

Abstract
Objective:
Spinocerebellar ataxia type 3 (SCA3) is the most common dominantly inherited ataxia and biomarkers are needed to noninvasively monitor disease progression and treatment response. Anti-ATXN3 antisense oligonucleotide (ASO) treatment has been shown to mitigate neuropathology and rescue motor phenotypes in SCA3 mice. Here, we investigated if repeated ASO administration reverses brainstem and cerebellar neurochemical abnormalities by magnetic resonance spectroscopy (MRS).
Methods:
Symptomatic SCA3 mice received intracerebroventricular treatment of ASO or vehicle and were compared to wild-type vehicle-treated littermates. To quantify neurochemical changes in treated mice, longitudinal 9.4T MRS of cerebellum and brainstem was performed. Acquired MR group means were analyzed by two-way ANOVA mixed-effects sex-adjusted analysis with post-hoc Sidak correlation for multiple comparisons. Pearson correlations were used to relate SCA3 pathology and behavior.
Results:
MR spectra yielded 15–16 neurochemical concentrations in the cerebellum and brainstem. ASO treatment in SCA3 mice resulted in significant total choline rescue and partial reversals of taurine, glutamine, and total N-acetylaspartate across both regions. Some ASO-rescued neurochemicals correlated with reduction in diseased protein and nuclear ATXN3 accumulation. ASO-corrected motor activity correlated with total choline and total N-acetylaspartate levels early in disease.
Interpretation:
SCA3 mouse cerebellar and brainstem neurochemical trends parallel those in patients with SCA3. Decreased total choline may reflect oligodendrocyte abnormalities, decreased total N-acetylaspartate highlights neuronal health disturbances, and high glutamine may indicate gliosis. ASO treatment fully or partially reversed select neurochemical abnormalities in SCA3 mice, indicating the potential for these measures to serve as non-invasive treatment biomarkers in future SCA3 gene silencing trials.
Keywords: ATXN3, polyglutamine disease, magnetic resonance spectroscopy
INTRODUCTION
Spinocerebellar ataxia type 3 (SCA3) is a polyglutamine disease that results from the expansion of a CAG triplet repeat within the coding region of the ataxin-3 (ATXN3) gene and the most common dominantly inherited ataxia in the world, affecting an estimated 1 in 50,000 people1–3. In this neurodegenerative disease, patients with SCA3 generally exhibit widespread degenerative and neuropathological changes including neuronal loss and gliosis in the deep cerebellar nuclei, pontine nuclei, many brainstem nuclei, spinocerebellar tracts, substantia nigra, thalamus, and globus pallidus4,5. Affected persons progressively lose motor control, leading to death within 10 to 20 years.
While there currently is no effective treatment for this relentlessly progressive and fatal disease, therapies that target the disease gene or mRNA transcript offer hope. Our group and others have found that antisense oligonucleotide (ASO) mRNA silencing therapy is particularly effective in the treatment of SCA3 models6–9. Briefly, ASOs are designed to complimentarily bind a target gene of interest, allowing for cleavage of the transcript by an endogenous RNase H mechanism, thus preventing downstream expression of the encoded Ataxin-3 protein. Specifically, we have shown that the ATXN3 disease mRNA is readily and safely targetable by anti-ATXN3 ASOs, leading to correction of disease features in animal models6–9. With the strong scientific premise and preclinical animal data supporting disease-modifying therapy for SCA3, preparation for efficacy clinical trials is of utmost importance, such as for the ongoing evaluation of the efficacy of anti-ATXN3 ASO in SCA3 patients (ClinicalTrials.gov Identifier: NCT05160558).
Because SCA3 progression often spans two decades, it has been challenging to develop sensitive and specific disease biomarkers. Success of disease-modifying interventions will depend on early detection of CNS abnormalities prior to major cell loss. Conventional imaging techniques primarily detect structural damage10,11, however assessments prior to cell death that can also measure cellular dysfunction could facilitate both early detection of cellular abnormalities and assessment of their reversal with therapies. Such criteria can be fulfilled using advances in magnetic resonance imaging technologies, which are becoming more widely available. Studies by our group and others have detected biochemical abnormalities in patients with SCA3 through high field magnetic resonance spectroscopy (MRS)12–15. We further compared MRS biomarkers in the cerebellum in two SCA3 animal models in late disease, which recapitulated many of these neurochemical changes, albeit only at a single end-stage timepoint16.
Direct assessment of vulnerable brain pathology will not be feasible in future SCA3 human ASO therapy trials, therefore a non-invasive imaging measure that reflects the ASO effects on disease pathology will be crucial for the success of these trials. As SCA3 also significantly affects the brainstem, which is faithfully reproduced in this SCA3 mouse model, we sought to expand our previous MRS study16 to longitudinally study metabolic changes in both the brainstem and cerebellum and to investigate proof-of-concept biomarker reversibility in repeat ASO-treated SCA3 mice. Using the Q84 SCA3 mouse model that expresses the full-length human mutant ATXN3 gene under the endogenous human promoter and demonstrates human pathology7,17, our study aims to investigate the potential of neurochemical MRS biomarkers in future anti-ATXN3 ASO efficacy trials in two highly affected SCA3 brain regions.
MATERIALS AND METHODS
Experimental design:
This study assesses the longitudinal cerebellar and brainstem MRS neurochemical profiles of the homozygous YACMJD84.2 (Jackson Laboratories; Strain #012705), referred to as SCA3 mice throughout this manuscript17. This transgenic mouse model is frequently used in SCA3 preclinical therapeutic trials, as it expresses the full-length human mutant ATXN3 disease gene. All animal procedures were approved by the University of Michigan (Protocol #PRO00008445) and University of Minnesota (Protocol #1708–35065A) Institutional Animal Care and Use Committee. Study groups included SCA3 Vehicle-treated, SCA3 ASO-treated, and their littermate non-transgenic (WT) Vehicle-treated mice on the C57Bl6/J background. Antisense oligonucleotide intracerebroventricular (ASO ICV) treatment was administered every 12 weeks beginning at 7–12 weeks of age (Figure 1A). Activity chamber motor assessment was completed 1 week prior to first treatment and 1 week after subsequent magnetic resonance spectroscopy (MRS) scanning at +8, +26, and +32 weeks of age relative to first injection. Interim tissue was collected after +9 weeks testing (n=6 per group) and endpoint tissue was collected after +33 weeks testing (n=4–6 per group). Mice were housed in cages with corncob bedding with a maximum of five animals (range 3–5) and maintained in a standard 12-hours light/dark cycle with food and water ad libitum. Mice were shipped from the University of Michigan to the University of Minnesota for MR scanning, at approximately 12–14 weeks of age. After MR scanning, mice designated for tissue collection were deeply anesthetized with a lethal dose of ketamine/xylazine, sacrificed by transcardial perfusion with pH 7.4 phosphate buffered saline; tissue samples were sent to the University of Michigan for molecular analysis.
Figure 1.
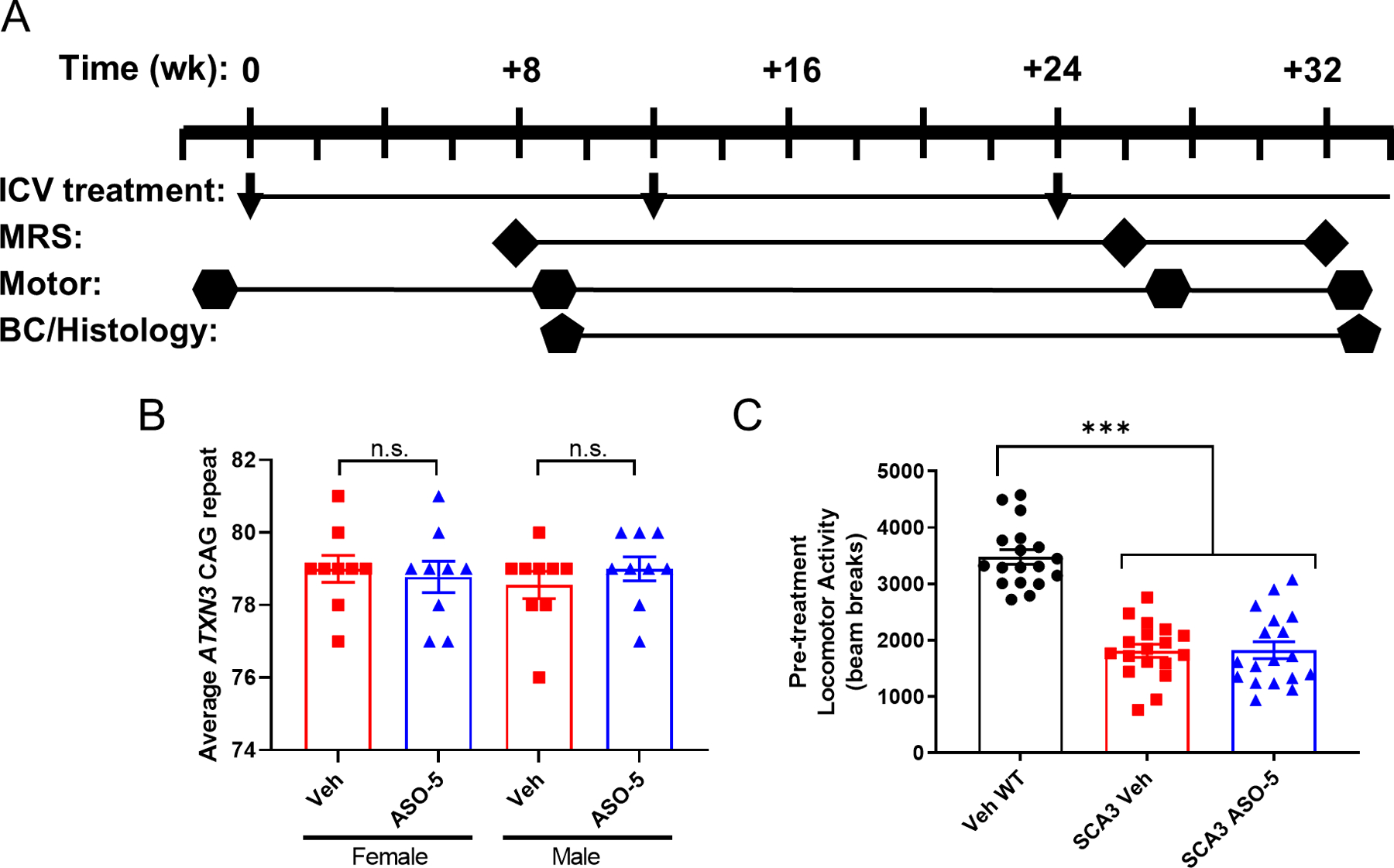
Study overview. (A) Antisense oligonucleotide intracerebroventricular (ASO ICV) treatment was administered every 12 weeks beginning at 7–12 weeks of age. Activity chamber motor assessment was completed 1 week prior to first treatment (n=18 per condition, 9 male/9 female) and repeated 1 week after subsequent magnetic resonance spectroscopy (MRS) scanning at +8 weeks, +26 weeks, and +32 weeks. Interim biochemical (BC) and histological tissue was collected after +9-week motor testing for n=6 mice per condition. Endpoint tissue was collected after last motor testing for n≥5 mice per condition. (B) All SCA3 mice enrolled in the study had an average CAG repeat size of 76 or more, with average CAG repeats not significantly different between vehicle control and ASO-5 treatment groups. Data reported as mean ± SEM. Student’s t-test. n.s.=not significant. (C) All SCA3 mice enrolled in the study exhibited significant motor impairment as defined by open field activity chamber total beam breaks one week prior to first ICV treatment. Data reported as mean ± SEM. One-way ANOVA with post-hoc Sidak test. *** p < 0.001.
Mouse genotyping and CAG repeat size assessment:
Genotyping was performed using tail biopsy DNA isolated prior to weaning and confirmed postmortem, as previously described9. Human ATXN3 CAG-trinucleotide repeat lengths in SCA3 mice were analyzed by gene fragmentation analysis as previously described17, with SCA3 mouse average 76–81 CAG repeats enrolled in this study (Figure 1B). The mean CAG repeats in either vehicle-treated SCA3 (78.78 ± 1.10 standard deviation) versus ASO-treated SCA3 (78.89 ± 1.13 standard deviation) group were not statistically different (students t-test, p=0.7684).
Antisense Oligonucleotides:
The anti-ATXN3 candidate ASO (ASO-5; GCATCTTTTCATTACTGGC) used in this study was previously described7,9 and targets the 3’-untranslated region of both human and mouse ATXN3. Briefly, the ASOs utilize a gapmer design with a central 8 unmodified deoxynucleotides with the native sugar phosphate backbone, flanked on either side by 5 ribonucleotides with a phosphonothioate backbone and 2’-O-methoxyethylribose modifications. Oligonucleotides were synthesized as previously described18,19 and solubilized in phosphate-buffered saline (without Ca2+ or Mg2+).
Intracerebroventricular (ICV) Injections:
Mice 7–12 weeks-of-age, under vaporized isoflurane anesthesia, received their first right lateral ICV bolus injection of 700 µg ASOs diluted in 10 µL phosphate-buffered saline or 10 µL of vehicle (phosphate-buffered saline) using established protocols7,9,20, with repeated injections of 700 µg ASOs or vehicle administered to the same location every 12 weeks until endpoint. Injections were performed with a 26G needle (7758–04; Hamilton). All treatment groups were blinded to the experimenter prior to injection. Following surgery, weight, grooming activity, and home cage activity was recorded for up to 10 days according to ICUCA guidelines.
Animal Preparation for MR Scanning:
Animals were induced with 3% isoflurane in a 1:1 mixture of O₂:N₂O. Mice were secured in a custom-built holder and physiological status was monitored (SA Instruments) and recorded. Anesthesia was maintained with 1.5–2% isoflurane to achieve a respiration rate of 70–100 breaths per minute. Body temperature was maintained at 36–37ºC with a circulating warm water system and a heating fan controlled by feedback received from a fiber-optic rectal thermometer. The scanning time was approximately 1 hour for each animal.
Magnetic Resonance Spectroscopy:
All experiments were performed on a 9.4T/31 cm scanner (Agilent), as described previously21,22. A quadrature surface radio frequency (RF) coil with two geometrically decoupled single turn coils (14 mm diameter) was used as the MR transceiver. The MR operator and data analyst was blinded to the mouse group during data acquisition and analysis. Mice were positioned in the magnet and coronal and sagittal multislice images were obtained using a rapid acquisition with relaxation enhancement (RARE) sequence23 [repetition time (TR)= 4 s, echo train length= 8, echo time (TE)= 60 ms, slice thickness= 1 mm, 7 slices]. Two volumes of interest (VOIs) were studied; one centered on the midline of the cerebellum (6.8 µl, 2.1 × 1.8 × 1.8 mm³) and one centered on the brainstem (11.5 µl, 4.0 × 1.8 × 1.6 mm³). The VOIs were positioned consistently in follow-up scans by using anatomical landmarks. First- and second-order shims were adjusted using FASTMAP with echo-planar readout24. Localized ¹H MR spectra were acquired with a short-echo localization by adiabatic selective refocusing (LASER) sequence [TE= 15 ms, TR= 5 s, 128 transients]25 combined with VAPOR (variable power RF pulses with optimized relaxation delays) water suppression26. Spectra were acquired and saved as single scans. Unsuppressed water spectra acquired from the same VOIs were used as a metabolite quantification reference.
Metabolite quantification:
Single shots were eddy current, frequency, and phase corrected using MRspa software (http://www.cmrr.umn.edu/downloads/mrspa/) before averaging. The contributions of individual metabolites to the averaged spectra were quantified using LCModel27 as described previously22. The following metabolites were included in the basis set: alanine (Ala), ascorbate/vitamin C (Asc), aspartate (Asp), glycerophosphocholine (GPC), phosphocholine (PCho), creatine (Cr), phosphocreatine (PCr), gamma-aminobutyric acid (GABA), glucose (Glc), glutamine (Gln), glutamate (Glu), glutathione (GSH), glycine (Gly), myo-inositol (Ins), lactate (Lac), N-acetylaspartate (NAA), N-acetylaspartylglutamate (NAAG), phosphoethanolamine (PE), scyllo-inositol (Scyllo), taurine (Tau), and macromolecules (MM). The MM spectra were experimentally obtained from a VOI that covered the cerebellum and brainstem using an inversion recovery technique [VOI = 3.5 × 4.3 × 1.8 mm³, TE = 15 ms, TR = 1.5 s, inversion time (TIR) = 579 ms, 1920 transients, N=3]. The model metabolite spectra were generated using density matrix simulations28 with the MATLAB software (MathWorks) based on previously reported chemical shifts and coupling constants29,30. Concentrations with mean Cramér-Rao lower bounds ≤20%22 at +8, +26, or +32 weeks for a given treatment group are reported. If the correlation between two metabolites was consistently high (correlation coefficient <−0.7), their sum is reported rather than the individual values22. Strong negative correlation was found in two cases, so that Cr+PCr (denoted tCr for total creatine) and GPC+PCho (denoted tCho for total choline) are reported.
Motor Assessments:
Pretreatment motor assessments were performed at the University of Michigan in a photobeam activity system (PAS) open field apparatus and post-treatment motor assessments were performed using an AnyMaze activity tracking system at the University of Minnesota Mouse Behavior Core. All motor assessment tasks were assessed for 30-minute trials on days when no other testing occurred, by an experimenter blinded to mouse grouping. Total activity on the PAS open field apparatus was assessed by the total x/y-axis beam breaks. Total activity on the AnyMaze activity tracking system was calculated in total meters traveled over time. Body weights were recorded prior to behavior testing and injection time points.
Immunoblotting:
Protein lysates from macrodissected brainstem and cerebellar tissue were processed as previously described7. Total protein lysates (40 μg) were resolved in 4–20% gradient sodium dodecyl sulfate–polyacrylamide electrophoresis gels and transferred to 0.45 μm nitrocellulose membranes for overnight incubation at 4°C with either mouse anti-ATXN3 (1H9; 1:1,000, MAB5360; Millipore, Billerica, MA) or mouse anti-GAPDH (1: 10,000), MAB374; Millipore, Billerica, MA). Bound primary antibodies were visualized by incubation with peroxidase-conjugated anti-mouse secondary antibody (1:10,000; Jackson ImmunoResearch Laboratories, West Grove, PA), followed by treatment with ECL-plus reagent (Western Lighting; PerkinElmer, Waltham, MA) and exposure to autoradiography films. Band intensities were quantified using ImageJ analysis software (NIH, Bethesda, MD).
Immunohistochemistry:
Whole brains perfused with PBS were processed as previously described9. Left hemisphere ATXN3 nuclear histological evaluation was completed in ASO-treated samples relative to controls using the mouse anti-ATXN3 (1H9; 1:1,000, MAB5360, Millipore) primary antibody. Imaging was performed using a Nikon-A1 confocal microscope (Melville, NY) in the basilar pontine nuclei (denoted as pons) and deep cerebellar nuclei (DCN). Nuclear ATXN3 accumulation was quantified as previously described7.
Statistical Methods:
Repeated measures ANOVAs for group by time, also adding in a random litter effect, were implemented using linear mixed models for each outcome of interest. Tests of interest include all pairwise comparisons among the 3 groups at each time point, and comparisons across time points within group. Sidak step-down procedures were used to correct type I error for multiple comparisons. Pearson correlation coefficients were reported to define strength of a relationship between notable quantitative variables.
RESULTS
Select cerebellum and brainstem neurochemical abnormalities are fully or partially reversed in SCA3 mice after ASO treatment.
In this study, we sought to define early and progressive metabolite changes in vulnerable SCA3 brain regions. We compared the neurometabolic signatures of SCA3 anti-ATXN3 ASO-treated mice to sex- and age-matched SCA3 vehicle-treated and wildtype vehicle-treated control littermates (n=17–19 mice/group). Of the 54 mice scanned, two mice were identified as having the abnormally high-glutamine phenotype, a consequence of portosystemic shunting that was previously characterized in C57BL/6 mice31, and were excluded from analysis. As noted in the methods, mice received their first stereotaxic ICV injection to the right lateral ventricle at 7–12 weeks old and were repeat treated every 12 weeks until the study endpoint (Fig 1). Our previously published anti-ATXN3 ASO studies established the greatest reduction in ATXN3 mRNA expression 8 weeks after treatment. Therefore, we scanned the mice +8, +26 and +32 weeks after the first treatments (Fig 1), allowing for two MRS analyses captured at 8-weeks after ASO treatment and one MRS analysis captured 2-weeks after ASO treatment. Overall, MR spectra yielded 15–16 reliably quantified metabolites in cerebellum (Table 1) and brainstem (Table 2). Spectral quality, as assessed by signal-to-noise ratio, linewidth (Tables 1 & 2), and water/artifact suppression, was high and comparable across groups (Fig 2 & 3, B-D). Multislice T2-weighted images (Fig 2A and 3A) did not indicate overt atrophy in SCA3 (ASO-5 and Veh) mice compared to WT Veh mice, hence the MRS VOI did not contain additional cerebrospinal fluid, which would have reduced all metabolite concentrations in SCA3 vs. WT mice. Therefore, the group differences observed in the temporal and regional neurochemical profiles in this study could not be attributed to the group differences in age, sex, CAG repeat size, atrophy, or spectral quality.
Table 1:
Neurochemical concentrations (µmol/g, mean ± SD) and spectral quality measures estimated by LCModel (mean ± SD) in the cerebellum of mice. Group means were compared with one-way ANOVA with Sidak adjustment for multiple comparisons. Bolded p < 0.05. SNR*= signal-to-noise ratio.
| CEREBELLUM +8 WEEKS | Sidak p-value | |||||
|---|---|---|---|---|---|---|
| SCA3 Veh (n=18) | SCA3 ASO-5 (n=18) | WT Veh (n=18) | SCA3 Veh v. WT Veh | SCA3 ASO-5 v. SCA3 Veh | SCA3 ASO-5 v. WT Veh | |
| Asc | 2.20 ± 0.39 | 2.13 ± 0.27 | 1.96 ± 0.41 | 0.470 | 1.000 | 0.723 |
| GABA | 1.87 ± 0.34 | 1.92 ± 0.33 | 1.96 ± 0.45 | 1.000 | 1.000 | 0.999 |
| Glc | 2.73 ± 0.85 | 2.94 ± 0.97 | 2.78 ± 1.13 | 1.000 | 0.998 | 0.998 |
| Gln | 4.28 ± 0.48 | 3.75 ± 0.39 | 2.87 ± 0.42 | 0.000 | 0.039 | 0.000 |
| Glu | 7.55 ± 0.42 | 7.68 ± 0.47 | 7.67 ± 0.39 | 0.998 | 1.000 | 1.000 |
| GSH | 1.07 ± 0.18 | 1.03 ± 0.20 | 1.08 ± 0.22 | 1.000 | 1.000 | 0.988 |
| Ins | 6.49 ± 0.36 | 6.31 ± 0.41 | 5.95 ± 0.45 | 0.006 | 0.950 | 0.223 |
| Lac | 2.87 ± 0.36 | 2.66 ± 0.51 | 2.63 ± 0.61 | 0.686 | 0.790 | 1.000 |
| NAA | 6.67 ± 0.24 | 6.86 ± 0.27 | 7.17 ± 0.26 | 0.000 | 0.428 | 0.013 |
| NAAG | 0.57 ± 0.14 | 0.58 ± 0.14 | 0.53 ± 0.19 | 1.000 | 1.000 | 0.993 |
| Tau | 7.47 ± 0.37 | 7.96 ± 0.44 | 7.84 ± 0.52 | 0.053 | 0.022 | 1.000 |
| PCho+GPC | 1.53 ± 0.18 | 1.73 ± 0.19 | 1.70 ± 0.16 | 0.021 | 0.012 | 1.000 |
| Cr+PCr | 11.40 ± 0.30 | 11.04 ± 0.46 | 10.68 ± 0.61 | 0.000 | 0.024 | 0.426 |
| NAA+NAAG | 7.24 ± 0.27 | 7.44 ± 0.30 | 7.71 ± 0.31 | 0.001 | 0.353 | 0.108 |
| Glu+Gln | 11.83 ± 0.67 | 11.43 ± 0.63 | 10.54 ± 0.51 | 0.000 | 0.144 | 0.001 |
| Glc+Tau | 10.19 ± 0.98 | 10.90 ± 0.91 | 10.62 ± 1.39 | 0.900 | 0.228 | 0.986 |
| SNR* | 25.67 ± 2.28 | 26.28 ± 2.47 | 23.67 ± 2.85 | |||
| Linewidth (Hz) | 16.28 ± 1.41 | 15.06 ± 0.86 | 15.86 ± 0.88 | |||
| CEREBELLUM +26 WEEKS | Sidak p-value | |||||
| SCA3 Veh (n=11) | SCA3 ASO-5 (n=10) | WT Veh (n=11) | SCA3 Veh v. WT Veh | SCA3 ASO-5 v. SCA3 Veh | SCA3 ASO-5 v. WT Veh | |
| Asc | 2.03 ± 0.45 | 1.81 ± 0.40 | 2.10 ± 0.58 | 0.996 | 0.916 | 0.398 |
| GABA | 2.01 ± 0.22 | 1.80 ± 0.24 | 1.69 ± 0.33 | 0.130 | 0.852 | 0.884 |
| Glc | 3.20 ± 1.27 | 3.57 ± 0.82 | 3.34 ± 1.12 | 1.000 | 1.000 | 0.998 |
| Gln | 4.74 ± 0.93 | 4.23 ± 0.45 | 2.88 ± 0.38 | 0.000 | 0.346 | 0.000 |
| Glu | 7.63 ± 0.63 | 8.04 ± 0.38 | 7.87 ± 0.34 | 0.993 | 0.595 | 0.968 |
| GSH | 0.96 ± 0.17 | 1.01 ± 0.17 | 1.07 ± 0.23 | 0.999 | 1.000 | 1.000 |
| Ins | 5.98 ± 0.46 | 5.84 ± 0.82 | 5.89 ± 0.35 | 0.988 | 0.932 | 1.000 |
| Lac | 2.52 ± 0.60 | 1.92 ± 0.53 | 2.70 ± 0.46 | 0.992 | 0.052 | 0.025 |
| NAA | 6.60 ± 0.28 | 6.78 ± 0.25 | 7.14 ± 0.35 | 0.008 | 0.778 | 0.325 |
| NAAG | 0.56 ± 0.16 | 0.66 ± 0.17 | 0.59 ± 0.08 | 1.000 | 0.850 | 0.998 |
| Tau | 7.44 ± 0.46 | 8.10 ± 0.62 | 8.26 ± 0.54 | 0.000 | 0.024 | 0.880 |
| PCho+GPC | 1.39 ± 0.17 | 1.77 ± 0.14 | 1.66 ± 0.15 | 0.030 | 0.000 | 0.576 |
| Cr+PCr | 11.03 ± 0.37 | 10.59 ± 0.48 | 10.83 ± 0.55 | 0.985 | 0.016 | 0.228 |
| NAA+NAAG | 7.16 ± 0.27 | 7.44 ± 0.20 | 7.73 ± 0.40 | 0.018 | 0.327 | 0.767 |
| Glu+Gln | 12.37 ± 0.99 | 12.27 ± 0.44 | 10.74 ± 0.37 | 0.000 | 1.000 | 0.000 |
| Glc+Tau | 10.64 ± 1.22 | 11.66 ± 0.97 | 11.60 ± 1.17 | 0.440 | 0.275 | 1.000 |
| SNR* | 22.91 ± 3.65 | 23.80 ± 2.25 | 24.36 ± 4.06 | |||
| Linewidth (Hz) | 17.44 ± 1.19 | 15.82 ± 0.97 | 16.10 ± 0.67 | |||
| CEREBELLUM +32 WEEKS | Sidak p-value | |||||
| SCA3 Veh (n=11) | SCA3 ASO-5 (n=10) | WT Veh (n=11) | SCA3 Veh v. WT Veh | SCA3 ASO-5 v. SCA3 Veh | SCA3 ASO-5 v. WT Veh | |
| Asc | 1.64 ± 0.44 | 1.78 ± 0.14 | 1.87 ± 0.46 | 1.000 | 1.000 | 1.000 |
| GABA | 1.94 ± 0.28 | 1.97 ± 0.21 | 1.89 ± 0.18 | 1.000 | 1.000 | 0.999 |
| Glc | 2.55 ± 0.42 | 2.69 ± 0.69 | 2.77 ± 0.46 | 1.000 | 1.000 | 1.000 |
| Gln | 4.95 ± 0.40 | 5.63 ± 0.80 | 2.82 ± 0.29 | 0.000 | 0.625 | 0.000 |
| Glu | 7.38 ± 0.50 | 7.71 ± 0.75 | 8.18 ± 0.36 | 0.458 | 0.321 | 1.000 |
| GSH | 1.04 ± 0.08 | 0.84 ± 0.15 | 1.22 ± 0.14 | 0.865 | 0.625 | 0.075 |
| Ins | 5.48 ± 0.57 | 4.79 ± 0.43 | 5.71 ± 0.22 | 1.000 | 0.320 | 0.179 |
| Lac | 2.28 ± 0.25 | 1.64 ± 0.47 | 2.86 ± 0.60 | 0.278 | 0.632 | 0.010 |
| NAA | 6.47 ± 0.25 | 6.81 ± 0.47 | 7.22 ± 0.21 | 0.006 | 0.061 | 0.965 |
| NAAG | 0.54 ± 0.18 | 0.66 ± 0.11 | 0.53 ± 0.12 | 1.000 | 0.967 | 0.998 |
| Tau | 7.22 ± 0.65 | 7.77 ± 0.33 | 7.90 ± 0.31 | 0.388 | 0.452 | 1.000 |
| PCho+GPC | 1.29 ± 0.09 | 1.61 ± 0.29 | 1.78 ± 0.16 | 0.003 | 0.007 | 1.000 |
| Cr+PCr | 11.09 ± 0.38 | 10.30 ± 0.31 | 10.70 ± 0.66 | 0.615 | 0.013 | 0.594 |
| NAA+NAAG | 7.01 ± 0.17 | 7.47 ± 0.42 | 7.75 ± 0.24 | 0.017 | 0.022 | 1.000 |
| Glu+Gln | 12.34 ± 0.81 | 13.34 ± 0.85 | 11.00 ± 0.50 | 0.025 | 0.031 | 0.000 |
| Glc+Tau | 9.77 ± 1.02 | 10.46 ± 0.96 | 10.66 ± 0.75 | 0.965 | 0.981 | 1.000 |
| SNR* | 27.00 ± 2.00 | 25.20 ± 3.77 | 24.40 ± 2.97 | |||
| Linewidth (Hz) | 16.71 ± 0.63 | 15.53 ± 0.50 | 16.90 ± 0.98 | |||
Table 2:
Neurochemical concentrations (µmol/g, mean ± SD) and spectral quality measures estimated by LCModel (mean ± SD) in the brainstem of mice. Group means were compared with one-way ANOVA with Sidak adjustment for multiple comparisons. Bolded p < 0.05. SNR*= signal-to-noise ratio.
| BRAINSTEM +8 WEEKS | Sidak p-value | |||||
|---|---|---|---|---|---|---|
| SCA3 Veh (n=18) | SCA3 ASO-5 (n=18) | WT Veh (n=18) | SCA3 Veh v. WT Veh | SCA3 ASO-5 v. SCA3 Veh | SCA3 ASO-5 v. WT Veh | |
| Asp | 3.11 ± 0.89 | 3.22 ± 1.16 | 2.61 ± 0.85 | 0.784 | 0.997 | 0.999 |
| GABA | 2.10 ± 0.40 | 2.15 ± 0.41 | 2.15 ± 0.46 | 1.000 | 1.000 | 1.000 |
| Gln | 3.18 ± 0.48 | 2.99 ± 0.64 | 2.09 ± 0.59 | 0.000 | 0.976 | 0.001 |
| Glu | 5.37 ± 0.48 | 5.54 ± 0.76 | 5.33 ± 0.47 | 1.000 | 0.927 | 0.991 |
| Gly | 3.73 ± 0.54 | 3.99 ± 0.60 | 3.93 ± 0.44 | 0.820 | 0.925 | 1.000 |
| Ins | 6.10 ± 0.61 | 6.19 ± 0.79 | 5.75 ± 0.68 | 0.961 | 0.986 | 0.486 |
| Lac | 3.68 ± 0.49 | 4.53 ± 0.84 | 4.70 ± 0.85 | 0.020 | 0.017 | 1.000 |
| NAA | 7.48 ± 0.31 | 7.75 ± 0.50 | 7.81 ± 0.58 | 0.579 | 0.636 | 1.000 |
| NAAG | 0.80 ± 0.22 | 1.07 ± 0.18 | 0.90 ± 0.34 | 0.994 | 0.026 | 0.401 |
| Tau | 2.98 ± 0.51 | 2.85 ± 0.33 | 2.54 ± 0.49 | 0.047 | 0.993 | 0.073 |
| PCho+GPC | 1.01 ± 0.17 | 1.40 ± 0.19 | 1.20 ± 0.21 | 0.008 | 0.000 | 0.020 |
| Cr+PCr | 6.98 ± 0.30 | 7.09 ± 0.46 | 6.74 ± 0.72 | 0.671 | 0.986 | 0.299 |
| NAA+NAAG | 8.28 ± 0.42 | 8.82 ± 0.59 | 8.71 ± 0.63 | 0.395 | 0.077 | 1.000 |
| Glu+Gln | 8.55 ± 0.73 | 8.53 ± 1.17 | 7.42 ± 0.76 | 0.004 | 1.000 | 0.025 |
| Glc+Tau | 5.35 ± 1.20 | 5.60 ± 1.52 | 5.08 ± 1.28 | 0.999 | 0.999 | 0.907 |
| SNR* | 18.44 ± 2.38 | 17.88 ± 2.23 | 16.24 ± 2.41 | |||
| Linewidth (Hz) | 16.51 ± 1.65 | 16.94 ± 1.15 | 17.27 ± 0.88 | |||
| BRAINSTEM +26 WEEKS | Sidak p-value | |||||
| SCA3 Veh (n=11) | SCA3 ASO-5 (n=10) | WT Veh (n=11) | SCA3 Veh v. WT Veh | SCA3 ASO-5 v. SCA3 Veh | SCA3 ASO-5 v. WT Veh | |
| Asp | 2.81 ± 1.14 | 3.19 ± 1.24 | 3.46 ± 1.29 | 1.000 | 1.000 | 1.000 |
| GABA | 1.98 ± 0.46 | 2.19 ± 0.37 | 2.44 ± 0.88 | 0.994 | 0.750 | 1.000 |
| Gln | 3.60 ± 0.56 | 2.89 ± 0.39 | 2.00 ± 0.77 | 0.000 | 0.076 | 0.253 |
| Glu | 5.32 ± 0.60 | 5.41 ± 0.50 | 5.08 ± 0.78 | 0.951 | 1.000 | 0.979 |
| Gly | 3.95 ± 0.60 | 3.96 ± 0.51 | 3.88 ± 0.47 | 0.934 | 1.000 | 0.756 |
| Ins | 5.25 ± 0.51 | 6.13 ± 0.50 | 5.85 ± 0.48 | 0.407 | 0.054 | 0.969 |
| Lac | 2.80 ± 0.52 | 3.48 ± 0.74 | 4.15 ± 1.30 | 0.038 | 0.712 | 0.956 |
| NAA | 7.60 ± 0.47 | 7.92 ± 0.44 | 8.19 ± 0.84 | 0.816 | 0.489 | 1.000 |
| NAAG | 0.88 ± 0.20 | 1.03 ± 0.21 | 0.90 ± 0.35 | 1.000 | 0.491 | 0.840 |
| Tau | 3.10 ± 0.72 | 2.88 ± 0.36 | 2.99 ± 0.52 | 0.859 | 0.960 | 1.000 |
| PCho+GPC | 1.03 ± 0.22 | 1.17 ± 0.18 | 1.33 ± 0.16 | 0.017 | 0.297 | 0.960 |
| Cr+PCr | 7.01 ± 0.39 | 6.87 ± 0.53 | 6.95 ± 0.63 | 0.990 | 1.000 | 1.000 |
| NAA+NAAG | 8.47 ± 0.46 | 8.95 ± 0.54 | 9.01 ± 0.86 | 0.820 | 0.232 | 1.000 |
| Glu+Gln | 8.93 ± 0.68 | 8.30 ± 0.79 | 7.08 ± 1.06 | 0.001 | 0.642 | 0.440 |
| Glc+Tau | 6.12 ± 1.34 | 5.72 ± 1.23 | 6.56 ± 0.91 | 1.000 | 1.000 | 1.000 |
| SNR* | 18.27 ± 3.38 | 18.10 ± 2.18 | 16.00 ± 3.03 | |||
| Linewidth (Hz) | 15.78 ± 0.72 | 16.39 ± 0.86 | 16.64 ± 0.67 | |||
| BRAINSTEM +32 WEEKS | Sidak p-value | |||||
| SCA3 Veh (n=11) | SCA3 ASO-5 (n=10) | WT Veh (n=11) | SCA3 Veh v. WT Veh | SCA3 ASO-5 v. SCA3 Veh | SCA3 ASO-5 v. WT Veh | |
| Asp | 2.26 ± 0.85 | 3.36 ± 0.48 | 2.82 ± 0.83 | 0.999 | 0.691 | 0.978 |
| GABA | 1.99 ± 0.20 | 2.14 ± 0.50 | 2.01 ± 0.40 | 1.000 | 1.000 | 1.000 |
| Glc | 2.69 ± 1.33 | 3.03 ± 0.59 | 2.98 ± 0.97 | 1.000 | 1.000 | 1.000 |
| Gln | 3.44 ± 0.33 | 4.05 ± 0.50 | 1.84 ± 0.57 | 0.010 | 0.775 | 0.000 |
| Glu | 5.28 ± 0.32 | 5.64 ± 0.70 | 4.82 ± 0.52 | 0.865 | 0.994 | 0.524 |
| Gly | 3.75 ± 0.39 | 4.14 ± 0.75 | 3.76 ± 0.20 | 1.000 | 0.592 | 0.399 |
| Ins | 5.70 ± 1.04 | 5.48 ± 0.51 | 5.53 ± 0.46 | 0.990 | 0.992 | 1.000 |
| Lac | 2.86 ± 0.29 | 3.41 ± 0.94 | 3.75 ± 1.15 | 0.178 | 0.570 | 1.000 |
| NAA | 7.69 ± 0.22 | 7.96 ± 0.29 | 7.54 ± 0.69 | 1.000 | 0.997 | 1.000 |
| NAAG | 0.74 ± 0.23 | 1.21 ± 0.50 | 1.05 ± 0.16 | 0.471 | 0.046 | 0.992 |
| Tau | 2.93 ± 0.54 | 2.53 ± 0.39 | 2.71 ± 0.40 | 1.000 | 0.851 | 0.891 |
| PCho+GPC | 0.91 ± 0.08 | 1.28 ± 0.10 | 1.34 ± 0.17 | 0.037 | 0.024 | 1.000 |
| Cr+PCr | 7.26 ± 0.40 | 7.04 ± 0.45 | 6.96 ± 0.45 | 0.999 | 0.927 | 1.000 |
| NAA+NAAG | 8.43 ± 0.43 | 9.17 ± 0.72 | 8.59 ± 0.68 | 0.989 | 0.415 | 0.993 |
| Glu+Gln | 8.73 ± 0.49 | 9.69 ± 0.90 | 6.66 ± 0.91 | 0.013 | 0.832 | 0.003 |
| Glc+Tau | 5.62 ± 1.24 | 5.57 ± 0.49 | 5.70 ± 0.89 | 1.000 | 1.000 | 1.000 |
| SNR* | 21.75 ± 2.06 | 19.40 ± 1.14 | 18.20 ± 2.86 | |||
| Linewidth (Hz) | 15.78 ± 1.27 | 16.98 ± 0.87 | 17.75 ± 0.88 | |||
Figure 2.
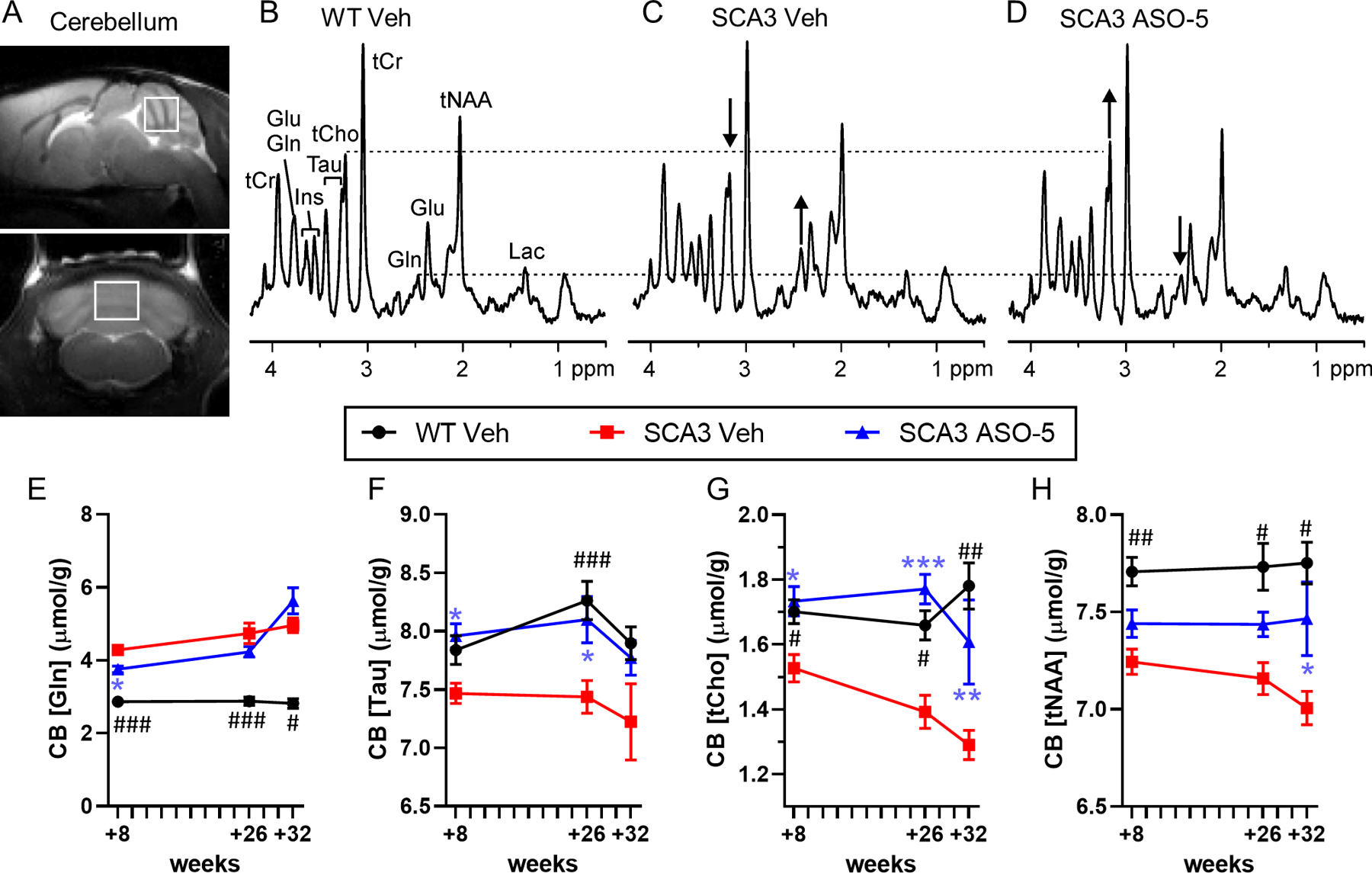
Select cerebellar neurochemical abnormalities are fully or partially reversed towards wild type (WT) concentrations in SCA3 mice after ASO-5 treatment. (A) Representative +26 week midsagittal and coronal T2-weighted images showing voxel position for acquired spectra. Localized proton magnetic resonance spectra [LASER sequence, TE= 15 ms, TR= 5 s, 128 transients, 9.4T scanner] from a (B) WT vehicle-treated mouse, (C) SCA3 vehicle-treated mouse, and (D) SCA3 ASO-5 treated mouse all at +26 weeks. Differences in glutamine (Gln) and total choline (tCho) concentrations in the SCA3 vehicle-treated relative to WT vehicle-treated mouse and the reversal of these in the SCA3 ASO-5 treated mouse are shown with arrows. Time course of (E) glutamine, (F) taurine, (G) total choline, and (H) total NAA in the cerebellum of WT vehicle-treated (circle), SCA3 vehicle-treated (square), and SCA3 ASO-5 treated (triangle) mice. Group size per genotype ranged from n=17–18 at +8 weeks (n≥8 male, n≥8 female), n=10–11 at +26 weeks (n≥4 male, n≥4 female), and n=4–5 at +32 weeks (n≥2 male, n≥2 female). Data reported as mean ± SEM. Two-way ANOVA mixed-effects analysis. Post-hoc Sidak multiple comparisons p-value denoted as # between WT and SCA3 vehicle-treated mice and * between SCA3 ASO-5 and SCA3 vehicle-treated mice. # or * p < 0.05; ## or ** p < 0.01; ### or *** p < 0.001.
Figure 3.
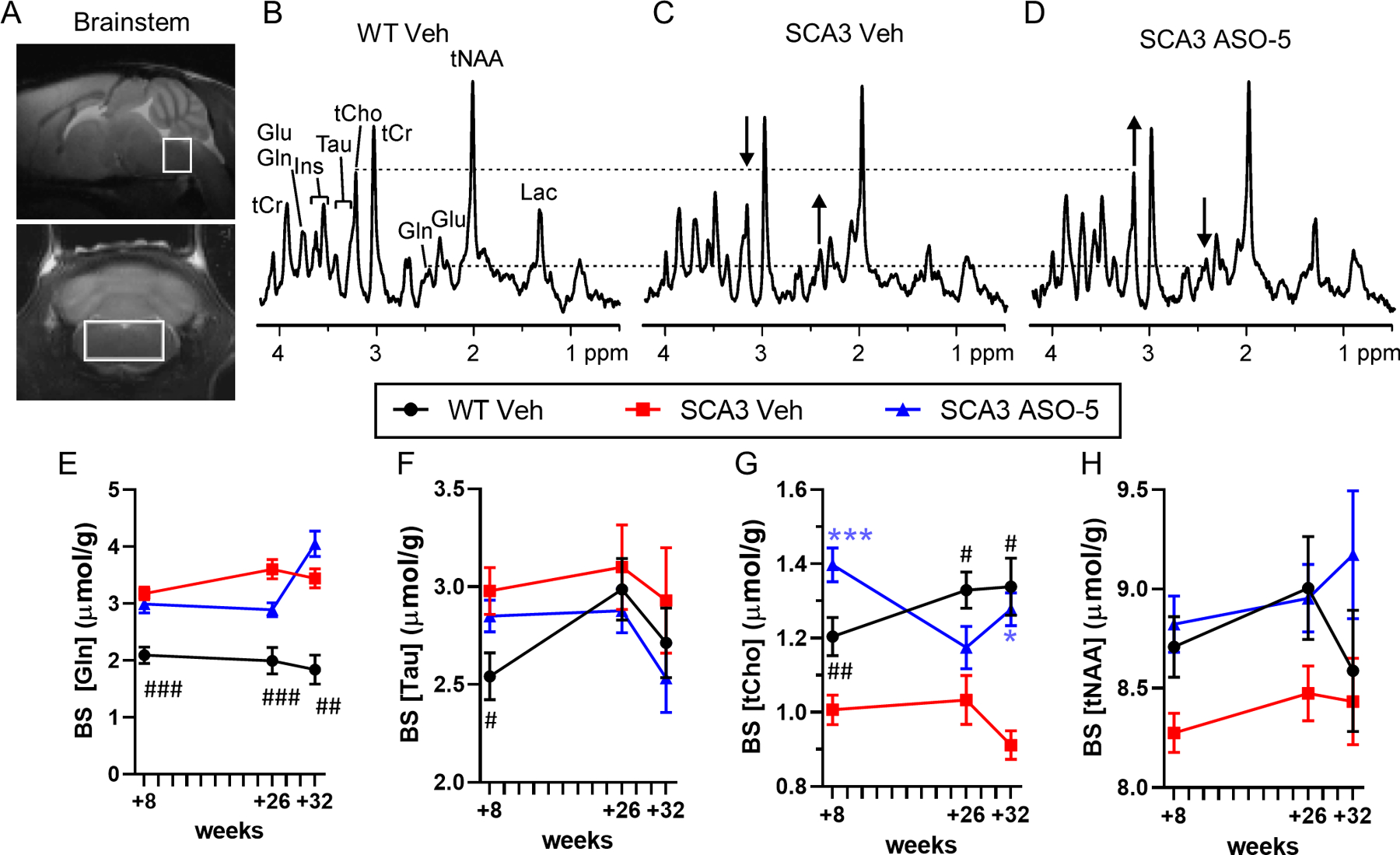
Select brainstem neurochemical abnormalities are fully or partially reversed towards wild type (WT) concentrations in SCA3 mice after ASO-5 treatment. (A) Representative +26-weekmidsagittal and coronal T2-weighted images showing voxel position for acquired spectra. Localized proton magnetic resonance spectra [LASER sequence, TE= 15 ms, TR= 5 s, 128 transients, 9.4T scanner] from a (B) WT vehicle-treated mouse, (C) SCA3 vehicle-treated mouse, and (D) SCA3 ASO-5 treated mouse all at +26 weeks. Differences in glutamine (Gln) and total choline (tCho) concentrations in the SCA3 vehicle-treated relative to WT vehicle-treated mouse and the reversal of these in the SCA3 ASO-5 treated mouse are shown with arrows. Time course of (E) glutamine, (F) taurine, (G) total choline, and (H) total NAA in the brainstem of WT vehicle-treated (circle), SCA3 vehicle-treated (square), and SCA3 ASO-5 treated (triangle) mice. Group size per genotype ranged from n=17–18 at +8 weeks (n≥8 male, n≥8 female), n=10–11 at +26 weeks (n≥4 male, n≥4 female), and n=4–5 at +32 weeks (n≥2 male, n≥2 female). Data reported as mean ± SEM. Two-way ANOVA mixed-effects analysis. Post-hoc Sidak multiple comparisons p-value denoted as # between WT and SCA3 vehicle-treated mice and * between SCA3 ASO-5 and SCA3 vehicle-treated mice. # or * p < 0.05; ## or ** p < 0.01; ### or *** p < 0.001.
MR spectra in the cerebellar vermis of SCA3 vehicle-treated mice yielded significantly higher glutamine compared to WT vehicle-treated mice that was partially rescued by ASO-5 treatment at +8 weeks, but not +26 or +32 weeks (Fig 2E). Cerebellar vermis taurine levels were lower in SCA3 vehicle-treated mice compared to WT mice at the +26-week time point, but not statistically different at either the +8- or +32-weeks timepoint (Fig 2F). SCA3 vehicle-treated mice exhibited significantly lower tCho (Fig 2G) and tNAA (Fig 2H) levels compared to WT vehicle-treated mice at all time points. SCA3 ASO-5 treated mice showed significant rescue of cerebellar taurine (Fig 2F) and tCho at +8 and +26 weeks and partial rescue of tCho at the +32-week timepoint (Fig 2G). The lower levels of tNAA in SCA3 vehicle-treated mice were significantly rescued in ASO-5 treated SCA3 mice at +26 weeks and partially rescued at +32 weeks (Fig 2H).
MR spectra in the brainstem of SCA3 vehicle-treated mice yielded significantly higher glutamine (Fig 3E) and significantly lower tCho (Fig 3G) compared to WT vehicle-treated mice at all time points. SCA3 vehicle-treated mice additionally exhibited higher taurine and lower tNAA levels than WT vehicle-treated mice, with taurine significantly higher at the +8-week timepoint (Fig 3F). SCA3 ASO-5 treated mice showed significant rescue of brainstem tCho at +8 and +32 weeks (Fig 3G) as well as partial rescue at +26 weeks for glutamine (Fig 3E) and full rescue of tNAA at +8- and +26-weeks (Fig 3H).
MRS correlations with regional ATXN3 protein expression
Because the gapmer anti-ATXN3 ASO directly targets the transcript to degrade it through an RNase-H cleavage mechanism, we confirmed the efficacy of repeat ASO-treatment in this study by immunoblotting for ATXN3 protein expression in cohort sacrificed at +9-weeks and at +33-weeks, both time points 9-weeks after ICV injection of Veh or ASO-treatment (Fig 1A, Fig 4). Consistent with previous reports of anti-ATXN3 ASO in vivo treatment in SCA3 mice7, we see striking reduction of high molecular weight ATXN3 (HMW ATXN3) in brainstem tissue at +9 weeks that is mirrored after repeat ASO treatment in +33-week tissue compared to SCA3 vehicle-treated controls (Fig 4A, 4B). Repeat ASO treatment also partially rescued HMW ATXN3 levels in cerebellum, although to a lesser extent than seen in the brainstem (Fig 4A, 4B). Quantification of the brainstem and cerebellar soluble mutant ATXN3 (mutATXN3) monomer was also reduced to ~50% of the SCA3 vehicle-treated animals (Fig 4C). Additionally, there is a significant reduction of endogenous ATXN3 levels upon ASO treatment in both brain regions and across both timepoints assessed (Fig 4D). When comparing the HMW ATXN3 and mutATXN3 protein knockdown levels to the neurochemical changes, only tCho was statistically significant by Pearson correlation coefficient in brainstem tissue at +33 weeks (Fig 4E, 4F).
Figure 4.
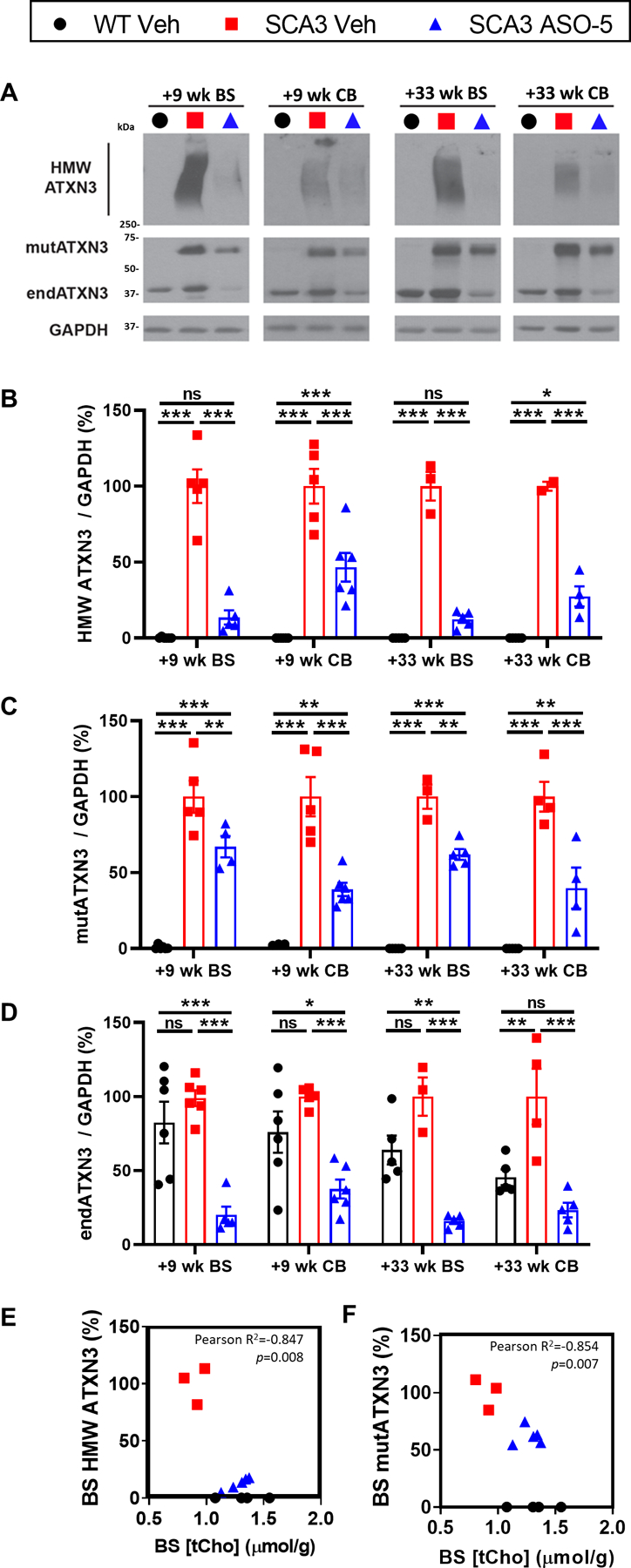
Brainstem ASO silencing of ATXN3 protein in SCA3 treated mice correlates with brain-stem total Choline neurochemical changes. (A) Representative immunoblots and quantification of (B) high molecular weight ATXN3, (C) soluble mutant ATXN3, and (D) endogenous mouse ATXN3 in the brainstem and cerebellum at +9 weeks and +33 weeks relative to first treatment. Data (mean ± SEM) are reported relative to vehicle-treated SCA3 samples. Group size per genotype ranged from n=5–6 (n≥2 male, n≥2 female) at +9 weeks and n=2–5 (n≥1 male, n≥1 female) at +33 weeks. Two-way ANOVA with a post hoc Sidak multiple comparisons test (*p<0.05; **p<0.01; ***p<0.001). Plots showing Pearson cor-relations of levels of +32-week brainstem total choline levels with timepoint-corresponding (E) brainstem HWM ATXN3 or (F) brainstem mutant ATXN3 expression in SCA3 Veh and SCA3 ASO-5Veh WT samples plotted for total choline expression reference levels. Pearson correlation p<0.05 bolded.
MRS correlations with regional nuclear accumulation in the cerebellum
Another hallmark of SCA3 disease is the neuropathological accumulation of ATXN3 in nuclei. We previously showed that anti-ATXN3 ASO therapy can reduce ATXN3 nuclear accumulation in the SCA3 vulnerable nuclei of both the DCN and pons7. Consistent with these previous findings, immunofluorescence quantification revealed that SCA3 vehicle-treated mice exhibited significantly increased total nuclear integrated ATXN3 accumulation in both the pons (Fig 5A, 5B) and DCN (Fig 5A, 5C), which was rescued by ASO-5 treatment. To correlate these findings with the neurometabolite changes found by MRS, we plotted tNAA and tCho against nuclear ATXN3 accumulation levels at corresponding timepoints. Total choline levels in the brainstem were not significantly correlated with nuclear ATXN3 accumulation in the pons (Fig 5D). tNAA levels in the brainstem (Fig 5E) and cerebellum (Fig 5G) showed no significant correlation with nuclear ATXN3 levels in the pons and DCN, respectively. Only a significant negative correlation between tCho levels in the cerebellum and nuclear ATXN3 accumulation in the DCN was found (Fig 5F).
Figure 5.
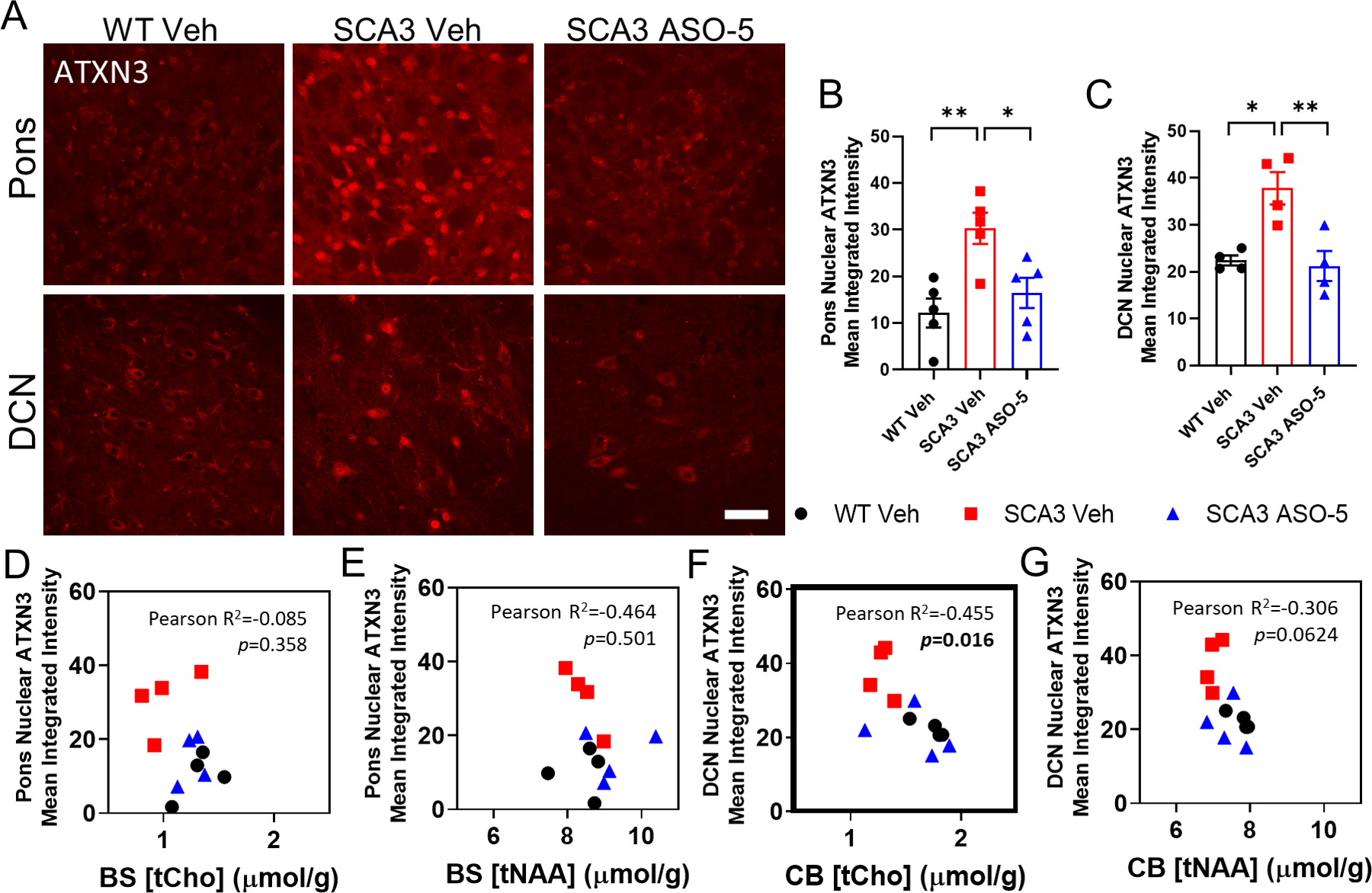
Longitudinal ASO silencing reduces ATXN3 nuclear accumulation in SCA3 treated mice. (A) Representative anti-ATXN3 immunofluorescent images in pons and DCN of vehicle-treated WT, ve-hicle- and ASO-treated SCA3 mice at +33 weeks after initial treatment. Scale bar, 25 μm. Quantification of total ATXN3 nuclear accumulation in (B) pons and (C) DCN at +33 weeks. Data (mean ± SEM) are reported (n=4–5 mice per condition, n≥2 male and n≥2 female 2–3 images per mouse). One-way ANOVA with a post-hoc Sidak test (*p<0.05; **p<0.01; ***p<0.001). Plots showing Pearson correlations of corre-sponding timepoints of either pons or DCN nuclear ATXN3 mean integrated intensity with (D, F) total choline or (E, G) total NAA, respectively (n=4 mice per condition). Pearson correlation p<0.05 bolded.
MRS correlations with motor impairment in cerebellum and brainstem
SCA3 disease is characterized by progressive loss of motor control, a phenotype that is recapitulated in mouse models of SCA3 and a key clinical target and indicator of anti-ATXN3 ASO treatment efficacy. Here, we assessed the motor phenotype of WT and SCA3 mice via an open-field activity test, with motor activity measured by total distance traveled (Fig 6). SCA3 vehicle-treated mice traveled significantly less distance compared to WT vehicle-treated mice at both the pre-treatment stage (Fig 1C) and at all study timepoints, and exhibited significantly progressive motor impairments with age (Fig 6A). Treatment with ASO-5 significantly rescued motor activity of SCA3 mice to WT levels at all study timepoints (Fig 6A). As both motor impairment and MRS neurochemical abnormalities characterize SCA3 disease and could serve as clinical markers of ASO therapy success in patients, correlations between motor activity and neurometabolite levels were assessed. At the +26-week time point, tCho in both the brainstem (Fig 6B) and cerebellum (Fig 6C) was correlated with corresponding 1-week subsequent motor activity. Additionally, at corresponding timepoints, total distance traveled was positively correlated with tCho levels in the brainstem at +32 weeks (Fig 6B) and with tNAA levels in the cerebellum at +8 and +26 weeks (Fig 6E).
Figure 6.
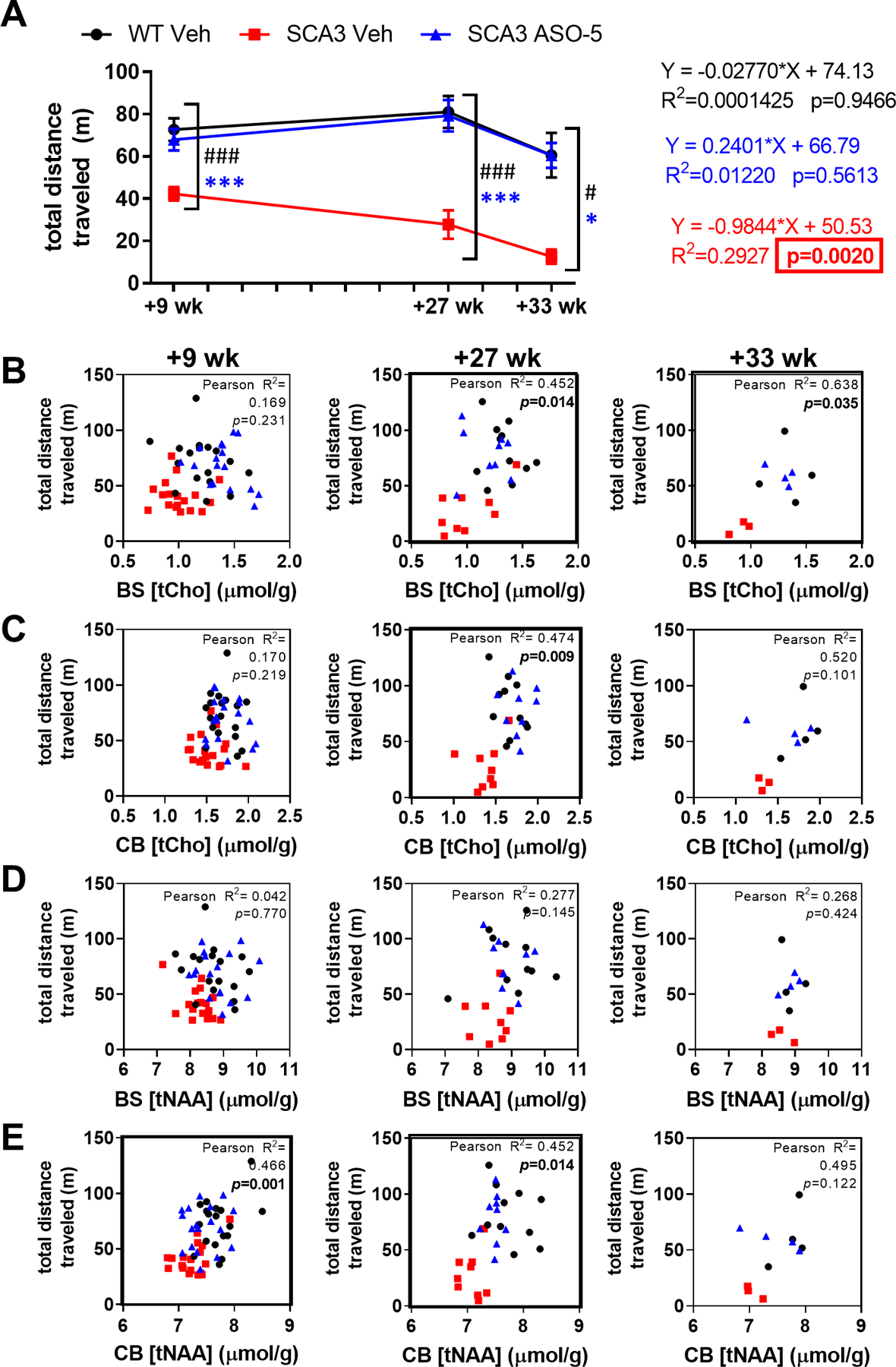
Longitudinal rescue of motor behavior in SCA3 ASO-treated mice correlates with brainstem and cerebellar neurochemical rescue. (A) Longitudinal open field activity at +9, +27, +33 weeks after first injection. Test group size used in motor tests ranged from n=17–18 (n≥8 male, n≥8 female) at +9 weeks, n=9–11 (n≥4 male, n≥4 female) at +27 weeks, and n=3–5 (n≥1 male, n≥1 female) at +33 weeks. Two-way ANOVA mixed-effects analysis with a post-hoc Sidak multiple comparisons test, graphs represent statistical data comparisons of SCA3 Veh relative to SCA3 ASO-5 as * and SCA3 Veh relative to WT Veh as # (*or # p<0.05; *** or # # # p<0.001). Plots show Pearson correlations of total distance traveled with timepoint-corresponding (B, D) brainstem and (C, E) cerebellum total choline (tCho) or total NAA (tNAA) levels over time, respectively. Pearson correlation p<0.05 bolded.
DISCUSSION
Progressive ataxia assessments in patients with SCA has historically relied on clinical rating scales such as the Scale for Assessment and Rating of Ataxia (SARA) and International Cooperative Ataxia Rating Scale (ICARS) for measurement of disease severity32,33. While useful for assessing motor deficits, these scores are not quantitative of molecular pathology progression and require very large sample sizes in prospective trials34. MRS conducted in patients with polyglutamine expansion diseases has revealed strong evidence for specific neurochemical abnormalities12,13,21,22,32,33,35–37. Longitudinal MRS patient data exists for multiple SCA types in natural history studies relative to age- and sex-matched controls38,39. Similar natural history studies have demonstrated motor impairment progression in SCA3 mouse models that exhibit onset of motor deficits as early as 4 weeks of age, with homozygous SCA3 mice exhibiting significantly decreased distance traveled in open-field assessments40,41, increased duration of beam walking assays41, abnormal gait, and hindlimb clasping17. Furthermore, we have previously demonstrated success of ASOs in reversing motor deficits in pre-clinical mouse models of SCA37, and with this therapy now in phase I clinical trials (ClinicalTrials.gov Identifier: NCT05160558) it is imperative that informative biomarkers of ASO therapeutic efficacy be established. In this study, we provide proof-of-concept of neurochemical changes in a SCA3 pre-clinical disease model that demonstrates correlations between neurometabolite rescue and disease phenotype rescue upon ASO treatment.
Our investigation focused on the SCA3 mouse cerebellum and brainstem, as prior research demonstrated large structural degeneration and volume decreases within these regions in patients4,32,36,37,42. Though the pons and cerebellum have previously been studied via MRS in patients12,13,15,36 and the cerebellum in SCA3 mouse models16,37, to our knowledge, this is the first study to investigate SCA3 mouse brainstem neurochemical abnormalities. Our study reveals specific neurometabolite changes in SCA3 mice, including increased glutamine and decreased tNAA and tCho levels in both the brainstem and cerebellum. These trends parallel neurometabolite changes in patients with SCA3, in which decreased tNAA has been observed in the cerebellum12,13,32,36,37 and pons12,13,32. While tNAA levels exhibit common trends in decreased abundance across patients with SCA1, SCA2, SCA3, and SCA712,13,32, additional metabolite changes differ between disease types43, suggesting differences in regional disease pathology and affected cell types. Specifically, changes in levels of glutamine, NAA, and tCho have been reported to reflect glial, neuronal, and oligodendrocyte contributions to disease, respectively.
Though SCA3 and other polyglutamine expansion diseases have historically been studied through a neuron-centric focus, evidence of glial contributions to these diseases is gaining prominence in the literature. A role for gliosis has been demonstrated by increased GFAP staining in vulnerable brain regions in SCA3 mice17,44,45 and higher serum GFAP levels in patients with SCA332. Here, we show mice exhibit significant increases in glutamine levels in the cerebellum and brainstem across all MRS-assessed time points. These findings are consistent with increased glutamine in the cerebellum of patients with SCA313. Additionally, patients with SCA1 demonstrate increased glutamine in the cerebellum and pons46, supporting evidence for gliosis in multiple SCAs. Notably, ASO rescue of motor phenotype in SCA3 mice was observed despite a lack of significant ASO rescue of cerebellar and brainstem glutamine levels, suggesting that glutamine rescue may not be imperative to ASO efficacy on the motor task. Previous studies have evidenced efficient osmoregulation in rodents, demonstrating responsive shifts in metabolite levels following glutamine increase in the brain47. Though we observe inverse relationships between glutamine and responsive osmolytes in SCA3 mice, ASOs failed to rescue glutamine levels yet consistently rescued cerebellar tCho and taurine levels. This result leads us to conclude the observed changes in these two metabolites have an alternative, likely neurodegenerative, mechanistic driver. Previous MRS studies in patients with SCA1, SCA2, SCA3, and SCA7 have additionally implicated increased myo-inositol as reflective of gliosis12,13,32,33,37,46. Changes in myo-Ins are primarily observed at early stages of SCA disease, leading some researchers to suggest a role for reactive astrocytosis and microglial-mediated inflammation as a neuroprotective or compensative mechanism during the onset of neurodegeneration12, including in SCA332,37. Such changes in cerebellar tNAA/myo-Ins ratios have additionally been found to distinguish patients from controls at early disease13,46, and pons myo-Ins levels in patients with SCA3 correlate with SARA and quality of life scores12,13, further emphasizing a role for glial disturbances in the motor phenotype of patients.
Levels of tNAA are known to reflect neuronal heath and axonal abundance13,32,33,36,46. Decreased tNAA levels can be detected in the cerebellum of patients with SCA at pre-manifest stages, distinguishing patients from controls13,37, and negatively correlating with disease severity in patients with SCA135,46 and SCA337. Large tNAA decreases in SCA3 occur in vulnerable regions of the brain32,36,37, as indicated by neuronal ATXN3 accumulation4,17 and structural atrophy in the pons and cerebellum4,32,37. Consistent with findings in patients12, we found a significant decrease in tNAA levels in SCA3 mice, which was partially or fully rescued with ASO treatment. Parallels between human and mouse neurochemical abnormalities were previously reported for lower cerebellar and brainstem tNAA levels in mouse models of SCA122,48. Our results reveal a compelling correlation between cerebellar tNAA and motor activity at early and middle study timepoints; with a similar magnitude of correlation at the end-stage timepoint, low sample numbers may explain lack of significance. Together with prior work, our findings of altered neurometabolites in SCA3 highlight the substantial impact of cerebellar neurodegeneration on the motor phenotype in SCA3 mice and point towards cerebellar tNAA levels as informative of efficacy of ASOs for treatment of motor deficits.
White matter abnormalities have been reported in patients with SCA1, SCA2, SCA3, SCA6, and SCA7, with specific degeneration of the heavily myelinated axons of cerebellar grey matter tracts4,37. Findings of early white matter changes in the cerebellum of pre-symptomatic patients with SCA3 have led some researchers to hypothesize that oligodendrocyte and myelination impairments occur early in disease37, prior to extensive axonal degradation, neuronal death37, and peripheral neuropathy4,33. This study reveals significantly decreased tCho levels in the cerebellum and brainstem of SCA3 mice throughout longitudinal assessments, with ASO treatment demonstrating partial to full rescue to wildtype levels in both brain regions. Altered tCho levels have been previously linked to oligodendrocyte disturbances, due to the role of choline in cell membrane synthesis and energy metabolism32,37. Patients with SCA3 exhibit decreased cerebellar vermis NAA/Cr ratios49, and patients with SCA1 exhibit decreased pons Cho/Cr ratios35 that are paralleled by mouse models, with rescue of cerebellar tCho levels under ASO treatment22. We found a pronounced correlation between brainstem tCho levels and reduction in aggregated high-molecular weight and mutant monomeric ATXN3, suggesting a noteworthy correlation between oligodendrocyte function and mutant protein levels. This is further bolstered by our finding of a strong correlation between intranuclear ATXN3 inclusion in the DCN and cerebellar tCho levels. We have previously demonstrated that ASOs are targeted to both neuronal and glial cells7, suggesting that ASO reduction of oligodendrocyte ATXN3 accumulation may be contributing to this observed correlation. Consistent with this, our recent work has highlighted a historically understudied contribution of oligodendrocytes in SCA3 pathogenesis, revealing cell autonomous impairments of oligodendrocyte maturation in the pons and DCN of SCA3 mice50,51. Assessing motor impairment and tCho revealed a robust correlation between +26-week tCho levels in the brainstem and cerebellum and +32-week brainstem tCho levels relative to timepoint-corresponding motor activity timepoints. This suggests correlative ASO-mediated rescue of oligodendrocyte-associated neurometabolites and motor activity. Cumulatively, these findings provide further support for oligodendrocyte contributions to SCA3 and advise tCho as a key metabolite for informing timeline and monitoring regional impact of ASO treatment in patients with SCA3.
The promising preclinical development and demonstrated efficacy of ASOs for treatment of SCA3 has evoked a need for non-invasive biomarkers. Anti-ATXN3 ASOs are actively being evaluated for safety in patients with SCA3 in randomized, placebo-controlled phase I clinical trials (ClinicalTrials.gov Identifier: NCT05160558). If anti-ATXN3 ASOs are found to be safe and tolerable in a small patient cohort, it will be imperative to have established readouts of therapeutic efficacy to assess success of ASOs for treatment of SCA3 in a phase 3 efficacy clinical trial. MRS has been used in prior patient studies as a reproducible, non-invasive biomarker of neurodegeneration, leading to interest in the applicability of MRS in clinical trials. The present study displays quantitative neurometabolic changes for monitoring of anti-ATXN3 ASO therapeutic efficacy in vulnerable regions of the SCA3 brain. We demonstrate prominent correlations between neurometabolite rescue and rescue of motor impairments in animal models of disease. This evidence supports the application of MRS as a non-invasive measure of therapeutic efficacy that could be related to patient SARA scores in clinical trial endpoint design. As prior findings have suggested neurometabolite changes to be detectable as early as 10 years prior to symptom onset in mutation carriers and correlated with age of onset and motor impairments12,13,32,46, the implementation of MRS as a biomarker may allow for treatment efficacy prior to onset of motor impairments. We have yet to demonstrate the pre-symptomatic predictive value of neurometabolites as this study was limited to symptomatic SCA3 mouse models, but our findings warrant future investigation into pre-ataxic SCA3 mice. Echoing results in patients12,13,32,33,35,46, our findings emphasize spatiotemporal differences in neurometabolite changes in the cerebellum and brainstem. Furthermore, trends in particular metabolites suggest involvement of neuronal as well as glial cell types within SCA3 pathogenesis. Our findings of progressive metabolic changes in vulnerable brain regions of SCA3 mice parallel those seen in patients12,32,37. With existing data on neurochemical abnormalities in SCA1, SCA2, SCA3, SCA6, and SCA712,13,15,35,37,46, and prior studies demonstrating use of MRS for monitoring of ASO treatment in SCA1 mice, it is feasible that future work in the field may identify unifying signatures for different polyglutamine diseases. Our findings in the present study underscore the value of longitudinal regional neurometabolite data and taken in context with patient MRS data, provide support for the use of MRS as a biomarker tool for SCA3.
Summary for Social Media If Published.
1. If you and/or a co-author has a Twitter handle that you would like to be tagged, please enter it here. (format: @AUTHORSHANDLE).
@McLoughlinLab
2. What is the current knowledge on the topic? (one to two sentences)
Prior studies in patients with neurodegenerative diseases, including polyglutamine repeat expansion diseases, have demonstrated abnormalities in levels of neurochemicals quantified by magnetic resonance spectroscopy (MRS) in affected brain regions. Altered levels of specific neurochemicals are evidenced to serve as a non-invasive readout of neurodegeneration, making MRS a promising biomarker for clinical trials.
3. What question did this study address? (one to two sentences)
Do levels of neurochemicals serve as reliable disease biomarkers in pre-clinical mouse models of spinocerebellar ataxia type 3 (SCA3)? Can altered neurometabolites in vulnerable brain regions be related to neuropathological hallmarks of disease and rescued by antisense oligonucleotides in SCA3 mice?
4. What does this study add to our knowledge? (one to two sentences)
This study reveals the YACQ84 SCA3 mouse to exhibit significant neurochemical abnormalities, indicative of neurodegeneration as well as glial contributions to disease. Treatment with anti-ATXN3 ASOs rescues select neurochemical abnormalities, demonstrating correlations between neurochemical rescue and reversal of the ataxic behavioral phenotype.
5. How might this potentially impact on the practice of neurology? (one to two sentences)
These findings validate the use of MRS as a non-invasive measure of disease progression in the most relevant pre-clinical model of SCA3, revealing the timeline of neurochemical changes and reversal during disease course and supporting the use of neuroimaging as a biomarker for therapeutic assessment in upcoming clinical trials for SCA3 ASOs.
Acknowledgements
The authors would like to thank Ionis Pharmaceuticals for the use of the anti-ATXN3 ASOs and Dr. Dinesh Deelchand for guidance in data analysis and assistance in LCModel basis set generation. This work was funded by NIH/NINDS R21 NS111154 to H.S.M. and G.Ö. and NIH/NINDS R01 NS122751 and NIH/NINDS U01 NS106670 to H.S.M. The Center for Magnetic Resonance Research is supported by the National Institute of Biomedical Imaging and Bioengineering (NIBIB) grant P41 EB027061, NIH/NINDS the Institutional Center Cores for Advanced Neuroimaging award P30 NS076408 and the W.M. Keck Foundation.
Footnotes
Potential Conflicts of interest
Dr. Öz consults for IXICO Technologies Limited, which provides neuroimaging services and digital biomarker analytics to biopharmaceutical firms conducting clinical trials for SCAs, and receives research support from Biogen, which develops therapeutics for SCA3. All other authors declare no conflicts of interest.
References:
- 1.McLoughlin HS, Moore LR, Paulson HL. Pathogenesis of SCA3 and implications for other polyglutamine diseases. Neurobiol Dis Feb 2020;134:104635. doi: 10.1016/j.nbd.2019.104635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Durr A Autosomal dominant cerebellar ataxias: Polyglutamine expansions and beyond. The Lancet Neurology 2010;9:885–894. doi: 10.1016/S1474-4422(10)70183-6 [DOI] [PubMed] [Google Scholar]
- 3.Gardiner SL, Boogaard MW, Trompet S, et al. Prevalence of Carriers of Intermediate and Pathological Polyglutamine Disease-Associated Alleles Among Large Population-Based Cohorts. JAMA neurology Apr 1 2019;doi: 10.1001/jamaneurol.2019.0423 [DOI] [PMC free article] [PubMed]
- 4.Rub U, Schols L, Paulson H, et al. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Progress in neurobiology May 2013;104:38–66. doi: 10.1016/j.pneurobio.2013.01.001 [DOI] [PubMed] [Google Scholar]
- 5.Rosenberg RN. Machado-Joseph disease: an autosomal dominant motor system degeneration. Mov Disord 1992;7(3):193–203. doi: 10.1002/mds.870070302 [DOI] [PubMed] [Google Scholar]
- 6.Bushart DD, Zalon AJ, Zhang H, et al. Antisense Oligonucleotide Therapy Targeted Against ATXN3 Improves Potassium Channel-Mediated Purkinje Neuron Dysfunction in Spinocerebellar Ataxia Type 3. Cerebellum Aug 13 2020;doi: 10.1007/s12311-020-01179-7 [DOI] [PMC free article] [PubMed]
- 7.McLoughlin HS, Moore LR, Chopra R, et al. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann Neurol Jul 2018;84(1):64–77. doi: 10.1002/ana.25264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore LR, Keller L, Bushart DD, et al. Antisense oligonucleotide therapy rescues aggresome formation in a novel spinocerebellar ataxia type 3 human embryonic stem cell line. Stem Cell Res Aug 2019;39:101504. doi: 10.1016/j.scr.2019.101504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore LR, Rajpal G, Dillingham IT, et al. Evaluation of Antisense Oligonucleotides Targeting ATXN3 in SCA3 Mouse Models. Mol Ther Nucleic Acids Jun 16 2017;7:200–210. doi: 10.1016/j.omtn.2017.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piccinin CC, Rezende TJR, de Paiva JLR, et al. A 5-Year Longitudinal Clinical and Magnetic Resonance Imaging Study in Spinocerebellar Ataxia Type 3. Mov Disord Jun 9 2020;doi: 10.1002/mds.28113 [DOI] [PubMed]
- 11.Arruda WO, Meira AT, Ono SE, et al. Volumetric MRI Changes in Spinocerebellar Ataxia (SCA3 and SCA10) Patients. Cerebellum Aug 2020;19(4):536–543. doi: 10.1007/s12311-020-01137-3 [DOI] [PubMed] [Google Scholar]
- 12.Adanyeguh IM, Henry PG, Nguyen TM, et al. In vivo neurometabolic profiling in patients with spinocerebellar ataxia types 1, 2, 3, and 7. Mov Disord Apr 15 2015;30(5):662–70. doi: 10.1002/mds.26181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joers JM, Deelchand DK, Lyu T, et al. Neurochemical abnormalities in premanifest and early spinocerebellar ataxias. Ann Neurol Apr 2018;83(4):816–829. doi: 10.1002/ana.25212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krahe J, Binkofski F, Schulz JB, Reetz K, Romanzetti S. Neurochemical profiles in hereditary ataxias: A meta-analysis of Magnetic Resonance Spectroscopy studies. Neurosci Biobehav Rev Jan 2020;108:854–865. doi: 10.1016/j.neubiorev.2019.12.019 [DOI] [PubMed] [Google Scholar]
- 15.Chandrasekaran J, Petit E, Park Y-W, et al. Clinically meaningful MR endpoints sensitive to preataxic SCA1 and SCA3. Annals of Neurology (published online ahead of print 13 December). Availible at: 10.1002/ana.26573 (Accessed 2 February 2023)(n/a)doi: 10.1002/ana.26573 [DOI]
- 16.Costa MDC, Radzwion M, McLoughlin HS, et al. In Vivo Molecular Signatures of Cerebellar Pathology in Spinocerebellar Ataxia Type 3. Mov Disord Jul 4 2020;35(10):1744–1786. doi: 10.1002/mds.28140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cemal CK, Carroll CJ, Lawrence L, et al. YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum Mol Genet May 1 2002;11(9):1075–94. [DOI] [PubMed] [Google Scholar]
- 18.Cheruvallath ZS, Kumar RK, Rentel C, Cole DL, Ravikumar VT. Solid phase synthesis of phosphorothioate oligonucleotides utilizing diethyldithiocarbonate disulfide (DDD) as an efficient sulfur transfer reagent. Nucleosides, nucleotides & nucleic acids Apr 2003;22(4):461–8. doi: 10.1081/NCN-120022050 [DOI] [PubMed] [Google Scholar]
- 19.McKay RA, Miraglia LJ, Cummins LL, Owens SR, Sasmor H, Dean NM. Characterization of a potent and specific class of antisense oligonucleotide inhibitor of human protein kinase C-alpha expression. J Biol Chem Jan 15 1999;274(3):1715–22. [DOI] [PubMed] [Google Scholar]
- 20.DeVos SL, Miller TM. Direct intraventricular delivery of drugs to the rodent central nervous system. J Vis Exp May 12 2013;(75):e50326. doi: 10.3791/50326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oz G, Kittelson E, Demirgoz D, et al. Assessing recovery from neurodegeneration in spinocerebellar ataxia 1: Comparison of in vivo magnetic resonance spectroscopy with motor testing, gene expression and histology. Neurobiol Dis Feb 2015;74:158–66. doi: 10.1016/j.nbd.2014.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedrich J, Kordasiewicz HB, O’Callaghan B, et al. Antisense oligonucleotide-mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight Nov 2 2018;3(21)doi: 10.1172/jci.insight.123193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hennig J, Nauerth A, Friedburg H. RARE imaging: a fast imaging method for clinical MR. Magnetic resonance in medicine Dec 1986;3(6):823–33. doi: 10.1002/mrm.1910030602 [DOI] [PubMed] [Google Scholar]
- 24.Gruetter R, Tkac I. Field mapping without reference scan using asymmetric echo-planar techniques. Comparative Study Research Support, U.S. Gov’t, P.H.S. Magnetic resonance in medicine Feb 2000;43(2):319–23. doi: [DOI] [PubMed] [Google Scholar]
- 25.Garwood M, DelaBarre L. The return of the frequency sweep: designing adiabatic pulses for contemporary NMR. J Magn Reson Dec 2001;153(2):155–77. doi: 10.1006/jmre.2001.2340 [DOI] [PubMed] [Google Scholar]
- 26.Tkac I, Starcuk Z, Choi IY, Gruetter R. In vivo 1H NMR spectroscopy of rat brain at 1 ms echo time. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Magnetic resonance in medicine Apr 1999;41(4):649–56. doi: [DOI] [PubMed] [Google Scholar]
- 27.Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magnetic resonance in medicine Dec 1993;30(6):672–9. [DOI] [PubMed] [Google Scholar]
- 28.Deelchand DK, Henry PG, Ugurbil K, Marjanska M. Measurement of transverse relaxation times of J-coupled metabolites in the human visual cortex at 4 T. Research Support, N.I.H., Extramural. Magnetic resonance in medicine Apr 2012;67(4):891–7. doi: 10.1002/mrm.23080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Govindaraju V, Young K, Maudsley AA. Proton NMR chemical shifts and coupling constants for brain metabolites. Research Support, U.S. Gov’t, P.H.S. NMR Biomed May 2000;13(3):129–53. doi: [DOI] [PubMed] [Google Scholar]
- 30.Tkac I Refinement of simulated basis set for LCModel analysis. presented at: Paper presented at: 16th Scientific Meeting of the ISMRM2008; Toronto, Canada. [Google Scholar]
- 31.Cudalbu C, McLin VA, Lei H, et al. The C57BL/6J mouse exhibits sporadic congenital portosystemic shunts. PLoS One 2013;8(7):e69782. doi: 10.1371/journal.pone.0069782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen ML, Lin CC, Rosenthal LS, Opal P, Kuo SH. Rating scales and biomarkers for CAG-repeat spinocerebellar ataxias: Implications for therapy development. J Neurol Sci May 15 2021;424:117417. doi: 10.1016/j.jns.2021.117417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brooker SM, Edamakanti CR, Akasha SM, Kuo SH, Opal P. Spinocerebellar ataxia clinical trials: opportunities and challenges. Ann Clin Transl Neurol Jul 2021;8(7):1543–1556. doi: 10.1002/acn3.51370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobi H, du Montcel ST, Bauer P, et al. Long-term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: a longitudinal cohort study. Lancet Neurol Nov 2015;14(11):1101–8. doi: 10.1016/S1474-4422(15)00202-1 [DOI] [PubMed] [Google Scholar]
- 35.Mascalchi M, Tosetti M, Plasmati R, et al. Proton magnetic resonance spectroscopy in an Italian family with spinocerebellar ataxia type 1. Ann Neurol Feb 1998;43(2):244–52. doi: 10.1002/ana.410430215 [DOI] [PubMed] [Google Scholar]
- 36.D’Abreu A, Franca M Jr, Appenzeller S, Lopes-Cendes I, Cendes F. Axonal dysfunction in the deep white matter in Machado-Joseph disease. J Neuroimaging Jan 2009;19(1):9–12. doi: 10.1111/j.1552-6569.2008.00260.x [DOI] [PubMed] [Google Scholar]
- 37.Miranda CO, Nobre RJ, Paiva VH, et al. Cerebellar morphometric and spectroscopic biomarkers for Machado-Joseph Disease. Acta neuropathologica communications Mar 19 2022;10(1):37. doi: 10.1186/s40478-022-01329-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deelchand DK, Joers JM, Ravishankar A, et al. Sensitivity of Volumetric Magnetic Resonance Imaging and Magnetic Resonance Spectroscopy to Progression of Spinocerebellar Ataxia Type 1. Mov Disord Clin Pract Sep 2019;6(7):549–558. doi: 10.1002/mdc3.12804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin CC, Ashizawa T, Kuo SH. Collaborative Efforts for Spinocerebellar Ataxia Research in the United States: CRC-SCA and READISCA. Front Neurol 2020;11:902. doi: 10.3389/fneur.2020.00902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schuster KH, DiFranco DM, Putka AF, et al. Disease-associated oligodendrocyte signatures are spatiotemporally dysregulated in Spinocerebellar Ataxia Type 3. Front Neurosci 2023. 2023;17doi: 10.3389/fnins.2023.1118429 [DOI] [PMC free article] [PubMed]
- 41.Costa MC, Luna-Cancalon K, Fischer S, et al. Toward RNAi therapy for the polyglutamine disease Machado-Joseph disease. Mol Ther Oct 2013;21(10):1898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Faber J, Schaprian T, Berkan K, et al. Regional Brain and Spinal Cord Volume Loss in Spinocerebellar Ataxia Type 3. Mov Disord Oct 2021;36(10):2273–2281. doi: 10.1002/mds.28610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oz G, Iltis I, Hutter D, Thomas W, Bushara KO, Gomez CM. Distinct neurochemical profiles of spinocerebellar ataxias 1, 2, 6, and cerebellar multiple system atrophy. Cerebellum Jun 2011;10(2):208–17. doi: 10.1007/s12311-010-0213-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silva-Fernandes A, Costa Mdo C, Duarte-Silva S, et al. Motor uncoordination and neuropathology in a transgenic mouse model of Machado-Joseph disease lacking intranuclear inclusions and ataxin-3 cleavage products. Neurobiol Dis Oct 2010;40(1):163–76. doi: 10.1016/j.nbd.2010.05.021 [DOI] [PubMed] [Google Scholar]
- 45.Switonski PM, Szlachcic WJ, Krzyzosiak WJ, Figiel M. A new humanized ataxin-3 knock-in mouse model combines the genetic features, pathogenesis of neurons and glia and late disease onset of SCA3/MJD. Neurobiol Dis Jan 2015;73:174–88. doi: 10.1016/j.nbd.2014.09.020 [DOI] [PubMed] [Google Scholar]
- 46.Öz G, Hutter D, Tkac I, et al. Neurochemical alterations in spinocerebellar ataxia type 1 and their correlations with clinical status. Mov Disord Jul 15 2010;25(9):1253–61. doi: 10.1002/mds.23067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cordoba J, Gottstein J, Blei AT. Glutamine, myo-inositol, and organic brain osmolytes after portocaval anastomosis in the rat: implications for ammonia-induced brain edema. Hepatology Oct 1996;24(4):919–23. doi: 10.1002/hep.510240427 [DOI] [PubMed] [Google Scholar]
- 48.Oz G, Nelson CD, Koski DM, et al. Noninvasive detection of presymptomatic and progressive neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci Mar 10 2010;30(10):3831–8. doi: 10.1523/JNEUROSCI.5612-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Y, Jin Y, Hu Z, et al. Association Between Serum Neurofilament Light Chain and Neurochemistry Deficits in Patients with Spinocerebellar Ataxia Type 3. Cerebellum Jan 4 2023;(published online ahead of print 4 January). Availible at: doi.org/ 10.1007/s12311-022-01507-z (Accessed 2 February 2023)doi: 10.1007/s12311-022-01507-z [DOI] [PubMed]
- 50.Schuster KH, Zalon AJ, Zhang HJ, et al. Impaired oligodendrocyte maturation is an early feature in SCA3 disease pathogenesis. Journal of Neuroscience Feb 23 2022;42(8)doi: 10.1523/Jneurosci.1954-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schuster KH, Putka AF, McLoughlin HS. Pathogenetic Mechanisms Underlying Spinocerebellar Ataxia Type 3 Are Altered in Primary Oligodendrocyte Culture. Cells 2022;11(16):2615. [DOI] [PMC free article] [PubMed] [Google Scholar]