

Abstract
The Hippo pathway is a signaling pathway that controls organ size in animals by regulating cell proliferation and apoptosis. Yes-associated protein 1 (YAP1), an oncogene associated with the development and progression of breast cancer, is downregulated by the Hippo pathway and is associated with the development and progression of breast cancer. Yippee-like 3 (YPEL3) is a target gene of the tumor suppressor protein p53, and its activation has been shown to inhibit cell growth, induce cellular senescence, and suppress tumor cell metastasis. In this study, we found that YAP1 inhibits the expression of YPEL3 expression in breast cancer cells. Furthermore, a decrease in lamin B1, a marker protein of cellular senescence, coupled with the activation of senescence-associated β-galactosidase indicated that upregulating YPEL3 levels through YAP1 downregulation can induce cellular senescence. Additionally, elevated YPEL3 levels resulted in higher levels of oxygen consumption rate in mitochondria, thus promoting apoptosis. This suggests that YPEL3 plays a crucial role in regulating oxidative stress and cell apoptosis in breast cancer cells. Therefore, the interaction between YAP1 and YPEL3 represents a novel mechanism of cellular senescence mediated by the Hippo signaling pathway. Collectively, our findings suggest that the Hippo signaling pathway plays an important role in regulating cellular senescence, which could have implications for the development of new therapeutic strategies for diseases such as cancer.
Keywords: Hippo signaling pathway, YAP1, YPEL3, Cellular senescence
Introduction
The Hippo signaling pathway is a crucial regulator of cell proliferation and apoptosis in various cancer cells. This pathway encompasses several key components, including mammalian Ste20-like kinases 1/2 (MST1/2), large tumor suppressor 1/2 (LATS1/2), and the yes-associated protein 1 (YAP1) and/or its paralog PDZ-binding motif (TAZ) [1]. In its inactive state, the Hippo pathway leads to the dephosphorylation and nuclear translocation of YAP1. In the nucleus, YAP1 interacts with TEA domain (TEAD) transcription factors to promote the expression of genes associated with cell growth, proliferation, and survival [2, 3].
Yippee-like 3 (YPEL3) is an intriguing protein in the context of the Hippo signaling pathway, as it belongs to a protein family that shares homology with the Yippee gene of Drosophila. Previous studies have identified YPEL3 as a novel target of the tumor suppressor protein p53 [4, 5]. Notably, overexpression of YPEL3 has been found to inhibit the proliferation, migration, and invasion of colon adenocarcinoma cells [6]. These findings emphasize the importance of investigating the potential interplay between YAP1, a promoter of cancer, and YPEL3, which exhibits cancer-suppressing properties.
Both YAP1 and YPEL3 have been implicated in the regulation of cellular senescence. YAP1 deficiency has been shown to increase senescence, whereas YPEL3 has been identified as a protein that induces senescence [4, 7]. Therefore, it is crucial to explore the potential interplay between YAP1 and YPEL3 in the context of cellular senescence. Cellular senescence is an irreversible cell cycle arrest that can be triggered by various types of cellular stress [8]. This process is characterized by the upregulation of cell cycle inhibitors such as p21 and p16, and can also be identified by positive staining for senescence-associated β-galactosidase (SA-β-gal), morphological changes, DNA damage, and telomere attrition [9–12]. Mitochondrial dysfunction has been identified as both a cause and a consequence of cellular senescence [13]. Moreover, previous studies have identified an association between lamin B1 loss and cellular senescence [14, 15], which highlights the need to study the role of lamin B1 in the context of cellular senescence. Furthermore, the induction of cellular senescence has been suggested as a promising strategy to permanently halt the proliferation of cancer cells [8]. Numerous researchers have explored the potential antitumor effects of cellular senescence in cancer treatment [16]. However, the link between senescence and apoptosis is still not fully understood.
In this study, we investigated the effects of YAP1 deficiency on YPEL3 expression and observed an increase in YPEL3 expression in breast cancer cells in response to YAP1 deficiency. We also found that YPEL3 upregulation resulting from YAP1 downregulation leads to cellular senescence and triggers cell apoptosis. Collectively, our findings not only provide novel insights into the relationship between YAP1 and YPEL3 within the Hippo signaling pathway but also shed light on a new molecular mechanism underlying cellular senescence.
Materials and methods
Chemicals and reagents
YPEL3 polyclonal antibodies were purchased from Boster Bio (Pleasanton, CA, USA), YAP1 and lamin B1 mouse monoclonal antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and α-tubulin antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). Human epidermal growth factor (EGF) was purchased from PeproTech (London, UK), and RPMI 1640 medium and fetal bovine serum (FBS) were obtained from Hyclone (Logan, VI, USA). The BCA protein assay and ECL kit were obtained from Thermo Scientific (Waltham, MA, USA), and the D-Plus™ ECL solution was purchased from Dongin LS (Seoul, Korea). RNase inhibitor and M-MLV reverse transcriptase were obtained from Promega (Madison, WI, USA), and Ex Taq DNA polymerase was obtained from TaKaRa Bio (Shiga, Japan). SYBR blue was obtained from QIAGEN (Hilden, Germany). All other chemicals and reagents used in this study were of the highest quality available.
Cell culture
MCF-7 human breast cancer cells were obtained from the Korea Cell Line Bank (KCLB, Korea) and cultured in RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2.
Transient transfection with plasmid DNA and siRNA
Transfection was performed using the Neon™ transfection system (Invitrogen, Carlsbad, CA, USA) and 1.2 × 106 cells were transfected with 70 nM siRNAs of YAP1 or 4 μg of pRP _YPEL3 vector (Vectorbuilder, Korea). The YAP1 and YPEL3 siRNA target sequences were 5′-GGU GAU ACU AUC AAC CAA Att-3′ and 5′-CGG AUC UAG CUC CUG UAU Att-3′, respectively. The cells were cultured in RPMI 1640 medium containing 10% FBS without antibiotics for 24 h, then maintained in the same medium with antibiotics for 24 h after transfection.
RNA isolation, reverse transcription, and RT-PCR
Total RNA was extracted using the GeneALL Ribospin™ kit (Seoul, Korea). A volume of 20 μL containing 5 × RT buffer, 10 mM dNTPs, 40 units of RNase inhibitor, 200 units of M-MLV reverse transcriptase, and 100 pmol of oligo-dT primer was used to transcribe 1000 ng of total RNA at 37 °C for 1 h. Next, 0.8 μL of the reaction mixture of each sample was amplified to a final volume of 25 μL using 10 pmol of each oligonucleotide primer pair, 0.2 mM dNTPs, 1.5 mM MgCl2, and 1.25 units of Ex Taq DNA polymerase. RT-PCR was conducted using the Rotor-Gene Q machine (QIAGEN, Netherlands) according to the manufacturer’s instructions and analyzed using the QIAGEN Rotor-Gene Q Series software. Each reaction contained 10 μL of SYBR Blue 2 × qPCR Master Mix, 1 μM oligonucleotide primers, and 20 ng of cDNA in a final volume of 20 μL. The following primer sets were used for qPCR: YAP1, 5′-CGC TCT TCA ACG CCG TCA-3′ and 5′-AGT ACT GGC TGT CGG AGT-3′; YPEL3, 5′-GTG CGG ATT TCA AAG CCC AAG-3′ and 5′ CCC ACG TTC ACC ACT GAG TT-3′; lamin B1, 5′-GAA AAA GAC AAC TCT CGT CGC A-3′ and 5′-GTA AGC ACT GAT TTC CAT GTC CA-3′; 18S rRNA, 5′-GTA ACC CGT TGA ACC CCA TT-3′ and 5′-CCA TCC AAT CGG TAG TAG CG-3′. The thermal profile for amplification consisted of an initial denaturation cycle at 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 15 s and annealing/extension at 55 °C for 45 s.
Western blotting
To extract proteins, the cells were harvested and solubilized with an ice-cold lysis buffer consisting of 50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.5% sodium deoxycholate, 1 mM PMSF, 2 mM EDTA, and 10 mM NaF. The concentrations of the extracted proteins were determined using BCA Protein Assay Reagents (Thermo Scientific). Afterward, 20–40 μg of the extracted proteins were heated at 99 °C for 5 min and separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) on 10–15% polyacrylamide gels and then electrophoretically transferred to PVDF membranes. The PVDF membranes were then blocked with 5% skim milk in Tris-buffered saline with Tween 20 at 4 °C for 2 h, after which they were incubated overnight at 4 °C with specific primary antibodies diluted at a 1:1000 ratio. After overnight incubation with a secondary antibody diluted at a 1:1000 ratio, the protein samples were visualized using the D-Plus™ ECL solution and subsequently analyzed using the ChemiDoc XRS system (Bio-Rad, Hercules, CA).
Confocal microscopy
The samples were fixed with 4% (w/v) formaldehyde for 1 h at 25 °C and then blocked with a solution containing 10% goat serum, 0.2% Triton X-100, and PBS for 30 min. Afterward, the samples were incubated with primary antibodies (1:200) overnight at 4 °C and then stained with specific secondary antibodies (1:200) overnight. After an additional washing step, the coverslips were mounted onto glass slides with 3 μL of UltraCruz™ mounting medium containing DAPI. The fluorescent signals were then analyzed with an LSM800 confocal laser scanning microscope (Carl Zeiss, Jena, Germany).
Cellular OCR assay
Cells (5 × 103 per well) transfected with YAP1 siRNA or pRP_YPEL3 plasmid were seeded in XF24-well plates (Agilent Technologies, Santa Clara, CA, USA) to measure mitochondrial respiration. After 48 h, the medium was replaced with Seahorse XF RPMI medium (without Phenol Red), supplemented with 10 mM glucose, 2 mM glutamine, and 1 mM pyruvate. The cells were then equilibrated at 37 °C without CO2 for 1 h to ensure a stable baseline. After sequential treatment with oligomycin (0.5 μM), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP, 0.25 μM), and a combination of rotenone and antimycin A (0.5 μM), oxygen consumption was determined using the Seahorse XFe24 Extracellular Flux Analyzer (Agilent). All data analyses were conducted using the Seahorse Wave software. The cellular oxygen consumption rate was normalized according to the protein concentrations measured with the BCA protein assay kit (Thermo).
Apoptosis assay
Cells were seeded in a 60-mm culture dish at a specific concentration and incubated for 48 h. Following incubation, the cells were harvested using 0.1% trypsin–EDTA and washed with PBS to remove residual media or cellular debris. The cells were then stained with Muse™ Annexin V & Dead Cell Reagent. Finally, the cells were examined using a Muse™ Cell Analyzer (Merck Millipore).
Statistical analysis
One-way analysis of variance (ANOVA) was conducted to identify differences between the experimental groups, followed by Dunnett’s multiple comparison t-test using the GraphPad Prism 7 software (GraphPad Software Inc., CA, USA). Differences were considered statistically significant when p < 0.05.
Results
Kaplan–Meier analysis of YAP1 and YPEL3 expression in breast cancer patients
Kaplan–Meier analyses were conducted using the KMplot database (https://kmplot.com/analysis/) to examine the correlation between the expression of YAP1 and YPEL3 and the survival rates of breast cancer patients. The analysis included assessments of overall survival (OS) and recurrence-free survival (RFS). Our results indicated that patients with low YAP1 expression exhibited a higher survival rate (Fig. 1a). Similarly, patients with high YPEL3 expression presented a higher survival rate (Fig. 1b).
Fig. 1.

Kaplan–Meier analysis of YAP1 and YPEL3 expression in breast cancer patients. a Kaplan–Meier analysis of overall survival and recurrence-free survival in breast cancer patients showing high or low YAP1 expression. b Kaplan–Meier analysis of overall survival and recurrence-free survival in breast cancer patients showing high or low YPEL3 expression. The data were obtained from the KMplot database
YAP1 deficiency induces the expression of YPELs in MCF-7 cells
To investigate the impact of YAP1 deficiency on the expression of YPEL3 in the MCF-7 cells, we used a specific siRNA targeting YAP1, which resulted in a substantial reduction in YAP1 mRNA and protein levels. Interestingly, the downregulation of YAP1 using siRNA led to a significant increase in both mRNA and protein expression of YPEL3 (Fig. 2a and b). Confocal microscopy analysis further confirmed that YAP1 suppression by siRNA induced the expression of YPEL3 (Fig. 2c).
Fig. 2.

YAP1 deficiency induces the expression of YPEL3 in MCF-7 cells. a MCF-7 cells were transfected with YAP1 siRNA (70 nM) for 48 h. Real-time qPCR was performed to measure the expression of YAP1 and YPEL3 mRNA. The data represent the mean ± SD (n = 3). *p < 0.05. b YAP1 and YPEL3 protein levels were measured using western blot analysis. α-Tubulin was used as a loading control. Western blot analyses were performed in triplicate. c Confocal analysis was performed to measure the YPEL3 expression. Microscopy scale bar = 20 μm
To further elucidate the role of YAP1 in regulating YPEL3 expression, verteporfin, a specific inhibitor of the Hippo-YAP pathway, was used to disrupt YAP-TEAD interactions in the nucleus [17]. MCF-7 cells were treated with varying concentrations of verteporfin (0, 0.625, 1.25, and 2.5 μM) for 48 h. Our findings demonstrated that treatment with verteporfin significantly enhanced the protein expression of YPEL3 by inhibiting YAP1 activity (Fig. 3).
Fig. 3.
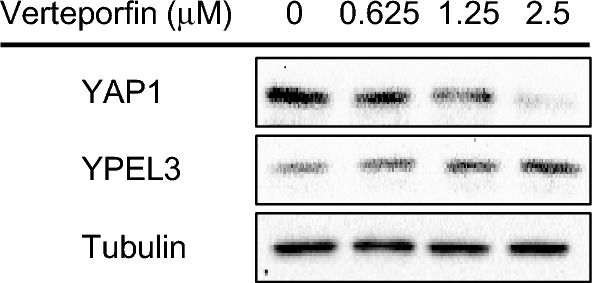
Verteporfin induces the expression of YPEL3. MCF-7 cells were treated with verteporfin (0, 0.625, 1.25, or 2.5 μM) for 48 h. YAP1 and YPEL3 protein levels were measured via western blot analysis. α-Tubulin was used as a loading control
EGF decreases YPEL3 by upregulating the expression of YAP1
Previous studies have demonstrated that EGF inhibits LATS1 within the Hippo pathway, resulting in YAP1 upregulation [18]. In this study, cells were treated with 100 ng/mL of EGF for 15, 30, or 60 min to determine whether EGF suppresses YPEL3 expression by inactivating the Hippo pathway. Our findings revealed that EGF treatment for up to 60 min led to a significant suppression of YPEL3 expression, whereas YAP1 and its target protein Cyr61 exhibited a marked upregulation (Fig. 4a and b). These findings suggest that YPEL3 may be a target protein within the signaling cascade of the Hippo pathway.
Fig. 4.
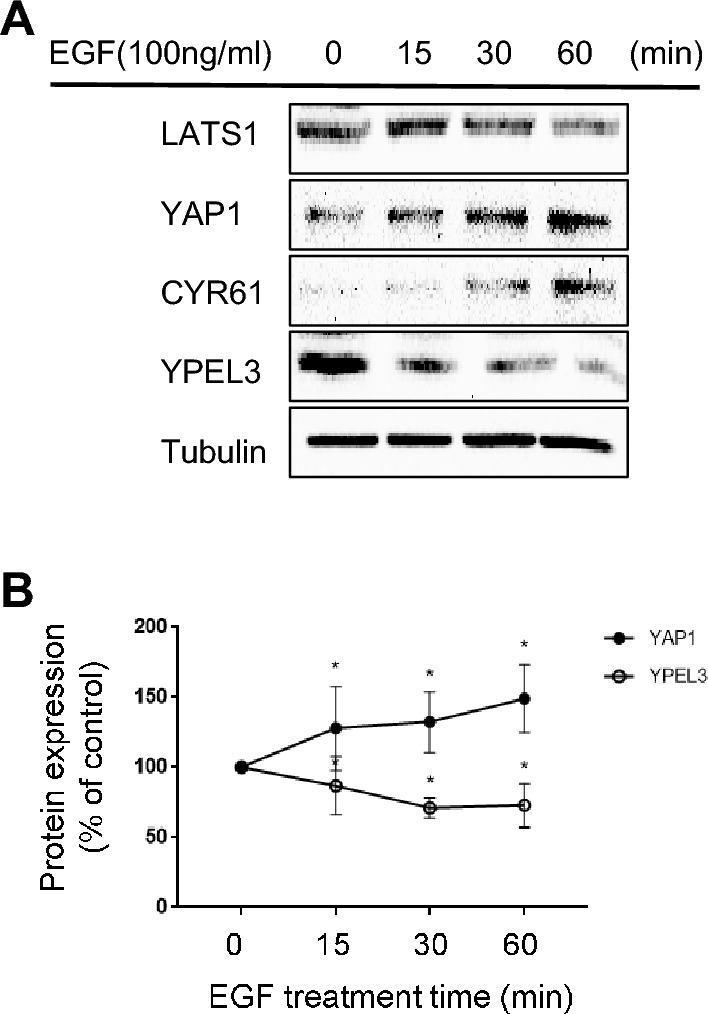
EGF suppresses YPEL3 expression through YAP1 activation. a MCF-7 cells were treated with EGF (100 ng/mL) for 15, 30, or 60 min. YAP1, CYR61, LATS1, and YPEL3 protein levels were measured via western blot analysis. α-Tubulin was used as a loading control. b Western blot was performed in triplicate. The data represent the mean ± SD (n = 3). *p < 0.05
Increased YPEL3 suppresses lamin B1 expression
Previous studies have demonstrated that YPEL3 can induce cellular senescence in MCF-7 breast cancer cells and U2OS osteosarcoma cells [19]. Furthermore, lamin B1 downregulation has been recognized as a useful biomarker for cellular senescence in various human and murine fibroblast cells [20]. To explore the relationship between YAP1, YPEL3, and lamin B1, specific siRNAs were used to knock down the expression of YAP1 and YPEL3. Our findings indicated that YPEL3 downregulation led to an increase in lamin B1 expression (Fig. 5a). To further confirm the relationship between YPEL3 and lamin B1, cells were transfected with a YPEL3 expression vector (pRP_YPEL3), and subsequent experiments including qRT-PCR, western blotting, and confocal microscopy were conducted 48 h after transfection (Fig. 5b–d). Our findings demonstrated that the overexpression of YPEL3 resulted in a significant reduction in lamin B1 expression. This decrease in lamin B1 expression may contribute to cellular senescence.
Fig. 5.

Increased YPEL3 suppresses lamin B1 expression. a MCF-7 cells were transfected with YAP1 siRNA and YPEL3 siRNA (70 nM) for 48 h. YAP1, YPEL3, and lamin B1 protein levels were measured via western blot analysis. α-Tubulin was used as a loading control. b MCF-7 cells were transfected with the pRP_YPEL3 vector (4 μg) for 48 h. Real-time qPCR was performed to measure the expression of YPEL3 and lamin B1 mRNA. The data represent the mean ± SD (n = 3). *p < 0.05. c YPEL3 and lamin B1 protein levels were measured via western blot analysis. α-Tubulin was used as a loading control. d Confocal analysis was performed to measure the YPEL3 and lamin B1 expression. Microscopy scale bar = 20 μm
Increased YPEL3 induces cellular senescence and apoptosis
To investigate the roles of YPEL3 induced by YAP1 deficiency in MCF-7 cells, senescence-associated β-galactosidase (SA-β-gal) activity was evaluated. The SA-β-gal assay is a widely used method for measuring cellular senescence, as it enables the detection of increased expression and activity of lysosomal β-galactosidase in senescent cells using X-gal as a chromogenic substrate [21]. After transfection with YAP1 and YPEL3 siRNAs, β-galactosidase staining was carried out for 24 h, and the extent of staining was analyzed using an inverted microscope. The cells treated with YAP1 siRNA exhibited higher β-galactosidase activity compared to the control cells. In contrast, the knockdown of YPEL3 prevented cellular senescence in cells treated with YAP1 siRNA, as shown in Fig. 6a.
Fig. 6.

Increased YPEL3 causes cellular senescence and apoptosis. a MCF-7 cells were transfected with YAP1 siRNA and YPEL3 siRNAs (70 nM) for 48 h. Cellular senescence was measured using the CST-senescence β-galactosidase staining kit. b MCF-7 cells were transfected with YAP1 and YPEL3 siRNAs (70 nM) for 48 h. The cells were treated with Muse™ annexin V & Dead Cell reagent. After incubation for 30 min at 37 °C, cell viability, and cell density were measured using a Muse™ cell analyzer. c The cellular OCR was estimated to evaluate mitochondrial respiration capacity in response to YAP1 siRNA and YPEL3 overexpression using a Seahorse XFe24 analyzer
Cellular senescence, which is an irreversible cell cycle arrest, is associated with various genes, including YAP1 and YPEL3 [4, 5, 7]. Therefore, apoptosis was measured to investigate the effect of YAP1 and YPEL3 knockdown on cellular senescence and cell death. Flow cytometry analysis revealed that YAP1 siRNA treatment induced apoptosis compared to the control cells (Fig. 6b). However, treatment with YPEL3 siRNA (70 nM) appeared to prevent apoptosis caused by YAP1 knockdown in MCF7 cells. These results suggest that YPEL3 plays a crucial role as both an important apoptosis-promoting factor and a novel marker protein for cellular senescence.
Reactive oxygen species (ROS) are produced during the process of energy generation. Due to their high reactivity, ROS can cause damage to DNA and proteins, leading to cellular senescence [22]. Furthermore, senescent cells have been found to exhibit a higher rate of oxygen consumption, as measured by the oxygen consumption rate (OCR) during pyruvic acid respiration [23]. Our results demonstrated that cells transfected with YAP1 siRNA or the YPEL3 expression vector exhibited higher OCR levels compared to the control cells (Fig. 6c). This finding suggests that YPEL3 overexpression induces senescence in cells with elevated oxygen consumption.
Discussion
The Hippo signaling pathway plays a crucial role in regulating cell growth, tissue homeostasis, and organ size. Central to this pathway is the YAP1, which functions as a transcriptional co-activator of target genes involved in cell proliferation and survival [24]. Although previous gene targeting studies have shed light on various aspects of the function of YAP1, many of its functions are still being discovered. Moreover, previous studies have demonstrated that YPEL3 induces permanent growth arrest and inhibits metastasis in various cancer cells [4, 5]. In this context, the relationship between the oncogene YAP1 and the tumor suppressor YPEL3 is an intriguing discovery with significant implications.
First, the KMplot database was used to assess the survival rate of breast cancer patients based on the expression levels of YAP1 and YPEL3 genes. The analysis revealed that patients with lower expression of the YAP1 gene had a higher survival rate, whereas those with higher expression of the YPEL3 gene also showed increased survival [25]. Afterward, YAP1 siRNA and verteporfin were used to downregulate YAP1 and examine its impact on YPEL3 expression. Treatment with YAP1 siRNA resulted in increased mRNA and protein levels of YPEL3. Similarly, verteporfin treatment, which inhibits YAP1 activity, led to a concentration-dependent upregulation of the YPEL3 protein. To induce YAP1 expression, MCF-7 cells were treated with EGF. EGF is known to suppress LATS1 expression and activate YAP1 activity [26]. LATS1 inactivation was identified through the reduced phosphorylation of its hydrophobic motif (T1079) upon EGF treatment. Consequently, EGF treatment resulted in decreased phosphorylation of YAP1 and increased the expression of YAP1 target genes including CTGF, CYR61, or AMOTL2 [27]. Our study also demonstrated that EGF induces YAP1 and CYR61 expression through the downregulation of LATS1. Taken together, our findings demonstrated that EGF treatment suppressed YPEL3 expression through the inactivation of Hippo signaling.
YPEL3 and YAP1 are known to be associated with cellular senescence [4, 7]. Cellular senescence is classically defined as an irreversible cell cycle arrest in the G1 phase of the cell cycle. However, previous studies have revealed that senescence can also be induced during prolonged G2 arrest, with the involvement of p21 in G2/M regulation [28]. ROS are byproducts generated during ATP generation via oxidative phosphorylation in mitochondria [23]. Senescent cells often exhibit elevated mitochondrial respiration [29]. The increased OCR levels and apoptosis caused by YAP1 siRNA in this study suggest that YPEL3 upregulation in response to YAP1 deficiency may induce cellular senescence. To further confirm the induction of cellular senescence by YPEL3, the expression of senescence biomarker protein lamin B1 and β-galactosidase activity were determined. Induction of YPEL3 led to a decrease in lamin B1 expression and increased SA-β-gal activity. Many senescence-associated factors activate the p53 pathways. In MCF-7 cells, YPEL3 exhibited a gene expression profile associated with the p53 pathway, which is consistent with the YPEL3-mediated p53 activation in human tumor cells carrying wild-type p53 [5]. Lamin B1 is also reduced upon p53 activation [15]. Therefore, our findings demonstrated that YPEL3 upregulation suppresses lamin B1 via p53 pathway.
In a previous study using human foreskin fibroblasts, the authors reported that an increased number of cell passages resulted in several senescence-associated changes, including increased DNA damage, morphological alterations, an increase in the number of mitochondria, and a significant increase in mitochondrial respiration. Moreover, it was found that apoptosis occurs in senescent cells at a later stage, which was potentially due to heightened oxidative stress. This suggests a potential link between mitochondrial function and apoptosis in senescent cells [23]. Additionally, YPEL3 has been identified as a small unstable apoptotic protein that is associated with p53 [5]. Our study revealed that the upregulation of YPEL3 led to an increase in apoptotic cells. This finding suggests that YPEL3 promotes cellular senescence and contributes to oxidative stress, which in turn can trigger apoptosis.
To the best of our knowledge, this study is the first to demonstrate that YPEL3 is a novel target protein of YAP1. However, the precise molecular mechanism through which YAP1 inactivates YPEL3 expression remains unclear. The regulation likely occurs at the transcriptional level, given that YAP1 siRNA can induce YPEL3 mRNA expression. To elucidate the specific mechanism of this interaction, it will be necessary to determine whether the YAP1-TEAD complex is involved in the inhibition of YPEL3 transcription. Recent studies have shown that the YAP1-mediated pathway can inhibit the transcription of the estrogen receptor α (ESR1) gene. Among the differentially expressed genes targeted by YAP1-TEAD, vestigial-like protein 3 (VGLL3) acts as an inhibitor of YAP1 by competitively inhibiting YAP1/TEAD interaction. Additionally, VGLL3 can recruit the corepressor NCOR2 to form the TEAD-VGLL3-NCOR2 complex, which enhances the transcriptional silencing of the ESR1 gene [30]. Additionally, a previous study reported that estrogen represses YPEL3 expression in an estrogen receptor α-dependent manner [19]. Based on these findings, it can be speculated that the formed TEAD-VGLL3-NCOR2 complex may transcriptionally inhibit YPEL3 expression. However, further studies are required to fully characterize the relationship between YAP1 activation and YPEL3 downregulation.
Funding
This research was supported by the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (Grant no. 2021R1A2C201239513, 2022R1A5A600076012) and the Chung-Ang University Graduate Research Scholarship in 2021. The funding agency had no role in the study design, data collection or analysis, the decision to publish, or the preparation of the manuscript.
Data availability
The Kaplan–Meier plot data that supports this study are available on the Kaplan–Meier Plotter website (https://kmplot.com/analysis/). Further information about our study can be provided by the corresponding author upon reasonable request.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Yeonju Kwon and Hyein Lee contributed equally to this study.
References
- 1.Han Y. Analysis of the role of the Hippo pathway in cancer. J Transl Med. 2019;17:116. doi: 10.1186/s12967-019-1869-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2014;13:63–79. doi: 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang J, Ji JY, Yu M, et al. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat Cell Biol. 2009;11:1444–1450. doi: 10.1038/ncb1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tuttle R, Simon M, Hitch DC, et al. Senescence-associated gene YPEL3 is downregulated in human colon tumors. Ann Surg Oncol. 2011;18:1791–1796. doi: 10.1245/s10434-011-1558-x. [DOI] [PubMed] [Google Scholar]
- 5.Kelley KD, Miller KR, Todd A, Kelley AR, Tuttle R, Berberich SJ. YPEL3, a p53-regulated gene that induces cellular senescence. Cancer Res. 2010;70:3566–3575. doi: 10.1158/0008-5472.CAN-09-3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong X, Li Y, Zhang X. Increased expression of the YPEL3 gene in human colonic adenocarcinoma tissue and the effects on proliferation, migration, and invasion of colonic adenocarcinoma cells in vitro via the Wnt/β-catenin signaling pathway. Med Sci Monit. 2018;24:4767–4775. doi: 10.12659/MSM.908173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie Q, Chen J, Feng H, et al. YAP/TEAD–mediated transcription controls cellular senescence. Cancer Res. 2013;73:3615–3624. doi: 10.1158/0008-5472.CAN-12-3793. [DOI] [PubMed] [Google Scholar]
- 8.Lee S, Lee J. Cellular senescence: a promising strategy for cancer therapy. BMB Rep. 2019;52:35–41. doi: 10.5483/BMBRep.2019.52.1.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumari R, Jat P. Mechanisms of cellular senescence: cell cycle arrest and senescence-associated secretory phenotype. Front Cell Dev Biol. 2021;9:645593. doi: 10.3389/fcell.2021.645593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He L, Chen Y, Feng J, Sun W, Li S, Ou M, Tang L. Cellular senescence regulated by SWI/SNF complex subunits through p53/p21 and p16/pRB pathway. Int J Biochem Cell Biol. 2017;90:29–37. doi: 10.1016/j.biocel.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Zeng S, Shen WH, Liu L. Senescence and cancer. Cancer Transl Med. 2018;4:70–74. doi: 10.4103/ctm.ctm_22_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim H, Chang J, Shao L, et al. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell. 2017;16:693–703. doi: 10.1111/acel.12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiley CD, Velarde MC, Lecot P, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23:303–314. doi: 10.1016/j.cmet.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vergnes L, Péterfy M, Bergo MO, Young SG, Reue K. Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci USA. 2004;101:10428–10433. doi: 10.1073/pnas.0401424101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freund A, Laberge R, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012;23:2066–2075. doi: 10.1091/mbc.e11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campisi J, Kim SH, Lim CS, Rubio M. Cellular senescence, cancer, and aging: the telomere connection. Exp Gerontol. 2001;36:1619–1637. doi: 10.1016/S0531-5565(01)00160-7. [DOI] [PubMed] [Google Scholar]
- 17.Wang C, Zhu X, Feng W, et al. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am J Cancer Res. 2015;6:27–37. [PMC free article] [PubMed] [Google Scholar]
- 18.Guo Q, Quan M, Xu L, et al. Enhanced nuclear localization of YAP1-2 contributes to EGF-induced EMT in NSCLC. J Cell Mol Med. 2022;26:1013–1023. doi: 10.1111/jcmm.17150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tuttle R, Miller KR, Maiorano JN, Termuhlen PM, Gao Y, Berberich SJ. Novel senescence associated gene, YPEL3, is repressed by estrogen in ER+ mammary tumor cells and required for tamoxifen-induced cellular senescence. Int J Cancer. 2012;130:2291–2299. doi: 10.1002/ijc.26239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang AS, Ong PF, Chojnowski A, Clavel C, Dreesen O. Loss of lamin B1 is a biomarker to quantify cellular senescence in photoaged skin. Sci Rep. 2017;7:15678. doi: 10.1038/s41598-017-15901-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (β)-galactosidase reflects an increase in lysosomal mass during replicative aging of human endothelial cells. J Cell Sci. 2000;113:3613–3622. doi: 10.1242/jcs.113.20.3613. [DOI] [PubMed] [Google Scholar]
- 22.Olga M, Véronique B, Antoine R, Xavier D-S, Gerardo F. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol. 2009;29:4495–4507. doi: 10.1128/MCB.01868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang D, Liu Y, Zhang R, et al. Apoptotic transition of senescent cells accompanied with mitochondrial hyper-function. Oncotarget. 2016;7:28286–28300. doi: 10.18632/oncotarget.8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan D. The Hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo L, Chen Y, Luo J, Zheng J, Shao G. YAP1 overexpression is associated with poor prognosis of breast cancer patients and induces breast cancer cell growth by inhibiting PTEN. FEBS Open Bio. 2019;9:437–445. doi: 10.1002/2211-5463.12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, You H, Li Y, Xu Y, He Q, Harris RC. EGF receptor–dependent YAP activation is important for renal recovery from AKI. J Am Soc Nephrol. 2018;29:2372–2385. doi: 10.1681/ASN.2017121272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ando T, Arang N, Wang Z, et al. EGFR regulates the hippo pathway by promoting the tyrosine phosphorylation of MOB1. Commun Biol. 2021;4:1237. doi: 10.1038/s42003-021-02744-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gire V, Dulic V. Senescence from G2 arrest, revisited. Cell Cycle. 2015;14:297–304. doi: 10.1080/15384101.2014.1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S, Mehta HH, Wan J, et al. Mitochondrial peptides modulate mitochondrial function during cellular senescence. Aging. 2018;10:1239–1256. doi: 10.18632/aging.101463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma S, Tang T, Probst G, et al. Transcriptional repression of estrogen receptor alpha by YAP reveals the Hippo pathway as therapeutic target for ER+ breast cancer. Nat Commun. 2022;13:1061. doi: 10.1038/s41467-022-28691-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The Kaplan–Meier plot data that supports this study are available on the Kaplan–Meier Plotter website (https://kmplot.com/analysis/). Further information about our study can be provided by the corresponding author upon reasonable request.