

Abstract
Adenosine receptor (AR) ligands are being developed for metabolic, cardiovascular, neurological, and inflammatory diseases and cancer. The ease of drug discovery is contingent on the availability of pharmacological tools. Fluorescent antagonist ligands for the human A2A and A3ARs were synthesized using two validated pharmacophores, 1,3-dipropyl-8-phenylxanthine and triazolo[1,5-c]quinazolin-5-yl)amine, which were coupled to eight reporter fluorophores: AlexaFluor, JaneliaFluor (JF), cyanine, and near infrared (NIR) dyes. The conjugates were first screened using radioligand binding in HEK293 cells expressing one of the three AR subtypes. The highest affinities at A2AAR were Ki 144–316 nM for 10, 12, and 19, and at A3AR affinity of Ki 21.6 nM for 19. Specific binding of JF646 conjugate MRS7774 12 to the HEK293 cell surface A2AAR was imaged using confocal microscopy. Compound 19 MRS7535, a triazolo[1,5-c]quinazolin-5-yl)amine containing a Sulfo-Cy7 NIR dye, was suitable for A3AR characterization in whole cells by flow cytometry (Kd 11.8 nM), and its bitopic interaction mode with an A3AR homology model was predicted. Given its affinity and selectivity (11-fold vs. A2AAR, ~ 50-fold vs. A1AR and A2BAR) and a good specific-to-nonspecific binding ratio, 19 could be useful for live cell or potentially a diagnostic in vivo NIR imaging tool and/or therapy targeting the A3AR.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11302-022-09873-3.
Keywords: Adenosine receptor, Antagonist, Receptor binding, Fluorescent ligands, Drug discovery, Flow cytometry
Introduction
Fluorescent ligands of the adenosine receptor (AR) family have provided insights into the orthosteric and allosteric ligand binding sites and receptor dimerization that conventional radioligands could not achieve [1–3]. The first high affinity fluorescent ligand of an adenosine receptor 1 (Fig. 1) was for the A2AAR, based on the ability of the terminal carboxylate group of selective agonist CGS21680 to be extended through amides without loss of binding affinity [4]. Other agonist fluorescent ligands in the same structural class, such as 2, were used to detect A2AAR heterodimerization with the D2 dopamine receptor [5]. A fluorescent N6-substituted A3AR agonist, 3, was used to characterize receptor-receptor interactions in A3AR homodimers [6]. Antagonist fluorescent ligands have been reported for the A1AR, A2AAR, A2BAR, and A3AR [7–14]. These probes, such as triazolo[1,5-c]quinazolin-5-yl)amine MRS5449 5, facilitate drug screening using flow cytometry of whole cells and other techniques to discover novel antagonists [8]. The bitopic nature of these fluorescent-tethered ligands often causes deviation from the simple bimolecular interaction between ligand and target receptor, as was shown for various A2AAR antagonists [13]. The binding of the fluorophore moiety can allosterically modulate the pharmacophore affinity at the orthosteric site, depending on precise distal modification of the functionalized chain. For example, xanthine derivative XAC-X-BY630 (CA200634) 4 and pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine derivative MRS7416 6 each displayed negative cooperativity in A2AAR binding [13]. A covalently binding fluorescent antagonist derived from A2AAR antagonist ZM241385 was recently reported [15].
Fig. 1.

Structures of six reported fluorescent agonists (1–3) and antagonists (4–6) for the ARs
Since AR ligands are in development for various metabolic, neurological, inflammatory, and malignant conditions [16–21], additional AR fluorescent ligands are needed to expand the range of tool compounds for drug discovery. Here we conjugated known fluorophores to two known AR antagonist chemotypes, i.e., 1,3-dipropylxanthines aryl-functionalized at the C8 position and triazolo[1,5-c]quinazolin-5-yl)amines functionalized through acylation at the N5 position [8, 22, 23]. These versatile scaffolds can be directed toward multiple AR subtypes based on their functionalization, and also dependent on the species being studied [24]. For example, 1,3-dialkyl-8-phenylxanthines, although originally reported as selective rat A1AR antagonists [22], have been modified to achieve selectivity for the human (h) A2AAR, A2BAR, or A3AR [24]. N-(2-Aminoethyl)-2-(4-(2,6-dioxo-1,3-dipropyl-2,3,6,7-tetrahydro-1H-purin-8-yl)phenoxy)acetamide (xanthine amine congener, XAC, 7, Scheme 1) was designed to serve as a functionalized congener for coupling to reporter groups and carrier moieties [22] and served as a precursor here. A triazolo[1,5-c]quinazolin-5-yl)amine CGS15943 8 (Scheme 2) binds to both A2AAR and A3AR and when acylated at the N5 position can be highly selective for the hA3AR (Scheme 2) [24]. In this study, we focus on the hA2AAR and hA3AR, using a wide range of tethered fluorophores, including the AlexaFluor (AF), JaneliaFluor (JF), cyanine, and near infrared (NIR) dyes [25, 26]. JF dyes, which are useful for super resolution microscopy and live cell imaging, do not photobleach as easily as other dyes, and some can be used for fluorescence detection of target protein binding without removing the supernatant [25]. The FNIR-Tag fluorophore was recently reported by Schnermann and coworkers as a tag for in vivo detection of NIR fluorescence [26].
Scheme 1.

Synthesis of XAC-fluorophore conjugates. Reagents and conditions: (a) functionalized fluorophore active ester (TFP or NHS), DMSO, DIPEA/TEA, rt in dark/wrapped in Al foil, 3–18 h, 40% to quantitative yields. Compound 9 was prepared as reported [13]
Scheme 2.

Synthesis of CGS15943-derived fluorescent conjugate 19 by two routes. Reagents and conditions: (a) 6-azidohexan-1-amine, DMF, sodium ascorbate, CuSO4.5H2O, rt, 18 h, 6%; (b) DMSO, triethylamine, Sulfo-Cy7-NHS ester 20, rt, 4 h, 24%. (c) 6-azidohexan-1-amine, DMF, rt, 4 h; (d) DMF, sodium ascorbate, CuSO4.5H2O, rt, 18 h, 5.3%. Compound 17 was prepared as reported [8]
Materials and methods
Chemistry
Materials and methods
AF-dyes were purchased from Thermo Fisher Scientific (New York, USA); CGS15943 and JF dyes were from Tocris (Bio-techne, Minneapolis, MN, USA). Cyanine dyes were purchased from Lumiprobe (Cockeysville, MD, USA). All other chemicals and solvents were from Sigma-Aldrich (St. Louis, MO, USA). Anhydrous solvents were obtained directly from commercial sources. All reactions were carried out under argon atmosphere using anhydrous solvents. Room temperature or rt refers to 25 ± 2 °C. NMR spectra were recorded on a Bruker 400 MHz spectrometer. Chemical shifts are given in ppm (δ), calibrated to the residual solvent or TMS signals for hydrogen, carbon, and internally calibrated by solvent frequency for other nuclei (MestReNova 10.0.2). Exact mass measurements were performed on a proteomics optimized Q-TOF-2 (Micromass, Waters, Milford, MA, USA) mass spectrometer equipped with a standard electrospray ionization (ESI) and modular LockSpray TM interface. The RP-HPLC was performed using Luna 5 µm C18(2)100A, AXIA, 21.2 × 250 mm column (Phenomenex, Torrance, CA, USA). Purity was determined using C18-XDB, 5 µm, 4.6 × 250 mm column (Agilent, Santa Clara, CA, USA), and a 0 → 100% linear gradient of acetonitrile/10 mM triethylammonium acetate (TEAA) as mobile phase at flow rate of 1.0 mL/min. Purity of all the tested compounds was > 95% at 254 nm and/or the respective absorption wavelength in nm, unless noted otherwise.
Radioligands [3H]cyclopentyl-1,3-dipropylxanthine ([3H]DPCPX, 164 Ci/mmol), [3H]-2-[p-(2-carboxyethyl)phenylethylamino]-5′-N-ethylcarboxamido-adenosine ([3H]CGS21680, 30.5 Ci/mmol), and [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide ([125I]I-AB-MECA, 2170 Ci/mmol) for A1, A2A, and A3 receptors, respectively, were purchased from PerkinElmer (Waltham, MA, USA). DMEM medium and 1 M Tris–HCl (pH 7.5) were purchased from Mediatech, Inc. (Herndon, VA, USA). Adenosine deaminase was from Worthington Biochemical Corp. (Lakewood, NJ, USA). AR ligands 9-chloro-2-(2-furanyl)-[1,2,4]triazolo[1,5-c]quinazolin-5-amine (CGS15943) and CGS21680 were from Tocris (Ellisville, MO). XAC was synthesized at NIDDK, National Institutes of Health (Bethesda, MD). All other materials were from Sigma-Aldrich (St. Louis, MO, USA) or other standard commercial sources and of analytical grade. 6-Amino-N-(5-((3-((2-(2-(4-(2,6-dioxo-1,3-dipropyl-2,3,6,7-tetrahydro-1H-purin-8-yl)phenoxy)acetamido)ethyl)amino)-3-oxopropyl)amino)-5-oxopentyl)hexanamide (XAC245, 9) was synthesized in the TFA salt form, as described in Gao et al. [13].
General procedure for the synthesis of XAC-fluorophore analogues (10–16)
A stock solution of XAC245.TFA (9, 6.0 mg, 7.26 µmol) and DIPEA (4.0 µL, 21.79 µmol) in anhydrous DMSO (596 µL) was prepared. To the TFP (2,3,5,6-tetrafluorophenyl)/NHS (N-hydroxysuccinimide)-ester of the fluorophore (1.0 eq.) was added XAC245.TFA-DIPEA-DMSO stock solution (1.5 eq.) under argon atmosphere at room temperature. The reaction vial was covered with Al foil and stirred at room temperature for 18 h. The product was purified by C18-RP-HPLC.
10, XAC245-AF488 triethylamine salt
Reacting AF488-5-TFP ester (1.0 mg, 1.13 µmol) with XAC245 stock solution (150 µL, 1.70 µmol) gave the product as an orange solid (0.88 mg, 59%, purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O (v/v) 05/95 → 45/55 in 40 min, flow rate = 5.0 mL/min, Rt = 39.14 min). 1H NMR (400 MHz, D2O) δ 8.20 (d, J = 1.9 Hz, 1H), 7.92 (dd, J = 7.9, 1.9 Hz, 1H), 7.72 (d, J = 8.5 Hz, 2H), 7.24 (d, J = 7.9 Hz, 1H), 6.98 (d, J = 9.3 Hz, 2H), 6.96–6.91 (m, 2H), 6.77 (d, J = 9.3 Hz, 2H), 4.53 (s, 2H), 3.98 (t, J = 7.5 Hz, 2H), 3.87 (t, J = 7.7 Hz, 2H), 3.47–3.33 (m, 5H), 3.29 (t, J = 6.8 Hz, 2H), 3.07 (t, J = 6.5 Hz, 2H), 2.32 (t, J = 6.8 Hz, 2H), 2.21 (t, J = 7.0 Hz, 2H), 2.05 (t, J = 7.2 Hz, 2H), 1.62 (dp, J = 15.8, 7.7 Hz, 8H), 1.51–1.33 (m, 7H), 0.89 (td, J = 7.4, 4.1 Hz, 6H). HRMS m/z [M-H]− for C56H65O17N11S2 calculated 1226.3923, found 1226.3918.
11, XAC245-AF647 bis-triethylamine salt
Reacting AF647-NHS ester (1.0 mg, 0.80 µmol) with XAC245 stock solution (130 µL, 1.60 µmol) gave the product as a blue-violet solid (1.80 mg, quantitative, purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O (v/v) 20/80 → 35/65 in 40 min, flow rate = 5.0 mL/min, Rt = 26.12 min). 1H NMR (400 MHz, D2O) δ 8.04–7.86 (m, 2H), 7.86–7.72 (m, 5H), 7.31 (d, J = 8.8 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 6.95 (d, J = 8.8 Hz, 2H), 6.42 (t, J = 12.4 Hz, 1H), 6.24 (d, J = 13.5 Hz, 1H), 6.13 (d, J = 13.5 Hz, 1H), 4.57 (s, 2H), 4.18 (s, 2H), 4.12 (t, J = 7.9 Hz, 2H), 3.97 (t, J = 7.6 Hz, 2H), 3.82 (t, J = 7.7 Hz, 2H), 3.45–3.28 (m, 6H), 3.06–2.89 (m, 9H), 2.36 (t, J = 6.8 Hz, 2H), 2.17 (dt, J = 19.4, 7.4 Hz, 2H), 2.11–2.04 (m, 3H), 2.01 (t, J = 7.2 Hz, 2H), 1.68 (q, J = 7.6 Hz, 2H), 1.65–1.56 (m, 9H), 1.56–1.49 (m, 1H), 1.40 (dp, J = 21.8, 7.3 Hz, 4H), 1.14 (dt, J = 17.1, 8.0 Hz, 3H), 0.89 (dt, J = 18.6, 7.4 Hz, 6H). HRMS m/z [M-H]− for C71H97O20N11S4 calculated 1550.5716, found 1550.5736.
12, XAC245-JF646
Reacting JF646-NHS ester (1.0 mg, 1.70 µmol) with XAC245 stock solution (210 µL, 2.5 µmol) gave the product as a light blue solid (1.0 mg, 50%, purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O (v/v) 50/50 → 100/00 in 40 min, flow rate = 5.0 mL/min, Rt = 32.32 min). 1H NMR (400 MHz, DMSO-d6) δ 8.73 (t, J = 5.6 Hz, 1H), 8.17 (t, J = 5.6 Hz, 1H), 8.11–7.97 (m, 3H), 7.93 (t, J = 5.5 Hz, 1H), 7.77 (t, J = 5.7 Hz, 1H), 7.70 (t, J = 5.6 Hz, 1H), 7.66 (s, 1H), 7.01 (d, J = 8.3 Hz, 2H), 6.71 (d, J = 2.7 Hz, 2H), 6.62 (d, J = 8.7 Hz, 2H), 6.32 (dd, J = 8.7, 2.7 Hz, 2H), 4.50 (s, 2H), 3.98 (t, J = 7.3 Hz, 2H), 3.83 (q, J = 7.0 Hz, 10H), 3.18 (ddt, J = 18.0, 11.3, 6.3 Hz, 7H), 2.97 (q, J = 6.5 Hz, 2H), 2.42 (q, J = 7.1 Hz, 2H), 2.29 (p, J = 7.3 Hz, 4H), 2.20 (t, J = 7.0 Hz, 2H), 2.01 (td, J = 7.4, 2.4 Hz, 4H), 1.72 (q, J = 7.4 Hz, 2H), 1.55 (q, J = 7.4 Hz, 2H), 1.43 (dq, J = 14.5, 7.2 Hz, 5H), 1.36–1.15 (m, 8H), 0.97–0.79 (m, 9H), 0.60 (s, 3H), 0.49 (s, 3H). HRMS m/z [M + H]+ for C64H79O10N11Si calculated 1190.5859, found 1190.5859.
13, XAC245-JF549
Reacting JF549-NHS ester (1.15 mg, 2.10 µmol) with XAC245 stock solution (260 µL, 3.15 µmol) gave the product as a dark pink solid (0.83 mg, 40%, purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O (v/v) 05/95 → 95/05 in 40 min, flow rate = 5.0 mL/min, Rt = 30.10 min). 1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 1H), 8.15 (d, J = 9.9 Hz, 2H), 8.07–7.89 (m, 3H), 7.78 (s, 1H), 7.70 (s, 1H), 7.63 (s, 1H), 6.98 (s, 2H), 6.56 (s, 1H), 6.50 (d, J = 8.6 Hz, 1H), 6.21 (d, J = 2.2 Hz, 2H), 6.19–6.10 (m, 2H), 4.48 (s, 2H), 3.97 (s, 2H), 3.85 (t, J = 7.3 Hz, 9H), 3.67 (d, J = 11.0 Hz, 1H), 3.27–3.06 (m, 40H), 2.96 (d, J = 6.6 Hz, 2H), 2.42 (q, J = 7.1 Hz, 2H), 2.37–2.24 (m, 3H), 2.19 (t, J = 7.0 Hz, 2H), 2.00 (d, J = 6.5 Hz, 4H), 1.72 (d, J = 7.8 Hz, 2H), 1.54 (d, J = 7.1 Hz, 1H), 1.43 (s, 5H), 1.36–1.11 (m, 6H), 1.00–0.77 (m, 10H). HRMS m/z [M + H]+ for C62 H73N11O11 calculated 1148.5569, found 1148.5585.
14, XAC245-Sulfo-Cy7
Reacting Sulfo-Cy7-NHS ester (1.06 mg, 1.26 µmol) with XAC245 stock solution (156 µL, 1.90 µmol) gave the product as a blue solid (1.61 mg, 91%, purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O (v/v) 05/95 → 100/00 in 40 min, flow rate = 5.0 mL/min, Rt = 27.36 min). 1H NMR (400 MHz, DMSO-d6) δ 8.22 (s, 1H), 8.11–8.04 (m, 2H), 7.97 (d, J = 5.7 Hz, 1H), 7.83–7.66 (m, 4H), 7.62 (ddd, J = 9.7, 5.5, 2.7 Hz, 1H), 7.28 (dd, J = 13.1, 8.3 Hz, 1H), 7.09 (d, J = 8.9 Hz, 2H), 6.53 (s, 1H), 6.14 (dd, J = 14.2, 6.8 Hz, 2H), 4.54 (s, 2H), 4.10 (s, 2H), 4.01 (t, J = 7.3 Hz, 2H), 3.92–3.81 (m, 2H), 3.61 (s, 3H), 3.25–3.07 (m, 5H), 2.96 (q, J = 6.4 Hz, 4H), 2.20 (t, J = 7.1 Hz, 2H), 2.10–1.95 (m, 6H), 1.88–1.70 (m, 3H), 1.65 (d, J = 3.2 Hz, 10H), 1.56 (dq, J = 14.0, 7.2 Hz, 2H), 1.48–1.38 (m, 2H), 1.25–1.10 (m, 2H), 1.04 (q, J = 7.8, 7.3 Hz, 6H), 0.88 (dt, J = 11.3, 7.4 Hz, 6H). HRMS m/z [M + H]+ for C72H95O14N11S2 calculated 1402.6580, found 1402.6597.
15, XAC245-Sulfo-Cy7.5
Reacting Sulfo-Cy7.5-NHS ester (1.4 mg, 1.20 µmol) with XAC245 stock solution (150 µL, 1.80 µmol) gave the product as a blue solid (1.52 mg, 69%, purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O (v/v) 05/95 → 45/55 in 40 min, flow rate = 5.0 mL/min, Rt = 38.58 min). 1H NMR (400 MHz, D2O) δ 8.72 (dd, J = 14.2, 9.1 Hz, 2H), 8.56 (d, J = 11.4 Hz, 2H), 8.31 (d, J = 8.4 Hz, 1H), 7.48 (dt, J = 30.3, 14.5 Hz, 3H), 7.34 (d, J = 8.2 Hz, 2H), 7.06 (s, 1H), 6.57 (d, J = 8.3 Hz, 2H), 5.79 (dd, J = 28.7, 13.8 Hz, 2H), 4.34 (s, 2H), 3.92–3.65 (m, 6H), 3.53–3.28 (m, 9H), 3.03 (t, J = 6.8 Hz, 2H), 2.73 (d, J = 12.4 Hz, 2H), 2.40 (t, J = 6.8 Hz, 2H), 2.27–2.04 (m, 8H), 1.96 (t, J = 7.5 Hz, 2H), 1.74 (s, 12H), 1.46 (s, 8H), 1.39–1.31 (m, 1H), 1.12 (s, 1H), 0.94 (d, J = 8.5 Hz, 1H), 0.80 (dt, J = 15.2, 7.2 Hz, 8H). HRMS m/z [M + H]+ for C80H97O20N11S4 calculated 1660.5872, found 1660.5852.
16, XAC245-FNIR-Tag
Reacting FNIR-Tag-NHS ester (0.5 mg, 0.43 µmol) with XAC245 stock solution (55 µL, 0.65 µmol) gave the product as a green solid (0.53 mg, 70%, purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O (v/v) 05/95 → 65/35 in 40 min, flow rate = 5.0 mL/min, Rt = 36.35 min). 1H NMR (400 MHz, DMSO-d6) δ 8.17 (s, 1H), 7.95 (t, J = 13.0 Hz, 5H), 7.80 (s, 2H), 7.74 (t, J = 5.6 Hz, 1H), 7.63 (d, J = 8.1 Hz, 1H), 7.33 (d, J = 8.4 Hz, 2H), 6.94 (d, J = 8.6 Hz, 2H), 6.28 (d, J = 14.1 Hz, 2H), 4.46 (s, 2H), 4.36 (d, J = 6.7 Hz, 3H), 3.99 (d, J = 25.1 Hz, 3H), 3.80 (dt, J = 10.4, 6.3 Hz, 5H), 3.65 (s, 1H), 3.51 (dd, J = 5.9, 3.5 Hz, 4H), 3.22 (s, 4H), 3.18 (s, 3H), 3.10–3.02 (m, 1H), 2.97 (d, J = 6.2 Hz, 1H), 2.58 (s, 3H), 2.42 (q, J = 7.1 Hz, 14H), 2.21 (q, J = 7.1 Hz, 3H), 2.05–1.96 (m, 5H), 1.86 (s, 13H), 1.68 (s, 10H), 1.53 (d, J = 7.1 Hz, 1H), 1.43 (s, 4H), 1.34–1.16 (m, 5H), 0.93 (t, J = 7.1 Hz, 20H), 0.86 (dd, J = 14.1, 7.2 Hz, 6H). HRMS m/z [M + H]+ for C88H128O21N12S2 calculated 1753.8837, found 1753.8864.
18, CGS15943-3T6-amine
To a suspension of compound 17 [8] (20 mg, 0.053 mmol) in DMF (1.0 mL) was added sequentially a freshly prepared solution of sodium ascorbate (1 M, 132 µL, 0.132 mmol), 6-azidohexane-1-amine [27] (15 mg, 0.106 mmol) and CuSO4.5H2O (1 M, 27 µL, 0.027 mmol) and the mixture stirred at room temperature for 18 h. The solvent was evaporated under high vacuum, and the residue was purified by silica-gel column chromatography to afford the 18 as a minor product (1.5 mg, 6%; Rf = 0.3, TLC eluent = 10% MeOH in CH2Cl2 + 1% Et3N). 1H NMR (400 MHz, DMSO) δ 8.16 (dd, J = 8.0, 2.6 Hz, 1H), 7.97–7.78 (m, 2H), 7.75–7.63 (m, 1H), 7.56 (d, J = 8.8 Hz, 1H), 7.21 (d, J = 3.8 Hz, 1H), 6.71 (s, 1H), 4.33–4.17 (m, 2H), 2.69 (t, J = 8.7 Hz, 2H), 2.63–2.51 (m, 2H), 1.92 (t, J = 7.4 Hz, 2H), 1.82–1.65 (m, 2H), 1.44–1.11 (m, 6H). HRMS m/z [M + H]+ for C25H28O2N9Cl calculated 522.2133, found 522.2134.
19, CGS15943-3T6-Sulfo-Cy7
First route
A mixture of compound 18 (1.5 mg, 2.87 µmol) and triethylamine (2.0 µL, 14.0 µmol) in anhydrous DMSO (150 µL) was added to Sulfo-Cy7-NHS ester 20 (1.0 mg, 1.20 µmol) under insert atmosphere and stirred at room temperature for 4 h. The product was purified immediately by RP-HPLC to afford compound 19 as a dark-blue solid (0.34 mg, 24%; purified using Agilent C18-XDB 4.6 × 250 mm column and linear gradient of CH3CN/10 mM aq. TEAA (v/v) 20/80 → 55/45 in 20 min at 1.00 mL/min, Rt = 17.34 min). 1H NMR (400 MHz, DMSO) δ 8.39 (s, 1H), 8.00 (s, 1H), 7.91 (s, 2H), 7.75 (s, 3H), 7.61–7.33 (m, 4H), 7.25–7.34 (m, 3H), 6.77 (s, 1H), 6.52 (s, 1H), 6.11–6.19 (m, 2H), 4.28 (t, J = 7.4 Hz, 2H), 4.09 (m, 2H), 3.61 (s, 3H), 2.94 (m, 6H), 2.67–2.77 (m, 4H), 1.91–2.02 (m, 4H), 1.78 (m, 2H), 1.65 (s, 8H), 1.52 (m, 2H), 1.13–1.31 (m, 6H), 1.12 (t, J = 7.1 Hz, 4H). HRMS m/z [M-H]− for C62H70O9N11S2Cl calculated 1210.4410, found 1210.4409. Purity—94.69% at 750 nm; 88.20% at 254 nm.
Second route
To a solution of compound 20 (100 mM, 13 µL, 0.013 mmol) in DMF was added 6-azidohexan-1-amine (100 mM, 15 µL, 0.015 mmol) and the mixture stirred at room temperature for 4 h. The reaction mixture was evaporated under a stream of nitrogen gas. Freshly prepared solution of sodium ascorbate (50 mM, 158 µL, 0.079 mmol), CuSO4∙5H2O (50 mM, 30 µL, 0.015 mmol), and compound 17 (100 mM, 32 µL, 0.032 mmol) were added and the mixture stirred at room temperature for 18 h. The product was purified by RP-HPLC, using the same conditions as above, to afford compound 19 as a dark-blue solid (1.96 mg, 5.3%).
To avoid freeze–thaw cycles, compound 19 was dissolved in EtOH and divided into small aliquots that were dried and stored at − 80 °C for use in binding assays. Radioligand binding was performed as described in Gao et al. [13].
Flow cytometry
Saturation assay: determination of dissociation constant
Flow cytometry was run using a CytoFLEX B4-RO-VO System (Beckman Coulter, Brea, CA, USA). The instrument includes 13 band pass filters which can be repositioned as needed, and it is available with different configurations to provide application flexibility. Violet side scatter resolution: (VSSC) < 200 nm blue laser wavelength: 488 nm; power: 50 mw; beam spot size: 5 × 80 μm; red laser wavelength: 638 nm; power: 50 mw; beam spot size: 5 × 80 μm; violet laser wavelength: 405 nm; power: 80 mw; beam spot size: 5 × 80 μm; flow cell dimensions: 420 μm × 180 μm internal diameter; signal processing: fully digital system with a 7-decade data display; carryover: single tube format < 1.0%; plate loader format < 0.5%.
HEK293 cells expressing either human A3AR or human A2AAR were plated in black 96-well plates overnight in complete DMEM media (DMEM, FBS (10%), penicillin/steptomycin (100 U/mL)). Media was then aspirated, and fresh complete media containing a range of concentrations of fluorescent ligand was added to appropriate cells. Non-specific binding was prepared through the addition of XAC (14 µM final concentration) at each concentration of fluorescent ligand. Cells were incubated for 1 h at 37 °C. Media was then aspirated, and cells were detached with non-enzymatic Cellstripper and resuspended in 200 µL PBS (without Ca2+, Mg2+). Cell fluorescence was measured on a CytoFLEX B4-RO-VO system. For the detection of 19 and 12, a 638-nm laser was employed and a 780/60 bandpass filter and a 660/20 bandpass filter were used, respectively. The data were analyzed using CellQuest Software. Specific binding was calculated by subtracting non-specific binding from total binding.
Competitive binding assay
For competitive binding experiments, chemically transfected HEK293 cells expressing either human A3AR or human A2AAR were plated in black 96-well plates overnight in complete DMEM media (DMEM, FBS (10%), penicillin/streptomycin (100 U/mL)). The media was then aspirated and increasing concentrations of ligand in complete DMEM media were incubated with 12 (250 nM) for hA2AAR or 19 (100 nM) for hA3AR expressing cells at 37 °C for 1 h. XAC (14 uM) was used to determine non-specific binding. Media was then aspirated, and cells were detached with non-enzymatic Cellstripper and resuspended in PBS (without Ca2+, Mg2+). Cell fluorescence was measured on a Beckman Coulter CytoFLEX B4-RO-VO system. For the detection of 19 and 12, a 638-nm laser was employed and a 780/60 bandpass filter and a 660/20 bandpass filter were used, respectively. The data were analyzed using CellQuest Software. Specific binding was calculated by subtracting non-specific binding from total binding. The inhibition constants (Ki), dissociation constants (Kd), and kinetic constants were calculated using Prism, version 9.0.0 (GraphPad, San Diego, CA).
Determination of off-rate
Chemically transfected HEK293 cells expressing human A3AR were plated in black 96-well plates overnight in complete DMEM media (DMEM, FBS (10%), penicillin/streptomycin (100 U/mL)). Media was then aspirated, and fresh complete media containing a saturating concentration of 19 (400 nM) was added to each well. Cells were then incubated for 1 h at 37 °C. Media was then aspirated and PBS (without Ca2+, Mg2+) containing a saturating concentration of XAC (100 µM) was added to each well. Upon addition of XAC, data collection immediately began and continued for various timepoints using a Beckman Coulter CytoFLEX B4-RO-VO system with a 638-nm red laser and a 780/60 bandpass filter. Dissociation of 19 was followed by the change in mean fluorescence intensity (MFI) vs time. Data were analyzed using CellQuest Software.
The laser wavelengths of the longpass/bandpass filters used for the flow cytometry experiment were not optimal for compound 19. The λmax of the Sulfo-Cy7 fluorophore of 19 is 750 nm. Therefore, the 638-nm laser used to excite 19 resulted in low fluorescence emission consistent with non-optimal excitation. However, the emission signal resulting from the 638-nm excitation was sufficient to obtain a signal.
Confocal microscopy
HEK293 cells transiently expressing human A2AAR were plated in 8-well Ibibi µ-slides and grown overnight in complete DMEM media (DMEM, FBS (10%), containing penicillin/streptomycin (100 U/mL)). The media was removed, and total binding was determined by treatment of cells with 500 nM 12 in complete DMEM media. Non-specific binding was determined by cotreatment of cells with 500 nM 12 and 50 µM XAC. Cells were incubated with treatments for 1 h. Media was removed and DPBS (w/Ca2+ & Mg2+) was added.
Cell fluorescent imaging
A Zeiss LSM 700 confocal scanhead (Carl Zeiss Microscopy, LLC, Thornwood, NY, USA) mounted on a Zeiss AxioObserver.Z1 microscope, running ZEN 2.3 software, was used to simultaneously collect brightfield with fluorescence overlay images. The objective lens used was the Zeiss Plan-Apo 63 × /1.4 Oil DIC. The 639-nm laser was used for illumination/excitation, at 28.4% power. Emission was filtered by a 640-nm longpass filter. Imaging was set at a scan field of 203.12 µm × 203.12 µm with a pinhole of 61 µm, which corresponds to a confocal sectioning thickness of 1.0 µ.
Molecular modeling
The model of hA3AR was retrieved from a previous work [28], where it was obtained through homology modeling using an antagonist-bound inactive structure of hA1AR (PDB ID: 5UEN [29]) as a template, minimization of non-conserved residues and induced fit docking of the high affinity A3AR antagonist N-[9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-yl]benzeneacetamide (MRS1220).
The structure was prepared with the Protein Preparation Wizard tool [30] (Schrödinger suite, Maestro 2021–2 [31], New York, NY, USA). The predicted tautomeric state of histidines was HIE (hydrogen at Nε nitrogen) for H79, H95, H124, H158 and H304, and HID (hydrogen at Nδ nitrogen) for H272.
Compound 19 was docked at the receptor orthosteric binding site using Glide-XP [32–34], employing a grid centered on the centroid of F168 and N250, and with inner and outer box dimensions of 10 Å and 46 Å, respectively. The conformational search was restricted constraining the scaffold of compound 9 to the reference pose of MRS1220, using the maximum common substructure option and an RMSD tolerance of 2 Å. A maximum of 20 poses was set as output, and a post-docking minimization phase was included. One pose was generated.
Results
Chemistry
To prepare various fluorescent compounds for this study, derivatives of XAC 7 and CGS15943 8 with a linker containing a primary amine group were prepared and coupled with respective chromophores (Schemes 1 and 2). XAC245-amine TFA salt 9 was synthesized by successive addition of N-Boc-amino acids as reported earlier [13]. The pharmacologically active XAC-fluorophore conjugates 10–16 were synthesized by the reaction of corresponding amines with activated fluorophore esters under basic conditions (Scheme 1). Cyanine dyes appear in compound 11 and NIR probes 14–16. The two JF dye conjugates containing azetidine moieties are 12 (JF649) and 13 (JF549). Compound 16 contains a net neutral heptamethine cyanine with a stable C4′-O-alkyl linker as reported [26]. This fluorophore has a λmax of 765 nm, with emission at 788 nm. Sulfo-Cy7 conjugate 19 has a λmax of ~ 750 nm, with emission at ~ 773 nm. The spectral characteristics of each fluorophore are given in Supporting Information.
For the synthesis of an 8-derived fluorescent compound 19, a click reaction between alkyne 17 [8] and an azide [27] gave the product 18 in low yields (Scheme 2). The reaction had unreacted starting material 17 and the major by-product 8, both of which were recovered. An amide bond formation by the amino group in compound 18 with activated carbonyl of Sulfo-Cy7-NHS ester 20 gave the desired product CGS15943-3T6-Sulfo-Cy7 19 in acceptable yields. A second, more efficient route involved the coupling of the fluorophore as an active ester 20 to 6-azidohexan-1-amine, followed by click reaction of the resulting azide 21 to the alkyne-functionalized pharmacophore 17. The overall yield of the second route (compound 19 compared to the amount of compound 17) was ~ tenfold higher than the first route.
Affinity determination
The AR affinity of the fluorescent derivatives was initially determined using standard radioligand binding methods [10, 13] in membranes of HEK293 cells overexpressing each receptor: A1, A2A, and A3 (Table 1). The highest affinity was observed for the fluorescent conjugates 10, 12, and 19 at the A2AAR (Ki 144–316 nM). The A1AR affinity was generally low (Ki > 1 µM) and only percent inhibition is indicated. The A3AR and A2AAR affinities were consistently higher than at the A1AR with the highest A3AR affinity (Ki 21.6 nM) observed for 19. Compounds 10 and 13–15 were inactive at the A3AR. Thus, compound 12 was highly selective for the A2AAR compared to the A1AR, but only 3.4-fold selective compared to A3AR. Compounds 13–15 were > two-fold selective for the A2AAR compared to the A3AR. Binding affinity was measured for two compounds at the hA2BAR, using [3H]8-cyclopentyl-1,3-dipropylxanthine ([3H]DPCPX) as radioligand in membranes of transfected HEK293 cells [28]. At a concentration of 1 µM, compounds 12 and 19 inhibited binding by only 57.1% and 38.5%, respectively. Thus, these two fluorescent ligands were selective for their respective target receptors compared to the A2BAR.
Table 1.
Fluorescent ligand binding affinitiesa at three AR subtypes
| No | Compound |
hA1, Ki (nM) or % inhib. at 1 µM |
hA2A Ki (nM) or % inhib. at 1 µM |
hA3 Ki (nM) or % inhib. at 1 µM |
|---|---|---|---|---|
| XAC 7 and its conjugates | ||||
| 7 | XACb | 8.3 ± 1.1 | 32.1 ± 5.9 | 38.3 ± 6.7 |
| 9 | XAC-245b | 35% | 198 ± 33 | 220 ± 36 |
| 10 | XAC245-AF488 | 19% | 316 ± 24 | 40 ± 3% |
| 11 | XAC245-AF647 | 18% | 37 ± 21% | 41 ± 5% |
| 12 | XAC245-JF646c | 53% | 144 ± 24 | 495 ± 89 |
| 13 | XAC245-JF549 | 29% | 882 ± 215 | 36.9 ± 4.3% |
| 14 | XAC245-Sulfo-Cy7 (750 nm)d | 20% | 1290 ± 470 | 49.7 ± 11.8% |
| 15 | XAC245-Sulfo-Cy7.5 (778 nm)d | 27% | 1120 ± 310 | 30.8 ± 3.5% |
| 16 | XAC245-FNIR-Tag (765 nm)d | 27% | 956 ± 232 | 232 ± 53 |
| CGS15943 8 and its conjugates | ||||
| 8 | CGS15943e | 3.5 | 1.2 | 35 |
| 5 | MRS5449e | 87 ± 24 | 73 ± 8 | 6.4 ± 2.5 |
| 17 | Alkyne deriv.e | 170 ± 40 | 51 ± 10 | 33.5 ± 10.1 |
| 19 | CGS15943-3T6-Sulfo-Cy7c | 53% | 244 | 21.6 ± 4.0 |
aDetermined with membranes of HEK293 cell expressing each receptor and using the following radioligands (final concentration in incubation medium), unless noted: A1, [3H]DPCPX (0.7 nM); A2A, [3H]CGS21680 (8.1 nM); A3, [.125I]I-AB-MECA (0.15 nM). Results are expressed as mean ± SEM from at least 3 independent experiments
bA2A and A3 AR affinities reported in Gao et al. [13]. A1AR affinity of XAC (using [3H]R-PIA) is from Kecskés et al. [46]
c12, MRS7774; 19, MRS7535
dExcitation wavelength
eAR affinities reported in Kozma et al. [8]
Application to flow cytometry and fluorescence microscopy
The objective was to determine the suitability of these fluorescent conjugates for use in binding assays by flow cytometry and in fluorescence microscopy. The conjugate with the highest A3AR affinity was compound 19 , with a radioligand binding Ki value of 21.6 nM. The fluorophore of 19 was a NIR dye with a maximum excitation wavelength of 750 nm, which could be sufficiently exited with a red laser (638 nm) to obtain significant emission due to the high quantum yield (0.88) of the dye. A saturation experiment (Fig. 2) using human embryonic kidney (HEK) 293 cells overexpressing the A3AR indicated an acceptably low level of non-specific binding (determined using 14 µM XAC). Non-specific binding was linear with respect to the fluorescent ligand concentration (up to 200 nM). The specific binding of 19 was saturable up to a concentration of 200 nM. A Kd value of 11.8 ± 0.7 nM was determined by nonlinear regression analysis using a one-site specific binding model (Eq. 1: y = Bmax*x/(Kd + x). In non-AR-expressing control HEK293 cells, specific binding of 19 was absent.
Fig. 2.

Representative saturation binding assay with 19 using FCM in HEK293 cells expressing hA3AR (A, B) or non-transfected HEK293 cells (C, D), N = 2 for all experiments. Fluorescence intensity was measured using a Beckman Coulter CytoFLEX B4-RO-VO System with a 638-nm laser and a 780/60 bandpass filter. (A) Total binding of HEK293 cells expressing hA3AR was determined by treating cells with a range of concentrations of 19, and non-specific binding was determined in the presence of XAC (14 µM). (B) Specific binding of 19 at HEK293 cells expressing hA3AR, calculated as total binding MFI minus non-specific binding MFI. The dissociation constant (Kd) was calculated to be 11.8 ± 0.7 nM. Results reported as mean ± SEM. Note that the Y-axis in (B) has a different scale compared to other plots. (C) Total and nonspecific binding of 19 with non-transfected HEK293 cells. (D) Lack of specific binding of 19 with non-transfected HEK293 cells
Kinetic experiments showed the off-rate to be 0.0118 nM−1 min−1 (Fig. 3), while the on-rate calculated from binding saturation Kd and koff was 0.334 min−1 (N = 1).
Fig. 3.
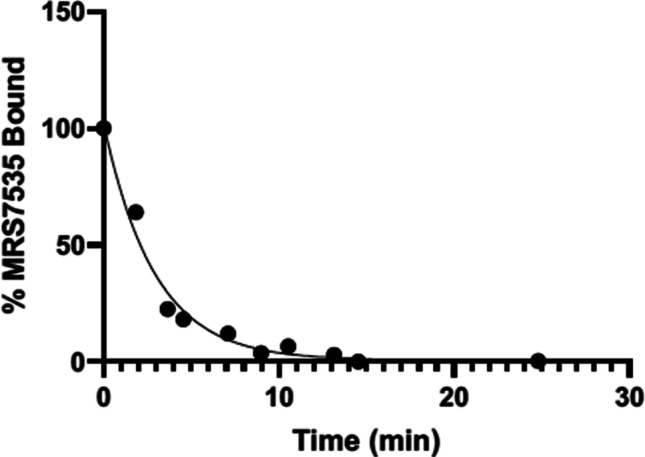
Kinetic determination of koff for 19 at hA3AR in HEK293 cells expressing hA3AR (N = 2). Cells were treated with a saturating concentration of 19 (400 nM) in complete DMEM media and incubated for 1 h at 37 °C. Media was removed, and PBS (without Ca2+, Mg2+) containing XAC (100 µM) was added to the cells. MFI was measured over time. Data were fit to a monophasic decay curve. Apparent koff was measured to be 0.334 min−1 (N = 1). Results reported as mean ± SEM
Fluorescent binding inhibition (Fig. 4) by known A3AR agonist IB-MECA and AR antagonist XAC was performed using a fixed concentration of 19 (100 nM) resulting in sigmoidal inhibition curves with Hill coefficients ~ 1. This high concentration (~ 10X the Kd of 19) was needed to obtain an adequate signal. The Ki values of 71 and 171 nM for IB-MECA and XAC, respectively, were of the same rank order, but roughly an order of magnitude higher than radioligand binding Ki values obtained for the same ligands at the hA3AR. Their reported Ki values using an A3AR agonist radioligand were 1.8 and 13.8, respectively [8].
Fig. 4.
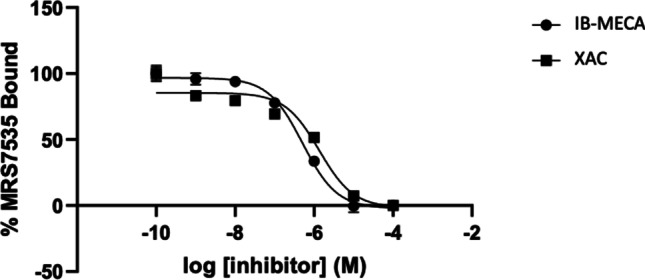
Competitive binding of 19 with a non-selective antagonist, XAC, and an A3AR-selective agonist, IB-MECA, in HEK293 cells expressing hA3AR determined using FCM. Fluorescence intensity was measured using a Beckman Coulter CytoFLEX B4-RO-VO System with a 638-nm laser and a 780/60 bandpass filter. A Ki of 171 ± 29 nM (N = 2) was calculated for XAC and 71 ± 17 nM (N = 2) for IB-MECA based on a Kd of 11.8 nM for 19 at hA2AAR
A2AAR flow cytometry (Fig. 5 and Fig. 6) and microscopic cell imaging data (Fig. 7) were determined using the conjugate 12 showing the highest A2AAR affinity. A FCM binding saturation experiment demonstrated a Kd value of 222 nM at the hA2AAR by Scatchard analysis, which is close to its binding Ki value in Table 1. Competition for A2AAR binding by non-selective AR antagonist, XAC, and A2AAR-selective agonist, CGS21680, was determined using FCM. The sigmoidal inhibition curves indicated Ki values of 53 and 783, respectively, compared to Ki values of 38.3 and 16.3 nM using an agonist radioligand [13]. Thus, the affinity determined for antagonist XAC was similar to previous values, but not the affinity of agonist CGS21680, using this antagonist fluorescent ligand. By comparison, the inhibition of the hA2AAR binding in whole cells using fluorescent antagonist 6 by agonists was complex, possibly due to multiple affinity states of this GPCR for agonists [10]. The cell images (Fig. 7B, C) show specific binding on the outer cell membrane, which disappeared when 50 µM XAC was present to block the fluorescent binding (Fig. 7E, F). Some cells showed no binding, which is reasonable considering that a transient transfection of the HEK293 cells was used.
Fig. 5.

Representative saturation binding assay with 12 using FCM in HEK293 cells expressing hA2AAR. Fluorescence intensity was measured using a Beckman Coulter CytoFLEX B4-RO-VO System with a 638-nm laser and a 660/20 bandpass filter. (A) Total binding of HEK293 cells expressing hA2AAR was determined by treating cells with a range of concentrations of 12 and non-specific binding determined in the presence of XAC (14 µM). (B) Specific binding of 12 at HEK293 cells expressing hA2AAR, calculated as total binding MFI minus non-specific binding MFI. The dissociation constant (Kd) was determined to be 222 ± 64 nM (N = 3)
Fig. 6.
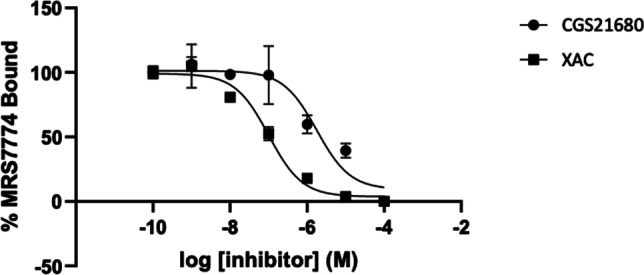
Competitive binding of 12 with antagonist, XAC and A2AAR-selective agonist, CGS21680 in HEK293 cells expressing hA2AAR. Fluorescence intensity was measured using a Beckman Coulter CytoFLEX B4-RO-VO System with a 638-nm laser and a 660/20 bandpass filter. A Ki of 53 ± 6 nM (N = 2) was calculated for XAC and 783 ± 52 nM (N = 2) for CGS21680 based on a Kd of 222 nM for 12 at hA2AAR
Fig. 7.

Confocal microscopy images of HEK293 cells transfected with hA2AAR (transient). (A) and (D) are bright field images, (B) and (E) are bright field images with fluorescent overlay, and (C) and (F) are fluorescent images. The cells in images (A), (B), and (C) were treated with 12 (500 nM) for 1 h at 37 °C to measure the total binding of 12. The cells in images (D), (E), and (F) were treated with both 12 (500 nM) and XAC (50 µM) to measure the non-specific binding of 12
Computational studies
The hA3AR interaction with antagonists was previously modeled using computational docking based on X-ray crystallographic structures of an antagonist-bound hA2AAR (PDB ID: 4EIY) [35, 36]. Here, a hypothetical binding mode was predicted for compound 19 at hA3AR orthosteric binding site using molecular docking based on a structurally similar template (Fig. 8). The structure of hA3AR was obtained through homology modeling, using as template an antagonist-bound hA1AR structure in the inactive state (PDB ID: 5UEN [29]), as recently reported [28]. The model was previously optimized by induced fit docking of the known hA3AR antagonist MRS1220 (Ki ≈ 0.7 nM), which shares with compound 19 the 9-chloro-2-(2-furanyl)-[1,2,4]triazolo[1,5-c]quinazolin-5-amido scaffold [28]. The maximum common substructure between compound 19 and MRS1220 was used to constrain the docking search of compound 19. The predicted pose of the ligand interacts with F168 (EL2) through a π-π stacking interaction, with N250 (TM6) through a bidentate H-bond engaging N3 and the amido group at position 5. The carbonyl oxygen of the 5-amido group can be stabilized by a H-bond with Q167 on EL2, and further interactions of extracellular residues may involve the Sulfo-Cy7 group: in particular, in the predicted poses, the two sulfonic groups are interacting through a H-bond with the amido group of V259 on EL3 and through an ionic H-bond with the side chain of R173 in EL2. Since the linker connecting the fluorophore to the triazoloquinazoline scaffold is flexible and its interactions are solvent exposed, we expect them just to be transient and not to contribute extensively to the ligand stabilization. The flexibility and lack of stable interactions of a fluorophore-conjugated moiety is pointed out in a recently determined X-ray structure of a stabilized hA2AAR bound to a BODIPY-conjugated pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine high affinity antagonist, which was the first X-ray structure of a GPCR bound to a fluorophore-conjugated ligand [37]. The chain linking the fluorophore to the N7-position of the pyrazolotriazolopyrimidine scaffold folds back upon the ELs of the receptor, pointing out of the orthosteric pocket, but the fluorophore could not be unambiguously solved because of the high flexibility of the linker. Instead, the triazolopyrimidine scaffold assumes the typical pattern of interactions at the hA2AAR involving the bidentate hydrogen bond with N253 (TM6) and π-π interaction with F168(EL2), resembling the predicted binding mode of the triazolopyrimidine substructure of compound 19 at hA3AR binding site.
Fig. 8.
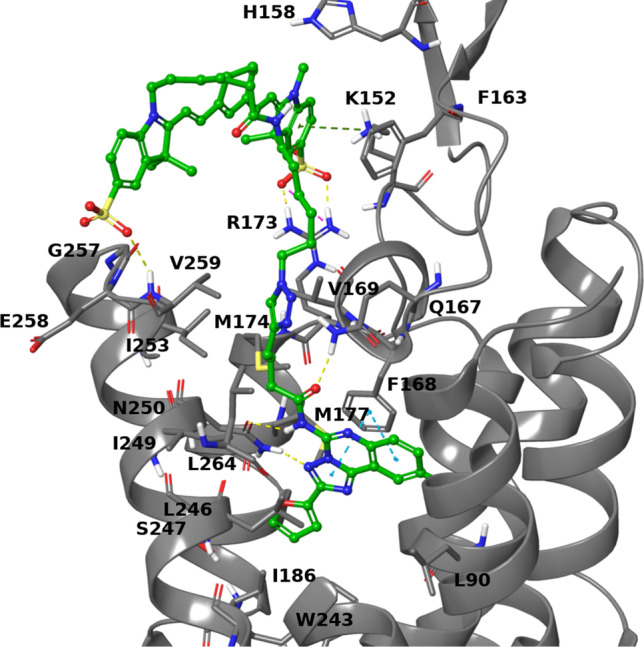
Docking pose of compound 19 (C atoms in green) at the hA3AR (gray) orthosteric binding site. Residues within 3 Å of the ligand are shown as sticks
Discussion
Since the first instance of fluorescence observed for a chemical compound, i.e., quinine in 1845, much of the innovation of new specialized fluorophores has been driven by the proven utility of fluorescent compounds as biomolecular labels, cellular stains, and mechanistic probes [25, 38–40]. Part of the impetus to develop high affinity fluorescent ligands for ARs also arose from the cost and regulatory restrictions associated with the use of radioligands. Widespread availability of robust fluorescent AR ligands would make this research more economical and accessible to researchers worldwide.
Currently, rhodamine dyes are one of the most useful classes of fluorescent small molecules due to their brightness, the ease by which a large library of spectrally diverse dyes can be made through structural modification, and their characteristic chemical transformation from the non-fluorescent lactone form to the fluorescent zwitterionic form upon binding to their molecular target [25, 40]. Upon fluorophore binding, the lactone-zwitterion equilibrium is shifted toward the fluorescent zwitterion form, such that bound ligand fluoresces while non-bound ligand remains colorless [40]. This property of modified rhodamine dyes potentially eliminates the need for washing excess ligand when conducting cell imaging and binding studies [40]. One such rhodamine fluorophore is JF646, which can be easily coupled via the NHS ester to a wide variety of agonists and antagonists for live cell imaging and pharmacological characterization of different cellular receptors [40].
Fluorescent A2AAR antagonist 12, which contains the JF646 fluorophore, was effective as a high-affinity flow cytometry probe for the A2AAR (Kd 222 ± 64 nM). The success of 12 in flow cytometric competitive binding assays with canonical antagonist XAC suggested that 12 is a suitable A2AAR fluorescent ligand for antagonist drug screening. Competitive binding of 12 with agonist CGS21680 did follow monophasic, sigmoidal inhibition. Thus, agonist screening using 12 appears to be more complex than antagonist screening. The potential of 12 as an A2AAR dye for fluorescent confocal microscopy was also confirmed, with 12 efficiently labeling the hA2AAR in a specific manner. The selective activation of JF646 fluorescent activity only upon binding of the ligand to its molecular target, allowed for wash free imaging of hA2AAR in live cells.
Fluorescent A3AR antagonist 19 (Kd 11.8 ± 0.7 nM), containing the NIR Sulfo-Cy7, displayed high affinity and modest selectivity (11-fold vs. A2AAR, roughly 50-fold vs. A1AR and A2BAR) and a good specific-to-nonspecific binding ratio. While 19 was demonstrated to be an effective flow cytometric probe for the A3AR, the high excitation wavelength required to obtain a strong fluorescent emission signal presents a challenge since many flow cytometers are not equipped with far-red lasers. The greatest potential of 19 could be its application for in vivo imaging as a diagnostic NIR agent for imaging and/or therapy targeting the A3AR [41, 42], although extensive pharmacokinetic characterization and additional control experiments would be required for in vivo studies. Near infrared light is particularly well suited for in vivo imaging because of the low levels of biomolecular absorption and general cell autofluorescence in this wavelength range [43]. NIR fluorophore-tagged GPCR ligands have been used for live-cell imaging experiments, for example, the human orexin type 2 receptor [44]. A Cy5.5 fluorophore conjugated to a peripherally selective, high affinity agonist of long half-life was used to image the oxytocin receptor in living mice [45].
Conclusions
We have introduced novel fluorescent conjugates of known AR pharmacophore functionalized congeners in two chemical series, 8-aryl-1,3-dialkylxanthines and triazolo[1,5-c]quinazolin-5-yl)amine. Among the conjugates synthesized and characterized are a JF probe 12, which binds and can be imaged at the A2AAR in whole cells, and an A3AR NIR antagonist probe 19, containing a Sulfo-Cy7 NIR dye, of even higher affinity, which can be used as a screening tool in flow cytometry for drug discovery. We also predicted a bitopic interaction mode of 19 with an A3AR homology model.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Dr. Martin J. Schnermann, NCI, Frederick, for a generous supply of FNIR-Tag-NHS ester. We thank Tocris for the gift of JF dyes. We thank the NIDDK Advanced Light Microscopy & Image Analysis Core (ALMIAC) for the use of its resources and its director Jeff Reece for helpful discussions.
Abbreviations
- AN
Acetonitrile
- AR
Adenosine receptor
- BY
BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene)
- CGS15943
9-Chloro-2-(furan-2-yl)-[1,2,4]triazolo[1,5-c]quinazolin-5-amine
- CGS21680
2-[p-(2-Carboxyethyl)phenylethylamino]-5′-N-ethylcarboxamido-adenosine
- DIPEA
Diisopropylethylamine
- DMEM
Dulbecco’s modified Eagle medium
- DMF
N,N-Dimethylformamide
- DPCPX
Cyclopentyl-1,3-dipropylxanthine
- EL
Extracellular loop
- IE
Interaction energy
- FBS
Fetal bovine serum
- GPCR
G protein-coupled receptor
- HEK
Human embryonic kidney
- I-AB-MECA
N6-(4-Amino-3-iodobenzyl)adenosine-5′-N-methyluronamide
- MD
Molecular dynamics
- MFI
Mean fluorescence intensity
- NHS
N-Hydroxysuccinimide
- NIR
Near infrared
- RMSD
Root mean squared deviation
- TEAA
Triethylammonium acetate
- TFA
Trifluoroacetic acid
- TFP
2,3,5,6-Tetrafluorophenyl
- TM
Transmembrane helical domain
- XAC
N-(2-aminoethyl)-2-[4-(2,6-dioxo-1,3-dipropyl-2,3,6,7-tetrahydro-1H-purin-8-yl)phenoxy]acetamide
- ZM241385
4-[2-[7-Amino-2-(2-furyl)-1,2,4-triazolo[1,5-a][1,3,5]triazin-5-yl-amino]ethyl]phenol
Kiran S. Toti,
was a post-doctoral fellow in Dr Jacobson’s research group at NIH, USA, from 2014-2020. He holds a PhD degree from Ghent University, Belgium, for the dissertation titled ‘Synthesis and Biological Evaluation of 4-Hydroxymethyl Deleted, Transposed and Modified Nucleosides’ under the guidance of Prof. Serge Van Calenbergh. Kiran has 10+ years medicinal chemistry research experience working on GPCR, kinase and polymerase small molecule modulators involving nucleosi(t)ides and heterocycles. At present, he is an Assoc. Scientist in the group of Prof. Dennis Liotta at the Chemistry Department, Emory University.

Funding
National Institute of Diabetes and Digestive and Kidney Diseases, Intramural Research Program (grant no. ZIADK031117).
Data availability
The original data from this study will be made available upon reasonable request.
Declarations
Conflict of interest
Kiran S. Toti declares that he has no conflict of interest. Ryan G. Campbell declares that she has no conflict of interest. Hobin Lee declares that she has no conflict of interest. Veronica Salmaso declares that she has no conflict of interest. R. Rama Suresh declares that he has no conflict of interest. Zhan-Guo Gao declares that he has no conflict of interest. Kenneth A. Jacobson declares that he has no conflict of interest.
Ethical approval
Not applicable; no animal studies are included.
Consent to participate
Not applicable; no patient studies are included.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Baker JG, Middleton R, Adams L, May LT, Briddon SJ, Kellam B, Hill SJ. Influence of fluorophore and linker composition on the pharmacology of fluorescent adenosine A1 receptor ligands. Br J Pharmacol. 2010;159:772–786. doi: 10.1111/j.1476-5381.2009.00488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciruela F, Fernández-Dueñas V, Jacobson KA. Lighting up G protein-coupled purinergic receptors with engineered fluorescent ligands. Neuropharmacology. 2015;98:58–67. doi: 10.1016/j.neuropharm.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Comeo E, Kindon ND, Soave M, Stoddart LA, Kilpatrick LE, Scammells PJ, Hill SJ, Kellam B. Subtype-selective fluorescent ligands as pharmacological research tools for the human adenosine A2A receptor. J Med Chem. 2020;63:2656–2672. doi: 10.1021/acs.jmedchem.9b01856. [DOI] [PubMed] [Google Scholar]
- 4.McCabe RT, Skolnick P, Jacobson KA. 2-[2-[4-[2-[2-[1,3-Dihydro-1,1-bis (4-hydroxyphenyl)-3-oxo-5-isobenzofuranthioureidyl]ethylaminocarbonyl]ethyl]phenyl]-ethylamino]-5′-N-ethylcarboxamidoadenosine (FITC-APEC): a fluorescent ligand for A2-adenosine receptors. J Fluoresc. 1992;2:217–223. doi: 10.1007/BF00865279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernández-Dueñas V, Gómez-Soler M, Jacobson KA, Kumar TS, Fuxe K, Borroto-Escuela DO, Ciruela F. Molecular determinants of the adenosine A2AR-dopamine D2 receptor-receptor allosterism: role of the intracellular loop 3 of the dopamine D2 receptor. J Neurochem. 2012;123:373–384. doi: 10.1111/j.1471-4159.2012.07956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.May LT, Bridge LJ, Stoddart LA, Briddon SJ, Hill SJ. Allosteric interactions across native adenosine-A3 receptor homodimers: quantification using single-cell ligand-binding kinetics. FASEB J. 2011;25:3465–3476. doi: 10.1096/fj.11-186296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briddon SJ, Middleton RJ, Cordeaux Y, Flavin FM, Weinstein JA, George MW, Kellam B, Hill SJ. Quantitative analysis of the formation and diffusion of A1-adenosine receptor-antagonist complexes in single living cells. Proc Natl Acad Sci USA. 2004;101:4673–4678. doi: 10.1073/pnas.0400420101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kozma E, Kumar TS, Federico S, Phan K, Balasubramanian R, Gao ZG, Paoletta S, Moro S, Spalluto G, Jacobson KA. Novel fluorescent antagonist as a molecular probe in A3 adenosine receptor binding assays using flow cytometry. Biochem Pharmacol. 2012;83:1552–1561. doi: 10.1016/j.bcp.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vernall AJ, Stoddart LA, Briddon SJ, Ng HW, Laughton CA, Doughty SW, Hill SJ, Kellam B. Conversion of a non-selective adenosine receptor antagonist into A3-selective high affinity fluorescent probes using peptide-based linkers. Org Biomol Chem. 2013;11:5673–5682. doi: 10.1039/c3ob41221k. [DOI] [PubMed] [Google Scholar]
- 10.Duroux R, Ciancetta A, Mannes P, Yu J, Boyapati S, Gizewski E, Yous S, Ciruela F, Auchampach JA, Gao ZG, Jacobson KA. Bitopic fluorescent antagonists of the A2A adenosine receptor based on pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine functionalized congeners. Med Chem Commun. 2017;8:1659–1667. doi: 10.1039/C7MD00247E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouzo-Lorenzo M, Stoddart LA, Xia L, IJzerman AP, Heitman LH, Briddon SJ, Hill SJ. A live cell NanoBRET binding assay allows the study of ligand-binding kinetics to the adenosine A3 receptor. Purinergic Signal. 2019;15:139–153. doi: 10.1007/s11302-019-09650-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Federico S, Margiotta E, Moro S, Kozma E, Gao ZG, Jacobson KA, Spalluto G. Conjugable A3 adenosine receptor antagonists for the development of functionalized ligands and their use in fluorescent probes. Eur J Med Chem. 2020;186:11188. doi: 10.1016/j.ejmech.2019.111886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao ZG, Toti KS, Campbell R, Suresh RR, Yang H, Jacobson KA. Allosteric antagonism of the A2A adenosine receptor by a series of bitopic ligands. Cells. 2020;9:1200. doi: 10.3390/cells9051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Köse M, Gollos S, Karcz T, Fiene A, Heisig F, Behrenswerth A, Kieć-Kononowicz K, Namasivayam V, Müller CE. Fluorescent-labeled selective adenosine A2B receptor antagonist enables competition binding assay by flow cytometry. J Med Chem. 2018;61:4301–4316. doi: 10.1021/acs.jmedchem.7b01627. [DOI] [PubMed] [Google Scholar]
- 15.Stoddart LA, Kindon ND, Otun O, Harwood CR, Patera F, Veprintsev DB, Woolard J, Briddon SJ, Franks HA, Hill SJ, Kellam B. Ligand-directed covalent labelling of a GPCR with a fluorescent tag in live cells. Commun Biol. 2020;3:722. doi: 10.1038/s42003-020-01451-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobson KA, Tosh DK, Jain S, Gao ZG. Historical and current adenosine receptor agonists in preclinical and clinical development. Frontiers Cell Neurosci. 2019;13:124. doi: 10.3389/fncel.2019.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boison D, Rho JM. Epigenetics and epilepsy prevention: The therapeutic potential of adenosine and metabolic therapies. Neuropharmacology. 2020;167:107741. doi: 10.1016/j.neuropharm.2019.107741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martí Navia A, Dal Ben D, Lambertucci C, Spinaci A, Volpini R, Marques-Morgado I, Coelho JE, Lopes LV, Marucci G, Buccioni M (2020) Adenosine receptors as neuroinflammation modulators: role of A1 agonists and A2A antagonists. Cells 9:1739 [DOI] [PMC free article] [PubMed]
- 19.Sacramento JF, Martins FO, Rodrigues T, Matafome P, Ribeiro MJ, Olea E, Conde SV. A2 adenosine receptors mediate whole-body insulin sensitivity in a prediabetes animal model: primary effects on skeletal muscle. Frontiers Endocrinol. 2020;11:262. doi: 10.3389/fendo.2020.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu F, Zhu C, Xie Q, Wang Y. Adenosine A2A receptor antagonists for cancer immunotherapy. J Med Chem. 2020;63:12196–12212. doi: 10.1021/acs.jmedchem.0c00237. [DOI] [PubMed] [Google Scholar]
- 21.de Oliveira M, Mathias LS, de Sibio MT, Noronha-Matos JB, Costa MA, Nogueira CR, Correia-de-Sá P. Pitfalls and challenges of the purinergic signaling cascade in obesity. Biochem Pharmacol. 2020;182:114214. doi: 10.1016/j.bcp.2020.114214. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson KA, Ukena D, Kirk KL, Daly JW. [3H]Xanthine amine congener of 1,3-dipropyl-8-phenylxanthine: an antagonist radioligand for adenosine receptors. Proc Natl Acad Sci USA. 1986;83:4089–4093. doi: 10.1073/pnas.83.11.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim YC, de Zwart M, Chang L, Moro S, von Frijtag Drabbe Künzel JK, Melman N, IJzerman AP, Jacobson KA (1998) Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) having high potency at the human A2B and A3 receptor subtypes. J Med Chem 41:2835–2841 [DOI] [PubMed]
- 24.Jacobson KA, IJzerman AP, Müller CE (2021) Medicinal chemistry of P2 and adenosine receptors: common scaffolds adapted for multiple targets, Biochem. Pharmacol 187:114311 [DOI] [PMC free article] [PubMed]
- 25.Grimm JB, Muthusamy AK, Liang Y, Brown TA, Lemon WC, Patel R, Lu R, Macklin JJ, Keller PJ, Ji N, Lavis LD. A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nature Meth. 2017;14:987–994. doi: 10.1038/nmeth.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luciano MP, Crooke SN, Nourian S, Dingle I, Nani RR, Kline G, Patel NL, Robinson CM, Difilippantonio S, Kalen JD, Finn MG, Schnermann MJ. A nonaggregating heptamethine cyanine for building brighter labeled biomolecules. ACS Chem Biol. 2019;14:934–940. doi: 10.1021/acschembio.9b00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inverarity IA, Viguier RFH, Cohen P, Hulme AN. Biotinylated anisomycin: a comparison of classical and "click" chemistry approaches. Bioconjugate Chem. 2007;18:1593–1603. doi: 10.1021/bc070085u. [DOI] [PubMed] [Google Scholar]
- 28.Suresh RR, Gao ZG, Salmaso V, Chen E, Campbell RG, Poe RB, Liston TE, Jacobson KA. Selective A3 adenosine receptor antagonist radioligand for human and rodent species. ACS Med Chem Lett. 2022;13(4):623–631. doi: 10.1021/acsmedchemlett.1c00685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glukhova A, Thal DM, Nguyen AT, Vecchio EA, Jörg M, Scammells PJ, May LT, Sexton PM, Christopoulos A. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell. 2017;168:867–877. doi: 10.1016/j.cell.2017.01.042. [DOI] [PubMed] [Google Scholar]
- 30.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 31.Schrödinger Release 2021–2: Maestro; Schrödinger, LLC: New York, NY, 2021.
- 32.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 33. Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47:1750–1759 10.1021/jm030644s. [DOI] [PubMed]
- 34. Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49:6177–6196 10.1021/jm051256ox [DOI] [PubMed]
- 35.Yu J, Mannes P, Jung YH, Ciancetta A, Bitant A, Lieberman DI, Khaznadar S, Auchampach JA, Gao ZG, Jacobson KA. Structure activity relationship of 2-arylalkynyl-adenine derivatives as human A3 adenosine receptor antagonists. Med Chem Commun. 2018;9:1920–1932. doi: 10.1039/C8MD00317C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tosh DK, Salmaso V, Rao H, Bitant A, Fisher CL, Lieberman DI, Vorbrüggen H, Reitman ML, Gavrilova O, Gao ZG, Auchampach JA, Jacobson KA. Truncated (N)-methanocarba nucleosides as partial agonists at mouse and human A3 adenosine receptors: Affinity enhancement by N6-(2-phenylethyl) substitution. J Med Chem. 2020;63:4334–4348. doi: 10.1021/acs.jmedchem.0c00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Claff T, Klapschinski TA, Tiruttani Subhramanyam UK, Vaaßen VJ, Schlegel JG, Vielmuth C, Voß JH, Labahn J, Müller CE (2022) Single stabilizing point mutation enables high-resolution co-crystal structures of the adenosine A2A receptor with preladenant conjugates. Angew Chem Int Ed e202115545 [DOI] [PMC free article] [PubMed]
- 38.Herschel JFW. On a case of superficial colour presented by a homogeneous liquid internally colourless. Phil Trans R Soc. 1845;135:143–145. doi: 10.1098/rstl.1845.0004. [DOI] [Google Scholar]
- 39.Lu L, Wu Zy, Li X, Han F (2019) State-of-the-art: functional fluorescent probes for bioimaging and pharmacological research. Acta Pharmacol Sin 40:717–723. [DOI] [PMC free article] [PubMed]
- 40.Zheng Q, Ayala AX, Chung I, Weigel AV, Ranjan A, Falco N, Grimm JB, Tkachuk AN, Wu C, Lippincott-Schwartz J, Singer RH, Lavis LD. Rational design of fluorogenic and spontaneously blinking labels for super-resolution imaging. ACS Central Sci. 2019;5:1602–1613. doi: 10.1021/acscentsci.9b00676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan C, Zhang Y, Guo Z. Recent progress on molecularly near-infrared fluorescent probes for chemotherapy and phototherapy. Coordination Chem Rev. 2021;427:213556. doi: 10.1016/j.ccr.2020.213556. [DOI] [Google Scholar]
- 42.Baues M, Klinkhammer BM, Ehling J, Gremse F, van Zandvoort MAMJ, Reutelingsperger CPM, Daniel C, Amann K, Bábíčková J, Kiessling F, Floege J, Lammers T, Boor P. A collagen-binding protein enables molecular imaging of kidney fibrosis in vivo. Kidney Internat. 2020;97:609–614. doi: 10.1016/j.kint.2019.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weissleder RA. Clearer vision for in vivo imaging. Nature Biotechnol. 2001;19:316–317. doi: 10.1038/86684. [DOI] [PubMed] [Google Scholar]
- 44.Lesiak L, Zhou X, Fang Y, Zhao J, Beck JR, Stains CI. Imaging GPCR internalization using near-infrared Nebraska red-based reagents. Org Biomolec Chem. 2020;18:2459–2467. doi: 10.1039/D0OB00043D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Esteoulle L, Daubeuf F, Collot M, Riché S, Durroux T, Brasse D, Marchand P, Karpenko JA, Klymchenko SA, Bonnet D. A near-infrared fluorogenic dimer enables background-free imaging of endogenous GPCRs in living mice. Chemical Sci. 2020;11:6824–6829. doi: 10.1039/D0SC01018A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kecskés A, Tosh DK, Wei Q, Gao ZG, Jacobson KA. GPCR ligand dendrimer (GLiDe) conjugates: Adenosine receptor interactions of a series of multivalent xanthine antagonists. Bioconjugate Chem. 2011;22:1115–1127. doi: 10.1021/bc1005812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original data from this study will be made available upon reasonable request.