

Abstract
Hutchinson–Gilford progeria syndrome (HGPS) is a rare genetic disorder characterized by features reminiscent of marked premature ageing1,2. Here, we present evidence of mutations in lamin A (LMNA) as the cause of this disorder. The HGPS gene was initially localized to chromosome 1q by observing two cases of uniparental isodisomy of 1q–the inheritance of both copies of this material from one parent–and one case with a 6-megabase paternal interstitial deletion. Sequencing of LMNA, located in this interval and previously implicated in several other heritable disorders3,4, revealed that 18 out of 20 classical cases of HGPS harboured an identical de novo (that is, newly arisen and not inherited) single-base substitution, G608G(GGC > GGT), within exon 11. One additional case was identified with a different substitution within the same codon. Both of these mutations result in activation of a cryptic splice site within exon 11, resulting in production of a protein product that deletes 50 amino acids near the carboxy terminus. Immunofluorescence of HGPS fibroblasts with antibodies directed against lamin A revealed that many cells show visible abnormalities of the nuclear membrane. The discovery of the molecular basis of this disease may shed light on the general phenomenon of human ageing.
Premature senescence is the hallmark of HGPS (Online Mendelian Inheritance in Man (OMIM) 176670), both clinically and in cell culture1,2. Children affected with HGPS typically appear normal at birth, but within a year the characteristic features of failure to thrive, delayed dentition, alopecia and sclerodermatous skin changes begin to appear. Death occurs on average at age 13, and at least 90% of the patients die from progressive atherosclerosis of the coronary and cerebrovascular arteries2. Although the prevailing hypothesis of the inheritance of HGPS has been sporadic autosomal dominant5,6, the possibility of autosomal recessive inheritance7,8 led us to conduct a genome-wide scan searching for evidence of homozygosity9. A whole-genome scan including 403 polymorphic microsatellite markers with an average spacing of 9.2 cM was performed on 12 DNA samples derived from individuals considered by one of us (W.T.B.) to represent classical HGPS. Although no evidence of homozygosity was identified in the overall sample set, two HGPS samples were found to have uniparental isodisomy (UPD) of chromosome 1q (Fig. 1a). Spectral karyotyping and G-banding of one of the two UPD cases showed a normal karyotype (data not shown).
Figure 1.
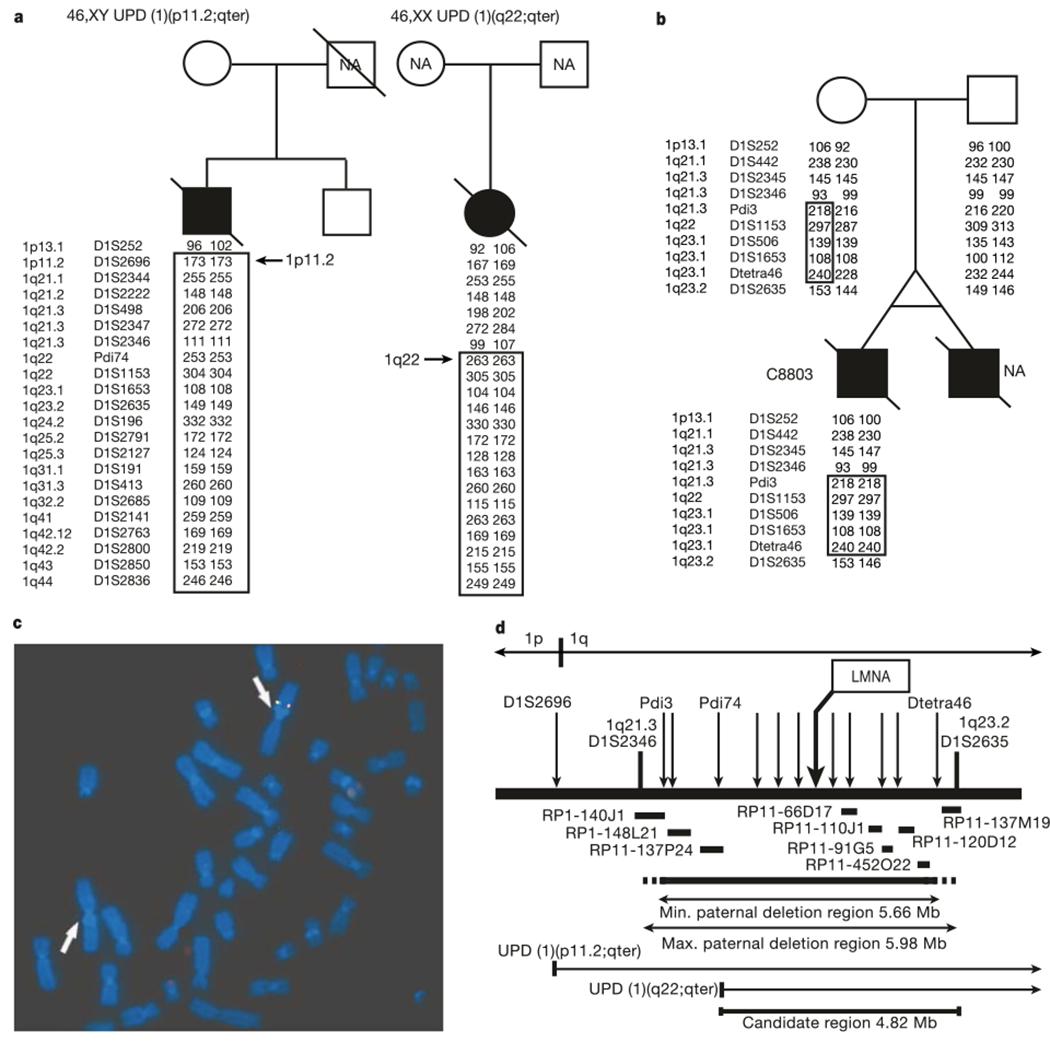
Localization of the HGPS gene to a 4.82-Mb interval on chromosome 1q. a, Uniparental isodisomy of chromosome 1q in fibroblasts from two patients with HGPS (filled symbols). A subset of markers and their genotypes are shown. NA, not available b, The boxed interval is the region in proband C8803 that has been inherited exclusively from the mother. c, FISH analysis of a metaphase spread from C8803 fibroblasts hybridized with BAC probes RP11-110J1 (green) and RP11-91G5 (red). d, Summary map of the candidate region. Microsatellite markers are indicated with arrows; horizontal bars indicate BAC probes that were used for FISH on sample C8803. LMNA is one of the approximately 80 known genes in the 4.82-Mb candidate interval.
An earlier report10 described an abnormal karyotype in a monozygotic twin with HGPS. That report described a mosaic cell population in which 70% of the cells contained a balanced inverted insertion (46,XY, inv ins (1;1)(q32;q44q23)), whereas the rest of the cells had an apparently normal karyotype. We obtained a fibroblast culture from the same individual (sample identification C8803), as well as his parents. Unexpectedly, genotyping of microsatellite markers identified a roughly 6-megabase (Mb) interval where all tested paternal alleles were completely missing (Fig. 1b). We confirmed that this deletion was also present in the cells that had an apparently normal karyotype, using fluorescent in situ hybridization (FISH) with bacterial artificial chromosomes (BACs) that map throughout this interval (Fig. 1c). Putting all of this information together with genotypes from a total of 44 additional microsatellite markers, we determined that the HGPS gene must lie in an interval of 4.82 Mb on proximal chromosome 1q (Fig. 1d).
The candidate interval contains roughly 80 known genes. Our attention was immediately drawn to one of them, the LMNA gene that encodes two protein products (lamin A and lamin C), representing major constituents of the inner nuclear membrane lamina. Mutations in LMNA have been found to be the cause of six different recessive and dominant disorders, including Emery–Dreifuss muscular dystrophy type 2, a form of dilated cardiomyopathy, the Dunnigan type of familial partial lipodystrophy, limb girdle muscular dystrophy type 1B, Charcot–Marie–Tooth disorder type 2B1, and mandibuloacral dysplasia (see reviews of laminopathies in refs 3, 4). The gene contains 12 exons and covers about 25 kilobases of genomic DNA. Lamin A is coded by exons 1–12 and lamin C by exons 1–10. A splice site within exon 10, located just upstream of the stop codon for lamin C, splices together with exons 11 and 12 to code for lamin A11,12.
Polymerase chain reaction (PCR) amplification of all of the exons of the LMNA gene (including exon–intron boundaries), followed by direct sequencing, was carried out in 23 samples from patients with classical HGPS. In 18 of these samples a heterozygous base substitution (G608G(GGC > GGT)) within exon 11 of the LMNA gene was identified (Fig. 2a). In HGPS sample AG10801, a different heterozygous base substitution was identified within the same codon (G608S(GGC > AGC)) (Fig. 2a). In HGPS sample AG10677, we identified a heterozygous base substitution within exon 2 (E145K(GAG > AAG)). In the eight cases where DNA from both parents was available, the G608G mutation was absent in the parents, confirming that these are de novo mutations. Similarly, the G608S and E145K mutations were not found in parents of AG10801 or AG10677, respectively. Thus, of the 23 classical HGPS cases studied, there were only three in which no LMNA mutations were found (Table 1): the two UPD cases (AG10578 and HGADFN005) and the sample with the 6-Mb paternal deletion (C8803).
Figure 2.
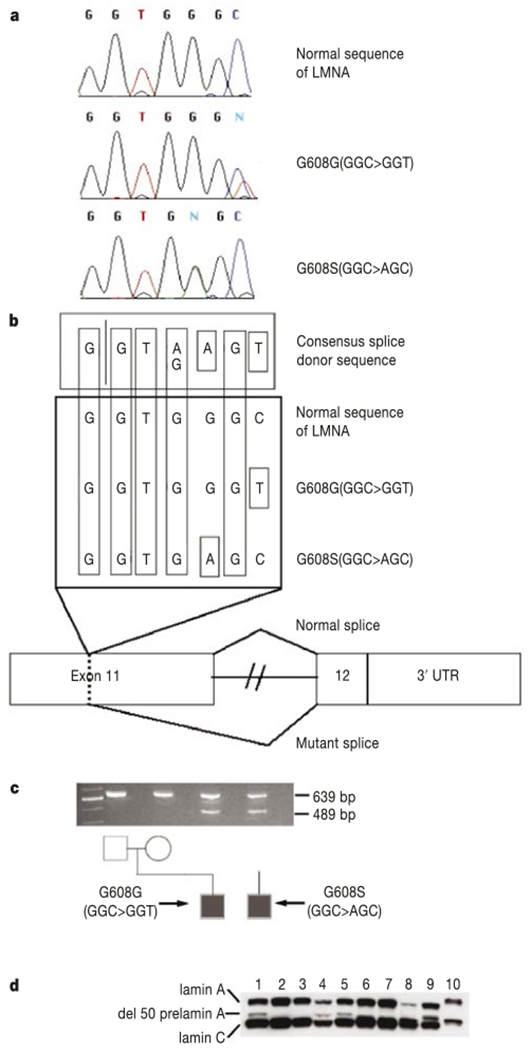
Point mutations in exon 11 of LMNA cause HGPS. a, Sequence traces from a normal control and two HGPS patients. b, Hypothesis for activation of a cryptic splice donor site in exon 11. c, Demonstration of the abnormal splice product using RT–PCR, showing an abnormal product of 489 bp in the two HGPS probands due to activation of the cryptic splice site. Alternative lanes to the right lack reverse transcriptase. d, Western blot using a monoclonal antibody against lamin A/C. Lanes 1, 5, 8 and 9 are from AG03506, AG03344, AG11498 and AG01972; they all carry G608G(GGC > GGT). Lane 4 is from AG10801, carrying G608S(GGC > AGC); lanes 2 and 3 are from parents of AG03506; lanes 6 and 7 are from father of AG03259 and mother of AG06917, respectively. Lanes 1–5 are from lymphoblastoid cell lines; lanes 6–9 are from fibroblasts. A protein sample from HeLa cells is in lane 10.
Table 1.
LMNA mutations in samples from classical HGPS
| Codon 608 nucleotide sequence |
||||||
|---|---|---|---|---|---|---|
| Sample ID (classical HGPS) | Codon 608 nucleotide sequence | Mutation | Comment | Mother | Father | Sibling(s) |
| AG01972 | GGC/T | G608G | – | NA | NA | NA |
| AG06297 | GGC/T | G608G | – | NA | NA | NA |
| AG11498 | GGC/T | G608G | – | NA | NA | NA |
| AG11513 | GGC/T | G608G | – | NA | NA | NA |
| AG03506 | GGC/T | G608G | – | Normal | Normal | Normal |
| AG03344 | GGC/T | G608G | – | Normal | Normal | Normal |
| AG03259 | GGC/T | G608G | – | Normal | Normal | Normal |
| AG06917 | GGC/T | G608G | – | Normal | Normal | NA |
| AG10579 | GGC/T | G608G | – | NA | NA | NA |
| AG10587 | GGC/T | G608G | – | Normal | NA | Normal |
| HGADFN001 | GGC/T | G608G | – | NA | NA | NA |
| HGADFN003 | GGC/T | G608G | – | NA | NA | NA |
| HGALBV009 | GGC/T | G608G | – | Normal | Normal | NA |
| HGALBV011 | GGC/T | G608G | – | Normal | Normal | NA |
| HGALBV057 | GGC/T | G608G | – | Normal | Normal | NA |
| HGADFN008 | GGC/T | G608G | – | NA | NA | NA |
| HGADFN014 | GGC/T | G608G | – | NA | NA | NA |
| HGALBV071 | GGC/T | G608G | – | NA | NA | NA |
| AG10801 | A/GGC | G608S | – | Normal | Normal | NA |
| AG10578 | GGC | – | UPD | Normal | NA | Normal |
| HGADFN005 | GGC | – | UPD | NA | NA | NA |
| C8803 | GGC | – | Deletion | Normal | Normal | NA |
| AG10677 | GGC | E145K | – | Normal* | Normal* | NA |
Additional sequence variants, presumed to be polymorphisms, were identified in exon 3 (L240L(CTG > CTA)), intron 4 (IVS4 + 61C > T), exon 5 (A287A(GCT > GCC)), exon 7 (D446D(GAT > GAC)), intron 8 (IVS8 – 41C > T) and exon 10 (H566H(CAC > CAT)). The variants in exons 5, 7 and 10 have been reported previously4,28.
NA, not available.
Normal at codon 145.
The de novo recurrence of the same exact point mutation in 18 out of 20 cases of classical HGPS is a surprising finding, but is not without precedent. The common HGPS mutation is a C to T in the context of a CpG dinucleotide, which represents the most mutable base in the vertebrate genome, as a methylated C can be deaminated to T and miscopied. In the E145K case and in four out of four G608G HGPS cases where parental DNA was available and silent polymorphisms in exon 10 (H566H) or exon 7 (D446D) were informative, we were able to determine that the new mutation was paternal in origin (data not shown). A very similar phenomenon occurs in achondroplasia13, where nearly all sporadic cases are due to paternal CpG to TpG mutations in the FGFR3 gene, resulting in an apparent gain-of-function mutation.
The most common mutation, G608G(GGC > GGT), is a silent substitution. The second mutation in the same codon, G608S(GGC > AGC), results in a conservative substitution of serine for glycine. How is it possible that these bland-appearing de novo mutations could cause HGPS? Inspection of the normal sequence surrounding codon 608 reveals that both of the observed HGPS mutations improve the match to a consensus splice donor (Fig. 2b), suggesting that they might activate a cryptic splice site. To test this hypothesis, PCR with reverse transcription (RT) was performed using a forward primer spanning the junction of exons 7 and 8, and a reverse primer in exon 12. In RNA from unaffected individuals, the expected product appears (Fig. 2c). In RNA samples from cell lines harbouring HGPS mutations, an additional smaller RT–PCR product appears. Sequence of these fragments shows that 150 nucleotides within exon 11 are missing. As the reading frame is maintained, this abnormal transcript would be expected to code for a protein with an internal deletion of 50 amino acids near the C terminus of lamin A. Lamin C would be unaffected. Cloning and sequencing of the normal full-length fragment obtained from RT–PCR on RNA extracted from primary fibroblasts containing the more common codon 608 mutation revealed that 7 out of 23 clones carry the mutant sequence. Thus, activation of the cryptic splice site within exon 11 is not complete. To determine whether the mutant messenger RNA is actually translated into protein, a western blot was performed using a monoclonal antibody against lamin A/C (Fig. 2d). In addition to the normal bands for lamin A and lamin C, an additional band is present in four of the lanes corresponding to samples from classical HGPS cases, but not in their parents. The abnormal band is not visible in the lane that contains the protein sample from classical HGPS patient AG11498 (which carries G608G(GGC > GGT)), but this is probably due to the very small amount of lamin A being expressed in this particular fibroblast line.
Although the major cause of HGPS appears to be the creation of an abnormal splice donor in exon 11, the finding of a de novo point mutation in exon 2 in a single patient (AG10677) is of interest. In retrospect, this patient14 had atypical clinical features (including persistence of coarse hair over the head, ample subcutaneous tissue over the arms and legs, and severe strokes beginning at age 4) that may subtly distinguish this phenotype from classical HGPS.
The two UPD cases present an interesting dilemma: if the LMNA gene sequence is normal in both cases, why do they have HGPS? The possibility of imprinting must be considered; however, previous cases of both paternal and maternal complete isodisomy of chromosome 1 do not support the presence of any imprinted loci on this chromosome15,16. In the case where DNA samples were available from the mother and sibling (Fig. 1a), we can conclude that this phenomenon resulted in a chromosome that has partly paternal and partly maternal alleles. This must have arisen by a post-fertilization event, probably a mitotic crossover between homologues. We postulate that these cases actually represent ‘somatic rescue’ events of the premature senescence phenotype of HGPS. Under this hypothesis, the individuals from whom these fibroblasts were derived originally harboured typical codon 608 HGPS mutations in LMNA. Perhaps as an in vivo event, or perhaps as an in vitro event in the fibroblast culture, a mitotic crossover occurred, generating a cell with segmental UPD that had duplicated the wild-type allele of LMNA and lost the HGPS mutation. Such a cell would probably then have a growth advantage over its neighbours. Proof of this hypothesis would require access to multiple tissues of the deceased UPD patients, which unfortunately are not available.
We also did not identify any mutation in the LMNA gene in the patient with the 6-Mb deletion (Fig. 1b). We believe that this might also be due to a somatic rescue event— specifically, we hypothesize that this patient was originally heterozygous for a codon 608 LMNA mutation, but in this instance the ‘rescue’ involved an internal deletion of 6 Mb containing the mutant allele, associated with a more complex mosaic rearrangement of chromosome 1. A recent study reported on a case of classical HGPS in which an apparent interstitial deletion of chromosome 1q23 was seen17. We obtained cells and DNA from this patient, and to our surprise the typical heterozygous G608G mutation is present (data not shown). Furthermore, we have not been able to confirm the presence of an interstitial deletion by high-resolution chromosome banding or by FISH with BACs spanning the 1q23–1q24 region. This may be another example of somatic rescue, involving an interstitial deletion in the clone of cells analysed in the original report, but not present in other samples from the same patient.
Extensive prior study of the biochemical function of lamin A suggests a possible mechanism for disease causation. Lamin A is normally synthesized as a precursor molecule (prelamin A). At the C terminus is a CAAX-box motif that is subject to farnesylation. After that, an internal proteolytic cleavage occurs, removing the last 18 coding amino acids18–20, to generate mature lamin A. We predict that the codon 11 HGPS mutations and consequent abnormal splicing would produce a prelamin A that still retains the CAAX box, but is missing the site for endoproteolytic cleavage. There is also evidence that cell-cycle-dependent phosphorylation of lamin A is important for its normal function, and at least one site for phosphorylation (Ser 625) is deleted in the abnormal HGPS protein21. As lamin A forms a multiprotein complex within the inner nuclear membrane, this incompletely processed prelamin A might well act as a dominant negative.
Notably, defective prelamin A processing has recently been identified in a mouse knockout of the Zmpste24 metalloproteinase22,23. Zmpste24 is believed to be involved in proteolytic processing of prelamin A, and may represent the actual endoprotease. The homozygote knockout of Zmpste24 has a phenotype resembling the clinical features observed in HGPS patients, including growth retardation, premature death (20 weeks) from cardiac dysfunction, and alopecia. However, additional features such as pronounced osteoporosis are also present23. Immunofluorescence experiments on cells from these animals show elongated and irregular nuclei with herniation of the nuclear membrane. A mouse knockout of the Lmna gene has also been reported previously24, showing severe postnatal growth retardation, muscular dystrophy and similar nuclear abnormalities.
To pursue the possibility of structural nuclear membrane abnormalities in HGPS, immunofluorescence studies with two different monoclonal antibodies against lamin A/C were performed on primary fibroblasts from five classical HGPS cases with the common G608G mutation, as well as unaffected parents. Representative data from an affected patient (AG11498) and an unaffected parent (AG06299) are shown in Fig. 3. In 48% of the HGPS cells we noted an irregular shape of the nuclear envelope (Fig. 3e–h). Cells from the unaffected control (Fig. 3a–d) showed significantly fewer cells of this phenotype (<6%). To be certain that this result was not an artefact of secondary differences in the status of the HGPS and control fibroblast cultures, cells originating from the same flasks as the cells used for the immunofluorescence studies were monitored for differences in cell cycle (by fluorescent-activated cell analysis) and degree of apoptosis. No significant differences between the cells derived from the classical HGPS patients and the unaffected parents were noted.
Figure 3.
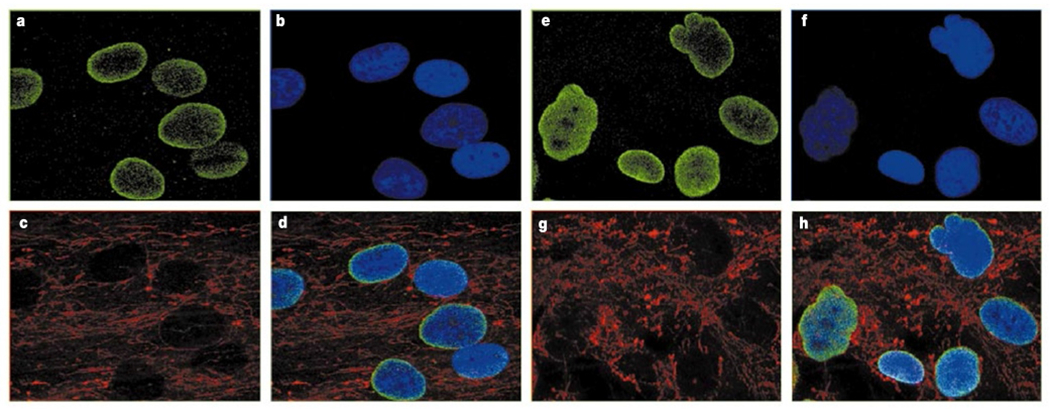
Immunofluorescence on primary dermal fibroblasts from an unaffected mother and a child with classical HGPS, using antibody JOL2 against lamin A/C. Identical results were obtained with antibody XB10 (data not shown). a–d, Results from an unaffected mother, AG06299. e–h, Results from a classical HGPS patient, AG11498. a, e, Antibody against lamin A/C. b, f, DAPI staining showing location of the nuclei. c, g, Antibody staining mitochondria, showing distribution of the cytosol. d, h, Merged image of a–c (d) and e–g (h).
HGPS can now be added to the long list of human genetic disorders shown to be due to mutations in the LMNA gene3,4. As most cases of HGPS appear to have a de novo mutation in the same codon, molecular diagnostics should be almost immediately feasible. This will be particularly useful in making the diagnosis in a young child before the full clinical phenotype has appeared. Molecular diagnostic methods may also provide reassurance in the prenatal arena, where the possibility of parental somatic mosaicism and recurrence of disease in future pregnancies can now be addressed. Finally, the discovery of the molecular basis of HGPS suggests a possible role for LMNA in aspects of the normal ageing process. It will be important to look for common variants in this gene that might show association with exceptional longevity, and perhaps also to explore whether somatic mutations in LMNA, accumulated over a lifetime, have some role in senescence. □
Methods
Subjects and DNA/RNA preparation
This study was approved through the NIH Human Subjects review process. Primary dermal fibroblast cell cultures and EBV-transformed lymphoblastoid cell lines from individuals diagnosed as classical HGPS, and their first-degree relatives (when available), were obtained from the Aging Repository of the Coriell Cell Repository (CCR), and the Progeria Research Foundation Cell and Tissue Bank. DNA was prepared using the Puregene DNA isolation kit (Gentra Systems). The genome scan for homozygosity included samples derived from 12 classical HGPS patients (samples AG01972, AG03259, AG03344, AG03506, AG06297, AG06917, AG10578, AG10579, AG10587, AG10801, AG11498 and AG11513) and 16 unaffected first-degree relatives (samples AG03258, AG03260, AG03262, AG03263, AG03343, AG03342, AG03345, AG03346, AG03504, AG03505, AGO3507, AGO3508, AG06298, AG06299, AG10585 and AG10588). Additional samples used in this study were samples from classical HGPS (samples AG10677, HGADFN001, HGADFN003, HGALBV009, HGALBV011, HGALBV057, HGADFN005, HGADFN008, HGADFN014, HGALBV071 and C8803 (also known as AG10548 in the CCR)) and samples derived from their unaffected first-degree relatives (samples HGMLBV010, HGFLBV021, HGMLBV023, HGFLBV031, HGMLBV066, HGFLBV067, HGMLBV013, HGFLBV050, HGMLBV058, HGSLBV059, HGMLBV078 HGFLBV079, HGFLBV082 and HGMLBV081). Total RNAwas extracted from cells with TRIzol reagent (Invitrogen).
Genotyping
The whole-genome scan included 403 highly polymorphic microsatellite markers with an average spacing of 9.2 cM and average heterozygosity of about 0.8 (E. Gillanders, M. Jones, D. Freas-Lutz, R. Sood and J. Trent, manuscript in preparation). Pedigree checking was performed using PedCheck (J. O’Connell, http://watson.hgen.pitt.edu/register/docs/pedcheck.html) and any identified genotype errors were removed. We carried out homozygosity mapping assuming various degrees of inbreeding for the HGPS cases25. Additional microsatellite repeats on chromosome 1q were identified using the Sputnik program (C. Abajian, http://rast.abajian.com/sputnik/) (see Supplementary Table 1) and were used to further investigate the UPD cases and the paternal deletion region in C8803. Microsatellite markers were analysed using a 3100 genetic analyser (PE Biosystems). Genotypes were analysed using GeneScan 3.7 and Genotyper 2.5 Software (PE Biosystems).
FISH analysis
Single- and two-colour FISH was performed on metaphases from sample C8803 following previously published procedures26, using a subset of BACs in the region of the paternal deletion. BACs used as probes for the FISH were RP1-140J1, RP1-148L21, RP1-178F15, RP11-137P24, RP11-66D17, RP11-110J1, RP11-91G5, RP11-120D12, RP11-101J8, RP11-81N17, RP11-144L1, RP11-317F9, RP11-452O22 and RP11-137M19.
Mutation analysis of LMNA
Direct sequencing of LMNA was performed primarily using previously described primer sequences for the LMNA exons 1–12 (ref. 27). Sequencing reactions were performed at quarter strength reaction volumes with the Big Dye Terminator chemistry kit (Applied Biosystems), and electrophoresed on an ABI 3700 DNA Analyser (Applied Biosystems). Multiple sequence alignment was performed with Sequencher (Genecodes Inc.).Attempts were made to sequence all PCR products in both directions, but approximately 7% of exons failed to yield readable sequence.
RT–PCR on exon 11
For all RNA samples, 20 μg of total RNA was treated with RQ1 RNase-Free DNase according to manufacturers recommendations (Promega). A total of 800 ng of DNase-treated total RNA was used for first-strand complementary DNA synthesis with random hexamers (Superscript, Invitrogen). Control samples without reverse transcriptase were processed at the same time for each sample. PCR primers for the lamin A/C gene were designed in exon 7/8 and exon 12. Primer sequences were 5′-GCAACAAGTCCAATGAGGACCA-3′ and 5′-GTCCCAGATTACATGATGC-3′. PCR fragments were gel-purified or cloned (TOPO TA-cloning kit, Invitrogen) and sequenced. PCR with GAPDH-specific primers was performed on all samples as control.
Western analysis
Whole cells (1 × 106) were collected and washed twice in PBS, the pellets were resuspended in RIPA buffer and included a cocktail of proteinase inhibitors (Roche). A total of 20 μg of protein was loaded and electrophoresed on an 8% Tris-Glycine mini gel (Invitrogen). Primary monoclonal lamin A/C antibody was clone JOL2 (Chemicon International).
Immunofluorescence
Cultivated fibroblasts were fixed (3.2% paraformaldehyde), permeabilized (1% NP-40), blocked (0.1% Brij58 and 5% serum) and labelled for lamin A/C (monoclonal antibodies JOL2, Chemicon International, and clone XB10, CRP Inc.) mitochondria (HMS-0100, Immunovision Inc.) and DNA (using 4,6-diamidine-2-phenylindole (DAPI) dihydrochloride mixed in at 1 μg ml−1 with the second antibody). Analysis was done by confocal microscopy using a Biorad 1024 and Leica SP2 systems.
Cell cycle and apoptosis
Cells were collected and washed in PBS one day after the immunofluorescence experiments. Duplicate experiments were performed on each cell culture. A total of 5 × 105 cells were resuspended in 0.5 ml of NuCycl propidium iodide (NuCycl PI kit, Exalpha) and processed as recommended by the manufacturer. The total DNA content was measured by DNA flow cytometry. Cells were also assayed for viability using annexin V-fluorescein isothiocyanate and propidium iodide according to standardized procedures (BD Biosciences).
Supplementary Material
Acknowledgements
We are grateful to all of the families with HGPS who donated blood and skin samples, without which this study would not have been possible. We would also like to express our gratitude to the entire Progeria Research Consortium, especially B. Toole, for their encouragement during the course of this work. We also thank E. Gillanders for providing the linkage marker set; P. Hollstein for assistance with genotyping; P. Chines for the identification of additional microsatellite markers on chromosome 1q; and M. Kirby for assistance with the flow cytometry. Support from the Progeria Research Foundation (L.B.G., M.W.G. and T.W.G.) and the Bedminster Foundation (W.T.B.) is gratefully acknowledged.
Footnotes
Supplementary Information accompanies the paper on www.nature.com/nature.
Competing interests statement The authors declare that they have no competing financial interests.
References
- 1.DeBusk FL The Hutchinson-Gilford progeria syndrome. J. Pediat 80, 697–724 (1972). [DOI] [PubMed] [Google Scholar]
- 2.Baker PB, Baba N & Boesel CP Cardiovascular abnormalities in progeria. Case report and review of the literature. Arch. Pathol. Lab. Med 105, 384–386 (1981). [PubMed] [Google Scholar]
- 3.Burke B & Stewart CL Life at the edge: the nuclear envelope and human disease. Nature Rev. 3, 575–585 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Genschel J & Schmidt H-J Mutations in the LMNA gene encoding lamin A/C. Hum. Mutat 16, 451–459 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Brown WT Human mutations affecting aging—a review. Mech. Aging Dev 9, 325–336 (1979). [DOI] [PubMed] [Google Scholar]
- 6.Brown WT, Zebrowser M & Kieras FJ in Genetic Effects on Aging II (ed. Harrison DE) 521–542 (Telford, Caldwell, New Jersey, 1990). [Google Scholar]
- 7.Trevas-Maciel A Evidence for autosomal recessive inheritance of progeria (Hutchinson Gilford). Am. J. Med. Genet 31, 483–487 (1988). [DOI] [PubMed] [Google Scholar]
- 8.Khalifa MM Hutchinson-Gilford progeria syndrome: report of a Libyan family and evidence of autosomal recessive inheritance. Clin. Genet 35, 125–132 (1989). [DOI] [PubMed] [Google Scholar]
- 9.Lander ES & Botstein D Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 236, 1567–1570 (1987). [DOI] [PubMed] [Google Scholar]
- 10.Brown WT Progeria: a human disease model of accelerated aging. Am. J. Clin. Nutr 55, 1222S–1224S (1992). [DOI] [PubMed] [Google Scholar]
- 11.Fisher DZ, Chaudhary N & Blobel G cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl Acad. Sci. USA 83, 6450–6454 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin F & Worman HJ Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem 268, 16321–16326 (1993). [PubMed] [Google Scholar]
- 13.Shiang R et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 78, 335–342 (1994). [DOI] [PubMed] [Google Scholar]
- 14.Smith AS, Wiznitzer M, Karaman BA, Horwitz SJ &Lanzieri CF MRA detection of vascular occusion in a child with progeria. Am. J. Neuroradiol 14, 441–443 (1993). [PMC free article] [PubMed] [Google Scholar]
- 15.Pulkkinen L, Bullrich F, Czarnecki P, Weiss L & Uitto J Maternal uniparental disomy of chromosome 1 with reduction to homozygosity of the LAMB3 locus in a patient with Herlitz junctional epidermolysis bullosa. Am. J. Hum. Genet 61, 611–619 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gelb BD et al. Paternal uniparental disomy for chromosome 1 revealed by molecular analysis of a patient with pycnodysostosis. Am. J. Hum. Genet 62, 848–854 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delgado-Luengo W et al. Del(1)(q23) in a patient with Hutchinson-Gilford progeria. Am. J. Med. Genet 113, 298–301 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Lutz RJ, Trujillo MA, Denham KS, Wenger L & Sinensky M Nucleoplasmic localization of prelamin A: implications for prenylation-dependent lamin A assembly into the nuclear lamina. Proc. Natl Acad. Sci. USA 89, 3000–3004 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinensky M et al. The processing pathway of prelamin A. J. Cell Sci 107, 61–67 (1994). [DOI] [PubMed] [Google Scholar]
- 20.Hennekes H &Nigg EA The role of isoprenylation in membrane attachment of nuclear lamins, a single point mutation prevents the proteolytic cleavage of the lamin A precursor and confers membrane binding properties. J. Cell Sci 107, 1019–1029 (1994). [DOI] [PubMed] [Google Scholar]
- 21.Eggert M et al. Identification of novel phosphorylation sites in murine A-type lamins. Eur. J. Biochem 213, 659–671 (1993). [DOI] [PubMed] [Google Scholar]
- 22.Pendas AM et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nature Genet. 31, 94–99 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Bergo MO et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl Acad. Sci. USA 99, 13049–13054 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sullivan T et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol 147, 913–920 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith C The detection of linkage in human genetics. J. R. Stat. Soc. B 15, 153–184 (1953). [Google Scholar]
- 26.Casper AM, Nghiem P, Arlt MF & Glover TW ATR regulates fragile site stability. Cell 111, 779–789 (2002). [DOI] [PubMed] [Google Scholar]
- 27.De Sandre-Giovannoli A et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am. J. Hum. Genet 70, 726–736 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Speckman RA et al. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am. J. Hum. Genet 66, 1192–1198 (2000); erratum 67, 775 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.