

Abstract
Mutations in the bone morphogenetic protein receptor type 2 (bmpr2) gene and signaling pathway impairment are observed in heritable and idiopathic pulmonary arterial hypertension (PAH). In PAH, endothelial dysfunction is currently handled by drugs targeting the endothelin‐1 (ET‐1), nitric oxide (NO), and prostacyclin (PGI2) pathways. The role of angiogenesis in the disease process and the effect of PAH therapies on dysregulated angiogenesis remain inconclusive. We aim to investigate in vitro whether (i) bmpr2 silencing can impair angiogenic capacity of human lung microvascular endothelial cells (HLMVECs) and (ii) PAH therapies can restore them. The effects of macitentan (ET‐1), tadalafil (NO), and selexipag (PGI2), on BMPRII pathway activation, endothelial barrier function, and angiogenesis were investigated in bmpr2‐silenced HLMVECs. Stable bmpr2 silencing resulted in impaired migration and tube formation in vitro capacity. Inhibition of ET‐1 pathway was able to partially restore tube formation in bmpr2‐silenced HLMVECs, whereas none of the therapies was able to restore endothelial barrier function, no deleterious effects were observed. Our findings highlight the potential role of BMPRII signaling pathway in driving pulmonary endothelial cell angiogenesis. In addition, PAH drugs display limited effects on endothelial function when BMPRII is impaired, suggesting that innovative therapeutic strategies targeting BMPRII signaling are needed to better rescue endothelial dysfunction in PAH.
Keywords: endothelial barrier function, endothelial bmpr2 silencing, in vitro angiogenesis, PAH drugs, pulmonary hypertension
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a severe and progressive disease characterized by a distal precapillary vasculopathy resulting in increased pulmonary vascular resistance, right ventricular hypertrophy, progressive right heart failure, and ultimately death if not treated. 1 PAH is a complex multifactorial disease involving abnormal vascular tone, endothelial dysfunction, inflammation, dysregulated angiogenesis, and enhanced thrombosis. Plexiform lesions, a hallmark of severe PAH, are typically defined as a dynamic network of vascular channels, with the presence of phenotypically distinct endothelial cells, for example, a quiescent phenotype lining the channels and a proliferating, apoptosis‐resistant phenotype at the core of the lesion 2 , 3 as well as inflammatory cells 4 ; they are mainly located at arterial branch points or at the origin of supernumerary arteries.
More than 70% of patients with heritable PAH and 20% of idiopathic PAH harbor heterozygous mutations in the bone morphogenetic protein type 2 receptor (bmpr2) gene. 5 , 6 , 7 bmpr2 transcodes for a transmembrane serine/threonine kinase receptor of the bone morphogenetic protein (BMP) pathway, which is essential for embryogenesis, development, and adult tissue homeostasis. 8 Upon ligand binding, a heterodimer complex, formed by a combination of two type I receptors and two type II receptors, propagates the signal through a canonical pathway by phosphorylation of the Smad1/5/8 transcription factors or a noncanonical pathway by phosphorylation of p38 MAPK. 9 To date, about 500 distinct bmpr2 variants have been identified and most of them are pathogenic. 10 In addition, mutations in other genes including the activin receptor‐like kinase 1 (alk1), endoglin (eng), 11 small mothers against decapentaplegic homolog 1 (smad1), smad4 12 and smad9 13 have been found being associated to PAH, highlighting the crucial role of the BMP pathway in the pathogenesis of PAH. 14
Among the multifactorial processes associated with PAH, endothelial dysfunction and defective angiogenesis play a key role in initiating structural changes in the pulmonary vasculature. The pharmacological therapies currently used to treat PAH mainly display vasodilator effects by targeting 3 pathways: endothelin‐1 (ET‐1), nitric oxide (NO), and prostacyclin (PGI2) pathways, 15 but fail to reverse pulmonary vascular remodeling.
Expression of vascular endothelial growth factors (VEGFs), considered as the most potent angiogenic factors, and their receptors is upregulated in pulmonary tissue from PAH patients, potentially resulting in the formation of plexiform lesions. 16 , 17 Interestingly, rats exposed to hypoxia and who received a single injection of SU5416, an inhibitor of the VEGF receptor with potent antiangiogenic properties, can develop severe PAH and plexiform‐like lesions. 18 In addition, tyrosine kinase inhibitors (TKIs), used as chemotherapeutic and antiangiogenic drugs, such as imatinib, have proven efficacious in patients with severe PAH, but serious side effects (intracranial bleeding) have prevented further use to treat PAH. 19 By contrast, nine patients with chronic myelogenous leukemia treated with another TKI, dasatinib, developed severe PAH. 20 The so‐called angiogenic paradox remains a matter of debate in the PAH research field.
Induced pluripotent stem cells (iPSCs) of PAH patients carrying a bmpr2 mutation and further differentiated in endothelial cells display reduced migration and tube forming capacity in vitro compared with iPSCs from control subjects or healthy bmpr2 mutation carriers. 21 Eventually, we recently observed that stable bmpr2 silencing in human lung microvascular endothelial cells (HLMVECs) impairs endothelium barrier function. 22 The role of vascular endothelial (VE)‐cadherin, a highly endothelial‐specific adhesion molecule, in the BMPRII‐dependent loss of barrier function remains inconclusive.
Consequently, in addition to impaired BMPRII signaling, deficient endothelial barrier function, and impaired angiogenesis are thought to contribute to PAH progression. 23 , 24 Therefore, we aim to investigate whether stable bmpr2 silencing would worsen angiogenic capacities of HLMVECs and whether PAH drugs could restore endothelium barrier function and angiogenic capacities in a context of impaired BMPRII signaling, using in vitro functional assays.
MATERIALS AND METHODS
bmpr2 silencing in HLMVECs
bmpr2 was silenced in HLMVECs (Cell Applications Inc.) issued from one single donor, as previously described. 22 Briefly, simian immunodeficiency virus (SIV)‐based lentiviral encoding microRNA 30 (miR30)‐based knockdown hairpins siRNA (bmpr2 mRNA at position 1920 and firefly luciferase as control) were generated. The transfer plasmid constructs, pGAE SIV SFFV‐eGFP‐P2Azeo‐miRNA‐HsBMPR2−WPRE and pGAE‐SFFV‐eGFPP2A‐zeo‐miRNA‐fLuc‐WPRE, contain a zeocin resistance cassette driven from a spleen focus forming virus long terminal repeat promoter, followed by the respective miRs and the woodchuck hepatitis virus posttranscriptional regulatory element; the constructs also contain the cDNA for enhanced green fluorescent protein (eGFP) cassette, as a reporter gene. All lentiviral vector plasmids were designed and cloned, and vector production was performed at the Leuven Viral Vector Core. Stable BMPR2 knockdown and controls in HLMVECs were generated by lentiviral transduction. HLMVECs were seeded in a T75 flask at a density of 200,000 cells. When HLMVECs were 40% confluent, cells were transduced with a serial dilution series of lentiviral vectors. After 48 h, the medium was replaced with growth medium containing 200 mg/mL zeocin to select transduced cells. Transduction efficiency was evaluated by flow cytometry for eGFP expression. bmpr2 silencing efficiency was evaluated by qrt‐PCR and Western blot analysis resulting in an 89% and 87% decrease in BMPR2 mRNA and protein expression, respectively, in comparison with HLMVECs transduced with the control vector.
Cell culture
HLMVECs were cultured in microvascular endothelial cell growth medium (EGM; Cell Applications Inc.) containing 100 U/mL penicillin, 100 μg/mL streptomycin, and 1.25 μg/mL fungizone (ThermoFisher Scientific) and starved in microvascular endothelial cell basal medium ‐ EBM (Cell Applications Inc.) supplemented with 0.2% growth supplement.
Assessment of cell viability
Macitentan (ET‐1 receptor antagonist; Actelion), selexipag (PGI2 receptor agonist; Actelion), and tadalafil (phosphodiesterase‐5 [PDE5] inhibitor; Sigma‐Aldrich) solutions were prepared with serial dilutions from a stock solution (10 mM in dimethylsulfoxide; DMSO). The required concentrations were determined according to the manufacturer's datasheet and the maximum circulating concentration (Cmax) measured in PAH patients. Cmax values are as follows: macitentan, 1.35 µM; selexipag, 0.06 µM; tadalafil, 1.32 µM. Subsequently, a range of concentrations comprising Cmax was further used.
Drug toxicity was evaluated using the cell viability 2‐(2‐methoxy‐4‐nitrophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H‐tetrazolium (WST‐1) method. HLMVECs seeded in a 96‐well plate, at the density of 104 cells/well, were incubated in the presence of increasing drug concentrations ranging from 10−6 to 10−2 mM for 2, 4, and 6 h, or in the absence of drugs. Absorbance was measured at 550 nm and results were expressed as a percentage of values in the absence of drugs. Considering that respective Cmax values did not display any cytotoxic effects (Supporting Information: Figure S1) and to remain as close as possible to the in vivo situation, Cmax values were used as working concentrations in further experiments.
Western blot analysis
Confluent HLMVEC monolayers were starved overnight and homogenized in fresh, ice‐cold RIPA lysis buffer containing 50 mM Tris, pH 7.4, 1% octylphenoxypolyethoxyethanol (IGEPAL), 0.5% sodium‐deoxycholate, 0.1% sodium dodecyl sulfate, 150 mM NaCl, 1 mM ethylene glycol tetra‐acetic acid EGTA), 1% protease inhibitor cocktail 100x (Halt™), 1% phosphatase inhibitor cocktail 100x (Halt™). Homogenates were centrifuged at 12,000g for 15 min at 4°C to remove insoluble material and supernatants were collected. Protein concentrations were determined using the Pierce BCA Protein Assay Kit (ThermoFisher Scientific). Proteins were separated on a 10% acrylamide gel by SDS‐PAGE and further visualized as previously described. 22 Briefly, nonspecific binding sites were blocked and membranes were incubated with primary antibodies against p38 MAPK, phospho‐p38 MAPK, Smad1/5/8, phospho‐Smad1/5/8 and VE‐Cadherin (Cell Signaling Tech.) overnight at 4°C. Appropriate secondary antibodies were incubated for 1 h at room temperature, revealed with a chemiluminescence kit (GE Healthcare) and imaged using the Proxima 2850T imaging system (Isogen). A drug‐induced time‐response of activation of Smad1/5/8 and p38 MAPK was performed by Western blot analysis, showing that Smad1/5/8 and p38 MAPK activation was optimal between 30 min and 1 h. In further experiments, activation of BMPRII effectors was evaluated upon 1 h incubation with the different drugs.
Tube formation assay
Angiogenesis µ‐slides (Ibidi) were filled with Matrigel© (BD Bioscience) and incubated for 30 min at 37°C. HLMVECs were starved overnight and trypsinized, resuspended in growth medium in the absence or presence of macitentan (1.35 µM), selexipag (0.06 µM), or tadalafil (1.32 µM) and added on top of Matrigel©. After 4 h, images were taken using an Olympus IX71 inverted microscope (Olympus) and analyzed using the ImageJ software with Java version 1.8.0_231 (ImageJ, National Health Institute, Bethesda, MD, USA). Number of tubes, nodes, and meshes (Supporting Information: Figure S2A) were quantified using the software “Angiogenesis Analyzer” developed by Gilles Carpentier. 25
Migration scratch assay
HLVMECs were seeded in two‐well microdish culture inserts (Ibidi; Supporting Information: Figure S2B) and subconfluent cells were starved overnight. Inserts were removed, cells were washed twice with growth medium and incubated in growth medium containing vehicle, macitentan (1.35 µM), selexipag (0.06 µM), or tadalafil (1.32 µM). Pictures were taken every other hour using an inverted microscope (DMi1; Leica Microsystems GmbH). The distance of the gap was measured every consecutive hour up to 12 h using ImageJ. At each time point, the gap distance was measured and subtracted from the initial gap distance, further plotted over time, the area under the curve (AUC) was calculated and corresponds to migration velocity in µm/h (Supporting Information: Figure S4).
Assessment of HLMVEC barrier function
To assess HLMVEC barrier function, permeability assay was performed as previously described. 22 Briefly, cells were seeded onto 24‐well Transwell inserts (BD Bioscience; 100,000 cells/insert) to form a monolayer and starved overnight. Cells were first stimulated with macitentan, selexipag, or tadalafil for 1 h, then fluorescently labeled bovine serum albumin (BSA, Sigma‐Aldrich; 0.1 mg/mL) was added to the upper chamber. Leakage of labeled BSA into the lower chamber was assessed by collecting 50 µL from the lower chamber at baseline, after 30 min, 1, 2 and 4 h, as a measure for permeability. Absorbance was measured at 450 nm using a FLUOstar Omega microplate reader (BMG Labtech). Concentrations of BSA were calculated, plotted over time and the AUC was calculated for each condition.
Immunofluorescence
HLMVECs seeded onto fibronectin‐coated chamber slides. A confluent cell layer was stained by immunofluorescence using antibodies against VE‐cadherin and phospho‐VE‐cadherin (Cell Signaling; VE‐cadherin 1:800, phospho‐VE‐cadherin 1:50), as described previously. 22 Nuclei were counterstained using 4’,6‐diamino‐2‐phenylindole (DAPI; ThermoFisher Scientific). Quantification of immunofluorescent images was performed using Image J software by measuring staining intensity, corresponding to VE‐cadherin and phospho‐VE‐cadherin expression. The number of DAPI‐stained nuclei was calculated. The staining intensity of each image was related to the number of cells and expressed as arbitrary unit (AU).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9.0.0 (GraphPad Software Inc.). Differences between two groups were analyzed using paired or unpaired Student's t test. Differences between more than two groups were analyzed using analysis of variance (ANOVA) multiple comparisons followed by Tukey's post‐hoc tests. All p values are for two‐sided tests. A value of p < 0.05 was considered statistically significant and only significant p < 0.05 values were mentioned in the graphs. Data shown are expressed as median (25th–75th interquartile range) in graphs and as mean ± SD within the text.
RESULTS
Macitentan, selexipag, and tadalafil do not interfere with canonical or noncanonical BMPRII signaling
We previously observed that silencing of bmpr2 results in decreased canonical Smad1/5/8 and increased noncanonical p38 MAPK activation. 22 We therefore aim to investigate whether drugs targeting ET‐1 (macitentan), NO (tadalafil) and PGI2 (selexipag) pathways affect activation of BMPRII downstream effectors. We did not observe any effect of macitentan, selexipag, or tadalafil on the canonical Smad1/5/8 signaling (Figure 1) and on the noncanonical p38 MAPK protein activation (Figure 2) in control HLMVECs and in bmpr2‐silenced HLMVECs.
Figure 1.
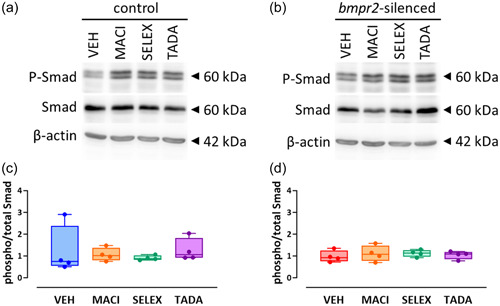
Effect of macitentan, selexipag, and tadalafil on BMPRII canonical signaling pathway in HLMVECs. Representative Western blots of phosphorylated (P‐Smad) and total Smad (1/5/8) proteins in control (a) and bmpr2‐silenced HLMVECs (b) incubated with vehicle (VEH), macitentan (MACI), selexipag (SELEX) and tadalafil (TADA) for 1 h. Quantitative expression of phosphorylated versus total Smad1/5/8 proteins in control (c) and bmpr2‐silenced HLMVECs (d). Results are presented as ratio of phosphorylated over total Smad (1/5/8) protein expression. Independent experiments were performed in four replicates for HLMVECs between passages 5 and 8 and expressed as median (25th–75th interquartile range). Comparisons by one‐way ANOVA and post‐hoc Tukey's multiple tests (c, d).
Figure 2.
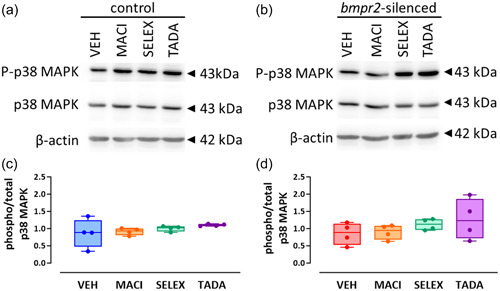
Effect of macitentan, selexipag, and tadalafil on BMPRII noncanonical signaling pathway in HLMVECs. Representative Western blots of phosphorylated (P‐p38 MAPK) and total p38MAPK in control (a) and bmpr2‐silenced HLMVECs (b) incubated with vehicle (VEH), macitentan (MACI), selexipag (SELEX) and tadalafil (TADA) for 1 h. Quantitative expression of phosphorylated versus total p38MAPK in control (c) and bmpr2‐silenced HLMVECs (d) in bmpr2‐silenced HLMVECs. Results are presented as ratio of phosphorylated over total p38 MAPK protein expression. Independent experiments were performed in four replicates for HLMVECs between passages 5 and 8 and expressed as median (25th–75th interquartile range). Comparisons by one‐way ANOVA and post‐hoc Tukey's multiple tests (c, d).
Macitentan, selexipag, and tadalafil do not restore altered barrier function in bmpr2‐silenced HLMVECs
We previously observed that bmpr2 silencing resulted in impaired endothelial barrier function in HLMVECs, without affecting VE‐cadherin content. 22 We therefore aim to investigate whether macitentan, selexipag, and tadalafil might restore the endothelial barrier function and/or affect VE‐cadherin content and phosphorylation. We did not observe any significant effects of macitentan, selexipag, or tadalafil on endothelial barrier function in control (Figure 3a) or bmpr2‐silenced HLMVECs (Figure 3b).
Figure 3.
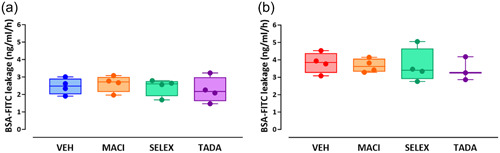
Effect of macitentan, selexipag, and tadalafil on endothelial barrier function of control (a) and bmpr2‐silenced (b) HLMVECs. Results are expressed as BSA‐FITC leakage (ng/mL/h), measured in four independent experiments performed in triplicate in HLMVECs between passages 5 and 8 and expressed as median (25th–75th interquartile range. Comparisons by one‐way ANOVA and post‐hoc Tukey's multiple tests (a, b). MACI, macitentan; SELEX, selexipag; TADA, tadalafil; VEH, vehicle.
The impairment of endothelial barrier function previously observed in bmpr2‐silenced HLMVECs, 22 was not associated with any change in VE‐cadherin expression or phosphorylation (Figure 4a–c). Similarly, bmpr2 silencing in HLMVECs did not alter VE‐cadherin expression and phosphorylation evaluated by immunofluorescent staining (Figure 4d–f). Interestingly, macitentan, selexipag, and tadalafil tend to increase phosphorylation of VE‐cadherin by 32%, 41%, and 49%, respectively, in bmpr2‐silenced HLMVECs (Supporting Information: Figure S3A,C), whereas they had no effect on VE‐cadherin phosphorylation in control HLMVECs (Supporting Information: Figure S3A,B).
Figure 4.
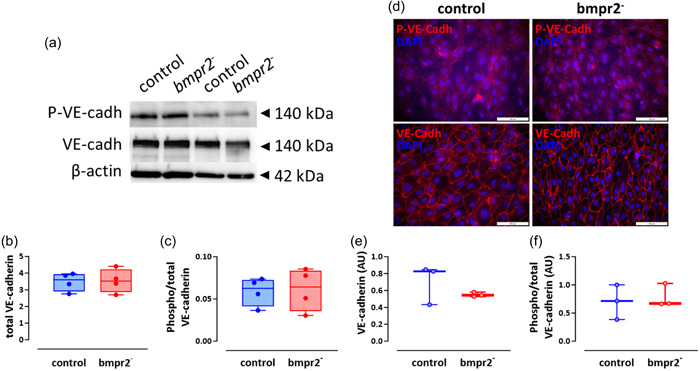
Effect of bmpr2 silencing on VE‐cadherin activation. Representative Western blots of phosphorylated VE‐cadherin (P‐VE‐cadh) and VE‐cadherin (VE‐cadh) protein expression. β‐actin was used as loading control (a). Quantification of VE‐cadherin expression (b) and phosphorylated over total VE‐cadherin expression (c) in control and bmpr2‐silenced (bmpr2 ‐) HLMVECs; results are presented as ratio of total VE‐cadherin over β‐actin protein expression (b) or phosphorylated over total VE‐cadherin protein expression (c), measured in four independent experiments. Representative pictures of P‐VE‐cadherin (P‐VE‐cadh) and total VE‐cadherin (VE‐cadh) immunofluorescent staining in control and bmpr2‐silenced (bmpr2 ‐) HLMVECs; nuclei were counterstained using 4’,6‐diamino‐2‐phenylindole (DAPI); Scale = 100 µm (d). Quantification of VE‐cadherin (e) and phosphorylated over total VE‐cadherin (f) immunofluorescent staining in tree independent experiments. Data are expressed as median (25th–75th interquartile range). Comparisons by unpaired Student's t test (b–f).
Angiogenic capacities are altered in bmpr2‐silenced HLMVECs
Angiogenic capacities were assessed in vitro by investigating migration capacities and tube formation in HLMVECs. Wound closure occurred faster in control than in bmpr2‐silenced HLMVECs (Supporting Information: Figure S4). Migration velocity of bmpr2‐silenced HLMVECs was significantly lower compared with control HLMVECs (25.1 ± 3.2 vs. 21.1 ± 2.3 µm/h; p = 0.0434; Figure 5a). Tube formation capacities were altered in bmpr2‐silenced compared with control HLMVECs (Figure 5b–d), as illustrated by 28%, 23% and 41% significant decrease in the number of tubes (p = 0.0189), nodes (p = 0.0056), and meshes (p = 0.0149), respectively.
Figure 5.
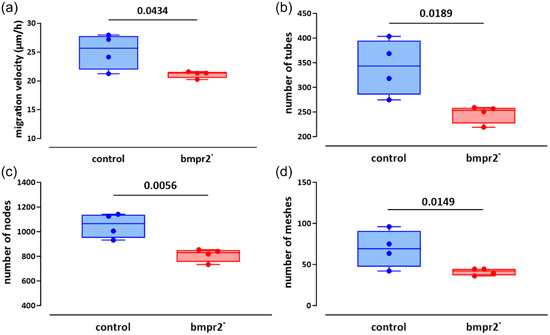
Effect of bmpr2 silencing on in vitro angiogenic capacities of HLMVECs. Migration capacities (a) expressed as migration velocity (µm/h) was evaluated in control and bmpr2‐silenced (bmpr2 ‐) HLMVECs. For tube forming capacity, the number of tubes (b) nodes (c), and meshes (d) was measured in control and bmpr2‐silenced (bmpr2 ‐) HLMVECs. Five independent experiments including one to two replicates were performed in HLMVECs between passages 5 and 8 and expressed as median (25th–75th interquartile range). Comparisons by paired Student's t test (a–d).
Macitentan partially restores altered angiogenic capacities in bmpr2‐silenced HLMVECs
Tadalafil significantly increased the migration velocity of bmpr2‐silenced HLMVECs (22.9 ± 2.1 µm/h vs. 20.8 ± 2.4, p = 0.0141, Figures 6 & 8e), whereas tadalafil (23.0 ± 2.7 µm/h vs. 24.7 ± 2.7, p = 0.0133) significantly decreased migration velocity of control HLMVECs (Figures 6 & 8a).
Figure 6.
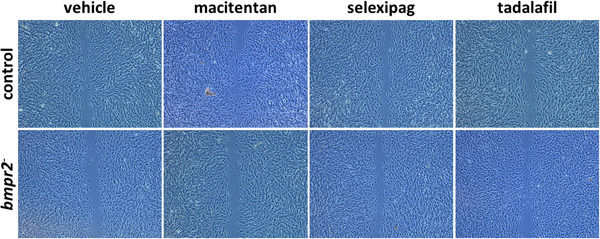
Effect of macitentan, selexipag, and tadalafil on migration capacities of control and bmpr2‐silenced (bmpr2 ‐) HLMVECs. Representative pictures of wound closure after 8 h are shown.
Figure 8.
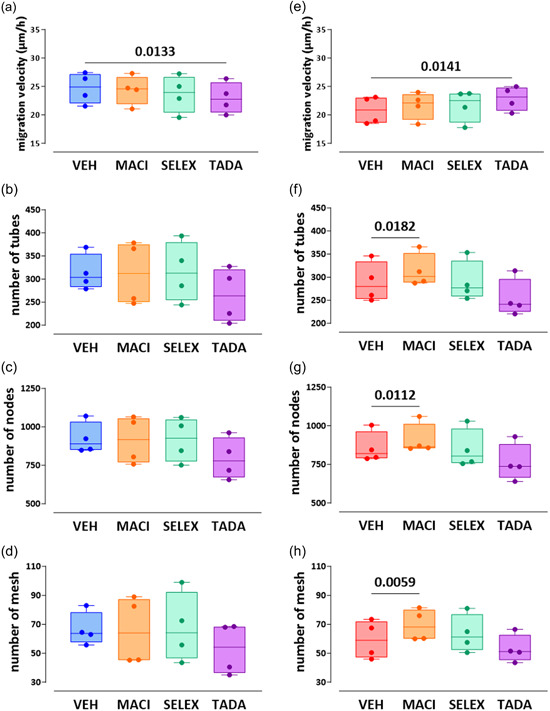
Effect of macitentan, selexipag, and tadalafil on in vitro angiogenic capacities of control and bmpr2‐silenced HLMVECs. Migration capacities (a, e) was expressed as migration velocity (µm/h). For tube forming capacity, the number of tubes (b, f) nodes (c, g), and meshes (d, h) was measured. Four independent experiments including at least two replicates were performed in bmpr2‐silenced HLMVECs between passages 5 and 8 and expressed as median (25th–75th interquartile range) and whiskers to min and max values. Comparisons by one‐way ANOVA and post‐hoc Tukey's multiple tests (a–h). MACI, macitentan; SELEX, selexipag; TADA, tadalafil; VEH, vehicle.
Regarding in vitro tube formation, we found that macitentan significantly enhanced tube formation capacities of bmpr2‐silenced HLMVECs by increasing the number of tubes, nodes, and meshes by 10% (p = 0.0182), 7% (p = 0.0112), and 19% (p = 0.0059), respectively (Figures 7 & 8f–h). By contrast, we did not observe any effects of macitentan on in vitro tube formation of control HLMVECs (Figures 7 & 8b–d).
Figure 7.
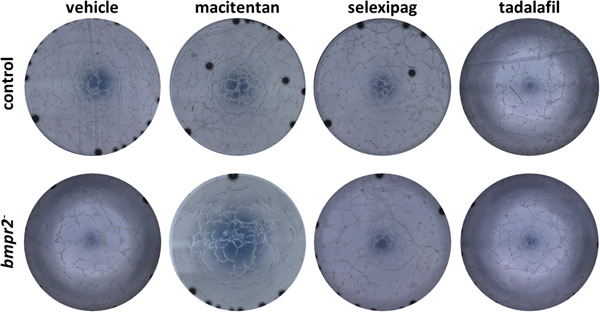
Effect of macitentan, selexipag, and tadalafil on tube forming capacity of control and bmpr2‐silenced (bmpr2 ‐) HLMVECs. Representative pictures of the tubular network formed 4 h after seeding cells on Matrigel© are shown.
An overview of effects of bmpr2 silencing and, macitentan, selexipag, and tadalafil on in vitro angiogenic capacities of control versus bmpr2‐silenced HLMVECs is shown in Supporting Information: Table S1.
DISCUSSION
In the present study, we observed that stable bmpr2 silencing in HLMVECs resulted in impaired angiogenic capacity in vitro, as illustrated by decreased migration capacity and loss of tube forming capabilities. We observed that macitentan can partially restore angiogenic capacity in bmpr2‐silenced HLMVECs. By contrast, we did not find any beneficial effects of drugs targeting ET‐1, NO, or PGI2 pathways on impaired endothelial barrier function in vitro.
bmpr2 silencing & angiogenesis
We found that stable bmpr2 silencing in HLMVECs resulted in impaired angiogenesis, as illustrated by reduced migration capacities and tube formation. Accordingly, IPSCs derived from bmpr2 mutation carriers with PAH display decreased migration capacity and tube forming capacity compared with IPSCs from healthy individuals or unaffected bmpr2 mutation carriers. 21 Moreover, rats expressing a bmpr2 mutant display a decreased pulmonary capillary network, in comparison with control rats, 26 suggesting that bmpr2 mutation is accompanied by a loss of angiogenic capacities. In agreement, loss of downstream BMPRII effectors in Smad8‐deficient mice resulted in impaired development of the pulmonary vasculature. 27 Whereas angiogenesis was previously considered deleterious in the early pathogenesis of PAH, recent findings have evidenced the potential beneficial effects of neovascularization in preventing PAH progression. In addition, extracellular ligands and activators of BMPRII can promote angiogenesis. Actually, BMP2 induced in vivo angiogenesis in mice, 28 , 29 whereas BMP4 induced migration of human microvascular ECs via the activation of the VEGF/VEGFR2 signaling. 28 , 29 BMP6 harbors similar effects as those of VEGF in inducing sprouting angiogenesis both in vivo and in vitro. 30 Ectopic expression of Id1, a downstream effector of BMPRII in ECs, mimicked BMP‐induced effects by inducing EC migration and tube formation. 31 Although the role of BMP9 remains debatable, Long et al. observed that administration of BMP9 to mice bearing a human bmpr2 mutation and in monocrotaline‐ and Sugen/hypoxia‐induced rat experimental PH, reversed PH by restoring tube formation and endothelial barrier function. 32 Finally, downregulation of BMP signaling in ECs by the natural compound codonolactone impairs tumor angiogenesis. 33 Interestingly, instillation of lentiviral vectors expressing angiopoietin‐1 in combination with hepatocyte growth factor or VEGF reduced vascular leakage, improved adherens junction integrity of vascular endothelial cells and promoted maturation of new blood vessels in monocrotaline rats. 34 Altogether, our results converge with other recent studies highlighting the key role of BMPRII signaling in promoting angiogenesis and preventing microvascular rarefaction in PAH.
Effect of PAH drugs on BMPRII signaling
Activation of the ET‐1, PGI2, or NO pathways by macitentan, selexipag, and tadalafil, respectively, did not modify canonical (Smad1/5/8) and noncanonical (p38 MAPK) BMPRII signaling, previously shown to be impaired by bmpr2 silencing in HLMVECs, 22 indicating that vasodilator drugs are not able to restore activation of downstream effectors of BMPRII. Our results are discrepant with previous studies mainly performed in pulmonary arterial smooth muscle cells (PASMCs) or fibroblast. Thus, bosentan, a dual ET‐1 receptor antagonist, was able to restore BMPRII expression and BMPRII downstream signaling in PASMCs. 35 Macitentan inhibits the TGFβ activity by preventing the formation of an ET‐1/TGFβ receptor complex in fibroblasts. 36 Sildenafil, a well‐characterized PDE5 inhibitor, can restore BMPRII signaling by enhancing canonical Smad signaling in PASMCs overexpressing a bmpr2 mutant. 37 , 38 Iloprost, an inhaled PGI2 analog, induces Id1 expression and in combination with treprostinil, a parenteral PGI2 analog, enhance Smad1/5 phosphorylation in response to BMP4 in PASMCs. 39 These differences can be attributed to potential differential effects of vasodilator drugs on BMPRII signaling in PAECs and PASMCs; whereas bmpr2 silencing results in enhanced PASMC proliferation, 40 it promotes PAEC apoptosis. 41 It also suggests that the target of PAH drugs harboring vasodilator effects, within the pulmonary vascular wall is rather PASMCs than PAECs. In vivo, administration of sildenafil in monocrotaline rats results in a mild increase in BMPRII expression in whole lung tissue, but specific effects of sildenafil on pulmonary vascular cells were not considered. 42 The absence of PAH drug effects on Smad signaling could be attributed to lateral Smad and mixed Smad complexes, previously observed in bmpr2‐deficient ECs. 43 Another aspect is the drug concentrations used. We have deliberately based the choice of drug working concentrations on respective drug Cmax, to reproduce the clinical situation observed in treated PAH patients. In addition, according to drug incubation duration, ranging from 1 to 24 h, different effects can be observed. 35 , 36 , 37 , 38 Hence, we performed a time‐response curve showing that 1‐h stimulation displayed the optimal response. Importantly, despite the absence of any effects of the drugs in restoring BMPRII signaling in HLMVECs, we did not experience any deleterious effect of PAH drugs.
Effect of PAH drugs on endothelial barrier function
Importantly, we previously reported that stable bmpr2 silencing in HLMVECs resulted in impaired endothelial barrier function 22 and ET‐1 secretion was higher in pulmonary microvascular ECs from bmpr2 mutation carriers compared with noncarriers or controls. 44 However, we were unable to restore the deficient bmpr2‐induced barrier function by blocking both ET‐1 receptors with macitentan. By contrast, ET‐1 receptor activation was previously demonstrated to reduce endothelial barrier function 45 and PGI2 analogs harbor endothelial barrier protective effects in vitro. 46 , 47 In the present in vitro model, the PGI2 pathway was activated through the IP receptor agonist, selexipag, whereas in the abovementioned studies, PGI2 analogs were used. Additionally, in the current study, the endothelial barrier function was measured using paracellular leakage of fluorescently labeled BSA through the endothelial monolayer in contrast with abovementioned studies, in which trans‐endothelial electrical resistance (TEER) was performed. Noteworthy, other PDE inhibitors without any vasodilator properties can modify brain microvascular endothelial cell barrier. 48 Interestingly, we did not report any harmful effects of vasodilator drugs on the endothelial barrier function.
Effect of PAH drugs on angiogenesis
Although dysregulated angiogenesis contributes to the pathophysiology of PAH, the potential effects of PAH drugs on angiogenic capacities remain poorly explored. In the present study, we observed limited, but beneficial effects of PAH vasodilator drugs, for example, macitentan and tadalafil, on bmpr2‐induced impaired angiogenesis. Macitentan did not display any effect on migration properties, but increased tube formation capacities of bmpr2‐silenced HLMVECs, in line with beneficial effects of macitentan in vitro on tube formation of microvascular pulmonary ECs from PAH patients and in vivo on microvessels density in the lungs of Sugen/hypoxia PAH rats. 49 However, ET‐1 is rather a proangiogenic factor for ECs issued from other vascular beds, 50 , 51 , 52 compatible with the differential vasoactive effects of ET‐1 depending on the vascular bed. Selexipag did not show any effect on angiogenic capacities of bmpr2‐silenced HLMVECs. Nevertheless, altered prostanoid metabolism results in impaired angiogenesis 53 and iloprost showed proangiogenic capacities in a corneal model of angiogenesis, 54 which can account for discrepancies between our present findings and those from previous studies. Tadalafil improved migration capacity, but did not display any effect on tube formation in bmpr2‐silenced HLMVECs. In comparison with iloprost (PGI2 analog) or bosentan, sildenafil is more potent in enhancing tube forming capacity, 55 in line with its effects on vascular density during chronic ischemia of the mouse hind limb 56 or capillary collateral formation in a mouse hind limb ischemia model and in vitro capillary‐like tube formation in response to vardenafil. 57 By contrast, tadalafil reduced migration velocity in control HLMVECs. Of note, PDE5 inhibitors can inhibit cancer progression and it was suggested to repurpose PDE5 inhibitors tadalafil and sildenafil as anticancer agents in hepatic cancer. 58 These contrasting findings could be attributed to different endothelial cell inner properties from different vascular beds, 59 but also to the various experimental models involved. For instance, in vitro angiogenesis assays imply the use of synthetic animal‐derived extracellular matrix, for example, matrigel, which does not fully reproduce human ECM and its disease‐driven alterations; in in vivo experimental animal models of pulmonary hypertension, microvascular rarefaction is observed as a result of altered angiogenesis, vascular obstruction, and pruning.
Limitations
Although we are aware that stable bmpr2 silencing in HLMVECs does not exactly reproduce bmpr2 mutations, it nicely overcomes the limited access to patient HLMVECs. 22 We, so far, explored the effects of each drug type separately and should consider combining them, in line with the current advent of combination therapy. 60 Although the endothelial phenotype can be maintained throughout subcultures, we cannot exclude any loss in the expression of surface markers and receptors, which may have biased our findings. Albeit more relevant to the physiopathological situation, using Cmax approaching drug concentrations may have mitigated in vitro effects.
CONCLUSION
In the present study, we highlighted the key role of BMPRII pathway in regulating angiogenesis of HLMVECs in the context of PAH pathogenesis. PAH drugs can partially restore bmpr2 silencing‐mediated impairment of migration and tube formation capacities, but not endothelial barrier function. Consequently, in addition to already existing PAH drugs, which did not display any detrimental effects on BMPRII‐mediated pulmonary endothelial function, innovative therapies deserve to be explored to reverse pulmonary vascular remodeling and offer a perspective to cure PAH.
AUTHOR CONTRIBUTIONS
Marion Delcroix and Rozenn Quarck conceived and designed the study. Birger Tielemans and Allard Wagenaar collected the data. Birger Tielemans and Rozenn Quarck performed the acquisition, analysis and interpretation of the data. Birger Tielemans and Rozenn Quarck drafted the manuscript. Catharina Belge and Marion Delcroix reviewed the manuscript. All authors approved the final version.
CONFLICT OF INTEREST STATEMENT
Marion Delcroix is holder of the Actelion chair for Pulmonary Hypertension at the KU Leuven ‐ University of Leuven.
ETHICS STATEMENT
None.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
The authors would like to thank Prof Rik Gijsbers (Department Pharmaceutical & Pharmacological Sciences, University of Leuven) for having designed hairpin lentiviral vectors for bmpr2 silencing, Actelion Benelux for having kindly provided macitentan and selexipag and the Belgian Association of Patients for Pulmonary Hypertension (Belgische Pulmonale Hypertensie Patiëntenvereniging) for its financial support. This work was funded by Internal Research Funds from the University of Leuven‐KU Leuven (C2 project_C24/17/079).
Tielemans B, Wagenaar A, Belge C, Delcroix M, Quarck R. Pulmonary arterial hypertension drugs can partially restore altered angiogenic capacities in bmpr2‐silenced human lung microvascular endothelial cells. Pulm Circ. 2023;13:e12293. 10.1002/pul2.12293
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS, Escribano‐Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke‐Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Vonk Noordegraaf A, Delcroix M, Rosenkranz S. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023;61:2200879. [DOI] [PubMed] [Google Scholar]
- 2. Galambos C, Sims‐Lucas S, Abman SH, Cool CD. Intrapulmonary bronchopulmonary anastomoses and plexiform lesions in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2016;193:574–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. St. Croix CM, Steinhorn RH. New thoughts about the origin of plexiform lesions. Am J Respir Crit Care Med. 2016;193:484–485. [DOI] [PubMed] [Google Scholar]
- 4. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF‐β receptor, cause familial primary pulmonary hypertension. Nature Genet. 2000;26:81–84. [DOI] [PubMed] [Google Scholar]
- 6. Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor‐II gene. Am J Hum Genet. 2000;67:737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thomson JR. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR‐II, a receptor member of the TGF‐beta family. J Med Genet. 2000;37:741–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tielemans B, Delcroix M, Belge C, Quarck R. TGFβ and BMPRII signalling pathways in the pathogenesis of pulmonary arterial hypertension. Drug Discov Today. 2019;24:703–716. [DOI] [PubMed] [Google Scholar]
- 9. Orriols M, Gomez‐Puerto MC, ten Dijke P. BMP type II receptor as a therapeutic target in pulmonary arterial hypertension. Cell Mol Life Sci. 2017;74:2979–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Machado RD, Southgate L, Eichstaedt CA, Aldred MA, Austin ED, Best DH, Chung WK, Benjamin N, Elliott CG, Eyries M, Fischer C, Gräf S, Hinderhofer K, Humbert M, Keiles SB, Loyd JE, Morrell NW, Newman JH, Soubrier F, Trembath RC, Viales RR, Grünig E. Pulmonary arterial hypertension: a current perspective on established and emerging molecular genetic defects. Hum Mutat. 2015;36:1113–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harrison RE. Molecular and functional analysis identifies ALK‐1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet. 2003;40:865–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nasim MT, Ogo T, Ahmed M, Randall R, Chowdhury HM, Snape KM, Bradshaw TY, Southgate L, Lee GJ, Jackson I, Lord GM, Gibbs JSR, Wilkins MR, Ohta‐Ogo K, Nakamura K, Girerd B, Coulet F, Soubrier F, Humbert M, Morrell NW, Trembath RC, Machado RD. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat. 2011;32:1385–1389. [DOI] [PubMed] [Google Scholar]
- 13. Shintani M, Yagi H, Nakayama T, Saji T, Matsuoka R. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J Med Genet. 2009;46:331–337. [DOI] [PubMed] [Google Scholar]
- 14. Guignabert C, Bailly S, Humbert M. Restoring BMPRII functions in pulmonary arterial hypertension: opportunities, challenges and limitations. Expert Opin Ther Targets. 2017;21:181–190. [DOI] [PubMed] [Google Scholar]
- 15. Seferian A, Simonneau G. Therapies for pulmonary arterial hypertension: where are we today, where do we go tomorrow? Eur Respir Rev. 2013;22:217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hirose S, Hosoda Y, Furuya S, Otsuki T, Ikeda E. Expression of vascular endothelial growth factor and its receptors correlates closely with formation of the plexiform lesion in human pulmonary hypertension. Pathol Int. 2000;50:472–479. [DOI] [PubMed] [Google Scholar]
- 17. Papaioannou AI, Zakynthinos E, Kostikas K, Kiropoulos T, Koutsokera A, Ziogas A, Koutroumpas A, Sakkas L, Gourgoulianis KI, Daniil ZD. Serum VEGF levels are related to the presence of pulmonary arterial hypertension in systemic sclerosis. BMC Pulm Med. 2009;9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frost AE, Barst RJ, Hoeper MM, Chang HJ, Frantz RP, Fukumoto Y, Galié N, Hassoun PM, Klose H, Matsubara H, Morrell NW, Peacock AJ, Pfeifer M, Simonneau G, Tapson VF, Torres F, Dario Vizza C, Lawrence D, Yang W, Felser JM, Quinn DA, Ghofrani HA. Long‐term safety and efficacy of imatinib in pulmonary arterial hypertension. J Heart Lung Transplant. 2015;34:1366–1375. [DOI] [PubMed] [Google Scholar]
- 19. Montani D, Bergot E, Günther S, Savale L, Bergeron A, Bourdin A, Bouvaist H, Canuet M, Pison C, Macro M, Poubeau P, Girerd B, Natali D, Guignabert C, Perros F, O'Callaghan DS, Jaïs X, Tubert‐Bitter P, Zalcman G, Sitbon O, Simonneau G, Humbert M. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125:2128–2137. [DOI] [PubMed] [Google Scholar]
- 20. Taraseviciene‐Stewart L, Kasahara Y, Alger L, Hirth P, Mahon GM, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death‐dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–438. [DOI] [PubMed] [Google Scholar]
- 21. Gu M, Shao N‐Y, Sa S, Li D, Termglinchan V, Ameen M, Karakikes I, Sosa G, Grubert F, Lee J, Cao A, Taylor S, Ma Y, Zhao Z, Chappell J, Hamid R, Austin ED, Gold JD, Wu JC, Snyder MP, Rabinovitch M. Patient‐specific iPSC‐derived endothelial cells uncover pathways that protect against pulmonary hypertension in BMPR2 mutation carriers. Cell Stem Cell. 2017;20:490–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tielemans B, Stoian L, Gijsbers R, Michiels A, Wagenaar A, Farre Marti R, Belge C, Delcroix M, Quarck R. Cytokines trigger disruption of endothelium barrier function and p38 MAP kinase activation in BMPR2‐silenced human lung microvascular endothelial cells. Pulm Circ. 2019;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chaudhary KR, Stewart DJ. Go with the (back) flow: what can retrograde perfusion teach us about arterial remodeling in pulmonary arterial hypertension? Am J Physiol Lung Cell Mol Physiol. 2018;314:L797–L798. [DOI] [PubMed] [Google Scholar]
- 24. Burton VJ, Ciuclan LI, Holmes AM, Rodman DM, Walker C, Budd DC. Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function. Blood. 2011;117:333–341. [DOI] [PubMed] [Google Scholar]
- 25. Carpentier G, Berndt S, Ferratge S, Rasband W, Cuendet M, Uzan G, Albanese P. Angiogenesis analyzer for ImageJ—A comparative morphometric analysis of “endothelial tube formation assay” and “fibrin bead assay”. Sci Rep. 2020;10:11568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hautefort A, Mendes‐Ferreira P, Sabourin J, Manaud G, Bertero T, Rucker‐Martin C, Riou M, Adão R, Manoury B, Lambert M, Boet A, Lecerf F, Domergue V, Brás‐Silva C, Gomez AM, Montani D, Girerd B, Humbert M, Antigny F, Perros F. Bmpr2 mutant rats develop pulmonary and cardiac characteristics of pulmonary arterial hypertension. Circulation. 2019;139:932–948. [DOI] [PubMed] [Google Scholar]
- 27. Huang Z, Wang D, Ihida‐Stansbury K, Jones PL, Martin JF. Defective pulmonary vascular remodeling in Smad8 mutant mice. Hum Mol Gen. 2009;18:2791–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Jesus Perez VA, Alastalo TP, Wu JC, Axelrod JD, Cooke JP, Amieva M, Rabinovitch M. Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt–β‐catenin and Wnt–RhoA–Rac1 pathways. J Cell Biol. 2009;184:83–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Suzuki Y, Montagne K, Nishihara A, Watabe T, Miyazono K. BMPs promote proliferation and migration of endothelial cells via stimulation of VEGF‐A/VEGFR2 and angiopoietin‐1/Tie2 signalling. J Biochem. 2008;143:199–206. [DOI] [PubMed] [Google Scholar]
- 30. Benn A, Hiepen C, Osterland M, Schütte C, Zwijsen A, Knaus P. Role of bone morphogenetic proteins in sprouting angiogenesis: differential BMP receptor‐dependent signaling pathways balance stalk vs. tip cell competence. FASEB J. 2017;31:4720–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Valdimarsdottir G, Goumans M, Rosendahl A, Brugman M, Itoh S, Lebrin F, Sideras P, ten Dijke P. Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein‐induced activation of endothelial cells. Circulation. 2002;106:2263–2270. [DOI] [PubMed] [Google Scholar]
- 32. Long L, Ormiston ML, Yang X, Southwood M, Gräf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, Moore SD, Drake KM, Aldred MA, Yu PB, Upton PD, Morrell NW. Selective enhancement of endothelial BMPR‐II with BMP9 reverses pulmonary arterial hypertension. Nature Med. 2015;21:777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang S, Cai R, Ma J, Liu T, Ke X, Lu H, Fu J. The natural compound codonolactone impairs tumor induced angiogenesis by downregulating BMP signaling in endothelial cells. Phytomedicine. 2015;22:1017–1026. [DOI] [PubMed] [Google Scholar]
- 34. Miao H, Qiu F, Zhu L, Jiang B, Yuan Y, Huang B, Zhang Y. Novel angiogenesis strategy to ameliorate pulmonary hypertension. J Thorac Cardiovasc Surg. 2021;161:e417–e434. [DOI] [PubMed] [Google Scholar]
- 35. Maruyama H, Dewachter C, Sakai S, Belhaj A, Rondelet B, Remmelink M, Vachiéry JL, Naeije R, Dewachter L. Bosentan reverses the hypoxia‐induced downregulation of the bone morphogenetic protein signaling in pulmonary artery smooth muscle cells. Life Sci. 2016;159:111–115. [DOI] [PubMed] [Google Scholar]
- 36. Cipriani P, Di Benedetto P, Ruscitti P, Verzella D, Fischietti M, Zazzeroni F, Liakouli V, Carubbi F, Berardicurti O, Alesse E, Giacomelli R. Macitentan inhibits the transforming growth factor‐β profibrotic action, blocking the signaling mediated by the ETR/TβRI complex in systemic sclerosis dermal fibroblasts. Arthritis Res Ther. 2015;17:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ogo T, Chowdhury HM, Yang J, Long L, Li X, Torres Cleuren YN, Morrell NW, Schermuly RT, Trembath RC, Nasim MT. Inhibition of overactive transforming growth factor–β signaling by prostacyclin analogs in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2013;48:733–741. [DOI] [PubMed] [Google Scholar]
- 38. Yang J, Li X, Al‐lamki RS, Wu C, Weiss A, Berk J, Schermuly RT, Morrell NW. Sildenafil potentiates bone morphogenetic protein signaling in pulmonary arterial smooth muscle cells and in experimental pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2013;33:34–42. [DOI] [PubMed] [Google Scholar]
- 39. Yang J, Li X, Al‐Lamki RS, Southwood M, Zhao J, Lever AM, Grimminger F, Schermuly RT, Morrell NW. Smad‐dependent and smad‐independent induction of id1 by prostacyclin analogues inhibits proliferation of pulmonary artery smooth muscle cells in vitro and in vivo. Circ Res. 2010;107:252–262. [DOI] [PubMed] [Google Scholar]
- 40. Maruyama H, Dewachter C, Belhaj A, Rondelet B, Sakai S, Remmelink M, Vachiery JL, Naeije R, Dewachter L. Endothelin‐Bone morphogenetic protein type 2 receptor interaction induces pulmonary artery smooth muscle cell hyperplasia in pulmonary arterial hypertension. J Heart Lung Transplant. 2015;34:468–478. [DOI] [PubMed] [Google Scholar]
- 41. Yang X, Long L, Reynolds PN, Morrell NW. Expression of mutant BMPR‐II in pulmonary endothelial cells promotes apoptosis and a release of factors that stimulate proliferation of pulmonary arterial smooth muscle cells. Pulm Circ. 2011;1:103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kuang T, Wang J, Zeifman A, Pang B, Huang X, Burg ED, Yuan JXJ, Wang C. Combination use of sildenafil and simvastatin increases BMPR‐II signal transduction in rats with monocrotaline‐mediated pulmonary. Pulm Circ. 2011;1:111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hiepen C, Jatzlau J, Hildebrandt S, Kampfrath B, Goktas M, Murgai A, Cuellar Camacho JL, Haag R, Ruppert C, Sengle G, Cavalcanti‐Adam EA, Blank KG, Knaus P. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLoS Biol. 2019;17:e3000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vengethasamy L, Hautefort A, Tielemans B, Belge C, Perros F, Verleden S, Fadel E, Van Raemdonck D, Delcroix M, Quarck R. BMPRII influences the response of pulmonary microvascular endothelial cells to inflammatory mediators. Pflug Arch Eur J. 2016;468:1969–1983. [DOI] [PubMed] [Google Scholar]
- 45. Hall SM, Davie N, Klein N, Haworth SG. Endothelin receptor expression in idiopathic pulmonary arterial hypertension: effect of bosentan and epoprostenol treatment. Eur Respir J. 2011;38:851–860. [DOI] [PubMed] [Google Scholar]
- 46. Meliton A, Meng F, Tian Y, Shah AA, Birukova AA, Birukov KG. Role of krev interaction trapped‐1 in prostacyclin‐induced protection against lung vascular permeability induced by excessive mechanical forces and thrombin receptor activating peptide 6. Am J Respir Cell Mol Biol. 2015;53:834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Birukova AA, Meng F, Tian Y, Meliton A, Sarich N, Quilliam LA, Birukov KG. Prostacyclin post‐treatment improves LPS‐induced acute lung injury and endothelial barrier recovery via Rap1. Biochim Biophys Acta Mol Basis Dis. 2015;1852:778–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu S, Yu C, Yang F, Paganini‐Hill A, Fisher MJ. Phosphodiesterase inhibitor modulation of brain microvascular endothelial cell barrier properties. J Neurol Sci. 2012;320:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nadeau V, Potus F, Boucherat O, Paradis R, Tremblay E, Iglarz M, Paulin R, Bonnet S, Provencher S. Dual ETA/ETB blockade with macitentan improves both vascular remodeling and angiogenesis in pulmonary arterial hypertension. Pulm Circ. 2018;8:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Salani D, Taraboletti G, Rosanò L, Di Castro V, Borsotti P, Giavazzi R, Bagnato A. Endothelin‐1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol. 2000;157:1703–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wu M‐H, Huang C‐Y, Lin J‐A, Wang SW, Peng CY, Cheng HC, Tang CH. Endothelin‐1 promotes vascular endothelial growth factor‐dependent angiogenesis in human chondrosarcoma cells. Oncogene. 2014;33:1725–1735. [DOI] [PubMed] [Google Scholar]
- 52. Wülfing P, Kersting C, Tio J, Fischer RJ, Wülfing C, Poremba C, Diallo R, Böcker W, Kiesel L. Endothelin‐1‐, endothelin‐A‐, and endothelin‐B‐receptor expression is correlated with vascular endothelial growth factor expression and angiogenesis in breast cancer. Clin Cancer Res. 2004;10:2393–2400. [DOI] [PubMed] [Google Scholar]
- 53. Mahajan CN, Afolayan AJ, Eis A, Teng RJ, Konduri GG. Altered prostanoid metabolism contributes to impaired angiogenesis in persistent pulmonary hypertension in a fetal lamb model. Pediatr Res. 2015;77:455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pola R. Comparative analysis of the in vivo angiogenic properties of stable prostacyclin analogs: a possible role for peroxisome proliferator‐activated receptors. J Mol Cell Cardiol. 2004;36:363–370. [DOI] [PubMed] [Google Scholar]
- 55. Doganci S, Yildirim V, Yesildal F, Erol G, Kadan M, Ozkan G, Avcu F, Ozgurtas T. Comparison of angiogenic and proliferative effects of three commonly used agents for pulmonary artery hypertension (sildenafil, iloprost, bosentan): is angiogenesis always beneficial? Eur Rev Med Pharmacol. 2015;19:1900–1906. [PubMed] [Google Scholar]
- 56. Senthilkumar A, Smith RD, Khitha J, Arora N, Veerareddy S, Langston W, Chidlow JH, Barlow SC, Teng X, Patel RP, Lefer DJ, Kevil CG. Sildenafil promotes ischemia‐induced angiogenesis through a PKG‐dependent pathway. Arterioscler Thromb Vasc Biol. 2007;27:1947–1954. [DOI] [PubMed] [Google Scholar]
- 57. Sahara M, Sata M, Morita T, Nakajima T, Hirata Y, Nagai R. A phosphodiesterase‐5 inhibitor vardenafil enhances angiogenesis through a protein kinase G–dependent hypoxia‐inducible factor‐1/vascular endothelial growth factor pathway. Arterioscler Thromb Vasc Biol. 2010;30:1315–1324. [DOI] [PubMed] [Google Scholar]
- 58. Chhonker SK, Rawat D, Koiri RK. Repurposing PDE5 inhibitor tadalafil and sildenafil as anticancer agent against hepatocellular carcinoma via targeting key events of glucose metabolism and multidrug resistance. J Biochem Mol Toxicol. 2022;36:e23100. [DOI] [PubMed] [Google Scholar]
- 59. Parab S, Setten E, Astanina E, Bussolino F, Doronzo G. The tissue‐specific transcriptional landscape underlines the involvement of endothelial cells in health and disease. Pharmacol Ther. 2023;246:108418. [DOI] [PubMed] [Google Scholar]
- 60. Galiè N, Barberà JA, Frost AE, Ghofrani HA, Hoeper MM, McLaughlin VV, Peacock AJ, Simonneau G, Vachiery JL, Grünig E, Oudiz RJ, Vonk‐Noordegraaf A, White RJ, Blair C, Gillies H, Miller KL, Harris JHN, Langley J, Rubin LJ. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373:834–844. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.