

Abstract
Population and genotype data are essential for genetic mapping. The multi-parent advanced generation intercross (MAGIC) population is a permanent mapping population used for precisely mapping quantitative trait loci. Moreover, genotyping-by-target sequencing (GBTS) is a robust high-throughput genotyping technology characterized by its low cost, flexibility, and limited requirements for information management and support. In this study, an 8-way MAGIC population was constructed using eight elite founder lines. In addition, GenoBaits Peanut 40K was developed and utilized for the constructed MAGIC population. A subset (297 lines) of the MAGIC population at the S2 stage was genotyped using GenoBaits Peanut 40K. Furthermore, these lines and the eight parents were analyzed in terms of pod length, width, area, and perimeter. A total of 27 single nucleotide polymorphisms (SNPs) were revealed to be significantly associated with peanut pod size-related traits according to a genome-wide association study. The GenoBaits Peanut 40K provided herein and the constructed MAGIC population will be applicable for future research to identify the key genes responsible for important peanut traits.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11032-023-01417-w.
Keywords: GenoBaits Peanut 40K, GBTS liquid chip, MAGIC population, Pod size, Peanut
Introduction
Peanut (Arachis hypogaea L.) is one of the most important oil crops worldwide (Lu et al. 2019). Developing high-quality and disease-resistant varieties with high yields has been a major goal of peanut breeding programs. Molecular marker-assisted selection (MAS) is one of the most effective plant breeding methods (Hasan et al. 2021). Elucidating the genetic basis of important peanut traits will help to improve specific characteristics of peanut cultivars through MAS. The development of next-generation sequencing technology and the availability of reference genomes for the cultivated groundnut and its ancestral species (Bertioli et al. 2016; Bertioli et al. 2019) have facilitated the mapping of genes mediating important peanut traits (Liu et al. 2020, Sun et al. 2022, Qi et al. 2022).
Single nucleotide polymorphism (SNP) arrays are robust high-throughput genotyping tools that are less expensive than next-generation sequencing platforms (Liu et al. 2022). Pandey et al. (2017) developed a high-density “Axiom_Arachis” genotyping array with 58K SNPs, which greatly promoted the mapping of genes related to key peanut characteristics. For example, this array was used to identify the genes mediating the resistance to late leaf spot (Moretzsohn et al. 2023). It has also been used along with a recombinant inbred line (RIL) population to reveal the genomic regions and candidate genes associated with the seed weight and shelling percentage of groundnut (Gangurde et al. 2023) as well as with the African core groundnut collection to detect novel loci for the resistance to groundnut rosette disease on the basis of a genome-wide association study (GWAS) (Achola et al. 2023). Additionally, the 58K SNP array was used to analyze the genetic diversity of Korean peanut germplasm (Nabi et al. 2021).
However, the targeted SNPs cannot be adjusted after the SNP probes are fixed in the routine chip SNP array (Liu et al. 2022). Therefore, Xu et al. (2020) developed genotyping-by-target sequencing (GBTS), which involves liquid chip technology and is characterized by its low cost, limited demands on facilities, highly flexible marker types, sharable and accumulative marker data, and limited requirements for information management and support. Moreover, this technology is widely applicable to the following areas including evaluating germplasm, constructing high-density genetic linkage maps, genetic mapping, and protecting intellectual property rights associated with crop varieties (Xu et al. 2020). To date, several GenoBaits marker panels have been developed for animals and plants, including GenoBaits Maize 20K (Guo et al. 2019), GenoBaits Rice 10K (Hussain et al. 2022), GenoBaits Soy40K (Liu et al. 2022), GenoBaits Wheat 16K (Huang et al. 2022), and GenoBaits Porcine SNP 50K (Wang et al. 2022).
Conventionally, populations used for quantitative trait locus (QTL) mapping have included RIL populations, doubled haploid backcrossed populations, or F2 populations derived from two parents, which can only be used to analyze two alleles and limits the genetic recombination and resolution for detecting QTLs (Bandillo et al. 2013). To overcome the limitations of bi-parental populations, a multi-parent advanced generation intercross (MAGIC) strategy was initially proposed for crops by Mackay and Powell (2007). This strategy can be used to analyze multiple alleles and to increase recombination rates and mapping resolutions (Cavanagh et al. 2008). Several MAGIC populations are available for diverse crops, including rice (Meng et al. 2016), maize (Dell'Acqua et al. 2015), and wheat (Stadlmeier et al. 2018). These populations have been used for the high-resolution dissection of the QTLs and genes responsible for complex agronomic traits.
To identify the genes associated with important peanut traits and develop useful markers for breeding, a MAGIC population was constructed and GenoBaits Peanut 40K was developed in this study. The objectives of this study were to (1) construct an 8-way MAGIC population using eight elite founder lines; (2) develop a liquid chip array GenoBaits Peanut 40K for peanut; (3) conduct a genetic analysis for the eight founder lines and the MAGIC population at the S2 stage; and (4) perform a GWAS for four traits related to peanut pod size using the MAGIC population at the S2 stage and its parents.
Materials and methods
Eight founder lines used for developing the MAGIC population
Eight elite peanut varieties or germplasms, namely, Yuanza9102 (N730), Zhonghua6 (N709), Yuhua15 (N734), Weihua8 (N743), Yueyou20 (N745), Fuhuasheng (N744), Silihong (N741), and NC94022 (N739) (Table 1), which were designated as A, B, C, D, E, F, G, and H, respectively, were used as the founder lines to develop the MAGIC population. These eight founder lines represent three of the five botanical varieties of peanut (i.e., ssp. fastigiata var. vulgaris, ssp. fastigiata var. fastigiata, and ssp. hypogaea var. hypogaea) and two kinds of irregular peanut types (i.e., irregular fastigiata-type and irregular hypogaea-type) (Table 1). Five of these lines (A–E), which were widely grown varieties developed by breeders in four major peanut-producing provinces in China, have high yield potentials and oil contents, are resistant to diseases, and exhibit other specific characteristics (e.g., wide adaptability, high shelling percentage, and deep pod mesh) (Table 1). Fuhuasheng and Silihong (F, G) are landraces originated from Shandong and Liaoning provinces in China, respectively (Table 1). Fuhuasheng is one of the most prominent parental varieties included in the pedigrees of most peanut varieties in China. Silihong, which produces pods that typically contain three or four seeds with red seed coat, is widely cultivated in northeastern China. NC94022 (H) is a late-maturing breeding line with a prostrate growth habit and originated in the USA (Shrestha et al. 2013).
Table 1.
Characteristics of the eight founder lines used for developing the MAGIC population
| ID | Germplasm/variety | Variant | Type | Origin | Characteristics |
|---|---|---|---|---|---|
| N730 | Yuanza9102 (A) | Irregular fastigiata-type | Breeding line | Henan, China | A variety, derived from an interspecific cross combination, with high oil content, bacterial wilt resistance, and wide adaption |
| N709 | Zhonghua6 (B) | subsp. fastigiata var. vulgaris | Breeding line | Hubei, China | Early maturity variety with small seed size, bacterial wilt resistance, and pale green leaves |
| N734 | Yuhua15 (C) | Irregular hypogaea-type | Breeding line | Henan, China | High yielding variety with good combining ability and high oil content, progenitor of many breeding lines |
| N743 | Weihua8 (D) | Irregular hypogaea-type | Breeding line | Shandong, China | High yielding variety with moderate pod size and thin pod shell |
| N745 | Yueyou20 (E) | subsp. fastigiata var. vulgaris | Breeding line | Guangdong, China | High resistance to leaves diseases, thick shell, and deep pod mesh on peanut shell |
| N744 | Fuhuasheng (F) | Irregular hypogaea-type | Landrace | Shandong, China | One of the main progenitors of Chinese peanut varieties, in the pedigrees of most varieties of China with deep pod waist |
| N741 | Silihong (G) | subsp. fastigiata var. fastigiata | Landrace | Liaoning, China | Multi-seeds in one pod with red seed coat and low number of branches |
| N739 | NC94022 (H) | subsp. hypogaea var. hypogaea | Breeding line | America | Late maturity with prostrate growth habit, small size seed, and light pod mesh |
SNP selection and array design for GenoBaits Peanut 40K
A diverse set comprising 353 peanut germplasms that underwent a whole-genome re-sequencing (20×) analysis was used to select SNPs. Approximately 0.93 million high-quality SNPs and insertions/deletions (Arachis hypogaea cv. Tifrunner version 1) were identified after the quality control and filtering: missing rate > 0.05 (any alleles with fewer than five supporting reads were marked as missing), minor allele frequency (MAF) < 0.01, and number of heterozygous alleles > 10 (Zheng et al. 2022). The SNP sites were selected according to the following criteria: (1) unique for each of the eight founder lines used as the parents of the MAGIC population (e.g., the genotype of one parent was A:A, whereas the genotype of the other seven parents was G:G); (2) evenly distributed across 20 chromosomes (as much as possible). The selected SNP sites were evaluated by MolBreeding Biotechnology Co., Ltd. (Shijiazhuang, China). Probes that were designed on the basis of the flanking sequences and targeted capture sequencing technology were subsequently synthesized. The effects of the selected SNPs on genes were predicted using SNPEff v5.0 (Cingolani et al. 2012).
Plant materials and phenotypes
The 297 S2 plants derived from one 8-way cross (A/E//D/G///B/C//F/H) and further two generation of single seed descent and the eight founder lines were used to evaluate GenoBaits Peanut 40K and the utility of the MAGIC population. The seeds of 297 S2 plants and eight founder lines were harvested and pod size-related characteristics (i.e., area, perimeter, length, and width) were measured using the SC-G software (Hangzhou Wanshen Detection Technology Co., Ltd., China). Because of the low reproduction efficiency of peanut, an average of approximately six pods was used for the phenotypic evaluation of each plant.
DNA isolation and genotyping with GenoBaits Peanut 40K
Genomic DNA was extracted from young unfolded leaves using the Plant Genomic DNA Extraction Kit (Tiangen Biotech, Beijing, China). The purity and integrity of the extracted DNA was evaluated by 1% agarose gel electrophoresis, whereas the DNA concentration was precisely determined using Qubit. The high-quality DNA samples were sequenced using GenoBaits Peanut 40K by MolBreeding Biotechnology Co., Ltd. (Shijiazhuang, China). The raw data were filtered for quality using the FASTQ software (Chen et al. 2018) and then aligned to the peanut reference genome (Arachis hypogaea cv. Tifrunner version 1) using the BWA software (Li and Durbin 2009). The standard pipeline of the GATK software (Poplin et al. 2018) was used to detect SNPs for genotyping. Finally, the SNP set was filtered according to the following parameters: missing rate < 0.3 and MAF > 0.05.
Diversity and population structure analyses and GWAS
The diversity of the 297 MAGIC lines and eight founder lines was analyzed using the UPGMA algorithm implemented in the TASSEL v5.0 software (Bradbury et al. 2007). The phylogenetic tree was drawn using the online program iTOL v6.7.3 (Letunic and Bork 2021). The population structure was deduced using ADMIXTURE v1.30 (K = 1–20) (Alexander and Lange 2011). The mixed linear model (MLM) implemented in TASSEL v5.0 (Bradbury et al. 2007) was used for the association analysis and the GWAS threshold was set as 0.05/n, with n representing the number of markers.
Results
Construction of a MAGIC population for peanut
A population was obtained from an 8-way cross involving the eight elite founder lines (Table 1). According to the method described by Bandillo et al. (2013), a half-allele mating system was used for the three stages required for the construction of the MAGIC population. At the first stage, 28 bi-parental crosses were conducted by inter-mating the eight founder lines. To obtain enough hybrids for the subsequent crosses, 30 seeds from each parent were sown. The resulting 28 F1 lines were inter-crossed for the 4-way cross (i.e., all 210 of the possible crosses). The combinations were set so that no parent was represented more than once in the 4-way cross. The 210 4-way F1 lines were inter-crossed for the 8-way cross (i.e., all 315 possible crosses were completed in the same manner).
For the 8-way cross, 4–36 confirmed hybrids were obtained from each of the crosses and advanced by selfing, with an average of approximately 250 seeds harvested per cross at the S2 stage. A subset of the 8-way cross consisting of 35 crosses with a population size of approximately 200 (or 500 for one cross) was selected and used for advancing generations via single seed descent. Thus, the target population comprised 7000 lines (i.e., 35 × 200). The subset was selected in such a manner that only one of the nine possible crosses was chosen (e.g., one of A/B//C/D///E/F//G/H, A/B//C/D///E/G//F/H, A/B//C/D///E/H//F/G, A/C//B/D///E/F//G/H, A/C//B/D///E/G//F/H, A/C//B/D///E/H//F/G, A/D//B/C///E/F//G/H, A/D//B/C///E/G//F/H, and A/D//B/C///E/H//F/G). The other crosses were stored at −20 °C for later use.
SNP selection and array design
To genotype the MAGIC population, GenoBaits Peanut 40K was developed using liquid chip technology. The 40,000 SNPs that were selected from the variation set of 353 peanut germplasms (Supplementary Table S1) were evenly distributed across the 20 peanut chromosomes (Fig. 1A). The number of SNPs per chromosome ranged from 1070 (Arahy.08) to 3029 (Arahy.14), with an average of one SNP per 63,457 bases (Table 2). In terms of their genomic positions, 48.83% of the SNPs were located in intergenic regions, but the SNPs were also present in the following locations: upstream_gene (19.31%), downstream_gene (18.05%), intron (5.58%), missense (3.13%), synonymous (1.57%), non_coding_transcript_extron (1.18%), 3_prime_UTR (0.90%), 5_prime_UTR (0.80%), and others (0.65%) including splice_region, stop_gained, 5_prime_UTR_premature_start_codon_gain, splice_accepter, splice_donor, start_lost, stop_lost, and stop_retained (Fig. 1B).
Fig. 1.

Distribution of the 40K SNPs on 20 chromosomes (A) and genomic positions of selected SNPs (B)
Table 2.
Number of variants on 20 chromosomes
| Chromosome | Length | Variants | Variants rate |
|---|---|---|---|
| arahy.Tifrunner.gnm1.Arahy.01 | 112,420,854 | 2009 | 55,958 |
| arahy.Tifrunner.gnm1.Arahy.02 | 102,981,163 | 1804 | 57,084 |
| arahy.Tifrunner.gnm1.Arahy.03 | 143,813,506 | 2570 | 55,958 |
| arahy.Tifrunner.gnm1.Arahy.04 | 128,801,742 | 1730 | 74,451 |
| arahy.Tifrunner.gnm1.Arahy.05 | 115,930,344 | 1933 | 59,974 |
| arahy.Tifrunner.gnm1.Arahy.06 | 115,504,342 | 1779 | 64,926 |
| arahy.Tifrunner.gnm1.Arahy.07 | 81,119,488 | 1763 | 46,012 |
| arahy.Tifrunner.gnm1.Arahy.08 | 51,897,010 | 1070 | 48,501 |
| arahy.Tifrunner.gnm1.Arahy.09 | 120,519,698 | 2463 | 48,932 |
| arahy.Tifrunner.gnm1.Arahy.10 | 117,088,237 | 1828 | 64,052 |
| arahy.Tifrunner.gnm1.Arahy.11 | 149,299,306 | 2788 | 53,550 |
| arahy.Tifrunner.gnm1.Arahy.12 | 120,579,088 | 1534 | 78,604 |
| arahy.Tifrunner.gnm1.Arahy.13 | 146,725,006 | 2614 | 56,130 |
| arahy.Tifrunner.gnm1.Arahy.14 | 143,237,272 | 3029 | 47,288 |
| arahy.Tifrunner.gnm1.Arahy.15 | 160,879,708 | 2543 | 63,263 |
| arahy.Tifrunner.gnm1.Arahy.16 | 154,808,347 | 1662 | 93,145 |
| arahy.Tifrunner.gnm1.Arahy.17 | 134,922,436 | 2249 | 59,992 |
| arahy.Tifrunner.gnm1.Arahy.18 | 135,150,084 | 1453 | 93,014 |
| arahy.Tifrunner.gnm1.Arahy.19 | 158,625,764 | 1441 | 110,080 |
| arahy.Tifrunner.gnm1.Arahy.20 | 143,980,330 | 1738 | 82,842 |
| Total | 2,538,283,725 | 40,000 | 63,457 |
To more precisely genotype the MAGIC population, 30,082 sites that were specific to one of the eight founder lines were designated as 1:7 (i.e., the genotype of one parent differed from that of the other seven parents), whereas 9918 sites were designated as 2:6 (i.e., the genotype of two parents differed from that of the other six parents) to ensure the 40,000 SNPs were evenly distributed on the 20 chromosomes (Supplementary Table S2). The founder line with the most unique sites was N741, followed by N745. The founder lines with the fewest unique sites were N734 and N743 (Supplementary Table S2). The number of polymorphic SNPs between each pair of the eight founder lines (28 combinations) ranged from 1815 (between N739 and N744) to 18,458 (between N741 and N745) (Supplementary Table S3). Probes were designed for each SNP in both the forward and reverse direction, but three primers were designed for nine SNP sites (Supplementary Table S4).
Accuracy of the GBTS technology
The accuracy of the GBTS technology was evaluated by comparing the genotypes of the eight founder lines revealed by GenoBaits Peanut 40K and the previously reported genotypes determined on the basis of whole-genome resequencing technology (Zheng et al. 2022). More specifically, the number of consistent SNPs between the two technologies was divided by the total number of SNP (i.e., 40,000). The accuracy ranged from 96.57 to 99.33% for N709, N730, N734, N743, N744, and N745, while those of the other two founder lines (N739 and N741) was only about 84% (Table 3). The lower accuracy for N739 and N741 was likely due to excessive heterozygous and missing sites, respectively (Table 3), which may be related to the differences between the genomes of these two lines and the reference genome.
Table 3.
Comparison of the genotypes of eight founder lines determined using liquid chip technology and sequencing data
| Variety | N709 | N730 | N734 | N739 | N741 | N743 | N744 | N745 |
|---|---|---|---|---|---|---|---|---|
| The number of SNPs different with sequencing | ||||||||
| Homozygous | 23 | 11 | 19 | 663 | 49 | 23 | 18 | 28 |
| Heterozygous | 61 | 39 | 42 | 4825 | 199 | 34 | 39 | 70 |
| Indel | 3 | 2 | 2 | 0 | 2 | 1 | 1 | 1 |
| Missing | 586 | 574 | 205 | 1232 | 6076 | 217 | 1315 | 582 |
| The number of SNPs consistent with sequencing | 39,327 | 39,374 | 39,732 | 33,280 | 33,674 | 39,725 | 38,627 | 39,319 |
| Accuracy | 98.32% | 98.44% | 99.33% | 83.20% | 84.19% | 99.31% | 96.57% | 98.30% |
Genetic analysis of the MAGIC population at the S2 stage
The 297 lines of the 8-way cross were genotyped at the S2 stage using GenoBaits Peanut 40K. A total of 18,816 filtered SNPs with a missing rate < 0.3 and MAF > 0.05 were used for the genetic analysis. A phylogenetic tree with the 297 lines and eight founder lines was constructed. The 305 lines were roughly divided into five clusters, which were differentiated by color in the phylogenetic tree (brown, red, blue, green, and purple) (Fig. 2). The parent N741 and line 216 were clustered into clade 1 and were far away from the other seven parents (clade 5) (Fig. 2), which due to that N741 is a landrace from ssp. fastigiata var. fastigiata and exhibits a huge genetic difference with other seven parents (i.e., 13,655 unique sites in Supplementary Table S2). Among the seven parents in clade 5, three parents from ssp. fastigiata (N709, N730, and N745) were grouped together and then clustered with the parents from ssp. hypogaea (N734, N743, N744, and N739) (Fig. 2). Except 216, the eight founder lines were not cluster together with the S2 lines, the reason may be that the S2 population is still highly heterozygous as well as the parents are homozygous.
Fig. 2.
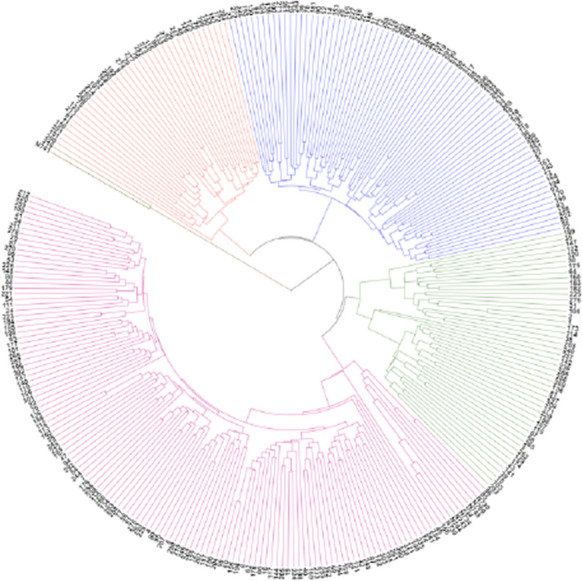
Phylogenetic tree comprising the 297 lines of the MAGIC population at the S2 stage and eight founder lines
Population structures were analyzed using ADMIXTURE v1.30, with K = 1–20. The 305 lines were grouped into nine clusters because the CV error reached the smallest when K = 9 (Fig. 3A–B). The eight founder lines were grouped into five clusters, with N709, N730, and N741 in separate clusters, N734, N739, and N745 in the same cluster, and N743 and N744 in another cluster.
Fig. 3.

CV error for K = 1–20 (A) and population structure for K = 9 (B)
Genome-wide association study for pod size-related traits
The 18,816 filtered SNPs were screened for SNPs significantly associated with the peanut pod area, perimeter, length, and width according to the MLM model. The Q file for K = 9 generated during the population structure analysis was used as the covariate (Q) in the MLM model. Kinship (K) was calculated using TASSEL v5.0. A total of 27 SNPs significantly associated with at least two of the four pod size-related traits were identified at the threshold of 5.50 [−log(0.05/18,186)] (Table 4, Fig. 4, and Supplementary Table S5). Of these SNPs, 10 were on chromosome 7, 16 were on chromosome 12, and one was on chromosome 17 (Table 4 and Supplementary Table S5).
Table 4.
Results for the GWAS of peanut pod size-related traits
| Chromosome | Position | −log(P) | add_effect | Trait |
|---|---|---|---|---|
| 7 | 130688 | 6.27–10.61 | − | Area, perimeter, width |
| 7 | 292285 | 6.27–10.70 | + | Area, perimeter, width |
| 7 | 492470 | 5.72–7.97 | + | Area, width |
| 7 | 586747 | 5.91–10.80 | + | Area, perimeter, width |
| 7 | 811108 | 5.63–9.89 | + | Area, perimeter, width |
| 7 | 848538 | 6.75–9.76 | − | Area, width |
| 7 | 890383 | 5.55–8.87 | − | Area, perimeter, width |
| 7 | 920314 | 5.74–9.24 | − | Area, perimeter, width |
| 7 | 1008387 | 6.30–8.40 | + | Area, width |
| 7 | 1056718 | 5.61–9.12 | + | Area, perimeter, width |
| 12 | 7302293 | 5.57–6.65 | + | Perimeter, length, width |
| 12 | 7423728 | 5.57–6.65 | − | Perimeter, length, width |
| 12 | 7926457 | 5.51–6.53 | − | Perimeter, length, width |
| 12 | 7964822 | 5.70–6.48 | − | Perimeter, length, width |
| 12 | 8555800 | 5.58–6.83 | + | Area, perimeter, length, width |
| 12 | 8695703 | 5.83–7.26 | − | Area, perimeter, length, width |
| 12 | 8746767 | 6.01–6.89 | + | Perimeter, length, width |
| 12 | 8817149 | 5.95–6.70 | − | Perimeter, length, width |
| 12 | 8913786 | 5.88–6.97 | + | Perimeter, length, width |
| 12 | 9170907 | 5.86–7.09 | − | Perimeter, length, width |
| 12 | 9387102 | 5.57–7.28 | − | Area, perimeter, length, width |
| 12 | 9486492 | 5.78–7.58 | + | Area, perimeter, length, width |
| 12 | 9610185 | 5.86–7.53 | − | Area, perimeter, length, width |
| 12 | 9723588 | 5.86–6.45 | − | Perimeter, width |
| 12 | 9837499 | 5.86–6.45 | − | Perimeter, width |
| 12 | 9911046 | 5.86–6.45 | − | Perimeter, width |
| 17 | 625720 | 6.10–10.61 | + | Area, perimeter, width |
Fig. 4.

Manhattan and QQ plots for peanut pod area (A), perimeter (B), length (C), and width (D)
The significant SNPs on chromosomes 7 and 12 were linked, respectively. The site Arahy17:625720 was linked with the identified SNPs on chromosome 7. There were significant phenotypic differences between the lines with the genotype of G:G and T:T at the site Arahy07:292285. Most lines had the same genotype at the significant sites on chromosome 12. For example, C:C was the common genotype at Arahy12:9486492, but 14 lines with small pods had a different genotype at this site (i.e., T:T). Notably, among the lines with C:C at Arahy12:9486492, the lines with G:G had significantly larger pods than those with T:T at Arahy07:292285 (Fig. 5). The genotypes of N734 and N739 differed from those of the other six parents at the significant SNP sites on chromosomes 7 and 17. Additionally, the genotype of N741 differed from those of the other seven parents at the significant SNP sites on chromosome 12.
Fig. 5.

Differences in the peanut pod area, perimeter, length, and width between lines with different genotypes (G:G or T:T) at Arahy07:292285, but the same genotype (C:C) at Arahy12:9486492
Candidate genes for pod size
The significant SNPs on chromosome 7 covered a physical region comprising 0.93 Mb (Table 4 and Supplementary Table S5) that contained 31 candidate genes, including those encoding a fasciclin-like arabinogalactan family protein (Arahy.P7DY53), a transcriptional regulator (STERILE APETALA-like; Arahy.5EZV1I), and a transcription factor subunit (NFYB/HAP3) (Supplementary Table S6). A total of 47 candidate genes were identified in the region (2.61 Mb) covered by the significant SNPs on chromosome 12. Some of the key genes in this region encoded a C6HC-type zinc finger RING/U-box protein (Arahy.UZLY68), a homeobox-leucine zipper protein (Arahy.V0IP08), and a MYB transcription factor (Arahy.7ML2J7) (Supplementary Table S6). The significant SNP on chromosome 17 was located in the exon of Arahy.5QP4QH, which encodes a rho GDP-dissociation inhibitor 1-like protein (Supplementary Table S6).
Discussion
Applicability of GenoBaits Peanut 40K
The greatest advantage of liquid chip technology-based marker panels over the alternatives is that the number of markers (e.g., 10, 20, and 40K) can vary depending on how the marker panels are being used. Moreover, they are useful for genotyping regardless of the number of samples (i.e., unlimited sample size) (Xu et al. 2020). Although GenoBaits Peanut 40K was designed for the MAGIC population, it has many other uses. For example, it is applicable for evaluating germplasm diversity, performing a linkage analysis of bi-parental populations, and conducting GWAS. The number of polymorphic SNPs between almost each pair of the two parents exceeded 3000 (approximately 9900 on average), ensuring to some extent its wide applicability (Supplementary Table S3).
The GenoBaits Peanut 40K panel may be used to analyze most peanut germplasms, although there are a few exceptions. The accuracy of the genotyping of N741 and N739 was relatively low (i.e., 84%) because of the number of missing or heterozygous sites in N741 (6076 missing sites) and N739 (4825 missing sites and 1232 heterozygous sites) (Table 3). This may be associated with the fact that N741 belonged to ssp. fastigiata var. fastigiata and one parent of N739 belonged to ssp. hypogaea var. hirsuta (Shrestha et al. 2013), the genome of which may differ substantially from the reference genome used in the present study. Therefore, an increase in sequencing depth may be required to capture the missing sites.
Benefits of the MAGIC population
Constructing MAGIC populations is a new approach for exploiting the diversity in plant genetic resources (Arrones et al. 2020). These populations are very useful for dissecting complex traits, selecting elite lines for breeding, and constructing genomic prediction models (Arrones et al. 2020; Puglisi et al. 2021). The identified region on chromosome 7 was consistent with the QTL reported by Alyr et al. (2020). The candidate gene identified on chromosome 17 was mapped to the significant region of chromosome 7 using the updated reference genome. To the best of our knowledge, the identified region on chromosome 12 has not been reported. The significant SNPs on chromosome 7 may resulted from the difference between two parents (N734 and N739) and the other six parents, whereas the sites identified on chromosome 12 may due to the difference between N741 and the other seven parents. In addition to pod size, the constructed MAGIC population may also be used to investigate other important peanut traits because the progenies of the population vary in terms of growth habit, seed coat color, pod shell type, oil content, and other characteristics.
Functions of the candidate genes
A total of 79 candidate genes influencing pod size were detected in the three significant regions (Supplementary Table S6). The fasciclin-like arabinogalactan family protein gene (Arahy.P7DY53) has been reportedly related to fundamental aspects of embryogenesis and seed development across angiosperms (Costa et al. 2019). A previous study showed that this gene is involved in the regulation of the Brassica napus L. silique length (Wang et al. 2019). The transcriptional regulator STERILE APETALA-like and the F-box domain encoded by Arahy.5EZV1I was reported to regulate the peanut pod and seed sizes (Alyr et al. 2020). Furthermore, Arahy.UZLY68 encodes a C6HC-type zinc finger RING/U-box protein that may modulate the peanut pod size via ubiquitination according to a recent report on rice (Yang et al. 2021). The homeobox-leucine zipper protein gene (Arahy.V0IP08) affects maize kernel size and weight (Sun et al. 2022). The MYB transcription factor gene (Arahy.7ML2J7) controls the size of Arabidopsis thaliana seeds (Zhang et al. 2013). Additionally, the possibility the gene encoding a rho GDP-dissociation inhibitor 1-like protein (Arahy.5QP4QH) may affect peanut pod size is supported by the findings of an earlier study, which revealed the Rho-family GTPase-encoding gene OsRac1 controls rice grain size and yield by regulating cell division (Zhang et al. 2019).
Supplementary information
(XLSX 9234 kb)
Acknowledgements
We thank Liwen (Edanz) (http://www.liwenbianji.cn) for editing the English text of a draft of this manuscript.
Author contribution
Z.S. wrote the manuscript and performed the genotype analysis. Z.Z. designed the GenoBaits Peanut 40K and the experiment. F.Q. prepared the figures. J.W., M.W., and R.Z. performed the experiment in laboratory. H.L., J.X., L.Q., W.D., B.H., and S.H. provided help in field experiment. X.Z. supervised this project, made the choice of founder lines, and revised the manuscript. All authors read and approved of its content.
Funding
This study was supported by the National Key Research and Development Program of China (2022YFD1200400), Major Science and Technology Projects of Henan Province (221100110300), Henan Union Fund (222301420026), Fund for Distinguished Young Scholars from Henan Academy of Agricultural Sciences (2023JQ02), China Agriculture Research System of MOF and MARA (CARS-13), Henan Provincial Agriculture Research System, China (S2012-5), and Special Fund for Scientific and Technological Innovation Team of Henan Academy of Agricultural Sciences (2023TD1).
Data Availability
All data generated or analyzed during this study are included in this published article and its supplementary information files. The clean resequencing data obtained in this study are available at the BioProject database at China National Center for Bioinformation under the BioProject ID: PRJCA019839 (https://ngdc.cncb.ac.cn/gsub/submit/bioproject/PRJCA019839). Materials used in this study are available from the corresponding authors.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors provided the consent for publication.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ziqi Sun and Zheng Zheng contributed equally to this work.
References
- Achola E, Wasswa P, Fonceka D, Clevenger JP, Bajaj P, Ozias-Akins P, Rami JF, Deom CM, Hoisington DA, Edema R, Odeny DA, Okello DK. Genome-wide association studies reveal novel loci for resistance to groundnut rosette disease in the African core groundnut collection. Theor Appl Genet. 2023;136(3):35. doi: 10.1007/s00122-023-04259-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DH, Lange K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinformatics. 2011;12:246. doi: 10.1186/1471-2105-12-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alyr MH, Pallu J, Sambou A, Nguepjop JR, Seye M, Tossim HA, Djiboune YR, Sane D, Rami JF, Fonceka D. Fine-mapping of a wild genomic region involved in pod and seed size reduction on chromosome A07 in peanut (Arachis hypogaea L.) Genes (Basel) 2020;11(12):1402. doi: 10.3390/genes11121402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrones A, Vilanova S, Plazas M, Mangino G, Pascual L, Díez MJ, Prohens J, Gramazio P. The dawn of the age of multi-parent MAGIC populations in plant breeding: novel powerful next-generation resources for genetic analysis and selection of recombinant elite material. Biology (Basel) 2020;9(8):229. doi: 10.3390/biology9080229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandillo N, Raghavan C, Muyco PA, Sevilla MA, Lobina IT, Dilla-Ermita CJ, Tung CW, McCouch S, Thomson M, Mauleon R, Singh RK, Gregorio G, Redoña E, Leung H. Multi-parent advanced generation inter-cross (MAGIC) populations in rice: progress and potential for genetics research and breeding. Rice (NY) 2013;6(1):11. doi: 10.1186/1939-8433-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertioli DJ, Cannon SB, Froenicke L, Huang G, Farmer AD, Cannon EK, Liu X, Gao D, Clevenger J, Dash S, Ren L, Moretzsohn MC, Shirasawa K, Huang W, Vidigal B, Abernathy B, Chu Y, Niederhuth CE, Umale P, Araújo AC, Kozik A, Kim KD, Burow MD, Varshney RK, Wang X, Zhang X, Barkley N, Guimarães PM, Isobe S, Guo B, Liao B, Stalker HT, Schmitz RJ, Scheffler BE, Leal-Bertioli SC, Xun X, Jackson SA, Michelmore R, Ozias-Akins P. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet. 2016;48(4):438–446. doi: 10.1038/ng.3517. [DOI] [PubMed] [Google Scholar]
- Bertioli DJ, Jenkins J, Clevenger J, Dudchenko O, Gao D, Seijo G, Leal-Bertioli SCM, Ren L, Farmer AD, Pandey MK, Samoluk SS, Abernathy B, Agarwal G, Ballén-Taborda C, Cameron C, Campbell J, Chavarro C, Chitikineni A, Chu Y, Dash S, El Baidouri M, Guo B, Huang W, Kim KD, Korani W, Lanciano S, Lui CG, Mirouze M, Moretzsohn MC, Pham M, Shin JH, Shirasawa K, Sinharoy S, Sreedasyam A, Weeks NT, Zhang X, Zheng Z, Sun Z, Froenicke L, Aiden EL, Michelmore R, Varshney RK, Holbrook CC, Cannon EKS, Scheffler BE, Grimwood J, Ozias-Akins P, Cannon SB, Jackson SA, Schmutz J. The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat Genet. 2019;51(5):877–884. doi: 10.1038/s41588-019-0405-z. [DOI] [PubMed] [Google Scholar]
- Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23(19):2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- Cavanagh C, Morell M, Mackay I, Powell W. From mutations to MAGIC: resources for gene discovery, validation and delivery in crop plants. Curr Opin Plant Biol. 2008;11:215–221. doi: 10.1016/j.pbi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Pereira AM, Pinto SC, Silva J, Pereira LG, Coimbra S. In silico and expression analyses of fasciclin-like arabinogalactan proteins reveal functional conservation during embryo and seed development. Plant Reprod. 2019;32(4):353–370. doi: 10.1007/s00497-019-00376-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Acqua M, Gatti DM, Pea G, Cattonaro F, Coppens F, Magris G, Hlaing AL, Aung HH, Nelissen H, Baute J, Frascaroli E, Churchill GA, Inzé D, Morgante M, Pè ME. Genetic properties of the MAGIC maize population: a new platform for high definition QTL mapping in Zea mays. Genome Biol. 2015;16(1):167. doi: 10.1186/s13059-015-0716-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangurde SS, Pasupuleti J, Parmar S, Variath MT, Bomireddy D, Manohar SS, Varshney RK, Singam P, Guo B, Pandey MK. Genetic mapping identifies genomic regions and candidate genes for seed weight and shelling percentage in groundnut. Front Genet. 2023;14:1128182. doi: 10.3389/fgene.2023.1128182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Wang H, Tao J, Ren Y, Xu C, Wu K, Zou C, Zhang J, Xu Y. Development of multiple SNP marker panels affordable to breeders through genotyping by target sequencing (GBTS) in maize. Mol Breed. 2019;39:37. doi: 10.1007/s11032-019-0940-4. [DOI] [Google Scholar]
- Hasan N, Choudhary S, Naaz N, Sharma N, Laskar RA. Recent advancements in molecular marker-assisted selection and applications in plant breeding programmes. J Genet Eng Biotechnol. 2021;19(1):128. doi: 10.1186/s43141-021-00231-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Zhang Y, Ren H, Li X, Zhang X, Zhang Z, Zhang C, Liu S, Wang X, Zeng Q, Wang Q, Singh RP, Bhavani S, Wu J, Han D, Kang Z. Epistatic interaction effect between chromosome 1BL (Yr29) and a novel locus on 2AL facilitating resistance to stripe rust in Chinese wheat Changwu 357-9. Theor Appl Genet. 2022;135(7):2501–2513. doi: 10.1007/s00122-022-04133-9. [DOI] [PubMed] [Google Scholar]
- Hussain I, Ali S, Liu W, Awais M, Li J, Liao Y, Zhu M, Fu C, Liu D, Wang F. Identification of heterotic groups and patterns based on genotypic and phenotypic characteristics among rice accessions of diverse origins. Front Genet. 2022;13:811124. doi: 10.3389/fgene.2022.811124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–W296. doi: 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Sun Z, Zhang X, Qin L, Qi F, Wang Z, Du P, Xu J, Zhang Z, Han S, Li S, Gao M, Zhang L, Cheng Y, Zheng Z, Huang B, Dong W. QTL mapping of web blotch resistance in peanut by high-throughput genome-wide sequencing. BMC Plant Biol. 2020;20(1):249. doi: 10.1186/s12870-020-02455-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu S, Zhang Z, Ni L, Chen X, Ge Y, Zhou G, Tian Z. GenoBaits Soy40K: a highly flexible and low-cost SNP array for soybean studies. Sci China Life Sci. 2022;65:1898–1901. doi: 10.1007/s11427-022-2130-8. [DOI] [PubMed] [Google Scholar]
- Lu Q, Hong Y, Li S, Liu H, Li H, Zhang J, Lan H, Liu H, Li X, Wen S, Zhou G, Varshney RK, Jiang H, Chen X, Liang X. Genome-wide identification of microsatellite markers from cultivated peanut (Arachis hypogaea L.) BMC Genomics. 2019;20(1):799. doi: 10.1186/s12864-019-6148-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay I, Powell W. Methods for linkage disequilibrium mapping in crops. Trends Plant Sci. 2007;12:57–63. doi: 10.1016/j.tplants.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Meng L, Guo L, Ponce K, Zhao X, Ye G. Characterization of three indica rice multiparent advanced generation intercross (MAGIC) populations for quantitative trait loci identification. Plant Genome. 2016;9:1–14. doi: 10.3835/plantgenome2015.10.0109. [DOI] [PubMed] [Google Scholar]
- Moretzsohn MC, Dos Santos JF, Moraes ARA, Custódio AR, Michelotto MD, Mahrajan N, Leal-Bertioli SCM, Godoy IJ, Bertioli DJ. Marker-assisted introgression of wild chromosome segments conferring resistance to fungal foliar diseases into peanut (Arachis hypogaea L.) Front Plant Sci. 2023;14:1139361. doi: 10.3389/fpls.2023.1139361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabi RBS, Cho KS, Tayade R, Oh KW, Lee MH, Kim JI, Kim S, Pae SB, Oh E. Genetic diversity analysis of Korean peanut germplasm using 48K SNPs ‘Axiom_Arachis’ array and its application for cultivar differentiation. Sci Rep. 2021;11(1):16630. doi: 10.1038/s41598-021-96074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey MK, Agarwal G, Kale SM, Clevenger J, Nayak SN, Sriswathi M, Chitikineni A, Chavarro C, Chen X, Upadhyaya HD, Vishwakarma MK, Leal-Bertioli S, Liang X, Bertioli DJ, Guo B, Jackson SA, Ozias-Akins P, Varshney RK. Development and evaluation of a high density genotyping ‘Axiom_Arachis’ array with 58 K SNPs for accelerating genetics and breeding in groundnut. Sci Rep. 2017;7:40577. doi: 10.1038/srep40577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Auwera G, Kling DE, Gauthier LD, Levy-Moonshine A, Roazen D, Shakir K, Thibault J, Chandran S, Whelan C, Lek M, Gabriel S, Daly MJ, Neale B, MacArthur DG, Banks E (2018) Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. 10.1101/201178
- Puglisi D, Delbono S, Visioni A, Ozkan H, Kara İ, Casas AM, Igartua E, Valè G, Piero ARL, Cattivelli L, Tondelli A, Fricano A. Genomic prediction of grain yield in a barley MAGIC population modeling genotype per environment interaction. Front Plant Sci. 2021;12:664148. doi: 10.3389/fpls.2021.664148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi F, Sun Z, Liu H, Zheng Z, Qin L, Shi L, Chen Q, Liu H, Lin X, Miao L, Tian M, Wang X, Huang B, Dong W, Zhang X. QTL identification, fine mapping, and marker development for breeding peanut (Arachis hypogaea L.) resistant to bacterial wilt. Theor Appl Genet. 2022;135(4):1319–1330. doi: 10.1007/s00122-022-04033-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha A, Srinivasan R, Sundaraj S, Culbreath AK, Riley DG. Second generation peanut genotypes resistant to thrips-transmitted tomato spotted wilt virus exhibit tolerance rather than true resistance and differentially affect thrips fitness. J Econ Entomol. 2013;106(2):587–596. doi: 10.1603/EC12430. [DOI] [PubMed] [Google Scholar]
- Stadlmeier M, Hartl L, Mohler V. Usefulness of a multiparent advanced generation intercross population with a greatly reduced mating design for genetic studies in winter wheat. Front Plant Sci. 2018;9:1–12. doi: 10.3389/fpls.2018.01825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Qi F, Liu H, Qin L, Xu J, Shi S, Zhang Z, Miao L, Huang B, Dong W, Wang X, Tian M, Feng J, Zhao R, Zheng Z, Zhang X. QTL mapping of quality traits in peanut using whole-genome resequencing. The crop journal. 2022;10(1):177–184. doi: 10.1016/j.cj.2021.04.008. [DOI] [Google Scholar]
- Wang H, Zaman QU, Huang W, Mei D, Liu J, Wang W, Ding B, Hao M, Fu L, Cheng H, Hu Q. QTL and candidate gene identification for silique length based on high-dense genetic map in Brassica napus L. Front Plant Sci. 2019;10:1579. doi: 10.3389/fpls.2019.01579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Shi S, Wang G, Luo W, Wei X, Qiu A, Luo F, Ding X. Using machine learning to improve the accuracy of genomic prediction of reproduction traits in pigs. J Anim Sci Biotechnol. 2022;13(1):60. doi: 10.1186/s40104-022-00708-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YB, Yang QN, Zheng HJ, Xu YF, Sang ZQ, Guo ZF, Peng H, Zhang C, Lan HF, Wang YB, Wu KS, Tao JJ, Zhang JN. Genotyping by target sequencing (GBTS) and its applications (in Chinese) Sci Agric Sin. 2020;53:2983–3004. [Google Scholar]
- Yang W, Wu K, Wang B, Liu H, Guo S, Guo X, Luo W, Sun S, Ouyang Y, Fu X, Chong K, Zhang Q, Xu Y. The RING E3 ligase CLG1 targets GS3 for degradation via the endosome pathway to determine grain size in rice. Mol Plant. 2021;14(10):1699–1713. doi: 10.1016/j.molp.2021.06.027. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liang W, Shi J, Xu J, Zhang D. MYB56 encoding a R2R3 MYB transcription factor regulates seed size in Arabidopsis thaliana. J Integr Plant Biol. 2013;55(11):1166–1178. doi: 10.1111/jipb.12094. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Liu R, Xue HW, Yang Z. The Rho-family GTPase OsRac1 controls rice grain size and yield by regulating cell division. Proc Natl Acad Sci U S A. 2019;116(32):16121–16126. doi: 10.1073/pnas.1902321116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Sun Z, Qi F, Fang Y, Lin K, Pavan S, Huang B, Dong W, Du P, Tian M, Shi L, Xu J, Han S, Liu H, Qin L, Zhang Z, Dai X, Miao L, Zhao R et al (2022) DNA sequencing sheds light on the evolutionary history of peanut and identifies genes associated with phenotypic diversification. Res Square. 10.21203/rs.3.rs-1776558/v1
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX 9234 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files. The clean resequencing data obtained in this study are available at the BioProject database at China National Center for Bioinformation under the BioProject ID: PRJCA019839 (https://ngdc.cncb.ac.cn/gsub/submit/bioproject/PRJCA019839). Materials used in this study are available from the corresponding authors.