

Abstract
B-cell cloning methods enable the analysis of antibody responses against target antigens and can be used to reveal the host antibody repertoire, antigenic sites (epitopes), and details of protective immunity against pathogens. Here, we describe improved methods for isolation of canine peripheral blood B cells producing antibodies against canine parvovirus (CPV) capsids by fluorescence-activated cell sorting, followed by cell cloning. We cultured sorted B cells from an immunized dog in vitro and screened for CPV-specific antibody production. Updated canine-specific primer sets were used to amplify and clone the heavy and light chain immunoglobulin sequences directly from the B cells by reverse transcription and PCR. Monoclonal canine IgGs were produced by cloning heavy and light chain sequences into antibody expression vectors, which were screened for CPV binding. Three different canine monoclonal antibodies were analyzed, including two that shared the same heavy chain, and one that had distinct heavy and light chains. The antibodies showed broad binding to CPV variants, and epitopes were mapped to antigenic sites on the capsid. The methods described here are applicable for the isolation of canine B cells and monoclonal antibodies against many antigens.
Keywords: B-cells, Canine, monoclonal, antibodies, Parvovirus
1.0. Introduction
Antibodies are a critical component of the humoral immune response that confers protection against viral infection or reinfection of vertebrates. Antiviral antibody titers are among the most frequently used correlates of protection, and are also used to determine previous exposures, or to reveal vaccine- or infection-induced immunity in outbreak situations (Earle et al., 2021; Khoury et al., 2021; Krammer, 2021). Single-specificity monoclonal antibodies (mAbs) have been widely used in research, clinical diagnostics, and as therapeutics. Studying viral antigenic structures and virus-specific antibody repertoires at a monoclonal level also greatly improves our understanding of immune responses and protection against viral infections or vaccines. This analysis can provide fundamental information on viral antigenic sites, the nature of the viral epitopes being targeted, the capacity of individual germline segments to form antibodies against a specific virus, and the roles of antibody pressure on viral evolution and the selection of new variants (Andrews et al., 2015; Clark et al., 2021; Dugan et al., 2020; Scheid et al., 2011; Schommers et al., 2020).
The dog is an important species that interacts closely with humans and other animals and is frequently infected by a variety of different viruses, including some that have caused new epidemics or pandemics in dogs. Those viruses include canine parvovirus (CPV) which caused a pandemic in dogs in the late 1970s, H3N2 canine influenza A virus (an epidemic that arose around 2004 from an avian reservoir), and H3N8 canine influenza A (epidemic initiated in ~1999 from horses) (Parrish et al., 1985; Parrish and Voorhees, 2019). Antiviral antibodies are an essential component of protective immune responses against these viruses. However, we currently lack well documented single B cell cloning methods for dogs that would allow us to study the virus-host interactions (or other canine antibody responses) in detail. The aim of this study was to develop a scalable method for production of CPV-specific antibodies, (which could also be applied to isolate and clone mAbs against other canine pathogens), and to identify the binding sites of the antibodies on the CPV capsid. In a follow up study, we will define the binding of the isolated canine antibodies to CPV in more detail in order to identify antibody-mediated protection and any selective pressures in the natural evolution of CPV.
Several approaches have been used to study the antigen-specific B cell and antibody repertoire, including production of monoclonal antibodies utilizing hybridoma technology where the host B cells are fused with myeloma cells (Smith and Crowe, 2015), immortalization of B cells by Epstein-Barr virus (Lanzavecchia et al., 2007), preparation and screening of phage-display libraries of Ig variable regions (Frenzel et al., 2016), single-cell B cell sorting and cloning (Smith et al., 2009; Tiller et al., 2008; Wardemann, 2003; Wrammert et al., 2008) in some cases in combination with B cell culture and screening (Huang et al., 2013), and single-cell RNA sequencing (Cao et al., 2020; Setliff et al., 2019). Each approach has different strengths and limitations, and challenges remain for developing high-throughput approaches to connect sequence information with antigen specificity.
The germline immunoglobulin gene repertoire available for variable (V), diversity (D) and joining (J) gene segment (V[D]J) recombination during B cell maturation to produce B cell-receptors (BCR) and antibodies (immunoglobulins, Ig) differs between species, so that the antigen recognition by their antibodies varies. In addition, the selection and Ig rearrangement process is skewed in many species (Martin et al., 2018). For example, antibody light chains can be formed either from gene segments of the kappa (κ) or of the lambda (λ) locus, and κ sequences are strongly favored in mice while λ sequences are commonly used in dogs (Arun et al., 1996; Haughton et al., 1978). Consequently, fully understanding host-pathogen interactions and immune protection requires characterization of the antibody responses in the natural host.
The canine immunoglobulin genes on chromosomes 8 (IGH), 17 (IGK) and 26 (IGL) have recently been fully characterized, and the annotated information made available on the international immunogenetics information system (IMGT) databases (Bao et al., 2010; Lefranc, 2014; Lefranc et al., 2015; Martin et al., 2018; Tang et al., 2001). The V(D)J gene segments encoded in the germline are essential for combinatorial antibody diversity and specificity, and the variable regions of antigen-specific B cells are fine-tuned by somatic hypermutation and affinity selection in ongoing immune responses.
Here, we adapt B cell isolation and cloning methods used in other animals to dog B cells and also leverage the newer data on canine immunoglobulin genes to establish techniques for amplification and cloning of CPV-specific monoclonal antibodies directly from peripheral blood B cells of a dog after CPV vaccination.
CPV is closely related to feline panleukopenia virus (FPV) and the ancestor of CPV jumped to dogs in the mid-1970s resulting in a pandemic in dogs during 1978 and 1979 (Miranda and Thompson, 2016; Parrish, 1990; Voorhees et al., 2019). Diseases seen included enteric disease in dogs older than about 6 weeks, or myocarditis after infection of neonatal puppies (Hayes et al., 1979; Nelson et al., 1979). CPV has a 26nm-diameter non-enveloped T=1 icosahedral capsid assembled from 60 copies of combinations of the overlapping virus proteins VP1 and VP2. The VP2 comprises ~90% of the protein, but VP1 appears to form equivalent structures in the assembled capsid, with the extra 143 extra residues on the N-terminal of VP1 being packed inside the capsid (Mietzsch et al., 2019; Reed et al., 1988; Tsao et al., 1991; Vihinen-Ranta et al., 2002). CPV continues to circulate and cause disease in dogs despite widespread use of effective vaccines (Parrish, 1999; Parrish et al., 1988; Voorhees et al., 2019). Antibody-mediated immunity to FPV or CPV protects animals after infection with attenuated vaccine or wildtype viruses. Maternal immunity transferred to young animals in colostrum shortly after birth both protects against infection, and also blocks vaccination in puppies during the first weeks of life (Carmichael et al., 1983; Gooding and Robinson, 1982; Pollock and Carmichael, 1982; Wilson et al., 2014a). During infection the virus circulates through the blood stream to infect susceptible tissues, including the small intestine which is the site of the most severe disease manifestations, and host antibodies intercept the capsids during this viremic circulation (Nelson et al., 1979; Robinson et al., 1980).
The original virus strain named CPV type-2 (CPV-2) that spread world-wide in 1978 was replaced during 1979 and 1980 by a new variant (named CPV-2a), and that virus is the ancestor of most of the recent CPV viruses in dogs since that time (Hoelzer and Parrish, 2010). Several variants of the CPV-2a strain have since been described that are distinguished by single or small numbers of amino acid substitutions, and some have been designated by specific names such as CPV-2b, CPV-2c or new-CPV-2a (Parrish, 1999; Parrish et al., 1988, 1985; Voorhees et al., 2019). Many of those viruses show antigenic variation in reactivity with rodent monoclonal antibodies (Nakamura et al., 2003; Parrish et al., 1991), and the roles of those changes in the success of the viruses are not well understood (Allison et al., 2016; Callaway et al., 2017; Parker and Parrish, 1997; Parrish and Carmichael, 1983; Strassheim et al., 1994; Voorhees et al., 2019). The antibodies used in previous studies were produced in mice or rats by immunization with purified viral capsids in the presence of Freund’s adjuvants and their binding footprints cover much of the capsid surface (Hafenstein et al., 2009; Organtini et al., 2016). Two general antigenic sites (designated A and B) were defined by antibody competitions, escape mutation analysis, and cryo-EM structural analysis of Fab:capsid complexes (Goetschius et al., 2021, p. 14; Hafenstein et al., 2009; Organtini et al., 2016; Parker and Parrish, 1997; Parrish et al., 1985; Parrish and Carmichael, 1983; Strassheim et al., 1994). Many antibody binding sites also overlap the receptor binding site on the capsid. The cellular receptor used for infection is the transferrin receptor type-1 (TfR), and differences in receptor structures control viral host ranges. Variation in the virus may alter both antibody- and receptor-binding sites, and there is a complex interplay between receptor binding, antibody recognition, virus evolution and host range (Lee et al., 2019; Parker et al., 2001; Voorhees et al., 2019).
Here flow cytometric sorting of canine primary B cells binding to fluorescent virus-like particles (VLPs) was followed by B cell culture. Screening for CPV-binding Ig in the supernatant was used to identify CPV-binding B cell clones, then Ig sequences were amplified using new PCR primer sets and variable regions were expressed from canine-specific heavy (H) and light (L) chain expression vectors and capsid binding confirmed by ELISA. This provides new approaches for the analysis of canine B cells and antibodies to this important viral pathogen.
2.0. Materials and methods
2.1. Cells and viruses.
HEK293 cells and NIH 3T3-msCD40L feeder cells (Kershaw et al., 2001) were grown in DMEM + 10% fetal calf serum (FCS) and NLFK cells were grown in 1:1 McCoys 5A-Lebovitz L15 medium (Corning) with 5% FCS in 5% CO2 at 37°C. Sf9 insect cells were grown in Grace’s insect medium (Invitrogen) with 10% FCS and High Five insect cells were grown in Express Five serum-free medium (Invitrogen) with 2.4% Glutamine at 28°C. Virus-like particles (VLPs) were produced by expression of CPV-2a VP2 using baculovirus infection in High Five cells (Bac-to-Bac Baculovirus Expression System, Life Technologies). CPV-2a VP2 containing M87L, I101T, A300G, D305Y, N375D and 426N was prepared from CPV-2 VP2 in the pFastBac1 plasmid (Callaway et al., 2018, 2017; Parrish et al., 1991) by Phusion and Q5 site-directed mutagenesis PCR and ligation with Quick Ligase and T4 PNK (NEB). The sequence containing the mutations was confirmed by Sanger sequencing of the pFastBac1 plasmid. The plasmid was transformed into Escherichia coli DH10Bac to generate the bacmid, which was transfected into Sf9 cells using TransIT Insect Transfection Reagent (Mirus) following the manufacturer’s instructions to generate infectious baculovirus. Passage 1 (P1) baculovirus was collected after 6 days and used to infect Sf9 cells for 6 days to generate P2 baculovirus. P2 was used to infect High Five cells for 3 days to generate CPV-2a VLPs. CPV-2, CPV-2a, CPV-2b, CPV-2 G224R and CPV-2 A300D viruses were produced from infectious plasmid clones in NLFK cells and purified using standard methods by cell lysis, PEG precipitation, chloroform extraction, spinning through sucrose cushion and banding on sucrose gradients to separate empty (light) and full capsids (DNA containing, [heavy]) (Agbandje et al., 1993; Parrish, 1991; Parrish et al., 1982; Parrish and Carmichael, 1983; Weichert et al., 1998).
2.2. VLP purification and labeling.
VLPs were purified as previously described (Callaway et al., 2017). Briefly, VLPs in supernatant were PEG 8,000 precipitated and cells were lysed followed by chloroform extraction of the cell pellet. Both fractions were pelleted through a 20% sucrose cushion and VLPs were banded in a continuous 10–40% sucrose gradient and then further purified by banding in an isopycnic gradient formed from 1.35g/cm3 CsCl.
A 900μg sample of CPV-2a VLPs in 0.1 M sodium carbonate/bicarbonate, pH8.3 were concentrated to 3.2 mg/ml on a 30 kD MWCO Amicon Centrifugal Filter Unit (Millipore Sigma). Alexa Fluor 647 succinimidyl Esters (NHS esters, Thermo Fisher Scientific,) were dissolved in anhydrous dimethylsulfoxide (DMSO) at 10 mg/ml and 20 μl were added to the VLPs. The mixture was incubated at 1 hour at room temperature with continuous stirring for conjugation. Free dye and conjugated VLPs were separated by running the mixture on a PD-10 Desalting Column with Sephadex G-25 Medium (GE Healthcare) according to the manufacturer’s instructions (gravity protocol) and further by running the VLP containing elution on a Sephacryl S-100-HR resin (Sigma) in 1x PBS, pH 7.2 in a gravity-flow column. VLP-containing elution was collected separately from free dye. Labeled VLPs were stored at 4°C protected from light. The degree of labeling was determined according to the manufacturer’s instructions and was 1.7 moles dye per mole of VP2. Absorbance was measured on a Nanodrop 2000 (Thermo Fisher Scientific).
2.3. Canine Ig primer design.
Heavy, lambda light- and kappa light chain primer sets were designed separately for the amplification of rearranged canine immunoglobulin mRNA. Annotated canine Ig reference and non-reference nucleotide sequences used as primer design templates were downloaded from the IMGT databases (Lefranc, 2014; Lefranc et al., 2015). To extract variable region nucleotide sequences, the IMGT/GENE-DB (Giudicelli et al., 2005) was queried for species “Canis lupus familiaris”, functionality “functional”, molecular component “IG” and IMGT group “IGHV”, “IGLV” or “IGKV”, and labels were extracted for the primer binding region “L-PART1+L-PART2” and the rearranged variable region including the leader sequences “L_PART1+V-EXON”. Likewise, IMGT/LIGM-DB (Giudicelli et al., 2006) was queried for species “Canis lupus familiaris”, functionality “functional, productive”, IMGT group “IGHV”, “IGLV” or “IGKV”, and labels were extracted for the primer binding region “L-Region” and the rearranged variable region including the leader sequences “L-V-REGION”.
To extract constant region nucleotide sequences, IMGT/GENE-DB was queried as for the variable region but with IMGT group classifications “IGHC”, “IGLC” or “IGKC” and labels “CH1”, “CL” or “CH1+CH2+CH3+CHS” were extracted. The query for variable regions from IMGT/GENE-DB returned 35 complete IGHV sequences, 18 complete IGKV sequences and 70 complete IGLV sequences, and the query from IMGT/LIGM-DB returned 121 complete IGHV sequences, 42 complete IGKV sequences and 29 complete IGLV sequences. The query for constant regions from IMGT/GENE-DB returned 5 IGHG CH1 sequences (IGHG1*01 – IGHG4*01 plus IGHG1*02), 2 IGKC sequences (IGKC*01 and IGKC*02) and 9 IGLC sequences (IGLC1*01 – IGLC9*01).
Fasta files from IMGT extracted sequences were combined for each heavy and light chain group and were uploaded as templates in openPrimeR (Kreer et al., 2020) to design optimal de novo primers for canine Ig amplification. openPrimerR was run inside a docker container (pulled on November 4, 2020 from mdoering88/openprimer) with R version 4.0.3 and parameters for primer design were as outlined in Supplementary Table 1. Optimal forward primer sets (init.algo = “tree”, opti.algo = “ILP”, max.degen = 1) were designed with the leader sequences assigned as forward binding regions, and nested reverse primers were designed within CH1 or CL. Primer sequences are listed in Table 1. Binding of the primer sets was evaluated on the original template sequences to determine the subset coverage and constraint fulfillments in openPrimeR with identical settings (Fig. 2, Supplementary Table 1).
Table 1.
Oligonucleotide sequences for amplification of rearranged canine immunoglobulin cDNA.
| Number | ID | Direction | Length [bps] | Primer sequence |
|---|---|---|---|---|
| H1 | IGHV_F>1|1–1|−30*0|14:39|_fw | F | 26 | ggagaatcttcttcctgctggcactg |
| H2 | IGHV_F>2|11–1|−35*0|8:31|_fw | F | 24 | ctgtgctctgctgggttttccttg |
| H3 | IGHV_F>3|15–1|−41*0|7:33|_fw | F | 27 | tctgtgctcagctgggtttttcttgtc |
| H4 | IGHV_F>4|156–1|_121|29:56|_fw | F | 28 | ttgtcgctatttcaaaaggtgtccaggg |
| H5 | IGHV_F>5|17–1|−47*0|11:31|_fw | F | 21 | tgctcggctgggttttccttg |
| H6 | IGHV_F>6|2–1|−10*0|12:33|_fw | F | 22 | gctcagctgggtgttccttgtc |
| H7 | IGHV_F>7|23–1|−61*0|8:35|_fw | F | 28 | ctgtgctctgcttgtttttccttttcgc |
| H8 | IGHV_F>8|25–1|−69*0|1:27|_fw | F | 27 | atggagtctttcgttggctgggttttc |
| H9 | IGHV_F>9|29–1|−76*0|4:27|_fw | F | 24 | gagtctgcactccatcgggtttcc |
| H10 | IGHV_F>10|3–1|−13*0|4:27|_fw | F | 24 | gagtctgtgcttggctgggttttc |
| H11 | IGHV_F>11|33–1|−82*0|6:27|_fw | F | 22 | gtccgtgcccacctgtgttttc |
| H12 | IGHV_F>12|34–1|−9*01|5:28|_fw | F | 24 | agtctgtgctcagctggcttttcc |
| H13 | IGHV_F>13|35–1|−1*01|1:20|_fw | F | 20 | atgccgtggtccctcctctg |
| H14 | IGHV_F>14|4–1|−16*0|2:28|_fw | F | 27 | tggagtctgtgctcagatggatttacc |
| H15 | IGHV_F>15|79–1|_44|2:28|_fw | F | 27 | tggaatctgtgctcggatggattttcc |
| H16 | >IGHG_R_OUTER_1|1–1|G1*01|75:99|_rev | R | 25 | agggcactgtcaccatgctgctgag |
| H17 | >IGHG_R_INNER_1|1–1|G1*01|150:173|_rev | R | 24 | tggtcaaggagccggaattccagg |
| L1 | IGLV_F>1|15–1|−151*|23:44|_fw | F | 22 | tcaccctcctcgctcacttcac |
| L2 | IGLV_F>2|3–1|−106*|37:55|_fw | F | 19 | cactgcacagggtcctggg |
| L3 | IGLV_F>3|41–1|−31*0|9:35|_fw | F | 27 | gactctggtcctcctcacctttctctc |
| L4 | IGLV_F>4|42–1|−11*0|32:55|_fw | F | 24 | tggctctctgcacaggttttgtgg |
| L5 | IGLV_F>5|48–1|−27*0|29:50|_fw | F | 22 | tgctcactctctgcacaggctc |
| L6 | IGLV_F>6|49–1|−28*0|32:54|_fw | F | 23 | tggctcactgcacaggttctgtg |
| L7 | IGLV_F>7|52–1|−30*0|1:19|_fw | F | 19 | atggcctggacccctctcc |
| L8 | IGLV_F>8|56–1|−22*0|20:47|_fw | F | 28 | acctcctgcccttcattttctctacagg |
| L9 | IGLV_F>9|6–1|−135*|20:41|_fw | F | 22 | tcctcaccctccttgctcactg |
| L10 | IGLV_F>10|60–1|−64*0|18:44|_fw | F | 27 | attcctcttgctcgtctttcactgcac |
| L11 | IGLV_F>11|61–1|−85*0|1:24|_fw | F | 24 | atggcctggattcctgtcatcgtc |
| L12 | IGLV_F>12|63–1|−128*|31:56|_fw | F | 26 | cttgcctatggctcaggagcagattc |
| L13 | >IGLC_R_OUTER_1|1–1|C1*01|60:85|_rev | R | 26 | agctgaagctgctgtgagatttccac |
| L14 | >IGLC_R_INNER_1|1–1|C1*01|101:127|_rev | R | 27 | ctcaggtagctgctggccgtgtacttg |
| K1 | IGKV_F>1|1–1|−10*0|29:50|_fw | F | 22 | tgctgatgctctggatcccagg |
| K2 | IGKV_F>2|1–1|−10*0|4:25|_fw | F | 22 | aggttcccatctcagctcctgg |
| K3 | IGKV_F>3|15–1|13*01|4:25|_fw | F | 22 | aggttccctgctcagctgcttg |
| K4 | IGKV_F>4|16–1|18*01|12:33|_fw | F | 22 | agctcgccttctctgccttctg |
| K5 | IGKV_F>5|18–1|−15*0|9:30|_fw | F | 22 | acagccccaagtcctcttcctg |
| K6 | IGKV_F>6|37–1|_19|1:27|_fw | F | 27 | aaattccagctctggatcccaggatcc |
| K7 | >IGKC_R_OUTER_1|1–1|KC*01|1:28|_rev | R | 28 | gtccactctctgacactcgctcctttgg |
| K8 | >IGKC_R_INNER_1|1–1|KC*01|138:165|_rev | R | 28 | aggtactgtccttgtcctgctctgtgac |
Figure 2. Coverage, binding region, and constraint fulfillment of canine Ig variable region forward primer sets.
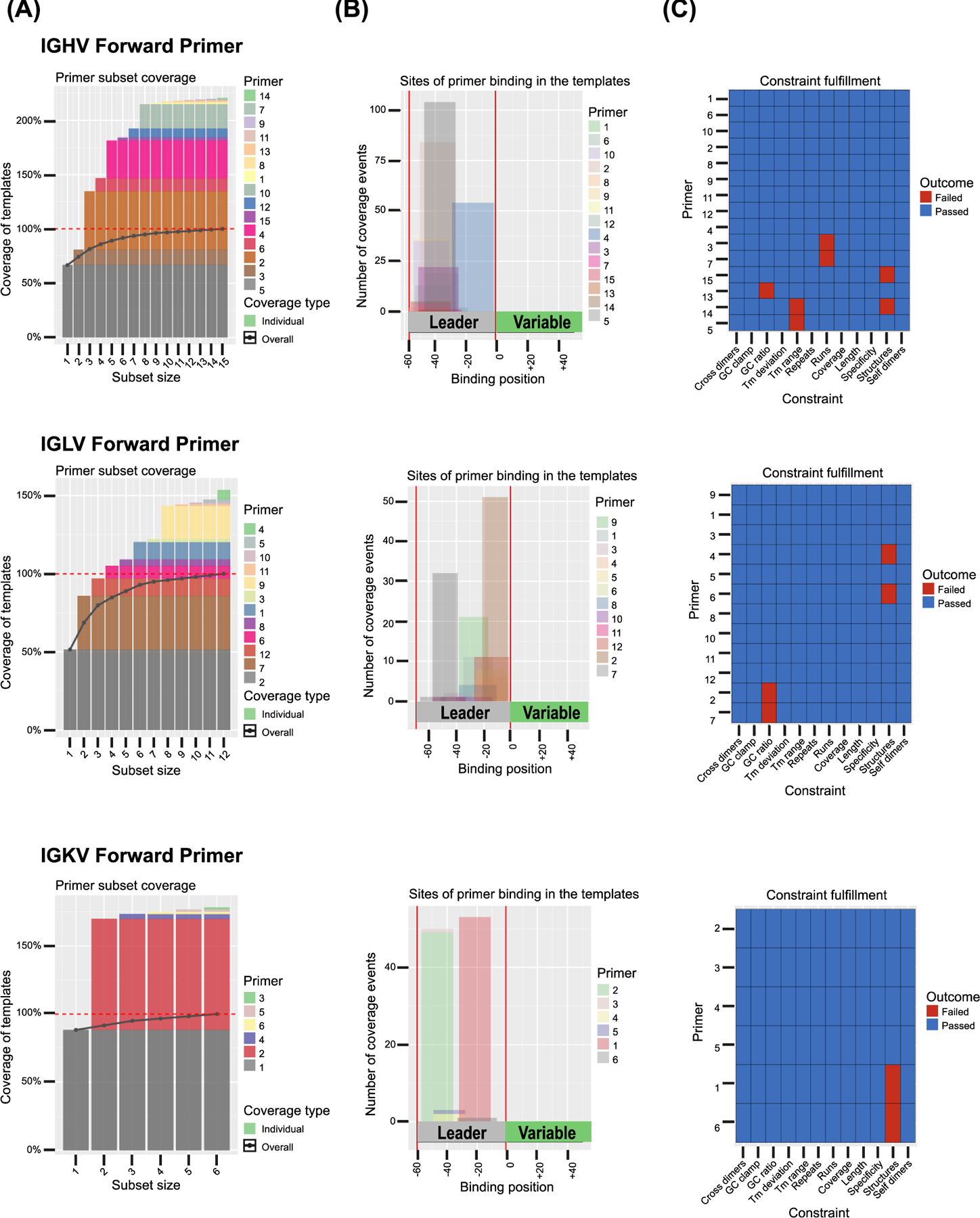
Multiplex primer sets were designed and evaluated with openPrimeR (Kreer et al., 2020) on 156 template sequences for the heavy chain variable region forward primer set (IGHV), 99 template sequences for the light chain λ variable region forward primer set (IGLV) and 60 template sequences for the light chain κ variable region forward primer set (IGKV). For each primer, (A) the individual percentage for the coverage of template sequences, (B) the binding site within the target region (leader sequence) relative to the template (variable region) and (C) the constraint fulfillment is shown. Blue color indicates fulfillment of primer design constraints, red color indicates that a constraint needed to be relaxed within user defined constraint limits (Supplementary Table 1) to achieve 100% coverage of template sequences.
2.4. RNA extraction and primer validation.
A small tissue sample of a male dog (Beagle) was collected within 30 minutes post-mortem from the spleen (euthanasia was performed for reasons unrelated to this study). Spleen cells were suspended by processing the tissue through a fine cell strainer using the plunger end of a syringe, incubated two times for 1 minute with ACK Lysing buffer (Thermo Fisher Scientific). Cells were washed twice by centrifugation and resuspension in DMEM, and flash frozen in freeze media (20% FCS +10% DMSO in DMEM) and stored at minus 80°C. For RNA extraction, spleen cells were thawed, pelleted at 500g for 5 minutes, supernatant was removed, and cells were resuspended in 1 ml Trizol reagent (Thermo Fisher Scientific). Chloroform based extraction and isopropanol RNA precipitation from Trizol was performed, and cDNA was generated from 5 μg RNA by first strand synthesis with random hexamers and SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific) according to the manufacturer’s instructions. The cDNA was diluted 1:5 to 100 μl and 1 μl was used as template for each reaction with standard Taq polymerase (NEB) PCR with 0.5 μl each of 10 μM canine forward and reverse primer, 30 s at 95°C, 30 cycles of 30 s at 95°C, 30 s at 57°C and 55 s at 68°C, and 5 minutes final extension at 68°C.
2.5. Canine blood sample.
Canine peripheral blood was collected from a male dog (Beagle) at ~12 weeks of age in K2 EDTA tubes 14 days after the last vaccination containing CPV MLV. Details about the vaccinations are provided in Supplementary Table 2. All procedures were approved by Cornell University Institutional Animal Care and Use Committee (IAUCUC) (Protocol ID: 2017–0085). PBMCs were isolated one day after blood collection. The blood sample was diluted 1:1 in 1x PBS and the mixture was layered on top of an equal volume of Ficoll-Paque Density Gradient Media (1.077 g/ml Density Max., GE Healthcare) and centrifugated at 400g for 30 minutes at room temperature. The layer containing the PBMCs was separated and washed twice by dilution and centrifugation with 1x PBS at 500g for 20 minutes. Cells were aliquoted at 8.8 x 106 cells/mL diluted in 10% DMSO in FCS and immediately frozen at −80°C in a freezing container.
2.6. Isolation of canine B cells by fluorescence activated cell sorting (FACS).
Aliquots with a total of 5 x 107 canine PBMCs were thawed in prewarmed IMDM/benzonase medium as described previously (Huang et al., 2013) and resuspended in 10% FBS in 1x PBS (sorting buffer). Cells were pelleted at 500g for 5 minutes at 4°C and resuspended in 1,200 μl 1xPBS, of which 50 μl were used for each staining control. Cells were incubated with LIVE/DEAD Aqua fluorescent reactive dye (Thermo Fisher Scientific), washed once with sorting buffer, filtered by pipetting through a 40 μm filter and incubated with Fc receptor binding inhibitor polyclonal antibody (Thermo Fisher Scientific). Cells were surface stained in 300 μl sorting buffer for 30 minutes on ice by incubation with fluorescently labeled monoclonal antibody PE-labeled rat anti-canine CD5 diluted 1:20 (YKIX322.3) (Thermo Fisher Scientific), polyclonal antibodies Fitc-labeled goat anti-dog IgM diluted 1:100 (BioRad) (Schaut et al., 2016) and DyLight405-labeled rabbit anti-dog IgG (H+L) diluted 1:100 (Jackson ImmunoResearch) (Panjwani et al., 2020) and Alexa Fluor 647-labeled CPV-2a VLP at 0.68 μg/ml. Cells were washed twice by dilution with sorting buffer and pelleting at 500g, and cells were filtered by pipetting through a 40 μm filter. Cells were re-filtered immediately prior to sorting by pipetting through a 40 μm filter. Cells were sorted using a 4 laser BD FACSAria Fusion (BD Biosciences) using an 85 μm nozzle at ~5,000 events per second and collected in sterile tubes containing sterile 1xPBS with 50% FCS at >95% purity. Lymphocytes in PBMCs were identified based on FSC and SSC characteristics and B cells were identified as live CD5-, surfaceIgM (sIgM)+ and/or surfaceIgG (sIgG)+ lymphocytes. Gates for VLP binding B cells were determined using a fluorescence-minus-one-control.
2.7. In vitro B cell stimulation.
NIH 3T3-msCD40L feeder cells (Kershaw et al., 2001) were grown in T75 cell culture flasks and irradiated with 5,000 rads following the procedures described by Huang et al. (Huang et al., 2013) and stored at 25 x 106 cells/ml in DMEM with 20% FCS and 10% DMSO at −80°C. Immediately after isolation of primary B cells, feeder cells were thawed, centrifuged, and resuspended as previously described (Huang et al., 2013). Nineteen 96 well plates were seeded with 100 μl of 2x feeder mixture containing 20,000 cells/well of irradiated NIH 3T3-msCD40L feeder cell fibroblasts, 200 U/ml recombinant human Interleukin (IL-) 2 (Preprotech), 50 ng/ml recombinant canine IL-4 (Kingfisher Biotech) and 50 ng/ml recombinant canine IL-21 (R&D) in complete IMDM (Iscove’s modified Dulbecco’s medium [IMDM] with GlutaMAX [Gibco] and 10% FBS, and MycoZap Plus-PR [Lonza] at 1 ml per 500 ml volume). 200 μl sterile water was added to columns 1 and 12 and rows A and H to prevent evaporation.
Primary sorted B cells were diluted in complete IMDM to 40 cells/ml and 100 μl/well were added to 1,020 wells containing feeder cells. 100 μl of complete IMDM with no primary B cells was added to wells in row 7 of each plate for a no-B-cell control. Plates were incubated at 37°C in 5% CO2 for 13 days. 100 μl of fresh cytokine mix containing recombinant human IL-2 at 200 U/ml and recombinant canine IL-21 at 50 ng/ml in complete DMEM was added to each well on day 6. 50 μl of supernatant was removed on day 12 for ELISA. Cells from CPV-specific wells and control cells were collected on day 13 by resuspension followed by separation of supernatant and cells by centrifugation at 500g for 10 minutes. Supernatant was stored at −80°C and cells were lysed in 50 μl of lysis solution containing 0.25 x RNase-free PBS (Thermo Fisher Scientific), 1 U/μl RNasin (Promega) and 1 U/μl RNaseOUT (Thermo Fisher Scientific) and immediately stored at −80°C.
2.8. ELISA for CPV-specific Ig.
96 well adsorption immunoassay plates (Thermo Fisher Scientific) were coated with 3 μg/ml CPV capsids in 0.05 M carbonate/bicarbonate, pH 9.6 at 4°C overnight, then washed three times with wash buffer (1x PBS, pH 7.2 containing 0.05% Tween 20) and blocked overnight with 5% (w/v) Bovine Serum Albumin in wash buffer at 4°C, or for two hours at 37°C. 50–100 μl of undiluted supernatant from B cell cultures, or a 2–3 fold dilution series of Ig transfection supernatant or purified mAb was added to ELISA plates and incubated for 1 hour at room temperature. Plates were washed three times with wash buffer and incubated for 1 hour at room temperature with horseradish peroxidase-conjugated rabbit anti-dog IgG (H+L) diluted 1:2,000 (Jackson ImmunoResearch). Plates were washed three times and incubated with 100 μl/well TMB ELISA substrate (Thermo Fisher Scientific) for 5 mins (for purified antibodies) or up to 25mins (B cell cultures) at room temperature. The reaction was stopped with 100 μl 2M sulfuric acid and absorbance at 450nm was immediately quantified on an infinite M200 pro TECAN ELISA plate reader (Tecan Group).
2.9. Ig RT-PCR and Ig variable region amplification.
For initial RT-PCR, individual reactions were set up manually for each clone. 150ng random hexamers, 1 μl of 10% NP-40, 1 μl of 10 mM dNTP mix (10 mM each) and 0.5 μl of RNaseOUT (all Thermo Fisher Scientific) were added to 4 μl of lysed B cells and the mixture was incubated at 65°C for 2 minutes and on ice for 2 minutes. Heavy chain, kappa light and lambda light chain variable regions were amplified separately for each B cell clone by semi-nested PCR using forward primer mixes (heavy = H, lambda = L, kappa = K) and outer reverse primers (Table 1). For each reaction 5 μl of 10x Standard Taq Reaction Buffer, 1 μl of 10mM dNTPs, 1 μl of 10 μM (H, L or K) Forward Primer Mix (all primers at equimolar ratios), 1 μl of 10 μM (H, L or K) outer reverse primer, 40.75 μl nuclease-free H2O and 0.25 μl Hot Start Taq DNA Polymerase (NEB) were added to 1 μl of cDNA. PCR1 was carried out in a thermocycler with an initial denaturation at 95°C for 30 s, followed by 50 cycles at 95°C for 30 seconds, 57°C for 30 s and 68°C for 55 s, followed by a final extension at 68°C for 5 mins. The second PCR (PCR2) was carried out with the same concentration of reagents and the inner reverse primer (H, L or K) with 0.5 μl template from PCR1 in a thermocycler at 95°C for 30 s, followed by 30 cycles at 95°C for 30 seconds, 57°C for 30 s and 68°C for 45 s and a final extension at 68°C for 5 mins. The amplified products from PCR2 were analyzed on a 2% Agarose gel. PCR2 products were purified using an E.Z.N.A. Cycle Pure Kit (Omega Bio-Tek) and submitted for Sanger sequencing.
2.10. Ig sequence analysis.
The Sanger sequencing reads were exported as FASTA files and Ig variable regions were automatically annotated using IMGT/V-QUEST program version 3.5.25 (Brochet et al., 2008; Giudicelli et al., 2011) with default settings and specified species “Canis lupus familiaris”, receptor type or locus “IG” and activated search for insertions and deletions in V-REGION.
2.11. DNA libraries preparation, deep sequencing and Data analysis.
From each individual clones selected for deep sequencing analysis, total RNA was isolated from 25μl lysed B-cells using an EZNA MicroElute Total RNA Kit (Omega Bio-Tek). 5μL of isolated RNA was used for cDNA synthesis following manufacturer’s instructions for random hexamer priming (Thermo Fisher Scientific). Obtained cDNA was used as DNA template for only one round of PCR (same as PCR1 as detailed above), with cycling and priming conditions kept the same except for a reduced number of cycles for each individual reaction (≤ 35 cycles). PCR products were purified using E.Z.N.A Cycle Pure Kit (Omega Bio-Tek), quantified using a Qubit 4 fluorometer (Thermo Fisher Scientific) and 1ng of input DNA (separately for each Ig chain) was used to prepare barcoded sequencing libraries with the Nextera XT DNA Library preparation Kit (Illumina). Libraries were multiplexed by MiSeq 2 x 300 Illumina sequencing.
Raw sequence reads were paired and trimmed using BBDuK on Geneious Prime v2022.2.2 (Dotmatics)” to remove adapters and low-quality end-reads form sequences. Standard NGS data cleanup (duplicate reads removal and error correct/normalize) was carried out using recommended default parameters from “Geneious Prime v.2022.2.2 (Dotmatics). De novo assembly was performed and consensus contigs obtained for each read were mapped to the 2C5 H and L reference sequences. Annotation of mapped contigs was performed using IMGT/V-QUEST as described above.
2.12. Expression Vector Cloning.
Cloning plasmids containing the canine IgG2 heavy chain (pFUSE-CHIg-dG2), canine lambda light chain (pFUSE-CHIg-dL) and canine kappa light chain (pFUSE-CHIg-dK) constant regions were acquired from Invivogen, USA. For each cloning plasmid the mouse Ig heavy chain V region 102 signal peptide sequence including a Kozac sequence (MGWSCIILFLVATATGVHS; nucleotide sequence: ATGGGATGGTCATGTATCATCCTTTTTCTAGTAGCAACTGCAACCGGTGTACATTCT) was inserted in frame with and immediately upstream of the constant region by Q5 site-directed mutagenesis PCR and ligation with Quick Ligase and T4 PNK (NEB). Ig variable regions FR1-CDR1-FR2-CDR2-FR3-CDR3-FR4 were inserted in frame into the respective heavy or light chain expression vector between the signal peptide and the constant region using NEBuilder HiFi DNA Assembly (NEB). Cloning vectors were linearized by PCR with Phusion Hot Start II polymerase (Thermo Fisher Scientific), and variable regions were amplified with Q5 High fidelity DNA polymerase (NEB) with ≥20bps vector-specific overhangs on the 5’ and 3’ ends of sequence-specific primers using purified PCR1 products as template (E.Z.N.A. Cycle Pure Kit, Omega Bio-Tek).
PCR fragments were separated on a 2% Agarose gel, purified using the Monarch DNA Gel Extraction Kit (NEB) according to the manufacturer’s instructions and purified fragments were assembled using the NEBuilder HiFi DNA Assembly Master Mix (NEB) according to the manufacturer’s instructions. Assembled products were transformed in DH10B competent cells for propagation and plasmids were purified with Wizard Plus Midiprep (Promega), or EZNA Plasmid DNA Mini kit I (Omega Bio-Tek). Correct sequences were confirmed by Sanger sequencing.
2.13. Antibody expression and Ig purification.
Heavy and light chain plasmids were co-transfected at a ratio of 2:3 (heavy:light) in HEK293 cells by Calcium Phosphate transfection or with TransIT-X2 transfection reagent (Mirus Bio) according to the manufacturer’s instructions. MAbs in cell culture supernatant were collected after 48 hours and were purified by Protein G or Protein A chromatography directly from cell culture supernatant, eluted with 0.1M Glycine, pH 3.0 and neutralized with 1M Tris-HCl, pH 9.0 and NaCl.
2.14. Antibody captures of fluorescent capsids.
96 well adsorption immunoassay plates (TFS) were coated with anti-CPV antibody 14 (Parrish et al., 1982) at 2.5 μg/ml in 0.05 M carbonate/bicarbonate, pH 9.6 at 4°C overnight, or in the same coating buffer without antibody 14 for the negative control. Plates were washed in wash buffer and blocked with 2% BSA in PBS containing 0.05% Tween 20 for 2 hours at 37°C. 50 μl each of a 2-fold dilution series (starting at 5 μg/ml) of fluorescent VLP was incubated in wells with or without coated antibody for 1 hour at room temperature. Plates were washed three times in wash buffer and fluorescence was read on an infinite M200 pro TECAN ELISA plate reader (Tecan Group) with an excitation wavelength of 635 nm (bandwidth 9 nm) and an emission wavelength of 680 nm (bandwidth 20 nm).
2.15. Statistical Analysis.
Statistical analysis was carried out using GraphPad Prism software (Version 6). Data were analyzed by Two-way ANOVA with Tukey’s multiple comparison test and were considered statistically significant with a P-value less than or equal to 0.05.
3.0. Results
3.1. Preparation of CPV VLPs and capsids.
Our initial goal was to use fluorescent parvovirus particles to bind and isolate antigen-specific B cells from dogs. CPV virions would infect canine B cells, and to avoid altering the capsid structure during activation, we used virus-like particles (VLP) as antigens in our screens. Those were prepared by expressing the canine parvovirus major capsid protein VP2 of the CPV 2a strain from baculovirus expression vectors (Jin et al., 2016; Saliki et al., 1992; Yuan and Parrish, 2001). Purified VLPs were labeled with Alexa Fluor 647 for use in flow cytometry (Fig. 1A). The labeling did not disrupt antigenicity as demonstrated by binding the labeled VLPs to the mouse mAb 14 that only binds assembled capsids (Fig. 1B).
Figure 1. Immunizations, VLP purification, isolation of VLP-binding B cells from canine peripheral blood, B cell culture and ELISA for CPV-specific Ig.
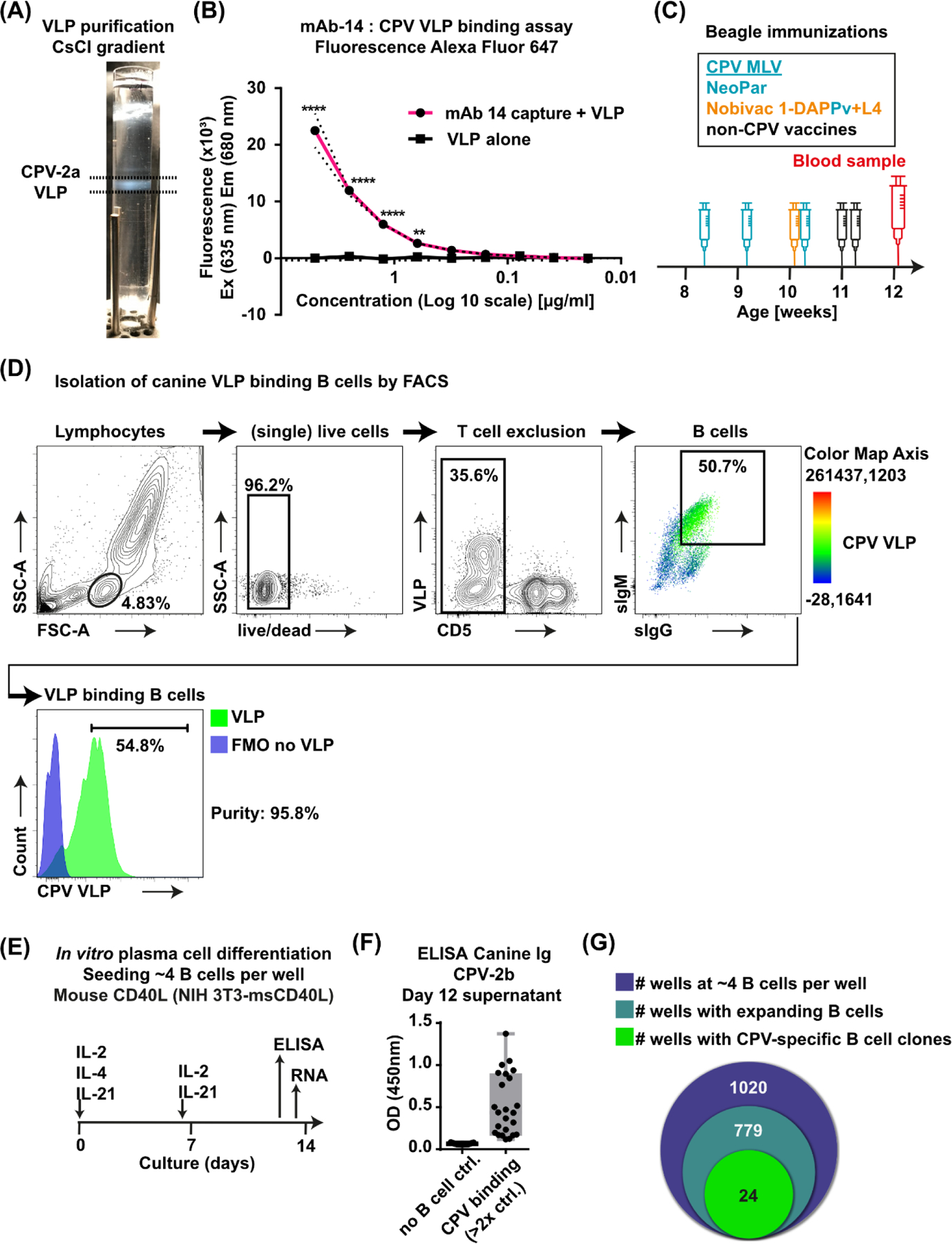
(A) Purification of VLPs in an isopycnic gradient. (B) Fluorescence of Alexa-Fluor 647 labeled VLPs binding to mouse monoclonal anti-CPV 14 compared to the negative control without mAb 14. (C) Timeline showing age, vaccination schedule with vaccine names of CPV-containing vaccines (indicated by blue color) and day of blood collection of the male dog (Beagle) analyzed in this study. Nobivac 2-DAPPv+L4 is a combination vaccine that contains CPV MLV. (D) FACS gating strategy for isolation of canine VLP+ B lymphocytes from PBMCs. Color in the B cell panel shows the intensity of VLP fluorescence detected for each cell according to the color map axis. (E) Sorted B cells were seeded on irradiated NIH 3T3 msCD40L feeder cells at 4 B cells/well in 96 well plates and stimulated with IL-2, IL-4 and IL-21. Fresh cytokines IL-2 and IL-21 were added on day 6 of culture. Aliquots of supernatants were removed on day 12 and cells lysates were collected on day 13. (F) CPV-2b-specific Ig in B cell culture supernatant was detected in 24 wells by ELISA (two-fold or higher OD [450 nm] than in no-B cell control wells of the same plate [feeder cells only]). (G) Venn diagram showing the number of seeded wells with primary B cells, the number of wells with expanded B cell colonies visible under a light microscope and the number of wells with CPV-specific Ig as determined by ELISA. Mean ± SD (dashed line); **, P ≤ 0.01; ****, P ≤ 0.0001; (B) n = 3 independent experiments; Mean ± SD (C) MLV = modified live vaccine; (D) FMO = fluorescence-minus-one control; (F) n=24 (CPV-binding); n=19 (no B cell control, shown in the graph is average of 6 control wells for each of the 19 plates)
3.2. Isolation of canine VLP-binding B cells, In vitro B cell culture and screening for CPV-specific Ig.
B cells were obtained from a male dog that had been inoculated four times with CPV modified live virus (MLV) vaccines between 8 and 11 weeks of age. Repeated vaccination is a standard protocol for dogs as it is unclear when the vaccine virus will infect due to the interference by maternal antibodies early in life (Pollock and Carmichael, 1982; Wilson et al., 2014b). A blood sample was collected 14 days after the last dose of CPV MLV vaccine (Fig. 1C, Supplementary Table 2). Lymphocytes in peripheral blood mononuclear cells (PBMCs) were analyzed for VLP binding. (Fig. 1D). A sIgM+sIgG+ B cell population bound more VLP than other lymphocytes, while the CD5+ T cells bound hardly any VLPs (Fig. 1D). A large population of sIgM+sIgG+ B cells in dogs has not been described and is unlikely due to the molecular mechanisms that occur during immunoglobulin class switch recombination in many species (Stavnezer et al., 2008). The “double positive” B cell population is likely due to cross-reactivity of the antibody reagent used to detect the surface immunoglobulin antibodies. Consequently, we did not rely on sIgM and sIgG staining to demarcate the two B cell populations but set the B cell gate guided by the signal in the third channel (VLP binding) to sort the B cells which bound most of the VLP. Overall, a high percentage of B cells were labelled by fluorescent VLP, indicating that the VLPs may bind generally to activated B cells likely due to increased expression of the host cell receptor TfR on those proliferating B cells (Ellebedy et al., 2016), The VLP-binding B cells in this dog may have been proliferating in response to CPV or to other viruses (canine adenovirus and canine distemper virus) in the vaccines. In any case, the VLP-binding live CD5- surfaceIgM (sIgM)+ and/or surfaceIgG (sIgG)+ B cells were isolated by fluorescence-activated cell sorting (FACS) and were cultured and screened for CPV-specific Ig (Fig. 1E). Sorted B cells were seeded at ~4 B cells per well in 96 well plates and, together with irradiated mouse CD40L-expressing mouse 3T3 feeder cells, stimulated for 13 days with cytokines Interleukin IL-2, IL-4, and IL-21 (Fig. 1E). Supernatants on day 12 were analyzed by enzyme-linked immunosorbent assays (ELISA) for antibodies binding to the CPV-2b strain frequently used in CPV MLV vaccines, which differs by only one residue from the VLP strain CPV-2a (VP2 N426D). Positive CPV reactivity was defined by ELISA OD at least double that seen for the feeder cell-only control wells of the same plate (Fig. 1F). Of 1,020 seeded wells, 76.4% contained visibly expanded B cell colonies (779 wells), and 2.4% (24 wells) contained CPV-specific antibodies (Fig. 1G). Cell lysates of wells with CPV-specific antibodies were collected on day 13.
3.3. Primers for analysis of antibody genes in canine B cells.
Comprehensive primer sets for the amplification of canine antibody heavy and light chain variable region sequences of antigen-specific B cells were based on the updated canine IGH, IGL and IGK Ig germline loci sequences (Martin et al., 2018). Those would amplify all annotated canine functional rearranged Ig sequences available on IMGT/GENE-DB (Giudicelli et al., 2005) and IMGT/LIGM-DB (Giudicelli et al., 2006). A total of 156 heavy chain variable (IGHV), 99 lambda light chain variable (IGLV) and 60 kappa light chain variable (IGKV) sequences were used as templates for the de novo design of optimal forward primer sets for each group, using the openPrimeR software (Kreer et al., 2020). The signal peptide required for antibody export and transport across membranes that is encoded by the Leader sequences L-PART1 and L-PART2 was defined as the primer binding region (Fig. 2B). These segments are joined to form the L-Region during somatic recombination and are immediately 5’ of the variable region in spliced Ig mRNA, and binding of primers to these relatively conserved sites enables isolation of highly mutated Ig sequences while avoiding primer-induced sequence changes in the variable region (Kreer et al., 2020; Scheid et al., 2011). A total of 15 IGHV, 12 IGLV and 6 IGKV forward primers was needed to cover all Ig template sequences, with only a small number of primers necessary to cover most sequences in each group (Table 1, Fig. 2A). Primer design constraints were fulfilled for most primers, and a maximum of two constraints needed to be relaxed for individual primers to achieve full coverage of all Ig template sequences (Fig. 2C). Outer and inner reverse primers for semi-nested amplification with forward primer sets were designed in openPrimeR with binding sites in conserved regions of the canine constant regions IgG1 – IgG4 (CH1), IGLC1 – IGLC9 and IGKC using the same primer design settings (Table 1 and Supplementary Table 1). Forward primers were tested individually by PCR using cDNA from canine splenic tissue as template, and all but one IGHV (ID H9), one IGLV (ID L10) and two IGKV (ID K4 and K5) produced amplicons of the expected size with the outer reverse primers (Fig. 3A–C). Two bands of 50–100 bp difference were observed by agarose gel electrophoresis for most heavy chain primers and for some IGL and IGK primers (light chain amplicons with smaller size difference). The significance of these larger bands is unclear, but they could potentially occur due to incomplete splicing of rearranged Ig in cDNA. When forward primers were tested together with the inner reverse primer, both bands were still present (data not shown for IGH) (Fig. 3B–C).
Figure 3. Strategy for amplification, sequencing and cloning of Ig heavy (H) and light (L) chain variable regions in H and L plasmids, and expression of monoclonal antibodies in cell culture.
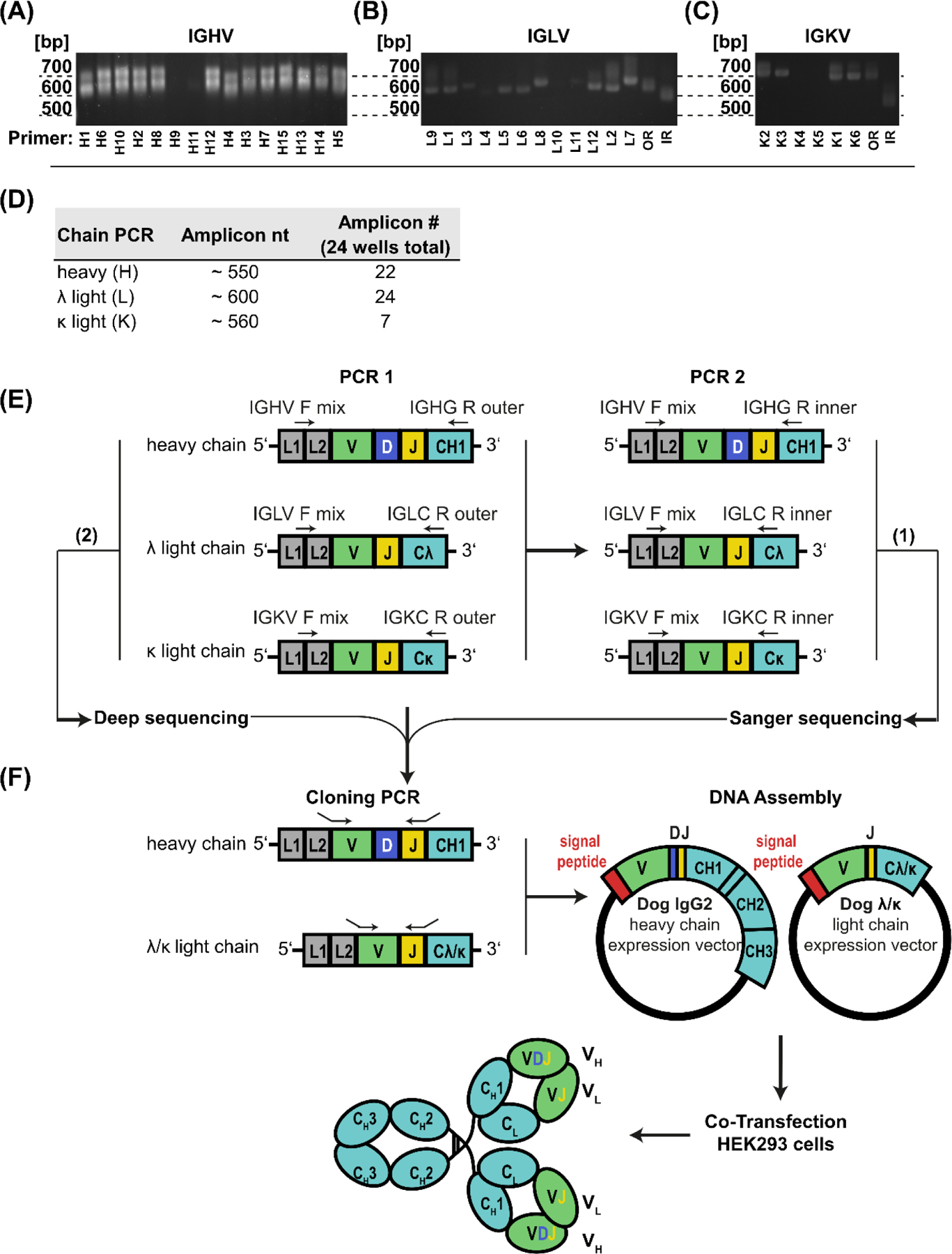
(A) IGHV, (B) IGLV and (C) IGKV forward primers were tested individually with the outer reverse primer for amplicons by PCR of cDNA generated from splenic tissue of a Beagle. (D) Approximate size and the number of successfully amplified heavy and light chains in PCR2 reactions from 24 CPV-specific wells (E) PCR amplification of canine Ig in separate reactions for H, L-λ and L-κ variable regions (V[D]J genes). PCR1 was performed on cDNA generated from canine (clonal) B cells in separate reactions with forward primers mixes (IGHV, IGLV or IGKV) and the corresponding outer reverse primer, and PCR2 was performed on PCR1 product in separate reactions with IGHV, IGLV or IGKV forward primer mixes and the corresponding nested reverse primers. (F) Sequence specific primers with overhang sequences complementary to the vector were used to clone variable regions in H and L expression vectors in frame with the signal peptide and the constant region sequences (separate L-λ and L-κ plasmids). mAbs were expressed by co-transfection of H and L plasmids in HEK293 cells. (A-C) bp = size of amplicon in base pairs, OR = complete forward primer set with outer reverse primer, IR = complete forward primer set with inner reverse primer (Table 1); (E-F) L1 = Leader Part 1, L2 = Leader Part 2, V = variable, D = diversity, J = joining, F = forward, R = reverse. CH1 – CH3 = constant heavy chain domain 1–3, C-λ/κ = light chain λ or κ constant domain, VH/L = heavy or light chain variable region: (1) Sanger sequencing of PCR2 amplicons, (2) Deep sequencing of selected PCR1 amplicons.
3.4. Gene cloning methods by RT-PCR.
Cloning strategies for the targeted amplification of canine B cell Ig mRNA used reverse transcription with random hexamers followed by semi-nested PCR amplification (Fig. 3E–F). The first of the two nested PCRs (PCR1) was used to amplify Ig variable regions directly from cDNA with separate reactions for IGHV, IGLV and IGKV primer sets (equimolar concentration), each with the corresponding outer reverse primer in the constant region. For the inner semi-nested PCR (PCR2), the same forward primer mixes were used to amplify PCR1 product as template with inner reverse primers (Figs. 3E). Amplicons were observed in 22 of the heavy, 24 of the λ light and 7 of the κ light chain reactions (Figure 3D). Sequences were initially determined by Sanger sequencing route (1) (Fig. 3E), and variable regions annotated using IMGT/V-QUEST (Brochet et al., 2008; Giudicelli et al., 2011) for V(D)J germline genes and analysis of somatic hypermutation. Canine-specific heavy (H) and light chain (L) expression vectors were then prepared for cloning and expression of heavy and light chain variable regions to test for antigen-specificity (Fig. 3F).
3.5. Plasmid vectors for antibody expression.
Canine IgG2 heavy and λ and κ light chain antibody expression vectors were used as that appears functionally similar to human IgG1, based on Fc receptor binding and the initiation of antibody-dependent cell-mediated cytotoxicity and complement binding (Bergeron et al., 2014; Tang et al., 2001). The mouse Ig heavy chain V region 102 signal peptide sequence including a Kozac translation initiation sequence, and the Ig variable regions were then cloned in frame with the constant region (Fig. 3F) (Smith et al., 2009; Vazquez-Lombardi et al., 2018). Heavy and light chain expressing plasmids were then co-transfected in mammalian cells (HEK293 cells) and the secreted IgGs were analyzed or purified from the cell culture supernatant (Fig. 3F)
3.6. Antibody Expression and Analysis.
Sanger sequencing revealed that multiple B cell clones were likely present in many wells, so we deep sequenced the PCR products from PCR1, route (2) (Fig. 3E) from anti-CPV positive wells. This analysis confirmed the presence of 1–4 heavy chain and 1–9 light chain sequences in individual wells. Various sequences from 7 ELISA CPV antibody-positive wells were cloned (Supplementary Table 3) and H and L chains were expressed from HEK293 cells. One of the wells contained only a single H and L chain sequence, and that monoclonal was CPV binding (clone 2C5). Identical H and L chain sequences were also found in another well, indicating that this B cell clone was already clonally expanded in the original blood sample. Another well contained the same (2C5) H chain as well as different L chains. After testing six L chains along with the 2C5H chain, only one combination gave a CPV-binding IgG (mAb 3G6) which showed 100% and 93% amino acid identity to 2C5’s heavy and light chain variable genes, respectively. Distinct H and L chain sequences from another well showed ELISA binding to the CPV-2a capsid (mAb 7C8). In summary, we obtained four CPV-2 specific antibodies from different wells – two were 100% identical for both H and L chains (mAbs 2C5), another (3G6) that shared the H chain with 2C5, and 7C8 that was from a different origin.
3.7. Canine anti-CPV antibodies isolated bind the major viral epitopes.
Canine mAbs 2C5, 3G6 and 7C8 were annotated by IMGT/V-QUEST (Brochet et al., 2008; Giudicelli et al., 2011) (Fig. 4A–D) mAbs 2C5 and 3G6 were identical and had a very long HCDR3 of 20 amino acids compared to an average of ~13 HCDR3 residues expressed in canines (Steiniger et al., 2014), as was the HCDR3 of mAb 7C8. mAbs 2C5 and 3G6 are clonotypes arising from germline genes IGHV3–19 and IGLV1–141/162 (Fig. 4A–B). The V gene sequences of 2C5 were 95.09% (IGHV) and 97.54% (IGLV) identical with the probable germline segments of origin, while mAb 3G6 IGLV was 99.30% identical. The mAb 7C8 derived from a very distinct B cell clone was 94.44% and 99.65% identical to the IGHV3–5 and IGLV8–128 germline (Fig 4C). Heavy and light chain V gene sequences of mAbs 2C5, 3G6 and 7C8 were aligned next to each other (Fig. 4D)
Figure 4. Canine anti-CPV monoclonal antibodies.
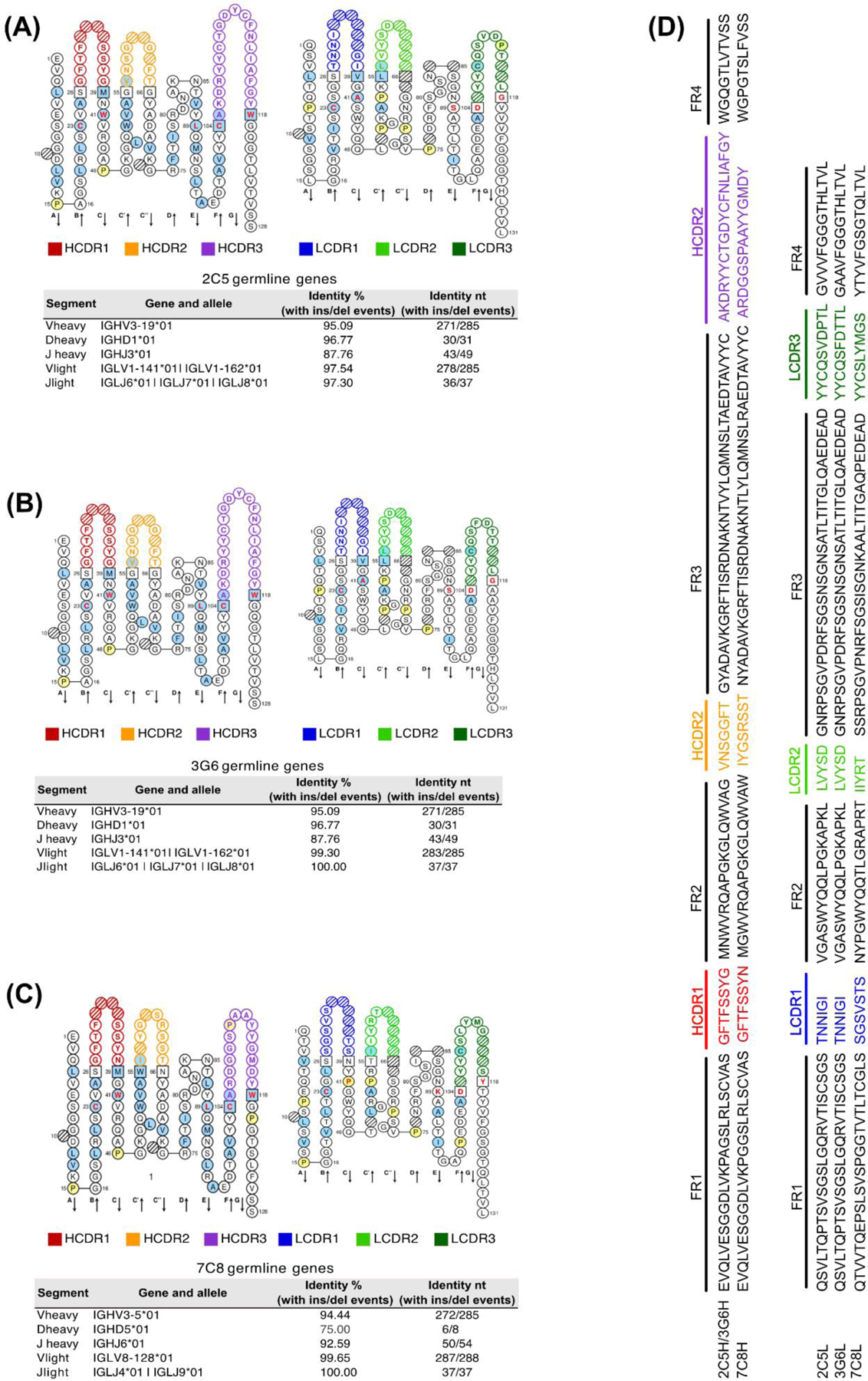
IMGT Collier de Perles (50% Hydrophobic positions) (Ehrenmann et al., 2011) and identity of canine mAbs V(D)J variable region genes compared to germline segments and alleles showing the percentage and number of identical nucleotides (annotated by IMGT/V-QUEST) (Brochet et al., 2008; Giudicelli et al., 2011) for (A) 2C5 (B) 3G6 and (C) 7C8 (D) Heavy and Light chain amino acid sequence for all three distinct canine mAbs. FR = Framework Region, CDR = Complementarity-determining region for H (heavy) and L (light) chains.
We conducted a preliminary mapping of mAbs 2C5, 3G6 and 7C8 binding to the capsids of the original CPV-2, natural CPV variants, or capsids with mutations influencing rodent mAb binding (Parker and Parrish, 1997; Strassheim et al., 1994; Voorhees et al., 2019). mAbs 2C5, 3G6 and 7C8 bound CPV variants CPV-2, CPV-2a and CPV-2b with similar binding curves (Fig. 5A–C). We then tested the binding of the three mAbs to two major antigenic determining residues G224 and A300. Our result (Fig. 5D–E) shows that all three mAbs bound the G224R CPV-2 mutant capsid; however, capsids with the A300D mutation abrogated mAbs 2C5 and 3G6 relative to mAb 7C8 binding. This suggest that mAbs 2C5 and 3G6 bind to antigenic site B, while mAb 7C8 may bind to the A antigenic site (Fig. 5F–G).
Figure 5. Canine mAbs bind CPV variants.
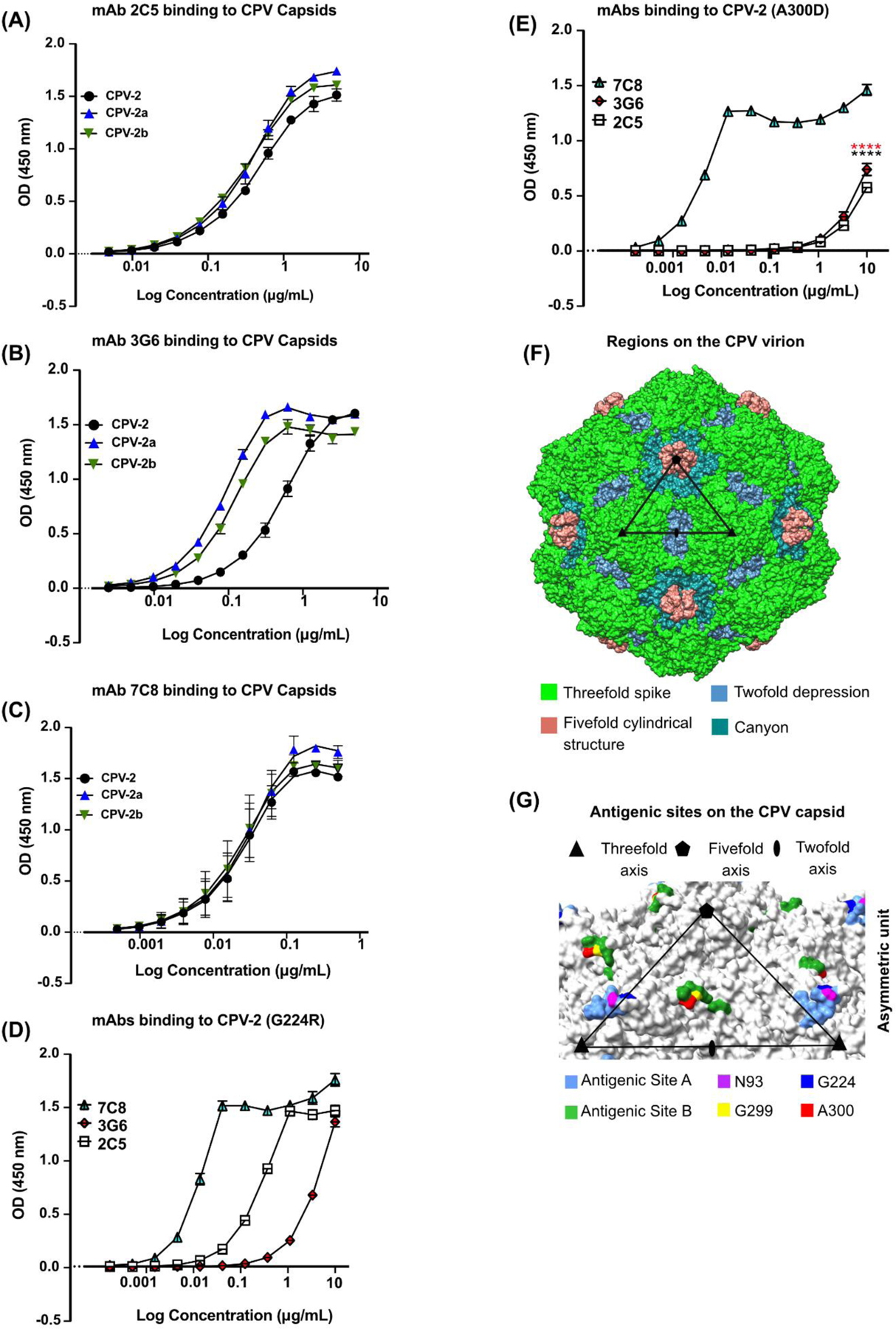
ELISA of purified canine mAbs binding to CPV variants. (A-C) Binding of (A) 2C5 (B) 3G6, and (C) 7C8 to CPV-2, CPV-2a and CPV-2b and (D-E) mAbs binding to (D) CPV-2 (G224R) and (E) CPV-2 (A300D). (F) CPV structure (PDB 6OAS) with color-coded structural features. One asymmetric unit (ASU) with the symmetry axis is outlined by a black triangle (adapted from (Tsao et al., 1991). (G) Common residues of antigenic sites A and B in cyan and green, respectively, (as defined in (Hafenstein et al., 2009)) highlighted on the CPV capsid relative to one asymmetric unit. Key antigenic residues 93 (purple), 224 (blue), 299 (yellow) and 300 (red) are highlighted. (A-F) n = 3 independent experiments; Results for each plot are shown as average OD values minus blank at 450nm against Log10 transformed concentrations; Mean ± SEM. ****, P < 0.0001; (A-F) n = 3 independent experiments
4.0. Discussion
Here we report new methods for isolation and cloning of Ig variable regions directly from canine B cells specific for target antigen and use those to identify and express three different canine mAbs against the capsid of CPV. The dog that was the source of the B cells was only 13 weeks of age and had been injected repeatedly with a commercial modified live vaccine, as is the standard of care for dogs to protect them against virus infection and disease, and the blood sample was likely collected only three to five weeks after the infection by the vaccine virus.
Direct cloning of mAbs from isolated B cells avoids several problems associated with other methods of monoclonal antibody production from B cells, including random pairing of heavy and light chain variable regions (phage display libraries), low efficiency (EBV transformation or hybridoma fusion), or high cost (single-cell sequencing). The method described here should be easily scalable, and thousands of B cell clones in culture can be screened for binding to target antigens by ELISA, so that more rare B cell clones may be identified. The B cell seeding density chosen in this study turned out to be >1 in many wells, so that cloning of multiple heavy or light chains from wells was necessary in some cases to identify the correct CPV-specific pairing. In future studies the seeding density would be reduced to ensure that there are single B cells in individual wells, so that heavy and light chain pairing can be revealed by Sanger sequencing. The approach described here can be used to isolate, screen and clone monoclonal antibodies against target antigen in a short timeframe without the need for de novo gene synthesis. Finally, the culture of single canine B cells for extended periods enables functional studies of B cell differentiation.
4.1. Development and validation of canine specific primer sets, and IgG expression methods.
The new IGH, IGL and IGK primer sets for Ig amplification and modified expression vectors allowed efficient antibody cloning, expression, and secretion (Fig. 3). CPV VLP-binding B cells were isolated by FACS and cultured using an adapted canine-specific protocol, then we identified wells expressing CPV-specific Ig after 12 days of culture and then collected the cell lysates for cloning.
It is likely that in this system the proliferating B cells were also displaying high levels of TfR (CD71), which bound the CPV-VLPs. In humans two main activated B cell subpopulations express CD71 and include CD38hiCD20-antibody secreting cells (ASCs or plasmablasts), and CD38int–loCD20hi activated B cells (ABCs) that are committed memory B cells (MBCs) (Ellebedy et al., 2016). Only ASCs secreted IgG directly ex vivo without further culture, in contrast to ABCs. Other frequently used B cell markers are expression of surface IgM+ or IgG+, with IgG identifying cells that have undergone activation and class-switch recombination (Sundling et al., 2021).
We screened for surface IgM and/or IgG expression to identify canine B lymphocytes and observed a large population of cells that expressed both markers (Fig. 1D). This pattern differs from that seen in mice, is unlikely to be a feature of canine B cells based on the molecular mechanism of class switch recombination and could potentially be due to cross-reactivity of the polyclonal IgG or IgM antibodies used. Importantly, the CD5-sIgM+sIgG+ B cells bound most of the VLP, and likely to the TfR rather than the surface IgG (Fig. 1D). We therefore needed to use B cell culture of sorted cells to screen for Ig secretion and specificity as a next step (Fig. 1E–G).
4.2. Isolation and characterization of canine monoclonal antibodies.
Many of the anti-CPV IgG positive wells appeared to contain more than one B cell clone, so we deep sequenced the Ig heavy and light variable regions of the antibody genes in ten wells, and then cloned and expressed those to screen for CPV binding. Of the four anti-CPV IgGs isolated, 2 were identical sequences, one shared the heavy chain with those first two antibodies, and one was quite distinct. The avidity to CPV viruses differed between the two antibodies with an identical heavy chain, indicating that the light chain also contacts the viral capsid. Our preliminary studies suggest that the 3 antibodies with the same H chain bind to the B antigenic site, while the 7C8 antibody may bind the A site, but we will confirm their binding sites using cryoEM structure analysis.
Overall, these studies provide new approaches for isolation and culture of canine B cells, and for cloning and expression of canine antibodies as IgGs. This initial testing of antibodies against CPV proved challenging due to the high binding of those capsids to the activated canine B cells, nevertheless, we were able to isolate a number of antibodies for analysis. In the future we will isolate more antibodies against CPV capsids from dogs of different ages and will also test the method for other canine viruses including canine distemper, canine adenovirus and canine parainfluenza virus, to refine the methods for sorting of B cells and isolation of specific antibodies.
Supplementary Material
Highlights.
-
➢
Here, we report the isolation of CPV-VLP binding B cells from peripheral blood cells obtained from a fully vaccinated dog
-
➢
We designed new primer sets to amplify the variable regions from rearranged canine Ig genes
-
➢
A modified and broadly applicable method for canine B cell culture and the production of cloned canine monoclonal IgGs was developed
-
➢
We identified and characterized the binding of three CPV-specific canine mAbs to CPV capsid variants.
Acknowledgments
We thank Dr. John Parker for generously providing the NIH 393-msCD40L cells and Dr. Neva Watson for her support with the irradiation of the cells. Supported by NIH grant R01-AI092571 to CRP, and by a Morris Animal Foundation fellowship to SF.
Footnotes
All procedures involving animals were approved by Cornell University Institutional Animal Care and Use Committee (IAUCUC) (Protocol ID: 2017–0085).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
References
- Agbandje M, McKenna R, Rossmann MG, Strassheim ML, Parrish CR, 1993. Structure determination of feline panleukopenia virus empty particles. Proteins 16, 155–171. 10.1002/prot.340160204 [DOI] [PubMed] [Google Scholar]
- Allison AB, Organtini LJ, Zhang S, Hafenstein SL, Holmes EC, Parrish CR, 2016. Single Mutations in the VP2 300 Loop Region of the Three-Fold Spike of the Carnivore Parvovirus Capsid Can Determine Host Range. J Virol 90, 753–767. 10.1128/JVI.02636-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews SF, Huang Y, Kaur K, Popova LI, Ho IY, Pauli NT, Henry Dunand CJ, Taylor WM, Lim S, Huang M, Qu X, Lee J-H, Salgado-Ferrer M, Krammer F, Palese P, Wrammert J, Ahmed R, Wilson PC, 2015. Immune history profoundly affects broadly protective B cell responses to influenza. Sci Transl Med 7, 316ra192. 10.1126/scitranslmed.aad0522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun SS, Breuer W, Hermanns W, 1996. Immunohistochemical Examination of Light-chain Expression (Λ/k Ratio) in Canine, Feline, Equine, Bovine and Porcine Plasma Cells. Journal of Veterinary Medicine Series A 43, 573–576. 10.1111/j.1439-0442.1996.tb00489.x [DOI] [PubMed] [Google Scholar]
- Bao Y, Guo Y, Xiao S, Zhao Z, 2010. Molecular characterization of the VH repertoire in Canis familiaris. Veterinary Immunology and Immunopathology 137, 64–75. 10.1016/j.vetimm.2010.04.011 [DOI] [PubMed] [Google Scholar]
- Bergeron LM, McCandless EE, Dunham S, Dunkle B, Zhu Y, Shelly J, Lightle S, Gonzales A, Bainbridge G, 2014. Comparative functional characterization of canine IgG subclasses. Veterinary Immunology and Immunopathology 157, 31–41. 10.1016/j.vetimm.2013.10.018 [DOI] [PubMed] [Google Scholar]
- Brochet X, Lefranc M-P, Giudicelli V, 2008. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res 36, W503–508. 10.1093/nar/gkn316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway HM, Feng KH, Lee DW, Allison AB, Pinard M, McKenna R, Agbandje-McKenna M, Hafenstein S, Parrish CR, 2017. Parvovirus Capsid Structures Required for Infection: Mutations Controlling Receptor Recognition and Protease Cleavages. Journal of Virology 91. 10.1128/JVI.01871-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway HM, Welsch K, Weichert W, Allison AB, Hafenstein SL, Huang K, Iketani S, Parrish CR, 2018. Complex and Dynamic Interactions between Parvovirus Capsids, Transferrin Receptors, and Antibodies Control Cell Infection and Host Range. J Virol 92, e00460–18. 10.1128/JVI.00460-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Su B, Guo X, Sun W, Deng Y, Bao L, Zhu Q, Zhang X, Zheng Y, Geng C, Chai X, He R, Li X, Lv Q, Zhu H, Deng W, Xu Y, Wang Y, Qiao L, Tan Y, Song L, Wang G, Du X, Gao N, Liu J, Xiao J, Su X, Du Z, Feng Y, Qin, Chuan, Qin, Chengfeng, Jin, R., Xie, X.S., 2020. Potent Neutralizing Antibodies against SARS-CoV-2 Identified by High-Throughput Single-Cell Sequencing of Convalescent Patients’ B Cells. Cell 182, 73–84.e16. 10.1016/j.cell.2020.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael LE, Joubert JC, Pollock RV, 1983. A modified live canine parvovirus vaccine. II. Immune response. Cornell Vet 73, 13–29. [PubMed] [Google Scholar]
- Clark SA, Clark LE, Pan J, Coscia A, McKay LGA, Shankar S, Johnson RI, Brusic V, Choudhary MC, Regan J, Li JZ, Griffiths A, Abraham J, 2021. SARS-CoV-2 evolution in an immunocompromised host reveals shared neutralization escape mechanisms. Cell 184, 2605–2617.e18. 10.1016/j.cell.2021.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan HL, Guthmiller JJ, Arevalo P, Huang M, Chen Y-Q, Neu KE, Henry C, Zheng N-Y, Lan LY-L, Tepora ME, Stovicek O, Bitar D, Palm A-KE, Stamper CT, Changrob S, Utset HA, Coughlan L, Krammer F, Cobey S, Wilson PC, 2020. Preexisting immunity shapes distinct antibody landscapes after influenza virus infection and vaccination in humans. Sci Transl Med 12, eabd3601. 10.1126/scitranslmed.abd3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earle KA, Ambrosino DM, Fiore-Gartland A, Goldblatt D, Gilbert PB, Siber GR, Dull P, Plotkin SA, 2021. Evidence for antibody as a protective correlate for COVID-19 vaccines. Vaccine 39, 4423–4428. 10.1016/j.vaccine.2021.05.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellebedy AH, Jackson KJL, Kissick HT, Nakaya HI, Davis CW, Roskin KM, McElroy AK, Oshansky CM, Elbein R, Thomas S, Lyon GM, Spiropoulou CF, Mehta AK, Thomas PG, Boyd SD, Ahmed R, 2016. Defining antigen-specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat Immunol 17, 1226–1234. 10.1038/ni.3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenzel A, Schirrmann T, Hust M, 2016. Phage display-derived human antibodies in clinical development and therapy. MAbs 8, 1177–1194. 10.1080/19420862.2016.1212149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudicelli V, Brochet X, Lefranc M-P, 2011. IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb Protoc 2011, 695–715. 10.1101/pdb.prot5633 [DOI] [PubMed] [Google Scholar]
- Giudicelli V, Chaume D, Lefranc M-P, 2005. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res 33, D256–261. 10.1093/nar/gki010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudicelli V, Duroux P, Ginestoux C, Folch G, Jabado-Michaloud J, Chaume D, Lefranc M-P, 2006. IMGT/LIGM-DB, the IMGT comprehensive database of immunoglobulin and T cell receptor nucleotide sequences. Nucleic Acids Res 34, D781–784. 10.1093/nar/gkj088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetschius DJ, Hartmann SR, Organtini LJ, Callaway H, Huang K, Bator CM, Ashley RE, Makhov AM, Conway JF, Parrish CR, Hafenstein SL, 2021. High-resolution asymmetric structure of a Fab–virus complex reveals overlap with the receptor binding site. PNAS 118. 10.1073/pnas.2025452118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooding GE, Robinson WF, 1982. Maternal antibody, vaccination and reproductive failure in dogs with parvovirus infection. Australian Veterinary Journal 59, 170–174. 10.1111/j.1751-0813.1982.tb15997.x [DOI] [PubMed] [Google Scholar]
- Hafenstein S, Bowman VD, Sun T, Nelson CDS, Palermo LM, Chipman PR, Battisti AJ, Parrish CR, Rossmann MG, 2009. Structural Comparison of Different Antibodies Interacting with Parvovirus Capsids. Journal of Virology 83, 5556–5566. 10.1128/JVI.02532-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughton G, Lanier LL, Babcock GF, 1978. The murine kappa light chain shift. Nature 275, 154–157. 10.1038/275154a0 [DOI] [PubMed] [Google Scholar]
- Hayes MA, Russell RG, Babiuk LA, 1979. Sudden death in young dogs with myocarditis caused by parvovirus. J Am Vet Med Assoc 174, 1197–1203. [PubMed] [Google Scholar]
- Hoelzer K, Parrish CR, 2010. The emergence of parvoviruses of carnivores. Vet Res 41, 39. 10.1051/vetres/2010011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Doria-Rose NA, Longo NS, Laub L, Lin C-L, Turk E, Kang BH, Migueles SA, Bailer RT, Mascola JR, Connors M, 2013. Isolation of human monoclonal antibodies from peripheral blood B cells. Nat Protoc 8, 1907–1915. 10.1038/nprot.2013.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Xia X, Liu B, Fu Y, Chen X, Wang H, Xia Z, 2016. High-yield production of canine parvovirus virus-like particles in a baculovirus expression system. Arch Virol 161, 705–710. 10.1007/s00705-015-2719-1 [DOI] [PubMed] [Google Scholar]
- Kershaw MH, Hsu C, Mondesire W, Parker LL, Wang G, Overwijk WW, Lapointe R, Yang JC, Wang R-F, Restifo NP, Hwu P, 2001. Immunization against Endogenous Retroviral Tumor-associated Antigens. Cancer Res 61, 7920–7924. [PMC free article] [PubMed] [Google Scholar]
- Khoury DS, Cromer D, Reynaldi A, Schlub TE, Wheatley AK, Juno JA, Subbarao K, Kent SJ, Triccas JA, Davenport MP, 2021. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat Med 27, 1205–1211. 10.1038/s41591-021-01377-8 [DOI] [PubMed] [Google Scholar]
- Krammer F, 2021. A correlate of protection for SARS-CoV-2 vaccines is urgently needed. Nat Med 27, 1147–1148. 10.1038/s41591-021-01432-4 [DOI] [PubMed] [Google Scholar]
- Kreer C, Döring M, Lehnen N, Ercanoglu MS, Gieselmann L, Luca D, Jain K, Schommers P, Pfeifer N, Klein F, 2020. openPrimeR for multiplex amplification of highly diverse templates. Journal of Immunological Methods 480, 112752. 10.1016/j.jim.2020.112752 [DOI] [PubMed] [Google Scholar]
- Lanzavecchia A, Corti D, Sallusto F, 2007. Human monoclonal antibodies by immortalization of memory B cells. Curr Opin Biotechnol 18, 523–528. 10.1016/j.copbio.2007.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Callaway HM, Cifuente JO, Bator CM, Parrish CR, Hafenstein SL, 2019. Transferrin receptor binds virus capsid with dynamic motion. Proc Natl Acad Sci USA 116, 20462–20471. 10.1073/pnas.1904918116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc M-P, 2014. Immunoglobulin and T Cell Receptor Genes: IMGT(®) and the Birth and Rise of Immunoinformatics. Front Immunol 5, 22. 10.3389/fimmu.2014.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc M-P, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, Carillon E, Duvergey H, Houles A, Paysan-Lafosse T, Hadi-Saljoqi S, Sasorith S, Lefranc G, Kossida S, 2015. IMGT®, the international ImMunoGeneTics information system® 25 years on. Nucleic Acids Res 43, D413–422. 10.1093/nar/gku1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J, Ponstingl H, Lefranc M-P, Archer J, Sargan D, Bradley A, 2018. Comprehensive annotation and evolutionary insights into the canine (Canis lupus familiaris) antigen receptor loci. Immunogenetics 70, 223–236. 10.1007/s00251-017-1028-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mietzsch, Pénzes, Agbandje-McKenna, 2019. Twenty-Five Years of Structural Parvovirology. Viruses 11, 362. 10.3390/v11040362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda C, Thompson G, 2016. Canine parvovirus: the worldwide occurrence of antigenic variants. Journal of General Virology 97, 2043–2057. 10.1099/jgv.0.000540 [DOI] [PubMed] [Google Scholar]
- Nakamura M, Nakamura K, Miyazawa T, Tohya Y, Mochizuki M, Akashi H, 2003. Monoclonal Antibodies That Distinguish Antigenic Variants of Canine Parvovirus. Clin Diagn Lab Immunol 10, 1085–1089. 10.1128/CDLI.10.6.1085-1089.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DT, Eustis SL, McAdaragh JP, Stotz I, 1979. Lesions of Spontaneous Canine Viral Enteritis. Vet Pathol 16, 680–686. 10.1177/030098587901600606 [DOI] [PubMed] [Google Scholar]
- Organtini LJ, Lee H, Iketani S, Huang K, Ashley RE, Makhov AM, Conway JF, Parrish CR, Hafenstein S, 2016. Near-Atomic Resolution Structure of a Highly Neutralizing Fab Bound to Canine Parvovirus. J. Virol 90, 9733–9742. 10.1128/JVI.01112-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panjwani MK, Atherton MJ, MaloneyHuss MA, Haran KP, Xiong A, Gupta M, Kulikovsaya I, Lacey SF, Mason NJ, 2020. Establishing a model system for evaluating CAR T cell therapy using dogs with spontaneous diffuse large B cell lymphoma. OncoImmunology 9, 1676615. 10.1080/2162402X.2019.1676615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JS, Murphy WJ, Wang D, O’Brien SJ, Parrish CR, 2001. Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J Virol 75, 3896–3902. 10.1128/JVI.75.8.3896-3902.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JS, Parrish CR, 1997. Canine parvovirus host range is determined by the specific conformation of an additional region of the capsid. J Virol 71, 9214–9222. 10.1128/jvi.71.12.9214-9222.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish CR, 1999. Host range relationships and the evolution of canine parvovirus. Vet Microbiol 69, 29–40. 10.1016/s0378-1135(99)00084-x [DOI] [PubMed] [Google Scholar]
- Parrish CR, 1991. Mapping specific functions in the capsid structure of canine parvovirus and feline panleukopenia virus using infectious plasmid clones. Virology 183, 195–205. 10.1016/0042-6822(91)90132-U [DOI] [PubMed] [Google Scholar]
- Parrish CR, 1990. Emergence, Natural History, and Variation of Canine, Mink, and Feline Parvoviruses. Adv Virus Res 38, 403–450. 10.1016/S0065-3527(08)60867-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish CR, Aquadro CF, Strassheim ML, Evermann JF, Sgro JY, Mohammed HO, 1991. Rapid antigenic-type replacement and DNA sequence evolution of canine parvovirus. J Virol 65, 6544–6552. 10.1128/JVI.65.12.6544-6552.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish CR, Carmichael LE, 1983. Antigenic structure and variation of canine parvovirus type-2, feline panleukopenia virus, and mink enteritis virus. Virology 129, 401–414. 10.1016/0042-6822(83)90179-4 [DOI] [PubMed] [Google Scholar]
- Parrish CR, Carmichael LE, Antczak DF, 1982. Antigenic relationships between canine parvovirus type 2, feline panleukopenia virus and mink enteritis virus using conventional antisera and monoclonal antibodies. Arch Virol 72, 267–278. 10.1007/BF01315223 [DOI] [PubMed] [Google Scholar]
- Parrish CR, Have P, Foreyt WJ, Evermann JF, Senda M, Carmichael LE, 1988. The global spread and replacement of canine parvovirus strains. J Gen Virol 69 (Pt 5), 1111–1116. 10.1099/0022-1317-69-5-1111 [DOI] [PubMed] [Google Scholar]
- Parrish CR, O’Connell PH, Evermann JF, Carmichael LE, 1985. Natural variation of canine parvovirus. Science 230, 1046–1048. 10.1126/science.4059921 [DOI] [PubMed] [Google Scholar]
- Parrish CR, Voorhees IEH, 2019. H3N8 and H3N2 Canine Influenza Viruses. Veterinary Clinics of North America: Small Animal Practice 49, 643–649. 10.1016/j.cvsm.2019.02.005 [DOI] [PubMed] [Google Scholar]
- Pollock RV, Carmichael LE, 1982. Maternally derived immunity to canine parvovirus infection: transfer, decline, and interference with vaccination. J Am Vet Med Assoc 180, 37–42. [PubMed] [Google Scholar]
- Reed AP, Jones EV, Miller TJ, 1988. Nucleotide sequence and genome organization of canine parvovirus. J Virol 62, 266–276. 10.1128/JVI.62.1.266-276.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson WF, Wilcox GE, Flower RLP, 1980. Canine Parvoviral Disease: Experimental Reproduction of the Enteric Form with a Parvovirus Isolated from a Case of Myocarditis. Vet Pathol 17, 589–599. 10.1177/030098588001700508 [DOI] [PubMed] [Google Scholar]
- Saliki JT, Mizak B, Flore HP, Gettig RR, Burand JP, Carmichael LE, Wood HA, Parrish CR, 1992. Canine parvovirus empty capsids produced by expression in a baculovirus vector: use in analysis of viral properties and immunization of dogs. J Gen Virol 73 (Pt 2), 369–374. 10.1099/0022-1317-73-2-369 [DOI] [PubMed] [Google Scholar]
- Schaut RG, Lamb IM, Toepp AJ, Scott B, Mendes-Aguiar CO, Coutinho JFV, Jeronimo SMB, Wilson ME, Harty JT, Waldschmidt TJ, Petersen CA, 2016. Regulatory IgDhi B Cells Suppress T Cell Function via IL-10 and PD-L1 during Progressive Visceral Leishmaniasis. The Journal of Immunology 196, 4100–4109. 10.4049/jimmunol.1502678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TYK, Pietzsch J, Fenyo D, Abadir A, Velinzon K, Hurley A, Myung S, Boulad F, Poignard P, Burton DR, Pereyra F, Ho DD, Walker BD, Seaman MS, Bjorkman PJ, Chait BT, Nussenzweig MC, 2011. Sequence and Structural Convergence of Broad and Potent HIV Antibodies That Mimic CD4 Binding. Science [DOI] [PMC free article] [PubMed]
- Schommers P, Gruell H, Abernathy ME, Tran M-K, Dingens AS, Gristick HB, Barnes CO, Schoofs T, Schlotz M, Vanshylla K, Kreer C, Weiland D, Holtick U, Scheid C, Valter MM, van Gils MJ, Sanders RW, Vehreschild JJ, Cornely OA, Lehmann C, Fätkenheuer G, Seaman MS, Bloom JD, Bjorkman PJ, Klein F, 2020. Restriction of HIV-1 Escape by a Highly Broad and Potent Neutralizing Antibody. Cell 180, 471–489.e22. 10.1016/j.cell.2020.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setliff I, Shiakolas AR, Pilewski KA, Murji AA, Mapengo RE, Janowska K, Richardson S, Oosthuysen C, Raju N, Ronsard L, Kanekiyo M, Qin JS, Kramer KJ, Greenplate AR, McDonnell WJ, Graham BS, Connors M, Lingwood D, Acharya P, Morris L, Georgiev IS, 2019. High-Throughput Mapping of B Cell Receptor Sequences to Antigen Specificity. Cell 179, 1636–1646.e15. 10.1016/j.cell.2019.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K, Garman L, Wrammert J, Zheng N-Y, Capra JD, Ahmed R, Wilson PC, 2009. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc 4, 372–384. 10.1038/nprot.2009.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SA, Crowe JE, 2015. Use of human hybridoma technology to isolate human monoclonal antibodies. Microbiol Spectr 3, AID-0027-2014. 10.1128/microbiolspec.AID-0027-2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, Guikema JEJ, Schrader CE, 2008. Mechanism and regulation of class switch recombination. Annu Rev Immunol 26, 261–292. 10.1146/annurev.immunol.26.021607.090248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiniger SCJ, Dunkle WE, Bammert GF, Wilson TL, Krishnan A, Dunham SA, Ippolito GC, Bainbridge G, 2014. Fundamental characteristics of the expressed immunoglobulin VH and VL repertoire in different canine breeds in comparison with those of humans and mice. Molecular Immunology 59, 71–78. 10.1016/j.molimm.2014.01.010 [DOI] [PubMed] [Google Scholar]
- Strassheim ML, Gruenberg A, Veijalainen P, Sgro J-Y, Parrish CR, 1994. Two Dominant Neutralizing Antigenic Determinants of Canine Parvovirus Are Found on the Threefold Spike of the Virus Capsid. Virology 198, 175–184. 10.1006/viro.1994.1020 [DOI] [PubMed] [Google Scholar]
- Sundling C, Lau AWY, Bourne K, Young C, Laurianto C, Hermes JR, Menzies RJ, Butt D, Kräutler NJ, Zahra D, Suan D, Brink R, 2021. Positive selection of IgG+ over IgM+ B cells in the germinal center reaction. Immunity 54, 988–1001.e5. 10.1016/j.immuni.2021.03.013 [DOI] [PubMed] [Google Scholar]
- Tang L, Sampson C, Dreitz MJ, McCall C, 2001. Cloning and characterization of cDNAs encoding four different canine immunoglobulin γ chains. Veterinary Immunology and Immunopathology 80, 259–270. 10.1016/S0165-2427(01)00318-X [DOI] [PubMed] [Google Scholar]
- Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H, 2008. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. Journal of Immunological Methods 329, 112–124. 10.1016/j.jim.2007.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao J, Chapman MS, Agbandje M, Keller W, Smith K, Wu H, Luo M, Smith TJ, Rossmann MG, Compans RW, Et A, 1991. The three-dimensional structure of canine parvovirus and its functional implications. Science 251, 1456–1464. 10.1126/science.2006420 [DOI] [PubMed] [Google Scholar]
- Vazquez-Lombardi R, Nevoltris D, Luthra A, Schofield P, Zimmermann C, Christ D, 2018. Transient expression of human antibodies in mammalian cells. Nat Protoc 13, 99–117. 10.1038/nprot.2017.126 [DOI] [PubMed] [Google Scholar]
- Vihinen-Ranta M, Wang D, Weichert WS, Parrish CR, 2002. The VP1 N-Terminal Sequence of Canine Parvovirus Affects Nuclear Transport of Capsids and Efficient Cell Infection. Journal of Virology 76, 1884–1891. 10.1128/JVI.76.4.1884-1891.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees IEH, Lee H, Allison AB, Lopez-Astacio R, Goodman LB, Oyesola OO, Omobowale O, Fagbohun O, Dubovi EJ, Hafenstein SL, Holmes EC, Parrish CR, 2019. Limited Intrahost Diversity and Background Evolution Accompany 40 Years of Canine Parvovirus Host Adaptation and Spread. J Virol 94, e01162–19. 10.1128/JVI.01162-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardemann H, 2003. Predominant Autoantibody Production by Early Human B Cell Precursors. Science 301, 1374–1377. 10.1126/science.1086907 [DOI] [PubMed] [Google Scholar]
- Weichert WS, Parker JS, Wahid AT, Chang SF, Meier E, Parrish CR, 1998. Assaying for structural variation in the parvovirus capsid and its role in infection. Virology 250, 106–117. 10.1006/viro.1998.9352 [DOI] [PubMed] [Google Scholar]
- Wilson S, Illambas J, Siedek E, Stirling C, Thomas A, Plevová E, Sture G, Salt J, 2014a. Vaccination of dogs with canine parvovirus type 2b (CPV-2b) induces neutralizing antibody responses to CPV-2a and CPV-2c. Vaccine 32, 5420–5424. 10.1016/j.vaccine.2014.07.102 [DOI] [PubMed] [Google Scholar]
- Wilson S, Siedek E, Thomas A, King V, Stirling C, Plevová E, Salt J, Sture G, 2014b. Influence of maternally-derived antibodies in 6-week old dogs for the efficacy of a new vaccine to protect dogs against virulent challenge with canine distemper virus, adenovirus or parvovirus. Trials in Vaccinology 3, 107–113. 10.1016/j.trivac.2014.06.001 [DOI] [Google Scholar]
- Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng N-Y, Mays I, Garman L, Helms C, James J, Air GM, Capra JD, Ahmed R, Wilson PC, 2008. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671. 10.1038/nature06890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Parrish CR, 2001. Canine Parvovirus Capsid Assembly and Differences in Mammalian and Insect Cells. Virology 279, 546–557. 10.1006/viro.2000.0734 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.