

Abstract
Neurofibrillary tangles (NFTs), which contain abnormally phosphorylated tau proteins spreading within components of the medial temporal lobe (MTL) memory circuit in Alzheimer’s disease (AD). Here, we used quantitative immunohistochemistry to determine the density of posttranslational oligomeric (TOC1 and TNT1), phosphorylated (AT8) and late truncated (TauC3) tau epitopes within the MTL subfields including EC layer II, subiculum, CA subfields and dentate gyrus (DG) in subjects who died with a clinical diagnosis of no cognitive impairment (NCI), mild cognitive impairment (MCI) and AD. We also examined whether alterations of the nuclear alternative splicing protein, SRSF2, are associated with tau pathology. Although a significant increase in TOC1, TNT1, and AT8 neuron density occurred in EC in MCI and AD, subicular, DG granule cells, CA1 and CA3 density was only significantly higher in AD. TauC3 counts were not different between connectome regions and clinical groups. SRSF2 intensity in AT8 positive cells decreased significantly in all regions independent of the clinical group. CA1 and subicular AT8, TauC3 and oligomeric densities correlated across clinical groups. EC AT8 counts correlated with CA subfields, subicular and DG values across clinical groups. Oligomeric and AT8 CA1, EC and subicular density correlated with Braak stage. Decreased nuclear SRSF2 in the presence of cytoplasmic phosphorylated tau suggests a dual hit process in NFT formation within the entorhinal hippocampal connectome during the onset of AD. Although oligomeric and phosphorylated tau follow a stereotypical pattern, clinical disease stage determined density of tau deposition and not anatomic location within the entorhinal-hippocampal connectome.
Keywords: Alzheimer’s disease, medial temporal lobe, mild cognitive impairment, neurofibrillary tangles, propagation, splicing, tau
Graphical Abstract
Introduction
Alzheimer’s disease (AD) is a progressive dementing disorder characterized early by clinically subtle changes in memory and verbal fluency terminating in frank dementia and alterations in several neurobiological systems including the inflammasome (Španić, Langer Horvat, Ilić, Hof, & Šimić, 2022), neurotransmitters (Mesulam, 2013), neurotrophins (Mufson et al., 2019), and oxidative stress (Scheff, Ansari, & Mufson, 2016). However, the two primary neuropathological hallmarks that define this neurodegenerative disorder are neuritic plaques (NP) containing the amyloid beta protein (Selkoe & Hardy, 2016) and neurofibrillary tangles (NFTs) composed of various posttranslational forms of tau (Binder, Guillozet-Bongaarts, Garcia-Sierra, & Berry, 2005). Although both lesions are found in the medial temporal lobe (MTL) (J. Chen et al., 2016; Mufson et al., 1999), NFTs develop prior to NPs (H. Braak & Braak, 1990). The medial temporal lobe (MTL) consists of connectionally related structures including the transentorhinal (TEC) and entorhinal (EC) cortex, hippocampal formation (HF) and subiculum (Berron, van Westen, Ossenkoppele, Strandberg, & Hansson, 2020; Hoesen & Pandya, 1975) that is essential for declarative memory and undergoes tau-based neuronal dysfunction during the early stages of Alzheimer’s disease (AD) (Berron et al., 2020; H. Braak & Braak, 1990; Mrdjen et al., 2019; Mufson et al., 2015). The MTL NFT pathology progresses in a stereotypical manner, first in the TEC and EC (stage I-II), then in the HF (stage III-IV) before spreading to isocortical regions (stage V-VI) (H. Braak & Braak, 1991; H. Braak & Del Tredici, 2018; Lace et al., 2009). However, others have reported NFT pathology in the brainstem prior to that seen in the MTL in AD (Grinberg et al., 2009; Simic et al., 2009). It’s well established that the evolution of pathological tau follows a linear temporal sequence of posttranslational events that includes phosphorylation, conformation and truncation that lead to the cellular accumulation of tau oligomers, proto-fibrils, fibrils, and paired-helical filaments, the main component of NFTs. These posttranslational tau events are visualized using antibodies directed against discrete tau epitopes (Gamblin et al., 2003; Garcia-Sierra, Ghoshal, Quinn, Berry, & Binder, 2003; Iqbal, Liu, Gong, & Grundke-Iqbal, 2010; Vana et al., 2011; Ward et al., 2013). For example, the conformational tau oligomeric complex 1 (TOC1) recognizes oligomers and short tau filaments in pre-tangles and neuropil threads (Ward et al., 2013), tau N-terminal 1 (TNT1) labels early conformational oligomeric tau species with an exposed phosphatase-activating domain (PAD) motif that disrupts fast axonal transport (FAT) (Combs, Hamel, & Kanaan, 2016). Aggregates of oligomeric tau (Wu et al., 2021), precedes NFT formation causing toxicity (Gyparaki et al., 2021) and RNA binding protein TIA1 promotes the generation of toxic oligomeric tau (Ash et al., 2021). Interestingly, exosomes have been suggested to be involved in the extracellular transport of tau oligomers underlying the intracellular neuron-to-neuron propagation of tau seeds in several neurogenerative diseases including AD (Dujardin et al., 2014; Polanco et al., 2016, 2021; Wang et al., 2017; Xiao et al., 2017). Together these findings provide a strong foundation for investigating the role that oligomeric tau species play in the spatial distribution of NFT pathology within the entorhinal-hippocampal connectome. AT8 recognizes tau phosphorylated at Ser202/Thr205, which identifies early and late degenerative changes (H. Braak & Braak, 1991; Goedert, Jakes, & Vanmechelen, 1995) and TauC3, an antibody generated against the C-terminus of the truncated form of tau at Asp421 (D421) by caspase-3, associated with late and more mature NFTs and ghost tangles (Gamblin et al., 2003; Nicholls et al., 2017). These tau epitopes were particularly useful in defining the evolution of NFTs within the cholinergic basal forebrain during the progression of AD (Tiernan et al., 2018; Vana et al., 2011) and within the entorhinal cortex in severe AD (Ghoshal et al., 2002; Lace et al., 2009). However, the field lacks a detailed analysis of the synaptic propagation (de Calignon et al., 2012; Pooler, Phillips, Lau, Noble, & Hanger, 2013) of different tau oligomers compared to phosphorylation and conformation tau epitopes in the entorhinal-hippocampal connectome during the onset of AD.
AD is considered a tauopathy and studies suggest that an imbalance in the ratio between the three (3R) and four (4R) microtubule-repeat tau isoforms is associated with the formation of NFTs in vulnerable brain networks in AD (Iqbal et al., 2010). In this regard, SRSF2 (aka SC35) a serine/arginine-rich splicing factor regulates the splicing of tau exon 10, which determines the generation of 3R and 4R tau isoforms (Qian, Iqbal, Grundke-Iqbal, Gong, & Liu, 2011). Disruption of the alternative splicing of tau exon 10 via SRSF2 results in an imbalance of 3R and 4R tau, which drives the formation of NFTs (C. Chen et al., 2014; Goedert, Spillantini, Jakes, Rutherford, & Crowther, 1989; Z. H. Wang et al., 2018). Functionally, tau mis-splicing and/or an imbalance contributes to isoform-specific impairments that enhance the recruitment of tau isoforms into a pathological process (Park, Ahn, & Gallo, 2016) that is postulated to be propagated as oligomers in neuronal networks in AD (Polanco et al., 2016, 2021; Takeda, 2019). However, whether alterations in nuclear SRSF2 are associated with tau isoforms involved in the evolution of NFTs (Bai et al., 2013) within the entorhinal-hippocampal memory circuit in AD remains unknown.
Evidence derived from patients that develop autosomal dominant familial AD (FAD), suggests that amyloid-beta (Aβ) acts as a stimulus for the pathological cascade underlying AD including the development of NFTs (Selkoe & Hardy, 2016). Although Aβ pathology presents within frontal isocortical regions early in AD (Arnold, Hyman, Flory, Damasio, & Van Hoesen, 1991; Thal, Rüb, Orantes, & Braak, 2002), compared to NFTs first appearing in the MTL, amyloid pathology does appear in the entorhinal cortex (Lacosta, Insua, Badi, Pesini, & Sarasa, 2017; Mufson et al., 1999). Moreover, an increase in the amyloid precursor protein (APP), a precursor of Aβ, was associated with a decrease of the U1–70K small nuclear ribonucleoprotein spliceosome component (Bai et al., 2013), which also plays a key role in tau splicing in AD. Therefore, we examined the relationship between amyloid and tau lesions using antibodies that recognize amino acids 1–42 of amyloid beta peptide (Aβ1–42) and MOAB-2 a pan-specific amyloid antibody that recognizes residues 1–4 of Aβ that differentiates intracellular Aβ from the APP (Youmans et al., 2012) to define their relationship with tau neuronal pathology within the entorhinal-hippocampal connectome during AD onset.
The present study combined bright-field and dual fluorescent immunohistochemistry with quantitative methods to compare alterations in cytoplasmic oligomeric vs phosphorylated/truncated tau and the nuclear splicing protein SRSF2 in relation to amyloid lesions within the entorhinal-hippocampal connectome using tissue samples obtained from individuals who died with a premortem clinical diagnosis of no cognitive impairment (NCI), mild cognitive impairment (MCI) and AD. Data was correlated with premortem cognitive performance scores, post-mortem neuropathological criteria, demographic and APOE allele status.
This special issue of The Journal of Comparative Neurology is dedicated to the memory of Dr. Deepak Pandya, whose classic neuroanatomical tract tracing studies provided the foundation for our understanding of the primate entorhinal-hippocampal memory circuit that is compromised AD.
Materials and Methods
Subjects
The study included 32 cases with either an antemortem clinical diagnosis of NCI (n=13, 9F/4M 85.15±10.64 years), MCI (n=6, 3F/3M, 89.17±5.5 years) or AD (n=12, 5F/7M, 87.83±9.6 years) from the University of Kentucky Alzheimer’s Disease Center (UKADC) brain repository (see Table 1).
Table 1.
Case demographic, clinical and neuropathological characteristics
| NCI (n=13) | MCI (n=6) | AD (n=12) | P-value | Group-wise Comparisons | |
|---|---|---|---|---|---|
| Age at Death | 85.1±10.6a [68–102]b | 89.2±5.6 [81–96] | 87.8±9.6 [67–100] | 0.7† | N/A |
| Sex (male/female) | 4/9 | 3/3 | 5/7 | 0.4‡ | N/A |
| Education | 15.9±2.4 [12–20] | 16.7±3.0 [12–20] | 14.7±4.00 [5–18] | 0.7† | N/A |
| MMSE | 29.00±1.1 [27–30] | 27.00±2.1 [24–30] | 21.1±6.0 [11–28] | <0.001† | NCI>AD |
| APOE ε4 (Carrier/Non-Carrier) | 11/2 | 4/2 | 9/1* | 0.5‡ | N/A |
| PMI (hours) | 3.8±3.5 [2.0–15.0] | 2.9±1.5 [1.8–5.8] | 2.8±1.0 [1.7–4.9] | 0.6† | N/A |
| Brain Weight (grams) | 1,122.5±148.1 [970–1455] | 1,174.7±180.6 [910–1429] | 1,146.3±122.8 [940–1380] | 0.5† | N/A |
| V-VI | 0 | 0 | 4 | ||
| Definite AD | 0 | 1 | 4 | ||
| High Likelihood |
Mean±standard deviation;
[range], AD, Alzheimer’s disease; APOE, apolipoprotein E; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; PMI, Postmortem Interval; MCI, mild cognitive impairment; MMSE, Mini-Mental State Examination; NCI, no cognitive impairment; NIA, National Institute on Aging; PMI – Post-mortem Interval;
APOE ε4 information was not available for two individuals; Braak stage was unavailable for two AD individuals;
Kruskal-Wallis test;
Chi-square
Clinical and Neuropathological Evaluation
Details of the clinical evaluation and criteria for diagnosis of AD, MCI and NCI in the UKADC cohort has been published elsewhere (Schmitt et al., 2000). Briefly, at a consensus conferences neurologists and neuropsychologists reviewed all clinical data, medical records, interviews with family members and then assigned a final clinical diagnosis for each case. For all subjects, cognitive test scores were available within the last year of life; the average interval from last evaluation to time of death was 7.0±3.6 months, with no differences among the three diagnostic groups (p<0.1). Only cases with Mini-Mental Status Examination (MMSE) scores within 2 years of death were included in this study. Here, the MMSE conducted closest to the date of death was the ‘severity metric’ for cognitive impairment. The MMSE score was chosen as it was the most consistently available measure obtained from both normal and demented subjects over the course of their evaluations. Neuropathological diagnosis was performed as described previously (Schmitt et al., 2012), which included NIA-Reagan criteria (Newell, Hyman, Growdon, & Hedley-Whyte, 1999), recommendations of the CERAD (Mirra et al., 1991) and Braak staging of NFTs (H. Braak & Braak, 1991). The UKADC neuropath core evaluation included information for each of these criteria. Therefore, the current cases were evaluated using criteria that do not differ greatly from the 2012 guidelines. The UKADC has not re-evaluated these cases using the revised NIA-Alzheimer’s Association neuropathological criteria (Montine et al., 2012) nor the inclusion of biomarkers with these guidelines (Jack et al., 2018). The later research framework should not be used to restrict alternative approaches to hypothesis testing that do not include biomarkers (Jack et al., 2018). The 2012 guidelines differ from previous consensus criteria due to the following: recognition that AD neuropathologic changes may occur in the apparent absence of cognitive impairment, an AD neuropathologic score that incorporates histopathologic assessments of amyloid β deposits (A), staging of NFTs (B), scoring of neuritic plaques (C), and more detailed approaches for assessing commonly co-morbid conditions. Exclusion criteria included stroke, Parkinson’s disease, dementia with Lewy bodies, primary age-related tauopathy (PART) (Jellinger et al., 2015) or limbic predominant age-related TDP-43 encephalopathy neuropathologic change (LATE-NC) (Nelson et al., 2019), significant trauma within 12 months before autopsy, seizure within the last 6 months before autopsy, on a respirator longer than 12 hours before death, in a coma greater than 12 hours immediately before death, or currently undergoing radiation therapy for a central nervous system tumor. Cases treated with anticholinesterase inhibitors were also excluded. Besides the range of interest for cognitive impairment and lack of concomitant pathologies, this is a convenience sample chosen randomly from autopsies performed at the UKADC. Since all cases were chosen based upon the last clinical evaluation without prior knowledge about the severity of neuropathology, the latter did not introduce selection bias that affected our primary outcome measures. APOE genotype was performed as previously reported (Mahady, He, Malek-Ahmadi, & Mufson, 2020; Vana et al., 2011). Human Research Committees of UKADC and Dignity Health approved this study, written informed consent for research and autopsy was obtained from the participants or their family/guardians and research protocols were approved by the UK internal review board (Mufson et al., 2012; Perez et al., 2015; Schmitt et al., 2012).
Immunohistochemistry
Tissue blocks containing the entorhinal-hippocampal complex were fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned at 8 μm onto charged slides and kept at room temperature until immunohistochemical processing. The antibodies used in this study were listed in Table 2.
Table 2.
Antibody characteristics
| Antibody | Description | Manufacturer | Dilution | RRID |
|---|---|---|---|---|
| TOC1 | Tau oligomeric complex 1 mouse monoclonal antibody | Gift from Dr. Lester ‘Skip’ Binder Available now from Dr. Kanaan | 1:500 | AB_2832939 |
| TNT1 | Tau phosphatase-activating domain (PAD) mouse monoclonal antibody | Gift from Dr. Lester ‘Skip’ Binder Available now from Dr. Kanaan | 1:3,000 | AB_2736930 |
| AT8 | Phospho-tau (Ser202, Thr205) mouse monoclonal antibody | Invitrogen, Carlsbad, CA, USA | 1:1,000; 1:100 IF | AB_223647 |
| TauC3 | Caspase-cleaved tau (cleaved Asp421, Asp422) mouse monoclonal antibody | Invitrogen, Carlsbad, CA, USA | 1:500 | AB_2536237 |
| MOAB-2 | Pan-Aβ (MOAB-2) mouse monoclonal antibody | Gift from M. J. LaDu | 1:5,000 | AB_2313888 |
| Aβ1–42 | Human Aβ1–42 rabbit polyclonal antibody | Millipore, Billerica, CA, USA | 1:100 | AB_11212712 |
| SRSF2 | Splicing factor SC35 rabbit monoclonal antibody | Abcam, Waltham, MA, USA | 1:100 IF | AB_2909393 |
IF, Immunofluorescence
Two paraffin embedded sections per case were deparaffinized in xylene, rehydrated in a series of concentrations of ethanol (100, 95, 70, and 50%) and pretreated with either heated citric acid (pH 6) for 10 min in a microwave, or with 88% formic acid for 20 min for antigen retrieval of Aβ1–42. Sections processed using the MOAB-2 antibody were pretreated with formic acid (75%, 5 min). After several washes in Tris-buffered saline (TBS), sections were incubated in sodium metaperiodate (0.01M) solution for 20 min to inactivate endogenous peroxidase activity, then rinsed in TBS, and placed in blocking solution containing 0.25% Triton X-100/3% goat serum for one hour at room temperature (RT) and then incubated with primary antibodies in a solution of TBS 0.25% Triton X-100/1% GS overnight at RT. Slides were incubated in a blocking solution consisting of 3% goat serum in TBST (Tris-buffered saline/TritonX-100) at RT for 1 hr. After overnight incubation with primary antibodies: TOC1 (1:500), AT8 (1:1,000), TauC3 (1:500), TNT1 (1:3,000), MOAB-2 (1:5,000), and Aβ1–42 (1:100) (see Table 2), sections were rinsed with TBS before applying the appropriate biotinylated secondary antibody (1:200, Vector laboratories, Newark, CA), diluted in blocking solution at RT for 1 hour. Rinsed sections were incubated in Vectastain ABC Elite reagent (1:200, Vector laboratories, Newark, CA) for 1 hour and visualized using acetate-imidazole 0.05% 3,3’-Diaminobenzidine tetrahydrochloride hydrate (DAB). Sections were counterstained with Mayer’s hematoxylin to aid with cytoarchitectural and laminar identification.
Immunofluorescence
A parallel set of sections were deparaffinized in xylene, rehydrated and antigen retrieval performed by incubating the sections in pH 9 target retrieval solution (Agilent Dako, Singapore) for 25 minutes in a commercial steamer. Sections were then incubated with a polyclonal antibody against SRSF2 (1:100; Abcam, Waltham, MA, USA see Table 2) in a solution containing TBS 0.25% Triton X-100/5% donkey serum for 48 hours at 4 °C and after several washes in TBS containing 1% donkey serum, reaction was visualized with a donkey anti-rabbit Cy3 conjugated affinity secondary antibody (1:200 for 1 hour; Jackson ImmunoResearch Laboratories, Inc., Chester County, PA, USA). Sections were then placed in a solution containing the monoclonal anti-AT8 antibody (1:100; Invitrogen, Carlsbad, CA, USA) in a TBS 0.25% Triton X-100/1% donkey serum overnight at RT and then incubated in a solution that contained donkey anti-mouse Cy5 conjugated secondary antibody (1:200 for 1 hour) (Jackson ImmunoResearch Laboratories, Inc., Chester County, PA, USA). Sections were then processed for nuclear visualization DAPI (1:2,000; Invitrogen, Eugene, OR USA)/TBS 0.25% solution for 15 minutes. Autofluorescence was blocked with autofluorescence eliminator reagent for 1 minute (Millipore-Sigma, St. Louis, MO) and sections were cover-slipped using Invitrogen ProLong Glass Antifade Mountant (Invitrogen, Carlsbad, CA, USA). Immunofluorescence was analyzed and photographed using an Echo Revolve fluorescence microscope (San Diego, CA, USA).
Tau Epitope and Amyloid Density Analyses
Neuronal tau and Aβ densities were analyzed in layer II of the EC, the Cornu Ammonis (CA) subregions CA3, CA2, CA1, polymorphic (Pl) and granule (Gl) cell layers of the dentate gyrus (DG) and subiculum (Figs. 1–5) according to the nomenclature of Insausti and Amaral, 2004. TEC was not analyzed due to absence of this region in the tissue blocks provided by UKADC. Tau positive cells were counted in five fields per region per section in two immunostained sections at 10x magnification and cell densities were determined by dividing the number of tau positive cells by the area of counting in each field. Density counts for MOAB-2 and Aβ1–42 positive plaques were assessed at 10x magnification per section covering an area of 1.10 mm2/field as previously described (Perez et al., 2019).
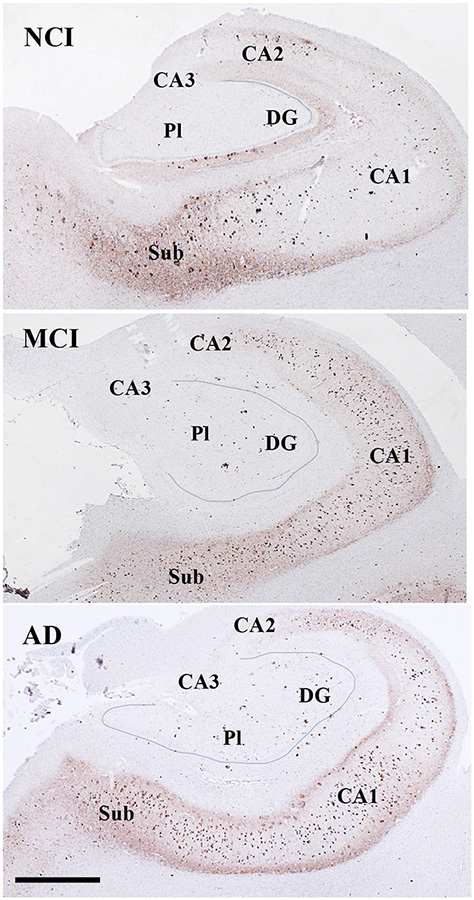
Figure 1.
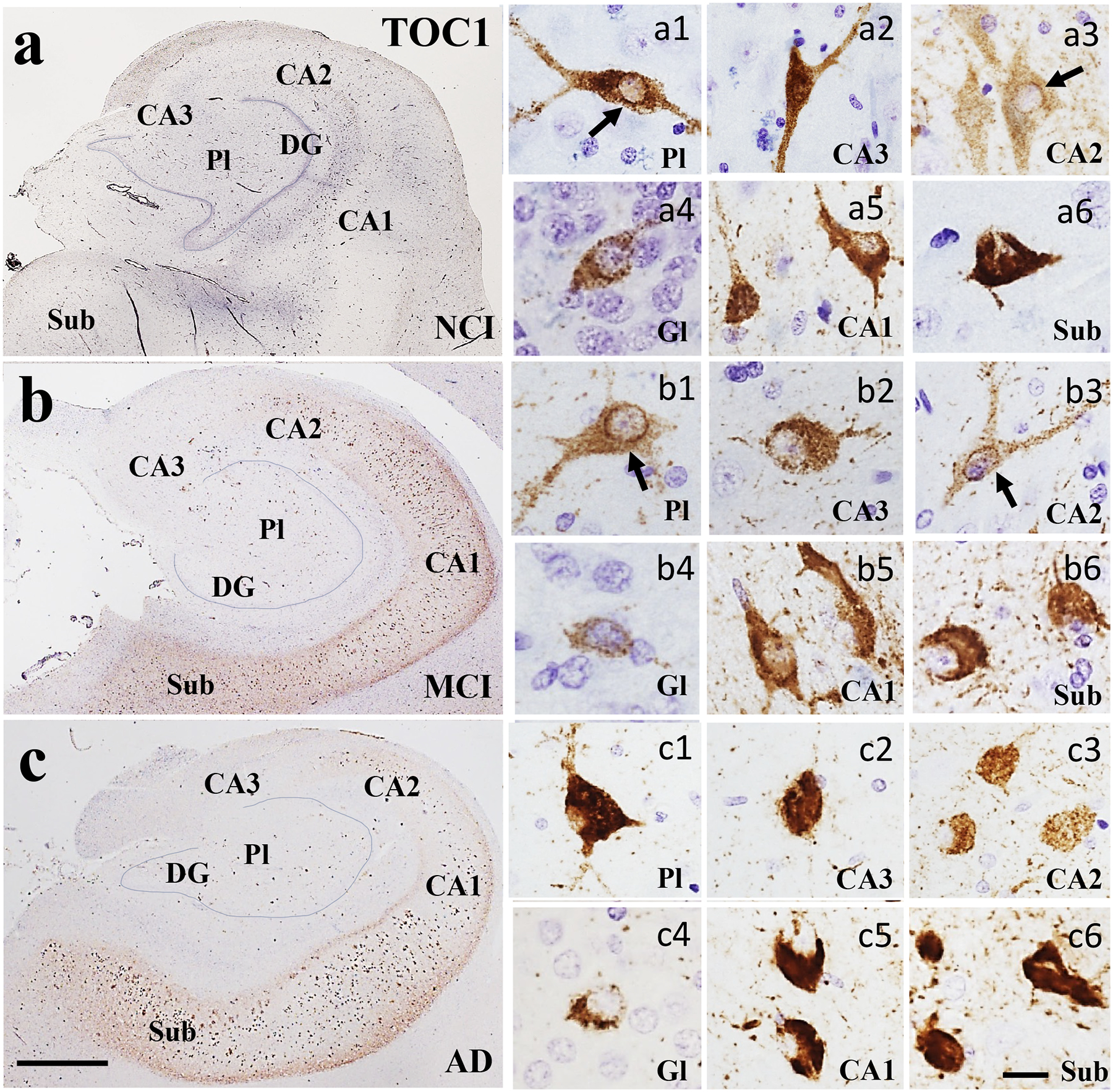
Low magnification photomicrographs showing TOC1 immunoreactivity in the hippocampus and subiculum in NCI (a), MCI (b), and AD (c) cases. Note the increase in TOC1 staining in CA2, CA1 and subiculum across disease stages. Images labeled with (a-b)1–6 show different TOC1 neuronal morphologies and immunostaining characteristics within each CA subfield and subiculum across clinical groups. Note the granular TOC1 cytoplasmic, perinuclear (arrows) and dendritic staining in the NCI and MCI cases compared to a denser aggregation in AD. Sections were counterstained with hematoxylin (light-blue nuclear staining). Abbreviations: CA1, hippocampal subfield CA1; CA2, hippocampal subfield CA2; CA3, hippocampal subfield CA3; DG, dentate gyrus; Gl, granule cell layer of the DG; Pl, polymorphic layer of the DG; Sub, subiculum. Scale bar in c=200 μm and applies to a and b, (a-c)6=25 μm applies to all (a-c)1–5 panels.
Figure 5.
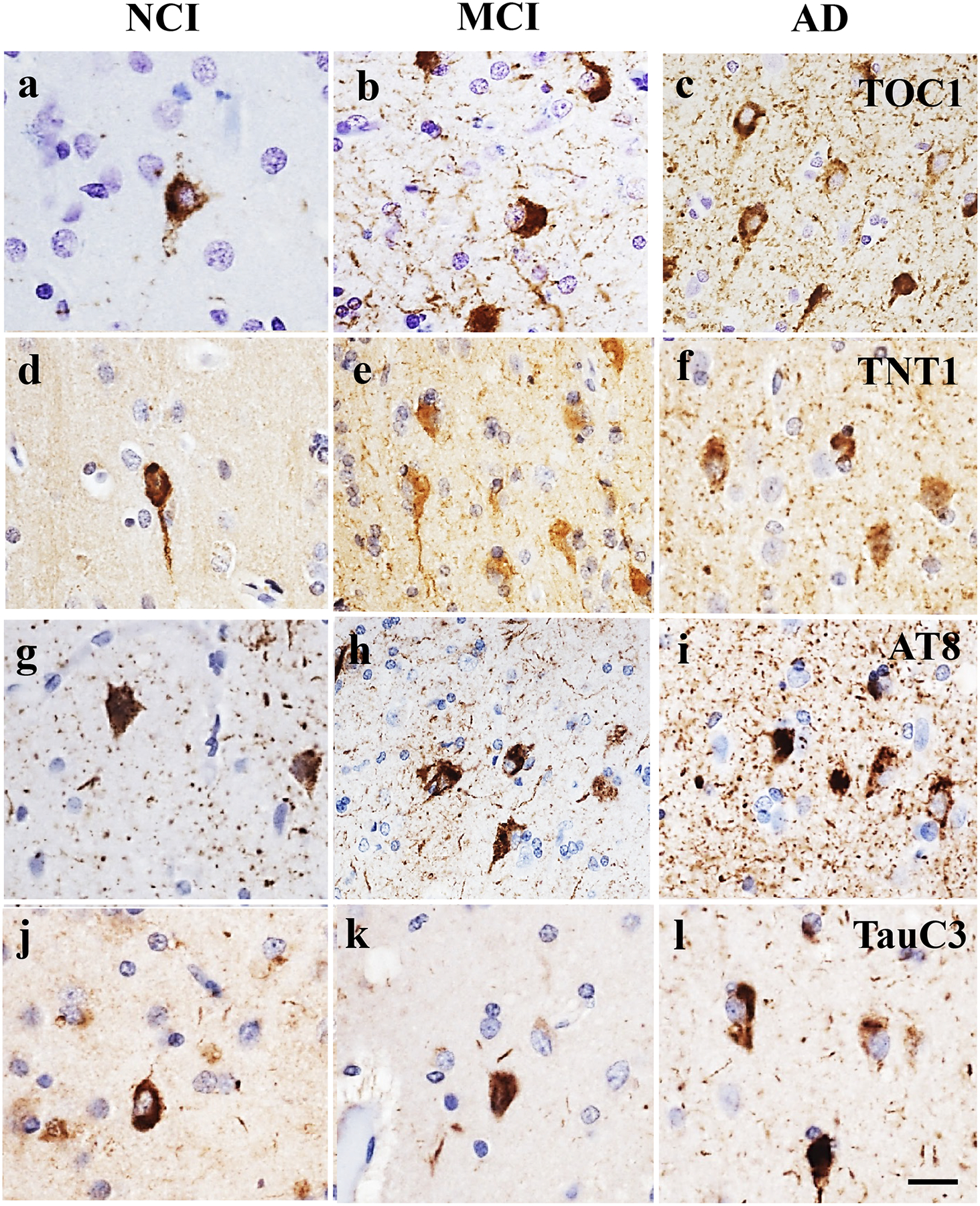
Photomicrographs showing EC layer II neurons immunolabeled for TOC1 (a-c), TNT1 (d-f), AT8 (g-i) and TauC3 (j-l) in NCI, MCI, and AD cases. Note the limited TOC1, TNT1 and AT8 positive cells and neuropile threads in NCI compared to MCI and AD. By contrast, the number of TauC3 positive cells was greater in AD compared to NCI and MCI. Sections were counterstained with hematoxylin (light-blue nuclear staining). Scale bar in l=25μm and applies to a-k.
Quantitation of SRSF2 Immunofluorescent Intensity
SRSF2 (SC35) nuclear immunofluorescent intensity was evaluated in layer II of the EC, hippocampal subfields CA1, CA3, CA2, DG polymorphic and granule cell layers and subiculum in sections fluorescently labelled with AT8. DAPI was used as a nuclear counterstain. Immunofluorescence images were acquired at the same light intensity, contrast, and exposure at 20x magnification using an Echo Revolve R4 microscope (San Diego, CA). Nuclear SRSF2 intensity was evaluated in ten images per subfield (2 slides per case) with the aid of ImageJ 1.53k thresholding option to highlight the structures counted. Background intensity was subtracted from SRSF2 measurements.
Statistical Analysis
Density measures were compared across clinical groups using the non-parametric Kruskal-Wallis, Wilcoxon-paired and Friedman repeated measurement test. Chi-square analyses determine significant differences in sex, APOE ε4, Braak stage, NIA Reagan, and CERAD diagnosis frequency between clinical groups. Mann-Whitney test compared APOE ε4 carriers and non-carriers for each tau epitope in all regions across clinical groups. Statistical significance was set at 0.05 (two-sided). Spearman rank correlation assessed associations between tau epitopes and demographic variables. Correlations of tau epitope densities and neuropathological criteria within the entorhinal-hippocampal complex and across clinical groups were adjusted using a Bonferroni test with significance levels set at α=0.007 and α=0.03, respectively.
Results
Case Demographics
There were no significant differences for age at death (p=0.71), sex (p=0.37), years of education (p=0.70), postmortem interval (p=0.64), or brain weight (p=0.54) at autopsy between clinical groups (Table 1). MMSE scores were significantly lower in the AD compared to NCI group (p<0.001; Table 1). Frequency of APOE genotypes did not differ significantly between groups (p=0.48). No differences were observed between Braak stage (p=0.08), CERAD diagnosis (p=0.16) or NIA Reagan diagnosis (p=0.17) between groups (Table 1).
Tau epitope staining characteristics within the entorhinal-hippocampal complex
Oligomeric TOC1 immunoreactivity appeared as granular deposits within the cytoplasm, dendrites and perinuclear in hippocampal CA subfields, DG polymorphic and granule cells, subicular neurons and layer II of the EC in NCI (Figs. 1a and 5a) and MCI cases (Figs. 2b and 5b). Granular TOC1 staining was tightly packed resulting in more intense appearance particularly in CA1 and subiculum in AD (Figs. 1c and 5c). TNT1 positive granules were found within the cytoplasm, perinuclear and dendrites in the entorhinal-hippocampal regions examined across clinical groups (Figs. 2 and 5e–f). Like TOC1, TNT1 labeled granules were densely aggregated within the cytoplasm in hippocampal neurons appearing more intensely stained in the AD group (Fig. 2c). Of particular interest was the observation of perinuclear oligomeric tau in CA2 and PL in NCI (Fig. 1a1) and MCI (Fig. 1b3), whereas the TNT1 appeared in CA2 in NCI (Fig. 2a3) and in CA2 and CA3 in MCI (Fig. 2b2) and AD (Fig. 2c) neurons. The pretangle/tangle marker, AT8, intensely labeled the neuronal cytoplasm in all regions and clinical groups (Figs. 3 and 5g–i). Unlike TOC1 and TNT1, AT8 immunostained CA1 pyramidal neurons displayed a flame-like shape in MCI and AD (Fig. 3b, c). TauC3 appeared mainly in the cytoplasm, and to a lesser extent in dendrites compared to the oligomeric and AT8 labeling, indicative of a further stage of NFT development (Guillozet-Bongaarts et al., 2005) (Figs. 4 and 5g–i). TauC3 immunostaining was minimal in DG polymorphic and granule cell layers and CA3 compared to CA1, CA2, subiculum and EC in the NCI cases (Figs. 4a and 5j). TauC3 labeled neurons displayed either a flame-like or globose morphology in MCI (Fig. 4b). Unlike TOC1 and TNT1, TauC3 positive profiles were heavily immunoreactive and displayed the classic flame-like NFT appearance in CA1, subiculum, and EC in AD (Figs. 4c and 5l).
Figure 2.
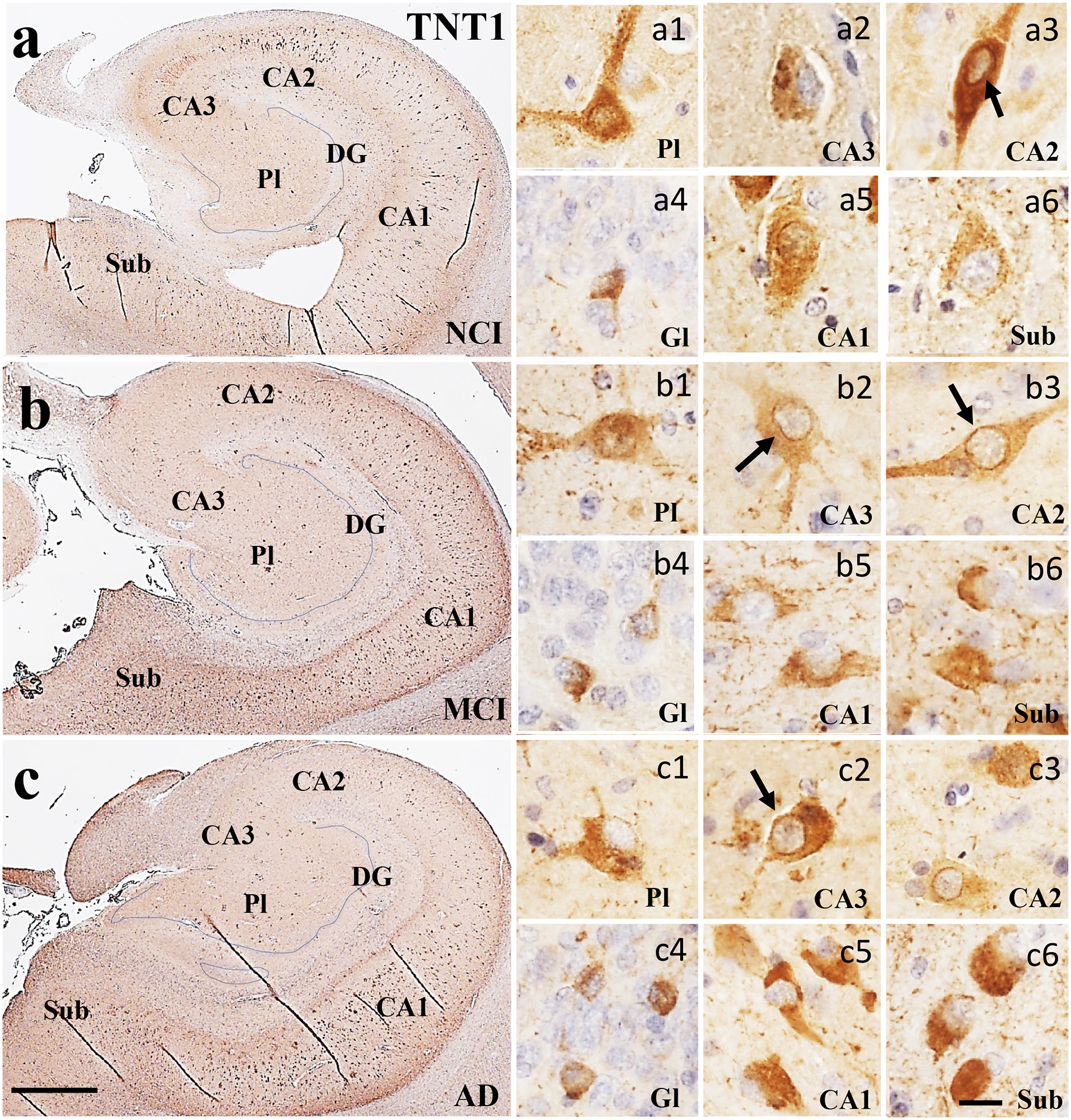
Low magnification photomicrographs of TNT1 immunoreactivity in the hippocampus and subiculum in NCI (a), MCI (b), and AD (c) cases which shows an increase of immunoreactivity in CA subfields and subiculum in MCI (b) and AD (c) compared to NCI (a). Images labeled (a-c)1–6 show different TNT1 neuronal morphologies and characteristics within each CA subfield and subiculum across clinical groups. TNT1 staining also appeared granular within the cytoplasm, perinuclear (arrows) and dendrites in multipolar neurons across regions and clinical groups. Note the increase of stain intensity and loss of multipolar cellular phenotype in AD compared to NCI and MCI. Sections were counterstained with hematoxylin (light-blue nuclear staining). Abbreviations: CA1, hippocampal subfield CA1; CA2, hippocampal subfield CA2; CA3, hippocampal subfield CA3; DG, dentate gyrus; Gl, granule cell layer of the DG; Pl, polymorphic layer of the DG; Sub, subiculum. Scale bar in c=200 μm and applies to a and b, (a-c)6=25 μm applies to all (a-c)1–5 panels.
Figure 3.
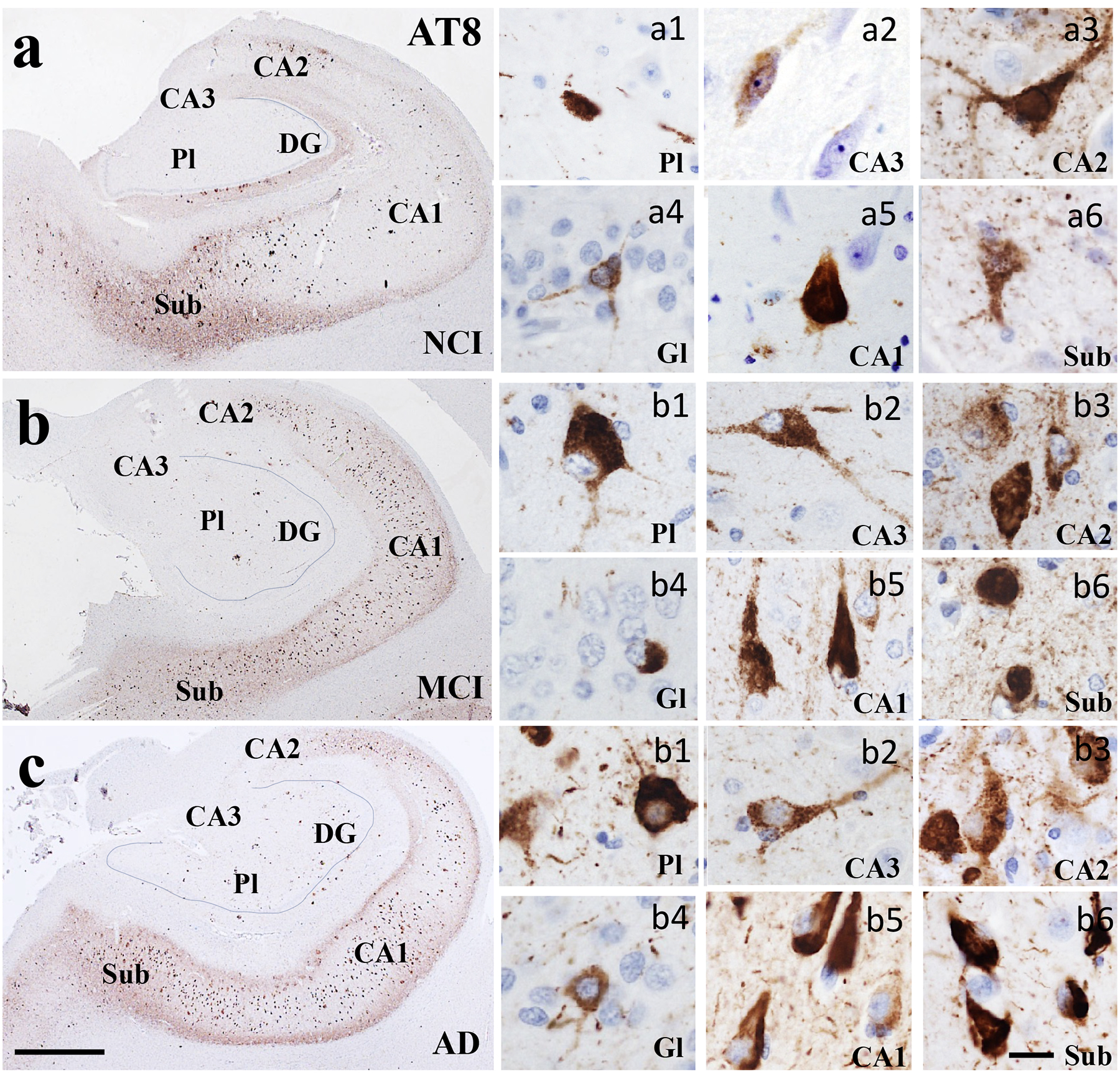
Low magnification photomicrographs of AT8-ir neurons in the hippocampus and subiculum in NCI (a), MCI (b), and AD (c) cases showing a marked increase in labeling within the CA1 across clinical groups. Images labeled with a (a-c)1–6 show different AT8 neuronal morphologies and characteristics within each CA subfield and subiculum across clinical groups. Note that AT8 immunoreactivity was more densely aggregated in each CA subfield and subiculum in comparison to the DG layers across clinical groups. Sections were counterstained with hematoxylin (light-blue nuclear staining). Abbreviations: CA1, hippocampal subfield CA1; CA2, hippocampal subfield CA2; CA3, hippocampal subfield CA3; DG, dentate gyrus; Gl, granule cell layer of the DG; Pl, polymorphic layer of the DG; Sub, subiculum. Scale bar in c=200 μm and applies to a and b, (a-c)6=25 μm and applies to panels (a-c)1-5.
Figure 4.
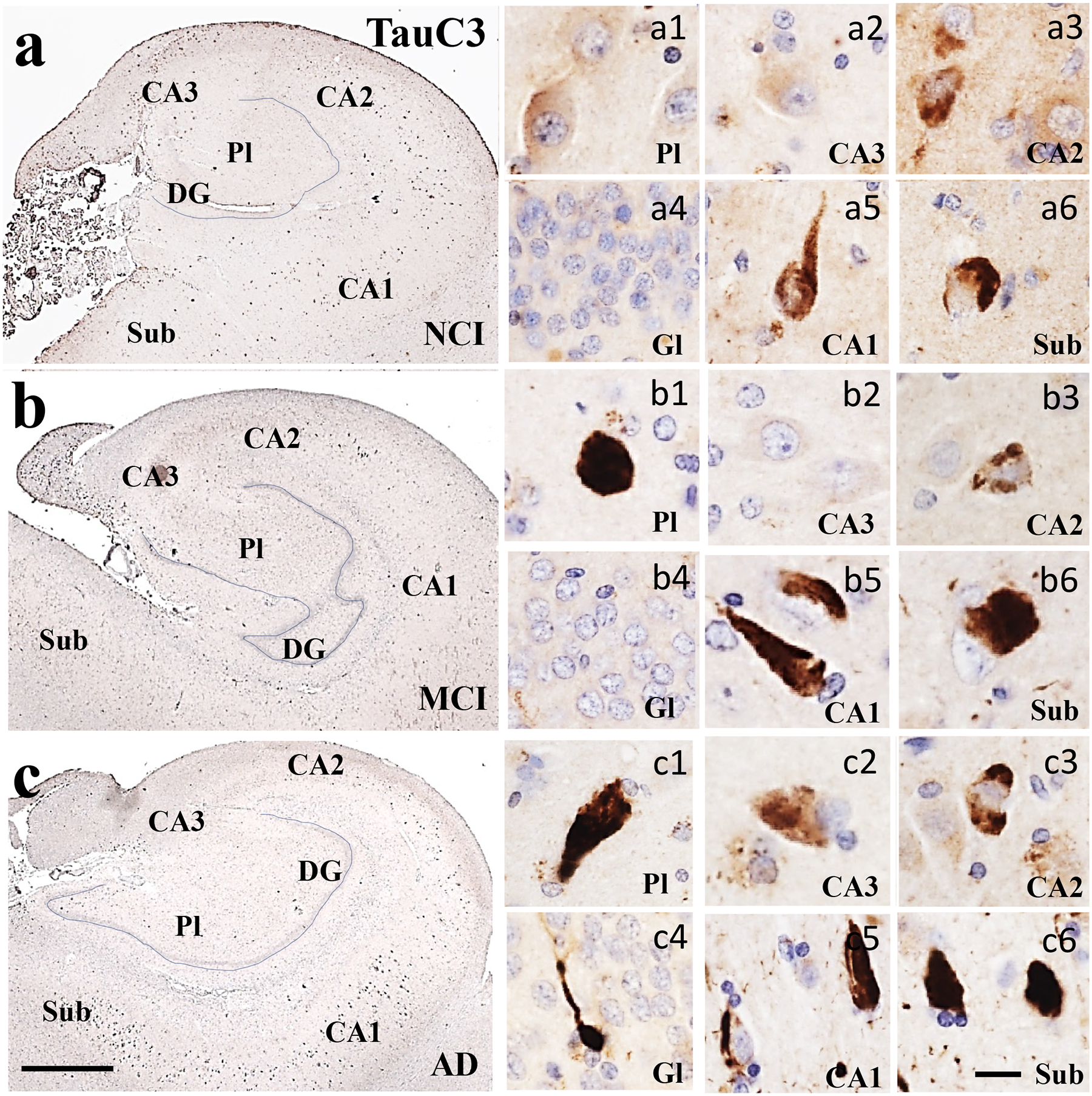
Low magnification photomicrographs of TauC3 immunostaining in the hippocampus and subiculum in NCI (a), MCI (b), and AD (c) showing limited staining within the CA subfields and subiculum across clinical groups. Images labeled (a-c)1–6 show different TauC3 neuronal morphologies and characteristics within each CA subfield and subiculum across clinical groups. Note the prevalence of TauC3 neurons displaying the classic flame-like NFT morphology in MCI and AD but to a lesser extent in NCI (a) compared to TOCI (Fig. 1) and TNT1 (Fig. 2) neuronal staining. Sections were counterstained with hematoxylin (light-blue nuclear staining). Abbreviations: CA1, hippocampal subfield CA1; CA2, hippocampal subfield CA2; CA3, hippocampal subfield CA3; DG, dentate gyrus; Gl, granule cell layer of the DG; Pl, polymorphic layer of the DG; Sub, subiculum. Scale bar in c=200 μm and applies to a and b, (a-c)6=25 μm and applies to all (a-c)1–5 panels.
Percentage of cases displaying tau epitope positive neurons within the entorhinal-hippocampal connectome within clinical groups
Oligomeric, phosphorylated AT8, and truncated TauC3 positive profiles were observed within the entorhinal-hippocampal complex in each clinical group. In general, a greater number of AD cases displayed NFT-bearing neurons in the entorhinal-hippocampal regions compared to NCI and MCI (Supplemental Table 1). TOC1, TNT1 and AT8 positive neurons in EC layer II, subiculum, CA1 and CA2 were found in one hundred percent of the AD cases, while the lowest percentage of these markers, particularly those containing the late truncated TauC3 epitope in the DG granule cell layer and CA3 were observed in the NCI cases examined. In MCI, TOC1 and AT8 positive cells were observed in EC layer II, CA1 and CA2 subfields in all cases, whereas TNT1 positive cells appeared only in EC and CA3 in all MCI cases. Unlike MCI, AT8 positive cells were detected in each entorhinal-hippocampal region in the AD cases. TOC1 and TNT1 positive cells were consistently found in the EC layer II, subiculum, DG granule cell layer, CA1 and CA2 subfields in AD, but not in DG polymorphic layer and the CA3 subfield. Although a great percentage of AD cases displayed TauC3 positive cells in the EC layer II, subiculum, CA1 and CA2 subfields, this tau isoform was not observed in all AD cases.
Tau epitope density in the entorhinal-hippocampal connectome during AD progression
Here we determined the density of NFTs containing different tau epitopes within specific regions of the entorhinal-hippocampal complex (e.g., EC layer II, hippocampal subfields CA2, CA3, CA1, DG polymorphic and granule cell layers and the subiculum) at different stages of AD (see Table 3). The density of TOC1, TNT1, AT8-ir cells in EC layer II was significantly greater in MCI and AD compared to NCI (Kruskal-Wallis, p<0.05; Fig. 6a), but not between MCI and AD. Density measurements of the oligomeric and AT8 positive neurons in CA1, CA3, DG granule cell layer and subiculum were significantly higher in AD (Kruskal-Wallis, p>0.05) compared to NCI, but not MCI (Kruskal-Wallis, p<0.05; Figs. 6b, c, d, e and 7). Moreover, the density of TOC1 neurons in CA2 and TNT1 and AT8-ir neurons in DG polymorphic layer were significantly increased in AD compared to NCI (Kruskal-Wallis, p>0.05; Fig. 6d and f). No significant differences in TauC3-bearing NFT densities were found in any of the entorhinal-hippocampal regions examined between clinical groups (Kruskal-Wallis, p>0.05; Figs. 6 and 7). However, when stratified for APOE genotype, ε4 carriers had significantly more TauC3 positive NFTs in EC layer II (Mann-Whitney, p=0.03) and subiculum (Mann-Whitney, p=0.04).
Table 3.
Groupwise comparisons of Tau and Aβ densities
| Tau cell density | EC Layer II | Subiculum | CA1 | CA2 | CA3 | DG polymorphic layer | DG granule layer |
|---|---|---|---|---|---|---|---|
| Oligomeric tau (TOC1) | p=0.04* MCI, AD>NCI | p=0.009 AD>NCI | p=0.006 AD>NCI | p=0.02 AD>NCI | p=0.04 AD>NCI | p=0.06 | p=0.003 AD>NCI |
| Oligomeric tau (TNT1) | p=0.03 MCI, AD>NCI | p=0.004 AD>NCI | p=0.02 AD>NCI | p=0.12 | p=0.02 AD>NCI | p=0.03 AD>NCI | p=0.007 AD>NCI |
| Phosphorylated tau (AT8) | p=0.007 MCI, AD>NCI | p=0.001 AD>NCI | p=0.03 AD>NCI | p=0.09 | p=0.007 AD>NCI | p=0.03 AD>NCI | p=0.001 AD>NCI |
| Truncated tau (TauC3) | p=0.16 | p=0.06 | p=0.10 | p=0.06 | p=0.40 | p=0.65 | p=0.63 |
| Aβ plaque density | EC Layer II | Subiculum | CA1 | CA2 | CA3 | DG polymorphic Layer | DG granule Layer |
| MOAB-2 DPs | p=0.30 | p=0.54 | p=0.59 | p=0.27 | p=0.21 | p=0.11 | N/A |
| MOAB-2 NPs | p=0.47 | p=0.26 | p=0.52 | p=0.54 | p=0.29 | p=0.09 | N/A |
| Aβ1–42 DPs | p=0.59 | p=0.97 | p=0.99 | p=0.57 | p=0.26 | p=0.01 MCI>NCI, AD | N/A |
| Aβ1–42 NPs | p=0.31 | p=0.13 | p=0.65 | p=0.39 | p=0.13 | p 0.12 | N/A |
Kruskal-Wallis test
AD, Alzheimer’s disease; MCI, mild cognitive impairment; NCI, no cognitive impairment. DPs, diffuse plaques; NPs, neuritic plaques.
Figure 6.
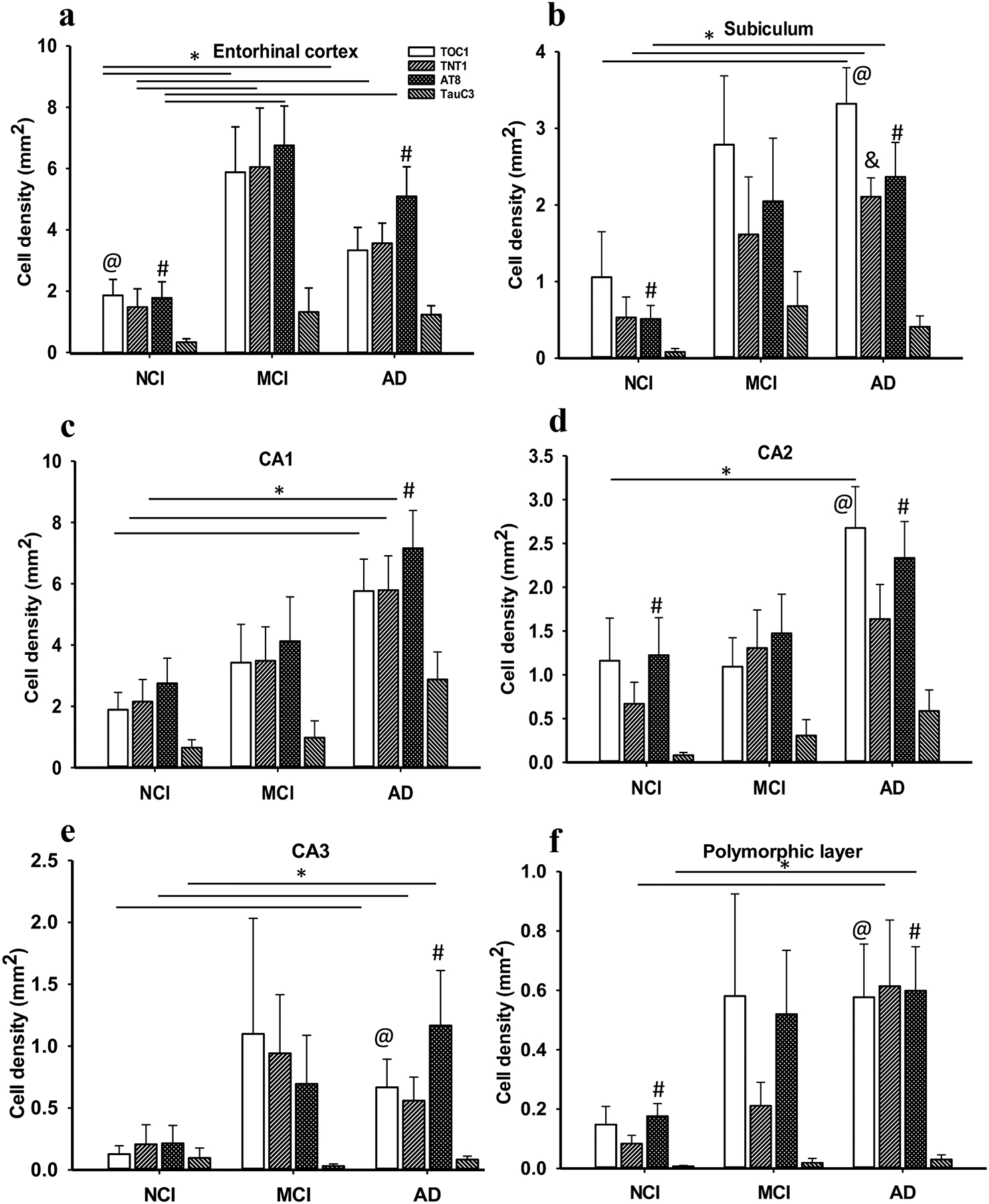
Bar graphs showing a significant increase in the density of TOC1, TNT1, AT8 reactivity in EC layer II in MCI and AD compared to NCI (a). TOC1, TNT1, AT8-ir cell densities in CA1 (c), CA3 (e), and subiculum (b) were significantly higher in AD compared to NCI, but not in MCI. CA2 TOC1 (d) and DG polymorphic layer TNT1 and AT8 (f) densities were significantly increased in AD compared to NCI. No significant differences in TauC3 density were found in any of the entorhinal-hippocampal regions (a-f) across clinical groups. Within group analysis revealed significantly higher TOC1 and AT8 cell densities compared to TauC3 in the EC in NCI (a), while only AT8 density was significantly higher compared to TauC3 in AD. Subicular (b) TOC1, TNT1 and AT8 positive cell densities were significantly higher than TauC3 in AD, whereas AT8 densities were greater than TauC3 in NCI. Although the density of AT8 cell labelling was significantly greater than TauC3 in CA1 (c), TOC1 and AT8 density in CA2 (d), CA3 (e) and DG polymorphic layer (f) was increased in AD. AT8 density was significantly greater compared to TauC3 in AD, whereas only AT8 density in CA2 (d) and DG polymorphic layer (f) were higher than TauC3 in NCI. No differences were found between the density of any tau epitope in MCI in the regions examined. Kruskal-Wallis, *, p<0.05; Significant comparisons: @, TOC1 vs TauC3, # AT8 vs TauC3 and &, TNT1 vs TauC3.
Figure 7.
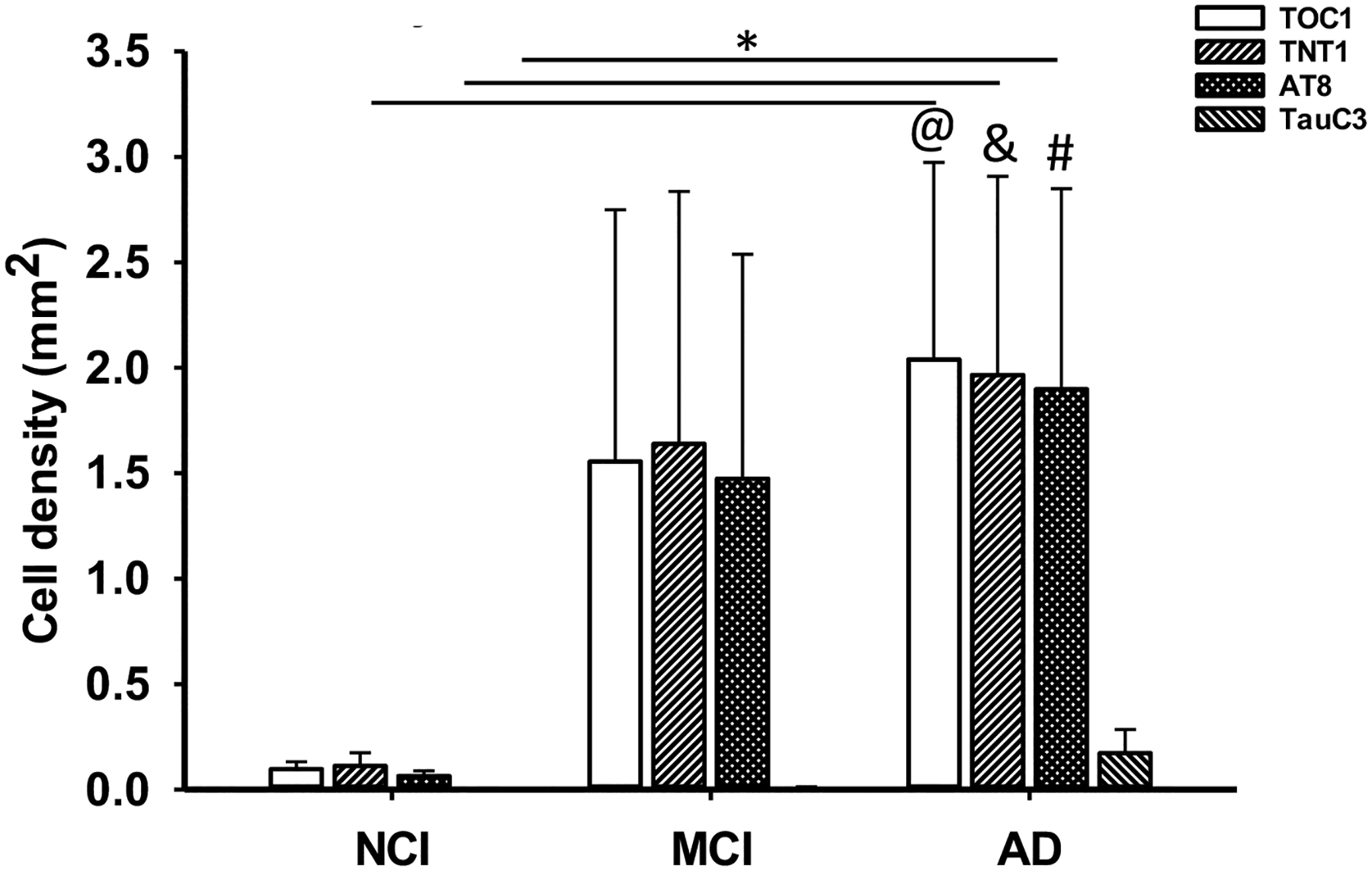
Bar graph showing a significant increase in TOC1, TNT1 and AT8 neuron densities in AD compared to NCI within the DG granule cell layer. TOC1, TNT1 and AT8 neuron densities were higher than TauC3 in the DG granule cell layer in AD. Kruskal-Wallis, *, p<0.05; Significant comparisons: @, TOC1 vs TauC3, # AT8 vs TauC3 and &, TNT1 vs TauC3.
Tau epitope prevalence within the entorhinal-hippocampal complex within clinical groups
We also evaluated the prevalence of different tau epitopes in each entorhinal-hippocampal region within each individual clinical group. Quantitation revealed a greater density of TOC1 (Kruskal-Wallis, p=0.008; Fig. 6a) and AT8 (Kruskal-Wallis, p=0.013; Fig. 6a) compared to TauC3-labeled NFTs in EC layer II in NCI. By contrast, there were no significant differences between NFT densities for TOC1, TNT1, AT8 and TauC3 in EC layer II in MCI (Kruskal-Wallis, p>0.05; Fig. 6a). AT8 cell density was only significantly higher compared to TauC3 in the EC in AD (Kruskal-Wallis, p=0.004; Fig. 6a). Density of TOC1 (Kruskal-Wallis, p<0.001), TNT1 (Kruskal-Wallis, p=0.006) and AT8 (Kruskal-Wallis, p=0.003) positive cells in the subiculum were significantly higher than TauC3 stained neurons in AD (Fig. 6b). In NCI, only AT8 densities were greater compared to TauC3 in the subiculum (Kruskal-Wallis, p<0.035; Fig. 6b). However, there were no density differences between tau epitopes in the subiculum in MCI cases (Kruskal-Wallis, p>0.05; Fig. 6b).
Density of AT8 positive cells were significantly greater than TauC3 (Kruskal-Wallis, p<0.001) in the hippocampal CA1 subfield in AD (Fig. 6c), but not between tau isoforms in NCI or MCI (Fig. 6c). Within CA2, TOC1 (Kruskal-Wallis, p=0.003) and AT8 (Kruskal-Wallis, p=0.008) cell densities were greater than TauC3 in AD (Fig. 6d), whereas only the density of AT8 labeled NFTs were greater than TauC3 in NCI (Kruskal-Wallis, p=0.015; Fig. 6d). There were no differences in cell densities between tau epitopes in the MCI group (Kruskal-Wallis, p>0.05; Fig. 6d). In CA3, TOC1 (Kruskal-Wallis, p=0.04; Fig. 6e) and AT8 (Kruskal-Wallis, p=0.003; Fig. 6e) cell densities were significantly higher than TauC3 in AD. No significant differences in cell density were observed between tau isoforms in this region in NCI or MCI.
In the DG polymorphic layer, the density of TOC1 (Kruskal-Wallis, p=0.002; Fig. 6f) and AT8 (Kruskal-Wallis, p=0.002; Fig. 6f) positive cells were greater than TauC3 in AD, while only the density of AT8 profiles was higher than TauC3 in NCI (Kruskal-Wallis, p<0.001; Fig. 6f). TOC1 (Kruskal-Wallis, p=0.001), TNT1 (Kruskal-Wallis, p=0.01) and AT8 (Kruskal-Wallis, p=0.007) cell densities were significantly higher than TauC3 in the DG granule cell layer in AD (Fig. 7). There were no differences between cell-densities for each tau epitope in the DG polymorphic and granule cells in MCI (Kruskal-Wallis, p>0.05; Figs. 6f and 7).
Tau epitope prevalence across the entorhinal-hippocampal connectome within a clinical group
The highest densities of oligomeric TOC1 positive cells were observed in the EC and hippocampal CA1 subfield compared to CA3 and DG granule cell layer in NCI across regions (Friedman repeated measures, p<0.05; Fig. 8a). Moreover, CA1 and subicular TOC1 positive cell densities were significantly greater than CA3 and DG polymorphic cells in AD (Friedman repeated measures, p<0.05; Fig. 8a). Although the highest number of TOC1 positive cells was found in the EC, there were no significant differences between regions in MCI (Friedman repeated measures, p>0.05; Fig. 8a).
Figure 8.
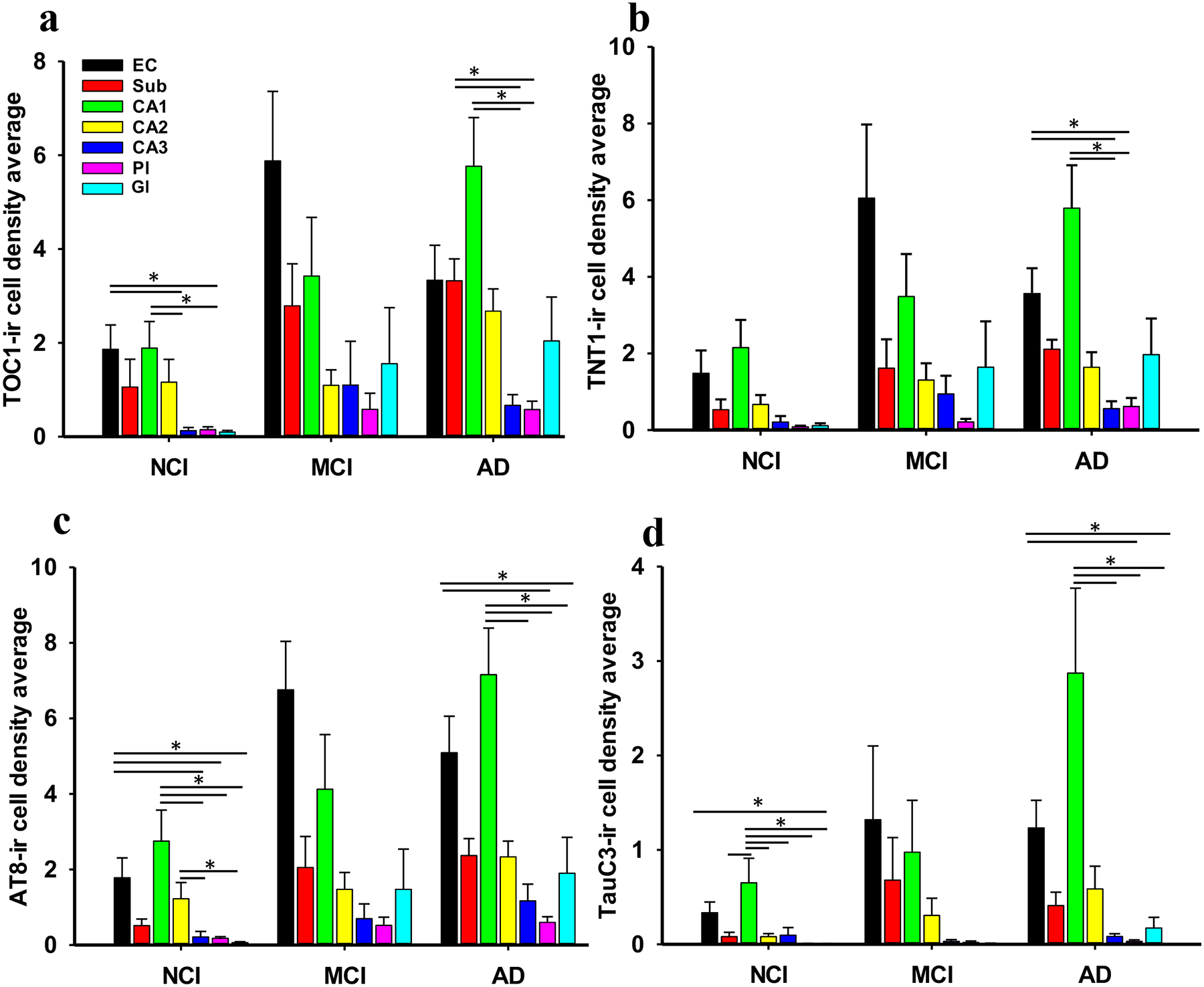
Bar graphs showing the density distribution of tau epitopes within regions of the entorhinal-hippocampal complex across clinical groups. (a) Highest densities of oligomeric TOC1 positive cells appeared in the EC and hippocampal CA1 field compared to CA3 and DG granule cells. CA1 and subicular TOC1 positive cell densities were significantly greater than CA3 and the DG polymorphic cell layer in AD. Despite the greatest number of TOC1 positive cells seen in the EC, no significant differences in TOC1 densities were found between brain regions in the MCI group. (b) Density of TNT1 profiles in the EC and CA1 subfield were significantly higher than either CA3 or DG polymorphic layer in AD. No significant differences in TNT1 positive densities were observed between the brain regions in MCI or NCI. (c) AT8-positive cell densities in EC and CA1 were greater than CA3, DG polymorphic and granule cell layers in the NCI group. The number of AT8-positive cells in CA2 were significantly higher than CA3 and the granule layer of the DG in NCI. Density of AT8 positive cells in CA1 was significantly greater than CA3 and the polymorphic and granule cell layers of the DG in AD. Density of EC AT8 positive cells was increased compared to the DG granule and polymorphic cell layers in AD. Despite the greater density of AT8 positive neurons in the EC of the MCI cases, no significant differences were found between brain regions in AD. (d) Density of NCI TauC3 labeled neurons revealed a significant increase in CA1 compared to CA3 and CA2 subfields, subiculum and DG granule and polymorphic layers, whereas the EC was only greater than the DG granule cell layer. AD TauC3 density within the CA1 was greater than CA3 and both layers of the DG, while the EC contained greater numbers compared to the DG polymorphic and granular cell layers. By contrast, no significant differences in any of the tau markers were observed between the different brain regions in the MCI group. Friedman’s test, *, p<0.05.
The density of TNT1 profiles in the EC (Friedman repeated measures, p<0.01) and CA1 subfield (Friedman repeated measures, p<0.001) were significantly higher than either CA3 or DG polymorphic cell layer in AD (Fig. 8b). However, no significant differences in TNT1 positive densities were observed between connectome regions in MCI and NCI (Friedman repeated measures, p>0.05; Fig. 8b).
AT8 positive cell densities within EC and CA1 were greater compared to CA3 and DG polymorphic and granule cell layers in the NCI group (Friedman repeated measures, p<0.01; Fig. 8c). The number of AT8 positive cells in CA2 were significantly higher than in CA3 and the DG granule layer in NCI (Friedman repeated measures, p<0.05). Density of AT8 positive cells in CA1 were significantly greater than CA3 and DG polymorphic and granule cell layers in AD (Friedman repeated measures, p<0.01; Fig. 8c). AT8 cell density in the EC was significantly increased compared to the DG granule and polymorphic cell layers in AD (Friedman repeated measures, p<0.05; Fig. 8c). Interestingly, despite a higher density of AT8 positive neurons in the EC in MCI, no significant differences were found between the other subregions.
In NCI, the density of TauC3 was significantly increased in CA1 compared to CA3 and CA2, subiculum and DG granular and polymorphic layers (Friedman repeated measures, p<0.05; Fig. 8d), while the EC was only greater than the DG granule cell layer (Friedman repeated measures, p<0.05; Fig. 8d). TauC3 density in CA1 was greater than CA3 and both layers of the DG (Friedman repeated measures, p<0.01; Fig. 8d), while the EC displayed greater numbers compared to the DG cell layers in AD (Friedman repeated measures, p<0.05; Fig. 8d). By contrast, no significant differences in TauC3 densities were observed between regions in MCI cases (Friedman repeated measures, p>0.05; Fig. 8d).
Nuclear SRSF2 intensity within AT8-bearing neurons in entorhinal-hippocampal connectome across clinical stages of AD
Dual immunofluorescence revealed no changes in the intensity of nuclear SRSF2 immunoreactivity in cells stained for AT8 in any of the entorhinal-hippocampal regions examined across clinical groups (Table 4). By contrast, SRSF2 nuclear intensity was significantly higher in AT8 immunonegative neurons in the EC (Kruskal-Wallis, p=0.02; Fig. 9a, Table 4) and CA2 (Kruskal-Wallis, p=0.04; Fig. 9b, Table 4) in NCI compared to AD, but not MCI. Interestingly, a within group analysis revealed that SRSF2 nuclear intensity was significantly decreased in AT8 positive compared to immunonegative cells in all entorhinal-hippocampal regions examined (Wilcoxon test, p<0.05; Table 4).
Table 4.
Intensity of SRSF2 nuclear immunoreactivity in AT8 tangle (+) and non-tangle (−) bearing neurons in the entorhinal-hippocampal complex
| NCI | MCI | AD | *P-value | Groupwise Comparisons | |
|---|---|---|---|---|---|
| EC layer II-lll SRSF2/AT8- | 494.90±155.12 | 272.37±71.15 | 296.56±151.82 | 0.02 | NCI>AD |
| EC layer II-III SRSF2/AT8+ | 236.66±135.96 | 178.05±80.96 | 202.38±77.92 | 0.49 | - |
| ** P-value | 0.02 | 0.03 | 0.002 | ||
| Subicular SRSF2/AT8- | 487.46±173.66 | 462.98±175.40 | 343.81±140.94 | 0.13 | - |
| Subicular SRSF2/AT8+ | 210.67±166.20 | 251.60±160.69 | 198.05±84.67 | 0.74 | - |
| P-value | 0.002 | 0.03 | 0.002 | ||
| CA1 SRSF2/AT8- | 424.16±196.00 | 496.77±190.05 | 326.78±146.30 | 0.08 | - |
| CA1 SRSF2/AT8+ | 209.21±104.70 | 269.46±149.16 | 172.23±84.37 | 0.17 | - |
| P-value | 0.002 | 0.03 | 0.002 | ||
| CA2 SRSF2/AT8- | 435.25±207.71 | 347.53±120.56 | 271.24±98.77 | 0.04 | NCI>AD |
| CA2 SRSF2/AT8+ | 161.39±157.39 | 219.31±115.82 | 162.24±53.16 | 0.09 | - |
| P-value | 0.002 | 0.03 | 0.002 | ||
| CA3 SRSF2/AT8- | 461.04±91.67 | 405.70±175.46 | 332.51±126.20 | 0.06 | - |
| CA3 SRSF2/AT8+ | 128.90±170.43 | 136.58±138.96 | 79.22±96.32 | 0.68 | - |
| P-value | 0.002 | 0.03 | 0.03 | ||
| DG Gl SRSF2/AT8- | 459.02±108.94 | 465.73±165.64 | 342.38±174.38 | 0.13 | - |
| DG Gl SRSF2/AT8+ | 229.68±164.79 | 241.25±126.29 | 158.26±92.66 | 0.26 | - |
| P-value | 0.002 | 0.03 | 0.002 | ||
| DG Pl SRSF2/AT8- | 414.58±127.91 | 325.54±90.43 | 368.48±160.78 | 0.39 | - |
| DG Pl SRSF2/AT8+ | 140.43±167.15 | 167.67±92.83 | 177.84±131.17 | 0.82 | - |
| P-value | 0.002 | 0.03 | 0.002 |
Kruskal-Wallis,
Wilcoxon test
AD, Alzheimer’s disease; MCI, mild cognitive impairment; NCI, no cognitive impairment
Figure 9.
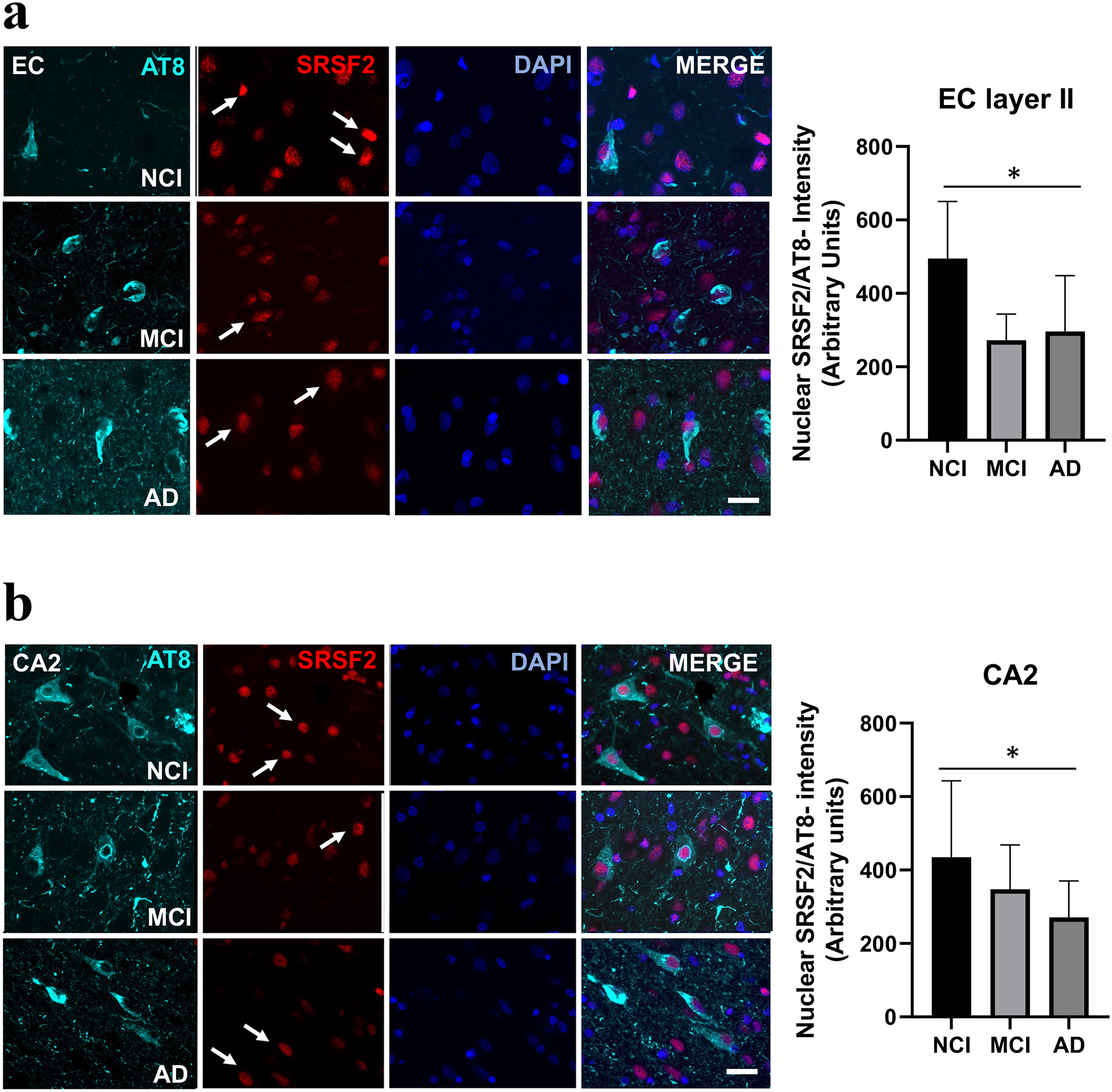
Immunofluorescence showing single stained AT8 (cyan), SRSF2 (red), DAPI (blue) and merged images in the EC layer II and CA2 in NCI, MCI and AD cases. Note a higher degree of SRSF2 fluorescence in AT8 immunonegative neurons in NCI compared to either AD or MCI (white arrows) within the EC layer II (a) and CA2 (b). Histograms showing a significant increase in SRSF2 nuclear intensity in AT8 immunonegative neurons in EC (a) and CA2 (b) in NCI compared to AD. Abbreviations: CA2, hippocampal subfield CA2; EC layer II, entorhinal cortex layer II. Scale bar=50 μm, *, p<0.05.
MOAB-2 and Aβ1–42 plaque density in the entorhinal-hippocampal connectome across clinical stages of AD
Neuritic and diffuse plaque densities were evaluated using a pan-amyloid (MOAB-2) and an Aβ1–42 antibody. We found no significant differences in MOAB-2 positive diffuse or neuritic plaque density in any of the entorhinal-hippocampal regions examined between clinical groups (Kruskal-Wallis, p>0.05; Table 3, Fig. 10a–c). Conversely, Aβ1–42 reactive diffuse plaque densities were significantly greater only in DG polymorphic layer in MCI compared to NCI and AD (Kruskal-Wallis, p=0.01; Table 3, Fig. 11a–c). Interestingly, APOE ε4 carrier analysis revealed significantly more Aβ1–42 neuritic plaques in CA3 compared to non-carriers (Kruskal-Wallis, p=0.03).
Figure 10.
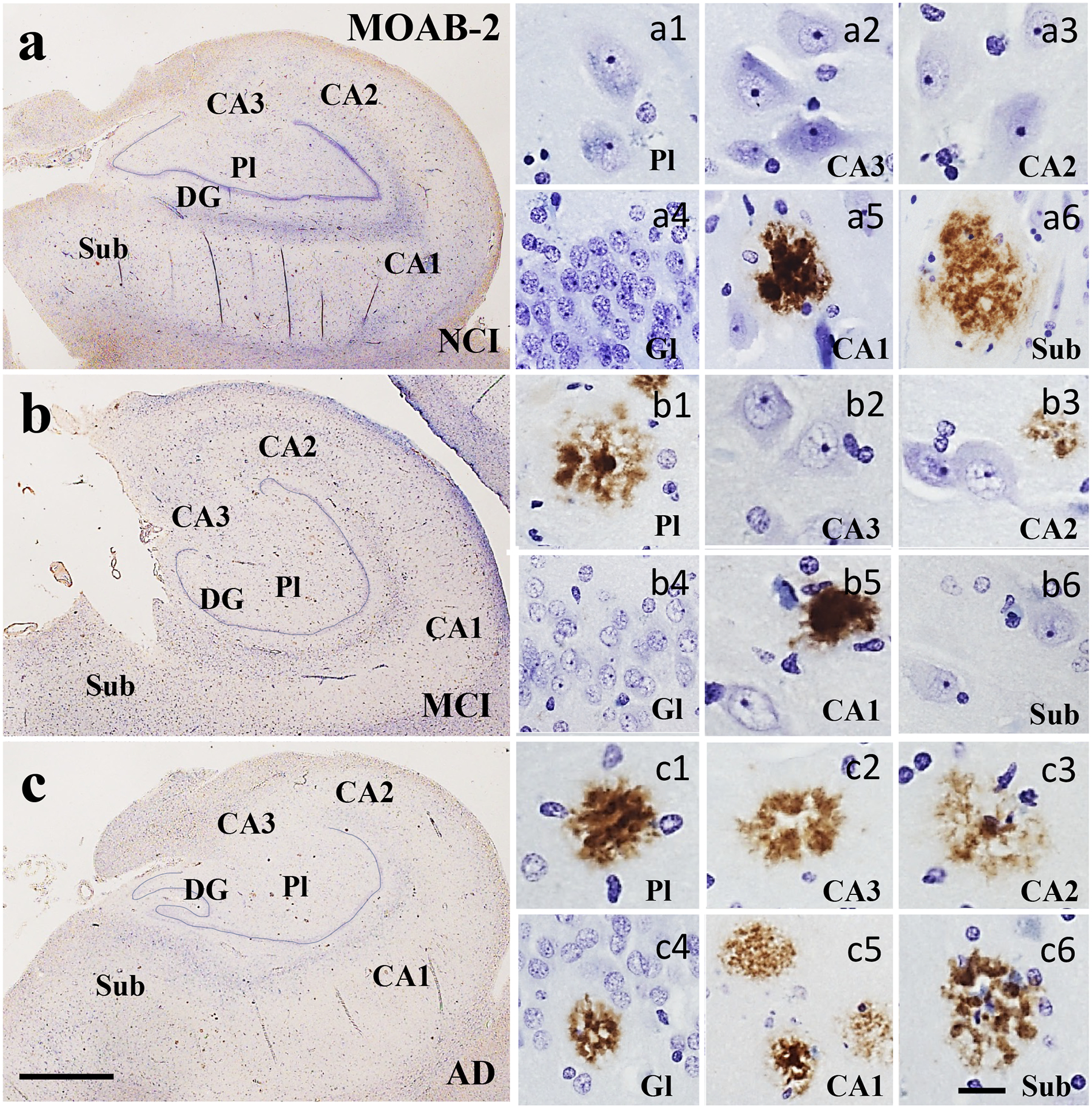
Low magnification photomicrographs showing limited pan Aβ (MOAB-2) immunoreactivity in the hippocampus and subiculum in NCI (a), MCI (b) and AD (c). Images labeled with (a-c)1-6 show Aβ reactive plaque pathology across clinical groups. In NCI (a), a few plaques were seen in the CA1 and subiculum. In MCI (b), plaques were seen in the DG polymorphic layer, CA2 and CA1, but not in the subiculum. By contrast, plaques were found in all hippocampal subfields and the subiculum in AD (c). Abbreviations: CA1, hippocampal subfield CA1; CA2, hippocampal subfield CA2; CA3, hippocampal subfield CA3; DG, dentate gyrus; Gl, granule cell layer of the DG; Pl, polymorphic layer of the DG; Sub, subiculum. Scale bar in c=200 μm and applies to a and b, (a-c)6=25 μm and applies to panels (a-c)1–5.
Figure 11.
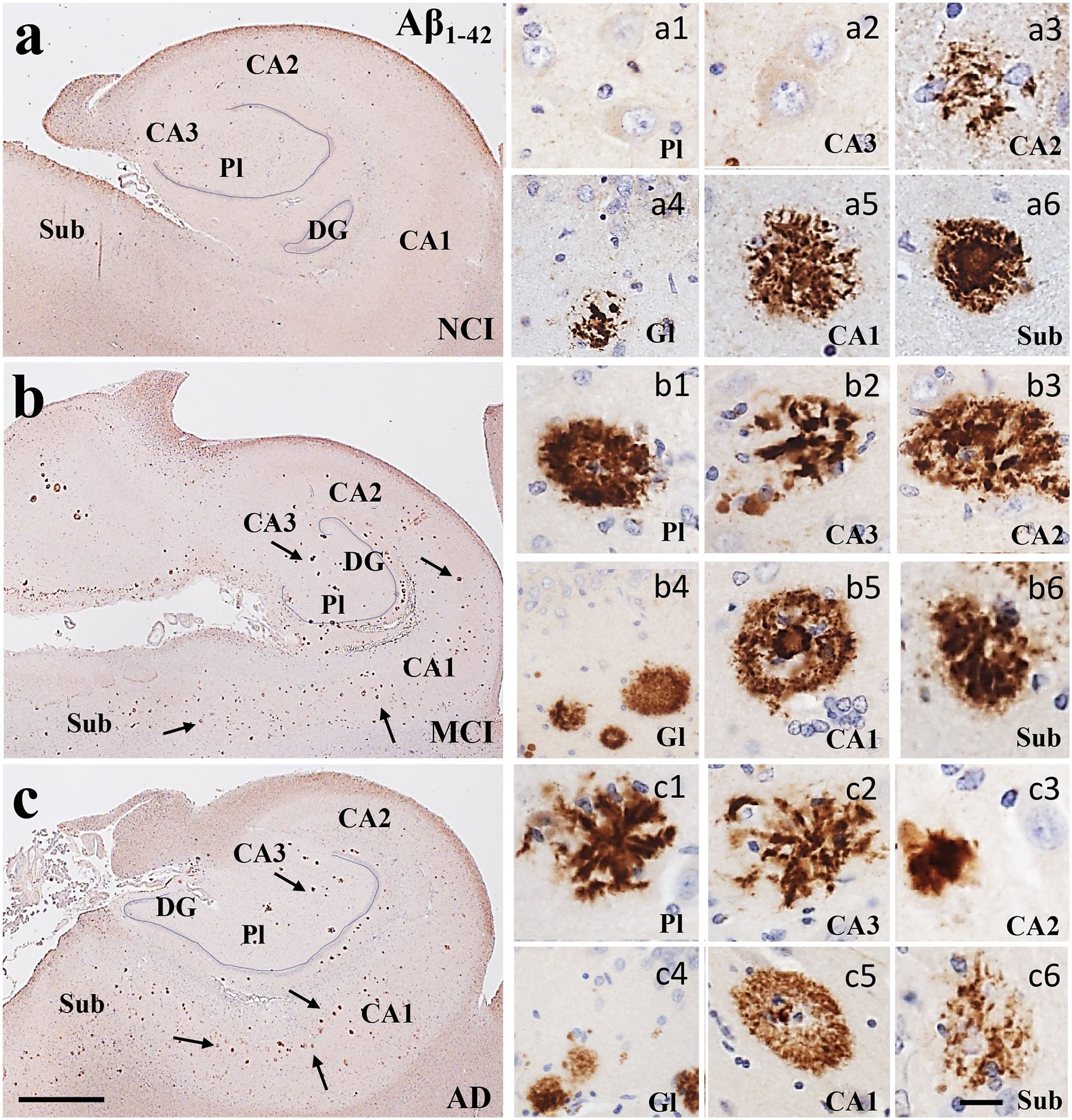
Low magnification photomicrographs of Aβ1–42 immunoreactive profiles (arrows) showing an increase in plaque load in the hippocampus and subiculum from NCI (a) to MCI (b) to AD (c). Images labeled with (a-c)1–6 show diffuse (a5, b1), neuritic (c1, c2) and cored plaques across regions and clinical groups. Sections were counterstained with hematoxylin (light-blue nuclear staining). Abbreviations: CA1, hippocampal subfield CA1; CA2, hippocampal subfield CA2; CA3, hippocampal subfield CA3; DG, dentate gyrus; Gl, granule cell layer of the DG; Pl, polymorphic layer of the DG; Sub, subiculum. Scale bar in c=200 μm and applies to aand b, (a-c)6=25 μm and applies to panels (a-c)1–5.
Correlations between tau and Aβ density, SRSF2 intensity, demographic, and neuropathology in the entorhinal-hippocampal connectome during the progression AD
Correlations between tau epitopes and Aβ plaque counts, nuclear SRSF2 intensity, demographic and pathological criteria were performed across clinical groups. However, we discuss only those showing the strongest significant correlations (Fig. 12 and Supplemental Table 2).
Figure 12.
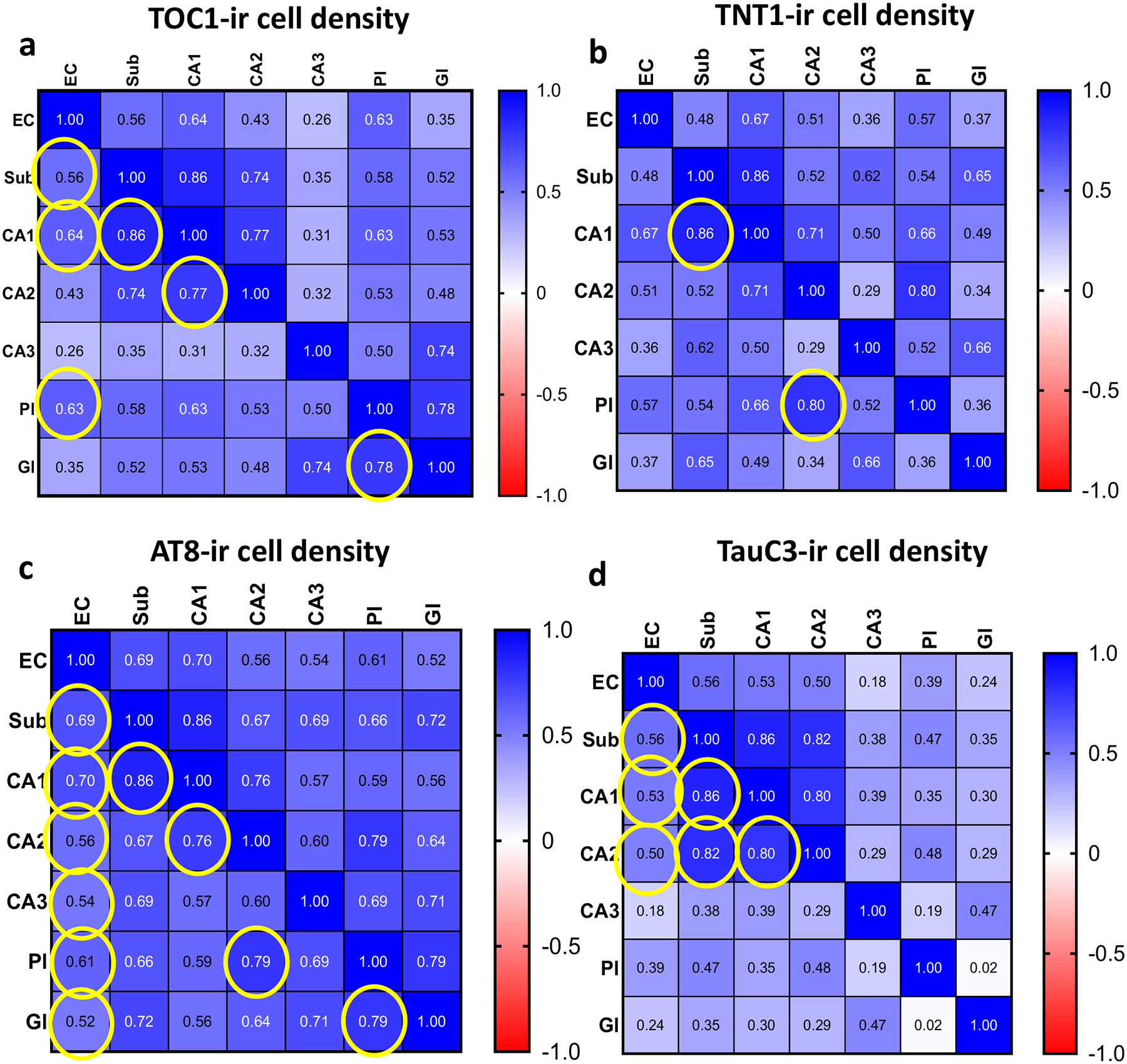
Heatmaps showing the spearman correlation coefficient values (r) obtained from the comparison of TOC1 (a), TNT1 (b), AT8 (c) and TauC3 (d) positive cell densities within the entorhinal-hippocampal regions across clinical groups. Yellow circles indicate significance, Bonferroni adjusted significance level α=0.03.
Within brain regions strong positive correlations were found between the density of TOC1 neurons in CA1, subiculum (Spearman test, r=0.86, p<0.0001) and CA2 (Spearman test, r=0.77, p<0.0001) across clinical groups (Fig. 12a). DG granular and polymorphic TOC1 cell values were highly correlated (Spearman test, r=0.78, p<0.0001; Fig. 12a). EC layer II TOC1 cell density correlated with CA1 (Spearman test, r=0.64, p=0.001; Fig. 12a), subiculum (Spearman test, r=0.56, p=0.006; Fig. 12a) and DG polymorphic (Spearman test, r=0.63, p=0.002; Fig. 12a) measurements. TNT1 density measurements in CA1 and CA2 were associated with subicular (Spearman test, r=0.86, p<0.0001) and DG polymorphic cell values (Spearman test, r=0.80, p<0.0001), respectively (Fig. 12b). Density of AT8 positive cells within CA1 were strongly associated with subicular (Spearman test, r=0.86, p<0.0001) and CA2 measurements (Spearman test, r=0.76, p<0.0001; Fig. 12c). Strong associations were found between the density of AT8 labeling in the DG granular and polymorphic cell layers (Spearman test, r=0.79, p<0.0001) and between DG polymorphic cell layer and CA2 (Spearman test, r=0.79, p<0.0001; Fig. 12c). In addition, the density of the AT8 positive cells in the EC strongly correlated with CA1 (Spearman test, r=0.70, p<0.0001) and subicular (Spearman test, r=0.69, p<0.0001) values, but weakly with CA2 (Spearman test, r=0.56, p=0.003), CA3 (Spearman test, r=0.54, p=0.005) and DG polymorphic (Spearman test, r=0.61, p=0.001) and granule (Spearman test, r=0.52, p<0.006) cells. Finally, the density of TauC3 labeled cells in the subiculum significantly correlated with CA1 (Spearman test, r=0.86, p<0.0001) and CA2 (Spearman test, r=0.82, p<0.0001) as well as between CA1 and CA2 (Spearman test, r=0.80, p<0.0001) values across clinical groups (Fig. 12d). EC TauC3 values correlated with CA1 (Spearman test, r=0.53, p=0.005), CA2 (Spearman test, r=0.50, p=0.009) and subicular (Spearman test, r=0.56, p=0.002) densities across clinical groups.
A strong correlation was found between TOC1, TNT1 and AT8 cell density in EC (Spearman test, r>0.85, p<0.0001) and subiculum (Spearman test, r> 0.88, p<0.0001) across clinical groups (Supplemental Table 1). Moreover, these tau epitopes strongly correlated with each other within CA1 (Spearman test, r>0.90, p<0.0001; Supplemental Table 1) and with CA1 TauC3 measurements (Spearman test, r=0.83–0.89, p<0.0001; Supplemental Table 1). AT8 densities highly correlated with TNT1, TOC1 and TauC3 (Spearman test, TNT1 r=0.83, TOC1 r=0.5, TauC3 r=0.76; Supplemental Table 1) in CA3. CA2 subfield AT8 measurements were associated with TOC1 (Spearman test, r=0.85, p<0.0001; Supplemental Table 1), and TNT1 and TauC3 (Spearman test, TNT1 r=0.76, TauC3 r=0.76, p<0.0001; Supplemental Table 1). In the DG polymorphic and granule cell layers, the density of AT8 NFTs were significantly associated with TOC1 (Spearman test, r>0.84, p<0.0001; Supplemental Table 1) and TNT1 (Spearman test, r>0.79, p<0.0001; Supplemental Table 1). However, only the density of these oligomeric tau epitopes within the DG granule cells strongly correlated across clinical groups (Spearman test, r=0.83, p<0.0001; Supplemental Table 1)
Density of TOC1 positive cells in CA1 (Spearman test, r=0.59, p<0.001; Supplemental Fig. 1a), subiculum (Spearman test, r=0.66, p<0.001; Supplemental Fig. 1b) and EC layer II (Spearman test, r=0.71, p<0.001; Supplemental Fig. 1c) correlated significantly with Braak stage. TOC1 cell density in the EC layer II correlated with NIA Reagan (Spearman test, r=0.71, p<0.001; Supplemental Fig. 1d) and CERAD (Spearman test, r=0.67, p<0.001; Supplemental Fig. 1e) scores. Density of TNT1 positive cells in CA1 (Spearman test, r=0.67, p<0.001; Supplemental Fig. 2a), CA2 (Spearman test, r=0.58, p=0.003; Supplemental Fig. 2b), subiculum (Spearman test, r=0.74, p<0.001; Supplemental Fig. 2c), and EC (Spearman test, r=0.74, p<0.001; Supplemental Fig. 2d) correlated strongly with Braak stage across groups. Furthermore, TNT1 NFT densities within the EC strongly correlated with CERAD (Spearman test, r=0.71, p<0.001; Supplemental Fig. 2e) and NIA Reagan scores (Spearman test, r=0.70, p<0.001; Supplemental Fig. 2f). Density of AT8 positive NFTs in CA1 (Spearman test, r=0.65, p<0.001; Supplemental Fig. 3a), CA2 (Spearman test, r=0.56, p=0.003; Supplemental Fig. 3b), CA3 (Spearman test, r= 0.58, p=0.001; Supplemental Fig. 3c), DG granular cell layer (Spearman test, r=0.49, p<0.007; Supplemental Fig. 3d), subiculum (Spearman test, r =0.72, p<0.001; Supplemental Fig. 3e) and EC (Spearman test, r=0.79, p<0.001; Supplemental Fig. 3f) strongly correlated with Braak stage. The density of AT8 cells in EC strongly correlated with NIA Reagan scores (Spearman test, r=0.62, p<0.001; Supplemental Fig. 4a). Conversely, none of the tau cell densities correlated with neuritic or diffuse plaque counts within the entorhinal-hippocampal regions across clinical groups.
Nuclear SRSF2 intensity in AT8 positive neurons in the DG polymorphic layer were significantly correlated with subicular TauC3 densities across groups (Spearman test, r=0.50, p=0.004; Supplemental Fig. 4b), likely resulting from transsynaptic activation of intrinsic hippocampal connections (Amaral, Scharfman, & Lavenex, 2007). Subicular nuclear SRSF2 intensity in cells lacking AT8 reactivity, negatively correlated with the density of Aβ1–42 diffuse plaques in the EC (Spearman test, r=−0.59, p=0.0008; Supplemental Fig. 4c). Nuclear SRSF2 intensity within AT8 negative cells in CA3 positively correlated with MMSE scores across clinical groups (r=0.58, p=0.007; Supplemental Fig. 4d). Nuclear SRSF2 density measurements in AT8 immunopositive or negative cells were not correlated with the specific Aβ marker MOAB-2 in the entorhinal cortex, hippocampus, and subiculum.
Discussion
The current study is the first quantitative immunocytochemical investigation of the relationship between oligomeric, phosphorylated and truncated tau isoforms and the SRSF2 nuclear splicing protein in NFT evolution within the entorhinal-hippocampal connectome during the clinical progression of AD. In the early 1970s, thioflavin NFT pathology was reported in layer II neurons of the EC, which innervate the hippocampus leading to the proposal that disconnection of this pathway plays an important role in the development of memory impairment in AD (Van Hoesen, Pandya, & Butters, 1972). About twenty years later, the presence of NFTs containing the abnormal phosphorylated microtubule-associated protein tau, that is involved in cytoskeletal maintenance, was reported within layer II of the EC and defined as one of the earliest site of NFTs followed by the hippocampal formation in AD (H. Braak & Braak, 1991). Here, we used tau specific antibodies against early conformational oligomeric TOC1 and TNT1, phosphorylated AT8 and late truncated TauC3 epitopes and the nuclear tau-splicing mediator factor SRSF2 to determine the regional deposition of NFT pathology within the entorhinal-hippocampal connectome during the clinical onset of AD.
Tau oligomeric TOC1, TNT1 and phosphorylated AT8: early pathological events in AD
The distribution and evolution of tangle pathology and the mechanisms underlying NFT toxicity in the entorhinal-hippocampal connectome across the clinical stages of AD remains elusive. It is well established that hyperphosphorylated tau detaches from microtubules undergoing post-translational conformation and truncation that lead to pretangle soluble tau oligomer/aggregate forms that assemble into insoluble paired helical filaments, the main component of NFTs. Animal models of AD and human-based tissue studies have demonstrated that pre-fibrillary soluble tau aggregates, specifically oligomeric species are required to induce neurodegeneration and cognitive decline (Lasagna-Reeves, Castillo-Carranza, Guerrero-Muoz, Jackson, & Kayed, 2010). Here, we found that oligomeric TOC1, TNT1 and phosphorylated AT8 immunoreactivity have a granular appearance in the cytoplasm and dendrites in neurons within the entorhinal-hippocampal complex mainly in NCI and MCI cases suggesting a pretangle stage. Both TOC1 and TNT1 are conformational dependent antibodies (Jeganathan et al., 2008; Kanaan et al., 2011; Patterson, Remmers, et al., 2011). TOC1 recognizes portions of the proline-rich region of the tau molecule (Patterson, Remmers, et al., 2011) and TNT1 reacts with the PAD portion of tau that is implicated in fast axonal transport disturbances (Combs et al., 2016). Oligomeric and conformational changes within the tau amino terminus identified by these antibodies are among the earliest alterations associated with tau pathology in AD (Combs et al., 2016; Koss et al., 2016; Patterson, Remmers, et al., 2011; Ward et al., 2013), supporting the concept that tau oligomerization and phosphorylation are events that precedes NFT formation during the pathogenesis of AD (Combs et al., 2016; Tiernan et al., 2018). Moreover, they have a close association with early phosphorylated tau events detected by AT8 and pS422 antibodies during tangle evolution in AD (Patterson, Ward, et al., 2011; Ward et al., 2013). Although the colocalization of the oligomeric and phosphorylated tau antigens were not possible due to tissue limitation and antibody host cross-reactivity, the strong associations between the densities of cells containing TOC1, TNT1 and phosphorylated AT8 in the EC layer II, subiculum and CA1 across clinical groups, suggest a co-occurrence of tau oligomerization and phosphorylation events in neurons within these structures in AD. Of particular interest was the observation of perinuclear accumulation of oligomeric tau in CA2 and CA3 neurons in NCI, MCI and AD and to a lesser degree following AT8 staining (Španić et al., 2022). The perinuclear accumulation of oligomeric tau proteins in CA3 and CA2 neurons further suggests a role in neuronal selective vulnerability during the onset of AD. Since tau directly interacts with nuclear pore components (NPC) resulting in mislocalization and putative dysfunction of the neuropore (Eftekharzadeh et al., 2018), perhaps perinuclear oligomer contributes to NPC tau-induced neurotoxicity in AD.
We found that the density of AT8 positive cells was consistently greater compared to TauC3 in most of the regions examined in NCI, while TOC1 and AT8 tau isoforms predominated in AD. The densities of TOC1 and AT8 positive cells were consistently higher in the EC and CA1 compared to DG layers in NCI and AD, but not in MCI. Unlike TNT1 and TOC1, phosphorylated AT8 positive neurons were observed in each region of the entorhinal-hippocampal complex in the AD cases. Previous studies have demonstrated that tau phosphorylation, including AT8, is a pre-requisite to conformational changes in AD (Luna-Muñoz, Chávez-Macías, García-Sierra, & Mena, 2007; Luna-Muñoz et al., 2005) and tau oligomerization (Ward et al., 2014). In fact, the AT8 antibody recognizes both pre- and mature NFTs, but not end-stage ghost tangles in AD (E. Braak, Braak, & Mandelkow, 1994). Here, tau phosphorylation at serine202/threonine205 (AT8) occurred prior to oligomeric tau, whereas TOC1 and TNT1 displayed no or minimal temporal overlap with the late-stage tau truncation markers MN423 and TauC3 in AD (Combs et al., 2016) in the entorhinal-hippocampal connectome. Together these findings support the concept that oligomeric tau is an early pathological event that is less prevalent in advanced NFTs including extracellular ghost tangles (Combs et al., 2016; Patterson, Ward, et al., 2011). It is interesting that TNT1, which recognizes the oligomeric and conformational disease-relevant modifications of tau amino acids 2–18, termed PAD, which inhibits FAT (Kanaan et al., 2011), was observed prior to frank NFT formation early in the pathogenesis of the entorhinal-hippocampal connectome (Kelley, Perez, & Mufson, 2019; Lace et al., 2009; Llamas-Rodríguez et al., 2022; Patterson, Remmers, et al., 2011; Swanson et al., 2017; Vogel et al., 2020). Perhaps biochemically heterogeneous modifications in tau (i.e., filament formation, truncation, hyperphosphorylation, etc.) increase PAD exposure resulting in FAT inhibition (Kanaan et al., 2012), which represents a possible mechanism for early synaptic dysfunction during preclinical stages of AD (Counts, Nadeem, Lad, Wuu, & Mufson, 2006).
Increase of oligomeric TOC1 and phosphorylated AT8 within the entorhinal-hippocampal connectome across clinical stages of AD
In the present study we found a significant increase in oligomeric and phosphorylated AT8 tau-bearing neurons within EC neurons in MCI and AD compared to NCI cases. By contrast, hippocampal densities of TOC1, TNT1 and AT8 positive neurons were significantly greater in the CA1 and CA3 subfields, subicular and granular cell layer of the DG in AD, but not MCI. These findings indicate a differential susceptibility to oligomeric and phosphorylated tau between EC and hippocampal neurons in prodromal AD. Perhaps these differences are, in part, due to the vulnerability to oxidative stress between EC and hippocampal neurons (Bartolome, Carro, & Alquezar, 2022), which has been implicated in tau oligomerization (Patterson, Ward, et al., 2011) and aggregation (Gamblin et al., 2003; Landino, Skreslet, & Alston, 2004). The greater density of oligomeric and phosphorylated tau positive neurons seen in the EC suggest that the buildup of these proteins triggers the anterograde propagation of tau epitopes via the perforant pathway, the major source of the glutamatergic excitatory input to the hippocampus (Hoesen & Pandya, 1975; Van Hoesen et al., 1972), but not to levels that distinguishes MCI from NCI. CA1, which innervates the subiculum (Rosene & Van Hoesen, 1977), compared to other hippocampal subfields, which consistently displayed increased densities of AT8, TauC3 and oligomeric tau in the AD cases. Others using antibodies against truncated TauC3 and MN423 and 3R and 4R tau isoforms have demonstrated that tau deposition followed the circuitry of the hippocampus in AD cases categorized by tau Braak staging criteria (Arezoumandan et al., 2022; Iseki et al., 2006; Lace et al., 2009). Here, our quantitative findings revealed that neuronal densities of TNT1, TOC1 and AT8 within the EC, hippocampus and subiculum significantly correlated with Braak staging across clinical groups. Previous studies using tissue sections from the EC, hippocampus, and superior temporal gyrus revealed that cellular TOC1 reactivity follows the characteristic pattern of Braak staging (Patterson, Remmers, et al., 2011). Others have reported strong correlations between TOC1 and AT8 protein levels derived from temporal cortex lysates (Koss et al., 2016) and phospho-tau antibody staining with Braak stages (Zhou et al., 2006). A recent tau-PET imaging study revealed some degree of overlap with Braak staging (Vogel et al., 2020), suggesting the former as biological marker for AD.
The connectional progression of tau pathology from EC to the hippocampus supports the suggestion that disconnection and/or deafferentation plays a pivotal role in the development of the dementia in patients with AD (E. Braak et al., 1994; Van Hoesen et al., 1972). Studies using animal models of AD combined with neuroanatomical tract tracing techniques have shown anterograde propagation of tau along the entorhinal-hippocampal connectome in rodents (de Calignon et al., 2012; Goedert, Eisenberg, & Crowther, 2017; Guo et al., 2016; McAllister, Lacoursiere, Sutherland, & Mohajerani, 2020; Ruan et al., 2021). A recent study revealed anterograde transport of tau and the cytoplasmic labeling of early and late tau epitopes in the hippocampus following an injection of a 4R tau using an adenovirus vector into the monkey EC (Beckman et al., 2021). These studies and others demonstrating the propagation of tau pathology along neuronal pathways (German, White, & Sparkman, 1987; Hyman et al., 1988; Pearson, 1996; Pearson, Esiri, Hiorns, Wilcock, & Powell, 1985; Rüb et al., 2000; Saper, Wainer, & German, 1987; Shukla & Bridges, 1999; Thal, Holzer, et al., 2000) led to the hypothesis that tau propagates along synaptically connected circuits using a “prion-like” trans-neuronal process (Fornari, Schäfer, Jucker, Goriely, & Kuhl, 2019; Jucker & Walker, 2011). Tau hyperphosphorylated oligomers are suggested to be seed-components underlying the circuit-based spread of tau in AD and other tauopathies (Dujardin et al., 2020). The present findings showing significant associations between the densities of phosphorylated AT8 bearing neurons in EC, CA hippocampal fields, subiculum and DG polymorphic and granule cell layer during AD progression lend support to this concept. The observation that the density of TOC1, but not TNT1, within the EC correlated with that seen in CA1, subicular and DG polymorphic layer suggests a prominent role for TOC1 in tau pathogenesis and its propagation in AD. We found that densities of TNT1, TOC1, AT8 and TauC3 in CA1, which innervates the subiculum (Rosene & Van Hoesen, 1977) were significantly correlated, providing support for trans-neuronal propagation of tau aggregates during the development of misfolded oligomeric tau species within the entorhinal-hippocampal circuit (DeVos et al., 2018). The spread of tau from the EC to the dentate gyrus was observed prior to synapse and neuronal loss (Pickett et al., 2017), suggesting that tau transport between synaptically interconnected neurons occurs prior to frank disease onset (DeVos et al., 2018). In this regard, a growing body of evidence suggests that transfer of toxic tau aggregates between neurons occurs via exosomes (Dujardin et al., 2014; Polanco et al., 2916, 2021, Wang et al., 2017, Xiao et al., 2017), which requires further investigation.
TauC3 densities are associated with APOE ε4 status
TauC3 recognizes a form of tau with a truncational change imposed by caspase-3-mediated cleavage at D421 (Fasulo et al., 2000), indicative of apoptotic cell death (Chung et al., 2001; Nicholls et al., 2017). Here, our quantitative analysis revealed very few TauC3 positive NFTs and no significant differences in TauC3 densities in any entorhinal-hippocampal subfield examined across clinical groups. However, a previous study found significantly more TauC3-positive NFTs in the hippocampus and EC when counts for each region were combined (Rissman et al., 2004). Others have reported TauC3-positive NFTs in the EC in severe AD, which inversely correlated with cognitive function (Gamblin et al., 2003; Guillozet-Bongaarts et al., 2005; Rissman et al., 2004). The present study utilized mild AD (mean MMSE: 21.08±6.05), while others examined “End-stage” AD cases with lower MMSE scores (mean MMSE: 6.50±4.69) (Rissman et al., 2004) and cases classified neuropathologically as severe AD cases (Braak stages V-VI) (Guillozet-Bongaarts et al., 2005), suggesting that both clinical and pathological severity is associated with more TauC3 pathology in AD.
When we stratified for APOE genotype, ε4 carriers had significantly more TauC3 positive NFTs in EC and subiculum. A previous investigation demonstrated a significant link between the expression the APOE ε4 allele and an increase of NFTs bearing TauC3 and MN423 truncated tau makers in the EC and subiculum in AD (Basurto-Islas et al., 2008). Although the biological mechanism(s) underlaying this association are unknown, other suggests that the link between APOE ε4 and tangle pathology is either indirect or dependent upon Aβ pathology (Farfel, Yu, De Jager, Schneider, & Bennett, 2016; Ramanan et al., 2019; Tosun et al., 2017). Tau-PET ligand imaging found that female APOE ε4 carriers showed a significantly greater tau burden in the hippocampus, EC, and parahippocampal cortices in aging and dementia cohorts, after accounting for age, educational attainment, clinical diagnosis and neocortical amyloid load (Y. T. Wang et al., 2021). Further studies are needed to investigate the effect of ApoE gene dosage upon the propagation of tau across clinical stages of AD.
SRSF2 nuclear intensity is associated with pretangle pathology in prodromal AD
The role of SRSF2 upon tau isoforms and NFT formation in AD is under investigated. Alternative splicing of tau exon 10 is a determinant factor in the generation of 3R and 4R tau isoforms. Phosphorylation of SRSF2 by glycogen 3 kinase (GSK3) favors the expression of 3R tau (Hernández et al., 2004), while the absence of SRSF2 favors exon 10 inclusion increasing 4R tau (K. L. Chen, Yuan, Hu, & Hsu, 2010), suggesting tau isoform modulation occurs through SRSF2. Studies have shown an increase in the ratio of the 3R/4R tau expression in cholinergic basal forebrain and hippocampal CA1 neurons in MCI and AD (Ginsberg, Che, Counts, & Mufson, 2006) and that the ratio of 3R tau-positive NFT was greater in hippocampal subfields with advanced NFT pathology in AD (Hara, Hirokawa, Kamei, & Uchihara, 2013; Iseki et al., 2006; Kitamura, Sugimori, Sudo, & Kobayashi, 2005; Lace et al., 2009; Uchihara, Hara, Nakamura, & Hirokawa, 2012) and a shift to higher numbers of 3Rtau to 4Rtau positive neurons in the brainstem in AD (Uematsu et al., 2018). In the current study, nuclear SRSF2 levels were decreased in AT8-positive neurons in EC layer II, CA subfields, subiculum and DG across all clinical groups, suggesting a link between tau pathology and loss of the protein for nuclear SRSF2 supporting studies demonstrating that tau acts as a mediator of spliceosome disruption in AD (Hsieh et al., 2019; Montalbano et al., 2021). We also found that nuclear SRSF2 density in AT8 immunonegative perikarya was significantly decreased in CA2 and EC in AD compared to NCI, but not MCI. This differential effect suggests that there are other processes involved with splicing dysregulation, which are not associated with the AT8 isoform. In this regard, the splicing factors U1–70K and U1A, which are associated with NFT formation have synergistic interactions with SRSF2 that may affect neuronal survival in AD (Bai et al., 2013; Diner et al., 2014; Hsieh et al., 2019; Zhu et al., 2020). Recently, a complex containing oligomeric tau and the splicing protein hnRNPA2B1 together with N6-methyladenosine was shown to contribute to tau neurodegeneration (Jiang et al., 2021). Therefore, future investigations are warranted to evaluate the interaction between different splicing proteins and tau epitopes in the formation of NFTs within the entorhinal-hippocampal circuit during the onset of AD.
Correlation of Aβ plaque density and SRSF2 intensity in the entorhinal-hippocampal connectome
Here, were found an increase in the density of MOAB-2 pan-amyloid and Aβ1–42 postiive neuritic and diffuse plaque densities. No significant differences in Aβ (MOAB-2) diffuse or neuritic plaques were found across the clinical groups examined. However, Aβ1–42 diffuse plaque density in the DG polymorphic layer was significantly increased in MCI compared to NCI and AD. Previous reports revealed low plaque numbers in prodromal AD, that plaques occur long after the emergence of NFTs in the EC and hippocampus, and amyloid and tau pathologies overlapped only in advanced AD (Thal, Rub, Orantes, & Braak, 2002; Thal, Rub, et al., 2000). This temporal divide between the onset of these pathological hallmarks questions the proposed link between Aβ and the downstream activation of NFT tau bearing lesions in AD. However, the present study did not examine oligomeric forms of Aβ, which may be present prior to insoluble amyloid or even pretangle pathology. Interestingly, subicular nuclear SRSF2-ir intensity negatively correlated with Aβ1–42 diffuse plaque burden in the EC. To our knowledge, no previous investigations have identified a link between SRSF2 and Aβ1–42 in the EC during the clinical progression of AD. Since SRSF2 and the RNA-binding protein hnRNPA1 may play a role in maintaining appropriate ratios of various forms of APP, changes in binding proteins could affect Aβ deposition (Donev, Newall, Thome, & Sheer, 2007). A study that crossed the N40K-tg mouse model of U1 snRNP dysfunction with the 5xFAD amyloidosis AD model demonstrated a synergy between a RNA splicing defect and amyloid resulting in a reorganization of the brain transcriptome and proteome, dysregulation of synaptic proteins and an increase in cognitive decline (K. L. Chen et al., 2022). In addition, this mouse model displayed a reduction of GABAergic synapse components that underlie entorhinal hippocampal neurotransmission, which is defective in AD.
Our results indicate that pretangle formation is concomitant with alterations in nuclear SRSF2 in cells containing NFT pathology across clinical groups, suggesting that alterations in nuclear SRSF2 occur independent of pretangle formation in the EC and hippocampal CA2 subfield in AD. Interestingly, studies have suggested the therapeutic use of aberrant splicing RNA to address tau and Aβ splicing defects in AD (Avale, Rodriguez-Martin, & Gallo, 2013; K. L. Chen et al., 2020; Garcia-Blanco, Baraniak, & Lasda, 2004). Therefore, understanding the complexity between splicing of tau propagation within the entorhinal-hippocampal circuit will provide novel information needed to develop drugs to slow the evolution of NFT pathology and the onset of dementia.
Study limitations
Limitations include the small number of MCI cases, which did not permit the subdivision into amnestic vs non-amnestic groups. Interestingly, a biochemical and biophysical investigation of tau aggregates revealed significant patient to patient heterogeneity in hyperphosphorylated species of soluble, oligomeric and seed components of tau (Dujardin et al., 2020). Here, the MCI group exhibited a large variation in tau densities suggesting that this clinical group did not contain a homogeneous tau signature (Arezoumandan et al., 2022) and that personalize medicine approaches are likely needed for each disease stage. Since the tissue examined in this study was not prepared for stereology, we used a non-stereological quantitative counting method, which is not inherently unbiased, but have been shown to produce similar outcomes (von Bartheld, 2002). Since only a few tissue blocks contained the TEC, this region was not examined. Although we did not examine the relationship between tau and amyloid within the different subdivisions of the entorhinal cortex, others have reported an increase in tau and amyloid in the lateral compared to the medial entorhinal cortex in AD (Lace et al., 2009) as well as an anterior to posterior increase in NFT accumulation and atrophy in the EC in preclinical AD cases (Llamas-Rodríguez et al., 2022), suggesting that more detailed analysis of EC subfields are needed to better define tau pathology during the progression of AD. Future studies should include the examination of the microstructure of white matter fiber tracts connecting the EC and hippocampus and the role that axonal disconnection plays in the ability of the diseased brain to display neuronal plasticity (DeKosky et al., 2002; Mufson et al., 2012; Mufson et al., 2015) and cognitive resilience (Kelley, Ginsberg, Liang, Counts, & Mufson, 2022; Mesulam, 1999; Mufson et al., 2016).
In summary, the present study revealed that the density of oligomeric TOC1, TNT1 and AT8 containing neurons in the EC was greater in both MCI and AD compared to NCI cases but no difference was found between the MCI and AD groups. Within the hippocampus, the density of these tau epitopes was increased in CA1, CA2, CA3, DG polymorphic and granular cell layers, and subiculum in AD compared to NCI, but not in MCI. There were no significant differences in TauC3 densities between the different MTL regions across the clinical groups. Strong correlations were found between TOC1, TNT1 and AT8 cell densities in CA1 and subiculum. Moreover, AT8 cell densities in the EC correlated with all CA subfields, subiculum, and DG polymorphic and granule cell layers. Nuclear SRSF2 levels were decreased in AT8-bearing neurons in EC layer II, hippocampal CA subfields and subiculum supporting a link between tau and the spliceosome that is associated with the stereotypic propagation of tau within the entorhinal-hippocampal connectome across the clinical stages of AD. Interestingly, we observed a stereotypic propagation of tau pathology, but clinical disease stage and not anatomical region affected the density of tau deposition within of the entorhinal-hippocampal connectome suggesting further investigation into clinical and molecular factors that modulate the anterograde transport of tau between synaptically connected brain regions during the onset of AD.
Supplementary Material
Supplemental Figure 1. Scatterplots showing significant correlations of SRSF2 nuclear intensity and tau epitope density with clinical and neuropathological variables (a-d). CA3 nuclear intensity of SRSF2 (a) significantly correlated with MMSE scores across clinical groups. TOC1 densities in the CA1 (b), subiculum (c), and EC layer II (d) were significantly correlated with Braak Stage. EC layer II TOC1 densities were correlated with NIA Reagan (e) and CERAD (f). Abbreviations: NCI, no cognitive impairment; MCI, mild cognitive impairment; AD, Alzheimer’s disease; CA1, hippocampal subfield CA1; CA3, hippocampal subfield CA3; EC, entorhinal cortex. Spearman correlations, Bonferroni adjusted α=0.007.
Supplemental Figure 2. Scatterplots showing significant correlations of TNT staining density with Braak stage (a-d). TNT densities in the CA1 (a), CA2 (b), subiculum (c), and EC layer II (d) were correlated strongly with Braak stage. Density of TNT labeling in EC layer II neurons strongly correlated with CERAD (e) and NIA Reagan score (f). Abbreviations: NCI, no cognitive impairment; MCI, mild cognitive impairment; AD, Alzheimer’s disease; CA1, hippocampal subfield CA1; CA2, hippocampal subfield CA2; EC, entorhinal cortex. Spearman correlations, Bonferroni adjusted α=0.007
Supplemental Figure 3. Scatterplots showing significant correlations of AT8 staining density with Braak stage (a-d). Densities of AT8-ir perikarya in the CA1 (a), CA2 (b), CA3 (c), DG (d), subiculum (e), and EC layer II (f) were strongly correlated with Braak stage. Abbreviations: NCI, no cognitive impairment; MCI, mild cognitive impairment; AD, Alzheimer’s disease; CA1, hippocampal subfield CA1; CA2, hippocampal subfield CA2; CA3, hippocampal subfield CA3; DG, dentate gyrus; EC, entorhinal cortex. Spearman correlations, Bonferroni adjusted α=0.007
Supplemental Figure 4. Scatterplots showing significant correlations between the intensity of SRSF2 nuclear immunoreactivity and tau epitope and Aβ1–42 density (a-c). A) AT8 positive neuron densities in EC layer II strongly correlated with NIA Reagan scores. SRSF2 nuclear intensity in AT8 labeled neurons in the DG polymorphic layer significantly correlated with TauC3 densities in the subiculum (b) Nuclear SRSF2 intensity in the subiculum negatively correlated with Aβ1–42 diffuse plaque densities in EC (c). Abbreviations: NCI, no cognitive impairment; MCI, mild cognitive impairment; AD, Alzheimer’s disease; EC layer II, entorhinal cortex layer II; Pl, polymorphic layer of the DG; CA3, hippocampal subfield CA3. Spearman correlations, Bonferroni adjusted α=0.007.
Acknowledgments
Support by NIH grants P01AG014449, R01AG061566, R01AG043375, P30AG028383, Fein Foundation, BrightFocus Foundation CA2018010 and Arizona Alzheimer’s disease consortium. Dr. Stephen Scheff provided the brain tissue samples and the late Dr. Lester (Skip) Binder at Northwestern University provided the TOC1 and TNT1 antibodies, now available through Dr. Nicholas Kanaan at Michigan State University (MSU).
Footnotes
Disclosures
The authors report no biomedical, financial interests or potential conflicts of interest.
Data availability statement
The data that support the findings of the study are available from the corresponding author upon reasonable request.
References
- Amaral DG, Scharfman HE, & Lavenex P (2007). The dentate gyrus: fundamental neuroanatomical organization (dentate gyrus for dummies). Prog Brain Res, 163, 3–22. doi: 10.1016/s0079-6123(07)63001-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arezoumandan S, Xie SX, Cousins KAQ, Mechanic-Hamilton DJ, Peterson CS, Huang CY, … Irwin DJ (2022). Regional distribution and maturation of tau pathology among phenotypic variants of Alzheimer’s disease. Acta Neuropathol, 144(6), 1103–1116. doi: 10.1007/s00401-022-02472-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Hyman BT, Flory J, Damasio AR, & Van Hoesen GW (1991). The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex, 1(1), 103–116. doi: 10.1093/cercor/1.1.103 [DOI] [PubMed] [Google Scholar]
- Ash PEA, Lei S, Shattuck J, Boudeau S, Carlomagno Y, Medalla M, … Wolozin B (2021). TIA1 potentiates tau phase separation and promotes generation of toxic oligomeric tau. Proc Natl Acad Sci U S A, 118(9). doi: 10.1073/pnas.2014188118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avale ME, Rodriguez-Martin T, & Gallo JM (2013). Trans-splicing correction of tau isoform imbalance in a mouse model of tau mis-splicing. Hum Mol Genet, 22(13), 2603–2611. doi: 10.1093/hmg/ddt108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, Hales CM, Chen PC, Gozal Y, Dammer EB, Fritz JJ, … Peng J (2013). U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc Natl Acad Sci U S A, 110(41), 16562–16567. doi: 10.1073/pnas.1310249110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolome F, Carro E, & Alquezar C (2022). Oxidative Stress in Tauopathies: From Cause to Therapy. Antioxidants, 11(8), 1421. Retrieved from https://www.mdpi.com/2076-3921/11/8/1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basurto-Islas G, Luna-Muñoz J, Guillozet-Bongaarts AL, Binder LI, Mena R, & García-Sierra F (2008). Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol, 67(5), 470–483. doi: 10.1097/NEN.0b013e31817275c7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman D, Chakrabarty P, Ott S, Dao A, Zhou E, Janssen WG, … Morrison JH (2021). A novel tau-based rhesus monkey model of Alzheimer’s pathogenesis. Alzheimers Dement, 17(6), 933–945. doi: 10.1002/alz.12318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berron D, van Westen D, Ossenkoppele R, Strandberg O, & Hansson O (2020). Medial temporal lobe connectivity and its associations with cognition in early Alzheimer’s disease. Brain, 143(4), 1233–1248. doi: 10.1093/brain/awaa068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, & Berry RW (2005). Tau, tangles, and Alzheimer’s disease. Biochim Biophys Acta, 1739(2–3), 216–223. doi: 10.1016/j.bbadis.2004.08.014 [DOI] [PubMed] [Google Scholar]
- Braak E, Braak H, & Mandelkow EM (1994). A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol, 87(6), 554–567. doi: 10.1007/bf00293315 [DOI] [PubMed] [Google Scholar]
- Braak H, & Braak E (1990). Alzheimer’s disease: striatal amyloid deposits and neurofibrillary changes. J Neuropathol Exp Neurol, 49(3), 215–224. [PubMed] [Google Scholar]
- Braak H, & Braak E (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol, 82(4), 239–259. doi: 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- Braak H, & Del Tredici K (2018). Spreading of Tau Pathology in Sporadic Alzheimer’s Disease Along Cortico-cortical Top-Down Connections. Cereb Cortex, 28(9), 3372–3384. doi: 10.1093/cercor/bhy152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Jin N, Qian W, Liu W, Tan X, Ding F, … Liu F (2014). Cyclic AMP-dependent protein kinase enhances SC35-promoted Tau exon 10 inclusion. Mol Neurobiol, 49(1), 615–624. doi: 10.1007/s12035-013-8542-3 [DOI] [PubMed] [Google Scholar]
- Chen J, Duan X, Shu H, Wang Z, Long Z, Liu D, … Zhang Z (2016). Differential contributions of subregions of medial temporal lobe to memory system in amnestic mild cognitive impairment: insights from fMRI study. Sci Rep, 6, 26148. doi: 10.1038/srep26148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KL, Han X, Shaw TI, Fu Y, Sun H, Niu M, … Peng J (2022). Alzheimer’s disease-associated U1 snRNP splicing dysfunction causes neuronal hyperexcitability and cognitive impairment. Nature Aging, 2(10), 923–940. doi: 10.1038/s43587-022-00290-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KL, Yuan R, Hu C, & Hsu C (2010). Amyloid-β peptide alteration of tau exon-10 splicing via the GSK3β-SC35 pathway. Neurobiol Dis, 40(2), 378–385. doi: 10.1016/j.nbd.2010.06.013 [DOI] [PubMed] [Google Scholar]
- Chen KL, Zhang P, Abe M, Aikawa H, Zhang L, Frank AJ, … Disney MD (2020). Design, Optimization, and Study of Small Molecules That Target Tau Pre-mRNA and Affect Splicing. Journal of the American Chemical Society, 142(19), 8706–8727. doi: 10.1021/jacs.0c00768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CW, Song YH, Kim IK, Yoon WJ, Ryu BR, Jo DG, … Jung YK (2001). Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis, 8(1), 162–172. doi: 10.1006/nbdi.2000.0335 [DOI] [PubMed] [Google Scholar]
- Combs B, Hamel C, & Kanaan NM (2016). Pathological conformations involving the amino terminus of tau occur early in Alzheimer’s disease and are differentially detected by monoclonal antibodies. Neurobiol Dis, 94, 18–31. doi: 10.1016/j.nbd.2016.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, Nadeem M, Lad SP, Wuu J, & Mufson EJ (2006). Differential Expression of Synaptic Proteins in the Frontal and Temporal Cortex of Elderly Subjects With Mild Cognitive Impairment. Journal of Neuropathology & Experimental Neurology, 65(6), 592–601. doi: 10.1097/00005072-200606000-00007 [DOI] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, … Hyman BT (2012). Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron, 73(4), 685–697. doi: 10.1016/j.neuron.2011.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA, … Mufson EJ (2002). Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol, 51(2), 145–155. doi: 10.1002/ana.10069 [DOI] [PubMed] [Google Scholar]
- DeVos SL, Corjuc BT, Oakley DH, Nobuhara CK, Bannon RN, Chase A, … Hyman BT (2018). Synaptic Tau Seeding Precedes Tau Pathology in Human Alzheimer’s Disease Brain. Front Neurosci, 12, 267. doi: 10.3389/fnins.2018.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner I, Hales CM, Bishof I, Rabenold L, Duong DM, Yi H, … Seyfried NT (2014). Aggregation properties of the small nuclear ribonucleoprotein U1–70K in Alzheimer disease. J Biol Chem, 289(51), 35296–35313. doi: 10.1074/jbc.M114.562959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donev R, Newall A, Thome J, & Sheer D (2007). A role for SC35 and hnRNPA1 in the determination of amyloid precursor protein isoforms. Mol Psychiatry, 12(7), 681–690. doi: 10.1038/sj.mp.4001971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin S, Commins C, Lathuiliere A, Beerepoot P, Fernandes AR, Kamath TV, … Hyman BT (2020). Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nature Medicine, 26(8), 1256–1263. doi: 10.1038/s41591-020-0938-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, … Hyman BT (2018). Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron, 99(5), 925–940.e927. doi: 10.1016/j.neuron.2018.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfel JM, Yu L, De Jager PL, Schneider JA, & Bennett DA (2016). Association of APOE with tau-tangle pathology with and without β-amyloid. Neurobiol Aging, 37, 19–25. doi: 10.1016/j.neurobiolaging.2015.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasulo L, Ugolini G, Visintin M, Bradbury A, Brancolini C, Verzillo V, … Cattaneo A (2000). The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J Neurochem, 2, 624–633. [DOI] [PubMed] [Google Scholar]
- Fornari S, Schäfer A, Jucker M, Goriely A, & Kuhl E (2019). Prion-like spreading of Alzheimer’s disease within the brain’s connectome. bioRxiv, 529438. doi: 10.1101/529438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, … Cryns VL (2003). Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci U S A, 100(17), 10032–10037. doi: 10.1073/pnas.1630428100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Blanco MA, Baraniak AP, & Lasda EL (2004). Alternative splicing in disease and therapy. Nat Biotechnol, 22(5), 535–546. doi: 10.1038/nbt964 [DOI] [PubMed] [Google Scholar]
- Garcia-Sierra F, Ghoshal N, Quinn B, Berry RW, & Binder LI (2003). Conformational changes and truncation of tau protein during tangle evolution in Alzheimer’s disease. J Alzheimers Dis, 5(2), 65–77. doi: 10.3233/jad-2003-5201 [DOI] [PubMed] [Google Scholar]
- German DC, White CL 3rd, & Sparkman DR (1987). Alzheimer’s disease: neurofibrillary tangles in nuclei that project to the cerebral cortex. Neuroscience, 21(2), 305–312. doi: 10.1016/0306-4522(87)90123-0 [DOI] [PubMed] [Google Scholar]
- Ghoshal N, García-Sierra F, Wuu J, Leurgans S, Bennett DA, Berry RW, & Binder LI (2002). Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease. Exp Neurol, 177(2), 475–493. doi: 10.1006/exnr.2002.8014 [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Counts SE, & Mufson EJ (2006). Shift in the ratio of three-repeat tau and four-repeat tau mRNAs in individual cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J Neurochem, 96(5), 1401–1408. doi: 10.1111/j.1471-4159.2005.03641.x [DOI] [PubMed] [Google Scholar]
- Goedert M, Eisenberg DS, & Crowther RA (2017). Propagation of Tau Aggregates and Neurodegeneration. Annu Rev Neurosci, 40, 189–210. doi: 10.1146/annurev-neuro-072116-031153 [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, & Vanmechelen E (1995). Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett, 189(3), 167–169. doi: 10.1016/0304-3940(95)11484-e [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Jakes R, Rutherford D, & Crowther RA (1989). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron, 3(4), 519–526. doi: 10.1016/0896-6273(89)90210-9 [DOI] [PubMed] [Google Scholar]
- Grinberg LT, Rüb U, Ferretti RE, Nitrini R, Farfel JM, Polichiso L, … Heinsen H (2009). The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer’s disease. A precocious onset? Neuropathol Appl Neurobiol, 35(4), 406–416. doi: 10.1111/j.1365-2990.2009.00997.x [DOI] [PubMed] [Google Scholar]
- Guillozet-Bongaarts AL, Garcia-Sierra F, Reynolds MR, Horowitz PM, Fu Y, Wang T, … Binder LI (2005). Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol Aging, 26(7), 1015–1022. doi: 10.1016/j.neurobiolaging.2004.09.019 [DOI] [PubMed] [Google Scholar]
- Guo JL, Narasimhan S, Changolkar L, He Z, Stieber A, Zhang B, … Lee VM (2016). Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med, 213(12), 2635–2654. doi: 10.1084/jem.20160833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyparaki MT, Arab A, Sorokina EM, Santiago-Ruiz AN, Bohrer CH, Xiao J, & Lakadamyali M (2021). Tau forms oligomeric complexes on microtubules that are distinct from tau aggregates. Proc Natl Acad Sci U S A, 118(19). doi: 10.1073/pnas.2021461118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Hirokawa K, Kamei S, & Uchihara T (2013). Isoform transition from four-repeat to three-repeat tau underlies dendrosomatic and regional progression of neurofibrillary pathology. Acta Neuropathol, 125(4), 565–579. doi: 10.1007/s00401-013-1097-6 [DOI] [PubMed] [Google Scholar]
- Hernández F, Pérez M, Lucas JJ, Mata AM, Bhat R, & Avila J (2004). Glycogen synthase kinase-3 plays a crucial role in tau exon 10 splicing and intranuclear distribution of SC35. Implications for Alzheimer’s disease. J Biol Chem, 279(5), 3801–3806. doi: 10.1074/jbc.M311512200 [DOI] [PubMed] [Google Scholar]
- Hoesen GV, & Pandya D (1975). Some connections of the entorhinal (area 28) and perirhinal (area 35) cortices of the rhesus monkey. I. Temporal lobe afferents. Brain Res, 95(1), 1–24. [DOI] [PubMed] [Google Scholar]
- Hsieh YC, Guo C, Yalamanchili HK, Abreha M, Al-Ouran R, Li Y, … Shulman JM (2019). Tau-Mediated Disruption of the Spliceosome Triggers Cryptic RNA Splicing and Neurodegeneration in Alzheimer’s Disease. Cell Rep, 29(2), 301–316 e310. doi: 10.1016/j.celrep.2019.08.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Wolozin BL, Davies P, Kromer LJ, & Damasio AR (1988). Alz-50 Antibody recognizes alzheimer-related neuronal changes. Annals of Neurology, 23(4), 371–379. doi: 10.1002/ana.410230410 [DOI] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong CX, & Grundke-Iqbal I (2010). Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res, 7(8), 656–664. doi: 10.2174/156720510793611592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseki E, Yamamoto R, Murayama N, Minegishi M, Togo T, Katsuse O, … Arai H (2006). Immunohistochemical investigation of neurofibrillary tangles and their tau isoforms in brains of limbic neurofibrillary tangle dementia. Neurosci Lett, 405(1–2), 29–33. doi: 10.1016/j.neulet.2006.06.036 [DOI] [PubMed] [Google Scholar]
- Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, … Sperling R (2018). NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement, 14(4), 535–562. doi: 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, & Mandelkow E (2008). Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem, 283(46), 32066–32076. doi: 10.1074/jbc.M805300200 [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Alafuzoff I, Attems J, Beach TG, Cairns NJ, Crary JF, … Wisniewski T (2015). PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol, 129(5), 757–762. doi: 10.1007/s00401-015-1407-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Lin W, Zhang C, Ash PEA, Verma M, Kwan J, … Wolozin B (2021). Interaction of tau with HNRNPA2B1 and N(6)-methyladenosine RNA mediates the progression of tauopathy. Mol Cell, 81(20), 4209–4227.e4212. doi: 10.1016/j.molcel.2021.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, & Walker LC (2011). Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol, 70(4), 532–540. doi: 10.1002/ana.22615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan NM, Morfini G, Pigino G, LaPointe NE, Andreadis A, Song Y, … Brady ST (2012). Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol Aging, 33(4), 826.e815–830. doi: 10.1016/j.neurobiolaging.2011.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan NM, Morfini GA, LaPointe NE, Pigino GF, P. KR, S. Y, … B. LI (2011). Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci, 27, 9858–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley C, Ginsberg S, Liang W, Counts S, & Mufson E (2022). Posterior cingulate cortex reveals an expression profile of resilience in cognitively intact elders. Brain Communications, 4(4). doi: 10.1093/braincomms/fcac162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley C, Perez SE, & Mufson E (2019). Tau pathology in the medial temporal lobe of athletes with chronic traumatic encephalopathy: a chronic effects of neurotrauma consortium study. Acta Neuropathol Commun, 7(1), 207. doi: 10.1186/s40478-019-0861-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Sugimori K, Sudo S, & Kobayashi K (2005). Relationship between microtubule-binding repeats and morphology of neurofibrillary tangle in Alzheimer’s disease. Acta Neurol Scand, 112(5), 327–334. doi: 10.1111/j.1600-0404.2005.00488.x [DOI] [PubMed] [Google Scholar]
- Koss DJ, Jones G, Cranston A, Gardner H, Kanaan NM, & Platt B (2016). Soluble pre-fibrillar tau and beta-amyloid species emerge in early human Alzheimer’s disease and track disease progression and cognitive decline. Acta Neuropathol, 132(6), 875–895. doi: 10.1007/s00401-016-1632-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lace G, Savva G, Forster G, Silva R. d., Brayne C, Matthews F, … Wharton S (2009). Hippocampal tau pathology is related to neuroanatomical connections: an ageing population-based study. Brain, 132, 1324–1334. [DOI] [PubMed] [Google Scholar]
- Lacosta A-M, Insua D, Badi H, Pesini P, & Sarasa M (2017). Neurofibrillary Tangles of Aβ x-40 in Alzheimer’s Disease Brains. Journal of Alzheimer’s Disease, 58, 661–667. doi: 10.3233/JAD-170163 [DOI] [PubMed] [Google Scholar]
- Landino LM, Skreslet TE, & Alston JA (2004). Cysteine oxidation of tau and microtubule-associated protein-2 by peroxynitrite: modulation of microtubule assembly kinetics by the thioredoxin reductase system. J Biol Chem, 279(33), 35101–35105. doi: 10.1074/jbc.M405471200 [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, & Kayed R (2010). Preparation and characterization of neurotoxic tau oligomers. Biochemistry, 49(47), 10039–10041. doi: 10.1021/bi1016233 [DOI] [PubMed] [Google Scholar]
- Llamas-Rodríguez J, Oltmer J, Greve DN, Williams E, Slepneva N, Wang R, … Augustinack JC (2022). Entorhinal Subfield Vulnerability to Neurofibrillary Tangles in Aging and the Preclinical Stage of Alzheimer’s Disease. J Alzheimers Dis, 87(3), 1379–1399. doi: 10.3233/jad-215567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna-Muñoz J, Chávez-Macías L, García-Sierra F, & Mena R (2007). Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer’s disease. J Alzheimers Dis, 12(4), 365–375. doi: 10.3233/jad-2007-12410 [DOI] [PubMed] [Google Scholar]
- Luna-Muñoz J, García-Sierra F, Falcón V, Menéndez I, Chávez-Macías L, & Mena R (2005). Regional conformational change involving phosphorylation of tau protein at the Thr231, precedes the structural change detected by Alz-50 antibody in Alzheimer’s disease. J Alzheimers Dis, 8(1), 29–41. doi: 10.3233/jad-2005-8104 [DOI] [PubMed] [Google Scholar]
- Mahady L, He B, Malek-Ahmadi M, & Mufson EJ (2020). Telomeric Alterations in the Default Mode Network during the Progression of Alzheimer’s disease: Selective Vulnerability of the Precuneus. Neuropath App Neurobio. doi: 10.1111/nan.12672 [DOI] [PubMed] [Google Scholar]
- McAllister BB, Lacoursiere SG, Sutherland RJ, & Mohajerani MH (2020). Intracerebral seeding of amyloid-β and tau pathology in mice: Factors underlying prion-like spreading and comparisons with α-synuclein. Neurosci Biobehav Rev, 112, 1–27. doi: 10.1016/j.neubiorev.2020.01.026 [DOI] [PubMed] [Google Scholar]
- Mesulam MM (1999). Neuroplasticity failure in Alzheimer’s disease: bridging the gap between plaques and tangles. Neuron, 24(3), 521–529. doi: 10.1016/s0896-6273(00)81109-5 [DOI] [PubMed] [Google Scholar]
- Mesulam MM (2013). Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer’s disease. J Comp Neurol, 521(18), 4124–4144. doi: 10.1002/cne.23415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, … Berg L (1991). The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology, 41(4), 479–486. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=2011243 [DOI] [PubMed] [Google Scholar]
- Montalbano M, Jaworski E, Garcia S, Ellsworth A, McAllen S, Routh A, & Kayed R (2021). Tau Modulates mRNA Transcription, Alternative Polyadenylation Profiles of hnRNPs, Chromatin Remodeling and Spliceosome Complexes. Front Mol Neurosci, 14, 742790. doi: 10.3389/fnmol.2021.742790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, … Alzheimer’s A (2012). National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol, 123(1), 1–11. doi: 10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrdjen D, Fox EJ, Bukhari SA, Montine KS, Bendall SC, & Montine TJ (2019). The basis of cellular and regional vulnerability in Alzheimer’s disease. Acta Neuropathol, 138(5), 729–749. doi: 10.1007/s00401-019-02054-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Chen EY, Cochran EJ, Beckett LA, Bennett DA, & Kordower JH (1999). Entorhinal cortex beta-amyloid load in individuals with mild cognitive impairment. Exp Neurol, 158(2), 469–490. doi: 10.1006/exnr.1999.7086 [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Counts SE, Ginsberg SD, Mahady L, Perez SE, Massa SM, … Ikonomovic MD (2019). Nerve Growth Factor Pathobiology During the Progression of Alzheimer’s Disease. Front Neurosci, 13, 533. doi: 10.3389/fnins.2019.00533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, He B, Nadeem M, Perez SE, Counts SE, Leurgans S, … Scheff SW (2012). Hippocampal proNGF signaling pathways and beta-amyloid levels in mild cognitive impairment and Alzheimer disease. J Neuropathol Exp Neurol, 71(11), 1018–1029. doi: 10.1097/NEN.0b013e318272caab [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Ikonomovic MD, Counts SE, Perez SE, Malek-Ahmadi M, Scheff SW, & Ginsberg SD (2016). Molecular and cellular pathophysiology of preclinical Alzheimer’s disease. Behav Brain Res, 311, 54–69. doi: 10.1016/j.bbr.2016.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Mahady L, Waters D, Counts SE, Perez SE, DeKosky ST, … Binder LI (2015). Hippocampal plasticity during the progression of Alzheimer’s disease. Neuroscience, 309, 51–67. doi: 10.1016/j.neuroscience.2015.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, … Schneider JA (2019). Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain, 142(6), 1503–1527. doi: 10.1093/brain/awz099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell KL, Hyman BT, Growdon JH, & Hedley-Whyte ET (1999). Application of the National Institute on Aging (NIA)-Reagan Institute criteria for the neuropathological diagnosis of Alzheimer disease. J Neuropathol Exp Neurol, 58(11), 1147–1155. doi: 10.1097/00005072-199911000-00004 [DOI] [PubMed] [Google Scholar]
- Nicholls SB, DeVos SL, Commins C, Nobuhara C, Bennett RE, Corjuc DL, … Hyman BT (2017). Characterization of TauC3 antibody and demonstration of its potential to block tau propagation. PLoS One, 12(5), e0177914. doi: 10.1371/journal.pone.0177914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SA, Ahn SI, & Gallo JM (2016). Tau mis-splicing in the pathogenesis of neurodegenerative disorders. BMB Rep, 49(8), 405–413. doi: 10.5483/bmbrep.2016.49.8.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson KR, Remmers C, Fu Y, B. S, K. NM, V. L, … B. LI (2011). Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J Biol Chem, 286(26), 23063–23076. doi: 10.1074/jbc.M111.237974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson KR, Ward SM, Combs B, Voss K, Kanaan NM, M. G, … B. LI (2011). Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry, 47, 10300–10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson RC (1996). Cortical connections and the pathology of Alzheimer’s disease. Neurodegeneration, 5(4), 429–434. doi: 10.1006/neur.1996.0058 [DOI] [PubMed] [Google Scholar]
- Pearson RC, Esiri MM, Hiorns RW, Wilcock GK, & Powell TP (1985). Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc Natl Acad Sci U S A, 82(13), 4531–4534. doi: 10.1073/pnas.82.13.4531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SE, He B, Nadeem M, Wuu J, Scheff SW, Abrahamson EE, … Mufson EJ (2015). Resilience of precuneus neurotrophic signaling pathways despite amyloid pathology in prodromal Alzheimer’s disease. Biol Psych, 77, 693–703. doi: 10.1016/j.biopsych.2013.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SE, Miguel J, He B, Malek-Ahmadi M, Abrahamson E, Ikonomovic M, … Mufson E (2019). Frontal cortex and striatal cellular and molecular pathobiology in individuals with Down syndrome with and without dementia. Acta Neuropathol, 137, 413–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett EK, Henstridge CM, Allison E, Pitstick R, Pooler A, Wegmann S, … Spires-Jones TL (2017). Spread of tau down neural circuits precedes synapse and neuronal loss in the rTgTauEC mouse model of early Alzheimer’s disease. Synapse, 71(6). doi: 10.1002/syn.21965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanco JC, Hand GR, Briner A, Li C, & Götz J (2021). Exosomes induce endolysosomal permeabilization as a gateway by which exosomal tau seeds escape into the cytosol. Acta Neuropathol, 141(2), 235–256. doi: 10.1007/s00401-020-02254-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanco JC, Scicluna BJ, Hill AF, & Götz JJ (2016). Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. Biol Chem, 291(24), 12445–12466. doi: 10.1074/jbc.M115.709485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Phillips EC, Lau DH, Noble W, & Hanger DP (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep, 14(4), 389–394. doi: 10.1038/embor.2013.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Iqbal K, Grundke-Iqbal I, Gong C, & Liu F (2011). Splicing factor SC35 promotes tau expression through stabilization of its mRNA. FEBS Lett, 585(6), 875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan VK, Castillo AM, Knopman DS, Graff-Radford J, Lowe VJ, Petersen RC, … Vemuri P (2019). Association of Apolipoprotein E ɛ4, Educational Level, and Sex With Tau Deposition and Tau-Mediated Metabolic Dysfunction in Older Adults. JAMA Netw Open, 2(10), e1913909. doi: 10.1001/jamanetworkopen.2019.13909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, … Cotman CW (2004). Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest, 114(1), 121–130. doi: 10.1172/jci20640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosene DL, & Van Hoesen GW (1977). Hippocampal efferents reach widespread areas of cerebral cortex and amygdala in the rhesus monkey. Science, 198(4314), 315–317. doi: 10.1126/science.410102 [DOI] [PubMed] [Google Scholar]
- Ruan Z, Pathak D, Venkatesan Kalavai S, Yoshii-Kitahara A, Muraoka S, Bhatt N, … Ikezu T (2021). Alzheimer’s disease brain-derived extracellular vesicles spread tau pathology in interneurons. Brain, 144(1), 288–309. doi: 10.1093/brain/awaa376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüb U, Del Tredici K, Schultz C, Thal DR, Braak E, & Braak H (2000). The evolution of Alzheimer’s disease-related cytoskeletal pathology in the human raphe nuclei. Neuropathol Appl Neurobiol, 26(6), 553–567. doi: 10.1046/j.0305-1846.2000.00291.x [DOI] [PubMed] [Google Scholar]
- Saper CB, Wainer BH, & German DC (1987). Axonal and transneuronal transport in the transmission of neurological disease: Potential role in system degenerations, including alzheimer’s disease. Neuroscience, 23(2), 389–398. doi: 10.1016/0306-4522(87)90063-7 [DOI] [PubMed] [Google Scholar]
- Scheff SW, Ansari MA, & Mufson EJ (2016). Oxidative stress and hippocampal synaptic protein levels in elderly cognitively intact individuals with Alzheimer’s disease pathology. Neurobiol Aging, 42, 1–12. doi: 10.1016/j.neurobiolaging.2016.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, & Markesbery WR (2000). “Preclinical” AD revisited: neuropathology of cognitively normal older adults. Neurology, 55(3), 370–376. doi: 10.1212/wnl.55.3.370 [DOI] [PubMed] [Google Scholar]
- Schmitt FA, Nelson PT, Abner E, Scheff S, Jicha GA, Smith C, … Kryscio RJ (2012). University of Kentucky Sanders-Brown healthy brain aging volunteers: donor characteristics, procedures and neuropathology. Curr Alzheimer Res, 9(6), 724–733. doi: 10.2174/156720512801322591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, & Hardy J (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med, 8(6), 595–608. doi: 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla C, & Bridges LR (1999). Regional distribution of tau, beta-amyloid and beta-amyloid precursor protein in the Alzheimer’s brain: a quantitative immunolabelling study. Neuroreport, 10(18), 3785–3789. doi: 10.1097/00001756-199912160-00012 [DOI] [PubMed] [Google Scholar]
- Simic G, Stanic G, Mladinov M, Jovanov-Milosevic N, Kostovic I, & Hof PR (2009). Does Alzheimer’s disease begin in the brainstem? Neuropathol Appl Neurobiol, 35(6), 532–554. doi: 10.1111/j.1365-2990.2009.01038.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Španić E, Langer Horvat L, Ilić K, Hof PR, & Šimić G (2022). NLRP1 Inflammasome Activation in the Hippocampal Formation in Alzheimer’s Disease: Correlation with Neuropathological Changes and Unbiasedly Estimated Neuronal Loss. Cells, 11(14). doi: 10.3390/cells11142223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson E, Breckenridge L, McMahon L, Som S, McConnell I, & Bloom GS (2017). Extracellular Tau Oligomers Induce Invasion of Endogenous Tau into the Somatodendritic Compartment and Axonal Transport Dysfunction. J Alzheimers Dis, 58(3), 803–820. doi: 10.3233/jad-170168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S (2019). Tau Propagation as a Diagnostic and Therapeutic Target for Dementia: Potentials and Unanswered Questions. Front Neurosci, 13, 1274. doi: 10.3389/fnins.2019.01274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Holzer M, Rub U, Waldmann G, Gunzel S, Zedlick D, & Schober R (2000). Alzheimer-related tau-pathology in the perforant path target zone and in the hippocampal stratum oriens and radiatum correlates with onset and degree of dementia. Exp Neurol, 163(1), 98–110. doi: 10.1006/exnr.2000.7380 [DOI] [PubMed] [Google Scholar]
- Thal DR, Rub, Orantes, & Braak H (2002). Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology, 58, 1791–1800. [DOI] [PubMed] [Google Scholar]
- Thal DR, Rüb U, Orantes M, & Braak H (2002). Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology, 58(12), 1791–1800. doi: 10.1212/wnl.58.12.1791 [DOI] [PubMed] [Google Scholar]
- Thal DR, Rub U, Schultz C, Sassin I, Ghebremedhin E, Del Tredici K, … Braak H (2000). Sequence of Abeta-protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol, 59(8), 733–748. doi: 10.1093/jnen/59.8.733 [DOI] [PubMed] [Google Scholar]
- Tiernan CT, Ginsberg SD, He B, Ward SM, Guillozet-Bongaarts AL, Kanaan NM, … Counts SE (2018). Pretangle pathology within cholinergic nucleus basalis neurons coincides with neurotrophic and neurotransmitter receptor gene dysregulation during the progression of Alzheimer’s disease. Neurobiol Dis, 117, 125–136. doi: 10.1016/j.nbd.2018.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosun D, Landau S, Aisen PS, Petersen RC, Mintun M, Jagust W, & Weiner MW (2017). Association between tau deposition and antecedent amyloid-β accumulation rates in normal and early symptomatic individuals. Brain, 140(5), 1499–1512. doi: 10.1093/brain/awx046 [DOI] [PubMed] [Google Scholar]
- Uchihara T, Hara M, Nakamura A, & Hirokawa K (2012). Tangle evolution linked to differential 3- and 4-repeat tau isoform deposition: a double immunofluorolabeling study using two monoclonal antibodies. Histochem Cell Biol, 137(2), 261–267. doi: 10.1007/s00418-011-0891-2 [DOI] [PubMed] [Google Scholar]
- Uematsu M, Nakamura A, Ebashi M, Hirokawa K, Takahashi R, & Uchihara T (2018). Brainstem tau pathology in Alzheimer’s disease is characterized by increase of three repeat tau and independent of amyloid β. Acta Neuropathologica Communications, 6(1), 1. doi: 10.1186/s40478-017-0501-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoesen GW, Pandya DN, & Butters N (1972). Cortical afferents to the entorhinal cortex of the Rhesus monkey. Science, 175(4029), 1471–1473. doi: 10.1126/science.175.4029.1471 [DOI] [PubMed] [Google Scholar]
- Vana L, Kanaan NM, Ugwu IC, Wuu J, Mufson EJ, & Binder LI (2011). Progression of tau pathology in cholinergic Basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. Am J Pathol, 179(5), 2533–2550. doi: 10.1016/j.ajpath.2011.07.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JW, Iturria-Medina Y, Strandberg OT, Smith R, Levitis E, Evans AC, … the Swedish BioFinder S (2020). Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nature Communications, 11(1), 2612. doi: 10.1038/s41467-020-15701-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bartheld C (2002). Counting particles in tissue sections: choices of methods and importance of calibration to minimize biases. Histol Histopathol, 17(2), 639–648. doi: 10.14670/hh-17.639 [DOI] [PubMed] [Google Scholar]
- Wang Y, Balaji V, Kaniyappan S, Krüger L, Irsen S, Tepper K, Chandupatla R, Maetzler W, Schneider A, Mandelkow E, & Mandelkow EM (2017). The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener, 12(1):5. doi: 10.1186/s13024-016-0143-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YT, Pascoal TA, Therriault J, Kang MS, Benedet AL, Savard M, … Rosa-Neto P (2021). Interactive rather than independent effect of APOE and sex potentiates tau deposition in women. Brain Commun, 3(2), fcab126. doi: 10.1093/braincomms/fcab126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZH, Liu P, Liu X, Yu SP, Wang JZ, & Ye K (2018). Delta-secretase (AEP) mediates tau-splicing imbalance and accelerates cognitive decline in tauopathies. J Exp Med, 215(12), 3038–3056. doi: 10.1084/jem.20180539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SM, Himmelstein DS, Lancia JK, Fu Y, Patterson KR, & Binder LI (2013). TOC1: characterization of a selective oligomeric tau antibody. J Alzheimers Dis, 37(3), 593–602. doi: 10.3233/JAD-131235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SM, Himmelstein DS, Ren Y, Fu Y, Yu XW, Roberts K, … Sahara N (2014). TOC1: a valuable tool in assessing disease progression in the rTg4510 mouse model of tauopathy. Neurobiol Dis, 67, 37–48. doi: 10.1016/j.nbd.2014.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Li L, Shi R, Zhou Y, Jin N, Gu J, … Chu D (2021). Dephosphorylation Passivates the Seeding Activity of Oligomeric Tau Derived From Alzheimer’s Brain. Front Mol Neurosci, 14, 631833. doi: 10.3389/fnmol.2021.631833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Zhang W, Jiao B, Pan CZ, Liu X, & Shen L (2017). The role of exosomes in the pathogenesis of Alzheimer’ disease. Transl Neurodegener, 26:3. doi: 10.1186/s40035-017-0072-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans KL, Tai LM, Kanekiyo T, Stine WB Jr., Michon SC, Nwabuisi-Heath E, … LaDu MJ (2012). Intraneuronal Abeta detection in 5xFAD mice by a new Abeta-specific antibody. Mol Neurodegener, 7, 8. doi: 10.1186/1750-1326-7-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XW, Li X, Bjorkdahl C, Sjogren MJ, Alafuzoff I, Soininen H, … Pei JJ (2006). Assessments of the accumulation severities of amyloid beta-protein and hyperphosphorylated tau in the medial temporal cortex of control and Alzheimer’s brains. Neurobiol Dis, 22(3), 657–668. doi: 10.1016/j.nbd.2006.01.006 [DOI] [PubMed] [Google Scholar]
- Zhu W, Wei X, Wang Y, Li J, Peng L, Zhang K, & Bai B (2020). Effects of U1 Small Nuclear Ribonucleoprotein Inhibition on the Expression of Genes Involved in Alzheimer’s Disease. ACS Omega, 5(39), 25306–25311. doi: 10.1021/acsomega.0c03568 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Scatterplots showing significant correlations of SRSF2 nuclear intensity and tau epitope density with clinical and neuropathological variables (a-d). CA3 nuclear intensity of SRSF2 (a) significantly correlated with MMSE scores across clinical groups. TOC1 densities in the CA1 (b), subiculum (c), and EC layer II (d) were significantly correlated with Braak Stage. EC layer II TOC1 densities were correlated with NIA Reagan (e) and CERAD (f). Abbreviations: NCI, no cognitive impairment; MCI, mild cognitive impairment; AD, Alzheimer’s disease; CA1, hippocampal subfield CA1; CA3, hippocampal subfield CA3; EC, entorhinal cortex. Spearman correlations, Bonferroni adjusted α=0.007.
Supplemental Figure 2. Scatterplots showing significant correlations of TNT staining density with Braak stage (a-d). TNT densities in the CA1 (a), CA2 (b), subiculum (c), and EC layer II (d) were correlated strongly with Braak stage. Density of TNT labeling in EC layer II neurons strongly correlated with CERAD (e) and NIA Reagan score (f). Abbreviations: NCI, no cognitive impairment; MCI, mild cognitive impairment; AD, Alzheimer’s disease; CA1, hippocampal subfield CA1; CA2, hippocampal subfield CA2; EC, entorhinal cortex. Spearman correlations, Bonferroni adjusted α=0.007
Supplemental Figure 3. Scatterplots showing significant correlations of AT8 staining density with Braak stage (a-d). Densities of AT8-ir perikarya in the CA1 (a), CA2 (b), CA3 (c), DG (d), subiculum (e), and EC layer II (f) were strongly correlated with Braak stage. Abbreviations: NCI, no cognitive impairment; MCI, mild cognitive impairment; AD, Alzheimer’s disease; CA1, hippocampal subfield CA1; CA2, hippocampal subfield CA2; CA3, hippocampal subfield CA3; DG, dentate gyrus; EC, entorhinal cortex. Spearman correlations, Bonferroni adjusted α=0.007
Supplemental Figure 4. Scatterplots showing significant correlations between the intensity of SRSF2 nuclear immunoreactivity and tau epitope and Aβ1–42 density (a-c). A) AT8 positive neuron densities in EC layer II strongly correlated with NIA Reagan scores. SRSF2 nuclear intensity in AT8 labeled neurons in the DG polymorphic layer significantly correlated with TauC3 densities in the subiculum (b) Nuclear SRSF2 intensity in the subiculum negatively correlated with Aβ1–42 diffuse plaque densities in EC (c). Abbreviations: NCI, no cognitive impairment; MCI, mild cognitive impairment; AD, Alzheimer’s disease; EC layer II, entorhinal cortex layer II; Pl, polymorphic layer of the DG; CA3, hippocampal subfield CA3. Spearman correlations, Bonferroni adjusted α=0.007.
Data Availability Statement
The data that support the findings of the study are available from the corresponding author upon reasonable request.