

SUMMARY
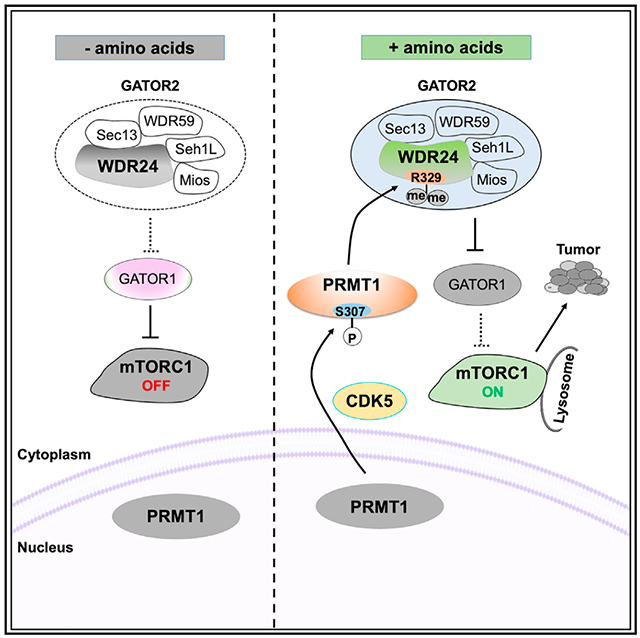
The mammalian target of rapamycin complex1 (mTORC1) is a central regulator of metabolism and cell growth by sensing diverse environmental signals, including amino acids. The GATOR2 complex is a key component linking amino acid signals to mTORC1. Here, we identify protein arginine methyltransferase 1 (PRMT1) as a critical regulator of GATOR2. In response to amino acids, cyclin-dependent kinase 5 (CDK5) phosphorylates PRMT1 at S307 to promote PRMT1 translocation from nucleus to cytoplasm and lysosome, which in turn methylates WDR24, an essential component of GATOR2, to activate the mTORC1 pathway. Disruption of the CDK5-PRMT1-WDR24 axis suppresses hepatocellular carcinoma (HCC) cell proliferation and xenograft tumor growth. High PRMT1 protein expression is associated with elevated mTORC1 signaling in patients with HCC. Thus, our study dissects a phosphorylation- and arginine methylation-dependent regulatory mechanism of mTORC1 activation and tumor growth and provides a molecular basis to target this pathway for cancer therapy.
Graphical Abstract
In brief
Yin et al. find that CDK5 serves as an upstream kinase and WDR24 acts as a downstream target of PRMT1 to promote mTORC1 pathway activation in response to amino acids. They further show that disruption of CDK5, PRMT1, and WDR24 suppresses tumor growth, implicating their therapeutic potential.
INTRODUCTION
The mammalian target of rapamycin complex1 (mTORC1) pathway is a key sensor of a variety of environmental clues, such as growth factors, amino acids, and energy. By integrating these inputs, the mTORC1 pathway regulates many fundamental cellular processes, including lipid synthesis, glucose metabolism, and autophagy, through which it controls cell growth and metabolism.1–3 Deregulation of the mTORC1 pathway is associated with numerous human pathologies, including cancers.4,5
Building on the breakthrough finding that the Rag GTPases act as crucial mediators of amino acid signals to mTORC1,6,7 extensive studies have therefore focused on the identification of key components involved in amino acid sensing and regulation of Rag GTPases in the past 15 years. The Ragulator complex, comprising MP1, p14, p18, HBXIP, and C7ORF59, functions as a guanine nucleotide exchange factor (GEF) to load GTP on Rag A/B and subsequently recruit mTORC1 to the lysosomal surface.8,9 In contrast, the GATOR1 complex, consisting of DEPDC5, NPRL2, and NPRL3, serves as a GTPase-activating protein (GAP) to hydrolyze GTP of RagA/B and suppress mTORC1 activation.10,11 Moreover, the GATOR2 complex, containing MIOS, WDR24, WDR59, SEH1L, and SEC13, is an inhibitor of GATOR1 and directly interacts with amino acid sensors.11–14 Furthermore, another complex termed KICSTOR, consisting of KPTN, ITFG2, C12orf66, and SZT2, was identified to regulate GATOR1/2 function.15,16 In addition, several amino acid sensors upstream of mTORC1 have been identified, including Sestrin2 (Leu sensor), SAMTOR (S-adenosylmethionine sensor), SLC38A9, and CASTOR1 (Arg sensors).12,13,17,18 Despite these advances on the mTORC1 network, it remains largely unknown how these complexes, particularly the GATOR2 complex, are regulated and contribute to aberrant mTORC1 signaling and cancers.
Protein arginine methylation is another widespread posttranslational modification (PTM), which is as abundant as phosphorylation and ubiquitination.19,20 In mammals, nine members of the protein arginine methyltransferase (PRMT) family can be classified into three categories based on their catalytic activity. Type I PRMTs, including PRMT1, PRMT2, PRMT3, PRMT4/CARM1, and PRMT6, catalyze asymmetric dimethylarginine (aDMA).21,22 Type II enzymes, including PRMT5 and PRMT9,generate symmetric DMA (sDMA).23,24 PRMT7 is the only member of the type III enzyme that promotes monomethylarginine (MMA).25,26 PRMTs methylate histone and non-histone substrates to control many cellular processes, including transcription,27 DNA damage response,28,29 and signal transduction.30 Deregulation of PRMTs is frequently observed in various human cancers, and thus they are considered promising targets for cancer therapy.31–34
Here, we identify that CDK5 acts an upstream kinase to phosphorylate PRMT1 at S307, leading to cytoplasmic translocation and enhanced activity of PRMT1 in response to amino acids. We also characterize that WDR24 serves as a downstream substrate of PRMT1 to promote amino-acid-induced mTORC1 activation. Finally, we dissect the role of the CDK5-PRMT1-WDR24 axis in cell proliferation and tumorigenesis, providing insights into targeting this pathway for cancer treatment.
RESULTS
PRMT1 is required for mTORC1 activation in response to amino acids
To determine whether arginine methylation is involved in mTORC1 regulation, we employed the CRISPR-Cas9 genome editing approach to deplete individual PRMT genes in HEK293 cells and examined mTORC1 signaling in response to amino acids. PRMT8 was excluded in our study because it displayed brain-specific expression.35 Depletion of PRMT1, but not other PRMTs, blocked amino-acid-induced mTORC1 activation in HEK293 cells, as evidenced by phosphorylation of the well-characterized mTORC1 substrates, including S6 kinase 1 (S6K1),36 eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1),37,38 Unc-51-like autophagy activating kinase 1 (ULK1),39 and ribosomal protein S6 (S6), which is a substrate of S6K1 (Figures 1A and S1A–S1G). This finding was further confirmed in PRMT1-depleted HeLa cells and Huh7 cells (Figures 1B and S1H). Moreover, treating cells with PRMT1 inhibitors, furamidine (FD) and TC-E 5003 (TC-E),40,41 also attenuated mTORC1 activation by amino acids (Figure S1I). Furthermore, Leu, Arg, and Met, which are key amino acids upstream of mTORC1,42 failed to induce mTORC1 pathway activation in PRMT1-depleted cells (Figures S1J–S1L). Given that recruitment of mTORC1 to the lysosomal surface is required for amino-acid-induced mTORC1 activation,9,11 we performed immunofluorescence staining and found that accumulation of mTOR to LAMP2-positive lysosomes was significantly decreased in PRMT1-depleted cells even in the presence of amino acids (Figures 1C and 1D). In contrast, ectopic expression of PRMT1 enhanced mTORC1 signaling in response to amino acid stimulation (Figure 1E). Notably, PRMT1 is frequently overexpressed in various cancers, such as hepatocellular carcinoma (HCC), breast cancer, and lung cancer.43,44 We found that in specimens of patients with HCC, high PRMT1 expression was positively correlated with elevated mTORC1 activation as assessed by the phosphorylation of S6 (Figures 1F and 1G). These results demonstrate that PRMT1 is required for amino-acid-induced mTORC1 activation, and pathologically, the expression of PRMT1 is associated with the deregulation of mTORC1 signaling in HCC.
Figure 1. PRMT1 is required for mTORC1 activation in response to amino acids.
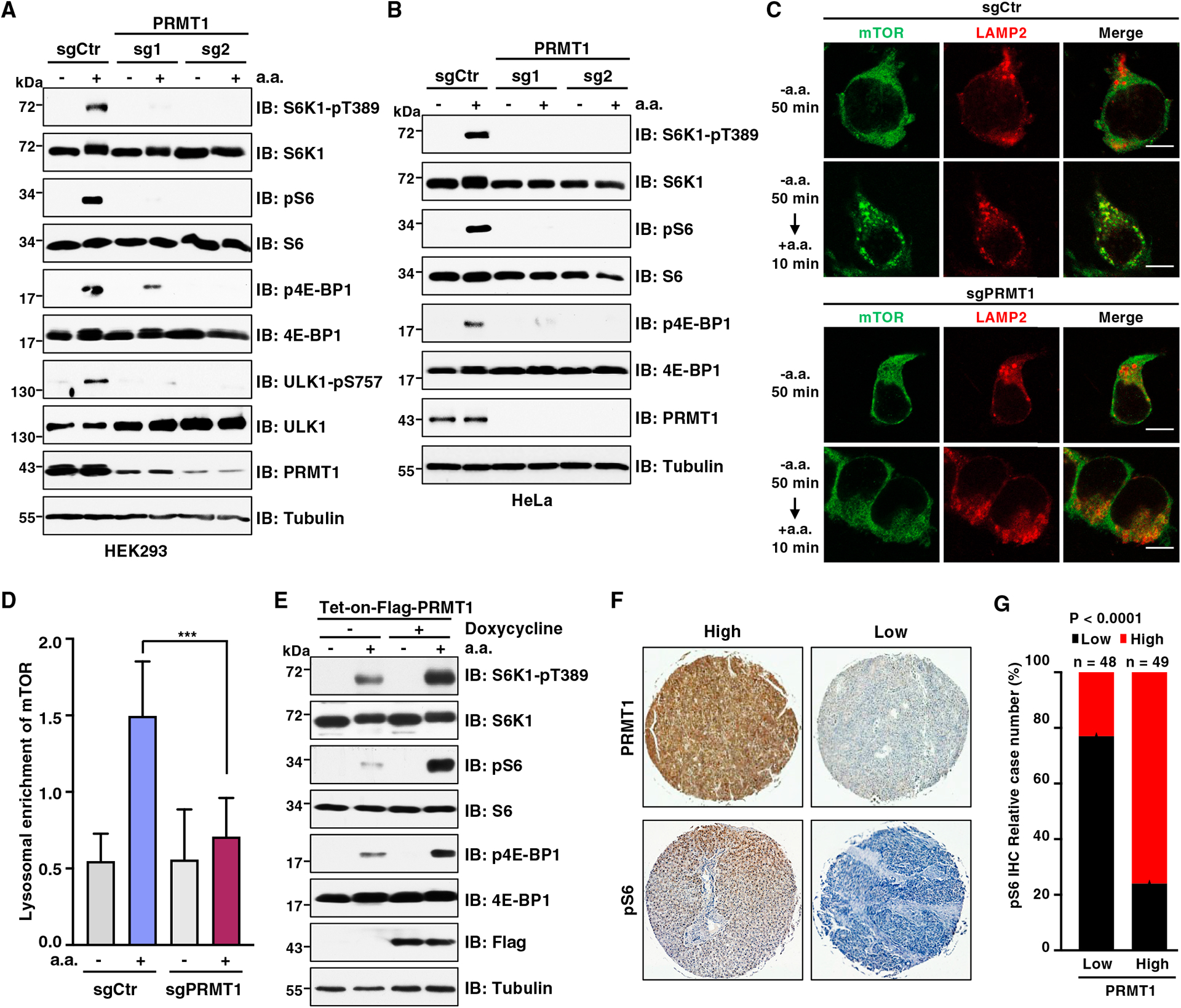
(A) Immunoblot (IB) analysis of whole-cell lysates (WCLs) derived from HEK293 cells infected with lentivirus expressing single guide RNAs (sgRNAs) of PRMT1. Cells were starved of amino acids (aa) for 50 min or starved for 50 min followed by restimulation with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
(B) IB analysis of WCLs derived from HeLa cells infected with lentivirus expressing sgRNAs of PRMT1. Cells were starved of aa for 50 min or starved for 50 min followed by restimulation with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
(C) Immunofluorescence (IF) analysis of mTOR and LAMP2 localization in control (sgCtr) and PRMT1-depleted (sgPRMT1) HEK293T cells. Cells were starved of aa for 50 min or starved for 50 min followed by restimulation with aa for 10 min. Scale bar, 10 μm. Similar results were obtained in three independent experiments.
(D) Quantification of lysosomal enrichment of mTOR in (C). Data are represented as mean ± SD. ***p < 0.001, two-tailed Student’s t test.
(E) IB analysis of WCLs derived from HEK293T infected with lentivirus expressing Tet-on-FLAG-PRMT1. Cells were treated with 1 μg/mL doxycycline for 48 h to induce PRMT1 expression. Then cells were starved of aa for 50 min or starved for 50 min followed by restimulation with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
(F) Representative images of immunohistochemistry (IHC) staining for PRMT1 and pS6 in a microarray of HCC tissues. Scale bar, 50 μm.
(G) Quantification of cases with PRMT1 and pS6 staining (n = 97 tissue specimens). p value was analyzed using χ2 test.
Amino acids promote PRMT1 activity and translocation to cytoplasm and lysosome
PRMT1 is the main type I enzyme responsible for the formation of approximately 85% of aDMA in histones and no-histone substrates.45 It was first identified as a partner of BTG1 and TIS21, which negatively regulate its enzymatic activity.46 hCAF1 has also been reported to interact with PRMT1 and regulate PRMT1 activity in a substrate-dependent manner.47 In addition, phosphorylation and ubiquitination are involved in modulating PRMT1 activity, substrate specificity, and protein stability.48–50 Interestingly, PRMT1 can shuttle between nucleus and cytoplasm, which also plays a role in controlling PRMT1 activity and substrate specificity.43,51 However, the upstream signals and regulators of PRMT1 nucleocytoplasmic shuttling remain unknown.
To investigate whether amino acids affect PRMT1 activity, we assessed total aDMA levels in cells starved for or stimulated with amino acids using a pan anti-aDMA antibody,52 Notably, aDMA signals gradually decreased during the extension of amino acid starvation (Figure 2A), whereas aDMA formation elevated upon amino acid stimulation (Figures 2B and S2A). To determine whether amino acids also regulate sDMA formation, we examined aDMA and sDMA on histone H4R3 (termed H4R3me2a and H4R3me2s), which are catalyzed by PRMT1 and PRMT5, respectively.53,54 Interestingly, amino acids only enhanced H4R3me2a, but not H4R3me2s, in multiple cell lines (Figures 2C, S2B, and S2C). It is possible that deprivation of all amino acids disrupts the Met cycle, leading to a reduction of the methyl donor S-adenosylmethionine (SAM) levels and subsequent PRMT1 inactivation.55 To test this possibility, we starved, or starved and then restimulated, cells with Leu, which is not involved in the Met cycle and unlikely to affect SAM levels. Met was used as a control. Strikingly, resupplementation of Leu or Met promoted total aDMA levels at a comparable level (Figures S2D and S2E), indicating that amino acids likely regulate PRMT1 activity in a SAM-independent mechanism in our experimental conditions. To further support this idea, in vitro arginine methylation assays showed that PRMT1 immunopurified from cells that were starved of amino acids failed to methylate H4R3, even adding sufficient SAM in the reaction (Figure 2D).
Figure 2. aa promote PRMT1 activation and cytoplasm accumulation.
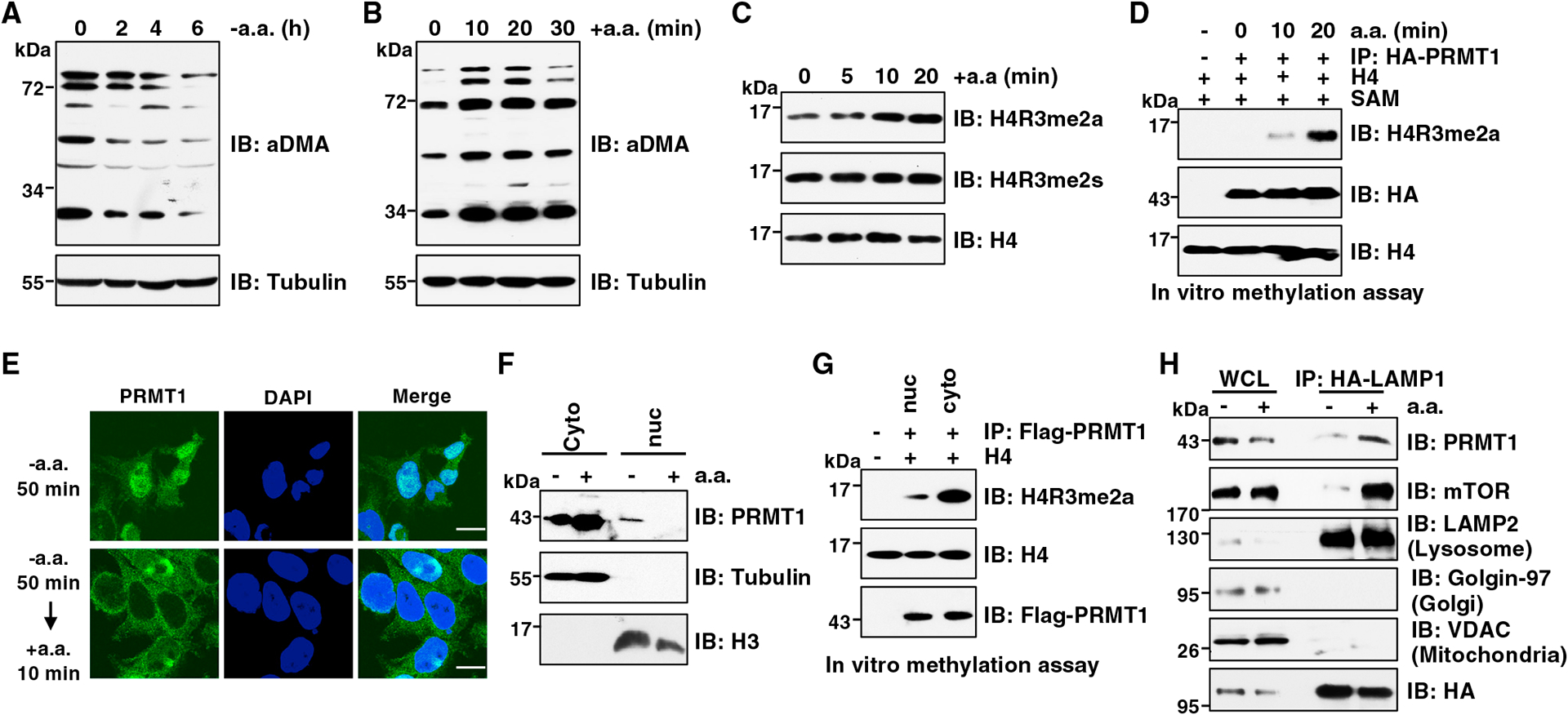
(A) IB analysis of WCLs derived from Huh7 cells starved of aa for indicated time periods. Similar results were obtained in three independent experiments.
(B) IB analysis of WCLs derived from Huh7 cells starved of aa for 2 h followed by restimulation with aa for indicated time periods. Similar results were obtained in three independent experiments.
(C) IB analysis of histone extraction derived from HEK293T cells starved of aa for 50 min followed by restimulation with aa for indicated time periods. Similar results were obtained in three independent experiments.
(D) In vitro arginine methylation assays using H4 protein as the substrate. HA-PRMT1 was immunopurified from HEK293T cells starved of aa for 50 min followed by restimulation with aa for indicated time periods. The reaction products were subjected to IB analysis. Similar results were obtained in three independent experiments.
(E) IF analysis of PRMT1 localization in PRMT1FLAG knockin HEK293T cells. Cells were starved of aa for 50 min or starved for 50 min followed by restimulation with aa for 10 min. Scale bar, 10 μm. Similar results were obtained in three independent experiments.
(F) IB analysis of cell fractionations derived from HEK293T cells. Cells were starved of aa for 50 min or starved for 50 min followed by restimulation with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
(G) In vitro arginine methylation assays using H4 proteins as substrate. FLAG-PRMT1 was immunopurified from nuclear or cytoplasm of PRMT1FLAG knockin HEK293T cells. The reaction products were subjected to IB analysis. Similar results were obtained in three independent experiments.
(H) IB analysis of WCLs and lysosomal enriched fraction. HEK293T cells were starved of aa for 50 min or starved for 50 min and then restimulated with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
See also Figure S2.
Next, we sought to explore whether amino acids regulate PRMT1 subcellular localization. Strikingly, immunofluorescence staining analysis showed that PRMT1 is predominantly located in the nucleus in amino-acid-starved HEK293T cells, whereas it primarily localizes in the cytoplasm upon amino acid stimulation (Figure 2E). We further confirmed this finding using cell fractionation assays (Figures 2F and S2F). In line with previous report,43 cytoplasmic PRTM1 displayed much higher enzymatic activity than nuclear PRMT1 toward methylating H4R3 in vitro (Figure 2G). Interestingly, like mTORC1, PRMT1 was accumulated on immunopurified lysosomes in response to amino acids (Figure 2H).
Taken together, the above results demonstrate that amino acids promote PRMT1 activity and translocation to cytoplasm and lysosome, where PRMT1 may activate mTORC1.
CDK5-dependent phosphorylation of PRMT1 at S307 promotes its cytoplasmic localization and mTORC1 signaling in response to amino acids
Since phosphorylation is a key mechanism through which environmental stimuli regulate protein subcellular localization,56–59 we explored whether phosphorylation is involved in the regulation of PRMT1 nucleocytoplasmic shuttling in response to amino acids. A search for kinases using NetPhos 3.1 (http://www.cbs.dtu.dk/services/NetPhos/) identified seven putative kinases that phosphorylate PRMT1 at Ser/Thr residues, including PKA, CDK5, and others. CDK5 was selected for further study because the CDK5 phosphorylation sites of PRMT1, S304 and S307, are proximal to its predicted nuclear export signal (aa 350–355). Moreover, CDK5-mediated phosphorylation has been reported to regulate its substrate localization.60,61 Notably, CDK5 depletion blocked PRMT1 cytoplasmic translocation in response to amino acids (Figure 3A). We also found that total aDMA levels and H4R3me2a are markedly decreased in CDK5-depleted cells (Figures 3B and S3A), indicating that CDK5 plays a critical role in regulation of PRMT1 activity.
Figure 3. CDK5-dependent phosphorylation of PRMT1 at S307 promotes its cytoplasmic localization and mTORC1 signaling.
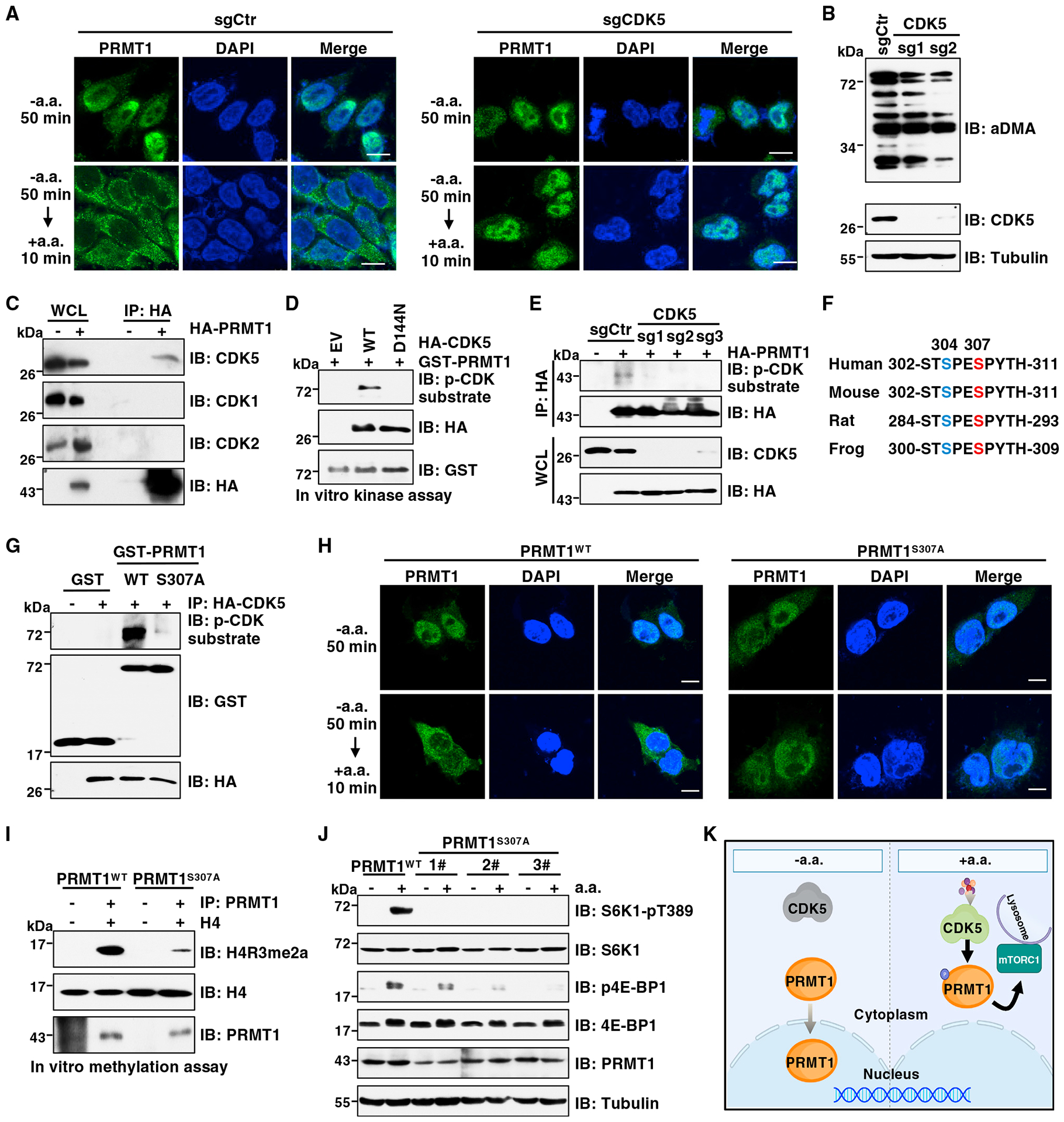
(A) IF analysis of PRMT1 localization in PRMT1FLAG knockin HEK293T cells depleted of CDK5. Cells were starved of aa for 50 min or starved for 50 min and then restimulated with aa. for 10 min. Scale bar, 10 μm. Similar results were obtained in three independent experiments.
(B) IB analysis of WCLs derived from Huh7 cells depleted of CDK5. Similar results were obtained in three independent experiments.
(C) IB analysis of WCLs and anti-hemagglutinin (HA) immunoprecipitates (IPs) derived from HEK293T cells stably expressing HA-PRMT1. Similar results were obtained in three independent experiments.
(D) In vitro kinase assays using recombinant GST-PRMT1 protein as the substrate. HA-CDK5-WT and HA-CDK5-D144N were immunopurified from HEK293T cells. The reaction products were subjected to IB analysis. Similar results were obtained in three independent experiments.
(E) IB analysis of WCLs and anti-HA IPs derived from HEK293T cells depleted of CDK5 and transfected with HA-PRMT1. Similar results were obtained in three independent experiments.
(F) A schematic presentation of the evolutionarily conserved S307 of PRMT1.
(G) In vitro kinase assays using recombinant GST-PRMT1-WT or GST-PRMT1-S307A proteins as substrate. HA-CDK5 protein purified from HEK293T cells was used as the kinase source. The reaction products were subjected to IB analysis. Similar results were obtained in three independent experiments.
(H) IF analysis of PRMT1 localization in PRMT1WT and PRMT1S307A cells. Cells were starved of aa for 50 min or starved for 50 min then restimulated with aa for 10 min. Scale bar, 10 μm. Similar results were obtained in three independent experiments.
(I) In vitro arginine methylation assays using H4 protein as the substrate. PRMT1 protein immunopurified from PRMT1WT and PRMT1S307A cells was used as the methyltransferase. The reaction products were subjected to IB analysis. Similar results were obtained in three independent experiments.
(J) IB analysis of WCL derived from PRMT1WT or PRMT1S307A cells. Cells were starved of aa for 50 min or starved for 50 min then restimulated with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
(K) A model depicting CDK5-mediated phosphorylation of PRMT1 at S307 controls as PRMT1 subcellular localization and mTORC1 activation.
To elucidate the molecular mechanism by which CDK5 regulates PRMT1, we found that PRMT1 specifically interacts with CDK5 but not with CDK1 and CDK2 (Figure 3C), which share 60% sequence identity and identical substrate specificity with CDK5.62 Moreover, their interactions were enhanced by amino acids (Figure S3B). As a proline-directed kinase, CDK5 phosphorylates Ser/Thr residues in the consensus sequence of (S/T)PX(K/H/R) or S/TP.63 This phosphorylation event can be putatively detected by two antibodies: the phosphorylated (phospho)-CDK substrate motif antibody that recognizes phosphoserine in the (K/H)SP or other SP motifs64–67 and the phospho-MAPK/CDK substrate antibody that recognizes phosphoserine in the PXSP or SPX(R/K) motif.68–71 By using these two antibodies, we found that CDK5 wild type (WT), but not the kinase-defective D144N mutant,72 catalyzed PRMT1 phosphorylation (Figures 3D and S3C), which was enhanced by amino acids (Figures S3D and S3E). In contrast, CDK5 depletion abolished PRMT1 phosphorylation (Figures 3E and S3F). To determine whether the predicted evolutionally conserved S304 and S307 residues are phosphorylation sites (Figure 3F), PRMT1 was immunopurified from cells and subjected to mass spectrometric analysis. Phosphorylation of S307, but not S304, was detected (Figure S3G). Moreover, in vitro kinase assays showed that mutation of S307 to an Ala (S307A) abolished PRMT1 phosphorylation detected by both antibodies and radioactive isotope, phosphorus-32 (32P) (Figures 3G, S3H, and S3I). These results together demonstrate that CDK5 is a kinase responsible for phosphorylation of PRMT1 at S307.
Next, we sought to explore the role of CDK5-mediated S307 phosphorylation in regulating PRMT1 localization and activity under physiological conditions. To this end, we introduced the S307A mutation into the endogenous PRMT1 gene (termed PRMT1S307A) using the CRISPR-Cas9 genome editing approach and validated by DNA sequencing (Figure S3J). Unlike the cytoplasmic localization of PRMT1WT, phospho-deficient mutant PRMT1S307A was predominantly located in the nucleus under normal cultured conditions or upon amino acid stimulation (Figures 3H, S3K, and S3L). A possible mechanism is that S307A mutation markedly decreased its interaction with exportin 1 (Figure S3M), which is the major nuclear protein export receptor.73 Compared with PRMT1WT, the PRMT1S307A mutant displayed much lower enzymatic activity as evidenced by a decrease of H4R3me2a signal in vitro and total aDMA levels in cells (Figures 3I and S3N). Furthermore, amino acids failed to promote H4R3me2a in PRMT1S307A cells (Figure S3O). These results demonstrate that CDK5-mediated phosphorylation of PRMT1 at S307 is critical for PRMT1 nucleocytoplasmic shuttling and activation in response to amino acids. We also attempted to generate the PRMT1S307D knockin cells to mimic constitutive S307 phosphorylation under physiological conditions. Unfortunately, we could not obtain such knockin cell lines. Thus, we ectopically expressed the PRMT1-S307D mutant in HEK293T cells. In vitro methylation assays showed that compared with PRMT1-WT, PRMT1-S307D significantly increased H4R3me2a (Figure S3P), supporting that phosphorylation of S307 enhances PRMT1 methyltransferase activity.
Given that PRMT1 promotes amino-acid-induced mTORC1 signaling (Figures 1A–1E), we were interested to examine whether CDK5-mediated phosphorylation of PRMT1 plays a role in mTORC1 pathway regulation. Notably, CDK5 depletion markedly attenuated mTORC1 activation in response to amino acids (Figure S3Q). Moreover, amino-acid-induced mTORC1 signaling was severely decreased in PRMT1S307A cells compared with PRMT1WT cells (Figure 3J). Importantly, ectopic expression of PRMT1-WT restored the mTORC1 signaling in PRMT1S307A cells (Figure S3R), which excludes the off-target effects. To further confirm this finding, reexpression of PRMT1-WT, but not the PRMT1-S307A mutant, led to robust activation of the mTORC1 pathway in endogenous PRMT1-depleted cells in response to amino acids (Figure S3S).
Taken together, these results demonstrate that in the presence of amino acids, active CDK5 phosphorylates PRMT1 and retains it in the cytoplasm and lysosome to activate the mTORC1 pathway, whereas in the absence of amino acids, the CDK5-PRMT1 axis was abrogated, leading to PRMT1 nuclear translocation (Figure 3K). However, it warrants further study to understand how CDK5 is regulated by amino acids.
PRMT1 promotes mTORC1 activation upstream of Rag GTPases
Having demonstrated that amino acids act as an input to promote PRMT1 function and subsequent mTORC1 activation, we next performed epistasis analysis to position PRMT1 within the mTORC1 pathway (Figure 4A). Rag GTPases are obligate heterodimers, which directly interact with and recruit mTORC1 to the lysosomal surface for activation. Their active form is composed of GTP-loaded RagA or RagB and GDP-loaded RagC or RagD.7 Notably, stable expression of the GTP-loaded RagBQ99L completely restored mTORC1 signaling in PRMT1-depleted cells (Figure 4B). The negative regulator, GATOR1, localizes to the lysosomal surface through interaction with the KICSTOR complex, then acts as a GAP to stimulate GTP hydrolysis of RagA/B, leading to inactivation of RagA/B and mTORC1.11,15,16 We found that depletion of GATOR1 or KICSTOR rendered mTORC1 signaling insensitive to PRMT1 depletion (Figures 4C, 4D, S4A, and S4B). In contrast, GATOR2 functions upstream of GATOR1 to inhibit its GAP function, thus positively regulating mTORC1 signaling.11 We found that depletion of GATOR2 suppressed PRMT1-induced mTORC1 activation (Figures 4E, 4F, and S4C). These results suggest that PRMT1 acts upstream of Rag GTPases to positively control mTORC1 pathway activation by either inhibiting GATOR1 and KICSTOR or promoting GATOR2 in response to amino acids.
Figure 4. PRMT1 regulates mTORC1 activation upstream of Rag GTPases.
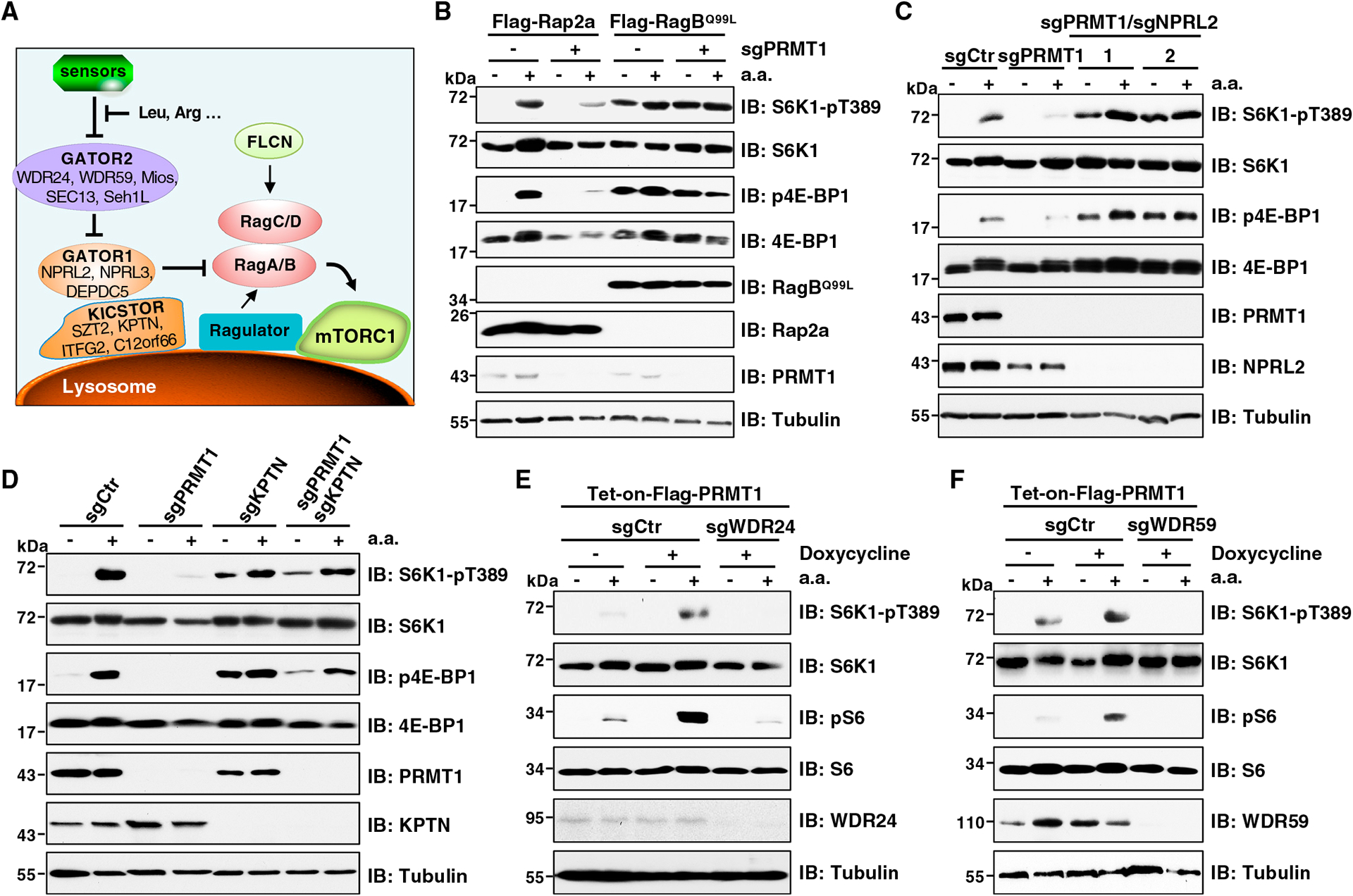
(A) The key complexes of the mTORC1 pathway in amino-acid-sensing branch.
(B) IB analysis of WCLs derived from HEK293T cells stably expressing FLAG-Rap2a or FLAG-RagBQ99L with or without PRMT1 depletion. Cells were starved of aa for 50 min or starved for 50 min and then restimulated with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
(C) IB analysis of WCL derived from HEK293T cells depleted of PRMT1 or PRMT1 and NPRL2. Cells were starved of aa for 50 min or starved for 50 min and then restimulated with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
(D) IB analysis of WCL derived from HEK293T cells depleted of PRMT1, KPTN, or both. Cells were starved of aa for 50 min or starved for 50 min and then restimulated with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
(E and F) IB analysis of WCLs derived from cells stably expressing Tet-on-FLAG-PRMT1 and depleted of WDR24 (E) or WDR59 (F). 1 μg/mL doxycycline was added for 48 hours to induce PRMT1 expression. Cells were starved of aa for 50 min or starved for 50 min and then restimulated with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
PRMT1 interacts with and methylates WDR24
To define the complex through which PRMT1 promotes amino-acid-induced mTORC1 signaling, we explored their interactions by transiently co-expressing PRMT1 with the components of the GATOR1/2 and KICSTOR complexes. FLAG-tagged PRMT1 co-immunoprecipitated with WDR24, and to a lesser extent, MIOS, DEPDC5, and NPRL3, but not KPTN (Figures S5A–S5C). Since GATOR1 and GATOR2 form an octamer,11 we further distinguished their interactions with PRMT1. Notably, DEPDC5 failed to interact with PRMT1 in WDR24-depleted cells (Figure S5D). In contrast, the binding between WDR24 and PRMT1 was not affected upon DEPDC5 depletion (Figure S5E). These results demonstrate that PRMT1 interacts with WDR24, through which it indirectly associates with the GATOR1 complex. In further support of this finding, endogenous PRMT1 co-immunoprecipitated with WDR24, but not DEPDC5, in modified HEK293T cells that express a FLAG epitope tag in the N terminus of the endogenous PRMT1 (Figure 5A). Reciprocally, Myc-tagged WDR24 co-immunoprecipitated endogenous PRMT1 (Figure S5F). Moreover, amino acid stimulation enhanced the binding between PRMT1 and WDR24 (Figure S5G).
Figure 5. PRMT1 interacts with WDR24 and methylates WDR24 at R329.
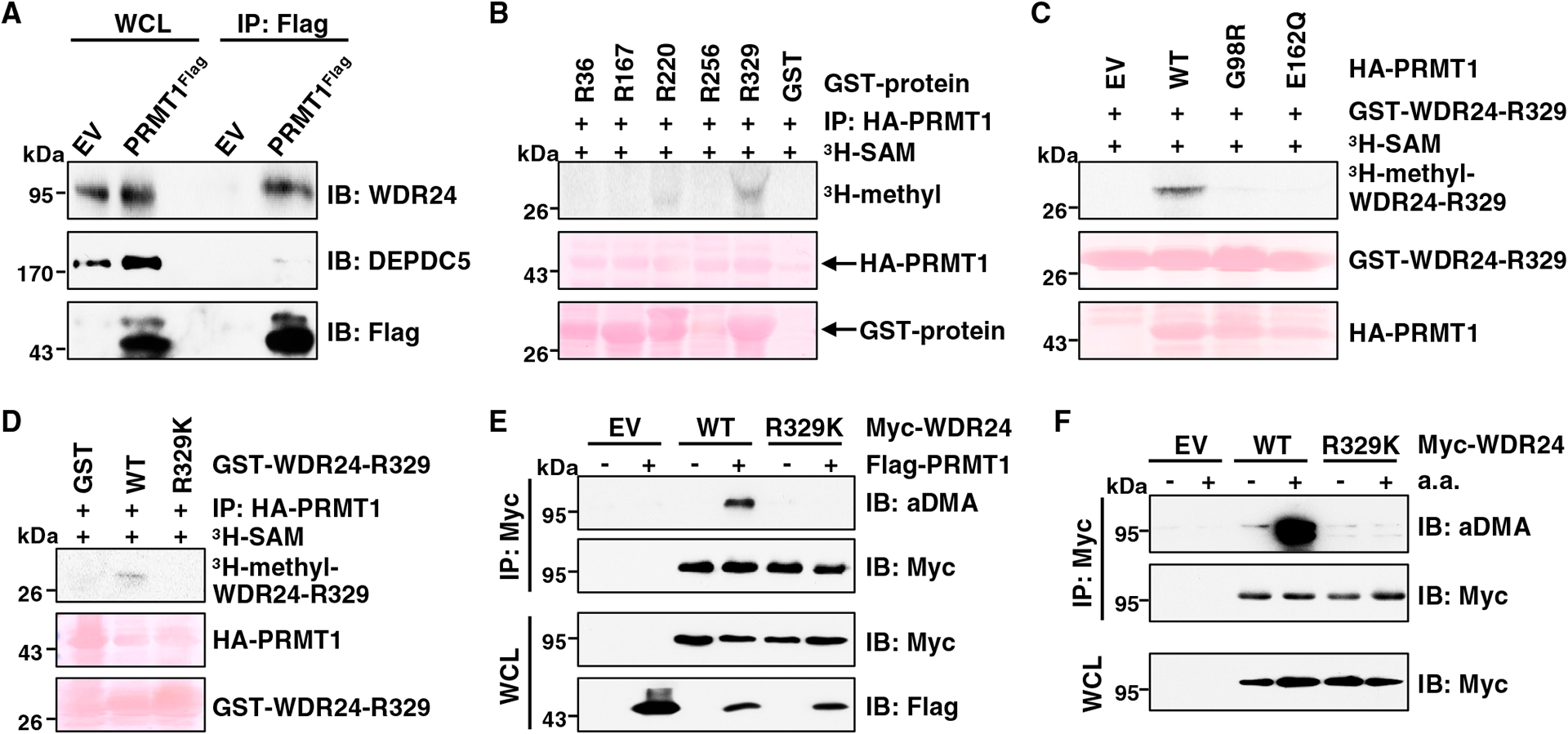
(A) IB analysis of WCLs and anti-FLAG IPs derived from HEK293T cells (EV, empty vector) and PRMT1FLAG knockin HEK293T cells. Similar results were obtained in three independent experiments.
(B) In vitro arginine methylation assays using recombinant GST-WDR24 truncated proteins purified from E. coli as substrates. HA-PRMT1 proteins immunopurified from HEK293T cells were used as the methyltransferase. Similar results were obtained in three independent experiments.
(C) In vitro arginine methylation assays using recombinant GST-WDR24-R329 protein as the substrate. HA-WT-PRMT1 or enzymatic-dead PRMT1 proteins derived from HEK293T cells were used as the methyltransferase. Similar results were obtained in three independent experiments.
(D) In vitro arginine methylation assays using recombinant GST-WDR24-R329-WT and GST-WDR24-R329K proteins as substrates. HA-WT-PRMT1 protein derived from HEK293T cells was used as the methyltransferase. Similar results were obtained in three independent experiments.
(E) IB analysis of WCLs and anti-Myc IPs derived from HEK293T cells transfected with indicated constructs. Similar results were obtained in three independent experiments.
(F) IB analysis of WCLs and anti-Myc IPs derived from HEK293T cells transfected with indicated constructs. Cells were starved of aa for 50 min or starved for 50 min and then restimulated with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
See also Figure S5.
We next determined whether WDR24 is a substrate of PRMT1. In vitro methylation experiments showed that only the fragment containing aa 298–383 of WDR24, but not other fragments, was methylated by PRMT1 in an enzymatic activity-dependent manner (Figures 5B and 5C). Mutating the evolutionarily conserved R329 residue to a Lys (R329K) blocked PRMT1-mediated methylation of WDR24 in vitro (Figures 5D and S5H). To determine R329 methylation in cells, we found that PRMT1 depletion or WDR24-R329K mutation inhibited aDMA formation of WDR24 (Figures 5E and S5I). Moreover, aDMA of WDR24 was significantly decreased in PRMT1S307A cells compared with PRMT1WT cells (Figure S5J). In keeping with the finding that amino acids enhance WDR24 interaction with PRMT1 (Figure S5G), aDMA of WDR24-WT, but not the WDR24-R329K mutant, was enhanced by amino acids (Figure 5F).
Taken together, these results demonstrate that PRMT1 functions as an upstream methyltransferase to catalyze the methylation of WDR24 at R329.
Methylation of WDR24 at R329 by PRMT1 promotes mTORC1 activation by amino acids
To demonstrate the importance of WDR24-R329 methylation on mTORC1 signaling under physiological condition, we again use the CRISPR-Cas9 genome editing approach to introduce the R329K mutation into the endogenous WDR24 gene (termed WDR24R329K) and validated by DNA sequencing (Figure S6A). In response to amino acids, the mTORC1 signaling was largely inhibited in WDR24R329K cells compared with WDR24WT cells (Figure 6A). To exclude the possibility that this observation was caused by the off-target effect of the CRISPR-Cas9 system, we performed rescue experiments and found that reintroduction of WDR24-WT restored mTORC1 activation in WDR24R329K cells (Figure S6B). Consistently, amino-acid-induced mTORC1 translocation to the lysosomal surface was impaired in cells expressing WDR24-R329K (Figures 6B–6D). In further support of the critical role of R329 methylation in mTORC1 activation, reconstitution of WDR24-WT, but not the methylation-deficient mutant WDR24-R329K, promoted mTORC1 signaling in WDR24-depleted Huh7 and HepG2 cells stimulated with amino acids (Figures S6C and S6D). Importantly, ectopic expression of PRMT1 enhanced mTORC1 activation in WDR24WT cells but not in WDR24R329K cells (Figure 6E). These results demonstrate that WDR24-R329 methylation is critical for mTORC1 pathway activation by amino acids.
Figure 6. WDR24-R329K mutation inhibits amino-acid-induced mTORC1 activation.
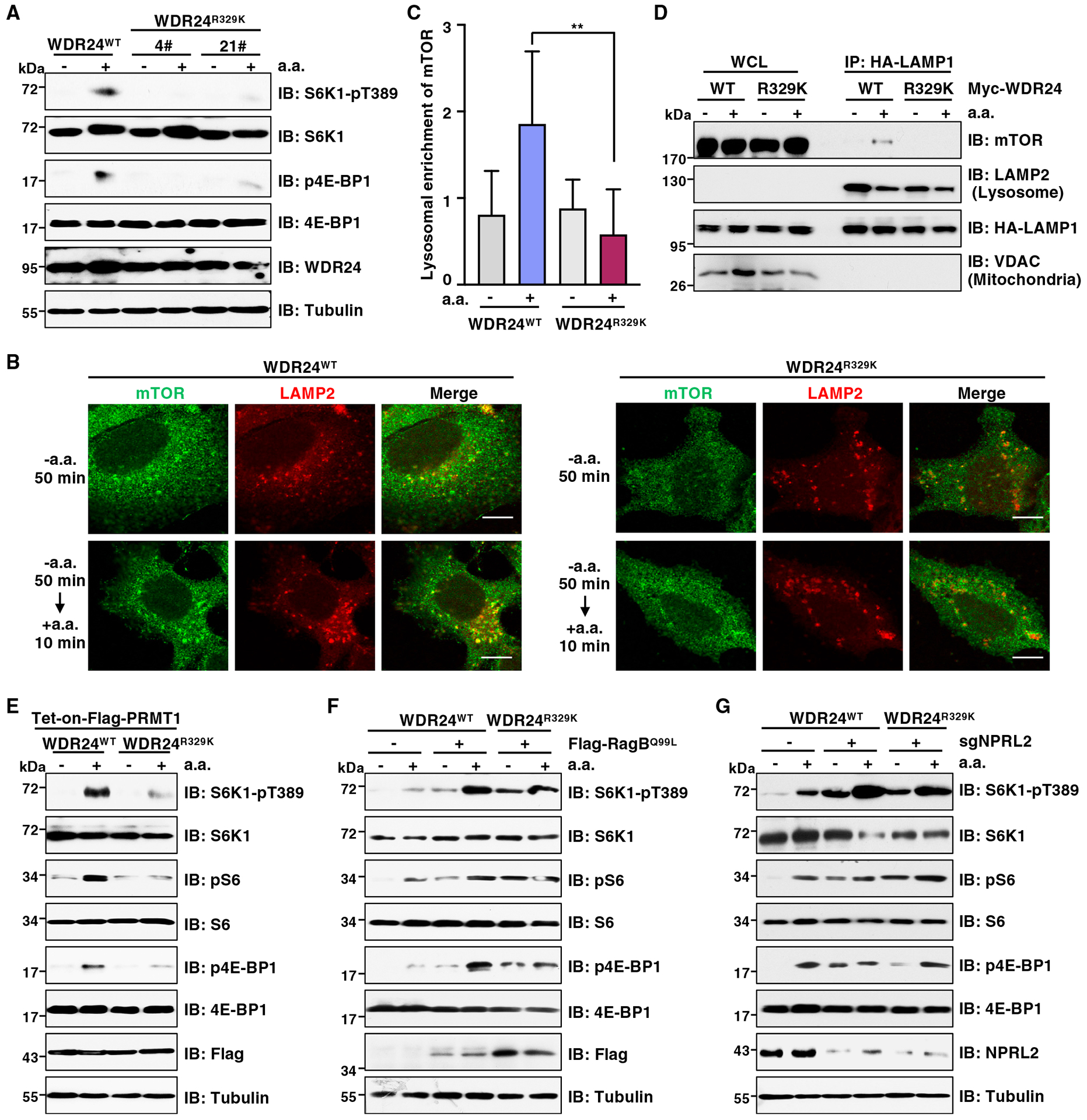
(A) IB analysis of WCLs derived from WDR24WT or WDR24R329K knockin Huh7 cells. Cells were starved of aa for 2 h or starved for 2 h and then restimulated with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
(B) IF analysis of mTOR lysosomal localization in WDR24WT or WDR24R329K knockin Huh7 cells. LAMP2 was a lysosomal marker. Cells were starved of aa for 2 h or starved for 2 h and then restimulated with aa for 10 min. Scale bar, 10 μm. Similar results were obtained in three independent experiments.
(C) Quantification of lysosome enrichment of mTOR in (B). Data are represented as mean ± SD. **p < 0.01, two-tailed Student’s t test.
(D) IB analysis of WCL and lysosomal enriched fraction. Huh7 cells stably expressing HA-LAMP1 were transfected with Myc-WDR24-WT or Myc-WDR24-R329K mutant. Cells were starved of aa for 2 h or starved for 2 h and then restimulated with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
(E) IB analysis of WCLs derived from WDR24WT or WDR24R329K knockin Huh7 cells stably expressing Tet-on-FLAG-PRMT1. Cells were treated with doxycycline for 48 h to induce PRMT1 expression. Cells were then starved of aa for 2 h or starved for 2 h and then restimulated with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
(F) IB analysis of WCLs derived from WDR24WT or WDR24R329K knockin Huh7 cells stably expressing FLAG-RagBQ99L. Cells were starved of aa for 2 h or starved for 2 h and then restimulated with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
(G) IB analysis of WCLs derived from WDR24WT or WDR24R329K knockin Huh7 cells depleted of NPRL2. Cells were starved of aa for 2 h or starved for 2 h and then restimulated with aa for 10 min before harvesting. Similar results were obtained in three independent experiments.
We next intended to dissect the molecular mechanisms by which PRMT1 promotes GATOR2-dependent mTORC1 activation via WDR24-R329 methylation. Arginine methylation can regulate protein function through multiple mechanisms, including protein-protein interactions.74–78 Since GATOR2 forms a complex with GATOR1, we examined whether PRMT1-mediated methylation of WDR24 affects their interactions. Intriguingly, neither overexpression nor depletion of PRMT1 affected GATOR2 complex integrity and its interaction with GATOR1 (Figures S6E and S6F), as well as Sestrin2 (Figures S6G and S6H) and CASTOR1 (Figures S6I and S6J). Consistently, both WDR24-WT and the WDR24-R329K mutant displayed a similar capacity to interact with GATOR1/2, Sestrin2, and CASTOR1 (Figures S6K–S6M). It has been reported that GATOR2 serves as an inhibitor of GATOR1 to impair GATOR1 interaction with Rag GTPases, although its exact function is unknown.11,79 We found that in response to amino acids, depletion of PRMT1 prevented GATOR1 dissociation from RagA (Figure S6N), whereas overexpression of PRMT1 decreased the interaction between GATOR1 and RagA (Figure S6O). These results suggest that PRMT1-mediated methylation of WDR24 at R329 promotes mTORC1 activation in part through enhancing GATOR2’s inhibitory role on GATOR1 and consequently weakening GATOR1 interaction with Rag GTPases. In further support of this idea, manipulation of the GATOR2 downstream effectors, either overexpression of RagBQ99L or depletion of GATOR1, rendered mTORC1 insensitive to amino acids in WDR24R329K cells (Figures 6F and 6G).
The CDK5-PRMT1-WDR24 axis is critical for HCC cell proliferation and tumor growth
Altered amino acid metabolism is a characteristic of liver diseases, including HCC.80,81 Given amino acids serve as an upstream signal of CDK5, PRMT1, and WDR24, we reasoned that this axis plays a role in HCC. Analysis of The Cancer Genome Atlas (TCGA) RNA sequencing (RNA-seq) dataset of HCC, which contains 50 healthy liver tissues and 371 HCC samples, revealed that the mRNA levels of CDK5, PRMT1, and WDR24 are significantly higher in HCC samples than healthy liver tissues (Figure S7A). Moreover, patients with high expression of CDK5 or PRMT1 had shorter survival than patients with low/medium expression (Figure S7B). There was no significant correlation between WDR24 expression and the survival of patients with HCC (Figure S7B), suggesting a possibility that the actual activation of WDR24 controlled by its upstream regulators, such as PRMT1, but not its expression levels, is critical for its role in HCC. In support of the clinical significance of the CDK5-PRMT1-WDR24 pathway, we found that cell proliferation and colony formation are significantly decreased in cells depleted of these genes compared with control cells (Figures S7C–S7I).
We next investigate the biological significance of PRMT1-S307 phosphorylation and WDR24-R329 methylation. Notably, cell proliferation and colony formation were significantly decreased in PRMT1S307A cells compared with PRMT1WT cells (Figures S7J–S7L). Similarly, compared with WDR24WT cells, WDR24R329K cells displayed a dramatic reduction of cell proliferation, colony formation, and anchorage-independent growth (Figures 7A–7C), which could be largely rescued by ectopic expression of WDR24-WT (Figures S7M–S7O). We also subcutaneously injected WDR24WT and WDR24R329K Huh7 cells into immunodeficient nude mice. Compared with WDR24-WT, WDR24-R329K mutation significantly suppressed tumor growth (Figures 7D–7F). Moreover, compared with tumors derived from the WDR24WT group, tumors derived from the WDR24R329K group displayed lower mTORC1 signaling and a reduction of cell proliferation as evidenced by Ki67 immunohistochemical staining (Figures 7G and 7H). These data demonstrate that PRMT1-mediated methylation of WDR24 at R329 promotes HCC tumor growth in part by activating the mTORC1 pathway.
Figure 7. Deficiency in WDR24-R329 methylation suppresses cell proliferation and xenograft tumor growth.
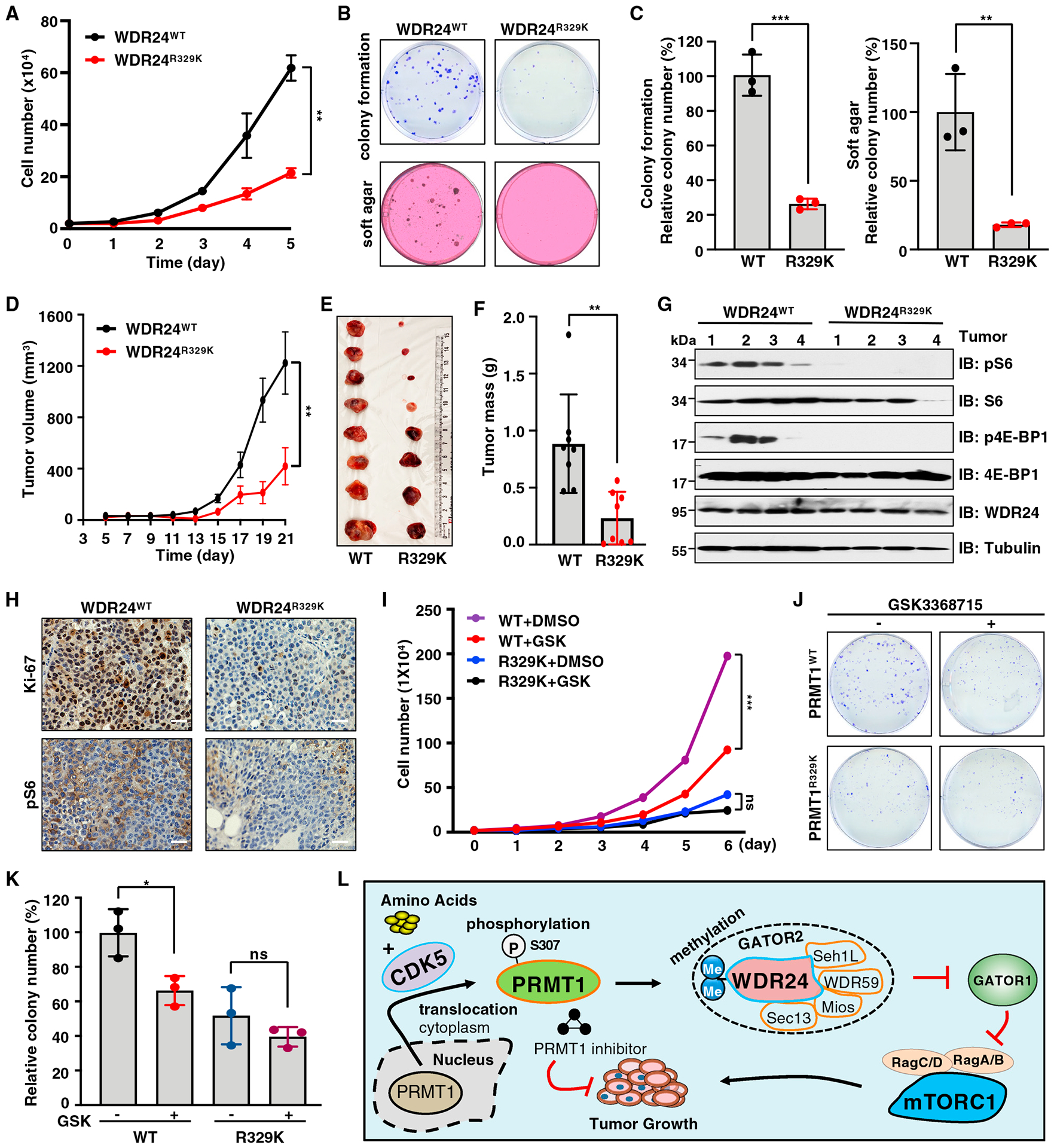
(A) WDR24WT and WDR24R329K knockin Huh7 cells were subjected to cell proliferation assays. Data are shown as mean ± SD of n = 3 independent experiments. **p < 0.01, two-way ANOVA.
(B and C) WDR24WT and WDR24R329K knockin Huh7 cells were subjected to colony formation and soft agar assays. Representative images are shown in (B) and quantification of colonies in (C). Data are shown as the mean ± SD of n = 3 independent experiments. **p < 0.01, ***p < 0.001, two-tailed Student’s t test.
(D) WDR24WT and WDR24R329K knockin Huh7 cells were subjected to mouse xenograft assays. Tumor growth was monitored. Data are shown as mean ± SEM of n = 8 tumors for each group. **p < 0.01, two-way ANOVA.
(E and F) Dissected tumors were weighed. Data are shown as the mean ± SD of n = 8 tumors for each group. **p < 0.01, two-tailed Student’s t test.
(G) IB analysis of lysates derived from tumor tissues in (E).
(H) IHC staining of Ki-67 and pS6 in tumor tissues. Scale bar, 50 μm.
(I) WDR24WT and WDR24R329K knockin Huh7 cells were treated with GSK3368715 and subjected to cell proliferation assays. Data are shown as mean ± SD of n = 3 independent experiments. ***p < 0.001, two-way ANOVA. ns, non-significant.
(J and K) WDR24WT and WDR24R329K knockin Huh7 cells were treated with GSK3368715 and subjected to colony-formation assays. Representative images are shown in (J) and quantification of colonies in (K). Data are shown as mean ± SD of n = 3 independent experiments. *p < 0.05, two-tailed Student’s t test. ns, nonsignificant.
(L) A model depicting the detailed molecular mechanism underlying the critical role of CDK5-PRMT1-WDR24 signaling axis in regulation of mTORC1 pathway activation and tumor growth.
Several type I/PRMT1 inhibitors have been developed and have shown anti-proliferation/tumor activity in preclinical studies,33,40,41,82 including GSK3368715, which has been evaluated in clinical trial for solid tumors and diffuse large B cell lymphoma (ClinicalTrials.gov: NCT03666988). Treating Huh7 cells with GSK3368715 decreased cell viability in a time- and dose-dependent manner, an effect similar to rapamycin (Figure S7P). Notably, GSK3368715 treatment significantly suppressed cell proliferation and colony formation of WDR24WT cells but not WDR24R329K cells (Figures 7I–7K). These results indicate that targeting PRMT1/WDR24 may be a potential strategy to suppress HCC.
In conclusion, our study revealed a detailed molecular mechanism underlying the critical role of the CDK5-PRMT1-WDR24 axis in regulation of mTORC1 signaling and tumorigenesis (Figure 7L). Specifically, in response to amino acid stimulation, active CDK5 phosphorylates PRMT1 at S307 to promote PRMT1 nucleocytoplasmic translocation and activation. Subsequently, PRMT1 methylates WDR24 at R329, an essential component of the GATOR2 complex, to potentiate the GATOR2’s inhibitory role on GATOR1, leading to activation of Rag GTPases and mTORC1. Consequently, the CDK5-PRMT1-WDR24 axis promotes cell proliferation and tumor growth. We have also shown that PRMT1 inhibitors block mTORC1 pathway activation, HCC cell proliferation, and colony formation, providing a molecular basis for targeting PRMT1 to combat cancer.
DISCUSSION
Our study identified amino acids as an upstream input regulating PRMT1 localization. Specifically, PRMT1 localizes in the nucleus under amino-acid-deficient conditions, whereas it translocates to cytoplasm upon amino acid stimulation, depending on CDK5-mediated phosphorylation at S307. Of note, phosphorylation plays an important role in regulating protein interaction with importins, exportins, and 14-3-3 proteins, through which it controls protein localization.83,84 Our result demonstrates that phosphorylation of PRMT1 at S307 is critical for its interaction with exportin 1, which may control PRMT1 nuclear export. Interestingly, we also observed an enrichment of PRMT1 in the lysosomes in response to amino acids, and it subsequently methylates WDR24 to promote mTORC1 signaling. Previous studies showed that in response to Wnt3a stimulation, PRMT1 is sequestered together with GSK3 into lysosomes through microautophagy via the ESCRT/Vps4 machinery and subsequently methylates many proteins to promote Wnt signal transduction.85–87 A recent study found that in response to cisplatin treatment, active DNA-dependent protein kinase (DNA-PK) phosphorylates and recruits PRMT1 to chromatin to methylate H4R3 and other chromatin-associated proteins.88 These findings suggest that various upstream signals dictate PRMT1 translocation to the right subcellular compartments to control specific protein methylation programs and signaling pathways.
PRMT1 not only functions as a transcriptional co-activator in epigenetic regulation of gene expression by depositing aDMA on histone H4R3 (H4R3me2a) but also methylates numerous non-histone substrates to control numerous cellular processes, including transcription, DNA repair, and signal transduction. However, only a few studies demonstrated that PRMT1 substrate specificity and activity are subjected to regulation by posttranslational modifications, including phosphorylation49,88,89 and ubiquitination.50,90,91 Notably, the THW loop (aa 310–312) of PRMT1, which is structurally located near the active site, is critical for its substrate binding.92,93 Phosphorylation of Y291 (corresponding to Y309 in human PRMT1 isoform 1), which is adjacent to the THW loop, alters PRMT1 substrate specificity and protein-protein interactions.49 While S307 is adjacent to Y309, we would speculate that phosphorylation of S307 represents another regulatory mechanism of PRMT1 substrate specificity and activity. Proteomics profiling of arginine methylation in PRMT1WT and PRMT1S307A cells should provide further insights into these phosphorylation events. Moreover, it will be interested to investigate whether there is crosstalk between S307 and Y309 phosphorylation.
Different amino acids regulate mTORC1 activation through different mechanisms. Leu and Arg bind Sestrin2 and CASTOR1, respectively, to release their inhibition on GATOR2 complex.12,13 Met is indirectly sensed through SAM, which disrupts interaction between SAMTOR (SAM sensor) and GATOR1, leading to GATOR1 inactivation.17 We found that PRMT1 depletion blocks mTORC1 activation in response to Leu, Arg, or Met stimulation (Figures S1J–S1L). It is possible that PRMT1 regulates mTORC1 pathway activation in response to various amino acids through different mechanisms. PRMT1 promotes Leu- and Arg-induced mTORC1 activation through WDR24 methylation-mediated GATOR2 activation. However, given PRMT1 can directly bind SAM,94 it may activate the mTORC1 pathway in response to Met by regulating SAMTOR/GATOR1 complexes, which requires further investigation.
Limitations of the study
Using two phospho-CDK substrate motif antibodies, we identified S307 of PRMT1 as the major phosphorylation site by CDK5. It would be helpful to further validate this conclusion by generating an antibody that specifically recognizes S307 phosphorylation. We also characterized WDR24 as a key downstream substrate of PRMT1, through which PRMT1 promotes GATOR2-dependent mTORC1 signaling and HCC growth. A follow-up study is warranted to address how PRMT1-mediated WDR24-R329 methylation promotes GATOR2 activation because the exact molecular function of GATOR2 has yet to be determined. In addition, our study mechanically demonstrates the tumor-promoting function and therapeutic potential of the CDK5-PRMT1-WDR24 axis using HCC cell-based assays, including cell proliferation, colony formation, and xenograft tumor growth. It is warranted to further functionally investigate this pathway in HCC progression using primary HCC mouse models, such as mouse models with HCC induced by a high branched-chain amino acid diet that are highly relevant to mTORC1 signaling.95
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents may be directed to and will be fulfilled by the lead contact Wenjian Gan (ganw@musc.edu).
Materials availability
A list of critical reagents is included in the key resources table. Relevant plasmids are available to the academic community. For additional materials, please email the lead contact for requests. Material that can be shared will be released via a Material Transfer Agreement.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-S6K1-pT389 | Cell Signaling Technology | Cat# 9234; RRID:AB_2269803 |
| Rabbit monoclonal anti-S6K1 | Cell Signaling Technology | Cat# 9202; RRID:AB_331676 |
| Rabbit monoclonal anti-pS6 Ser235/236 | Cell Signaling Technology | Cat# 4858; RRID:AB_916156 |
| Rabbit monoclonal anti-S6 | Cell Signaling Technology | Cat# 2217; RRID: AB_331355 |
| Rabbit polyclonal anti-p4E-BP1 Ser65 | Cell Signaling Technology | Cat# 9451; RRID:AB_330947 |
| Mouse monoclonal anti-Myc-tag | Cell Signaling Technology | Cat# 2276; RRID:AB_331783 |
| Rabbit monoclonal anti-4E-BP1 | Cell Signaling Technology | Cat# 9644; RRID:AB_2097841 |
| Rabbit polyclonal anti-ULK1-pSer757 | Cell Signaling Technology | Cat# 6888; RRID:AB_10829226 |
| Rabbit monoclonal anti-ULK1 | Cell Signaling Technology | Cat# 8054; RRID:AB_11178668 |
| Rabbit polyclonal anti-PRMT1 | Cell Signaling Technology | Cat# 2449; RRID:AB_2237696 |
| Rabbit monoclonal anti-mTOR | Cell Signaling Technology | Cat# 2983; RRID:AB_2105622 |
| Rabbit monoclonal anti-NPRL2 | Cell Signaling Technology | Cat# 37344; RRID:AB_2799113 |
| Rabbit monoclonal anti-Ki-67 | Cell Signaling Technology | Cat# 9027; RRID:AB_2636984 |
| Rabbit monoclonal anti-Myc Tag | Cell Signaling Technology | Cat# 2278; RRID:AB_490778 |
| Mouse monoclonal anti-PRMT4 | Cell Signaling Technology | Cat# 12495; RRID:AB_2797935 |
| Rabbit monoclonal anti-PRMT5 | Cell Signaling Technology | Cat# 79998; RRID:AB_2451435 |
| Rabbit monoclonal anti-WDR59 | Cell Signaling Technology | Cat# 53385; RRID:AB_2799432 |
| Rabbit monoclonal anti-HA | Cell Signaling Technology | Cat# 3724; RRID:AB_1549585 |
| Rabbit monoclonal anti-Flag | Cell Signaling Technology | Cat# 14793; RRID:AB_2572291 |
| Rabbit monoclonal anti-MIOS | Cell Signaling Technology | Cat# 13557; RRID:AB_2798254 |
| Rabbit monoclonal anti-RagA | Cell Signaling Technology | Cat# 4357; RRID:AB_10545136 |
| Rabbit monoclonal anti-RagC | Cell Signaling Technology | Cat# 9480; RRID:AB_10614716 |
| Rabbit monoclonal anti-CDK5 | Cell Signaling Technology | Cat# 14145; RRID:AB_2773717 |
| Rabbit monoclonal phosphor-CDK substrate motif | Cell Signaling Technology | Cat# 9477; RRID:AB_2714143 |
| Phospho-MAPK/CDK Substrates | Cell Signaling Technology | Cat# 2325 |
| Mouse monoclonal anti-LAMP2 | Santa Cruz Biotechnology | Cat# sc-18822; RRID:AB_626858 |
| Mouse monoclonal anti-PRMT6 | Santa Cruz Biotechnology | Cat# sc-271744; RRID:AB_10715087 |
| Mouse monoclonal anti-Sp1 | Santa Cruz Biotechnology | Cat#sc-17824; RRID:AB_628272 |
| Mouse monoclonal anti-Histone H3 | Santa Cruz Biotechnology | Cat# sc-517576; RRID:AB_2848194 |
| Mouse monoclonal anti-Flag | Sigma Aldrich | Cat# F3165; RRID:AB_259529 |
| Rabbit polyclonal anti-NPRL3 | Sigma Aldrich | Cat# HPA011741; RRID:AB_1845577 |
| Rabbit polyclonal anti-Flag | Sigma Aldrich | Cat# F7425; RRID:AB_439687 |
| Anti-Mouse IgG | Sigma Aldrich | Cat# A4416; RRID:AB_258167 |
| Anti-Rabbit IgG | Sigma Aldrich | Cat# A4914; RRID:AB_258207 |
| Mouse monoclonal anti-Tubulin | Proteintech | Cat# 66240–1-lg; RRID:AB_2881629 |
| Rabbit polyclonal anti-WDR24 | Proteintech | Cat# 20778–1-AP; RRID:AB_10696183 |
| Rabbit polyclonal anti-VDAC | Proteintech | Cat# 10866–1-AP; RRID:AB_2257153 |
| Rabbit polyclonal anti-KPTN | Proteintech | Cat# 16094–1-AP; RRID:AB_2134007 |
| Rabbit polyclonal anti-Golgin-97 | Proteintech | Cat# 12640–1-AP; RRID:AB_2115315 |
| Rabbit polyclonal anti-GST Tag | Proteintech | Cat# 10000–0-AP; RRID:AB_11042316 |
| Rabbit polyclonal anti-Histone H4 | Proteintech | Cat# 16047–1-AP; RRID:AB_2118625 |
| Rabbit polyclonal anti-Sestrin 2 | Proteintech | Cat# 10795–1-AP; RRID:AB_2185480 |
| Rabbit polyclonal anti-DEPDC5 | Thermo Fisher | Cat# PA5–71618; RRID:AB_2717472 |
| Rabbit polyclonal anti-PRMT2 | Thermo Fisher | Cat# 720141; RRID:AB_2608486 |
| Rabbit monoclonal anti-PRMT3 | Abcam | Cat# ab191562 |
| Rabbit monoclonal anti-Seh1L | Abcam | Cat# ab218531 |
| Rabbit polyclonal anti-PRMT7 | ABclonal | Cat# A12159; RRID:AB_2759045 |
| Rabbit polyclonal anti-SEC13 | ABclonal | Cat# A11613; RRID:AB_27588636 |
| Rabbit polyclonal anti-CDK1 | ABclonal | Cat# A0220; RRID:AB_2757034 |
| Rabbit polyclonal anti-CDK2 | ABclonal | Cat# A0294; RRID:AB_2757106 |
| Rabbit polyclonal anti- Histone H4R3 Dimethyl Asymmetric (H4R3me2a) | Epigentek | Cat# A-3708 |
| Rabbit polyclonal anti- Histone H4R3 Dimethyl Symmetric (H4R3me2s) | Epigentek | Cat# A-3718 |
| Mouse monoclonal anti-HA | BioLegend | Cat# 901503; RRID:AB_2565005 |
| Rabbit polyclonal anti-PRMT9 | Bethyl Laboratories | Cat# A304–189A; RRID:AB_2620386 |
| Anti-aDMA | Dr. Mark Bedford lab | N/A |
| Bacterial and virus strains | ||
| XL10 Gold Escherichia coli | Agilent | Cat# 200314 |
| BL21(DE3) Escherichia coli | Sigma Aldrich | Cat# CMC0014 |
| Biological samples | ||
| Liver cancer tissue microarray | Biomax | LV1021a |
| Chemicals, peptides, and recombinant proteins | ||
| Amino acids free RPMI medium | United States Biological | Cat# R8999–04A |
| Furamidine dihydrochloride (FD) | Tocris Bioscience | Cat# 5202 |
| TC-E 5003 | Tocris Bioscience | Cat# 5099 |
| Anti-Flag agarose beads | Sigma Aldrich | Cat#A2220 |
| Anti-HA agarose beads | Sigma Aldrich | Cat# A2095 |
| Anti-Myc Tag affinity gel | BioLedgend | Cat# 658502 |
| Glutathione Sepharose beads | Cytiva | Cat# 17075605 |
| VECTASHIELD Antifade Mounting Media | Vector Laboratories | Cat# H-1400–10 |
| Histone H4 recombination protein | New England BioLabs | Cat# M2504S |
| GSK3368715 | MedChemExpress | Cat# HY-128717A |
| SHURMount media | General Data | Cat# 682188 |
| Adenosyl-L-Methionine, S-[methyl-3H] | PerkinElmer | Cat# NET155H250UC |
| ATP, [γ-32P] | PerkinElmer | Cat# NEG502Z250UC |
| DSP | Thermo Fisher | Cat# 22585 |
| Critical commercial assays | ||
| QuikChange XL site-directed mutagenesis kit | Agilent | Cat# 20518 |
| CellTiter-Glo Assay Kit | Promega | Cat# G9242 |
| ImmPRESS Excel Amplifiered Polymer Kit | Vector Laboratories | Cat# MP-7601 |
| NE-PER™ Nuclear and Cytoplasmic Extraction Reagents | Thermo Fisher | Cat# 78833 |
| EpiQuik Total Histone Extraction Kit | Epigentek | Cat# OP-0006–100 |
| Experimental models: Cell lines | ||
| Human: HEK293 | ATCC | Cat# CRL-1573 |
| Human: HEK293T | ATCC | Cat# CRL-3216 |
| Human: Huh7 | ATCC | Cat# PTA-4583 |
| Human: HepG2 | ATCC | Cat# HB-8065 |
| Human: HeLa | ATCC | Cat# CCL-2 |
| Experimental models: Organisms/strains | ||
| Nude mice | The Jackson Laboratory | Strain # 007850 |
| Oligonucleotides | ||
| PRMTs sgRNAs | Dr. Wenjian Gan laboratory | Yin et al., 2021 |
| CDK5-sg1: TGTGTTCAAGGCCAAAAACC | This paper | N/A |
| CDK5-sg2: GGCCTTGAACACAGTTCCGT | This paper | N/A |
| CDK5-sg3: GAGGCTGGATGACGATGATG | This paper | N/A |
| WDR59-sg1: CGTTTTCGCTGCTCCATCGC | This paper | N/A |
| WDR59-sg2: CTTTCGGTGACCTTCGAAAG | This paper | N/A |
| DEPDC5-sg1: TAATACTCTTTTCAGAGTGG | This paper | N/A |
| DEPDC5-sg2: CTTCATCCAGTATCCAGTGT | This paper | N/A |
| NPRL2-sg1: CACCTTCAAGTGGATGGTGT | This paper | N/A |
| NPRL2-sg2: GTCCAACACCATCCACTTGA | This paper | N/A |
| KPTN-sg1: GCGCAACGGACAAGGCCCCG | This paper | N/A |
| KPTN-sg2: GCAGAGCAATGTGTACGGGC | This paper | N/A |
| WDR24-sg: CACGAACTGTTCCTCCTCGA | This paper | N/A |
| ssODN for PRMT1 Flag tag knock-in: GAGGAGAAAGGGGGGGTCTTGGCGGCCGGAGGAGGAGTAGGTGCGGGTGAAGATGGACTACAAAGACCATGACGGTGATTATAAAGATCATGACATTGATTACAAGGATGACGATGACAAGGCGGCCGCAGGCGCGGCAGCCGAGGCTGCGAACTGCATCATGGAAGTGAGCGCTTGGAGCGCCGCCGTGGGCGGGAG | This paper | N/A |
| ssODN for WDR24-R329K knock-in: TGTCTGGCTCCAAGGACAGCTCGCTGTGCCAGCACCTGTTCAAGGACGCCAGTCAGCCCGTCGAGCGCGCCAACCCTGAGGGCCTC | This paper | N/A |
| Recombinant DNA | ||
| Flag-PRMT1 | This paper | N/A |
| HA-PRMT1 | This paper | N/A |
| Myc-WDR24 | This paper | N/A |
| pLenti-HA-WDR24 | This paper | N/A |
| pLenti-HA-LAMP1 | This paper | N/A |
| Tet-on-Flag-PRMT1 | This paper | N/A |
| pGEX-PRMT1 | This paper | N/A |
| pLJM1-Flag-RagBQ99L | Addgene | Cat# 19315 |
| pLJM1-Flag-Rap2a | Addgene | Cat# 19311 |
| HA-METAP2 | Addgene | Cat# 100512 |
| HA-DEPDC5 | Addgene | Cat# 46327 |
| HA-NPRL2 | Addgene | Cat# 99709 |
| HA-NPRL3 | Addgene | Cat# 46330 |
| HA-WDR59 | Addgene | Cat# 46328 |
| HA-WDR24 | Addgene | Cat# 46335 |
| HA-MIOS | Addgene | Cat# 46329 |
| HA-SEH1L | Addgene | Cat# 46331 |
| HA-SEC13 | Addgene | Cat# 46332 |
| Flag-CASTOR1 | Addgene | Cat# 84488 |
| HA-KPTN | Addgene | Cat# 87042 |
| lentiCRISPR v2 | Addgene | Cat# 52961 |
| lentiCRISPR V2-Blast | Addgene | Cat# 83480 |
| HA-CDK5 | Addgene | Cat# 1872 |
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
HEK293T, HEK293, HeLa, Huh7 cells and their derivatives were cultured in Dulbecco’s Modified Eagle Medium. HepG2 cells were cultured in Minimum Essential Medium. 10% Fetal Bovine Serum (FBS), 100 units/ml penicillin and 100 μg/ml streptomycin were supplemented in the medium. All cells were maintained at 37°C and 5% CO2.
Xenograft tumor growth
WDR24WT or WDR24R329K Huh7 cells (2 × 106) were injected into the flank of 5-week-old male nude mice (Foxn1nu/Foxn1nu, The Jackson Laboratory). Tumor size was measured every other day with an electronic caliper. The tumor volume was calculated using the formula: L × W2 × 0.52, where L is the longest diameter and W is the shortest diameter. After 21 days, mice were euthanized and the xenografted solid tumors were dissected and weighed. Tumors were used for IHC staining of Ki-67 and pS6, and for Western blot analysis of the mTORC1 signaling. All animal experiments were conducted under protocol #IACUC-2018–00604 approved by the MUSC Institutional Animal Care and Use Committee.
Patient tumor tissue samples and immunohistochemical staining
A tissue microarray containing 97 cases of liver cancer was purchased from Biomax (LV1021a). The sections were deparaffinized by xylene, rehydrated with graded ethanol and heated in boiled citrate buffer (pH 6.0) for 20 min for antigen retrieval. The ImmPRESS Excel Amplifiered Polymer Kit (Vector Laboratories, MP-7601) was used in the following steps. Sections were incubated with BLOXALL solution for 10 min, washed with PBST buffer for 5 min, blocked with 2.5% normal horse serum for 20 min and incubated with anti-pS6 (1:400) or anti-PRMT1 (1:1000) antibody diluted in normal horse serum overnight at 4°C. Sections were then washed twice with PBST for 5 min, incubated with goat anti-rabbit IgG for 15 min, followed by incubation with ImmPRESS Plolymer for 30 min. After wash with PBST, sections were incubated with DAB working solution for 1–3 min, counterstained with hematoxylin and mounted using SHURMount media (General Data, 682188). The levels of pS6 and PRMT1 were assessed by HSCORE using computerized image analysis.
METHOD DETAILS
Lentiviral packaging and infection
Target constructs containing sgRNA or cDNA were co-transfected with PMD2.G and pSPAX2 plasmids into HEK293T cells with polyethylenimine (PEI). 48 hours post-transfection, virus-containing supernatants were collected and filtered with 0.45 μM PES filter. The targeted cells were infected with virus for 24 hours and selected with 200 μg/ml hygromycin, 2 μg/ml puromycin or 10 μg/ml blasticidin for 3–5 days.
Amino acid starvation and restimulation
Cells were starved with amino acids free RPMI medium supplemented with 10% dialyzed FBS for 50 min or 2 hours, and then restimulated with RPMI medium containing amino acids and FBS.
Generation of knockout or knock-in cells
To generate WDR24 knockout cells, the sgRNA (CACGAACTGTTCCTCCTCGA) targeting WDR24 was inserted into lentiCRISPR v2-Blast vector (#83480, Addgene) and transfected into Huh7 cells. 48 hours post-transfection, the cells were treated with 10 μg/ml blasticidin for 3 days and then seeded into a 96-well plate at 1–2 cell per well. The positive knockout colonies were identified by Western blot analysis of WDR24.
To generate Flag-tag knock-in PRMT1 cells, the sgRNA (GGCCGCGAACTGCATCATGG) targeting PRMT1 around the ATG was inserted into lentiCRISPR v2 vector. The ssODN containing 3x Flag tag was used as a template (key resources table). The sgRNA vector and ssODN were co-transfected into HEK293T cells. 48 hours post-transfection, the cells were treated with 2 μg/ml puromycin for two days and then seeded into a 96-well plate at 1–2 cell per well. The positive Flag knock-in colonies were identified by Western blot analysis of Flag and further confirmed by Sanger DNA sequencing.
To generate PRMT1-S307A or WDR24-R329K knock-in cells, the sgRNA targeting PRMT1 around the S307 or sgRNA targeting WDR24 around the R329 was inserted into lentiCRISPR v2 vector. The ssODN containing S307A mutation or R329K mutation was used as a template (key resources table). The sgRNA vector and ssODN were co-transfected into HEK293T or Huh7 cells. 48 hours post-transfection, the cells were treated with 2 μg/ml puromycin for two days and then seeded into a 96-well plate at 1–2 cells per well. The positive S307A or R329K knock-in colonies were identified by PCR coupled with restriction enzyme digestion and confirmed by sequencing.
Immunoblot (IB) and immunoprecipitation (IP) analyses
Cells were rinsed with ice-cold PBS and lysed with Triton lysis buffer for IB (40 mM HEPES pH7.4, 150 mM NaCl, 2.5 mM MgCl2, 1 Mm EDTA and 1% Triton X-100) and lysis buffer for IP (1% Triton X-100, 10 mM β-glycerol phosphate, 10 mM pyrophosphate, 40 mM HEPES pH7.4 and 2.5 mM MgCl2) supplemented with protease inhibitor and phosphatase inhibitors. The whole cell lysates (WCL) were centrifuged at 13,200 r.p.m. for 10 min at 4°C. Protein concentrations were determined by Nanodrop using Bio-Rad protein assay reagent. Equal amounts of WCL were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. For IP, the WCL were incubated with agarose conjugated with antibody for 3–4 hours at 4°C. The beads were washed with Triton lysis buffer containing 500 mM NaCl for five times. Anti-Flag agarose beads (A2220) and anti-HA agarose beads (A2095) were purchased from Sigma. Anti-Myc Tag affinity gel (658502) was purchased from BioLedgend. To detect interactions between GATOR1 and Rag GTPases, cells were treated with 1 mg/ml DSP crosslinker (22585, Thermo Fisher) for 7 min at room temperature. The reactions were stopped by adding 100 mM Tris-HCl pH 8.5.
Immunofluorescence (IF) staining
Cells were seeded on coverslips at 60% confluence overnight. After starvation or stimulation with amino acids, cells were rinsed once with PBS and fixed with 4% paraformaldehyde for 30 minutes at room temperature. After twice washes with PBS, cells on the coverslips were permeabilized for 5 minutes with 0.05%-0.1% Triton X-100. Then, the coverslips were washed three times with PBS, blocked with 5% BSA in PBS for 30 minutes at room temperature, incubated with primary antibodies [PRMT1 (1:400), mTOR (1:200), and LAMP2 (1:100)] overnight at 4°C. The cells were then washed three times with PBST and incubated with secondary antibody at a dilution of 1:500 for 1 hour at room temperature. Following three times washes with PBST, the cells were incubated with DAPI for 5 min, then washed with PBS twice before mounting on slides using VECTASHIELD Antifade Mounting Media (H-1400–10, Vector Laboratories). Following the protocol previously described,15 fluorescence intensity of mTOR was quantified using Image J and the lysosome enrichment of mTOR was calculated using the Fiji software.
Purification of recombinant GST proteins
Recombinant GST-PRMT1 and GST-WDR24 truncated proteins were purified from the BL21(DE3) E. coli. The protein expression was induced by 0.1 mM IPTG (isopropyl-β-D-thiogalactoside) at 25°C for 16 h. The bacteria cells were collected and re-suspended in GST buffer (25 mM Tris pH 8.0, 5 mM dithiothreitol, and 150 mM NaCl) and sonicated. After centrifugation, the supernatant was incubated with Glutathione Sepharose beads (17075605, Cytiva) for 2–3 hours at 4°C, followed by three-times washes with GST buffer and eluted with elution buffer (10 mM L-Glutathione and 50 mM Tris-HCl pH 8.0).
Lysosome purification
Cells were infected with HA-LAMP1 lentivirus to stably express the lysosome marker LAMP1. After amino acids starvation or stimulation, cells were harvested with fraction buffer (50 mM KCl, 90 mM potassium gluconate, 1 mM EGTA, 5 mM MgCl2, 50 mM Sucrose, 20 mM HEPES pH 7.4, phosphatase inhibitor set I/II, and protease inhibitor) and lysed using 23G needle for 4–5 times. After centrifuged at 2000 g for 10 min, the supernatant was used for HA-IP.
Histone extraction
The histone proteins were collected use EpiQuik Total Histone Extraction Kit according to the manufacturer’s instructions (EPIGENTEK). Briefly, cells were rinsed with ice-cold PBS and lysed with Pre-Lysis Buffer for 10 min. After centrifuge at 3000 r.p.m for 5 min, the supernatant was removed. The pellet was resuspended in lysis buffer and incubated on ice for 30 min. After centrifuge at 12000 r.p.m for 5 min, the supernatant fraction was collected and 0.3 volumes of Balance DTT buffer was added to the supernatant.
In vitro kinase assays
HA-CDK5 were transfected into HEK293T cells for 48 h. CDK5 lysis buffer (5 mM Tris-HCl pH 7.5, 25 mM NaCl, and 0.1 mM EDTA pH 8.0) supplemented with protease inhibitor and phosphatase inhibitors were used to lyse cells. The lysate was incubated with anti-HA agarose beads for 3 hours at 4°C. The beads were washed with CDK5 lysis buffer containing 0.1% NP-40 for three times and with kinase buffer (6.7 mM MOPS pH 7.0, 1.67 mM MgCl2, 0.03 mM EDTA pH 8.0, and 0.03 mM EGTA pH 8.0) for two times. 5 μg recombinant GST-PRMT1 proteins were incubated with HA-CDK5 in reaction buffer (8 μM MOPS and 0.2 mM EDTA pH 8.0) at 30°C for 30 min. The reactions were stopped by 3xSDS loading buffer. The samples were resolved by SDS-PAGE and transferred to PVDF membrane for IB analysis. For the radioactive isotope-based kinase assay, 5 μg PRMT1 protein, 0.2 μg active CDK5, and 0.5 μL ATP [γ-32P] were mixed in the kinase buffer and incubated at 30°C for 30 min. The reactions were stopped by loading buffer and the samples were resolved by SDS-PAGE. The gel was dried and exposed to X-ray film.
In vitro methylation assays
Recombinant GST-WDR24 truncated proteins were purified from E. coli as the substrates, and HA-PRMT1 protein was immunopurified from HEK293T cells by IP as the methyltransferase. 10 μg GST-WDR24 truncated proteins were incubated with HA-PRMT1 protein in reaction buffer (50 mM Tris-HCl pH 8.5, 20 mM KCl, 10 mM MgCl2, 1mM β-mercaptoethanol, and 100 mM sucrose) with 2 μL of adenosyl-L-methionine, S-[methyl-3H] (1 mCi/ml stock solution, Perkin Elmer) at 30°C for 1 hour. The reactions were stopped by 3xSDS loading buffer. The samples were resolved by SDS-PAGE and transferred to PVDF membrane, which was then sprayed with EN3HANCE (Perkin Elmer) and exposed to X-ray film.
Mass spectrometric analysis of PRMT1-S307 phosphorylation
pRK5-HA-PRMT1 construct was transfected into HEK293T cells for 48 hours. Cells were lysed with Triton lysis buffer and immunoprecipitated with anti-HA agarose beads for 3 hours and washed with Triton lysis buffer containing 500 mM NaCl for three times. The immunoprecipitated proteins were resolved by SDS-PAGE and visualized using GelCode blue staining reagent. The protein band containing HA-PRMT1 was excised and digested with trypsin. Peptides were analyzed by LC-MS/MS as described previously.96
Cell proliferation assays
Cells were seeded into 6-well plate at 2 × 104 cells per well at day 0 and manually counted every day using hemacytometer under microscopy.
Colony formation assays
Cells were seeded into 6-well plate at 200 cells per well and incubated for 7–10 days until visible colonies formation. Cells were fixed with 10% ethanol and 10% acetic acid for 30 min and then stained with 0.4% crystal violent in 20% ethanol for 30 min, followed by wash with dH2O and manually counting.
Soft agar assays
1 × 104 or 2 × 104 cells were mixed with noble agar at a final concentration of 0.4% and layered over the bottom layer containing 0.8% noble agar. Completed DMEM medium (500 μl) was added to keep the top layer moist. The cells were then cultured for 3–4 weeks and stained with 1 mg/ml iodonitrotetrazolium chloride for counting manually.
Cell viability assays
Cells were seeded in 96-well plates (2000 cells/well) for 24 hours and treated with indicated doses of GSK3368715 or Rapamycin for 2–6 days. Cell viability was determined by the CellTiter-Glo Cell Viability kit according to the manufacturer’s instructions (Promega).
QUANTIFICATION AND STATISTICAL ANALYSIS
As indicated in the figure legends, all quantitative data are presented as the mean ± SD or mean ± SEM of three biologically independent experiments or samples. Statistical analyses were performed using GraphPad Prism 9 and Excel. Statistical significance was determined by two-tailed Student’s t-test or two-way ANOVA. P-value < 0.05 was considered significant.
Supplementary Material
Highlights.
PRMT1 is required for amino-acid-induced mTORC1 activation
CDK5 phosphorylates PRMT1 to promote its cytoplasmic localization and activity
PRMT1 methylates WDR24 to promote mTORC1 activation by amino acids
The CDK5-PRMT1-WDR24 axis is critical for cell proliferation and tumor growth
ACKNOWLEDGMENTS
This work was in part supported by an NIH grant (R35GM146749) and the American Cancer Society (RSG-22-068-01-TBE) to W.G., NIH grants (S10 OD025126, Orbitrap Fusion Lumos ETD MS) to L.E.B., an NIH grant (R37CA251165) to H.W., and MUSC COBRE in Digestive and Liver Disease (P20 GM130457) and MUSC DDRCC (P30 DK123704). M.T.B. is supported by an NIH R01 grant (GM126421). S.Y. and L.L. were supported by the Hollings Cancer Center Abney Postdoctoral Fellowship.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112316.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Kim J, and Guan KL (2019). mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol 21, 63–71. 10.1038/s41556-018-0205-1. [DOI] [PubMed] [Google Scholar]
- 2.Saxton RA, and Sabatini DM (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wullschleger S, Loewith R, and Hall MN (2006). TOR signaling in growth and metabolism. Cell 124, 471–484. 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 4.Liu GY, and Sabatini DM (2020). mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol 21, 183–203. 10.1038/s41580-019-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mossmann D, Park S, and Hall MN (2018). mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 18, 744–757. 10.1038/s41568-018-0074-8. [DOI] [PubMed] [Google Scholar]
- 6.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, and Guan KL (2008). Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol 10, 935–945. 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, and Sabatini DM (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501. 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bar-Peled L, Schweitzer LD, Zoncu R, and Sabatini DM (2012). Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208. 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, and Sabatini DM (2010). Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303. 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Panchaud N, Péli-Gulli MP, and De Virgilio C (2013). Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Sci. Signal 6, ra42. 10.1126/scisignal.2004112. [DOI] [PubMed] [Google Scholar]
- 11.Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, and Sabatini DM (2013). A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340, 1100–1106. 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, Wang T, Harper JW, Gygi SP, and Sabatini DM (2016). The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 165, 153–164. 10.1016/j.cell.2016.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, and Sabatini DM (2016). Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351, 43–48. 10.1126/science.aab2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen J, Ou Y, Luo R, Wang J, Wang D, Guan J, Li Y, Xia P, Chen PR, and Liu Y (2021). SAR1B senses leucine levels to regulate mTORC1 signalling. Nature 596, 281–284. 10.1038/s41586-021-03768-w. [DOI] [PubMed] [Google Scholar]
- 15.Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, Condon KJ, Petri S, Kedir J, Scaria SM, et al. (2017). KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 543, 438–442. 10.1038/nature21423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng M, Yin N, and Li MO (2017). SZT2 dictates GATOR control of mTORC1 signalling. Nature 543, 433–437. 10.1038/nature21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu X, Orozco JM, Saxton RA, Condon KJ, Liu GY, Krawczyk PA, Scaria SM, Harper JW, Gygi SP, and Sabatini DM (2017). SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 358, 813–818. 10.1126/science.aao3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, Yuan ED, Jones TD, Chantranupong L, Comb W, et al. (2015). Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347, 188–194. 10.1126/science.1257132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larsen SC, Sylvestersen KB, Mund A, Lyon D, Mullari M, Madsen MV, Daniel JA, Jensen LJ, and Nielsen ML (2016). Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci. Signal 9, rs9. 10.1126/scisignal.aaf7329. [DOI] [PubMed] [Google Scholar]
- 20.Zhang F, Kerbl-Knapp J, Rodriguez Colman MJ, Meinitzer A, Macher T, Vujić N, Fasching S, Jany-Luig E, Korbelius M, Kuentzel KB, et al. (2021). Global analysis of protein arginine methylation. Cell Rep. Methods 1, 100016. 10.1016/j.crmeth.2021.100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bedford MT, and Clarke SG (2009). Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13. 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blanc RS, and Richard S (2017). Arginine methylation: the coming of age. Mol. Cell 65, 8–24. 10.1016/j.molcel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Branscombe TL, Frankel A, Lee JH, Cook JR, Yang Z, Pestka S, and Clarke S (2001). PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J. Biol. Chem 276, 32971–32976. 10.1074/jbc.M105412200. [DOI] [PubMed] [Google Scholar]
- 24.Yang Y, Hadjikyriacou A, Xia Z, Gayatri S, Kim D, Zurita-Lopez C, Kelly R, Guo A, Li W, Clarke SG, and Bedford MT (2015). PRMT9 is a type II methyltransferase that methylates the splicing factor SAP145. Nat. Commun 6, 6428. 10.1038/ncomms7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zurita-Lopez CI, Sandberg T, Kelly R, and Clarke SG (2012). Human protein arginine methyltransferase 7 (PRMT7) is a type III enzyme forming omega-NG-monomethylated arginine residues. J. Biol. Chem 287, 7859–7870. 10.1074/jbc.M111.336271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miranda TB, Miranda M, Frankel A, and Clarke S (2004). PRMT7 is a member of the protein arginine methyltransferase family with a distinct substrate specificity. J. Biol. Chem 279, 22902–22907. 10.1074/jbc.M312904200. [DOI] [PubMed] [Google Scholar]
- 27.Lee DY, Teyssier C, Strahl BD, and Stallcup MR (2005). Role of protein methylation in regulation of transcription. Endocr. Rev 26, 147–170. 10.1210/er.2004-0008. [DOI] [PubMed] [Google Scholar]
- 28.Auclair Y, and Richard S (2013). The role of arginine methylation in the DNA damage response. DNA Repair 12, 459–465. 10.1016/j.dnarep.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 29.Brobbey C, Liu L, Yin S, and Gan W (2022). The role of protein arginine methyltransferases in DNA damage response. Int. J. Mol. Sci 23, 9780. 10.3390/ijms23179780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu J, and Richard S (2021). Cellular pathways influenced by protein arginine methylation: implications for cancer. Mol. Cell 81, 4357–4368. 10.1016/j.molcel.2021.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siu LL, Rasco DW, Vinay SP, Romano PM, Menis J, Opdam FL, Heinhuis KM, Egger JL, Gorman S, Parasrampuria R, et al. (2019). METEOR-1: A phase I study of GSK3326595, a first-in-class protein arginine methyltransferase 5 (PRMT5) inhibitor, in advanced solid tumours. Ann. Oncol 30, v159. [Google Scholar]
- 32.Gao G, Zhang L, Villarreal OD, He W, Su D, Bedford E, Moh P, Shen J, Shi X, Bedford MT, and Xu H (2019). PRMT1 loss sensitizes cells to PRMT5 inhibition. Nucleic Acids Res 47, 5038–5048. 10.1093/nar/gkz200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fedoriw A, Rajapurkar SR, O’Brien S, Gerhart SV, Mitchell LH, Adams ND, Rioux N, Lingaraj T, Ribich SA, Pappalardi MB, et al. (2019). Anti-tumor activity of the type I PRMT inhibitor, GSK3368715, synergizes with PRMT5 inhibition through MTAP loss. Cancer Cell 36, 100–114.e25. 10.1016/j.ccell.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 34.Jarrold J, and Davies CC (2019). PRMTs and arginine methylation: cancer’s best-kept secret? Trends Mol. Med 25, 993–1009. 10.1016/j.molmed.2019.05.007. [DOI] [PubMed] [Google Scholar]
- 35.Lee J, Sayegh J, Daniel J, Clarke S, and Bedford MT (2005). PRMT8, a new membrane-bound tissue-specific member of the protein arginine methyltransferase family. J. Biol. Chem 280, 32890–32896. 10.1074/jbc.M506944200. [DOI] [PubMed] [Google Scholar]
- 36.Burnett PE, Barrow RK, Cohen NA, Snyder SH, and Sabatini DM (1998). RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 95, 1432–1437. 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brunn GJ, Hudson CC, Sekulić A, Williams JM, Hosoi H, Houghton PJ, Lawrence JC Jr., and Abraham RT (1997). Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 277, 99–101. 10.1126/science.277.5322.99. [DOI] [PubMed] [Google Scholar]
- 38.Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, and Sonenberg N (1999). Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev 13, 1422–1437. 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim J, Kundu M, Viollet B, and Guan KL (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol 13, 132–141. 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bissinger EM, Heinke R, Spannhoff A, Eberlin A, Metzger E, Cura V, Hassenboehler P, Cavarelli J, Schüle R, Bedford MT, et al. (2011). Acyl derivatives of p-aminosulfonamides and dapsone as new inhibitors of the arginine methyltransferase hPRMT1. Bioorg. Med. Chem 19, 3717–3731. 10.1016/j.bmc.2011.02.032. [DOI] [PubMed] [Google Scholar]
- 41.Yan L, Yan C, Qian K, Su H, Kofsky-Wofford SA, Lee WC, Zhao X, Ho MC, Ivanov I, and Zheng YG (2014). Diamidine compounds for selective inhibition of protein arginine methyltransferase 1. J. Med. Chem 57, 2611–2622. 10.1021/jm401884z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahara T, Amemiya Y, Sugiyama R, Maki M, and Shibata H (2020). Amino acid-dependent control of mTORC1 signaling: a variety of regulatory modes. J. Biomed. Sci 27, 87. 10.1186/s12929-020-00679-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goulet I, Gauvin G, Boisvenue S, and Côté J (2007). Alternative splicing yields protein arginine methyltransferase 1 isoforms with distinct activity, substrate specificity, and subcellular localization. J. Biol. Chem 282, 33009–33021. 10.1074/jbc.M704349200. [DOI] [PubMed] [Google Scholar]
- 44.Yoshimatsu M, Toyokawa G, Hayami S, Unoki M, Tsunoda T, Field HI, Kelly JD, Neal DE, Maehara Y, Ponder BAJ, et al. (2011). Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int. J. Cancer 128, 562–573. 10.1002/ijc.25366. [DOI] [PubMed] [Google Scholar]
- 45.Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, Clarke S, and Herschman HR (2000). PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem 275, 7723–7730. 10.1074/jbc.275.11.7723. [DOI] [PubMed] [Google Scholar]
- 46.Lin WJ, Gary JD, Yang MC, Clarke S, and Herschman HR (1996). The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J. Biol. Chem 271, 15034–15044. 10.1074/jbc.271.25.15034. [DOI] [PubMed] [Google Scholar]
- 47.Robin-Lespinasse Y, Sentis S, Kolytcheff C, Rostan MC, Corbo L, and Le Romancer M (2007). hCAF1, a new regulator of PRMT1-dependent arginine methylation. J. Cell Sci 120, 638–647. 10.1242/jcs.03357. [DOI] [PubMed] [Google Scholar]
- 48.Hartley AV, and Lu T (2020). Modulating the modulators: regulation of protein arginine methyltransferases by post-translational modifications. Drug Discov. Today 25, 1735–1743. 10.1016/j.drudis.2020.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rust HL, Subramanian V, West GM, Young DD, Schultz PG, and Thompson PR (2014). Using unnatural amino acid mutagenesis to probe the regulation of PRMT1. ACS Chem. Biol 9, 649–655. 10.1021/cb400859z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhuripanyo K, Wang Y, Liu X, Zhou L, Liu R, Duong D, Zhao B, Bi Y, Zhou H, Chen G, et al. (2018). Identifying the substrate proteins of U-box E3s E4B and CHIP by orthogonal ubiquitin transfer. Sci. Adv 4, e1701393. 10.1126/sciadv.1701393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Herrmann F, and Fackelmayer FO (2009). Nucleo-cytoplasmic shuttling of protein arginine methyltransferase 1 (PRMT1) requires enzymatic activity. Gene Cell 14, 309–317. 10.1111/j.1365-2443.200801266.x. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Person MD, and Bedford MT (2022). Pan-methylarginine antibody generation using PEG linked GAR motifs as antigens. Methods 200, 80–86. 10.1016/j.ymeth.2021.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang H, Huang ZQ, Xia L, Feng Q, Erdjument-Bromage H, Strahl BD, Briggs SD, Allis CD, Wong J, Tempst P, and Zhang Y (2001). Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science 293, 853–857. 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- 54.Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, and Sif S (2004). Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell Biol 24, 9630–9645. 10.1128/MCB.24.21.9630-9645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Serefidou M, Venkatasubramani AV, and Imhof A (2019). The impact of one carbon metabolism on histone methylation. Front. Genet 10, 764. 10.3389/fgene.2019.00764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jans DA, and Hübner S (1996). Regulation of protein transport to the nucleus: central role of phosphorylation. Physiol. Rev 76, 651–685. 10.1152/physrev.1996.76.3.651. [DOI] [PubMed] [Google Scholar]
- 57.Moll T, Tebb G, Surana U, Robitsch H, and Nasmyth K (1991). The role of phosphorylation and the CDC28 protein kinase in cell cycle-regulated nuclear import of the S. cerevisiae transcription factor SWI5. Cell 66, 743–758. 10.1016/0092-8674(91)90118-i. [DOI] [PubMed] [Google Scholar]
- 58.Sbia M, Parnell EJ, Yu Y, Olsen AE, Kretschmann KL, Voth WP, and Stillman DJ (2008). Regulation of the yeast Ace2 transcription factor during the cell cycle. J. Biol. Chem 283, 11135–11145. 10.1074/jbc.M800196200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, and Amon A (1998). The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell 2, 709–718. 10.1016/s1097-2765(00)80286-5. [DOI] [PubMed] [Google Scholar]
- 60.Sang Y, Li Y, Zhang Y, Alvarez AA, Yu B, Zhang W, Hu B, Cheng SY, and Feng H (2019). CDK5-dependent phosphorylation and nuclear translocation of TRIM59 promotes macroH2A1 ubiquitination and tumorigenicity. Nat. Commun 10, 4013. 10.1038/s41467-019-12001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Asada A, Saito T, and Hisanaga SI (2012). Phosphorylation of p35 and p39 by Cdk5 determines the subcellular location of the holokinase in a phosphorylation-site-specific manner. J. Cell Sci 125, 3421–3429. 10.1242/jcs.100503. [DOI] [PubMed] [Google Scholar]
- 62.Meyerson M, Enders GH, Wu CL, Su LK, Gorka C, Nelson C, Harlow E, and Tsai LH (1992). A family of human cdc2-related protein kinases. EMBO J 11, 2909–2917. 10.1002/j.1460-2075.1992.tb05360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dhavan R, and Tsai LH (2001). A decade of CDK5. Nat. Rev. Mol. Cell Biol 2, 749–759. 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- 64.Tomimatsu N, Mukherjee B, Catherine Hardebeck M, Ilcheva M, Vanessa Camacho C, Louise Harris J, Porteus M, Llorente B, Khanna KK, and Burma S (2014). Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat. Commun 5, 3561. 10.1038/ncomms4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee JH, Shamanna RA, Kulikowicz T, Borhan Fakouri N, Kim EW, Christiansen LS, Croteau DL, and Bohr VA (2021). CDK2 phosphorylation of Werner protein (WRN) contributes to WRN’s DNA double-strand break repair pathway choice. Aging Cell 20, e13484. 10.1111/acel.13484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu H, Shamanna RA, de Freitas JK, Okur M, Khadka P, Kulikowicz T, Holland PP, Tian J, Croteau DL, Davis AJ, and Bohr VA (2017). Cell cycle-dependent phosphorylation regulates RECQL4 pathway choice and ubiquitination in DNA double-strand break repair. Nat. Commun 8, 2039. 10.1038/s41467-017-02146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang H, Liu H, Chen Y, Yang X, Wang P, Liu T, Deng M, Qin B, Correia C, Lee S, et al. (2016). A cell cycle-dependent BRCA1-UHRF1 cascade regulates DNA double-strand break repair pathway choice. Nat. Commun 7, 10201. 10.1038/ncomms10201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Romero-Pozuelo J, Figlia G, Kaya O, Martin-Villalba A, and Teleman AA (2020). Cdk4 and Cdk6 couple the cell-cycle machinery to cell growth via mTORC1. Cell Rep 31, 107504. 10.1016/j.celrep.2020.03.068. [DOI] [PubMed] [Google Scholar]
- 69.Gatta AT, Olmos Y, Stoten CL, Chen Q, Rosenthal PB, and Carlton JG (2021). CDK1 controls CHMP7-dependent nuclear envelope reformation. Elife 10, e59999. 10.7554/eLife.59999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gassaway BM, Cardone RL, Padyana AK, Petersen MC, Judd ET, Hayes S, Tong S, Barber KW, Apostolidi M, Abulizi A, et al. (2019). Distinct hepatic PKA and CDK signaling pathways control activity-independent pyruvate kinase phosphorylation and hepatic glucose production. Cell Rep 29, 3394–3404.e9. 10.1016/j.celrep.2019.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, Chim H, Lim JH, Ruan HB, Yang X, et al. (2014). Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature 510, 547–551. 10.1038/nature13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nikolic M, Dudek H, Kwon YT, Ramos YF, and Tsai LH (1996). The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev 10, 816–825. 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- 73.Hutten S, and Kehlenbach RH (2007). CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol 17, 193–201. 10.1016/j.tcb.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 74.Xu W, Chen H, Du K, Asahara H, Tini M, Emerson BM, Montminy M, and Evans RM (2001). A transcriptional switch mediated by cofactor methylation. Science 294, 2507–2511. 10.1126/science.1065961. [DOI] [PubMed] [Google Scholar]
- 75.Mostaqul Huq MD, Gupta P, Tsai NP, White R, Parker MG, and Wei LN (2006). Suppression of receptor interacting protein 140 repressive activity by protein arginine methylation. EMBO J 25, 5094–5104. 10.1038/sj.emboj.7601389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Côté J, and Richard S (2005). Tudor domains bind symmetrical dime-thylated arginines. J. Biol. Chem 280, 28476–28483. 10.1074/jbc.M414328200. [DOI] [PubMed] [Google Scholar]
- 77.Bedford MT, Frankel A, Yaffe MB, Clarke S, Leder P, and Richard S (2000). Arginine methylation inhibits the binding of proline-rich ligands to Src homology 3, but not WW, domains. J. Biol. Chem 275, 16030–16036. 10.1074/jbc.M909368199. [DOI] [PubMed] [Google Scholar]
- 78.Bedford MT, and Richard S (2005). Arginine methylation an emerging regulator of protein function. Mol. Cell 18, 263–272. 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 79.Shen K, Huang RK, Brignole EJ, Condon KJ, Valenstein ML, Chantranupong L, Bomaliyamu A, Choe A, Hong C, Yu Z, and Sabatini DM (2018). Architecture of the human GATOR1 and GATOR1-Rag GTPases complexes. Nature 556, 64–69. 10.1038/nature26158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wei Z, Liu X, Cheng C, Yu W, and Yi P (2020). Metabolism of amino acids in cancer. Front. Cell Dev. Biol 8, 603837. 10.3389/fcell.2020.603837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hou Y, Hu S, Li X, He W, and Wu G (2020). Amino acid metabolism in the liver: nutritional and physiological significance. Adv. Exp. Med. Biol 1265, 21–37. 10.1007/978-3-030-45328-2_2. [DOI] [PubMed] [Google Scholar]
- 82.Eram MS, Shen Y, Szewczyk M, Wu H, Senisterra G, Li F, Butler KV, Kaniskan HÜ, Speed BA, Dela Seña C, et al. (2016). A potent, selective, and cell-active inhibitor of human type I protein arginine methyltransferases. ACS Chem. Biol 11, 772–781. 10.1021/acschembio.5b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Okada N, Ishigami Y, Suzuki T, Kaneko A, Yasui K, Fukutomi R, and Isemura M (2008). Importins and exportins in cellular differentiation. J. Cell Mol. Med 12, 1863–1871. 10.1111/j.1582-4934.2008.00437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gardino AK, Smerdon SJ, and Yaffe MB (2006). Structural determinants of 14-3-3 binding specificities and regulation of subcellular localization of 14-3-3-ligand complexes: a comparison of the X-ray crystal structures of all human 14-3-3 isoforms. Semin. Cancer Biol 16, 173–182. 10.1016/j.semcancer.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 85.Albrecht LV, Bui MH, and De Robertis EM (2019). Canonical Wnt is inhibited by targeting one-carbon metabolism through methotrexate or methionine deprivation. Proc. Natl. Acad. Sci. USA 116, 2987–2995. 10.1073/pnas.1820161116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Albrecht LV, Ploper D, Tejeda-Muñoz N, and De Robertis EM (2018). Arginine methylation is required for canonical Wnt signaling and endolysosomal trafficking. Proc. Natl. Acad. Sci. USA 115, E5317–E5325. 10.1073/pnas.1804091115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tejeda-Muñoz N, Albrecht LV, Bui MH, and De Robertis EM (2019). Wnt canonical pathway activates macropinocytosis and lysosomal degradation of extracellular proteins. Proc. Natl. Acad. Sci. USA 116, 10402–10411. 10.1073/pnas.1903506116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Musiani D, Giambruno R, Massignani E, Ippolito MR, Maniaci M, Jammula S, Manganaro D, Cuomo A, Nicosia L, Pasini D, and Bonaldi T (2020). PRMT1 is recruited via DNA-PK to chromatin where it sustains the senescence-associated secretory phenotype in response to cisplatin. Cell Rep 30, 1208–1222.e9. 10.1016/j.celrep.2019.12.061. [DOI] [PubMed] [Google Scholar]
- 89.Bao X, Siprashvili Z, Zarnegar BJ, Shenoy RM, Rios EJ, Nady N, Qu K, Mah A, Webster DE, Rubin AJ, et al. (2017). CSNK1a1 regulates PRMT1 to maintain the progenitor state in self-renewing somatic tissue. Dev. Cell 43, 227–239.e5. 10.1016/j.devcel.2017.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hirata Y, Katagiri K, Nagaoka K, Morishita T, Kudoh Y, Hatta T, Naguro I, Kano K, Udagawa T, Natsume T, et al. (2017). TRIM48 promotes ASK1 activation and cell death through ubiquitination-dependent degradation of the ASK1-negative regulator PRMT1. Cell Rep 21, 2447–2457. 10.1016/j.celrep.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 91.Sanchez-Bailon MP, Choi SY, Dufficy ER, Sharma K, McNee GS, Gunnell E, Chiang K, Sahay D, Maslen S, Stewart GS, et al. (2021). Arginine methylation and ubiquitylation crosstalk controls DNA end-resection and homologous recombination repair. Nat. Commun 12, 6313. 10.1038/s41467-021-26413-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tewary SK, Zheng YG, and Ho MC (2019). Protein arginine methyltransferases: insights into the enzyme structure and mechanism at the atomic level. Cell. Mol. Life Sci 76, 2917–2932. 10.1007/s00018-019-03145-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang X, and Cheng X (2003). Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure 11, 509–520. 10.1016/s0969-2126(03)00071-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hu H, Luo C, and Zheng YG (2016). Transient kinetics define a complete kinetic model for protein arginine methyltransferase 1. J. Biol. Chem 291, 26722–26738. 10.1074/jbc.M116.757625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ericksen RE, Lim SL, McDonnell E, Shuen WH, Vadiveloo M, White PJ, Ding Z, Kwok R, Lee P, Radda GK, et al. (2019). Loss of BCAA catabolism during carcinogenesis enhances mTORC1 activity and promotes tumor development and progression. Cell Metab 29, 1151–1165.e6. 10.1016/j.cmet.2018.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yin S, Liu L, Brobbey C, Palanisamy V, Ball LE, Olsen SK, Ostrowski MC, and Gan W (2021). PRMT5-mediated arginine methylation activates AKT kinase to govern tumorigenesis. Nat. Commun 12, 3444. 10.1038/s41467-021-23833-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.