

Abstract
Objectives:
We hypothesized that polypathology is more severe in older than younger mice during the acute phase following repetitive mild traumatic brain injury (r-mTBI).
Methods:
Young and aged male and female mice transgenic for human tau (hTau) were exposed to r-mTBI or a sham procedure. Twenty-four hours post-last injury, mouse brain tissue was immunostained for alterations in astrogliosis, microgliosis, tau pathology, and axonal injury.
Results:
Quantitative analysis revealed a greater percent distribution of glial fibrillary acid protein and Iba-1 reactivity in the brains of all mice exposed to r-mTBI compared to sham controls. With respect to axonal injury, the number of amyloid precursor protein-positive profiles was increased in young vs aged mice post r-mTBI. An increase in tau immunoreactivity was found in young and aged injured male hTau mice.
Conclusions:
We report the first evidence in our model that r-mTBI precipitates a complex sequelae of events in aged vs young hTau mice at an acute time point, typified by an increase in phosphorylated tau and astroglisosis, and a diminished microgliosis response and axonal injury in aged mice. These findings suggest differential age-dependent effects in TBI pathobiology.
Keywords: Traumatic Brain Injury, neurodegeneration, tau, axonal injury, animal models, age, inflammation
Introduction
Traumatic brain injury (TBI) is the leading cause of mortality and morbidity in the world for individuals under the age of 45 (1). Mild TBI (mTBI) accounts for approximately 70–90% of all TBIs and is a major source of morbidity, with up to 15% of patients experiencing long-term symptoms (2–5). Since epidemiological data do not account for the substantial number of individuals who do not seek hospital treatment post-injury and the lack of a clinical consensus on diagnostic criteria for mTBI remains, the prevalence of mTBI and consequences on health are likely underestimated from the limited prospective studies conducted to date.
Studies on the epidemiology of mTBI have shown that age is a major factor influencing the clinicopathological outcomes following exposure to mTBI, with a bimodal distribution between young adults (13–20 years old) and older adults (>65 years old) recovering differently from injuries of a similar severity (6–8). However, a recent review on the chronic consequences of mild and moderate/severe TBI (9) reported that the association between mTBI and the long-term mortality (at least 5 years after mTBI) depended largely on the sample population. The role that age plays in the pathological response to mTBI remains controversial (10–12). Disparity in these findings may be related to environmental factors such as drug abuse, level of education, rehabilitation length and familial support (13).
Nevertheless, it has been accepted that age at injury also has a significant influence on dementia risk in patients >65 years of age following exposure to mTBI (14). Characterization of autopsy-acquired tissue from long-term survivors of repetitive mTBI reveals a complex neuropathology, best described as a ‘polypathology’, including abnormal tau and amyloid protein aggregation, neuroinflammation, white matter degradation and axonal degeneration (15). This pathology is masked by effect of normal ageing and may augment or accelerate pre-existing age-related pathologies. Therefore, it is important to develop a greater understanding of the age-dependent pathophysiological process following mTBI, to improve diagnostic and therapeutic interventions and recognize age-related risk factors for patients.
The present study investigated the ‘polypathology’ associated with repetitive mTBI in 3- and 12-month-old mice at an acute time point post-injury (24 h). To our knowledge, only a few pre-clinical studies have investigated the influence of age and injury mechanism after TBI (16–19). Therefore, our study aims to address the impact of TBI on acute neuropathology in the young adult vs aged brain. We hypothesized that polypathology is more severe in older mice than younger mice during the acute phase following repetitive mild traumatic brain injury (r-mTBI). Our second objective is to determine whether sex differences in the animals play a role in their recovery. The focus on pathogenic tau pathology has been well documented chronically after exposures to repetitive mTBI in postmortem human brain tissue of patients (20,21); however, the origin of this tau pathology and relationship to the inciting injury are undetermined. Therefore, we have chosen to assess the mouse brain tissue at early time points (24 h) following repetitive injuries. We utilized hTau mice that have been genetically modified to express all six isoforms of non-mutant human Microtubule-Associated Protein Tau (MAPT) in a murine Mapt knockout background. These mice start to express membranous tau redistribution at 3 months of age and present tau hyperphosphorylation and aggregation by 12 months of age (22–24). Here, we explore the influence of age at injury (young [3 months] and aged [12 months]) on acute neuropathological sequelae in hTau mice.
Materials and methods
Animals
Young (3 months old) and aged (12–13 months old) male and female mice, expressing all six isoforms of human tau (hTau) on a C57BL/6 and null murine tau background (Jackson Laboratories, Bar Harbor, ME), were housed singly under standard laboratory conditions (23°C ± 1°C, 50 ± 5% humidity, and 12-h light/dark cycle) with free access to food and water. All procedures were carried out under Institutional Animal Care and Use Committee approval and in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Injury groups and schedule
Forty-eight young and fifty-two aged hTau mice were randomly assigned to TBI or sham conditions. Unless otherwise noted, histochemical studies were performed with: young male TBI (n = 6), young female TBI (n = 8), young male sham (n = 6), young female sham (n = 7), aged male TBI (n = 11), aged female TBI (n = 8), aged male sham (n = 11), and aged female sham (n = 10). The rest of the brains were stored for additional future analyses. Mice assigned to r-mTBI conditions received five injuries over 9 days, with an inter-injury interval of 48 h. Sham (r-sham) animals were anaesthetized with the same frequency and exposure time as their r-mTBI counterparts, but without injury. As described in our original publication on this model (25), the inter-injury interval was chosen to accommodate repeated injuries occurring within an asymptomatic window of vulnerability from the previous injury that had been described in a rat model (26). We have extensively characterized this model in wild-type mice (25,27,28) and therefore have knowledge of the outcomes observed that can be used as point of reference.
Injury protocol
Mice were subjected to closed head mTBI as previously described (25). Prior to mTBI all mice were anaesthetized with 1.5 L/min of oxygen and 3% isoflurane, the top of their heads were shaved, and they were transferred to a stereotaxic frame (Just For Mice™ Stereotaxic, Stoelting, Wood Dale, IL) placed on a heating pad to maintain body temperature at 37°C and maintained under anaesthesia through a nose cone. A 5mm blunt metal impactor tip was retracted and positioned midway relative to the sagittal suture before each impact. Injury was triggered using the myNeuroLab controller at a strike velocity of 5 m/s, strike depth of 1.0 mm, and dwell time of 200 ms over the shaved area of the head. To be considered as a concussive injury, our injury paradigm should follow the following criteria: no skull fractures, hematomas or other gross signs of injury, and the presence of amyloid precursor protein (APP) immunoreactivity profiles in the corpus calossum as a sign of traumatic axonal injury. No mortality was observed with these mice during these experiments. At the end of the procedure, each animal was removed from the stereotaxic table, allowed to recover in its home cage resting on a heating pad until the animal was ambulatory. To control for the effects of repeated anaesthesia, sham animals underwent the same procedures and were exposed to anaesthesia for the same length of time as the mTBI animals, but were not exposed to head trauma.
Histology
All mice were euthanized 24 h after the last mTBI/sham injury by anaesthetization with isoflurane, followed by transcardial perfusion with heparinized Phosphate Buffered Saline (PBS) (pH 7.4) and PBS containing 4% paraformaldehyde. After perfusion, brains were post-fixed in 4% paraformaldehyde (4°C) for 48 h, embedded in paraffin using Tissue-Tek VIP (Sakura, Torrance, CA, USA), cut at 6 μm on a 2030 Biocut microtome (Reichert/Leica, Germany), and mounted on positively charged glass slides (Fisher, Superfrost Plus). Sections were deparaffinized in xylene and rehydrated in an ethanol-to-water gradient. Slides were analysed using a bright field microscope (BX60, Leica, Germany) and digital images were visualized and acquired using a MagnaFire SP camera (Olympus, Tokyo, Japan). Sets of adjacent sections were stained for glial fibrillary acid protein (GFAP, 1:20 000; Dako, Glostrup, Denmark, ZO334), ionized calcium binding adaptor molecule 1 (Iba1. 1:5000; Abcam, Cambridge, MA, ab5076), or amyloid precursor protein (APP, 1:20 000; Millipore, Billerica, MA, MAB348). Tau immunohistochemistry was performed using the following monoclonal antibodies at a 1:500 dilution: CP13 [pS202], PHF1 [pS396/404], and RZ3 [pThr231]. CP13, PHF1, and RZ3 were generously provided by Dr Peter Davies, The Feinstein Institute for Medical Research, Bronx, NY. As a negative control, for each antibody, a single section was processed for immunostaining without the inclusion of the primary antibody. Tissue sections were subjected to antigen retrieval with either heated trisethylene-diaminetetraacetic acid (EDTA) buffer (pH-8.0) or citrate buffer (pH-6.0) under pressure for 7 min. Endogenous peroxidase activity was quenched with a 15 min H2O2 treatment (3% in water). Each section was rinsed and incubated with the appropriate blocking buffer (ABC Elite kit, MOM kit, Vector Laboratories, CA) for 20 min, before applying the appropriate primary antibody overnight at 4°C. Then, the diluted biotinylated secondary antibody from the ABC Elite Kit was applied. Antibodies were detected using the avidin-peroxidase complex, after incubation with the chromogen 3,3-diaminobenzidine (DAB) peroxidase solution (0.05% DAB - 0.015% H2O2 in 0.01M PBS, pH 7.2) for 6–7 min and counterstained with hematoxylin. Immunofluorescence was performed with an antibody for p-tau RZ3 (1:500). Prior to immunostaining, samples were deparaffinized in xylene and rehydrated through a gradient of ethanol solutions of decreasing concentrations (2 × 100%, 95%, 70%). Antigen retrieval consisted of heating slides in a citrate solution (pH 6.0) under pressure, washing with PBS, and transferring into a Sudan black solution (EMD Millipore, MA) (15 min) to inhibit autofluorescence. Before primary antibody treatment, slides were blocked for 1 h with 10% donkey serum. The primary antibody for RZ3 was applied on the slides and left overnight at 4°C. The next day, donkey anti-Mouse IgG secondary antibody Alexa Fluor 488 was applied for RZ3. Slides were mounted with ProLong Gold Antifade DAPI Mount.
Quantitative immunohistochemistry
Mice from both age groups (n = 4) were euthanized 24 h post-injury, and sagittal sections were immunostained and then analysed by an observer blinded to experimental conditions using ImageJ software (US National Institutes of Health, Bethesda, MD). Images were separated into individual colour channels (red hematoxylin counter stain and DAB brown chromogen) using the colour deconvolution algorithm (29). Three non-overlapping areas of 100 μm (2) from each of two sagittal sections in the corpus callosum (CC) were randomly selected within which the area of GFAP or Iba-1 immunoreactivity was calculated and expressed as a percentage of the field of view as previously reported. The numbers of APP-positive profiles were manually counted in three non-overlapping areas of 100 μm (2) within the CC. The immunohistochemical outcomes were expressed as percent area of GFAP, Iba-1, and RZ3. Variables of interest included sex, injury group (mTBI vs sham), age (young vs aged), and their interactions. Descriptive statistics, including means and standard errors, were calculated from the percent area of GFAP and Iba-1 measurements for each age, injury group, and sex. Average percent areas were calculated within an animal (across sections), prior to calculating age, injury group, and sex averages and standard errors. Descriptive statistics, including medians and 25th and 75th percentiles, were calculated from the number of APP-positive profiles for each age, injury group, and sex. The raw percent area data were assessed for normality using the Shapiro–Wilk test as well as four alternative transformations (square root, base-10 logarithm, logit, and arcsine square root). The transformation that most closely approached normality was used for all subsequent analysis. The GFAP and Iba-1 data were analysed using a mixed ANOVA model with age, sex, and injury group, and their interactions, as explanatory variables. In addition to these model terms, a random variance component for mouse was included such that multiple observations on the same mouse were weighted together and not individually. This model was used to estimate the size and significance of the difference in percent area between ages (overall and within injury group and gender), injury groups (overall and within age and gender), and between genders (overall and within age and injury group).
For the APP data, no APP-positive profiles were observed in any of the Sham animals. The APP data were analysed using a mixed Poisson regression model with age, sex, and their interaction as explanatory variables. In addition to these model terms, a random variance component for mouse was included such that multiple observations on the same mouse were weighted together and not individually. This model was used to estimate the size and significance of the difference in the number of APP-positive profiles between ages (overall and within gender) and between genders (overall and within age). All statistical analyses were performed using SAS (ver. 9.4) and all results are reported using the 0.05 level of significance.
Results
Repetitive mTBI induces a stronger astrogliosis response in the CC in aged mice
For all groups, the entire CC (splenium, body, and genu) was assessed in GFAP-stained sections. Quantitative analysis revealed TBI-dependent differences in GFAP immunopositivity in the body of the CC among females and males and in the young and aged cohorts (Figure 1(b, d, f, h); p < 0.001). Interactions between age and injury group, age and gender, injury group and gender, and the three-way interaction between age, injury group, and gender were not significant. The quantification of GFAP immunostaining is summarized in Table 1.
Figure 1.

Repetitive mTBI increases astrogliosis in the corpus callosum at 24 hours post-injury. Black box (i) indicates the area of interest shown at higher magnification (a–h) in a sagittal section of a mouse brain. Sagittal sections of the mouse brain approximately 0.2 mm lateral to midline in the body of the CC with GFAP stained in male (a-d) and female (e-h). Graphs showing that the average percent area for the r-mTBI group was greater compared to the sham control group for females (averaged over age, p < 0.001), males (averaged over age, p < 0.001), aged (averaged over sex, p < 0.001), young (averaged over sex, p < 0.001), and overall (averaged over sex and age, p < 0.001) (j, k). Tissue sections were counterstained with hematoxylin; each symbol represents 1 mouse. Scale bar, 1.4mm and 100 μm, respectively. Blue symbols, F-yrsham: Female young repetitive sham; Black symbols, M-yr-sham: Male young repetitive sham; Green symbols, F-yr-mTBI: Female young repetitive mTBI; Red symbols, M-yr-mTBI: Male young repetitive mTBI.
Table 1.
Summary of GFAP quantification.
| Effect | Effect p-value | Variable comparison | Model estimated average percent area | Comparison p-value | |
|---|---|---|---|---|---|
| Injury (mTBI vs sham) | <0.001* | mTBI | Sham | ||
| Within young | 6.93 | 2.27 | <0.001* | ||
| Within aged | 9.20 | 3.52 | <0.001* | ||
| Within females | 7.99 | 3.02 | <0.001* | ||
| Within males | 8.07 | 2.71 | <0.001* | ||
| Age (young vs aged) | <0.001* | Young | Aged | ||
| Within mTBI | 6.93 | 9.20 | 0.034* | ||
| Within sham | 2.27 | 3.52 | 0.051 | ||
| Within females | 3.89 | 6.72 | 0.001* | ||
| Within males | 4.70 | 5.37 | 0.427 | ||
Significant at the alpha = 0.05 level of significance.
Repetitive mTBI induces a stronger microgliosis response in the CC in young mice
To gain insight into whether age influences the degree of inflammation following mTBI, we investigated Iba-1, a marker of microglia in young and aged animals. Microglial cell structures were similar across comparable groups displaying a primed morphology characteristic of an aged mouse brain in the 12 months cohort (with a more inflammatory microglia phenotype, e.g., increased major histocompatibility complex II [MHCII], IL-1β, CD68, complement receptor [CR]3) (30). Similar to the GFAP analysis, the entire CC was assessed for Iba-1 immunoreactivity. There were TBI-dependent quantitative difference in the level of Iba-1 immunopositivity detected in the body of the CC among females and males and in the young and aged cohorts (Figure 2(b, d, f, h); p < 0.001). The interaction between age and injury group was significant, but the interactions between age and gender, injury group and gender, and the three-way interaction between age, injury group, and gender were not significant. The quantification of Iba-1 immunostaining is summarized in Table 2.
Figure 2.
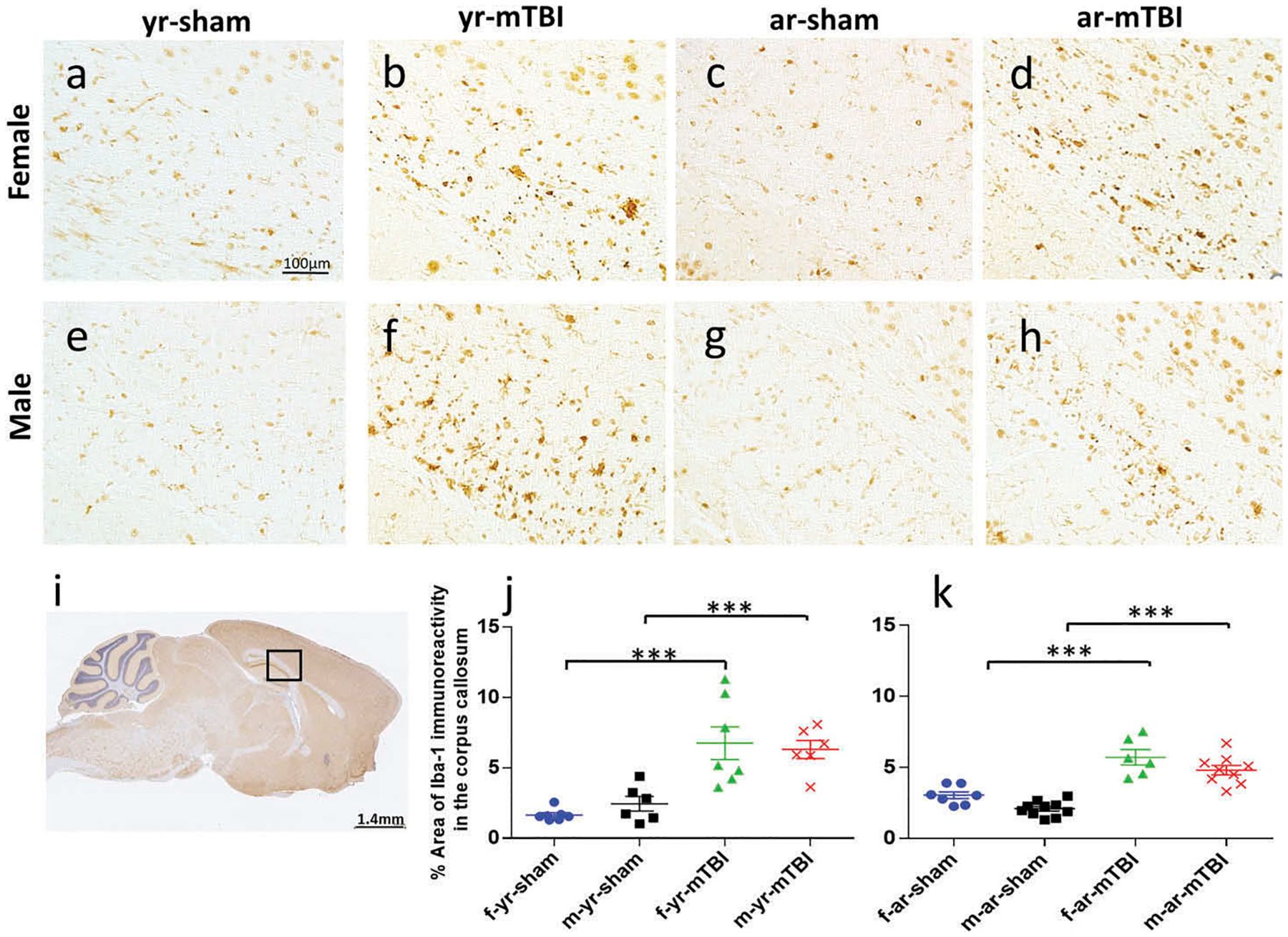
Corpus callosum Iba1 immunohistochemical anaylsis. Sagittal sections of the corpus callosum (± 0.4 mm lateral to midline) in female (a–d) and male (e–h) mice. Black box (i) indicates the area of interest shown at higher magnification (a–h) in a sagittal section of a mouse brain. There was no microglial activation in the sham groups (a, c, e, g). An increased area of anti-Iba1 immunoreactivity was observed in the corpus callosum at 24h post r-mTBI in young and aged animals (b, d, f, h).
Table 2.
Summary of Iba-1 quantification.
| Effect | Effect p-value | Variable comparison | Model estimated average percent area | Comparison p-value | |
|---|---|---|---|---|---|
| Injury (mTBI vs Sham) | <0.001* | mTBI | Sham | ||
| Within young | 6.27 | 1.73 | <0.001* | ||
| Within aged | 5.03 | 2.42 | <0.001* | ||
| Within females | 5.85 | 2.15 | <0.001* | ||
| Within males | 5.42 | 1.97 | <0.001* | ||
| Age (young vs aged) | 0.913 | Young | Aged | ||
| Within mTBI | 6.27 | 5.03 | 0.023* | ||
| Within sham | 1.73 | 2.42 | 0.034* | ||
| Within females | 3.55 | 4.00 | 0.327 | ||
| Within males | 3.74 | 3.24 | 0.242 | ||
Significant at the alpha = 0.05 level of significance.
APP immunoreactivity is reduced following r-mTBI in aged compared to young mice
APP-immunoreactive axonal profiles, a marker of axonal injury, were observed 24 h post-injury in the CC (Figure 3(i)) of both young and aged r-mTBI groups (Figure 3(b, d, f, h)) but not in controls (Figure 3(a, c, e, g)). These APP immunoreactive axonal profiles were observed as small, granular immunoreactive profiles within the CC (Figure 3(b, d, f, h)). The difference in the number of APP-immunoreactive profiles observed was greatest in the young r-mTBI vs aged r-mTBI comparison (Figure 3(j, k); p < 0.001), with no gender effects detected (p > 0.05). The quantification of APP immunostaining is summarized in Table 3.
Figure 3.
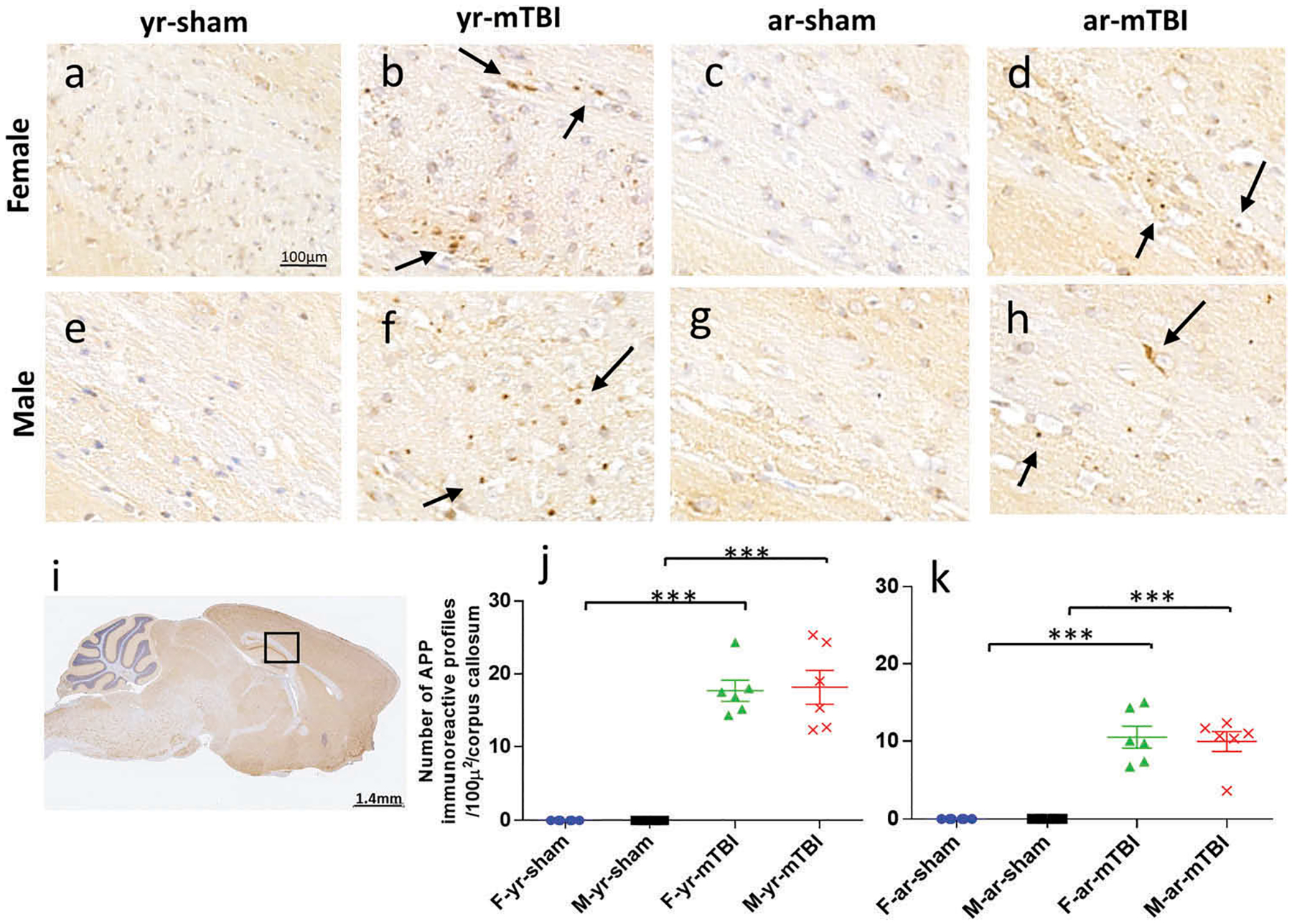
Amyloid Precursor protein (APP) immunohistochemistry of sagittal sections of the mouse brain at ± 0.4 mm lateral to midline in the corpus callosum in female (a–d) and male (e–h). Black box (i) indicates the area of interest shown at higher magnification (a–h). Young and aged sham tissue (a, c, e, g) was negative for APP immunostaining. Corpus callosum immunoreactive fragments appeared as discrete axonal profiles in all injured animals (b, d, f, h). The number of APP-positive profiles was greater in young compared to aged mice among females (p = 0.010) and males (p < 0.001) and overall (averaged over sex, p < 0.001) (j, k).
Table 3.
Summary of APP quantification.
| Effect | Effect p-value | Variable comparison | Model estimated average number of APP-positive profiles | Comparison p-value | |
|---|---|---|---|---|---|
| Age (young vs aged) | <0.001* | Young | Aged | ||
| Within females | 15 | 10 | 0.010* | ||
| Within males | 18 | 10 | <0.001* | ||
| Gender (female vs male) | 0.723 | Female | Male | ||
| Within young | 15 | 18 | 0.364 | ||
| With age | 10 | 10 | 0.745 | ||
Significant at the alpha = 0.05 level of significance.
Repetitive mTBI induces an elevation of hippocampal RZ3 p-tau 24 h post-injury
To investigate the effect of TBI on tau in our model, we performed a quantitative immunohistochemical analysis of RZ3, CP13, and PHF1 (an antibody that recognizes early and late tau pathology). The average percent area of RZ3 immunoreactivity in the hippocampal pyramidal layer (Figure 4(a, c, e, g, i, j)) was significantly increased in the mTBI group compared to the sham control group among male (averaged over age, p < 0.001), aged (averaged over gender, p = 0.002), young (averaged over gender, p = 0.015), and overall (averaged over gender and age, p < 0.001) mice. Additionally, the average percent area was reduced in young vs aged male mice (averaged over injury group, p = 0.026), within the mTBI group (averaged over gender, p = 0.034), and overall (averaged over gender and injury group, p = 0.009). The average percent area was also reduced in females vs males among aged mice (averaged over injury group, p = 0.038) and for the mTBI group (averaged over age, p = 0.002). There was no overall gender effect. The interactions between age and injury group, age and gender, and the three-way interaction between age, injury group, and gender were not significant, but the interaction between injury group and gender was significant (p = 0.008). Immunohistochemical assessment of soluble phosphorylated tau pSer-202 (CP13) was similar to RZ3 and none of the brains showed neurons positive for PHF1 (data not shown). The quantification of RZ3 immunostaining is summarized in Table 4.
Figure 4.

Immunohistochemical assessment of soluble phosphorylated tau pThr231 (RZ3) at approximately 0.5 mm lateral to midline in the CA1 region of the hippocampus (a, c, e, g) and in the neocortex (b, d, f, h) at 24h post injury in young female and male mice. Qualitatively, the r-mTBI group showed greater dendritic and membranous staining (green fluorescent signal) in both the CA1 and cortical neurons (arrows in Figure 4f, h) compared to their respective shams (a-d). An increase of RZ3 immunoreactivity was also observed in the hippocampus region of the injured animals for both genders and in the young and aged animals (Figure 4i, j). The red box indicates the region of interest, which is shown at a higher magnification. The average percent area of RZ3 immunoreactivity was increased in the mTBI group compared to the control group among male mice (averaged over age, p=0.001; Figure 4k, l). Additionally, the average percent area was significantly reduced in young vs. aged mice among male mice (averaged over injury group, p=0.0264; Figure 4k, l) and the mTBI injury group (averaged over gender, p=0.034; k, l). The average percent area was also observed to be significantly reduced in females vs. males among aged mice (averaged over injury group, p=0.038; k,l).
Table 4.
Summary of RZ3 quantification.
| Effect | Effect p-value | Variable comparison | Model estimated average percent area | Comparison p-value | |
|---|---|---|---|---|---|
| Injury (mTBI vs sham) | 0.0003* | mTBI | Sham | ||
| Within young | 10.30 | 3.16 | 0.0153* | ||
| Within aged | 17.35 | 7.69 | 0.0022* | ||
| Within females | 8.84 | 5.53 | 0.1800 | ||
| Within males | 19.38 | 4.83 | 0.0002* | ||
| Averaged over gender and age | 13.60 | 5.17 | 0.0003* | ||
| Age (young vs aged) | 0.0088* | Young | Aged | ||
| Within mTBI | 10.30 | 17.35 | 0.0339* | ||
| Within sham | 3.16 | 7.69 | 0.0836 | ||
| Within females | 5.36 | 9.06 | 0.1348 | ||
| Within males | 7.13 | 15.43 | 0.0264* | ||
| Averaged over injury and gender | 6.21 | 12.04 | 0.0088* | ||
Discussion
In the current study, we have examined the acute pathological outcome (24 h post-last injury) of r-mTBI in the brains of young and aged hTau mice. Our data support a TBI-dependent difference between young and aged animals, with increased astrogliosis and tau pathology in older animals, whereas an opposite pattern was observed for microgliosis and axonal degeneration. In addition, as we and others have previously reported (17,27,31), we observed age-dependent changes in astrogliosis, microgliosis, and axonal injury within the CC, an area of the white matter of the brain known to be particularly vulnerable to repetitive brain injuries in our model. This study also revealed a possible sex-dependent link between age at injury and a subsequent acute increase in phosphorylated tau species observed in pre-tangle neurons in both the hippocampus and cortex.
We previously identified an increase in PHF1 positive hyperphosphorylated tau in male TBI mice compared to females using a similar injury model at 15 days post-injury (17); herein, we now demonstrate that sex-dependent differences in p-tau pathology appear as early as 24 h post-last injury. The present study revealed an increase in RZ3 phosphorylated tau in the hippocampus without any appreciable increase in PHF1 levels, supporting time-dependent changes for different p-tau epitopes specific for pre-tangle structures. While p-tau protein is a key component of the pathology seen in neurodegenerative tauopathies (32–34), it also plays an important role in neuroplasticity, including dendritic/synaptic remodelling observed in the brain in response to environmental challenges, such as TBI (35,36) and hypothermia/hibernation (37,38). Therefore, the physiological changes reported in the brains of these mice may be the emergence of an insidious pathological process; however, they may also be part of an attempt by the brain to repair the structural damage caused by repeated head trauma. Regardless of the biological repercussions, our observations at 24 h, in addition to our previous work at 15 days post-injury, indicate that the levels of RZ3 and PHF1 phosphorylated tau are both increased in male mice, while no significant changes were observed in female mice at 24 h or 15 days post-injury. We cannot, however, rule out that these observations are unique to hTau mice, because no clinical studies to date have addressed the role of sex on tau pathology after TBI. It is worth noting that because it has been previously reported that male PS19 mice (mutant tau) develop tau pathology more consistently than females, almost all pre-clinical studies exclusively use male animals to reduce the variability of tau pathology (39). Similar to the conditions in pre-clinical models, the autopsied brain samples used for many of the current clinical histopathological reports of tauopathy following TBI are almost exclusively male in origin; thus, the speculation as to how sex influences the outcome on tau pathology has yet to be determined. Further work is necessary to address the gender difference in tau pathology in both, animals and most especially, in clinical studies.
Our study has several limitations. The first limitation is that the hTau mouse line has shown that naïve animals start to express signs of early tau pathology at 3 months of age and neurofibrillary tangles at 9 months of age (23,24). Given the increasing evidence that a disruption in the normal phosphorylation state of tau plays a key role in the pathogenic events that occur in other neurodegenerative conditions (40), our results may not reflect the pathology that would have been observed in wild-type animals. The second limitation of this study lies in its design as it cannot be determined whether the pathology observed is due to the first or last mTBI. However, the results observed from our previous work in wild-type animals suggest that our five injury paradigm exacerbates the pathology that would have been observed after a single injury (25). Another limitation is that even young mice may have early tau pathology and therefore could affect normal TBI pathology. Because we published and demonstrated a lack of injury effect of tau pathology in wild-type mice, we decided to use the hTau mice which expressed all six hTau isoforms and demonstrate age-related changes in tau pathology as observed in normal ageing. It is well established that age-related tauopathy is a normal feature of ageing (see progressive age-related tauopathy (41) and hyperphosphorylated tau in young and middle-aged subjects (42)). Therefore, using this model despite the progressive age-related increase in phospho-tau pathology between 3 months (approx. 14–21 years – humans) and 12 months (approx. 30–39 years – humans) of age, we consider to be related to the pattern of normal tau pathology observed with humans over time and in individuals exposed to injuries at these age groups. Finally, further studies with the inclusion of additional post-injury time points throughout the lifespan of the animal are required to understand how tau interacts with the polypathology resulting from the cumulative effects of repetitive mTBI. Multiple lines of evidence in pre-clinical (27,28,35) and clinical work (9) suggest that TBI is a chronic, evolving, and perhaps lifelong disorder. Such cohorts could serve as a platform and aid in the design and implementation of clinical trials of new therapies considering the different types of pathological markers present at acute and chronic time points post-injury. For example, exploring the long-term efficacy of different treatment regimens aimed at reducing potentially pathogenic tau species such as the use of an antibody against cis phospho tau conformations (43,44) or sodium selenate (45). Given the prominent changes in glial cells, a second possible treatment at chronic time points could target post-traumatic neuroinflammation by minimizing the detrimental neurotoxic effects and creating the optimal condition for regeneration.
Despite a growing body of clinical evidence suggesting that r-mTBI is an important risk factor for neurodegenerative diseases (14,15,21,46), the causal link and the role of tau as a common pathology remain unclear. Moreover, how the aged brain responds to repetitive mTBI compared to a younger brain remains unknown. Nonetheless, considering that older patients demonstrate worse outcomes despite sustaining less high energy impact (47,48), several studies have suggested that aged patients are more vulnerable to TBI (49–53). However, the particular relationship between mTBI and increased risk for dementia or morbidity is less clear (reviewed in Gardner and Yaffe, 2015 (54) and Wilson and colleagues 2017 (9)). To that end, we investigated whether an increased level of total tau and p-tau in the brains of aged hTau mice (12 months of age (22–24)) is associated with a stronger neuroinflammatory response after r-mTBI. We observed that increased astrogliosis and p-tau was more pronounced in aged mice when compared to young mice; however, this was not observed with respect to traumatic axonal injury and microgliosis, at 24 h post-injury. While our results highlight that older age at injury produces more pronounced astrogliosis and tau phosphorylation, these changes were relatively mild in nature (< two-fold change), suggesting that the polypathology resulting from the exposure of r-mTBI in mid-age animals is likely the result of normal ageing and the primed state of the resident glial cells (31). Another potential limitation is that our aged mice are only between 12 and 13 months old, and therefore are not representative of the clinical studies of 65+ year-old human’s mentioned in the introduction. Although mouse and human developmental stages are generally not a linear relationship, middle age is considered to be around 12–15 months in mice. In addition to increased astrogliosis, our results support a TBI-dependent increase in RZ3 p-tau observed in male hTau mice. Yet, this increase in p-tau pathology at 24 h post-injury was not associated with a more robust glial response in males when compared to their females’ counterparts, suggesting a diminished role for p-tau on acute neuroinflammation (24 h post-injury). Finally, we found that axonal injury was decreased in the aged injured group at 24 h post-injury. Whether this represents a true age-related effect on axonal injury will require future studies; it is noteworthy that APP immunostaining only captures a subpopulation of injured axons, and thus further measure of detecting the full extent of axonal injury is needed. Nonetheless, our results are consistent with our previous reports showing an attenuation of axonal swelling in older mice (17). Although the many differences in TBI models and experimental designs make direct comparison challenging, these results are consistent with the recent work of W.H. Cheng et al. that showed a robust decrease in axonal neurofilament pathology after mTBI in aged WT and transgenic mice harbouring the APP/PS1 mutations (55). Whether this pathology is unique to rodents remains to be determined. Further studies are necessitated in human autopsy cases.
Conclusion
This study shows that r-mTBI in young adult hTau mice induces age-dependent, sex-specific differences on pathological outcome at 24 h post-injury. Of particular interest here, we only found a sex-dependent difference for phosphorylated tau stained with RZ3 in young and aged male hTau mice. However, this increase in p-tau was not associated with an increase of Iba-1 and GFAP staining typically seen in this model of r-mTBI, suggesting a diminished role of phosphorylated tau in young and aged hTau mice at 24 h post-injury. Altogether, these findings suggest that future studies should incorporate both males and females to provide a greater understanding of injury prognosis and better inform clinical practice.
Declaration of interest
This work was supported by grant funding from Department of Defense, Chronic Effects of Neurotrauma Consortium (CENC) Award [W81XWH-13-2-0095] and Department of Veterans Affairs CENC [Award I01 CX001135]. Dr. Fiona Crawford is a VA Research Career Scientist. The authors report no conflicts of interest. The views, opinions, and/or findings contained in this article are those of the authors and should not be construed as an official Veterans Affairs or Department of Defense position, policy, or decision, unless so designated by other official documentation. Dr Stewart is supported by NIH grants [R01NS038104 and R01NS094], Department of Defense, and an NHS Research Scotland Career Researcher Fellowship.
References
- 1.Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99(1):4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- 2.Alexander MP. Mild traumatic brain injury: pathophysiology, natural history, and clinical management. Neurology. 1995;45(7):1253–60. [DOI] [PubMed] [Google Scholar]
- 3.Cassidy JD, Carroll LJ, Peloso PM, Borg J, Von Holst H, Holm L, Kraus J, Coronado VG, Injury W. Incidence, risk factors and prevention of mild traumatic brain injury: results of the WHO collaborating centre task force on mild traumatic brain injury. Journal of Rehabilitation Medicine. 2004;(43 Suppl):28–60. doi: 10.1080/16501960410023732. [DOI] [PubMed] [Google Scholar]
- 4.Ryu WH, Feinstein A, Colantonio A, Streiner DL, Dawson DR. Early identification and incidence of mild TBI in Ontario. Canadian Journal Neurological Sciences Le Journal Canadien Des Sciences Neurologiques. 2009;36(4):429–35. [DOI] [PubMed] [Google Scholar]
- 5.Vanderploeg RD, Curtiss G, Luis CA, Salazar AM. Long-term morbidities following self-reported mild traumatic brain injury. J Clin Exp Neuropsychol. 2007;29(6):585–98. doi: 10.1080/13803390600826587. [DOI] [PubMed] [Google Scholar]
- 6.Rapoport MJ, Feinstein A. Age and functioning after mild traumatic brain injury: the acute picture. Brain Injury. 2001;15(10):857–64. doi: 10.1080/02699050110065303. [DOI] [PubMed] [Google Scholar]
- 7.Taylor CA, Bell JM, Breiding MJ, Xu L. Traumatic brain injury-related emergency department visits, hospitalizations, and deaths - United States, 2007 and 2013. Morbidity Mortality Weekly Report Surveillance Summaries. 2017;66(9):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. Traumatic brain injury in the United States: a public health perspective. J Head Trauma Rehabil. 1999;14(6):602–15. [DOI] [PubMed] [Google Scholar]
- 9.Wilson L, Stewart W, Dams-O’Connor K, Diaz-Arrastia R, Horton L, Menon DK, Polinder S. The chronic and evolving neurological consequences of traumatic brain injury. Lancet Neurol. 2017;16(10):813–25. doi: 10.1016/S1474-4422(17)30279-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dams-O’Connor K, Pretz C, Billah T, Hammond FM, Harrison-Felix C. Global outcome trajectories after TBI among survivors and non-survivors: a national institute on disability and rehabilitation research traumatic brain injury model systems study. J Head Trauma Rehabil. 2015;30(4):E1–10. doi: 10.1097/HTR.0000000000000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McMillan TM, Weir CJ, Wainman-Lefley J. Mortality and morbidity 15 years after hospital admission with mild head injury: a prospective case-controlled population study. J Neurol Neurosurg Psychiatry. 2014;85(11):1214–20. doi: 10.1136/jnnp-2013-307279. [DOI] [PubMed] [Google Scholar]
- 12.Pretz CR, Dams-O’Connor K. Longitudinal description of the glasgow outcome scale-extended for individuals in the traumatic brain injury model systems national database: a national institute on disability and rehabilitation research traumatic brain injury model systems study. Arch Phys Med Rehabil. 2013;94(12):2486–93. doi: 10.1016/j.apmr.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown AW, Moessner AM, Mandrekar J, Diehl NN, Leibson CL, Malec JF. A survey of very-long-term outcomes after traumatic brain injury among members of a population-based incident cohort. J Neurotrauma. 2011;28(2):167–76. doi: 10.1089/neu.2010.1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardner RC, Burke JF, Nettiksimmons J, Kaup A, Barnes DE, Yaffe K. Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol. 2014;71(12):1490–97. doi: 10.1001/jamaneurol.2014.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Reviews Neurol. 2013;9(4):211–21. doi: 10.1038/nrneurol.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng WH, Stukas S, Martens KM, Namjoshi DR, Button EB, Wilkinson A, Bashir A, Robert J, Cripton PA, Wellington CL. Age at injury and genotype modify acute inflammatory and neurofilament-light responses to mild chimera traumatic brain injury in wild-type and app/ps1 mice. Exp Neurol. 2018;301(Pt A):26–38. doi: 10.1016/j.expneurol.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Ferguson SA, Mouzon BC, Lynch C, Lungmus C, Morin A, Crynen G, Carper B, Bieler G, Mufson EJ, Stewart W, et al. Negative impact of female sex on outcomes from repetitive mild traumatic brain injury in htau mice is age dependent: a Chronic Effects of Neurotrauma Consortium Study. Front Aging Neurosci. 2017;9:416. doi: 10.3389/fnagi.2017.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raghupathi R, Huh JW. Age-at-injury effects of microglial activation following traumatic brain injury: implications for treatment strategies. Neural Regeneration Research. 2017;12(5):741–42. doi: 10.4103/1673-5374.206639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rowe RK, Ziebell JM, Harrison JL, Law LM, Adelson PD, Lifshitz J. Aging with traumatic brain injury: effects of age at injury on behavioral outcome following diffuse brain injury in rats. Dev Neurosci. 2016;38(3):195–205. doi: 10.1159/000446773. [DOI] [PubMed] [Google Scholar]
- 20.McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68(7):709–35. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain: A Journal of Neurology, 2013;136(Pt 1):43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing non-mutant human tau isoforms. J Neuroscience: Official Journal Soc Neurosci. 2005;25(22):5446–54. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andorfer C, Kress Y, Espinoza M, De Silva R, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86(3):582–90. [DOI] [PubMed] [Google Scholar]
- 24.Polydoro M, Acker CM, Duff K, Castillo PE, Davies P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neuroscience: Official Journal Soc Neurosci. 2009;29(34):10741–49. doi: 10.1523/JNEUROSCI.1065-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mouzon B, Chaytow H, Crynen G, Bachmeier C, Stewart J, Mullan M, Stewart W, Crawford F. Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J Neurotrauma. 2012;29(18):2761–73. doi: 10.1089/neu.2012.2498. [DOI] [PubMed] [Google Scholar]
- 26.Longhi L, Saatman KE, Fujimoto S, Raghupathi R, Meaney DF, Davis J, Bsa M, Conte V, Laurer HL, Stein S, et al. Temporal window of vulnerability to repetitive experimental concussive brain injury. Neurosurgery. 2005;56(2):364–74. discussion 364–374. [DOI] [PubMed] [Google Scholar]
- 27.Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker CM, Davies P, Mullan M, Stewart W, Crawford F. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol. 2014;75(2):241–54. doi: 10.1002/ana.24064. [DOI] [PubMed] [Google Scholar]
- 28.Mouzon BC, Bachmeier C, Ojo JO, Acker CM, Ferguson S, Paris D, Ait-Ghezala G, Crynen G, Davies P, Mullan M, et al. Lifelong behavioral and neuropathological consequences of repetitive mild traumatic brain injury, Ann Clin Transl Neurol, 2018;5(1):64–80. doi: 10.1002/acn3.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Analytical Quantitative Cytology Histology. 2001;23(4):291–99. [PubMed] [Google Scholar]
- 30.Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP. Immune activation promotes depression 1 month after diffuse brain injury: A role for primed microglia. Biol Psychiatry. 2014;76(7):575–84. doi: 10.1016/j.biopsych.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging. 2013;34(5):1397–411. doi: 10.1016/j.neurobiolaging.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010;7(8):656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy, Acta Neuropathol, 2016;131(1):75–86. doi: 10.1007/s00401-015-1515-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Reviews Neurosci. 2016;17(1):5–21. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- 35.Ojo JO, Mouzon B, Algamal M, Leary P, Lynch C, Abdullah L, Evans J, Mullan M, Bachmeier C, Stewart W, et al. Chronic repetitive mild traumatic brain injury results in reduced cerebral blood flow, axonal injury, gliosis, and increased t-tau and tau oligomers, J Neuropathol Exp Neurol, 2016;75(7):636–55. doi: 10.1093/jnen/nlw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ojo JO, Mouzon BC, Crawford F. Repetitive head trauma, chronic traumatic encephalopathy and tau: challenges in translating from mice to men. Exp Neurol. 2016;275(Pt 3):389–404. doi: 10.1016/j.expneurol.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 37.Arendt T, Bullmann T. Neuronal plasticity in hibernation and the proposed role of the microtubule-associated protein tau as a “master switch” regulating synaptic gain in neuronal networks. American Journal Physiology Regulatory, Integrative Comparative Physiology. 2013;305(5):R478–489. doi: 10.1152/ajpregu.00117.2013. [DOI] [PubMed] [Google Scholar]
- 38.Arendt T, Stieler J, Strijkstra AM, Hut RA, Rudiger J, Van Der Zee EA, Harkany T, Holzer M, Hartig W. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neuroscience: Official Journal Soc Neurosci. 2003;23(18):6972–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang B, Carroll J, Trojanowski JQ, Yao Y, Iba M, Potuzak JS, Hogan AM, Xie SX, Ballatore C, Smith AB 3rd, et al. The microtubule-stabilizing agent, epothilone d, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice, J Neuroscience: Official Journal Soc Neurosci, 2012;32(11):3601–11. doi: 10.1523/JNEUROSCI.4922-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stoothoff WH, Johnson GV. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta. 2005;1739 (2–3):280–97. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 41.Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, et al. Primary age-related tauopathy (part): a common pathology associated with human aging, Acta Neuropathol, 2014;128(6):755–66. doi: 10.1007/s00401-014-1349-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elobeid A, Soininen H, Alafuzoff I. Hyperphosphorylated tau in young and middle-aged subjects. Acta Neuropathol. 2012;123(1):97–104. doi: 10.1007/s00401-011-0906-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, Yao Y, Lin YM, Driver JA, Sun Y, et al. Antibody against early driver of neurodegeneration cis p-tau blocks brain injury and tauopathy, Nature, 2015;523(7561):431–36. doi: 10.1038/nature14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, Li C, Herbert MK, Qiu J, Monuteaux M, Driver J, et al. Cis p-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae, Nat Commun, 2017;8(1):1000. doi: 10.1038/s41467-017-01068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan XL, Wright DK, Liu S, Hovens C, O’Brien TJ, Shultz SR. Sodium selenate, a protein phosphatase 2a activator, mitigates hyperphosphorylated tau and improves repeated mild traumatic brain injury outcomes. Neuropharmacology. 2016;108:382–93. doi: 10.1016/j.neuropharm.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 46.Johnson VE, Stewart W. Traumatic brain injury: age at injury influences dementia risk after TBI. Nat Reviews Neurol. 2015;11(3):128–30. doi: 10.1038/nrneurol.2014.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Vries R, Reininga IHF, Pieske O, Lefering R, El Moumni M, Wendt K. Injury mechanisms, patterns and outcomes of older polytrauma patients-an analysis of the Dutch trauma registry. PloS One. 2018;13(1):e0190587. doi: 10.1371/journal.pone.0190587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Susman M, DiRusso SM, Sullivan T, Risucci D, Nealon P, Cuff S, Haider A, Benzil D. Traumatic brain injury in the elderly: increased mortality and worse functional outcome at discharge despite lower injury severity. J Trauma. 2002;53(2):219–23. discussion 223–214. [DOI] [PubMed] [Google Scholar]
- 49.Hukkelhoven CW, Steyerberg EW, Rampen AJ, Farace E, Habbema JD, Marshall LF, Murray GD, Maas AI. Patient age and outcome following severe traumatic brain injury: an analysis of 5600 patients. J Neurosurg. 2003;99(4):666–73. doi: 10.3171/jns.2003.99.4.0666. [DOI] [PubMed] [Google Scholar]
- 50.Li CY, Karmarkar A, Adhikari D, Ottenbacher K, Kuo YF. Effect of age and sex on hospital readmission in traumatic brain injury. Arch Phys Med Rehabil. 2018; doi: 10.1016/j.apmr.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corrigan JD, Cuthbert JP, Whiteneck GG, Dijkers MP, Coronado V, Heinemann AW, Harrison-Felix C, Graham JE. Representativeness of the traumatic brain injury model systems national database. J Head Trauma Rehabil. 2012;27(6):391–403. doi: 10.1097/HTR.0b013e3182238cdd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walker W, Stromberg KA, Marwitz JH, Sima A, Agyemang AA, Graham KM, Harrison-Felix CL, Hoffman J, Brown AW, Kreutzer JS, et al. Predicting long-term global outcome after traumatic brain injury (TBI): development of a practical prognostic tool using the TBI model systems national database. J Neurotrauma. 2018; doi: 10.1089/neu.2017.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gardner RC, Dams-O’Connor K, Morrissey MR, Manley GT. Geriatric traumatic brain injury: epidemiology, outcomes, knowledge gaps, and future directions. J Neurotrauma. 2018; doi: 10.1089/neu.2017.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gardner RC, Yaffe K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol Cell Neurosci. 2015;66(PtB):75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng WH, Stukas S, Martens KM, Namjoshi DR, Button EB, Wilkinson A, Bashir A, Robert J, Cripton PA, Wellington CL. Age at injury and genotype modify acute inflammatory and neurofilament-light responses to mild chimera traumatic brain injury in wild-type and app/ps1 mice. Exp Neurol. 2017;301(Pt A):26–38. doi: 10.1016/j.expneurol.2017.12.007. [DOI] [PubMed] [Google Scholar]