | [−307, 84, 10] | −71 | 0.65 |
| HEAz (M121H/H46E) | [2.239, 2.062, 2.034] | [−462, 95, 70] | −99 | 0.74 |
| HESAz (M121H/H46E/N47S) | [2.250, 2.072, 2.025] | [−458, 70, 60] | −109 | 0.75 |
| HENAz (M121H/H46E/F114N) | [2.232, 2.058, 2.040] | [−414, 83, 31] | −101 | 0.69 |
| HENSAz (M121H/H46E/F114N/N47S) | [2.244, 2.057, 2.045] | [−420, 70, 28] | −107 | 0.71 |
| HEPAz (M121H/H46E/F114P) | [2.252, 2.056, 2.030] | [−482, 70, 51] | −120 | 0.78 |
α2 corresponds to the fractional metal character calculated by using κ = 0.30 for all variants. The assignment of signs was based on the assumption of a -dominated SOMO, where the dipolar hyperfine contribution (z, y, x) = (−4/7, 2/7, 2/7)Pd. Values for g and A for WTAz are reproduced from ref 14.
The CuII hyperfine coupling A|| can be calculated from the equation:72
| (1) |
where Pd = 396 × 10−4 cm−1 and κ is proportional to the Fermi contact term Aiso (typically 0.23–0.32 for CuII). Importantly, this equation can be rearranged to estimate α2, the percent character in the ground state wave function, using the experimentally determined values of A||, g||, and g⊥. Both κ and α2 are dependent on the covalency of Cu with the surrounding coordination environment; however, given the relatively low variation of Aiso, we can assume that κ does not vary significantly and further determine the relative differences in α2 for each HEAz variant. Using κ = 0.30, the fractional spin residing at CuII varies between 0.66 (HENAz) and 0.80 (HEPAz).
3.3. Characterization of the CuII Binding Environment.
To further understand the influence of the SCS in modulating the strength of the Cu–S interaction and S-nitrosylating activity in the HEAz series, Cu K-edge XAS and resonance Raman spectroscopies were employed to probe the primary coordination sphere of CuII. The Cu K-edge XANES region provides information regarding local CuII symmetry, while the EXAFS region may be simulated to yield structural insight regarding neighboring scattering atoms. Furthermore, resonance Raman spectroscopy provides a selective probe of the Cu–S vibrational modes. Combining the information gained from EXAFS and resonance Raman spectroscopy provides direct information regarding the strength of the Cu–S bond.
3.3.1. Cu K-Edge XAS.
The Cu K-edge XANES of the M121H/H46EAz variants is provided in Figure 5. The pre-edge feature appearing at ~8977.5 eV arises from the formally parity forbidden Cu(1s) → Cu(3dx2‑y2) transition. Given the constant CuII oxidation state between all samples, greater intensities are anticipated to arise through lowering of the local symmetry environment, enabling increased Cu 3d/4p mixing. HENAz displays the most intense pre-edge feature, implying an increased distortion in geometry, while HEAz, HESAz, and HENSAz all appear comparable. Meanwhile, reduced intensity is observed in HEPAz, implying greater local symmetry.
Figure 5.
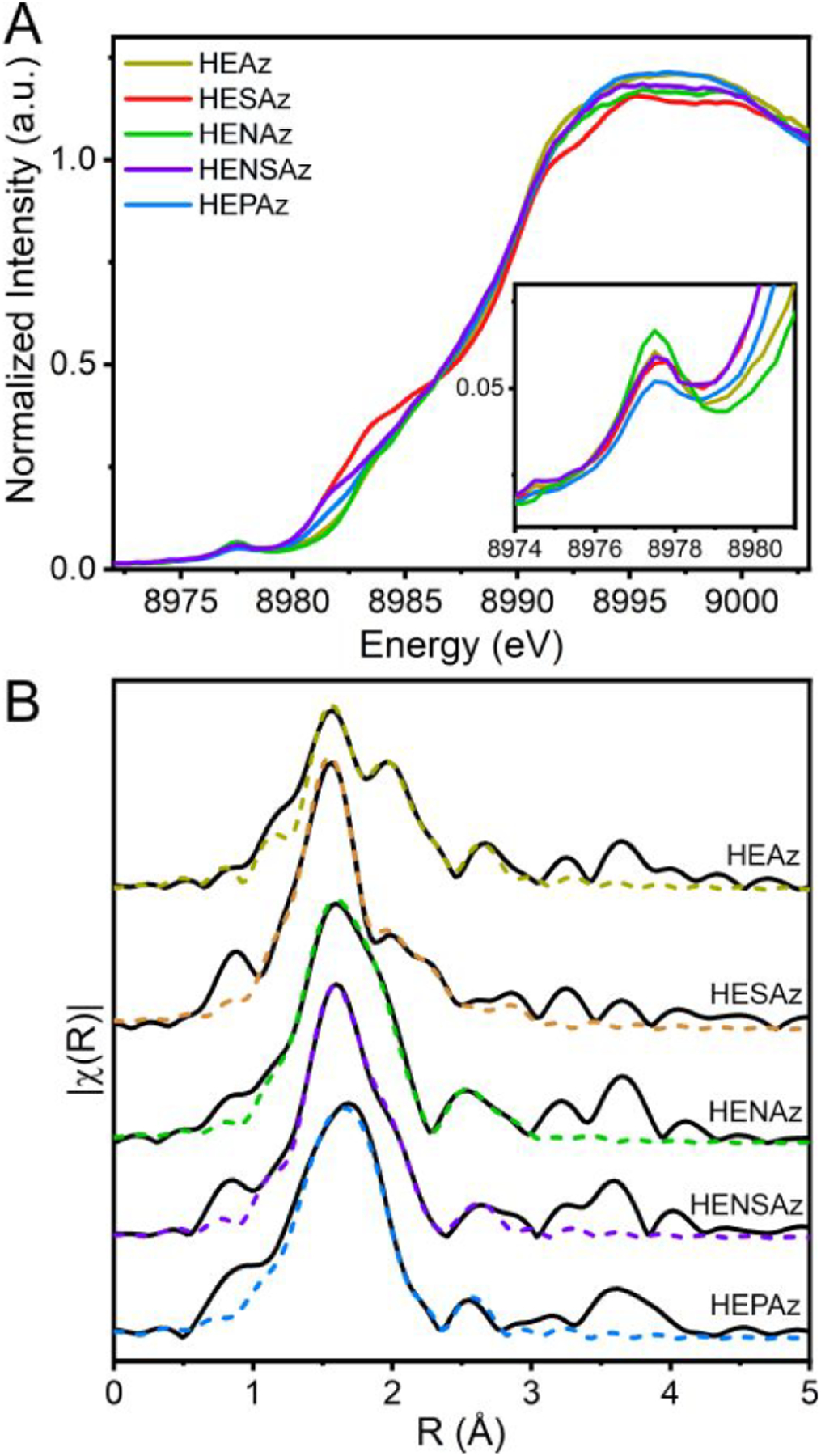
(A) Cu K-edge XANES of HEAz variants. (B) Comparison of Cu K-edge EXAFS spectra of HEAz variants as indicated by name. Fits corresponding to the parameters provided in Table 4 are shown as dashed colored lines. Spectra are k3-weighted, and FTs for all spectra were performed across a k-range of 2–13 Å–1. No phase shift has been applied.
In addition to a pre-edge feature, CuII can present “shakedown” transitions, corresponding to an effective Cu(1s) → Cu(4p) + LMCT excited state.73 These arise from the increase in Zeff resulting from the 1s core-hole bond, shifting the relative energy of the Cu(3d) shell below that of ligand p-orbitals. As a result, a final state containing both Cu(4p) and LMCT character is produced. Several features appear in the rising edge between 8979 and 8987 eV in HENAz, HEAz, HEPAz, HENSAz, and HESAz (with increasing intensity in that order); a band deconvolution analysis of this region is provided in Figure S6 and Table S2. Previous studies have suggested that decreased shakedown intensity may be indicative of increased covalency of the CuII site.74,75 In the present study, there is a good agreement between our S K-edge measurements and the integrated intensities of the observed shakedown transitions across the rising edge region of the Cu K-edge XAS of the HEAz variants with HENAz < HEAz < HENSAz < HESAz. Interestingly, HEPAz displays a lower shakedown intensity than anticipated, between HEAz and HENSAz; this may arise from a change in relative z vs x,y-polarized contributions to the Cu(1s) → Cu(4p) main and shakedown transitions resulting from a geometric change at Cu, as hinted by the decreased pre-edge intensity observed for HEPAz.
Cu K-edge EXAFS spectra were collected to further aid in structurally characterizing the PCS of the HEAz variants. In the Fourier-transformed R-space (Figure 5B), all spectra are dominated by a broad primary feature at R ≈ 1.55 Å, with a higher R feature shoulder ~1.95 Å. Weaker, broad scattering features are also found at R ≈ 2.55, 3.25, and 3.65 Å. The primary manifold changes significantly between different mutants, indicating multiple overlapping scattering paths.
Based on the previously determined crystal structure of HEAz (PDB 4WKX),51 the copper is coordinated by sulfur (from C112), two nitrogens (Nδ of H117 and H121), and one oxygen (E46). Unlike nitrosocyanin, the copper-bound H2O is not observed.76 Although the shortest scattering Cu-ligand distances reported for the HEAz crystal structure are 2.10 and 2.12 Å (Cu–Nδ(H117) and Cu–O(E46), respectively), our initial phase and amplitude parameters for HEAz determined using FEFF-calculated scattering paths revealed that the lowest R scattering paths appear at distances of ≤2.0 Å. Previous comparisons of crystallographic, EXAFS, and QM/MM calculations of WTAz have demonstrated that the exact geometry of the CuII-coordinating site varies significantly depending on methodology,77 which may be indicative of partial (or complete) photoreduction to CuI in reported crystallographic structures. Here, we find that QM/MM optimization of the CuI state of HEAz appears to be consistent with the reported crystal structure (see Figure SR-1 of the SI). Therefore, the FEFF calculations used to model the scattering paths of the HEAz variants were based on QM/MM optimized geometries for the CuII state. In these calculations, unique single scattering paths include sulfur (from C112, 2.26–2.32 Å), two nitrogens (Nδ of H117 and H121 at 1.92–1.95 and 2.05–2.23 Å), and two oxygens (E46, one at 2.02–2.08 Å and the second at 2.23–2.48 Å). Importantly, compared to the CuI-QM/MM and crystal structures of HEAz, these calculations show a contraction of the CuII–N distances and a change in denticity of E46, increasing the total coordination number of CuII to five. Additionally, a “short” Cu–C scatterer (Cu–C1) is calculated, corresponding to the Cδ of E46 (2.42 Å), as well as longer scatterers from Cε and Cγ of the more tightly coordinated His117. To minimize the number of required model parameters, the four unique light atom (O/N) scatterers were reduced to two groups with a total degeneracy of 4, this degeneracy was varied between the two paths in whole units. The least-squares refined EXAFS model parameters for the HEAz variants are summarized in Table 4. Individual contributing scattering paths are provided in Figures S7 and S8, while complementing fitting schemes are presented in SI, Table S3.
Table 4.
Summary of Representative Cu K-Edge EXAFS Fitting Parameters of the HEAz Variantsa
| Variant | Scattering Path | N | R (Å) | σ2 (10—3 Å2) | E0 (eV) | Red. X2 | R-factor |
|---|---|---|---|---|---|---|---|
| HEAz | Cu–(O/N)1 | 2 | 2.00 ± 0.02 | 2.1 ± 0.7 | 8994 ± 3 | 68 | 0.013 |
| (M121H/H46E) | Cu–(O/N)2 | 2 | 2.20 ± 0.05 | 1.5 ± 0.5 | |||
| Cu–S | 1 | 2.27 ± 0.01 | 2.3 ± 1.0 | ||||
| Cu–C1 | 1 | 2.59 ± 0.03 | 1.5 ± 0.3 | ||||
| Cu–C2 | 2 | 3.00 ± 0.03 | 5.9 ± 3.0 | ||||
| HESAz | Cu–(O/N)1 | 3 | 1.97 ± 0.01 | 6.0 ± 0.8 | 8991 ± 2 | 103 | 0.011 |
| (M121H/H46E/N47S) | Cu–(O/N)2 | 1 | 2.49 ± 0.07 | 10.0 ± 3.0 | |||
| Cu–S | 1 | 2.30 ± 0.01 | 9.0 ± 2.0 | ||||
| Cu–C1 | 1 | 2.51 ± 0.02 | 1.5 ± 0.1 | ||||
| Cu–C2 | 2 | 2.95 ± 0.03 | 4.1 ± 0.6 | ||||
| HENAz | Cu–(O/N)1 | 3 | 1.99 ± 0.01 | 6.7 ± 0.8 | 8992 ± 1 | 100 | 0.008 |
| (M121H/H46E/F114N) | Cu–(O/N)2 | 1 | 2.44 ± 0.01 | 1.8 ± 1.2 | |||
| Cu–S | 1 | 2.26 ± 0.01 | 4.7 ± 1.0 | ||||
| Cu–C1 | 1 | 2.64 ± 0.03 | 1.5 ± 0.3 | ||||
| Cu–C2 | 2 | 2.99 ± 0.02 | 3.0 ± 0.2 | ||||
| HENSAz | Cu–(O/N)1 | 3 | 1.98 ± 0.01 | 3.1 ± 0.6 | 8993 ± 1 | 99 | 0.008 |
| (M121H/H46E/F114N/N47S) | Cu–(O/N)2 | 1 | 2.55 ± 0.06 | 10.0 ± 1.0 | |||
| Cu–S | 1 | 2.28 ± 0.01 | 3.8 ± 1.0 | ||||
| Cu–C1 | 1 | 2.46 ± 0.07 | 10.0 ± 2.0 | ||||
| Cu–C2 | 2 | 2.97 ± 0.02 | 3.0 ± 1.0 | ||||
| HEPAz | Cu–(O/N)1 | 3 | 1.99 ± 0.01 | 8.7 ± 0.8 | 8990 ± 2 | 111 | 0.009 |
| (M121H/H46E/F114P) | Cu–(O/N)2 | 1 | 2.38 ± 0.04 | 4.3 ± 1.0 | |||
| Cu–S | 1 | 2.24 ± 0.01 | 5.0 ± 2.0 | ||||
| Cu–C1 | 1 | 2.68 ± 0.01 | 10.0 ± 2.0 | ||||
| Cu–C2 | 2 | 2.96 ± 0.03 | 6.7 ± 1.0 |
Plots of individual scattering path contributions are provided in Figure S6.
The best models of the HEAz variants HESAz, HENAz, HENSAz, and HEPAz support five-scattering paths with degeneracies of 3 in the shorter Cu–(O/N)1 scattering path (1.97–1.99 Å), and 1 in the longer Cu–(O/N)2 path, which varies from 2.35 to 2.55 Å. The observation of a 3-fold degenerate Cu–N/O scatterer at <2 Å is consistent with previous EXAFS models for both oxidized nitrosocyanin and HEPAz.51,70 The Cu K-edge EXAFS of HEAz is best fit using a degeneracy of 2 in each Cu–(O/N)1 and Cu–(O/N)2 scattering pathsuse of degeneracies 3 and 1, respectively, leads to a drastically poorer fit. This poorer fit may be indicative of a stronger bidentate interaction of CuII with Glu46.
3.3.2. Resonance Raman Spectroscopy.
To further investigate the Cu–SC112 interaction, resonance Raman spectroscopy was performed for the HEAz variants by excitation into the intense band at 415 nm. Resulting spectra are listed in Figure 6. While blue copper proteins, including variants of WTAz, typically display resonance enhanced features in the 325–475 cm–1 region, with ν(Cu–SCys) assigned ~400 cm−1,14,78–90 several enhanced resonance enhanced features of the HEAz variants arise in the 265–365 cm−1 region, consistent with previous reports of resonance Raman spectra for red copper proteins nitrosocyanin and BSco.70,71 As all of these features contain some CuII–SC112 stretching character, the Cu–S bond strength can be determined from the intensity-weighted average energy:89
| (2) |
where Ii and vi are the intensity and energy of the ith mode. The resulting intensity-weighted 〈ν(Cu–SC112)〉 for variants of HEAz is summarized in Table 5. HESAz exhibits the lowest energy at 306 cm−1, and HEPAz exhibits the highest at 315 cm−1.
Figure 6.
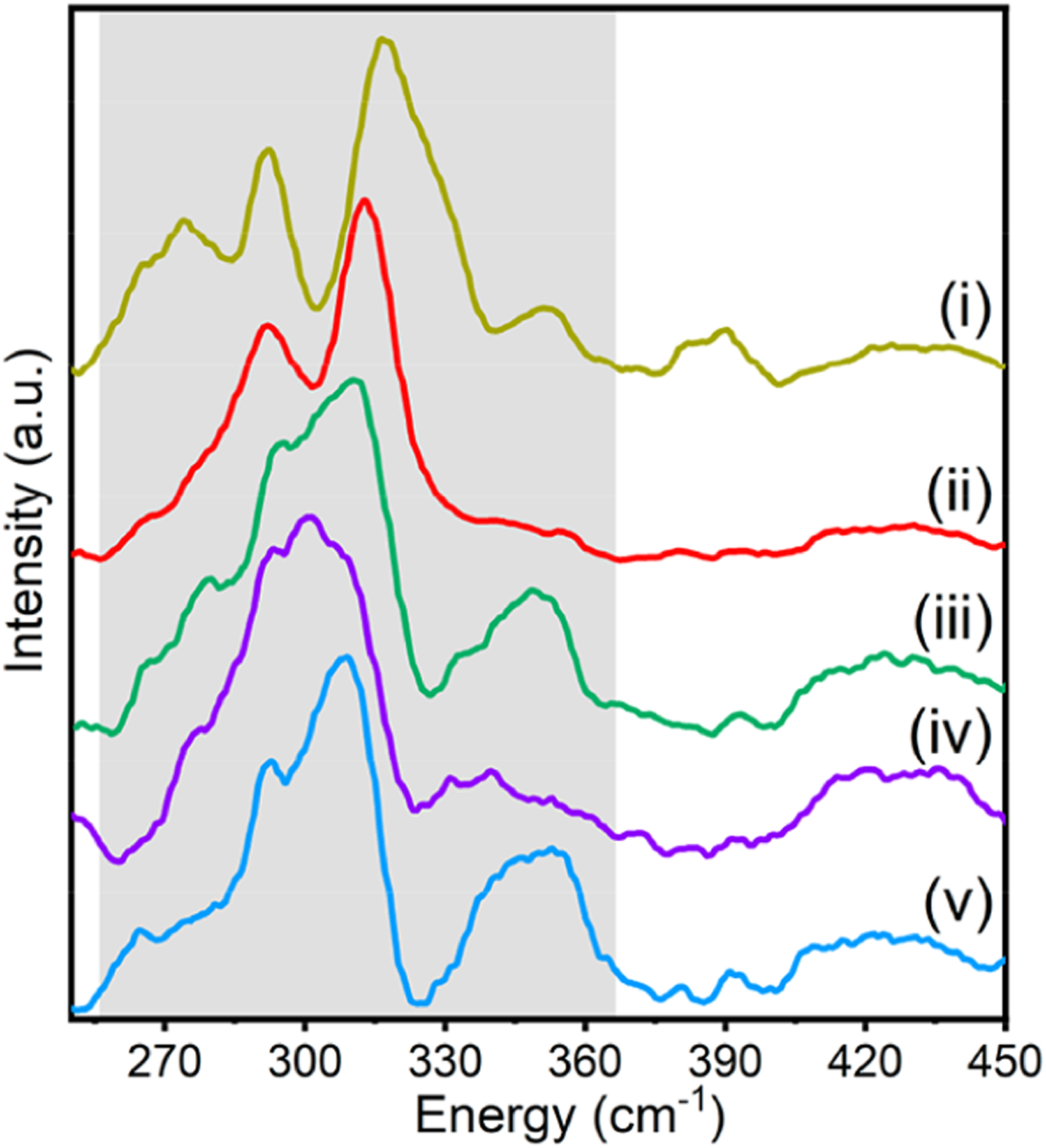
Resonance Raman spectra of HE Az variants: (i) HEAz, (ii) HESAz, (iii) HENAz, (iv) HENSAz, and (v) HEPAz. The dominant resonance-enhanced region is denoted by gray shading.
Table 5.
Summary of Intensity Weighted Energies for 〈ν(Cu–SC112)〉
| Variant | 〈v(Cu–SC112)〉 (cm−1) |
|---|---|
| HEAz (M121H/H46E) | 311 |
| HESAz (M121H/H46E/N47S) | 306 |
| HENAz (M121H/H46E/F114N) | 312 |
| HENSAz (M121H/H46E/F114N/N47S) | 309 |
| HEPAz (M121H/H46E/F114P) | 315 |
These intensity weighted averages can be cross-correlated with the EXAFS-determined Cu–S scattering distances using Badger’s rule, which relates the vibrational frequency, νe, to the equilibrium internuclear distance re:91
| (3) |
where Cij and dij are empirical constants for a pair of atoms i and j which can be determined by fitting a series of re and ve.92 From eq 3, R(Cu–S) should be linearly proportional to . Using the intensity-weighted average 〈ν(Cu–SC112)〉 from eq 2 as ve and Cu K-edge EXAFS determined Cu–S scattering distances as re, the observed frequencies and distances indeed follow Badger’s rule, with decreasing bond distance corresponding to an increase in the vibrational frequency for all HEAz variants (Figure 7).
Figure 7.
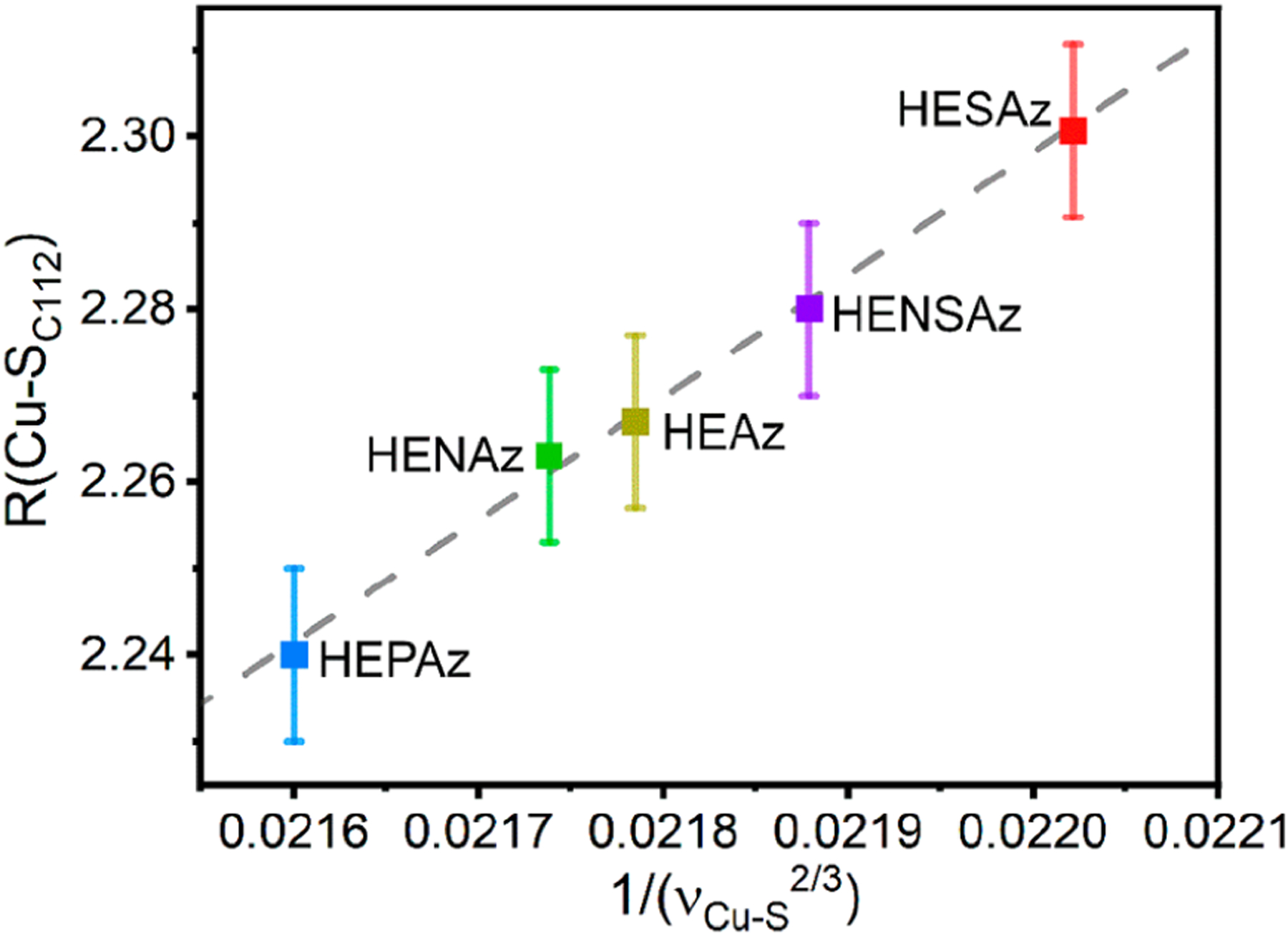
Cu K-edge EXAFS determined Cu–S scattering distances vs 1/(〈ν(Cu–SC112)〉)2/3. The dashed line represents the linear least-squares regression (R2 = 0.995). Error bars indicate the standard deviation of fit R(Cu–SC112) distances from Cu K-edge EXAFS.
Vibrational calculations using a numerical partial Hessian approach were employed to examine the Cu–S stretching frequencies. The results are summarized in SI, Table S4. All HEAz variants display a wide series of modes involving Cu and S displacement between ~260–350 cm−1, with clearer Cu–S stretching modes at ~290 and ~310 cm–1. These computational results support the intensity-weighted average approach employed to analyze our resonance Raman results.
4. DISCUSSION
4.1. Correlations of Spectroscopically Derived Properties with Reactivity.
Collectively, three pathways have been proposed for CuII-mediated S-nitrosylation, as summarized in Figure 8A: (i) Electrophilic attack of CuII to form {CuNO}10 followed by intramolecular attack of SC112 by NO+; (ii) Radical attack of SC112 by NO•; (iii) Direct attack of the Cu–S bond to form a Cu-(κ1-N(O))-SC112 intermediate.51,93 DFT-based calculations have favored the latter two mechanisms, with the largest calculated barrier among the three involving coupling of CuII to S = ½ NO• in the electrophilic attack mechanism.51
Figure 8.
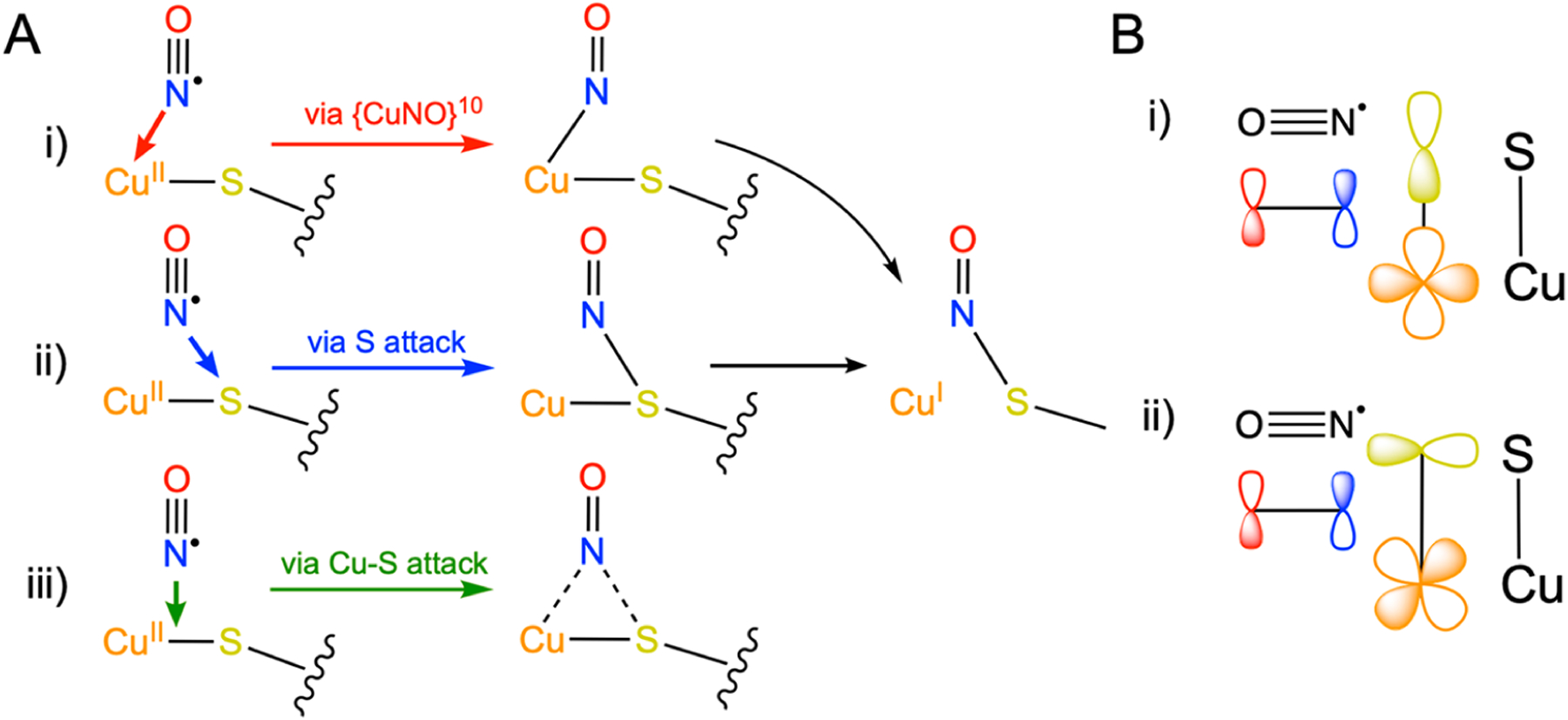
(A) Comparison of the three proposed mechanisms of CuII-catalyzed S-nitrosylation,51,92 involving (i) electrophilic addition to CuII, (ii) radical attack of S, and (iii) direct attack of the Cu–S bond. (B) Comparison of the orbital overlap requirements for NO attack of CuII–S via (i) S(pσ) vs (ii) S(pπ) interactions.
Importantly, each proposed mechanism of S-nitrosylation carries its own implications of which factors may promote or inhibit the forward S-nitrosylation reaction. The electrophilic attack mechanism requires the effective oxidation of NO• to NO+ by CuII, which would be facilitated by a more electron deficient CuII site. The radical attack mechanism requires significant spin density at S to directly couple with NO•; therefore, we anticipate an enhanced reaction rate would be correlate with increased spin density at S. Lastly, direct attack of the Cu–S bond would rely on maximized overlap between the partially occupied π* of NO• and the antibonding SOMO of the CuII active site, which would be optimized by increasing the effective strength of the Cu–S bond. In Figure 9, we compare the derived properties of Sp character (from S K-edge XAS measurements), both Cu–S distance (from Cu K-edge EXAFS) and intensity weighted 〈ν(Cu–SC112)〉 (from resonance Raman), and α2 (CuII character, from CW X-band EPR measurements) against the determined pseudo first-order rate constants.51,93
Figure 9.
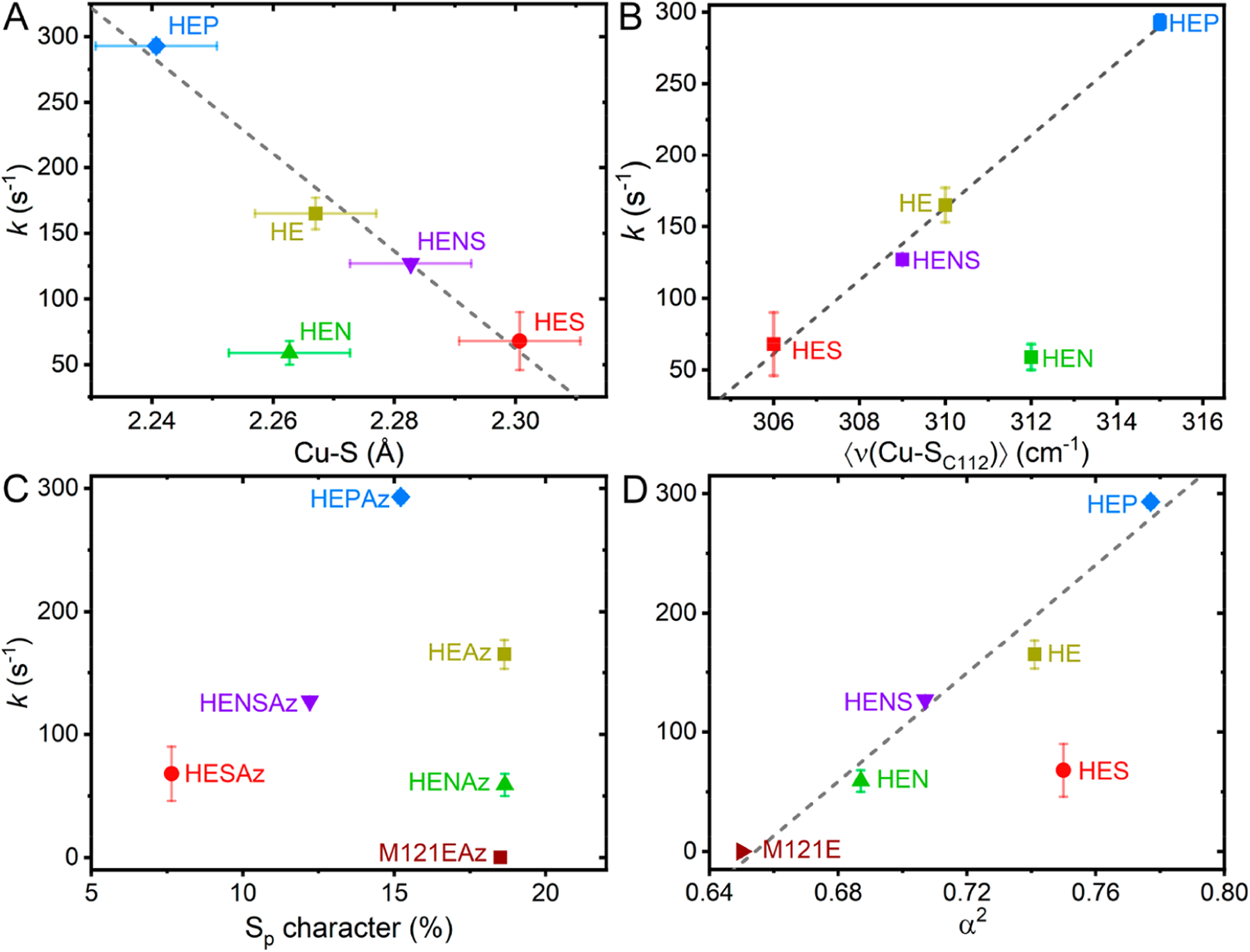
Comparisons of the pseudo first-order rate constant for S-nitrosylation (k) with spectroscopically derived parameters, including (A) the EXAFS-determined Cu–S scattering distance, (B) the resonance Raman determined intensity weighted average Cu–S stretching frequency 〈ν(Cu–SC112)〉, (C) the S K-edge determined Sp character, and (D) the CW X-band EPR derived metal character. Displayed error bars correspond to σ2. Dashed lines indicate linear least-squares regressions (R2 = 0.979 in A, 0.994 in B, and 0.979 in D), with outliers excluded as follows: HENAz in A and B, HESAz in D.
From Figure 9A and 9B, we can see that the reaction rate correlates well with the Cu–S distance and 〈ν(Cu–SC112)〉, where a decreasing Cu–S distance (and correspondingly increasing 〈ν(Cu–SC112)〉) results in an increase in the rate of S-nitrosylation. These results suggest that the Cu–S bond strength and therefore degree of Cu–S antibonding character are determining factors in S-nitrosylation. However, the HENAz variant appears to deviate significantly from this trend, where the observed rate of kf = 59 s–1 is far below the anticipated value of ~200 s−1 based on linear extrapolation.
To better understand the deviation observed in the reaction rate of HENAz, we compared k with the Sp character. Inspection of Figure 9C reveals no clear correlations between the total Sp character and S-nitrosylation reaction rate. This lack of correlation is perhaps most clearly highlighted by comparing M121EAz, HEAz, and HENAz. While the total Sp character of these three variants is virtually identical, the UV/vis measurements presented in Figure S9 highlight that the nature of this Sp character varies significantly, specifically in terms of S(pσ) and S(pπ) contributions. Here, decreasing S(pπ) character correlates with an increase in the S-nitrosylation reaction rate. While HEAz, HESAz, HENSAz, and HEPAz appear to have similar total S(pπ) contributions to the SOMO based on UV/vis, HENAz displays noticeably greater S(pπ) character. We propose that the deviation of HENAz from the Cu–S bond strength/S-nitrosylating activity correlation can be attributed to this increase in the S(pπ) character. Furthermore, the lack of correlation between total Sp character and reaction rate from those variants with similar S(pπ) character (HEAz, HESAz, HENSAz, and HEPAz) opposes the radical attack mechanism.
Lastly, we can turn to the CuII character of the SOMO based on the derived parameter α2. A linear relationship is observed among the HEAz variants, including HENAz, where an increasing CuII character leads to an enhanced reaction rate. These results imply that coupling of NO• proceeds through CuII rather than direct radical attack of S. However, HESAz now appears as an outlier, presenting a significantly lower rate than anticipated based on the CuII character. Notably, HESAz also presents the lowest degree of Sp character (~8%) and the weakest Cu–S bond. Meanwhile HEPAz, for example, exhibits the strongest Cu–S bond of the HE variants and nearly twice the Sp character of HESAz. These analyses support that increased spin density at CuII supports an increase in reaction rate only in-so-far as sufficient Cu(3dx2‑y2)/S(pσ) antibonding character is maintained.
4.2. Influence of Secondary Coordination Sphere on Blue vs Red Cu Az Variants.
A detailed study on the influence of the SCS on the electronic structure and reduction potential of blue copper Az has previously been presented by Hadt et al.14 through a comparison of WTAz, F114PAz, N47SAz, and F114NAz. This previous study allows a unique opportunity to draw parallels between how these mutations (F114P, N47S, F114N) modulate the electronic structure of CuII in red (HE) vs blue (WT) Az. The F114P mutation eliminates an H-bonding interaction between the backbone amide of residue 114, as well as modulates the flexibility of the loop. The N47S mutation both removes a negative carbonyl dipole in the vicinity of S, as well as disrupts a stabilizing H-bonding interaction between the Asn47 side chain and Thr113 amide backbone. Additionally, the F114N mutation adds positive amide/negative carbonyl dipoles in the vicinity of Cu (each ~5 Å away13).
Similar to its effect on blue copper, the F114N mutation has little impact on the covalency of the Cu–S bond in HEAz (19 vs 20% Sp character as determined by S K-edge XAS, Table 2) and a relatively small impact on bond strength (2.27 vs 2.26 Å Cu–S distance from Cu K-edge EXAFS, and 312 vs 311 cm−1 〈ν(Cu–SC112)〉 from resonance Raman analyses, Tables 4 and 5, respectively). Likewise, the N47S mutation results in a decrease in covalency, with changes of −11% in HESAz (relative to HE Az) and –8% in N47SAz (relative to WTAz), and decreases in 〈ν(Cu–SC112)〉 by ~5 cm−1. However, this trend deviates significantly in the F114P variants. While both HEPAz and F114PAz display an increase in Cu–S bond strength, as evidenced by increases in 〈ν(Cu–SC112)〉, HEPAz displays a decrease in Sp character (−7%) and increase in pre-edge energy (+0.4 eV), while F114PAz shows a significant increase in Sp character (+9%) and decrease in pre-edge energy (−0.2 eV). Additionally, α2 (CuII character) increases in HEPAz, while it decreases in F114PAz.
The clearest difference between the WTAz and HEAz series is the change in the orientation of the Sp orbital contributing to the -dominated SOMO from S(pπ) to S(pσ). As a result, the H-bonding interactions between S(C112) and the backbone amides of residues 47 and 114 are shifted from predominately active to predominately passive. Based on the previous computational study of Hadt et al., a shift to passive H-bonding is expected to have the same, albeit dampened, effect as active H-bonding.14 Furthermore, based on studies on small molecules, deletion of a passive H-bond in HEPAz is expected to give similar Sp character and a decrease in the S K-pre-edge energy with respect to the HEAz variant.94 Therefore, elimination of an active vs passive H-bond alone is not sufficient to explain the concomitant increase in Cu–S bond strength and Cu character with a decrease in Cu–S bond covalency observed for HEPAz. However, scans of the Cu–S–C angle of simplified square planar and trigonal models reveal an interesting trend (Figure S10). Specifically, as this angle deviates below or above ~100° in square planar CuII, a decrease in spin density at S is found together with an increase in spin density at Cu. Meanwhile, deviation of the Cu–S–C angle from ~100° in a trigonal CuII geometry displays the opposite trend, where the S spin density increases and the Cu spin density decreases. The F114P mutation removes a stabilizing H-bonding interaction with SC112, allowing greater conformational flexibility of the Cys112 side chain and feasibly variations in ∠Cu–SC112-Cβ, resulting in opposite variations in the %Sp character of F114PAz and HEPAz relative to WTAz and HEAz, respectively.
4.3. Why Red Cu in Az?
While the present study of engineered Az supports that S-nitrosylation proceeds via NO• attack of the Cu–S σ* interaction, a previous study of the blue copper model complex MesTpCu-SCPh3 has demonstrated that this process can still occur in systems where the SOMO is dominated by the Cu–S π* interaction.93 The juxtaposition of these results demonstrates that a red Cu center is not an intrinsic prerequisite for Cu-mediated S-nitrosylation.
To test whether •NO intrinsically favors attacking either the Cu–S σ* or the π* pathway, we further calculated the reaction coordinate of NO• with a simplified 3-coordinate planar T-shaped (σ Cu–S) and trigonal (π Cu–S) geometries of CuII with S, N, and O ligation with minimal steric factors. Both geometries form a bridging Cu-μ-N(O)-S intermediate on a broken symmetry singlet surface, where the trigonal geometry actually appears several kcal/mol lower in energy than that for the T-shaped (see Figure S11), demonstrating that there is not necessarily an intrinsic electronic requirement for S-nitrosylation to proceed via attack of the Cu–S σ* rather than π* SOMO. Therefore, we propose that this disparity arises largely from steric encumbrance of the Cu site. More specifically, the CuII–S(pπ) interaction requires the attack by •NO in the plane. Meanwhile, the CuII–S(pσ) interaction is isotropic with respect to the Cu–S bond, allowing for attack by •NO from any available direction. Therefore, we hypothesize that removing steric hindrance in the immediate vicinity of the plane would activate Cu-Az variants with significant S(pπ) contributions to efficient S-nitrosylation.
5. CONCLUSIONS
In this work, we have employed a multifaceted approach to investigate the factors influencing CuII-catalyzed intramolecular S-nitrosylation. To achieve the goal, we have examined the influence of the primary and secondary coordination spheres on the rate of CuII-catalyzed S-nitrosylation for a series of Az variants, targeting factors that influence S(pσ) vs S(pπ) character in the SOMO and modulate Cu–S bond through H-bonding and electrostatic interactions. Across this series, we have elucidated that the rate of S-nitrosylation is positively correlated with CuII spin density and Cu–S bond strength (rather than Cu–S bond covalency) and negatively correlated with the degree of S(pπ) contribution to the SOMO. Our results support a reaction mechanism involving direct attack of the Cu–S bond, rather than electrophilic attack of CuII or radical attack of S. We have also found that the negative rate dependence of CuII-catalyzed S-nitrosylation on S(pπ) character likely arises from steric hindrance imparted by the Az scaffold and that removal of this steric hindrance may enable S-nitrosylation at blue or green-type CuII sites, greatly expanding the breadth of potential Cu-containing enzymes capable of this reaction.
Supplementary Material
ACKNOWLEDGMENTS
We wish to acknowledge the U.S. National Science Foundation (CHE-2201279 to Y.L.), the U.S. National Institute of Health (F32GM145134 to C.V.S. and R01DK031450 to E.I.S.), and the Robert. A. Welch Foundation (F-0020 to Y.L.) for funding of research described in this work. C.V.S. also dearly thanks Dr. Ragnar Björnsson for his advice in QM/MM calculations, and Prof. Dr. Serena DeBeer for generously allowing use of computational resources for sections of this work, and for fruitful discussions regarding red copper sites. Drs. Leah Kelly, Matthew Latimer, and Erik Nelson are gratefully acknowledged for their technical assistance during XAS measurements on beamlines 7-3 and 4-3 of SSRL. The use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515.
ABBREVIATIONS
- Az
azurin
- CW
continuous wave
- DFT
density functional theory
- EPR
electron paramagnetic resonance
- EXAFS
extended X-ray absorption fine structure
- HEAz
M121H/H46E azurin
- HESAz
M121H/H46E/N47S azurin
- HENAz
M121H/H46E/F114N azurin
- HENSAz
M121H/H46E/F114N/N47S azurin
- HEPAz
M121H/H46E/F114P azurin
- PCS
primary coordination sphere
- QM/MM
quantum mechanics/molecular mechanics
- SCS
secondary coordination sphere
- SNO
S-nitrosylation
- WT
wild-type
- XANES
X-ray absorption near-edge spectroscopy
- XAS
X-ray absorption spectroscopy
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c07399.
Additional experimental details, supplementary discussion, figures, and tables. (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.3c07399
The authors declare no competing financial interest.
Contributor Information
Casey Van Stappen, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States.
Huiguang Dai, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States; Department of Chemistry, University of Urbana–Champaign, Champaign, Illinois 61801, United States.
Anex Jose, Department of Chemistry, Stanford University, Stanford, California 94305-5080, United States.
Shiliang Tian, Department of Chemistry, University of Urbana–Champaign, Champaign, Illinois 61801, United States; Present Address: Department of Chemistry, Purdue University, West-Lafayette, Indiana 47906, United States.
Edward I. Solomon, Department of Chemistry, Stanford University, Stanford, California 94305-5080, United States; Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Menlo Park, California 94025, United States
Yi Lu, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States; Department of Chemistry, University of Urbana–Champaign, Champaign, Illinois 61801, United States.
REFERENCES
- (1).Van Stappen C; Deng Y; Liu Y; Heidari H; Wang JX; Zhou Y; Ledray AP; Lu Y Designing Artificial Metalloenzymes by Tuning of the Environment beyond the Primary Coordination Sphere. Chem. Rev 2022, 122, 11974–12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Koebke KJ; Pinter TBJ; Pitts WC; Pecoraro VL Catalysis and Electron Transfer in De Novo Designed Metalloproteins. Chem. Rev 2022, 122, 12046–12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Schwizer F; Okamoto Y; Heinisch T; Gu Y; Pellizzoni MM; Lebrun V; Reuter R; Kohler V; Lewis JC; Ward TR Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- (4).Kerns SA; Biswas A; Minnetian NM; Borovik AS Artificial Metalloproteins: At the Interface between Biology and Chemistry. JACS Au 2022, 2, 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Rosati F; Roelfes G Artificial Metalloenzymes. ChemCatChem. 2010, 2, 916–927. [Google Scholar]
- (6).Peacock AF Incorporating metals into de novo proteins. Curr. Opin. Chem. Biol 2013, 17, 934–9. [DOI] [PubMed] [Google Scholar]
- (7).Chalkley MJ; Mann SI; DeGrado WF De novo metalloprotein design. Nat. Rev. Chem 2022, 6, 31–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lewis JC Beyond the Second Coordination Sphere: Engineering Dirhodium Artificial Metalloenzymes To Enable Protein Control of Transition Metal Catalysis. Acc. Chem. Res 2019, 52, 576–584. [DOI] [PubMed] [Google Scholar]
- (9).Hilvert D Design of protein catalysts. Annu. Rev. Biochem 2013, 82, 447–70. [DOI] [PubMed] [Google Scholar]
- (10).Carminati DM; Moore EJ; Fasan R Strategies for the expression and characterization of artificial myoglobin-based carbene transferases. Methods Enzymol. 2020, 644, 35–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Choi TS; Tezcan FA Design of a Flexible, Zn-Selective Protein Scaffold that Displays Anti-Irving-Williams Behavior. J. Am. Chem. Soc 2022, 144, 18090–18100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Miller AF Redox tuning over almost 1 V in a structurally conserved active site: lessons from Fe-containing superoxide dismutase. Acc. Chem. Res 2008, 41, 501–10. [DOI] [PubMed] [Google Scholar]
- (13).Marshall NM; Garner DK; Wilson TD; Gao YG; Robinson H; Nilges MJ; Lu Y Rationally tuning the reduction potential of a single cupredoxin beyond the natural range. Nature 2009, 462, 113–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hadt RG; Sun N; Marshall NM; Hodgson KO; Hedman B; Lu Y; Solomon EI Spectroscopic and DFT studies of second-sphere variants of the type 1 copper site in azurin: covalent and nonlocal electrostatic contributions to reduction potentials. J. Am. Chem. Soc 2012, 134, 16701–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hosseinzadeh P; Lu Y Design and fine-tuning redox potentials of metalloproteins involved in electron transfer in bioenergetics. Biochim. Biophys. Acta 2016, 1857, 557–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hosseinzadeh P; Marshall NM; Chacon KN; Yu Y; Nilges MJ; New SY; Tashkov SA; Blackburn NJ; Lu Y Design of a single protein that spans the entire 2-V range of physiological redox potentials. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 262–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Singha A; Sekretareva A; Tao L; Lim H; Ha Y; Braun A; Jones SM; Hedman B; Hodgson KO; Britt RD; Kosman DJ; Solomon EI Tuning the Type 1 Reduction Potential of Multicopper Oxidases: Uncoupling the Effects of Electrostatics and H-Bonding to Histidine Ligands. J. Am. Chem. Soc 2023, 145, 13284–13301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hess DT; Matsumoto A; Kim SO; Marshall HE; Stamler JS Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol 2005, 6, 150–66. [DOI] [PubMed] [Google Scholar]
- (19).Liu L; Yan Y; Zeng M; Zhang J; Hanes MA; Ahearn G; McMahon TJ; Dickfeld T; Marshall HE; Que LG; Stamler JS Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 2004, 116, 617–28. [DOI] [PubMed] [Google Scholar]
- (20).Baldelli S; Ciriolo MR Altered S-nitrosylation of p53 is responsible for impaired antioxidant response in skeletal muscle during aging. Aging 2016, 8, 3450–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Murad F Cyclic guanosine monophosphate as a mediator of vasodilation. J. Clin. Invest 1986, 78, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhang Y; Deng Y; Yang X; Xue H; Lang Y The Relationship Between Protein S-Nitrosylation and Human Diseases: A Review. Neurochem. Res 2020, 45, 2815–2827. [DOI] [PubMed] [Google Scholar]
- (23).Tan C; Li Y; Huang X; Wei M; Huang Y; Tang Z; Huang H; Zhou W; Wang Y; Hu J Extensive protein S-nitrosylation associated with human pancreatic ductal adenocarcinoma pathogenesis. Cell Death Dis. 2019, 10, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chanvorachote P; Nimmannit U; Stehlik C; Wang L; Jiang BH; Ongpipatanakul B; Rojanasakul Y Nitric oxide regulates cell sensitivity to cisplatin-induced apoptosis through S-nitrosylation and inhibition of Bcl-2 ubiquitination. Cancer Res. 2006, 66, 6353–60. [DOI] [PubMed] [Google Scholar]
- (25).Lim KH; Ancrile BB; Kashatus DF; Counter CM Tumour maintenance is mediated by eNOS. Nature 2008, 452, 646–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Leon-Bollotte L; Subramaniam S; Cauvard O; Plenchette-Colas S; Paul C; Godard C; Martinez-Ruiz A; Legembre P; Jeannin JF; Bettaieb A S-nitrosylation of the death receptor fas promotes fas ligand-mediated apoptosis in cancer cells. Gastroenterology 2011, 140, 2009–18. [DOI] [PubMed] [Google Scholar]
- (27).Guan W; Sha J; Chen X; Xing Y; Yan J; Wang Z S-Nitrosylation of mitogen activated protein kinase phosphatase-1 suppresses radiation-induced apoptosis. Cancer Lett. 2012, 314, 137–46. [DOI] [PubMed] [Google Scholar]
- (28).Rizza S; Montagna C; Cardaci S; Maiani E; Di Giacomo G; Sanchez-Quiles V; Blagoev B; Rasola A; De Zio D; Stamler JS; Cecconi F; Filomeni G S-nitrosylation of the Mitochondrial Chaperone TRAP1 Sensitizes Hepatocellular Carcinoma Cells to Inhibitors of Succinate Dehydrogenase. Cancer Res. 2016, 76, 4170–82. [DOI] [PubMed] [Google Scholar]
- (29).Gupta A; Anjomani-Virmouni S; Koundouros N; Dimitriadi M; Choo-Wing R; Valle A; Zheng Y; Chiu YH; Agnihotri S; Zadeh G; Asara JM; Anastasiou D; Arends MJ; Cantley LC; Poulogiannis G PARK2 Depletion Connects Energy and Oxidative Stress to PI3K/Akt Activation via PTEN S-Nitrosylation. Mol. Cell 2017, 65, 999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ghatak S; Nakamura T; Lipton SA Aberrant protein S-nitrosylation contributes to hyperexcitability-induced synaptic damage in Alzheimer’s disease: Mechanistic insights and potential therapies. Front. Neural Circuits 2023, 17, No. 1099467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ryu IH; Lee KY; Do SI Abeta-affected pathogenic induction of S-nitrosylation of OGT and identification of Cys-NO linkage triplet. Biochim. Biophys. Acta 2016, 1864, 609–21. [DOI] [PubMed] [Google Scholar]
- (32).Nakamura T; Lipton SA ‘SNO’-Storms Compromise Protein Activity and Mitochondrial Metabolism in Neurodegenerative Disorders. Trends Endocrinol. Metab 2017, 28 (12), 879–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Seneviratne U; Nott A; Bhat VB; Ravindra KC; Wishnok JS; Tsai LH; Tannenbaum SR S-nitrosation of proteins relevant to Alzheimer’s disease during early stages of neurodegeneration. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 4152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yin L; Xie Y; Yin S; Lv X; Zhang J; Gu Z; Sun H; Liu S The S-nitrosylation status of PCNA localized in cytosol impacts the apoptotic pathway in a Parkinson’s disease paradigm. PLoS One 2015, 10, No. e0117546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Calabrese V; Santoro A; Monti D; Crupi R; Di Paola R; Latteri S; Cuzzocrea S; Zappia M; Giordano J; Calabrese EJ; Franceschi C Aging and Parkinson’s Disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med 2018, 115, 80–91. [DOI] [PubMed] [Google Scholar]
- (36).Zhang Z; Liu L; Jiang X; Zhai S; Xing D The Essential Role of Drp1 and Its Regulation by S-Nitrosylation of Parkin in Dopaminergic Neurodegeneration: Implications for Parkinson’s Disease. Antioxid. Redox Signal 2016, 25, 609–622. [DOI] [PubMed] [Google Scholar]
- (37).Wilkaniec A; Lenkiewicz AM; Czapski GA; Jesko HM; Hilgier W; Brodzik R; Gassowska-Dobrowolska M; Culmsee C; Adamczyk A Extracellular Alpha-Synuclein Oligomers Induce Parkin S-Nitrosylation: Relevance to Sporadic Parkinson’s Disease Etiopathology. Mol. Neurobiol 2019, 56, 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Oh CK; Sultan A; Platzer J; Dolatabadi N; Soldner F; McClatchy DB; Diedrich JK; Yates JR 3rd; Ambasudhan R; Nakamura T; Jaenisch R; Lipton SA S-Nitrosylation of PINK1 Attenuates PINK1/Parkin-Dependent Mitophagy in hiPSC-Based Parkinson’s Disease Models. Cell Rep. 2017, 21, 2171–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wu B; Yu H; Wang Y; Pan Z; Zhang Y; Li T; Li L; Zhang W; Ge L; Chen Y; Ho CK; Zhu D; Huang X; Lou Y Peroxiredoxin-2 nitrosylation facilitates cardiomyogenesis of mouse embryonic stem cells via XBP-1s/PI3K pathway. Free Radic. Biol. Med 2016, 97, 179–191. [DOI] [PubMed] [Google Scholar]
- (40).Li J; Zhang Y; Zhang Y; Lu S; Miao Y; Yang J; Huang S; Ma X; Han L; Deng J; Fan F; Liu B; Huo Y; Xu Q; Chen C; Wang X; Feng J GSNOR modulates hyperhomocysteinemia-induced T cell activation and atherosclerosis by switching Akt S-nitrosylation to phosphorylation. Redox Biol. 2018, 17, 386–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Chen Y; Liu R; Zhang G; Yu Q; Jia M; Zheng C; Wang Y; Xu C; Zhang Y; Liu E Hypercysteinemia promotes atherosclerosis by reducing protein S-nitrosylation. Biomed. Pharmacother 2015, 70, 253–9. [DOI] [PubMed] [Google Scholar]
- (42).Fernando V; Zheng X; Walia Y; Sharma V; Letson J; Furuta S S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Stubauer G; Giuffre A; Sarti P Mechanism of S-nitrosothiol formation and degradation mediated by copper ions. J. Biol. Chem 1999, 274, 28128–33. [DOI] [PubMed] [Google Scholar]
- (44).Romeo AA; Capobianco JA; English AM Heme nitrosylation of deoxyhemoglobin by s-nitrosoglutathione requires copper. J. Biol. Chem 2002, 277, 24135–41. [DOI] [PubMed] [Google Scholar]
- (45).Luchsinger BP; Rich EN; Gow AJ; Williams EM; Stamler JS; Singel DJ Routes to S-nitroso-hemoglobin formation with heme redox and preferential reactivity in the beta subunits. Proc. Natl. Acad. Sci. U.S.A 2003, 100, 461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Bak DW; Pizzagalli MD; Weerapana E Identifying Functional Cysteine Residues in the Mitochondria. ACS Chem. Biol 2017, 12, 947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Chouchani ET; Hurd TR; Nadtochiy SM; Brookes PS; Fearnley IM; Lilley KS; Smith RA; Murphy MP Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem. J 2010, 430, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sun J; Morgan M; Shen RF; Steenbergen C; Murphy E Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ. Res 2007, 101, 1155–63. [DOI] [PubMed] [Google Scholar]
- (49).Piantadosi CA Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 712–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lee YI; Giovinazzo D; Kang HC; Lee Y; Jeong JS; Doulias PT; Xie Z; Hu J; Ghasemi M; Ischiropoulos H; Qian J; Zhu H; Blackshaw S; Dawson VL; Dawson TM Protein microarray characterization of the S-nitrosoproteome. Mol. Cell Proteomics 2014, 13, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tian S; Liu J; Cowley RE; Hosseinzadeh P; Marshall NM; Yu Y; Robinson H; Nilges MJ; Blackburn NJ; Solomon EI; Lu Y Reversible S-nitrosylation in an engineered azurin. Nat. Chem 2016, 8, 670–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Gorren AC; de Boer E; Wever R The reaction of nitric oxide with copper proteins and the photodissociation of copper-NO complexes. Biochim. Biophys. Acta 1987, 916, 38–47. [DOI] [PubMed] [Google Scholar]
- (53).Ehrenstein D; Filiaci M; Scharf B; Engelhard M; Steinbach PJ; Nienhaus GU Ligand binding and protein dynamics in cupredoxins. Biochemistry 1995, 34, 12170–7. [DOI] [PubMed] [Google Scholar]
- (54).Nar H; Messerschmidt A; Huber R; van de Kamp M; Canters GW Crystal structure analysis of oxidized Pseudomonas aeruginosa azurin at pH 5.5 and pH 9.0. A pH-induced conformational transition involves a peptide bond flip. J. Mol. Biol 1991, 221, 765–72. [DOI] [PubMed] [Google Scholar]
- (55).Stoll S; Schweiger A EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson 2006, 178, 42–55. [DOI] [PubMed] [Google Scholar]
- (56).Newville M Larch: An Analysis Package for XAFS and Related Spectroscopies. J. Phys. (Paris) 2013, 430, 012007. [Google Scholar]
- (57).Rehr JJ; Kas JJ; Prange MP; Sorini AP; Takimoto Y; Vila F Ab initio theory and calculations of X-ray spectra. Comp. Rend. Phys 2009, 10, 548–559. [Google Scholar]
- (58).Rehr JJ; Albers RC Theoretical approaches to X-ray absorption fine structure. Rev. Mod. Phys 2000, 72, 621–654. [Google Scholar]
- (59).Ankudinov AL; Ravel B; Rehr JJ; Conradson SD Real-space multiple-scattering calculation and interpretation of X-ray absorption near-edge structure. Phys. Rev. B 1998, 58, 7565–7576. [Google Scholar]
- (60).Bjornsson R ASH - a multiscale modelling program, 9.0; 2022.
- (61).Neese F The ORCA program system. WIREs Comp. Mol. Sci 2012, 2, 73–78. [Google Scholar]
- (62).Neese F Software update: the ORCA program system, version 4.0. WIREs Comp. Mol. Sci 2018, 8, No. e1327. [Google Scholar]
- (63).Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y; Beauchamp KA; Wang LP; Simmonett AC; Harrigan MP; Stern CD; Wiewiora RP; Brooks BR; Pande VS OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol 2017, 13, No. e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Gewirth AA; Solomon EI Electronic structure of plastocyanin: excited state spectral features. J. Am. Chem. Soc 1988, 110, 3811–3819. [Google Scholar]
- (65).Karlsson BG; Tsai LC; Nar H; Sanders-Loehr J; Bonander N; Langer V; Sjolin L X-ray structure determination and characterization of the Pseudomonas aeruginosa azurin mutant Met121Glu. Biochemistry 1997, 36, 4089–95. [DOI] [PubMed] [Google Scholar]
- (66).Karlsson BG; Nordling M; Pascher T; Tsai LC; Sjolin L; Lundberg LG Cassette mutagenesis of Met121 in azurin from Pseudomonas aeruginosa. Protein Eng. 1991, 4, 343–9. [DOI] [PubMed] [Google Scholar]
- (67).LaCroix LB; Shadle SE; Wang Y; Averill BA; Hedman B; Hodgson KO; Solomon EI Electronic Structure of the Perturbed Blue Copper Site in Nitrite Reductase: Spectroscopic Properties, Bonding, and Implications for the Entatic/Rack State. J. Am. Chem. Soc 1996, 118, 7755–7768. [Google Scholar]
- (68).Williams DLH The mechanism of nitric oxide formation from S-nitrosothiols (thionitrites). Chem. Commun 1996, 1085–1091. [Google Scholar]
- (69).Solomon EI; Hedman B; Hodgson KO; Dey A; Szilagyi RK Ligand K-edge X-ray absorption spectroscopy: covalency of ligand–metal bonds. Coord. Chem. Rev 2005, 249, 97–129. [Google Scholar]
- (70).Basumallick L; Sarangi R; DeBeer-George S; Elmore B; Hooper AB; Hedman B; Hodgson KO; Solomon EI Spectroscopic and density functional studies of the red copper site in nitrosocyanin: role of the protein in determining active site geometric and electronic structure. J. Am. Chem. Soc 2005, 127, 3531–44. [DOI] [PubMed] [Google Scholar]
- (71).Siluvai GS; Mayfield M; Nilges MJ; DeBeer-George S; Blackburn NJ Anatomy of a red copper center: spectroscopic identification and reactivity of the copper centers of Bacillus subtilis Sco and its Cys-to-Ala variants. J. Am. Chem. Soc 2010, 132, 5215–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).McGarvey BR Theory of the Spin Hamiltonian Parameters for Low Spin Cobalt(II) Complexes. Can. J. Chem 1975, 53, 2498–2511. [Google Scholar]
- (73).Kau LS; Spira-Solomon DJ; Penner-Hahn JE; Hodgson KO; Solomon EI X-ray absorption edge determination of the oxidation state and coordination number of copper. Application to the type 3 site in Rhus vernicifera laccase and its reaction with oxygen. J. Am. Chem. Soc 1987, 109, 6433–6442. [Google Scholar]
- (74).Lancaster KM; George SD; Yokoyama K; Richards JH; Gray HB Type-zero copper proteins. Nat. Chem 2009, 1, 711–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).DeBeer S; Kiser CN; Mines GA; Richards JH; Gray HB; Solomon EI; Hedman B; Hodgson KO X-ray Absorption Spectra of the Oxidized and Reduced Forms of C112D Azurin from Pseudomonas aeruginosa. Inorg. Chem 1999, 38, 433–438. [DOI] [PubMed] [Google Scholar]
- (76).Lieberman RL; Arciero DM; Hooper AB; Rosenzweig AC Crystal structure of a novel red copper protein from Nitrosomonas europaea. Biochemistry 2001, 40, 5674–81. [DOI] [PubMed] [Google Scholar]
- (77).Schulz CE; van Gastel M; Pantazis DA; Neese F Converged Structural and Spectroscopic Properties for Refined QM/MM Models of Azurin. Inorg. Chem 2021, 60, 7399–7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Siiman O; Young NM; Carey PR Resonance Raman studies of ″blue″ copper proteins. J. Am. Chem. Soc 1974, 96, 5583–5. [DOI] [PubMed] [Google Scholar]
- (79).Miskowski V; Tang SP; Spiro TG; Shapiro E; Moss TH The copper coordination group in ″blue″ copper proteins: evidence from resonance Raman spectra. Biochemistry 1975, 14, 1244–50. [DOI] [PubMed] [Google Scholar]
- (80).Siiman O; Young NM; Carey PR Resonance raman spectra of ″blue″ copper proteins and the nature of their copper sites. J. Am. Chem. Soc 1976, 98, 744–8. [DOI] [PubMed] [Google Scholar]
- (81).Ferris NS; Woodruff WH; Tennent DL; McMillin DR Native azurin and its Ni(II) derivative: a resonance Raman study. Biochem. Biophys. Res. Commun 1979, 88, 288–96. [DOI] [PubMed] [Google Scholar]
- (82).Thamann TJ; Frank P; Willis LJ; Loehr TM Normal coordinate analysis of the copper center of azurin and the assignment of its resonance Raman spectrum. Proc. Natl. Acad. Sci. U.S.A 1982, 79, 6396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Blair DF; Campbell GW; Lum V; Martin CT; Gray HB; Malmström BG; Chan SI A resonance raman investigation of perturbed states of tree and fungal laccase. J. Inorg. Biochem 1983, 19, 65–73. [Google Scholar]
- (84).Woodruff WH; Norton KA; Swanson BI; Fry HA Cryoresonance Raman spectroscopy of plastocyanin, azurin, and stellacyanin. A reevaluation of the identity of the resonance-enhanced modes. J. Am. Chem. Soc 1983, 105, 657–658. [Google Scholar]
- (85).Woodruff WH; Norton K; Swanson BI; Fry HA; Malmström BG; Pecht I; Blair DF; Cho W; Campbell GW; Lum V; Miskowski VM; Chan SI; Gray HB Cyro-vibrational spectroscopy of blue copper proteins. Inorg. Chim. Acta 1983, 79, 51–52. [Google Scholar]
- (86).Nestor L; Larrabee JA; Woolery G; Reinhammar B; Spiro TG Resonance Raman spectra of blue copper proteins: assignments from normal mode calculations and copper-63/copper-65 and H2O/D2O shifts for stellacyanin and laccase. Biochemistry 1984, 23, 1084–1093. [Google Scholar]
- (87).Woodruff WH; Norton KA; Swanson BI; Fry HA Temperature dependence of the resonance Raman spectra of plastocyanin and azurin between cryogenic and ambient conditions. Proc. Natl. Acad. Sci. U.S.A 1984, 81, 1263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Palmer AE; Randall DW; Xu F; Solomon EI Spectroscopic Studies and Electronic Structure Description of the High Potential Type 1 Copper Site in Fungal Laccase: Insight into the Effect of the Axial Ligand. J. Am. Chem. Soc 1999, 121, 7138–7149. [Google Scholar]
- (89).Blair DF; Campbell GW; Cho WK; English AM; Fry HA; Lum V; Norton KA; Schoonover JR; Chan SI Resonance Raman studies of blue copper proteins: effects of temperature and isotopic substitutions. Structural and thermodynamic implications. J. Am. Chem. Soc 1985, 107, 5755–5766. [Google Scholar]
- (90).Han J; Adman ET; Beppu T; Codd R; Freeman HC; Huq LL; Loehr TM; Sanders-Loehr J Resonance Raman spectra of plastocyanin and pseudoazurin: evidence for conserved cysteine ligand conformations in cupredoxins (blue copper proteins). Biochemistry 1991, 30, 10904–13. [DOI] [PubMed] [Google Scholar]
- (91).Badger RM A Relation Between Internuclear Distances and Bond Force Constants. J. Chem. Phys 1934, 2, 128–131. [Google Scholar]
- (92).Green MT Application of Badger’s rule to heme and non-heme iron-oxygen bonds: an examination of ferryl protonation states. J. Am. Chem. Soc 2006, 128, 1902–6. [DOI] [PubMed] [Google Scholar]
- (93).Zhang S; Melzer MM; Sen SN; Celebi-Olcum N; Warren TH A motif for reversible nitric oxide interactions in metalloenzymes. Nat. Chem 2016, 8, 663–9. [DOI] [PubMed] [Google Scholar]
- (94).Dey A; Green KN; Jenkins RM; Jeffrey SP; Darensbourg M; Hodgson KO; Hedman B; Solomon EI S K-edge XAS and DFT calculations on square-planar NiII-thiolate complexes: effects of active and passive H-bonding. Inorg. Chem 2007, 46, 9655–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.