

Abstract
In this review, we outline recent advancements in small molecule drug design from a structural perspective. We compare protein structure prediction methods and explore the role of the ligand binding pocket in structure-based drug design. We examine various structural features used to optimize drug candidates, including functional groups, stereochemistry, and molecular weight. Computational tools such as molecular docking and virtual screening are discussed for predicting and optimizing drug candidate structures. We present examples of drug candidates designed based on their molecular structure and discuss future directions in the field. By effectively integrating structural information with other valuable data sources, we can improve the drug discovery process, leading to the identification of novel therapeutics with improved efficacy, specificity, and safety profiles.
Keywords: small molecule drugs, drug design, three-dimensional protein structures, molecular docking, virtual screening
Introduction
The field of small molecule drug design is a dynamic and rapidly evolving discipline, with advancements in computational tools and methodologies significantly enhancing our ability to design and optimize small molecule drug candidates. This review provides a unique and comprehensive perspective on the current state-of-the-art small molecule drug design, with a particular focus on the role of molecular structure in determining the pharmacological properties of small molecule drugs. While the value of small molecule development is well known, our review highlights the novel approaches and insights that have emerged in recent years.
We delve into the importance of accurate three-dimensional (3D) protein structure prediction in small molecule drug discovery, comparing two distinct prediction methods: homology modeling and de novo modeling. We also highlight the critical role of the ligand binding pocket in the structure-based design of small molecule drugs. Furthermore, we explore the various structural features and properties that are commonly employed in the design and optimization of small molecule drug candidates, such as functional groups, stereochemistry, and molecular weight. We also discuss the computational tools and methods used to predict and optimize these structural properties, including molecular docking and structure-based virtual screening. In addition, we present recent examples of small molecule drug candidates that were designed based on their molecular structure, discussing their chemical structure, mechanism of action, and pharmacological properties. We also touch upon emerging trends and future directions in small molecule drug design, such as the advent of new computational tools, the identification of emerging targets, and the exploration of novel therapeutic applications.
This review is distinct in its approach to small molecule drug design, emphasizing the integration of structural information with other tools and data sources to achieve more efficient and effective small molecule drug discovery and development. In essence, this review offers a fresh perspective on small molecule drug design, underscoring the importance of molecular structure in drug development and highlighting the latest advancements and future directions in this exciting field.
Prediction of 3D protein structures in small molecule drug discovery
The determination of protein structure can be achieved through various methods such as X-ray diffraction of protein crystals, cryo-electron microscopy, nuclear magnetic resonance (NMR), and prediction techniques. The accurate prediction of 3D protein structures has become a pivotal aspect in the realm of drug design, garnering significant attention in recent years.1 Understanding the 3D structure of a protein is of paramount importance in elucidating the mechanisms underlying drug binding and facilitating the design of compounds with enhanced specificity and potency. Notably, the field of drug discovery has undergone a revolution due to remarkable advancements in computational techniques, facilitating the prediction of protein structures with exceptional precision.
One of the most widely used computational methods for predicting protein structures is homology modeling.2 Homology modeling, also known as comparative modeling, utilizes the known structures of related proteins to predict the structure of the target protein.3 This method assumes that evolutionarily related proteins have similar structures and that the structure of a protein is more conserved than its sequence.4 The process of homology modeling involves several steps including template selection, sequence alignment, model building, and model refinement.4 Each of these steps is essential for generating an accurate 3D model of a protein from its amino acid sequence (Figure 1).
FIGURE 1. Schematic diagram of the computational methods for predicting 3D protein structures in small molecule drug discovery.
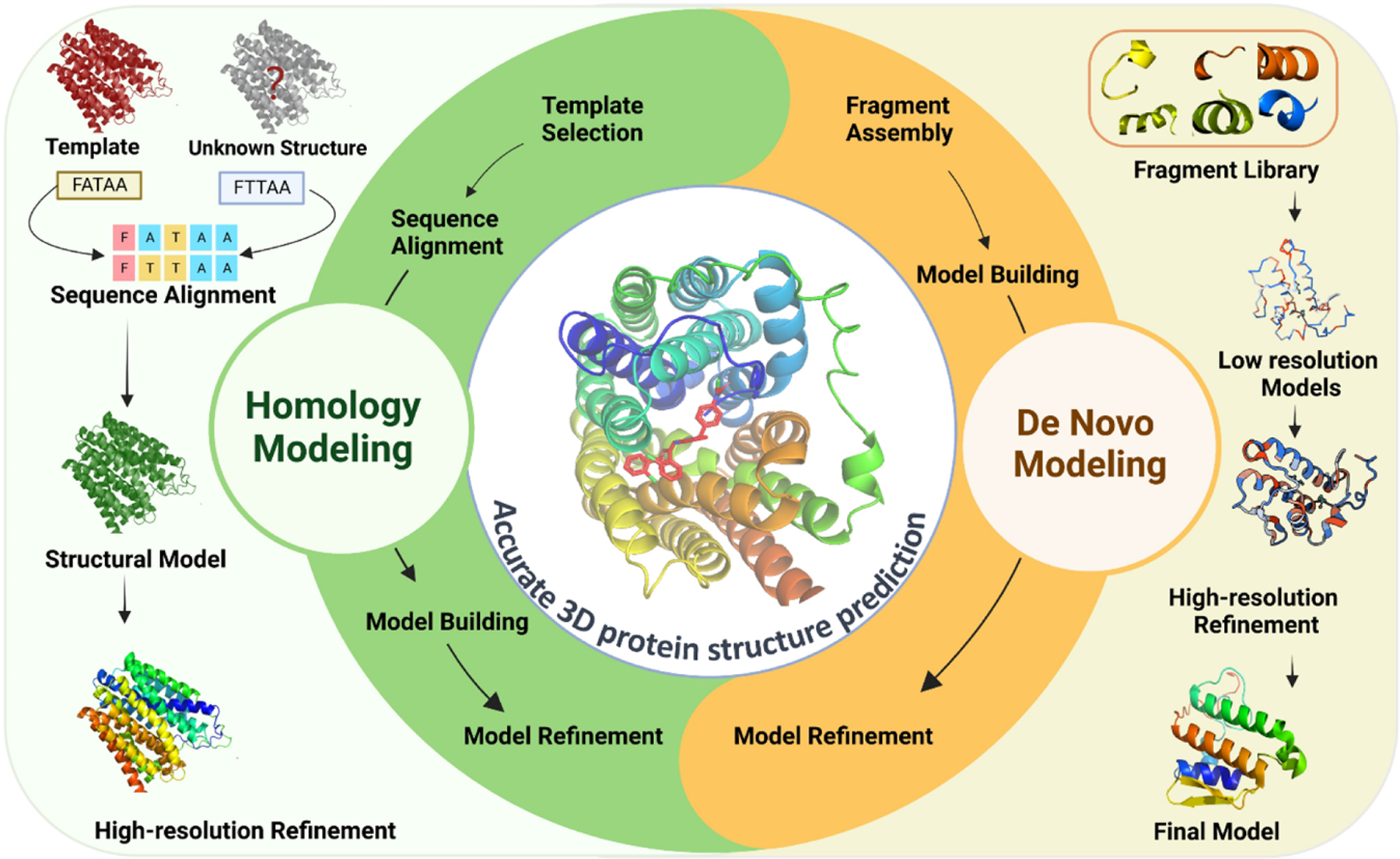
Homology modeling utilizes known structures of related proteins to predict the structure of the target protein. The process involves template selection, sequence alignment, model building, and model refinement. De novo protein structure prediction, on the other hand, involves predicting the structure of a protein from scratch without relying on known structures. The process involves fragment assembly, model building, and model refinement. Both methods are critical for understanding the mechanism of drug binding and designing drugs with improved specificity and potency.
Template selection: The first step is to select an appropriate template structure from the protein structure database. The template structure should have a high degree of homology with the target protein sequence, and should have a similar function or fold. Several methods, such as FASTA and BLAST, are used to identify homologous sequences in the protein structure database.5
Sequence alignment: The second step is to align the target protein sequence with the selected template structure. The alignment should be optimized to ensure that the conserved regions between the target and template structures are aligned correctly. Several algorithms, such as ClustalW and MUSCLE, are used to perform sequence alignment.6
Model building: In this step, a 3D model of the target protein is generated based on the aligned target and template sequences. The model is built by superimposing the target sequence onto the template structure and adjusting the side chains to fit the target sequence. Several software programs, such as MODELLER and SWISS-MODEL, are used for model building.7
Model refinement: Once the initial model is generated, it is refined to improve its accuracy. This step involves optimizing the side-chain orientations, minimizing the energy of the structure, and performing structural validation to ensure that the model is biologically plausible. Several software programs, such as CHARMM and GROMACS, are used for model refinement.8,9
Another method is de novo protein structure prediction (Figure 1), which involves predicting the structure of a protein from scratch without relying on known structures.10 This method is based on physical principles, such as energy minimization and free energy calculations, to predict the structure of a protein.11,12 De novo protein structure prediction is the algorithmic process of predicting the tertiary structure of a protein from its primary sequence of amino acids, without the use of templates from previously solved structures.13 The process involves several steps including fragment assembly, model building, and model refinement.14 This process can be guided by knowledge-based energy functions or machine learning (ML) algorithms.14,15
The first step in de novo protein structure prediction is fragment assembly. This involves breaking down the protein sequence into smaller fragments and predicting the relative orientations of these fragments in 3D space. Fragments can be assembled using either physics-based methods or statistical methods. Physics-based methods calculate the energy of the system to determine the optimal fragment orientation, whereas statistical methods use the likelihood of a particular fragment assembly based on known structures.16,17 Once fragments are assembled, the next step is model building, where a complete 3D model is constructed by joining the fragments together in a way that satisfies spatial constraints such as steric clashes and bond angles. The model building process can be guided by a knowledge-based energy function, which provides a score for the quality of the model, or by ML algorithms that learn from previous successful predictions.14,18 The final step in de novo protein structure prediction is model refinement, which involves optimizing the 3D model to improve its accuracy and overall quality. This can be achieved through molecular dynamics (MD) simulations, energy minimization algorithms, or ML methods.19,20
Homology modeling is generally more accurate than de novo protein structure prediction, as it relies on the availability of related protein structures.2 Homology modeling is considered to be the most accurate method for protein structure prediction, and is often used in drug design for screening of large libraries.2 Homology models contain sufficient information about the spatial arrangement of important residues in the protein, and accurate predictions can be obtained if the template and query sequences have high sequence similarity.21 However, homology modeling has limitations, especially for proteins with only remote homologs or for proteins with no known structural homologs.22 Accurate template-query alignment and template selection are still very challenging for these proteins, and it can be difficult to obtain accurate models. In such cases, de novo modeling can be useful as it does not rely on the availability of related protein structures.20 De novo modeling can also be used in cases where the available templates do not provide a good match for the query sequence or where the available templates have low sequence identity to the query.20 De novo modeling involves predicting the protein structure from scratch, and it can be challenging due to the complexity of the protein folding problem. However, recent advances in deep learning-based prediction have shown that more accurate models can be generated by extending deep learning-based prediction to inter-residue orientations in addition to distances, and the development of a constrained optimization by Rosetta.12
In conclusion, accurate protein structure prediction is crucial for designing small molecule drug candidates that bind specifically to the target protein, without causing off-target effects.23 The use of computational methods such as homology modeling and de novo structure prediction has enabled the prediction of protein structures with high accuracy, which has revolutionized the field of drug discovery. The ability to predict protein structures accurately has also led to the development of new drugs targeting proteins that were previously considered ‘undruggable’.24
The crucial role of ligand binding pocket in the structure-based design of small molecule drugs
The ligand binding pocket plays a crucial role in the structure-based design of small molecule drugs. This design approach starts with the assumption that a drug molecule exerts its biological activity through specific binding to a macromolecular target receptor, typically a protein. The binding pocket refers to a cavity or depression on the surface of the target protein where the ligand binds and interacts with the protein. The structural and chemical complementarity between the ligand and the receptor within the binding pocket is a prerequisite for strong and selective binding. By fitting precisely into the binding pocket, the ligand modulates the function of the protein, potentially leading to therapeutic effects. Therefore, understanding and characterizing the binding pocket is crucial for designing small molecule drugs.
One of the primary goals in structure-based drug design is to identify ligands that can bind with high affinity and specificity to the target protein’s binding pocket. This process involves several steps including target selection, molecular docking, and virtual screening. Molecular docking simulations use computational methods to predict the binding modes and binding affinities of small molecules within the binding pocket of the target protein. These simulations help identify potential drug candidates and optimize their interactions with the binding pocket. Advancements in structural biology techniques, such as X-ray crystallography and cryo-electron microscopy, have facilitated the determination of protein–ligand complex structures, providing valuable insights into the binding interactions.25 These structural insights allow medicinal chemists to design and optimize small molecules that can more effectively fit and interact with the binding pocket. Additionally, the druggability of the binding pocket is a crucial consideration in small molecule drug design. The druggability refers to the likelihood of successfully developing a drug that can bind to and modulate the target protein’s function. Factors such as the size, shape, and physicochemical properties of the binding pocket influence its druggability.25 Computational methods, including MD simulations and virtual screening, can aid in assessing the druggability of the binding pocket and guiding the design of small molecules with optimal properties for binding.
There are several key points highlighting the importance of the ligand binding pocket in structure-based drug design.
Specificity and affinity: The ligand binding pocket provides a 3D environment that accommodates the ligand with high specificity and affinity. The shape, electrostatic properties, and chemical composition of the pocket determine the ligand’s interactions and binding strength with the protein.
Rational ligand design: Knowledge of the ligand binding pocket’s structure allows for rational ligand design. By studying the pocket’s characteristics, such as its size, shape, and residues lining the pocket, researchers can design small molecules that fit optimally within the pocket and interact favorably with the surrounding protein residues.
Structure-based optimization: The ligand binding pocket serves as a target for structure-based optimization. By analyzing the interactions between the ligand and the pocket, medicinal chemists can modify the ligand’s chemical structure to improve its binding affinity, selectivity, and pharmacological properties. This optimization process can involve modifications such as introducing functional groups, adjusting molecular properties, or exploring different scaffolds.
Drug selectivity and off-target effects: Understanding the ligand binding pocket’s characteristics is crucial for achieving drug selectivity. By designing ligands that fit precisely into the binding pocket of the target protein, researchers can increase the specificity of the drug and reduce off-target effects. This specificity is essential for minimizing potential side effects and improving therapeutic outcomes.
In summary, the ligand binding pocket is of paramount importance in the structure-based design of small molecule drugs. Understanding the structural and chemical characteristics of the binding pocket, along with employing computational techniques and structural biology methods, enable the identification and optimization of ligands that can bind selectively and with high affinity, ultimately facilitating the development of effective therapeutic interventions.
Structural analysis of small molecule drugs
Molecular structure plays a crucial role in drug development, as the structural features of a molecule determine its ability to interact with biological targets and produce desired therapeutic effects. There are two main types of drugs: small molecules and biologics. Small molecules are typically synthesized chemically and are generally less complex than biologics. They are designed to target specific molecules involved in disease processes and often have a defined structure that allows them to bind to these targets with high specificity.26,27 By contrast, biologics are typically large, complex molecules, such as proteins, that are produced by living cells and are designed to interact with specific receptors or pathways in the body.27
One important approach to drug discovery is structure-based drug design (SBDD), which uses computational tools to predict the position of small molecules within a 3D representation of the protein structure and estimate the affinity of ligands to target protein with considerable accuracy and efficiency.28,29 SBDD can also help identify potential off-target effects of a drug candidate, which is important for minimizing unwanted side effects.
The molecular structure of a drug is a critical factor in its ability to interact with biological targets and produce therapeutic effects. Small molecules with specific structural features can be designed to bind to target proteins with high specificity, whereas biologics are typically large, complex molecules that are designed to interact with specific receptors or pathways in the body. For example, G protein-coupled receptors (GPCRs) are a family of cell membrane proteins that are involved in many physiological processes and are important drug targets. Approximately 350 non-olfactory members of the human GPCR family are considered druggable, of which 165 are validated drug targets.30 Small molecules that target GPCRs can have a variety of structural features, such as the presence of a benzene ring or a carboxyl group, which allow them to interact with specific binding sites on the receptor and modulate its activity. Computational tools such as SBDD have been used to design and optimize drug candidates targeting GPCRs, and demonstrating the structural features is important for drug efficacy.31–33
Functional groups are one of the key structural features that are commonly used in small molecule drug design (Figure 2). In particular, the presence of certain functional groups such as hydroxyl (-OH), carboxyl (-COOH), and amino (-NH2) groups can enable small molecules to interact with specific enzymes or receptors and confer specific pharmacological properties in the body.34,35 For example, the presence of hydroxyl groups in molecules such as ethanol can enable them to interact with receptors in the brain, leading to the well-known effects of alcohol consumption.36 Another critical aspect of small molecule drug design is stereochemistry (Figure 2), which refers to the 3D arrangement of atoms in a molecule and how it can affect the molecule’s interaction with biological targets.37 For example, the active form of the anti-inflammatory drug ibuprofen has a specific stereochemistry that allows it to bind to and inhibit the activity of cyclooxygenase enzymes.38 By contrast, the inactive form of ibuprofen lacks this stereochemistry and does not bind to cyclooxygenases.38 Overall, functional groups and stereochemistry are both key features in the design of small molecules. Understanding how different functional groups and stereoisomers can affect a molecule’s interaction with biological targets can help researchers develop more effective and specific drugs for treating a variety of diseases.
FIGURE 2. Key structural features considered in small molecule drug design.
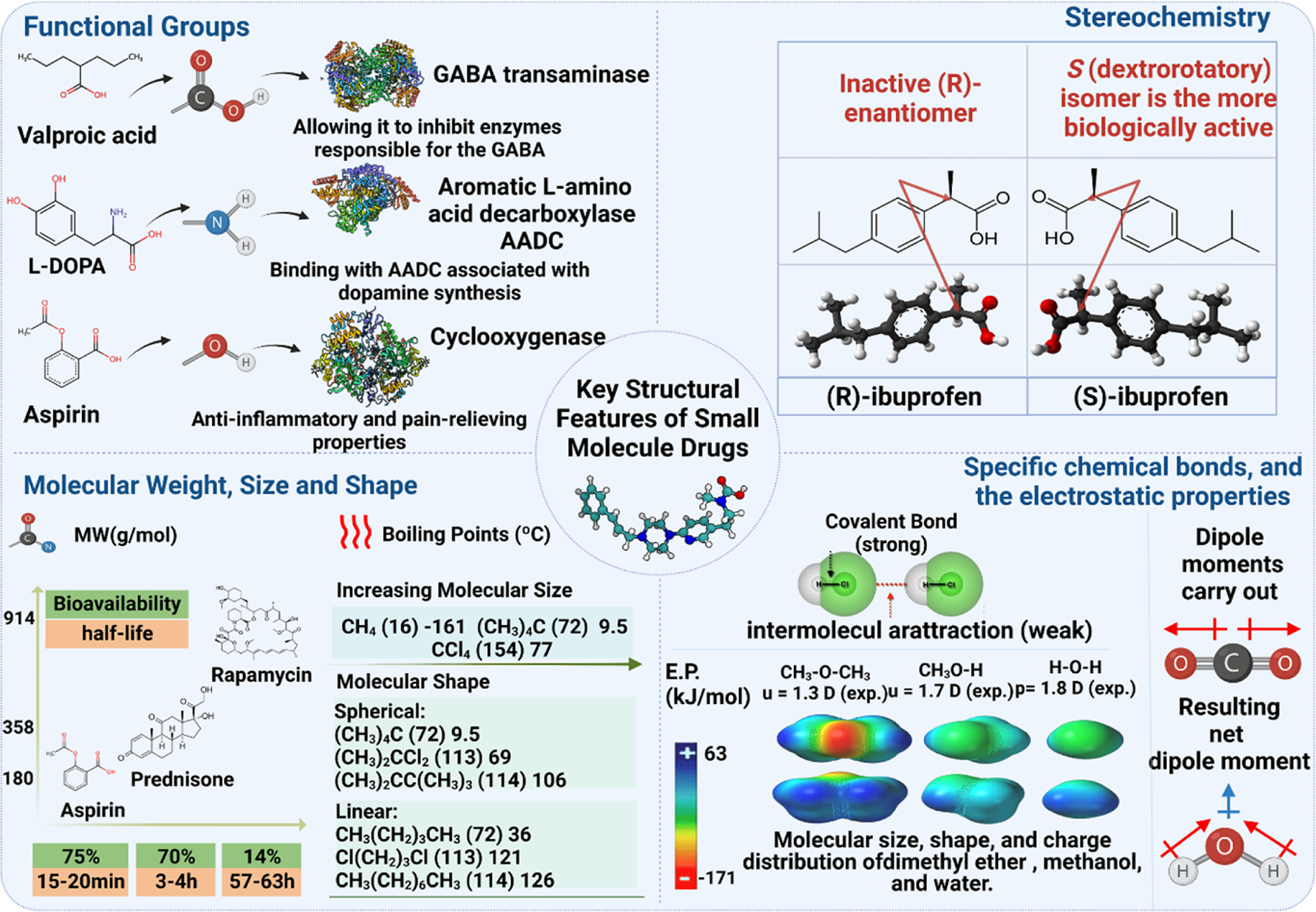
These features include functional groups (e.g., hydroxyl, carboxyl, and amino groups) that give drugs specific pharmacological properties, stereochemistry that can affect a drug’s interaction with biological targets, molecular weight, size and shape that impact pharmacokinetics and pharmacodynamics, and specific chemical bonds and electrostatic properties that influence a drug’s physical and chemical properties. Understanding these features is crucial for designing effective small molecule drugs with desired pharmacological properties.
Molecular weight is also an important consideration in small molecule drug design, as it can affect the pharmacokinetics and pharmacodynamics of a drug. For example, drugs with low molecular weight can be rapidly absorbed and excreted from the body, whereas drugs with high molecular weight may have a longer half-life and require lower dosages.39 Other structural features include the size and shape of the molecule, the presence of specific chemical bonds, and the electrostatic properties of the molecule (Figure 2). The presence of specific chemical bonds in a molecule can affect its structure and properties. For example, covalent bonds are formed when two atoms share a pair of valence shell electrons between them, and atoms of Groups IV through VII bond so as to complete an octet of valence shell electrons.40 The electrostatic properties of a molecule, such as its dipole moment and charge distribution, can affect its interactions with other molecules and its reactivity.40 The size and shape of a molecule can affect its physical and chemical properties. For example, larger molecules tend to have higher boiling points and melting points than smaller molecules.41 By understanding the importance of these structural features, researchers can design small molecule drug candidates with optimized pharmacological properties and reduced side effects.
Computational approaches to small molecule drug design
Computational methods play a crucial role in designing new drug candidates and optimizing their properties. The use of computational methods in drug design has increased rapidly in recent years, and numerous tools are available to support this process. Here, we discuss some of the most commonly used computational approaches in small molecule drug design (Figure 3).
FIGURE 3. The major computational approaches used in small molecule drug design.
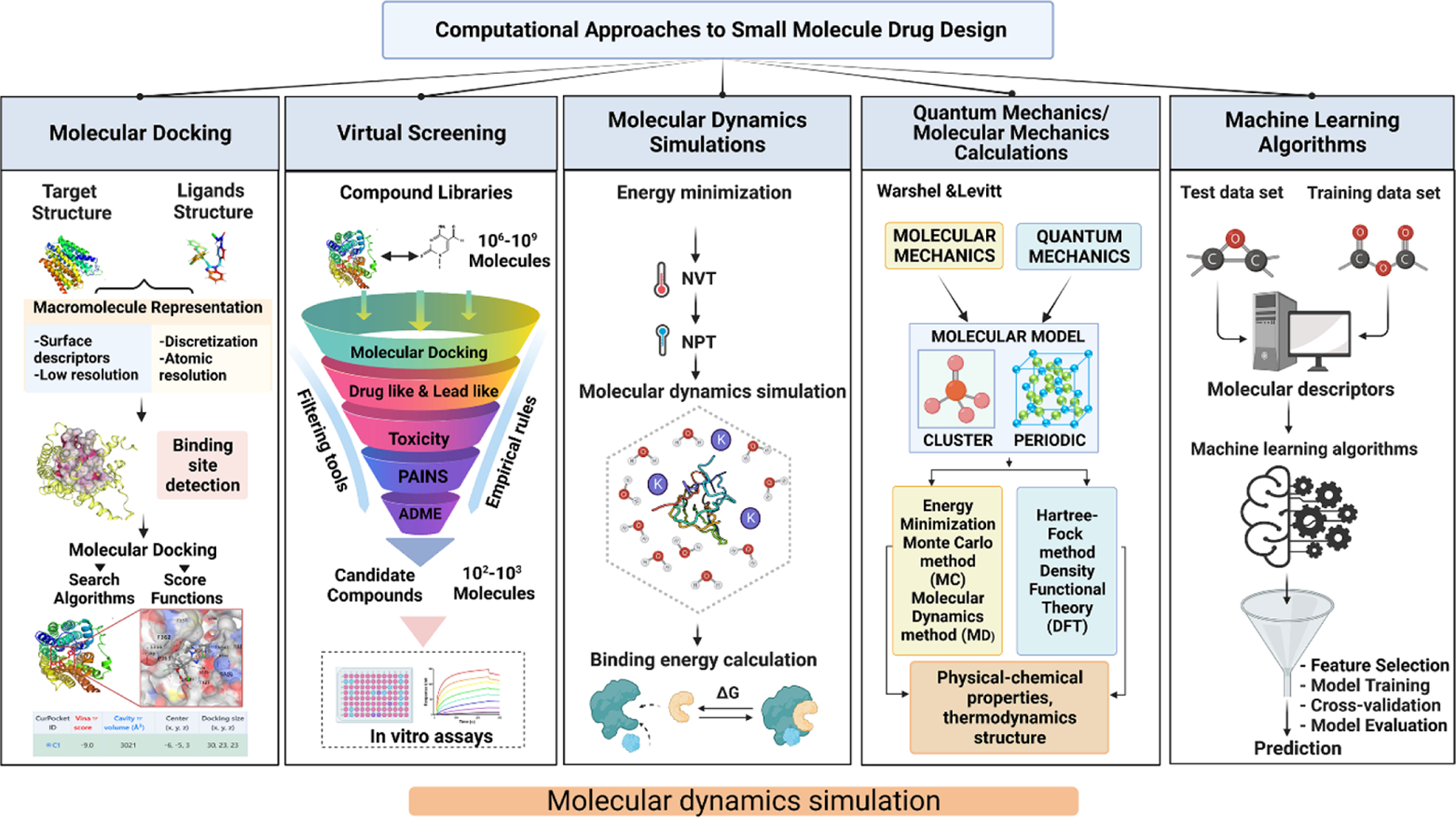
Molecular docking involves predicting the binding affinity and orientation of small molecules with larger macromolecular targets. Virtual screening is a cost-effective and time-saving method that helps researchers to narrow down the number of compounds for further experimental analysis. Molecular dynamics (MD) simulations provide detailed insights into the physical movements and interactions of atoms and molecules in a system. Quantum mechanics/molecular mechanics calculations combine the accuracy of quantum mechanical calculations with the speed of molecular mechanics simulations to study chemical and biochemical systems. Machine learning algorithms use computational approaches to predict the properties of potential small molecule drug candidates. These computational approaches are widely used in drug discovery and lead optimization to identify potential drug targets and predict molecular ligand-target interactions at the atomic level.
Molecular docking
One of the primary computational methods used in small molecule drug design is molecular docking. Molecular docking involves predicting the binding affinity and orientation of small molecules, known as ligands, with larger macromolecular targets, such as proteins or enzymes.42 Docking algorithms use energy minimization and scoring functions to evaluate the stability of the ligand–protein complex and rank potential drug candidates. The process of molecular docking helps researchers to gain insights into the molecular mechanisms of various biological processes such as drug–protein interactions, protein–protein interactions, and enzymatic reactions.43,44 In molecular docking, ligands are docked into the binding site of the protein, and the resulting complex is scored based on its fitness or complementarity.45 The docking process generates multiple possible conformations and orientations of the ligand with the binding site of the target protein.42 The goal of molecular docking is to identify the best-fit ligand–protein complex based on the calculated binding affinity and energy of the complex.42
Molecular docking has a wide range of applications in drug discovery, lead optimization, and structure-based drug design.42 It allows researchers to identify potential drug targets and predict molecular ligand–target interactions at the atomic level, providing crucial information for drug development.44 Moreover, molecular docking can also be used to study food proteins and bioactive peptides, providing insights into their structural and functional properties.43 Together, molecular docking is a powerful computational technique that is widely used in structural biology and drug discovery. It helps researchers gain insights into the molecular mechanisms of various biological processes and identify potential drug targets. The use of molecular docking is expected to continue to grow in the future as more researchers explore its potential applications in various fields.
Over the past few years, remarkable strides have been made in the field of molecular docking, resulting in substantial enhancements in the precision and effectiveness of this technique. For example, (i) Alchemical free energy methods, such as free energy perturbation and thermodynamic integration, have been developed to calculate the binding free energy of a ligand to a protein.46 These methods can provide a more accurate prediction of binding affinity than traditional docking methods. (ii) New docking tools, such as HADDOCK and RosettaDock, have been developed to incorporate protein flexibility into docking simulations.47,48 These tools can predict the conformational changes of a protein upon ligand binding, which can improve the accuracy of docking predictions. (iii) ML has been incorporated into docking tools to improve their predictive accuracy. For example, one study proposed a deep neural network model with an attention mechanism to improve the prediction accuracy of protein-ligand complex binding affinity.49 Another study investigated the use of random forest regression as an alternative to traditional linear regression methods and demonstrated improved prediction performance.50 These studies highlight the potential of ML techniques, including random forest regression, for predicting binding affinities in protein–ligand interactions. By training on known ligand–protein complexes with experimentally measured binding affinities, ML models can learn patterns and make predictions on new, unseen complexes.
Virtual screening
Another approach that has gained popularity in recent years is virtual screening, which involves screening large compound libraries for potential drug candidates using molecular docking or other techniques. It is a cost-effective and time-saving method that helps researchers narrow down the number of compounds for further experimental analysis. Virtual screening is used to identify small molecules that are likely to bind to a target protein.51 It involves the use of various software tools such as GOLD, grid-based ligand docking with energetics (GLIDE), and Autodock Vina. Autodock Vina is freely accessible and provides good results for screening different ligands.52 In the context of drug discovery, virtual screening can be used to identify chemical structures that have particular properties.53 This process helps researchers identify potential drug candidates that can selectively interact with a target protein while minimizing the side effects. Virtual screening has emerged as a groundbreaking technique that is helping to significantly improve and speed up the process of drug discovery.54 Research has shown that virtual screening is effective in scanning the potential affinity of millions of compounds to selected targets simultaneously.
The virtual screening process involves three main steps: preparation of the target protein structure, preparation of the compound library, and the screening of compounds against the target protein. During the preparation of the target protein structure, the protein is optimized by removing any water molecules or co-crystallized ligands. In the preparation of the compound library, a large database of small molecules is created from which potential lead compounds are identified. Finally, during the screening of compounds against the target protein, the compounds are ranked based on their predicted binding affinity to the target protein.55 Virtual screening is a powerful tool for identifying potential drug candidates, but it also has limitations. The accuracy of virtual screening results depends on the quality of the protein structure and the compound library. Additionally, the virtual screening process does not consider the pharmacokinetic and toxicological properties of the compounds. Therefore, compounds identified through virtual screening need to be validated experimentally to determine their efficacy and safety.56,57 In summary, virtual screening is a valuable computational technique that is helping to accelerate drug discovery. It involves the use of software tools to identify potential lead compounds from a large database of molecules. However, virtual screening results need to be validated experimentally, and the limitations of the method need to be considered.
MD simulations
MD simulations are a powerful computational tool used to study the physical movements and interactions of atoms and molecules in a system.58 MD simulation integrates Newton’s equations of motion over time to obtain the motion of the atoms/molecules in a system, which provides quantitative and qualitative information about the macroscopic behavior of the system at the atomic level.58 In other words, MD simulations provide a ‘movie’ of the dynamic ‘evolution’ of the system under investigation.
MD simulations consist of the numerical, step-by-step, solution of the classical equations of motion, which for a simple atomic system may be written as m_i * a_i = f_i, where m_i is the mass of particle i, a_i is its acceleration, and f_i is the force acting on particle i.59 The forces acting on each particle are determined by the interatomic interactions, which are typically represented by a mathematical function that describes the potential energy of the system as a function of the atomic positions. The interatomic potential function can be obtained using quantum mechanics, empirical potentials, or a combination of both.59 Figure 4 illustrates the basic steps and principle of working of MD simulations.
FIGURE 4. The basic steps and principle of working of molecular dynamics (MD) simulations used in new drug development.
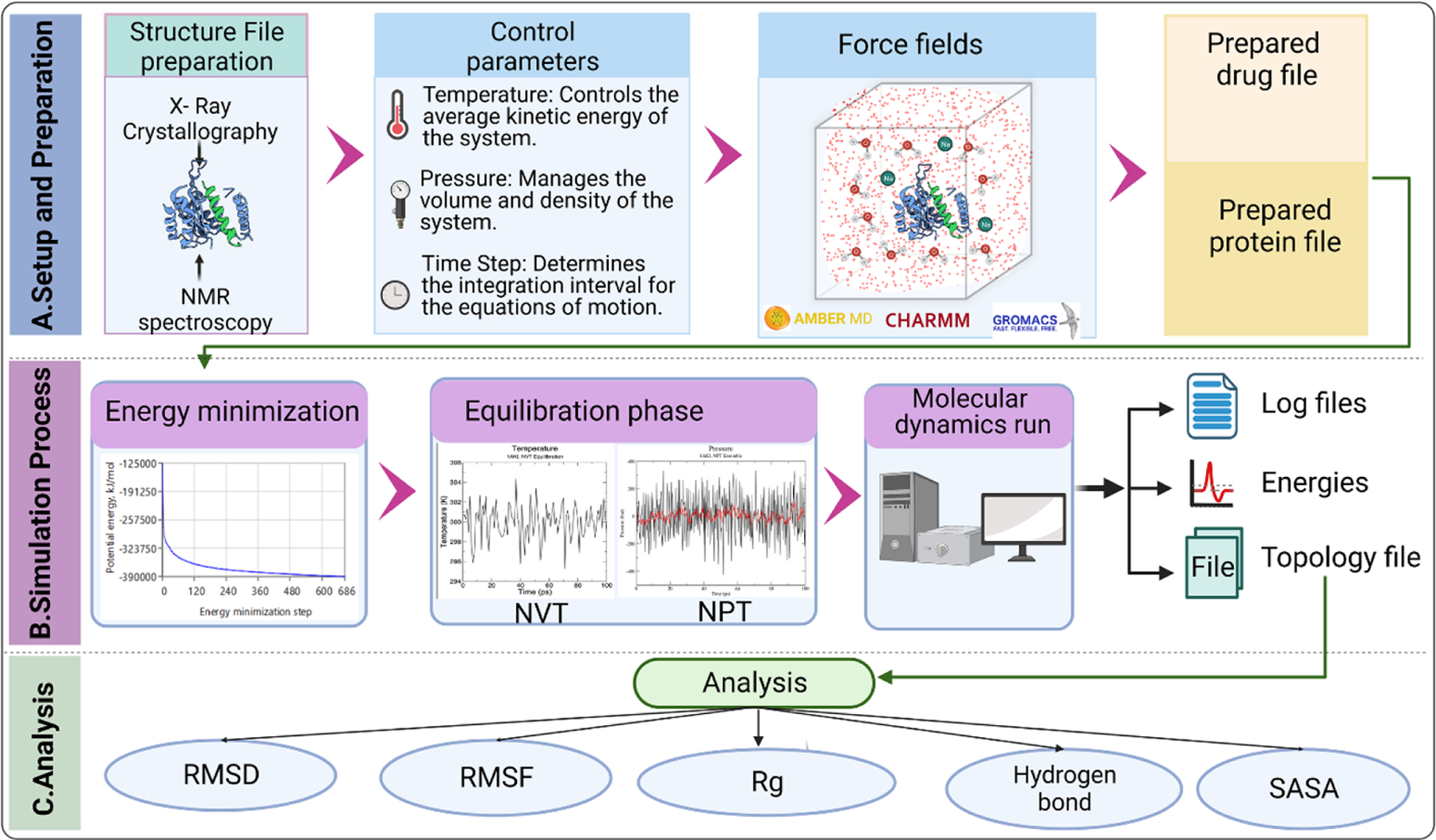
(a) Schematic representation of the steps involved in MD simulation for drug research. This figure illustrates the sequential steps in conducting MD simulations for drug research. The setup and preparation phase involves obtaining and converting the three-dimensional structures of protein and drug molecules into appropriate file formats. Essential simulation parameters, such as temperature, pressure, and time step, are defined. Additionally, an appropriate force field is chosen based on the system and research objectives. It is important to select simulation software that is compatible with the chosen force field. (b) During the simulation process, energy minimization is performed to alleviate steric clashes and minimize potential energy in the system. The equilibration phase allows the system to gradually relax by restraining specific atoms and allowing solvent molecules to adjust around the protein–drug complex. Subsequently, a time-dependent molecular dynamics run is carried out, where the equations of motion are numerically integrated to generate atom trajectories. (c) The analysis of the topology file includes various metrics. Root-mean-square deviation (RMSD) is used to measure the average deviation between different structures at different time points and a reference structure. Root-mean-square fluctuation (RMSF) determines the atomic fluctuations of the protein and drug, providing insights into their flexibility and stability. The radius of gyration (Rg) quantifies the compactness or spatial extent of the protein–drug complex. Hydrogen bonds are assessed to understand their formation and dynamics, indicating potential interactions. Finally, the solvent accessible surface area (SASA) is calculated to analyze the interactions of the protein and drug with the surrounding solvent molecules.
MD simulations can be used to study a wide range of systems, including biological macromolecules, materials science, and chemical physics.60 When applied to biological macromolecules, MD simulations can provide insights into the dynamic behavior of proteins and nucleic acids, including fluctuations in the relative positions of the atoms in a molecule.61 MD simulations are important in the theoretical study of food proteins and bioactive peptides, and can be used in conjunction with molecular docking to predict the binding of small molecules to proteins.43 MD simulations can be performed using different levels of detail, from atomistic simulations that simulate individual atoms to coarse-grained approaches that lump a number of atoms into pseudo-particles.62,63 The choice of simulation method depends on the level of detail required for the system under investigation.
Taken together, MD simulations are a powerful computational tool that can provide detailed insights into the physical movements and interactions of atoms and molecules in a system. The simulations integrate Newton’s equations of motion over time to obtain the motion of the atoms/molecules in a system and can provide quantitative and qualitative information about macroscopic behavior of the system at the atomic level. MD simulations can be used to study a wide range of systems, from biological macromolecules to materials science, and can be performed at different levels of detail depending on the system under investigation.
Recently, the following noteworthy advancements have emerged in the field of MD simulations. (i) Deep learning algorithms increasingly applied to MD simulations. For instance, Atomic Convolutional Networks have been used to predict the potential energy of a molecule directly from its atomic coordinates, which can significantly improve the accuracy of MD simulations.64 (ii) Enhanced sampling techniques, such as metadynamics, have been developed to overcome the limitations of traditional MD simulations.65,66 These techniques can explore a larger conformational space and provide a more accurate representation of the energy landscape of a system. (iii) Coarse-grained models, which simplify the representation of a system by grouping atoms together, have been developed to reduce the computational cost of MD simulations.67,68 These models can simulate larger systems and longer timescales than traditional all-atom models. (iv) GPU-accelerated tools, such as GROMACS and AMBER, have been developed to speed up MD simulations.69,70 These tools can perform simulations faster than CPU-based tools, enabling the study of larger and more complex systems. Together, these advancements have significantly improved the accuracy and efficiency of MD simulations. However, challenges remain, such as the accurate prediction of protein flexibility and the computational cost of large-scale simulations. The field is rapidly evolving, and we can expect further improvements in the coming years.
Quantum mechanics/molecular mechanics calculations
Quantum mechanics/molecular mechanics (QM/MM) calculations are a powerful computational approach used to study chemical and biochemical systems at the atomic and molecular level. The QM/MM approach combines the accuracy of quantum mechanical calculations with the speed of molecular mechanics simulations.71 This method allows for the calculation of thermodynamic properties and the characterization of reaction dynamics, making it a valuable tool for studying chemical and biochemical systems in solution or enzymes.72 QM/MM calculations can provide accurate calculations of reaction energies and reaction pathways.
In traditional molecular mechanics and quantum mechanics computations, inter- and intramolecular interactions are evaluated for a given frozen configuration of the system, often without explicit solvation.73 However, the QM/MM approach can take solvation effects into account by treating the QM region as a solute and the MM region as a solvent.74 The QM/MM approach was first introduced in 1976 by Warshel and Levitt.75 Since then, it has been widely used to study a variety of chemical and biochemical systems including enzymes, reaction mechanisms, and protein–ligand binding interactions. One of the advantages of the QM/MM approach is its ability to simulate larger systems than can be treated with quantum mechanical ab initio methods alone.74 The size of many systems of interest in chemistry and biochemistry prevents efficient and accurate treatment by quantum mechanical methods alone. Overall, the QM/MM approach is a valuable tool for studying the behavior of matter and light at the atomic and subatomic scale, making it a useful approach for understanding the properties of molecules and atoms and their constituents.76
ML algorithms
ML algorithms have become increasingly popular in predicting the properties of small molecules. These algorithms use computational approaches to predict the properties of potential small molecule drug candidates such as their solubility, toxicity, and binding affinity.77 Two main groups of ML methods are discussed in drug discovery: traditional ML methods (e.g., tree-based methods, latent variable methods) and deep learning methods.78 The characterization of molecular properties is a critical problem in drug discovery. Experimental methods have been widely used across the entire drug discovery process, including high-throughput in vitro screening and low-throughput in vivo testing. However, on average, one United States Food and Drug Administration (FDA) drug is approved for five compounds entering clinical trials, which shows the need for more efficient drug discovery methods.79
ML approaches vary in complexity and range from simple sum-over-atoms methods to more sophisticated approaches capable of describing collective interactions between many atoms or bonds.80 For example, ML models have been used to predict the binding of small molecules to RNA targets, and Lasso regression models were used to compare the performance of various ML algorithms to predict the binding scores of phenylthiazole-containing molecules.81
Solubility prediction, which can reduce waste and improve the crystallization process efficiency, has attracted increasing attention. However, there are still many urgent challenges, and several methods are being developed to address them.82 ML models, such as MegaTox, can be used to predict early-stage clinical compounds and recent FDA-approved drugs to identify potential drug-induced liver injury.83
Overall, ML algorithms have shown great promise in predicting the properties of small molecule drug candidates. These algorithms have the potential to revolutionize drug discovery by reducing the need for expensive experimental methods, making drug development faster and more efficient.
Computational approaches have made remarkable strides in advancing the field of small molecule drug design; however, these approaches are not without their inherent limitations. Within this context, several key challenges deserve careful consideration, including accounting for protein flexibility, improving scoring functions, addressing solvent effects, extending simulation time scales, managing computational costs, acquiring adequate experimental data, predicting induced fit, and accurately anticipating off-target effects.
Protein flexibility: Traditional docking algorithms often treat the protein as a rigid body, which is a significant oversimplification. Proteins are highly dynamic and can adopt a range of conformations. Accounting for this flexibility is a major challenge for docking algorithms.
Scoring functions: The scoring functions used in docking algorithms to predict the binding affinity of a ligand to a protein are often inaccurate. They are typically based on simplified models of molecular interactions and do not fully capture the complexity of these interactions.
Solvent effects: Many docking algorithms do not adequately account for the effects of the solvent. The solvent can play a crucial role interactions, and neglecting these effects can lead to inaccurate predictions.
Time scale: MD simulations can model the dynamic behavior of molecules, but they are limited by the time scale they can simulate. Most simulations are on the nanosecond to microsecond scale, while many biological processes occur on the millisecond to second scale.
Computational cost: Both docking and MD simulations are computationally intensive. This limits the size of the systems that can be studied and the length of the simulations that can be performed.
Lack of experimental data: There is often a lack of experimental data to validate the predictions made by docking and MD simulations. This makes it difficult to assess the accuracy of these methods.
Difficulty in predicting induced fit: Many docking algorithms struggle to predict the conformational changes that occur when a ligand binds to a protein, a phenomenon known as induced fit.
Limitations in predicting off-target effects: While computational methods are improving in predicting the interaction between a drug and its intended target, predicting potential off-target effects remain a significant challenge. Despite these challenges, the computational approaches used in small molecule drug design are continuously evolving. Emerging computational tools and techniques such as deep learning and artificial intelligence are likely to have a significant impact on drug design in the future.84 These tools can help researchers generate new hypotheses, analyze large data sets, and design new drug candidates with enhanced efficacy and safety profiles.
In conclusion, computational methods play a vital role in small molecule drug design. Molecular docking and virtual screening are commonly used methods for predicting ligand–protein interactions and screening large compound libraries. Other computational approaches such as MD simulations, quantum mechanics/molecular mechanics calculations, and ML algorithms have also been applied to drug design. Emerging computational tools and techniques are likely to further advance the field of small molecule drug design in the future.
Case studies
In recent years, there have been several notable case studies that highlight the importance of molecular structure in small molecule drug design. These case studies demonstrate the successful application of structure-based drug design and provide insights into the discovery process, mechanism of action, and pharmacological properties of the drugs. One such example is venetoclax, a small molecule drug used for the treatment of chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL).85 Venetoclax was designed based on the identification of the anti-apoptotic protein B-cell lymphoma 2 (BCL-2) as a therapeutic target.86 The drug’s chemical structure consists of a bicyclic scaffold with a 4-aminoindole core and two benzenesulfonamide moieties.85,87 Venetoclax binds selectively to BCL-2, leading to apoptosis of cancer cells by blocking the interaction between BCL-2 and pro-apoptotic proteins.85,88 Venetoclax has been shown to be effective in the treatment of CLL and SLL, with a favorable safety profile.86,89 Its pharmacological properties include a long half-life of approximately 26 h and linear pharmacokinetics with dose-proportional exposure.90 The discovery of venetoclax was made possible by the application of structure-based drug design, which involves the use of computational and experimental techniques to design small molecules that can selectively bind to a target protein.87,91,92 The structure of BCL-2 was determined by X-ray crystallography, which allowed for the identification of the binding site and the rational design of Venetoclax.93,94 Molecular docking and other computational methods were used to optimize the structure of venetoclax and predict its binding affinity.95
Another example is the development of the drug ivacaftor, which is used to treat cystic fibrosis. Ivacaftor was designed based on its ability to selectively bind to and enhance the activity of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, which is defective in patients with cystic fibrosis. The drug was designed to interact with specific amino acid residues in the CFTR protein, stabilizing the protein and increasing its activity.96 Once the drug target (CFTR) is chosen, the 3D structure of the protein is determined using techniques such as X-ray crystallography or NMR spectroscopy. The protein structure is then used to identify potential binding sites for small molecules.97 In the case of ivacaftor, the binding site was located in the intracellular domain of the CFTR protein. The next step is to design small molecules that can interact with the target protein and modify its function. This is done using computer-aided drug design (CADD) tools such as molecular docking and MD simulations.98 These tools allow researchers to predict the position of small molecules within a 3D representation of the protein structure and estimate the affinity of ligands to target protein with considerable accuracy and efficiency.29 After the small molecule is designed, it undergoes a series of tests to determine its efficacy and safety. This includes in vitro assays, in vivo studies, and toxicity tests. If the drug is found to be effective and safe, it proceeds to clinical trials.99,100 In brief, ivacaftor was developed using the structure-based drug design methodology. The process involves the choice of a drug target, determination of the protein structure, identification of potential binding sites, design of small molecules using CADD tools, and testing of the drug for efficacy and safety.
A recent case study involves the development of a novel metabolic reprogramming strategy (MRS) for the treatment of diabetes-associated breast cancer.101 As part of this strategy, we aimed to identify potential monocarboxylate transporter 4 (MCT4) inhibitors for combination therapy. The 3D model of human MCT4 structure was generated by using the I-TASSER On-line Server. Then, we used grid-based ligand docking with energetics (GLIDE) software to virtually screen seven commercial compound libraries and identify potential MCT4 inhibitors. The top-ranked compounds were visually inspected, and the most promising candidate CB-2 was pursued for further testing. Experimental testing confirmed the binding of CB-2 to MCT4, and further studies demonstrated its cytotoxic activity against cancer cells and its antitumor effects on animal models. The methodology used in this study highlights the importance of structure-based drug design in the development of novel small molecule drug candidates. At present, CB-2 has obtained a global patent and entered the preclinical study stage.
In addition to the above few examples, there are some classic case studies, such as the development of the drug olaparib for the treatment of breast and ovarian cancers, and the development of the drug sofosbuvir for the treatment of hepatitis C. Olaparib was designed based on its ability to selectively inhibit poly(ADP-ribose) polymerase (PARP), an enzyme that repairs damaged DNA in cancer cells. The drug was designed to mimic the structure of the substrate of PARP, allowing it to selectively bind to and inhibit the enzyme in cancer cells.102,103 Sofosbuvir was designed based on its ability to selectively inhibit the RNA polymerase of the hepatitis C virus. The drug was designed to mimic the structure of the natural substrate of the polymerase, allowing it to selectively bind to and inhibit the enzyme in the virus.104 Overall, these case studies emphasize the significance of molecular structure in drug design and its contribution to the development of effective therapeutic options.
Concluding remarks and future directions
Small molecule drug design is a rapidly growing field that has the potential to revolutionize the development of new pharmaceuticals. As advancements in structural biology and computational methods continue to expand our understanding of the relationship between molecular structure and drug efficacy, researchers are finding new and innovative ways to design small molecules that are more potent, selective, and safe than ever before. In this review, we explored some of the recent advancements in small molecule drug design from a structural perspective and discussed the emerging trends and future directions in this field.
One of the key areas of research in this field is the development of new computational tools and methods for predicting and optimizing the structural properties of small molecules. These tools are becoming increasingly important as the size and complexity of drug targets continue to grow, making it more difficult to design drugs using traditional methods. Some of the emerging computational methods that are being used in small molecule drug design include ML algorithms, MD simulations, and quantum mechanical calculations.105 Besides, other computational tools and methods such as statistical models have been developed for predicting toxicity and side effects of small molecules. These tools enable the identification and optimization of lead compounds, and the prediction of their pharmacological properties.87,106 Overall, the development of new computational tools and methods is crucial for the efficient and cost-effective discovery and development of small molecule drugs. These tools enable the prediction and optimization of the structural properties of small molecules, which is critical for their safety and efficacy. With the increasing use of ML and other computational methods in drug discovery, the future of small molecule drug design looks promising.84
Another important trend in small molecule drug design is the exploration of new therapeutic applications. While traditional drug design has focused primarily on treating common diseases such as cancer and cardiovascular disease, researchers are now beginning to investigate the potential of small molecule drugs in treating rare and neglected diseases. This has led to the development of new drug discovery programs focused on diseases such as rare genetic disorders, neglected tropical diseases, and emerging infectious diseases.107–109
Finally, the future of small molecule drug design is likely to be shaped by new technologies and innovations in the field of structural biology. For example, the recent development of cryo-electron microscopy has revolutionized the way that researchers study the structures of biological macromolecules, and has already led to the discovery of new drug targets and the development of new drugs.110,111 Other emerging technologies that are likely to have an impact on small molecule drug design include protein engineering, chemical genomics, and high-throughput screening.112,113
In conclusion, small molecule drug design is a rapidly evolving field that is driven by advancements in structural biology and computational methods. As researchers continue to explore the relationship between molecular structure and drug efficacy, new and innovative small molecule drug candidates are being developed that have the potential to revolutionize the treatment of a wide range of diseases. Looking to the future, the development of new computational tools and methods, the exploration of new therapeutic applications, and the continued evolution of structural biology are likely to be key drivers of innovation in this field.
Teaser:
Exploring the cutting-edge advancements in small molecule drug design, delving into the role of protein structures, computational tools, and optimization techniques. This review illuminates how integrating structural data can revolutionize drug discovery, enhancing efficacy, specificity, and safety.
Research Highlights:
Importance of accurate protein structure prediction for small molecule drug design.
Key structural features and properties used in small molecule drug design.
Computational tools and methods for predicting and optimizing small molecule structures.
Examples of successful small molecule drug design based on molecular structure.
Acknowledgments
This work was supported in part by NIH-NIMHD U54MD007598, NIH/NCI1U54CA14393, U56 CA101599–01; Department-of-Defense Breast Cancer Research Program grant BC043180, NIH/NCATS CTSI UL1TR000124 to J.V. Vadgama; Accelerating Excellence in Translational Science Pilot Grants G0812D05, NIH/NCI SC1CA200517 and 9 SC1 GM135050–05 to Y. Wu. We want to acknowledge Dr. Zhimin Huang, Dr. Albert Ngo and Hang Chee Erin Shum for their contributions to this review article. Their expertise and insights were instrumental in the development of the manuscript. We appreciate their valuable input and support throughout the process.
Abbreviations
- BCL-2
B-cell lymphoma 2
- CADD
computer-aided drug design
- CFTR
cystic fibrosis transmembrane conductance regulator
- CLL
chronic lymphocytic leukemia
- cryo-EM
cryo-electron microscopy
- GLIDE
grid-based ligand docking with energetics
- GPCRs
G protein-coupled receptors
- MCT4
monocarboxylate transporter 4
- MD
molecular dynamics
- ML
machine learning
- MRS
metabolic reprogramming strategy
- PARP
poly(ADP-ribose) polymerase
- QM/MM
quantum mechanics/molecular mechanics
- SBDD
structure-based drug design
- SLL
small lymphocytic lymphoma
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest
No interests are declared.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
No data were used for the research described in the article.
References
- 1.Lionta E, Spyrou G, Vassilatis DK, Cournia Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem 2014;14(16):1923–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muhammed MT, Aki-Yalcin E. Homology modeling in drug discovery: overview, current applications, and future perspectives. Chem Biol Drug Des 2019;93(1):12–20. [DOI] [PubMed] [Google Scholar]
- 3.Vyas VK, Ukawala RD, Ghate M, Chintha C. Homology modeling a fast tool for drug discovery: current perspectives. Indian J Pharm Sci 2012;74(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavasotto CN, Phatak SS. Homology modeling in drug discovery: current trends and applications. Drug Discov Today 2009;14(13):676–683. [DOI] [PubMed] [Google Scholar]
- 5.Pearson WR. An introduction to sequence similarity (“homology”) searching. Curr Protoc Bioinformatics 2013;Chapter 3:3.1.1–3.1.8. [DOI] [PMC free article] [PubMed]
- 6.Bramucci E, Paiardini A, Bossa F, Pascarella S. PyMod: sequence similarity searches, multiple sequence-structure alignments, and homology modeling within PyMOL. BMC Bioinformatics 2012;13(Suppl 4):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hameduh T, Haddad Y, Adam V, Heger Z. Homology modeling in the time of collective and artificial intelligence. Comput Struct Biotechnol J 2020;18:3494–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu Z, Kang L, Zhao W, et al. Identification of novel umami peptides from myosin via homology modeling and molecular docking. Food Chem 2021;344:128728. [DOI] [PubMed] [Google Scholar]
- 9.Rakhshani H, Dehghanian E, Rahati A. Enhanced GROMACS: toward a better numerical simulation framework. J Mol Model 2019;25(12):355. [DOI] [PubMed] [Google Scholar]
- 10.Mao W, Ding W, Xing Y, Gong H. AmoebaContact and GDFold as a pipeline for rapid de novo protein structure prediction. Nat Mach Intell 2020;2(1):25–33. [Google Scholar]
- 11.Cao S, Chen S-J. Physics-based de novo prediction of RNA 3D structures. J Phys Chem B 2011;115(14):4216–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D. Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci U S A 2020;117(3):1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheung NJ, Yu W. De novo protein structure prediction using ultra-fast molecular dynamics simulation. PLoS One 2018;13(11):e0205819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ngan S-C, Hung L-H, Liu T, Samudrala R. Scoring functions for de novo protein structure prediction revisited. In: Zaki MJ, Bystroff C, eds. Protein Structure Prediction Totowa, NJ: Humana Press; 2008:243–281. [DOI] [PubMed] [Google Scholar]
- 15.Abriata LA, Dal Peraro M. State-of-the-art web services for de novo protein structure prediction. Brief Bioinform 2020;22(3):bbaa139. [DOI] [PubMed] [Google Scholar]
- 16.Handl J, Knowles J, Vernon R, Baker D, Lovell SC. The dual role of fragments in fragment-assembly methods for de novo protein structure prediction. Proteins 2012;80(2):490–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng Q, Hou M, Liu J, Zhao K, Zhang G. Construct a variable-length fragment library for de novo protein structure prediction. Brief Bioinform 2022;23(3):bbac086. [DOI] [PubMed] [Google Scholar]
- 18.Ramachandran S, Kota P, Ding F, Dokholyan NV. Automated minimization of steric clashes in protein structures. Proteins 2011;79(1):261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baek M, Baker D. Deep learning and protein structure modeling. Nat Methods 2022;19(1):13–14. [DOI] [PubMed] [Google Scholar]
- 20.Du Z, Su H, Wang W, et al. The trRosetta server for fast and accurate protein structure prediction. Nat Protoc 2021;16(12):5634–5651. [DOI] [PubMed] [Google Scholar]
- 21.Gupta A, Pallavi M, Bhatnagar S. Protein structure prediction using homology modeling: methods and tools. In: Dastmalchi S, Hamzeh-Mivehroud M, Sokouti B, eds. Methods and Algorithms for Molecular Docking-Based Drug Design and Discovery 1st ed. IGI Global; 2016. [Google Scholar]
- 22.Zhang H, Shen Y. Template-based prediction of protein structure with deep learning. BMC Genomics 2020;21(11):878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borkakoti N, Thornton JM. AlphaFold2 protein structure prediction: implications for drug discovery. Curr Opin Struct Biol 2023;78:102526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He H, Liu B, Luo H, Zhang T, Jiang J. Big data and artificial intelligence discover novel drugs targeting proteins without 3D structure and overcome the undruggable targets. Stroke Vasc Neurol 2020;5(4):381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Payandeh J, Volgraf M. Ligand binding at the protein-lipid interface: strategic considerations for drug design. Nat Rev Drug Discov 2021;20(9):710–722. [DOI] [PubMed] [Google Scholar]
- 26.Ngo HX, Garneau-Tsodikova S. What are the drugs of the future? Medchemcomm 2018;9(5):757–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Makurvet FD. Biologics versus small molecules: drug costs and patient access. Med Drug Discov 2021;9:100075. [Google Scholar]
- 28.Batool M, Ahmad B, Choi S. A structure-based drug discovery paradigm. Int J Mol Sci 2019;20(11):2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Song K, Li L, Chen L. Structure-based drug design strategies and challenges. Curr Top Med Chem 2018;18(12):998–1006. [DOI] [PubMed] [Google Scholar]
- 30.Yang D, Zhou Q, Labroska V, et al. G protein-coupled receptors: structure- and function-based drug discovery. Signal Transduct Target Ther 2021;6(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wyss DF, Zartler ER. NMR studies of protein–small molecule interactions for drug discovery. In: Renaud J-P, ed. Structural Biology in Drug Discovery Wiley; 2020;325–345. [Google Scholar]
- 32.Ilatovskiy AV, Abagyan R. Computational structural biology for drug discovery. In: Renaud J-P, ed. Structural Biology in Drug Discovery Wiley; 2020;347–361. [Google Scholar]
- 33.Deflorian F, Mason JS, Bortolato A, Tehan BG. Impact of recently determined crystallographic structures of GPCRs on drug discovery. In: Renaud J-P, ed. Structural Biology in Drug Discovery Wiley; 2020;449–477. [Google Scholar]
- 34.Guvench O Computational functional group mapping for drug discovery. Drug Discov Today 2016;21(12):1928–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He Z, Zhang J, Shi X-H, et al. Predicting drug-target interaction networks based on functional groups and biological features. PLoS One 2010;5(3):e9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howard RJ, Slesinger PA, Davies DL, Das J, Trudell JR, Harris RA. Alcohol-binding sites in distinct brain proteins: the quest for atomic level resolution. Alcohol Clin Exp Res 2011;35(9):1561–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brooks WH, Guida WC, Daniel KG. The significance of chirality in drug design and development. Curr Top Med Chem 2011;11(7):760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orlando BJ, Lucido MJ, Malkowski MG. The structure of ibuprofen bound to cyclooxygenase-2. J Struct Biol 2015;189(1):62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuna M, Mahdi F, Chade AR, Bidwell GL. Molecular size modulates pharmacokinetics, biodistribution, and renal deposition of the drug delivery biopolymer elastin-like polypeptide. Sci Rep 2018;8(1):7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hutchinson JS. Concept Development Studies in Chemistry. 8: Molecular Structure and Physical Properties. LibreTexts; 2020. https://chem.libretexts.org/Bookshelves/General_Chemistry/Book%3A_Concept_Development_Studies_in_Chemistry_(Hutchinson)/08_Molecular_Structure_and_Physical_Properties. Accessed March 24, 2023.
- 41.Reusch W Boiling Points 2023. https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Fundamentals/Intermolecular_Forces/Boiling_Points. Accessed March 23, 2023.
- 42.Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 2011;7(2):146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vidal-Limon A, Aguilar-Toalá JE, Liceaga AM. Integration of molecular docking analysis and molecular dynamics simulations for studying food proteins and bioactive peptides. J Agric Food Chem 2022;70(4):934–943. [DOI] [PubMed] [Google Scholar]
- 44.Pinzi L, Rastelli G. Molecular docking: shifting paradigms in drug discovery. Int J Mol Sci 2019;20(18):4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan Y, Huang S-Y. Pushing the accuracy limit of shape complementarity for protein-protein docking. BMC Bioinformatics 2019;20(25):696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Chambers J, Abel R. Protein-ligand binding free energy calculations with FEP. Methods Mol Biol 2019;2022:201–232. [DOI] [PubMed] [Google Scholar]
- 47.Geng C, Narasimhan S, Rodrigues JP, Bonvin AM. Information-driven, ensemble flexible peptide docking using HADDOCK. Methods Mol Biol 2017;1561:109–138. [DOI] [PubMed] [Google Scholar]
- 48.Marze NA, Roy Burman SS, Sheffler W, Gray JJ. Efficient flexible backbone protein-protein docking for challenging targets. Bioinformatics 2018;34(20):3461–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seo S, Choi J, Park S, Ahn J. Binding affinity prediction for protein-ligand complex using deep attention mechanism based on intermolecular interactions. BMC Bioinformatics 2021;22(1):542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, Leung KS, Wong MH, Ballester PJ. Substituting random forest for multiple linear regression improves binding affinity prediction of scoring functions: Cyscore as a case study. BMC Bioinformatics 2014;15(1):291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vázquez J, López M, Gibert E, Herrero E, Luque FJ. Merging ligand-based and structure-based methods in drug discovery: an overview of combined virtual screening approaches. Molecules 2020;25(20):4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bauer MR, Ibrahim TM, Vogel SM, Boeckler FM. Evaluation and optimization of virtual screening workflows with DEKOIS 2.0 – a public library of challenging docking benchmark sets. J Chem Inf Model 2013;53(6):1447–1462. [DOI] [PubMed] [Google Scholar]
- 53.Ghiandoni GM, Caldeweyher E. Fast calculation of hydrogen-bond strengths and free energy of hydration of small molecules. Sci Rep 2023;13(1):4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luttens A, Gullberg H, Abdurakhmanov E, et al. Ultralarge virtual screening identifies SARS-CoV-2 main protease inhibitors with broad-spectrum activity against coronaviruses. J Am Chem Soc 2022;144(7):2905–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gimeno A, Ojeda-Montes MJ, Tomás-Hernández S, et al. The light and dark sides of virtual screening: what is there to know? Int J Mol Sci 2019;20(6):1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu KJ, Lei PM, Liu H, Wu C, Leung CH, Ma DL. Mimicking strategy for protein-protein interaction inhibitor discovery by virtual screening. Molecules 2019;24(24):4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walters WP, Wang R. New trends in virtual screening. J Chem Inf Model 2020;60(9):4109–4111. [DOI] [PubMed] [Google Scholar]
- 58.Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron 2018;99(6):1129–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Allen MP. Introduction to molecular dynamics simulation. In: Attig N, Binder K, Grubmuller H, Kremer K, eds. Computational Soft Matter: From Synthetic Polymers to Proteins Bonn, Germany: Winter School, Gustav-Stresemann-lnstitut; [Poster Abstracts]; 2004:1–28. [Google Scholar]
- 60.Filipe HAL, Loura LMS. Molecular dynamics simulations: advances and applications. Molecules 2022;27(7):2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karplus M, Petsko GA. Molecular dynamics simulations in biology. Nature 1990;347(6294):631–639. [DOI] [PubMed] [Google Scholar]
- 62.Ryu WH, Voth GA. Coarse-graining of imaginary time Feynman path integrals: inclusion of intramolecular interactions and bottom-up force-matching. J Phys Chem A 2022;126(35):6004–6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kern P, Brunne RM, Rognan D, Folkers G. A pseudo-particle approach for studying protein-ligand models truncated to their active sites. Biopolymers 1996;38(5):619–637. [DOI] [PubMed] [Google Scholar]
- 64.Lemm D, von Rudorff GF, von Lilienfeld OA. Machine learning based energy-free structure predictions of molecules, transition states, and solids. Nat Commun 2021;12(1):4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang YI, Shao Q, Zhang J, Yang L, Gao YQ. Enhanced sampling in molecular dynamics. J Chem Phys 2019;151(7):070902. [DOI] [PubMed] [Google Scholar]
- 66.O’Hagan MP, Haldar S, Morales JC, Mulholland AJ, Galan MC. Enhanced sampling molecular dynamics simulations correctly predict the diverse activities of a series of stiff-stilbene G-quadruplex DNA ligands. Chem Sci 2020;12(4):1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tan C, Jung J, Kobayashi C, Torre DU, Takada S, Sugita Y. Implementation of residue-level coarse-grained models in GENESIS for large-scale molecular dynamics simulations. PLoS Comput Biol 2022;18(4):e1009578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barnoud J, Monticelli L. Coarse-grained force fields for molecular simulations. Methods Mol Biol 2015;1215:125–149. [DOI] [PubMed] [Google Scholar]
- 69.Kohnke B, Kutzner C, Grubmuller H. A GPU-accelerated fast multipole method for GROMACS: performance and accuracy. J Chem Theory Comput 2020;16(11):6938–6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cruzeiro VWD, Manathunga M, Merz KM Jr, Gotz AW. Open-source multi-GPU-accelerated QM/MM simulations with AMBER and QUICK. J Chem Inf Model 2021;61(5):2109–2115. [DOI] [PubMed] [Google Scholar]
- 71.Woods CJ, Manby FR, Mulholland AJ. An efficient method for the calculation of quantum mechanics/molecular mechanics free energies. J Chem Phys 2008;128(1):014109. [DOI] [PubMed] [Google Scholar]
- 72.Shen L, Yang W. Molecular dynamics simulations with quantum mechanics/molecular mechanics and adaptive neural networks. J Chem Theory Comput 2018;14(3):1442–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koehler KF, Rao SN, Snyder JP. 7 - Modeling drug–receptor interactions. In: Cohen NC, ed. Guidebook on Molecular Modeling in Drug Design San Diego: Academic Press; 1996:235–336. [Google Scholar]
- 74.Sierka M, Sauer J. Hybrid quantum mechanics/molecular mechanics methods and their application. In: Yip S, ed. Handbook of Materials Modeling: Methods Dordrecht: Springer Netherlands; 2005:241–258. [Google Scholar]
- 75.Warshel A, Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J Mol Biol 1976;103(2):227–249. [DOI] [PubMed] [Google Scholar]
- 76.Groenhof G Introduction to QM/MM simulations. In: Monticelli L, Salonen E, eds. Biomolecular Simulations: Methods and Protocols Totowa, NJ: Humana Press; 2013:43–66. [Google Scholar]
- 77.Dara S, Dhamercherla S, Jadav SS, Babu CM, Ahsan MJ. Machine learning in drug discovery: a review. Artif Intell Rev 2022;55(3):1947–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pillai N, Dasgupta A, Sudsakorn S, Fretland J, Mavroudis PD. Machine learning guided early drug discovery of small molecules. Drug Discov Today 2022;27(8):2209–2215. [DOI] [PubMed] [Google Scholar]
- 79.Shen J, Nicolaou CA. Molecular property prediction: recent trends in the era of artificial intelligence. Drug Discov Today Technol 2019;32–33:29–36. [DOI] [PubMed]
- 80.Hansen K, Biegler F, Ramakrishnan R, et al. Machine learning predictions of molecular properties: accurate many-body potentials and nonlocality in chemical space. J Phys Chem Lett 2015;6(12):2326–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Grimberg H, Tiwari VS, Tam B, et al. Machine learning approaches to optimize small-molecule inhibitors for RNA targeting. J Cheminform 2022;14(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ma Y, Gao Z, Shi P, et al. Machine learning-based solubility prediction and methodology evaluation of active pharmaceutical ingredients in industrial crystallization. Front Chem Sci and Eng 2022;16(4):523–535. [Google Scholar]
- 83.Minerali E, Foil DH, Zorn KM, Lane TR, Ekins S. Comparing machine learning algorithms for predicting drug-induced liver injury (DILI). Mol Pharm 2020;17(7):2628–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jing Y, Bian Y, Hu Z, Wang L, Xie X-QS. Deep learning for drug design: an artificial intelligence paradigm for drug discovery in the big data era. AAPS J 2018;20(3):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tariq S, Tariq S, Khan M, Azhar A, Baig M. Venetoclax in the treatment of chronic lymphocytic leukemia: evidence, expectations, and future prospects. Cureus 2020;12(6):e8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Korycka-Wolowiec A, Wolowiec D, Kubiak-Mlonka A, Robak T. Venetoclax in the treatment of chronic lymphocytic leukemia. Expert Opin Drug Metab Toxicol 2019;15(5):353–366. [DOI] [PubMed] [Google Scholar]
- 87.Beck H, Härter M, Haß B, Schmeck C, Baerfacker L. Small molecules and their impact in drug discovery: a perspective on the occasion of the 125th anniversary of the Bayer Chemical Research Laboratory. Drug Discov Today 2022;27(6):1560–1574. [DOI] [PubMed] [Google Scholar]
- 88.Lew TE, Seymour JF. Clinical experiences with venetoclax and other pro-apoptotic agents in lymphoid malignancies: lessons from monotherapy and chemotherapy combination. J Hematol Oncol 2022;15(1):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Siddiqui M, Konopleva M. Keeping up with venetoclax for leukemic malignancies: key findings, optimal regimens, and clinical considerations. Expert Rev Clin Pharmacol 2021;14(12):1497–1512. [DOI] [PubMed] [Google Scholar]
- 90.Food and Drug Administration. Clinical Pharmacology and Biopharmaceuticals Review Center for Drug Evaluation and Research; 2015. [Google Scholar]
- 91.Wilson DM, Deacon AM, Duncton MAJ, et al. Fragment- and structure-based drug discovery for developing therapeutic agents targeting the DNA damage response. Prog Biophys Mol Biol 2021;163:130–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013;19(2):202–208. [DOI] [PubMed] [Google Scholar]
- 93.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta 2004;1644(2):83–94. [DOI] [PubMed] [Google Scholar]
- 94.Maveyraud L, Mourey L. Protein X-ray crystallography and drug discovery. Molecules 2020;25(5):1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Najafi V, Yoosefian M, Hassani Z. Development of venetoclax performance using its new derivatives on BCL-2 protein inhibition. Cell Biochem Funct 2023;41(1):58–66. [DOI] [PubMed] [Google Scholar]
- 96.Deeks ED. Ivacaftor: a review of its use in patients with cystic fibrosis. Drugs 2013;73(14):1595–1604. [DOI] [PubMed] [Google Scholar]
- 97.Ford RC, Birtley J, Rosenberg MF, Zhang L. CFTR Three-dimensional structure. In: Amaral MD, Kunzelmann K, eds. Cystic Fibrosis: Diagnosis and Protocols, Volume I: Approaches to Study and Correct CFTR Defects Totowa, NJ: Humana Press; 2011:329–346. [Google Scholar]
- 98.Laselva O, Qureshi Z, Zeng ZW, et al. Identification of binding sites for ivacaftor on the cystic fibrosis transmembrane conductance regulator. iScience 2021;24(6):102542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rosenfeld M, Wainwright CE, Higgins M, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. Lancet Respir Med 2018;6(7):545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nichols DP, Paynter AC, Heltshe SL, et al. Clinical effectiveness of elexacaftor/tezacaftor/ivacaftor in people with cystic fibrosis: a clinical trial. Am J Respir Crit Care Med 2022;205(5):529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hao Q, Huang Z, Li Q, et al. A novel metabolic reprogramming strategy for the treatment of diabetes-associated breast cancer. Adv Sci (Weinh) 2022;9(6):e2102303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Weil MK, Chen AP. PARP inhibitor treatment in ovarian and breast cancer. Curr Probl Cancer 2011;35(1):7–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Passeri D, Camaioni E, Liscio P, et al. Concepts and molecular aspects in the polypharmacology of PARP-1 inhibitors. ChemMedChem 2016;11(12):1219–1226. [DOI] [PubMed] [Google Scholar]
- 104.Cha A, Budovich A. Sofosbuvir: a new oral once-daily agent for the treatment of hepatitis C virus infection. P T 2014;39(5):345–352. [PMC free article] [PubMed] [Google Scholar]
- 105.Mudedla SK, Braka A, Wu S. Quantum-based machine learning and AI models to generate force field parameters for drug-like small molecules. Front Mol Biosci 2022;9. [DOI] [PMC free article] [PubMed]
- 106.Maurer TS, Edwards M, Hepworth D, Verhoest P, Allerton CMN. Designing small molecules for therapeutic success: a contemporary perspective. Drug Discovery Today 2022;27(2):538–546. [DOI] [PubMed] [Google Scholar]
- 107.Clare RH, Hall SR, Patel RN, Casewell NR. Small molecule drug discovery for neglected tropical snakebite. Trends Pharmacol Sci 2021;42(5):340–353. [DOI] [PubMed] [Google Scholar]
- 108.Mercorelli B, Palù G, Loregian A. Drug repurposing for viral infectious diseases: how far are we? Trends Microbiol 2018;26(10):865–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sun W, Zheng W, Simeonov A. Drug discovery and development for rare genetic disorders. Am J Med Genet A 2017;173(9):2307–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fernandez-Leiro R, Scheres SHW. Unravelling biological macromolecules with cryo-electron microscopy. Nature 2016;537(7620):339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vénien-Bryan C, Li Z, Vuillard L, Boutin JA. Cryo-electron microscopy and X-ray crystallography: complementary approaches to structural biology and drug discovery. Acta Crystallogr F Struct Biol Commun 2017;73(4):174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gurevich EV, Gurevich VV. Therapeutic potential of small molecules and engineered proteins. Handb Exp Pharmacol 2014;219:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen HY, Swaroop M, Papal S, et al. Reserpine maintains photoreceptor survival in retinal ciliopathy by resolving proteostasis imbalance and ciliogenesis defects. Elife 2023;12:e83205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data were used for the research described in the article.