

Abstract
Objective:
This study examined whether cerebrospinal fluid (CSF) baseline levels of the synaptic protein NPTX2 predict time to onset of symptoms of Mild Cognitive Impairment (MCI), both alone and when accounting for traditional CSF Alzheimer’s disease (AD) biomarker levels. Longitudinal NPTX2 levels were also examined.
Methods:
CSF was collected longitudinally from 269 cognitively normal BIOCARD Study participants (mean baseline age=57.7 years; mean follow-up=16.3 years; n=77 progressed to MCI/dementia). NPTX2 levels were measured from three correlated peptides using quantitative parallel reaction monitoring mass spectrometry. Levels of Aβ42/Aβ40, p-tau181, and t-tau were measured from the same CSF specimens using Lumipulse automated electrochemiluminescence assays.
Results:
In Cox regression models, lower baseline NPTX2 levels were associated with an earlier time to MCI symptom onset (HR=0.76, SE=0.09, p=0.023). This association was significant for progression within 7 years (p=0.036) and after 7 years from baseline (p=0.001). Baseline NPTX2 levels improved prediction of time to MCI symptom onset after accounting for baseline AD biomarker levels (p<0.01), and NPTX2 did not interact with the CSF AD biomarkers or APOE-ε4 genetic status. In linear mixed effects models, higher baseline p-tau181 and t-tau levels were associated with higher baseline levels of NPTX2 (both p<0.001) and greater rates of NPTX2 declines over time.
Interpretation:
NPTX2 may be a valuable prognostic biomarker during preclinical AD that provides additive and independent prediction of MCI onset among individuals who are cognitively normal. We hypothesize that NPTX2-mediated circuit homeostasis confers resilience during the early phase of AD.
Introduction
The neuropathological hallmarks of Alzheimer’s disease (AD), i.e., amyloid plaques and tau neurofibrillary tangles, begin to accumulate many years prior to the emergence of clinical symptoms of the disease.1, 2 Biomarker studies have shown that more abnormal cerebrospinal fluid (CSF) levels of amyloid and tau among individuals with normal cognition are associated with an increased risk of developing symptoms of Mild Cognitive Impairment (MCI) or dementia due to AD.3–6 However, the predictive accuracy of these markers alone is limited. Measures that can increase prediction, above and beyond the standard AD biomarkers, may not only improve prognosis and disease monitoring, but also serve to identify novel treatment targets.
Neuropathological studies indicate that synaptic loss is strongly correlated with cognitive impairment during the symptomatic phase of AD.7–9 Accordingly, recent efforts have focused on identifying potential biomarkers of synaptic damage in CSF.10 Neuronal pentraxin 2 (NPTX2) is a synaptic protein expressed in the cerebral cortex and medial temporal lobe. NPTX2 is involved in the homeostatic regulation of cortical network dynamics, synaptic plasticity, and memory.11–13 Prior cross-sectional studies have shown that CSF NPTX2 levels decline across the spectrum of symptomatic AD, with NPTX2 levels being lower among participants with MCI or dementia than among individuals with normal cognition.14–18 Lower NPTX2 levels have also been shown to be associated with an increased risk of progression from MCI to AD dementia.14, 19 Additionally, longitudinal declines in NPTX2 correlate with declines in cognition, particularly among individuals with MCI, and appear greater among participants with more abnormal levels of amyloid and tau.20 However, it is unclear whether NPTX2 levels among cognitively unimpaired individuals are associated with the onset of MCI and whether NPTX2 levels improve the prediction of time to MCI symptom onset after accounting for CSF levels of amyloid and tau.
To address this gap, we examined whether baseline levels of CSF NPTX2 predict time to onset of symptoms of MCI among 269 individuals who were cognitively normal at baseline and have undergone 16.3 years of clinical follow-up, on average. Participants were primarily middle-aged at baseline, which is particularly relevant for examining early AD-related changes, which may begin in mid-life.21 The long duration of follow-up also allowed us to examine whether NPTX2 is associated with risk of progression both proximally and more distally from baseline (i.e., within vs. after 7 years). Importantly, this study investigated the extent to which NPTX2 improves the prediction of MCI symptom onset when accounting for CSF biomarkers of AD pathology (amyloid and tau), and whether NPTX2 and CSF AD biomarkers have independent or synergistic effects on risk of progression. Lastly, to better understand the temporal relationship between changes in NPTX2 and CSF AD biomarkers, we also examined the inter-relationships between the baseline levels and trajectories of NPTX2 and CSF AD biomarkers over time (mean CSF follow-up = 10.4 years).
Methods
Study design and participant selection
Data were derived from the BIOCARD Study, an on-going longitudinal cohort study designed to examine the earliest features of AD. The study was initiated at the National Institutes of Health (NIH) in 1995. At baseline, participants completed a physical and neurological examination, neuropsychological testing, an electrocardiogram, and standard laboratory tests. Participants were excluded if they had cognitive impairment (based on cognitive testing or presence of clinical symptoms), chronic neurological or psychiatric disorders (e.g., epilepsy, schizophrenia), alcohol or drug abuse, or significant medical problems, including severe cardiovascular disease (e.g., atrial fibrillation). By design, about 75% of participants had a first degree relative with a history of dementia.
A total of 349 cognitively normal participants were enrolled at the NIH after providing written informed consent. Participants completed comprehensive clinical and cognitive assessments at the NIH on an annual basis; approximately every 2 years, participants provided blood and CSF specimens, and had MRI scans. In 2005, the study was stopped for administrative reasons and re-established at Johns Hopkins University (JHU) in 2009 to continue the annual clinical and cognitive evaluations and blood draws. In 2015, the biennial CSF and MRI collection was reinitiated and amyloid positron emission tomography (PET) imaging began. Tau PET imaging started in 2021. See Figure 1 for a timeline of the study. The BIOCARD study was approved by the Johns Hopkins Medicine Institutional Review Board. Written informed consent was obtained from all study participants.
Figure 1.
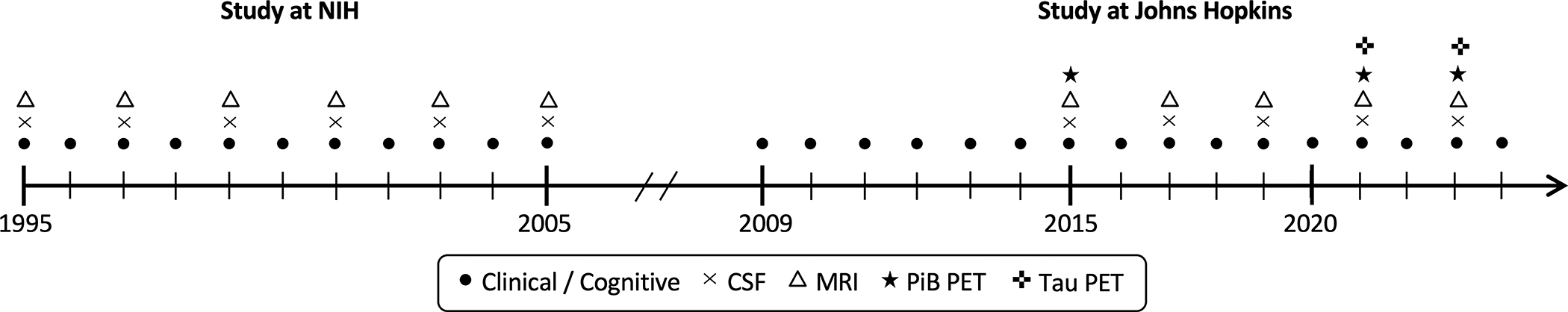
Approximate timeline of BIOCARD study indicating the types of data collected each year.
CSF specimens were obtained from 307 participants. Data from 38 of these participants were excluded because: (1) they have withdrawn or not yet re-enrolled in the study and not given permission to use their data (n=23); or (2) they had an estimated age of onset of clinical symptoms of MCI (the primary outcome measure) at or prior to their first CSF measure (n=15). Thus, this study included data from 269 participants who were cognitively normal at their baseline (i.e., first) CSF draw.
Clinical and cognitive assessments
The annual clinical and cognitive assessments conducted at both the NIH and at JHU include behavioral and mood assessments, medical history and record of medication use, family history of dementia, a physical and neurological exam, the Clinical Dementia Rating (CDR) scale22 and a comprehensive neuropsychological battery.23
At each study visit, participants received a consensus diagnosis by the BIOCARD Clinical Core at JHU. The diagnostic procedures are comparable to those used in the National Institute on Aging (NIA) Alzheimer’s Disease Centers program. Briefly, this first involves establishing a syndromic diagnosis (i.e., normal, MCI, dementia, or impaired-not-MCI) using three types of information: (1) clinical data relating to the neurological, psychiatric, and medical status of the participant; (2) evidence of cognitive decline (based on longitudinal testing and comparison to published norms); and (3) reports of changes in cognition by the subject and an informant (based on the CDR). Next, for participants with cognitive impairment, the likely syndromic etiology was determined based on the available neurologic, medical, and psychiatric information; more than one etiology could be endorsed. The diagnoses for MCI24 and dementia25 follow the recommendations by the NIA/Alzheimer’s Association working groups. Participants with contrasting information from the cognitive testing and the CDR received a diagnosis of “impaired not MCI”. In all analyses, these participants were included in the group with normal cognition (as they do not meet criteria for MCI). Sensitivity analyses were conducted excluding these participants. The few participants whose diagnosis changed from MCI to normal were considered normal in these analyses, if that was the diagnosis at their most recent visit. Consensus diagnoses were blinded to APOE genetic status and biomarker values (see23 for additional details regarding study design, recruitment, consensus diagnoses, and clinical and cognitive evaluations).
The primary outcome variable for this study was the time from baseline to onset of clinical symptoms of MCI. The age of MCI clinical symptom onset is based on the CDR interview (conducted with both the subject and the informant), and is confirmed on subsequent visits so that each participant with MCI or dementia has a single age of symptom onset that represents the consensus of the clinical team. The same diagnostic process was retrospectively applied to participants who developed cognitive impairment while the study was at the NIH.
APOE genetic status
APOE genotypes were determined by restriction endonuclease digestion of polymerase chain reaction amplified genomic DNA (performed by Athena Diagnostics). A binary indicator variable was used to code APOE-ε4 carrier status.
Cerebrospinal fluid assessments
CSF measures were derived from samples collected at the NIH (1995–2005) and at JHU (since 2015). CSF was obtained after an overnight fast. Immediately after collection via lumbar puncture, the CSF was aliquoted into polypropylene cryotubes, which were kept on dry ice, and transferred to a −80C freezer for long-term storage. Samples were thawed for the first time since collection to measure Aβ40, Aβ42, phosphorylated tau181 (p-tau181), and total tau (t-tau) using fully automated chemiluminescent enzyme immunoassays (Lumipulse G1200 platform; Fujirebio Diagnostics; for coefficients of variation, see4). To account for individual differences in total Aβ production and to reduce pre-analytic variability,26–28 we used the Aβ42/Aβ40 ratio instead of Aβ42 alone as a measure of amyloid. Details regarding the long-term trajectories of these measures have been published previously.29
CSF samples were thawed for a second time to measure NPTX2 levels using parallel reaction monitoring mass spectroscopy (PRM-MS), as previously described.30 Briefly, three 571.43 pM NPTX peptides (WPVETCEER, TESTLNALLQR, and AAVLQLR), labelled with stable isotopes on arginine residue, were synthesized and added to CSF for quantification. After enzyme digestions of CSF samples with trypsin, the abundances of both light and heavy peptides were measured by PRM-MS. The quantification of relative peptide abundance was performed using Skyline software.31 The PRM-MS measures were validated against NPTX2 measured with ELISA (see Figure 2). The amounts of each peptide were highly correlated with one another (r=0.87 to r=0.94, all p<0.0001, for baseline measures). We therefore combined the three peptides into an NPTX2 composite score by z-scoring and then averaging them. The NPTX2 composite score was used as the primary measure of NPTX2 to reduce the number of analyses. Sensitivity analyses examined whether the primary results were similar for the individual peptides. The NPTX2 peptides and composite score were log-transformed prior to analysis to correct for skewness.
Figure 2.
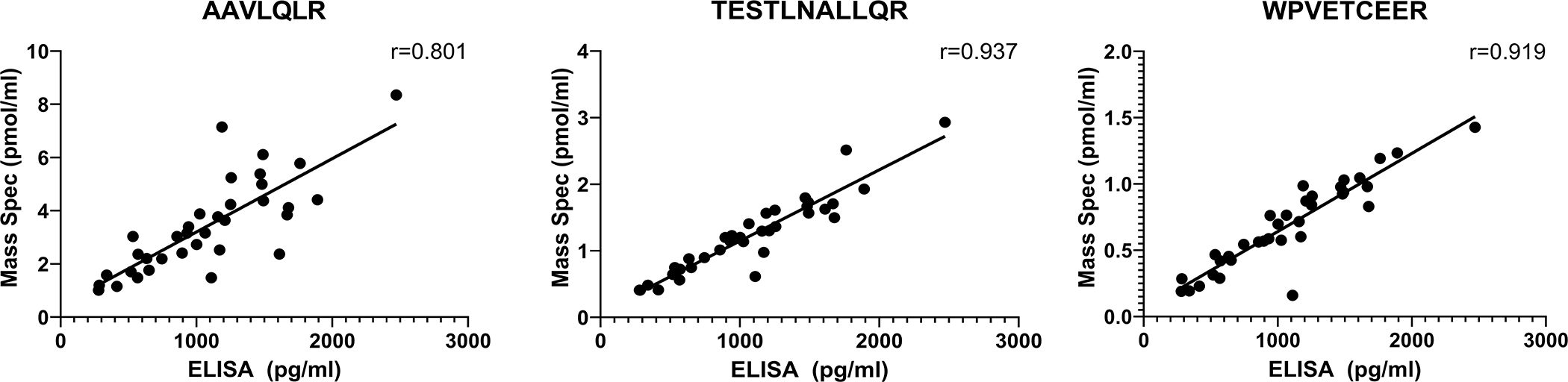
Scatterplots showing the correlation between CSF levels of the three quantotypic peptides for NPTX2 used in the current study, as measured by parallel reaction monitoring mass spectrometry (PRM-MS, y-axis, in pmol/ml) and levels of the same peptides measured by enzyme-linked immunosorbent assay (ELISA, x-axis, in pg/ml). The CSF samples used for the validation (N=35, including 11 with probable AD dementia and 24 cognitively normal participants, mean age=73.6 years, SD=6.4 years) were obtained from the University of California, San Diego Shirley Marcos Alzheimer’s Disease Research Center and the CSF collection methods have been described previously.14
Statistical analyses
To compare group differences in participants’ characteristics at baseline, t-tests were used for continuous variables and chi-square tests for dichotomous variables. In all analyses, ‘baseline’ was defined as time of first CSF measurement.
Analyses examining whether baseline NPTX2 levels (either alone or in combination with the CSF AD biomarker measures) were associated with time to MCI clinical symptom onset used Cox regression models (i.e., proportional hazard models), covarying age, sex, and years of education. Three sets of Cox models were conducted, where participants who remained cognitively normal over time were defined as being ‘event-free’, and participants who were diagnosed with MCI or dementia at their last follow-up were considered as having experienced an ‘event’ at the time of their estimated age of onset of symptoms. The last date of diagnosis was used as the censoring time. Hazard ratios (HRs; i.e., the relative hazard) and 95% confidence intervals (CI) were calculated for all variables in these models. Continuous variables were standardized before model fitting (mean=0, SD=1). Analyses were run in R (version 4.2.2). Statistical significance was set at p<0.05.
Model 1 included the NPTX2 composite score as a predictor of time to MCI symptom onset (in addition to the covariates). Given the long follow-up period in the study, Model 2 tested whether the relationship between the NPTX2 composite score and MCI clinical symptom onset was similar for progressions occurring closer in time to the baseline visit (i.e., within 7 years from baseline) vs. later in time after baseline (i.e., after 7 years). This was accomplished by including time-varying coefficients for the NPTX2 measure in the Cox models. Statistically, this is equivalent to treating the NPTX2 measure as a composition of two time-dependent variables, one having an effect on risk of progression within 7 years, the other after 7 years (see4, 32 for prior publications using this method). Seven years was selected as the cut-off as it reflected the median time to MCI clinical symptom onset. Model 3 investigated whether the relationship between the NPTX2 composite score and time to MCI symptom onset remained the same when accounting for baseline levels CSF Aβ42/Aβ40, p-tau181, or t-tau (using separate models for each CSF AD biomarker). In addition, interactions between the NPTX2 composite score and the AD biomarkers and with APOE-ε4 genetic status were tested by including interaction terms in each model (i.e., NPTX2 × AD biomarker [or APOE-ε4]).
The Akaike Information Criterion (AIC) was used to compare models to one another to determine whether models with NPTX2 provided a better fit compared to models including the traditional CSF AD biomarkers only. The AIC was selected because it provides an index of the relative balance of model fit (based on the partial likelihood function for the Cox proportional hazards model) and model parsimony (based on the number of parameters in the model). A smaller AIC value indicates a better balance between fit and parsimony,33 with a difference in AIC of >=2 indicating a meaningful difference in fit.34
Lastly, to gain a better understanding of the interrelationships between NPTX2 and changes in CSF AD biomarkers, and vice versa, we also examined whether a) baseline AD biomarkers levels were associated with the rate of change in NPTX2 over time (Model 4); and whether b) baseline NPTX2 was associated with the rate of change in the AD biomarkers over time (Model 5). For Model 4, the outcome variable was the NPTX2 composite score over time and CSF Aβ42/Aβ40, p-tau181, or t-tau were predictors (in separate models). For Model 5, the CSF-AD biomarkers were the outcome and the baseline NPTX2 composite score was the predictor. These analyses used longitudinal mixed effects models that included random intercepts and slopes with unstructured covariance. All models covaried baseline age and sex, and their interactions (i.e., cross-products) with time; sensitivity analyses further adjusted for years of education and APOE-ε4 status and their interactions with time. If significant (at p<0.05), time × time terms were included to model non-linear trajectories. Models also included a ‘collection site’ indicator, reflecting the location of CSF collection (NIH vs. JHU) to control for potential systematic site differences. For these models, all CSF measures were standardized (z-scored) separately for CSF collected at the NIH vs. JHU using the first available measure at each site, as described previously.29 Time was modeled in the unit of years (since baseline). Sensitivity analyses tested whether results changed when only including data collected at the NIH, as these are not subject to potentially confounding site differences.
Anonymized data pertaining to this report are available upon request by qualified investigators, as described on the BIOCARD Study website (www.biocard-se.org).
Results
Descriptive and baseline statistics
Baseline characteristics for the total sample and stratified by clinical outcome (i.e., remained cognitively normal [n=192] vs. progressed to MCI [n=43] or dementia [n=34]) are presented in Table 1. Participants who progressed were older at baseline and had less time between their baseline and last CSF measure (both p<0.05), but did not differ on other baseline characteristics (all p>0.10). Among the 77 participants who developed MCI or dementia over time, 88% were judged to have AD as the primary [n=49] or a secondary [n=19] etiology, as determined by consensus diagnosis.
Table 1.
Baseline characteristics of participants in analyses for total sample and stratified by clinical outcomes
| Baseline Characteristic | All participants in analysis (n = 269) | Remained Normal (n = 192) | Progressed to MCI/ Dementia (n = 77) |
|---|---|---|---|
|
| |||
| Age, mean years (SD) | 57.7 (10.4) | 55.6 (10.2) | 62.8 (9.1) ** |
| Age range (years) | 20.4 – 92.5 | 20.4 – 92.5 | 42.1 – 84.8 |
| Sex, N females (%) | 159 (59.1%) | 117 (60.9%) | 42 (54.6%) |
| Ethnicity, N White (%) | 262 (97.4%) | 187 (97.4%) | 75 (97.4%) |
| Education, mean years (SD) | 17.1 (2.4) | 17.2 (2.4) | 16.8 (2.4) |
| APOE-ε4 carriers, N (%) | 94 (34.9%) | 65 (33.9%) | 29 (37.7%) |
| MMSE score, mean (SD) | 29.6 (0.7) | 29.6 (0.7) | 29.6 (0.8) |
| Years between baseline CSF to most recent diagnosis, mean (SD) [range] | 16.3 (5.4) [0–24.5] | 16.2 (5.7) [0–24.5] | 16.4 (4.5) [4.3–24.2] |
| Years between baseline CSF to onset of clinical symptoms, mean (SD) [range] | -- | -- | 7.8 (4.8) [0.8–21.5] |
| Years between baseline and last CSF, mean (SD) [range] | 10.4 (7.3) [0–22.5 ] | 11.4 (7.3) [0–22.5] | 7.8 (6.6) ** [0–20.6] |
| Number of CSF measures over time, mean (SD) [range] | 3.6 (1.8) [1–9] | 3.7 (1.9) [1–9] | 3.3 (1.7) [1–7] |
| NPTX2 composite score, mean (SD) | 0.6 (0.5) | 0.6 (0.5) | 0.5 (0.4) |
| NPTX2 AAVLQLR peptide (pmol/ml), mean (SD) | 3.1 (1.5) | 3.2 (1.6) | 2.9 (1.2) |
| NPTX2 TESTLNALLQR peptide (pmol/ml), mean (SD) | 0.8 (0.4) | 0.9 (0.4) | 0.8 (0.4) |
| NPTX2 WPVETCEER peptide (pmol/ml), mean (SD) | 0.4 (0.2) | 0.4 (0.2) | 0.3 (0.1) |
| CSF Aβ42/Aβ40, mean (SD) | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) ** |
| CSF p-tau181 in pg/mL, mean (SD) | 34.5 (17.1) | 31.4 (13.6) | 42.1 (22.4) ** |
| CSF t-tau in pg/mL, mean (SD) | 258 (139) | 236 (109) | 314 (183) ** |
p < 0.05
p <= 0.01 assessing group difference between those who remained normal and those who progressed to MCI or dementia.
Relationship between baseline NPTX2 and time to onset of clinical symptoms of MCI
Model 1 showed that lower NPTX2 composite scores were associated with an earlier time of onset of MCI clinical symptoms (p=0.023; see Table 2). Results were the same when including APOE-ε4 genetic status as an additional model predictor (HR for NPTX2 composite score = 0.77, CI=0.60–0.97, p=0.027). Results were also the same when limiting cases (i.e., outcomes) to participants who progressed to MCI or dementia due to AD as the primary etiology (HR=0.71, CI=0.53–0.95, p=0.02), and when using the individual NPTX2 peptides instead of the composite (Table 2). There were no interactions between the baseline NPTX2 composite score and sex (HR=0.89, CI=0.56–1.42, p=0.62) or APOE-ε4 genetic status (HR=0.91, CI=0.56–1.48, p=0.71) with respect to time to MCI symptom onset, suggesting a similar relationship of lower NPTX2 to risk of progression in males and females, and in ε4 carriers and non-carriers.
Table 2.
Hazard ratios for the baseline NPTX2 composite score and individual NPTX2 peptides in relation to MCI clinical symptom onset for participants progressing to MCI or dementia (Model 1) and stratified by progression to MCI or dementia within 7 years vs. after 7 years from baseline (Model 2).
| Model 1 | Model 2 | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Risk of Progression | Risk of Progression ≤ 7 years from baseline | Risk of Progression > 7 years from baseline | ||||
|
| ||||||
| Model Predictor | HR (95% CI) | p-value | HR (95% CI) | p-value | HR (95% CI) | p-value |
|
| ||||||
| NPTX2 composite score | 0.76 (0.60, 0.96) | 0.023 | 0.78 (0.61, 0.98) | 0.036 | 0.62 (0.46, 0.83) | 0.001 |
| NPTX2 (AAVLQLR) | 0.79 (0.62, 1.01) | 0.058 | 0.84 (0.70, 1.00) | 0.055 | 0.68 (0.52, 0.88) | 0.004 |
| NPTX2 (TESTLNALLQR) | 0.75 (0.60, 0.95) | 0.015 | 0.50 (0.22, 1.10) | 0.084 | 0.19 (0.07, 0.57) | 0.003 |
| NPTX2 (WPVETCEER) | 0.75 (0.59, 0.94) | 0.012 | 0.13 (0.02, 0.83) | 0.031 | 0.02 (0.00, 0.31) | 0.006 |
Note: Models were adjusted by baseline age, sex, and years of education. Each NPTX2 measure was tested in a separate model.
Among the 77 participants who became cognitively impaired over time, 39 became symptomatic within 7 years of baseline and 38 more than 7 years from baseline. There were no significant differences between these two subgroups in baseline age, sex, education, APOE-ε4 carrier status, or MMSE score (all p>0.08; data not shown). Results for Model 2 demonstrated that lower NPTX2 composite scores were associated with time to MCI symptom onset for progression within 7 years of baseline (p=0.036) and for progression after 7 years from baseline (p=0.001), see Table 2. This association was the same when additionally covarying APOE-ε4 status (HR for NPTX2 composite score=0.78, CI=0.62–0.99, p=0.04 for progression within 7 years and HR=0.62, CI=0.46–0.83, p=0.002 for progression after 7 years). Results were also similar when using the individual NPTX2 peptide measures (Table 2).
Model 3 showed that lower baseline NPTX2 composite scores remained associated with an earlier time of MCI symptom onset when adding one of the traditional CSF AD biomarkers to the Cox regression model (all p<0.05), with both the AD biomarker and NPTX2 showing a significant association with risk of progression (see Table 3 for model results and Figure 3 for Kaplan-Meier plots). Adding NPTX2 to p-tau181 (AIC=739.4) produced a better model fit (for Model 3) than adding NPTX2 to Aβ42/Aβ40 (AIC=742.6) or t-tau (AIC=747.5), as indicated by a difference in AIC of >2.
Table 3.
Hazard ratios from Cox models including the NPTX2 composite score and CSF biomarkers of amyloid and/or tau in relation to MCI clinical symptom onset.
| Model predictors | Model 3 | |
|---|---|---|
|
| ||
| HR (95% CI) | p-value | |
|
| ||
| NPTX2 composite | 0.77 (0.61, 0.97) | 0.024 |
| Aβ42/Aβ40 | 0.52 (0.41, 0.68) | <0.001 |
|
| ||
| NPTX2 composite | 0.53 (0.41, 0.70) | <0.001 |
| p-tau181 | 1.93 (1.56, 2.40) | <0.001 |
|
| ||
| NPTX2 composite | 0.63 (0.50, 0.80) | <0.001 |
| t-tau | 1.66 (1.37, 2.02) | <0.001 |
|
| ||
| NPTX2 composite | 0.61 (0.45, 0.81) | 0.001 |
| Aβ42/Aβ40 | 0.72 (0.52, 1.00) | 0.050 |
|
| ||
| p-tau181 | 1.55 (1.14, 2.11) | 0.005 |
| NPTX2 composite | 0.67 (0.53, 0.85) | 0.001 |
| Aβ42/Aβ40 | 0.62 (0.48, 0.82) | 0.001 |
| t-tau | 1.41 (1.12, 1.78) | 0.004 |
Note: Models were adjusted by baseline age, sex, and years of education. The NPTX2 × CSF biomarker interaction term was not significant in any model (all p>0.12, see text for details).
Figure 3.
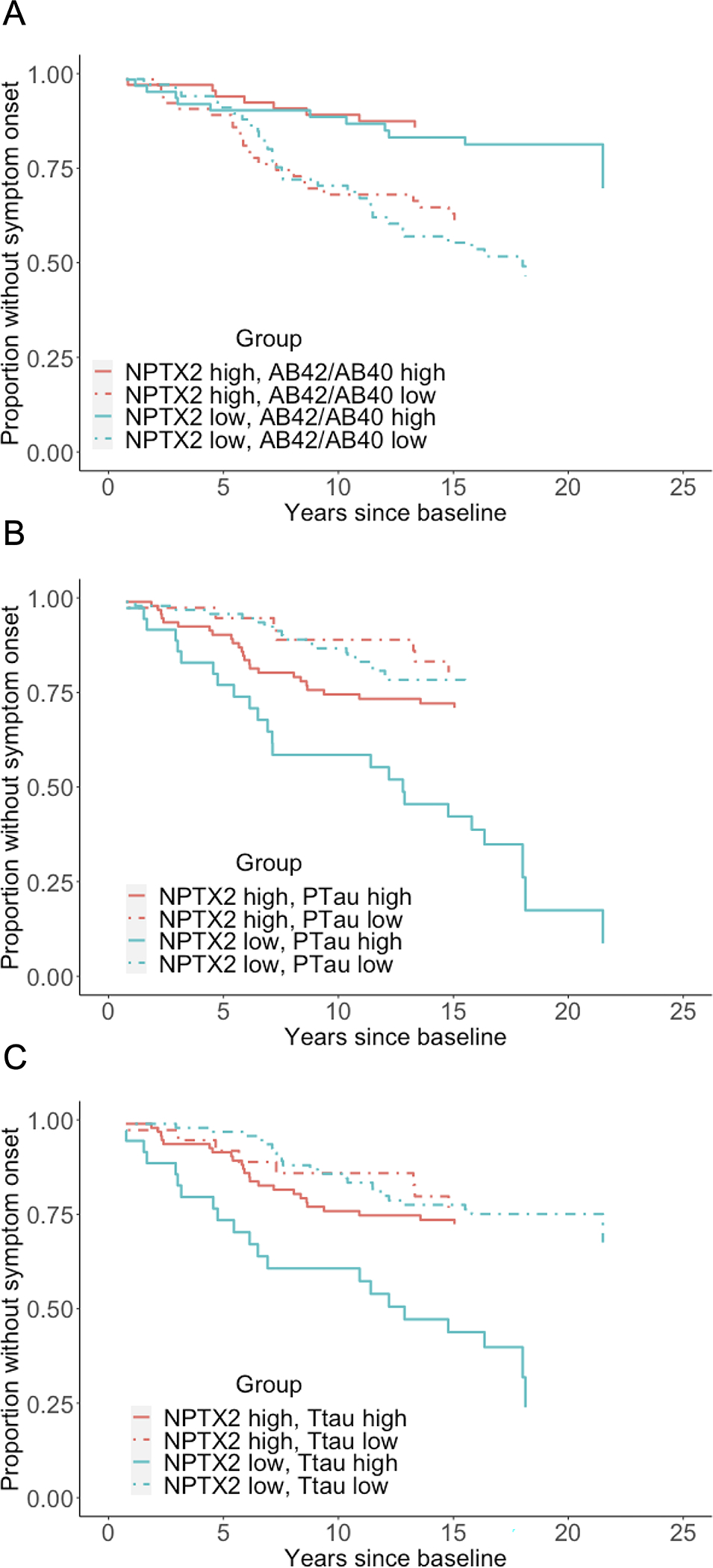
Kaplan-Meier plots showing the proportion of participants who remain MCI symptom-free as a function of baseline level of CSF NPTX2 (as measured by the NPTX2 composite score) and baseline levels of (A) CSF Aβ42/Aβ40; (B) CSF p-tau181; or (C) CSF t-tau. High vs. low levels of each CSF measure were based on the median.
There were no interactions between the NPTX2 composite score and Aβ42/Aβ40 (HR=0.95, CI=0.76–1.18, p=0.65), ptau181 (HR=0.85, CI=0.70–1.04, p=0.12), and t-tau (HR=0.97, CI=0.84–1.12, p=0.65) in relation to MCI symptom onset. Likewise, binary indicators based on cutpoints35 for high vs. low AD pathology (defined as abnormal Aβ42/Aβ40 and abnormal p-tau181) and absence of AD pathology (defined as normal Aβ42/Aβ40 and normal p-tau181) did not interact with NPTX2 in relation to symptom onset (p’s >0.3, data not shown). In models that included NPTX2, Aβ42/Aβ40, and either p-tau181 or t-tau, all three biomarkers were significantly associated with time to MCI symptom onset (see Table 3). Model fit statistics indicated that adding NPTX2 to a model that included Aβ42/Aβ40 and p-tau181 (and covariates) improved model fit (AIC without NPTX2 = 746.8; AIC with NPTX2 = 737.3) and the difference in model fits was significant (chi-square=11.5, p<0.01). Likewise, adding NPTX2 to a model including Aβ42/Aβ40 and t-tau improved model fit (AIC without NPTX2 = 745.6; AIC with NPTX2 =737.6, chi-square=10.0, p<0.01). Results remained the same when additionally covarying hypertension at baseline (i.e., presence vs. absence), data not shown.
Relationship of baseline CSF AD biomarkers to rate of change in NPTX2
As shown in Table 4, linear mixed effects models indicated that more abnormal (i.e., lower) levels of Aβ42/Aβ40 and more abnormal (i.e., higher) levels of p-tau181 and t-tau at baseline were associated with more negative trajectories of NPTX2 over both the short term (i.e., NIH data only, all p<=0.002; mean follow-up=2.2 years, max=8 years; Figure 4) and the long-term (NIH & JHU data, all p<=0.024; mean follow-up =10.4 years). Importantly, when both Aβ42/Aβ40 and one of the tau measures (and their interactions with time) were included as predictors, higher p-tau181 and t-tau remained significantly associated with more negative NPTX2 trajectories, while Aβ42/Aβ40 was not related to change in NPTX2 over the long-term (both p>=0.10), nor over the short-term when covarying p-tau181 (p=0.07; see Table 4). Additionally, higher levels of p-tau181 and t-tau at baseline were associated with higher levels of NPTX2 at baseline (all p<0.001), independent of Aβ42/Aβ40 levels. In contrast, lower baseline Aβ42/Aβ40 levels were related to lower baseline NPTX2 levels only after accounting for baseline p-tau181 levels. Results were similar when also adjusting for APOE-ε4 and education and when using dichotomous indicators (based on tertile cut-points) for high vs. low Aβ42/Aβ40 and the tau measures (data not shown). Baseline levels and longitudinal change in NPTX2 were unrelated to baseline age, sex, and years of education in models that did not include the CSF AD biomarkers and in models that covaried both Aβ42/Aβ40 and p-tau181 [or t-tau] (and their interactions with time), all p>0.06.
Table 4.
Results of linear mixed effects models with the NPTX2 composite score over time as the outcome and baseline AD biomarkers as predictors.
| Model predictors | Model 4 | |||
|---|---|---|---|---|
|
| ||||
| Short-term follow-up (NIH data only) | Long-term follow-up (NIH & JHU data) | |||
|
| ||||
| Estimate (95% CI) | p-value | Estimate (95% CI) | p-value | |
|
| ||||
| Aβ42/Aβ40 only model | ||||
| Aβ42/Aβ40 – Level | −0.04 (−0.16, 0.08) | 0.53 | −0.01 (−0.12, 0.09) | 0.81 |
| Aβ42/Aβ40 – Slope | 0.04 (0.02, 0.06) | <0.001 | 0.01 (0.00, 0.02) | 0.024 |
|
| ||||
| p-tau181 only model | ||||
| p-tau181 – Level | 0.54 (0.43, 0.66) | <0.001 | 0.44 (0.34, 0.53) | <0.001 |
| p-tau181 – Slope | −0.05 (−0.07, −0.02) | <0.001 | −0.02 (−0.03, −0.01) | <0.001 |
|
| ||||
| t-tau only model | ||||
| t-tau – Level | 0.40 (0.28, 0.52) | <0.001 | 0.31 (0.21, 0.41) | <0.001 |
| t-tau – Slope | −0.03 (−0.06, −0.01) | 0.002 | −0.01 (−0.02, −0.01) | 0.003 |
|
| ||||
| Aβ42/Aβ40 & p-tau model | ||||
| Aβ42/Aβ40 – Level | 0.26 (0.15, 0.38) | <0.001 | 0.24 (0.13, 0.34) | <0.001 |
| Aβ42/Aβ40 – Slope | 0.02 (−0.00, 0.05) | 0.07 | 0.01 (−0.00, 0.01) | 0.31 |
| p-tau181 – Level | 0.67 (0.55, 0.80) | <0.001 | 0.55 (0.44, 0.66) | <0.001 |
| p-tau181 – Slope | −0.03 (−0.06, −0.01) | 0.006 | −0.02 (−0.03, −0.01) | <0.001 |
|
| ||||
| Aβ42/Aβ40 & t-tau model | ||||
| Aβ42/Aβ40 – Level | 0.11 (−0.2, 0.23) | 0.09 | 0.10 (0.00, 0.21) | 0.06 |
| Aβ42/Aβ40 – Slope | 0.03 (0.01, 0.05) | 0.005 | 0.01 (−0.00, 0.02) | 0.10 |
| t-tau181 – Level | 0.43 (0.30, 0.55) | <0.001 | 0.34 (0.23, 0.45) | <0.001 |
| t-tau181 – Slope | −0.02 (−0.05, −0.00) | 0.027 | −0.01 (−0.02, −0.00) | 0.023 |
Note: All models were adjusted for baseline age, sex, and their interactions with time.
Figure 4.
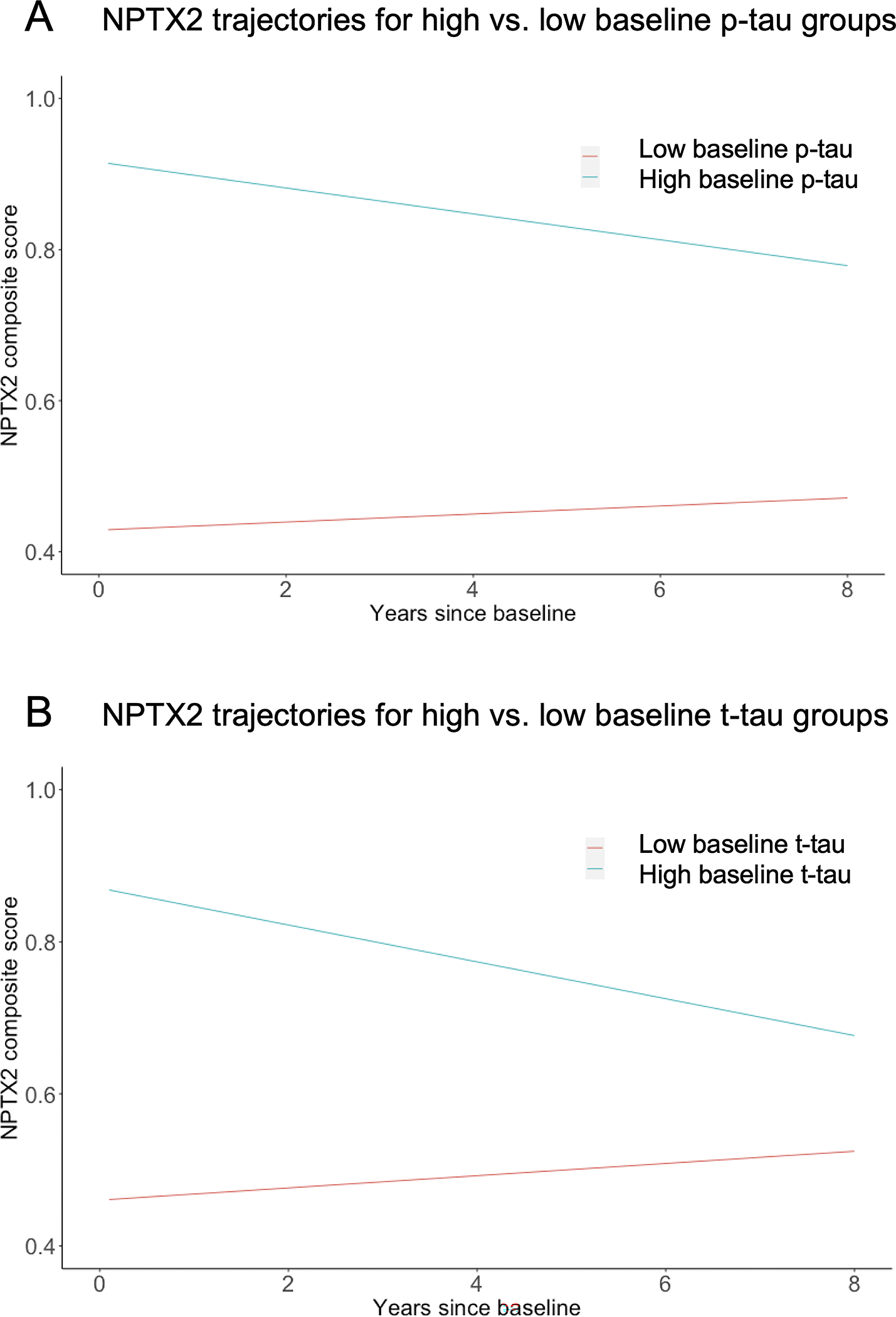
Adjusted estimates of longitudinal trajectories of NPTX2 composite score over time for (A) participants with high vs. low CSF p-tau181 (high = upper tertile of the distribution; low=bottom two tertiles of the distribution); and (B) participants with high vs. low t-tau (high = upper tertile of the distribution; low = bottom two tertiles of the distribution).
Relationship of CSF AD biomarkers to rate of change in NPTX2
Baseline NPTX2 composite scores were not associated with the rate of change in CSF Aβ42/Aβ40, p-tau181, or t-tau over time, both over the short term (NIH data only, all p>0.5) and over the long term (NIH & JHU data, all p>0.35; data not shown). Results were the same when additionally adjusting for APOE-ε4 and education.
All results from Cox models and linear mixed effects models were the same when excluding participants with a diagnosis of impaired-not-MCI (data not shown).
Discussion
The current study examined the relationship between levels of the synaptic protein NPTX2 – measured as a composite of three highly correlated peptides derived from mass spectrometry – to time to onset of symptoms of MCI among individuals with normal cognition at baseline. There are several findings of note. First, lower levels of NPTX2 were associated with an earlier time to MCI symptom onset (both for all-cause MCI and for MCI due to clinically diagnosed AD). Second, baseline NPTX2 levels were associated with time to MCI symptom onset for progression within 7 years from baseline and after 7 years from baseline, suggesting that low NPTX2 levels are an early marker for synaptic degeneration during preclinical AD. Third, baseline NPTX2 levels improved prediction of MCI symptom onset after accounting for CSF levels of Aβ42/Aβ40, p-tau181, and t-tau, and NPTX2 did not interact with the CSF AD biomarkers in relation to symptom onset. This suggests that NPTX2 and CSF biomarkers of amyloid and tau are independently associated with clinical progression. Taken together, these results provide strong evidence for NPTX2 as a valuable prognostic biomarker of synaptic dysfunction during preclinical AD that may improve the prediction of AD clinical symptom onset after accounting for levels of amyloid and tau.
Our results are consistent with and expand on prior studies reporting that lower levels of CSF NPTX2 are associated with an increased risk of progression from MCI to AD dementia,19 and with greater cognitive decline among cognitively normal20 and mixed diagnosis groups.14, 16, 20 While prior studies have been conducted among individuals with an average baseline age of >=70 years, participants in this study were largely middle aged at baseline, indicating that changes in NPTX2 are observed in mid-life among participants who will subsequently develop MCI.
When examining the long-term trajectories of NPTX2, we found that higher (i.e., more abnormal) levels of CSF p-tau181 and t-tau at baseline were associated with higher baseline levels of NPTX2 and greater decline in NPTX2 over time, independent of CSF Aβ42/Aβ40 levels. These results are broadly consistent with the one prior longitudinal study of NPTX2, which found that higher (i.e., more abnormal) CSF levels of p-tau181/Aβ42 ratios at baseline were associated with greater declines in NPTX2 over time among non-demented participants,20 though the use of ratios did not allow for the examination of individual relationships of Aβ42 and p-tau181 with NPTX2. These results are also consistent with two prior cross-sectional studies finding positive associations between higher CSF NPTX2 and higher CSF p-tau17 and t-tau14, 17 levels among cognitively normal and impaired participants.
The finding that higher baseline CSF p-tau181 and t-tau levels are associated with higher baseline NPTX2 levels but greater rates of decline in NPTX2 over time may reflect an initial up-regulation in NPTX2 in response to increasing tau phosphorylation, neurofibrillary tangle formation, and/or release of tau to maintain synaptic integrity and network excitability.17 These findings suggest that continually elevated p-tau and t-tau levels and/or progression of other AD-related pathophysiological processes may then result in a decline in NPTX2, reflecting synaptic and/or neuronal loss. This interpretation would be consistent with the view of NPTX2 as a resilience factor.17 It is also consistent with the finding of a positive correlation between mean yearly increases in CSF p-tau181 and NPTX2 within a group that combined cognitively normal and MCI participants.20 Interestingly, NPTX2 levels did not appear to change with age, suggesting that potential resilience provided by NPTX2 is not age-dependent. An alternative possibility is that both NPTX2 and p-tau/t-tau levels initially increase in response to a common third factor associated with AD-pathophysiological processes, such as inflammation.36 Future studies are needed to test these hypotheses.
Interestingly, baseline Aβ42/Aβ40 was not associated with rates of change in NPTX2 when covarying p-tau181, suggesting that the association of low Aβ42/Aβ40 and rate of decline in NPTX2 was at least partially mediated by the presence of high p-tau181. Furthermore, when covarying p-tau181 levels, low baseline Aβ42/Aβ40 was associated with low baseline NPTX2 and this relationship was not evident when p-tau181 levels were not covaried. This suggests that NPTX2 levels may vary depending on the levels of Aβ42/Aβ40 and p-tau during the earliest stages of preclinical AD, with NPTX2 declining when Aβ42 first begins accumulating in the brain, and then increasing as p-tau levels start rising, and finally declining again as synapses and/or neurons are lost. Prior cross-sectional studies have reported somewhat mixed results on this topic, with two studies finding lower NPTX2 among participants low CSF Aβ42,14, 16 and one study not finding such an association.17 Additionally, a longitudinal study reported that within-subject decreases in Aβ42 were correlated with decreases in NPTX2, particularly among participants with MCI,20 though tau levels were not controlled in these analyses. Our results highlight the importance of examining NPTX2 and other synaptic markers in conjunction with AD biomarkers (rather than in isolation) to capture the potentially complex dynamic relationships among these measures. Future studies examining longitudinal trajectories of NPTX2 in relation to trajectories of both CSF AD biomarkers and MRI measures of brain atrophy, or tau PET imaging of neurofibrillary tangle burden will be important for delineating the time course of NPTX2 during preclinical AD. Additional investigations are also needed to determine if NPTX2 improves prediction of MCI independent of other comorbid pathologies that also affect synapses, (e.g., cerebrovascular disease37 and alpha-synuclein38).
The finding that baseline NPTX2 levels were not associated with changes in CSF Aβ42/Aβ40, p-tau181, or t-tau over time suggests that alterations in NPTX2 do not precede the initial accumulation of these AD biomarkers. However, because the synaptic trafficking of NPTX2 that underlies its presence in CSF can be altered by multiple factors, including protein expression and circuit dynamics,17, 39 further studies are needed to evaluate the role of NPTX2 in the evolution of AD.
NPTX2 regulates activity-dependent strengthening of excitatory synapses from pyramidal neurons onto inhibitory parvalbumin interneurons.13 As such, NPTX2 is important for balancing excitatory and inhibitory neural activity, maintaining rhythmic synchrony in cortical circuits17, 40, and has been linked to resting-state functional connectivity in cognitive brain networks.15 The circuit selectivity of NPTX2 is distinct from general excitatory synaptic markers, such as neurogranin,10, 11 and may explain why it provides additive prediction to the standard AD biomarkers.14
In light of the finding that changes in NPTX2 occur many years prior to the emergence of AD clinical symptoms, this raises the possibility of developing novel therapeutics that target NPTX2 and related synaptic proteins to maintain synaptic (and cognitive) function in the face of accumulating pathology.11 Given that NPTX2 does not appear to be an AD-specific marker, but a marker of synaptic degeneration across neurodegenerative diseases,11, 38, 41 therapeutics targeting NPTX2 may have broad applicability.
The current findings have several limitations. Participants were primarily White, highly educated, and have a strong family history of AD. Consequently, results may not generalize to the general population. Second, we did not examine how within-subject changes in NPTX2 relate to within-subject changes in CSF biomarkers of amyloid and tau, and the potential dependence of these changes on other factors. It is also unclear what variables may be associated with individual differences in NPTX2 levels prior to and after the accumulation of amyloid and tau. Important directions for future research include identifying potentially modifiable factors that modulate NPTX2 levels and developing sensitive plasma measures of NPTX2, which would facilitate its incorporation into more diverse cohorts and clinical trials.
Acknowledgements
This work was supported by the National Institutes of Health [grant numbers U19-AG033655, P30-AG005146, and ]. The BIOCARD Study consists of 7 Cores and two Projects with the following members: (1) the Administrative Core (Marilyn Albert); (2) the Clinical Core (Marilyn Albert, Anja Soldan, Corinne Pettigrew, Greg Pontone, Leonie Farrington, Jules Gilles, Nicole Johnson, Maura Grega, Gay Rudow, Scott Rudow); (3) the Imaging Core (Michael Miller, Susumu Mori, Tilak Ratnanather, Andrea Faria, Anthony Kolasny, Kenichi Oishi, Laurent Younes, Hanzhang Lu, Peter vanZijl); (4) the Biospecimen Core (Abhay Moghekar, Alexandria Lewis, Megha Patel, Sara Ho); (5) the Informatics Core (Ann Ervin, David Shade, Jennifer Jones, Hamadou Coulibaly, Tara Foley); (6) the Biostatistics Core (Mei-Cheng Wang, Yuxin (Daisy) Zhu, Jiangxia Wang); (7) the Neuropathology Core (Juan Troncoso, David Nauen, Javier Redding, Roberta Knox); (8) Project 1 (Paul Worley, Jeremy Walston), and (9) Project 2 (Mei-Cheng Wang, Yifei Sun). The authors are grateful to the members of the BIOCARD Scientific Advisory Board who provide continued oversight and guidance regarding the conduct of the study including: Drs. David Holtzman, William Jagust, David Knopman, Walter Kukull, and Kevin Grimm, and Drs. John Hsiao and Laurie Ryan, who provide oversight on behalf of the National Institute on Aging. The authors thank the members of the BIOCARD Resource Allocation Committee who provide ongoing guidance regarding the use of the biospecimens collected as part of the study, including: Drs. Constantine Lyketsos, Carlos Pardo, Gerard Schellenberg, Leslie Shaw, Madhav Thambisetty, and John Trojanowski. The authors acknowledge the contributions of the Geriatric Psychiatry Branch of the intramural program of NIMH who initiated the study (Principal investigator: Dr. Trey Sunderland). The authors are indebted to Dr. Karen Putnam, who provided documentation of the Geriatric Psychiatry Branch study procedures and the data files received from NIMH.
Footnotes
Potential Conflicts of Interest
Nothing to report.
Data Availability
Data used in these analyses are available through standard application procedures described on the BIOCARD website (biocard-se.org).
References
- 1.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jansen WJ, Ossenkoppele R, Knol DL, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA 2015;313:1924–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moghekar A, Li S, Lu Y, et al. CSF biomarker changes precede symptom onset of mild cognitive impairment. Neurology 2013;81:1753–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenberg BD, Pettigrew C, Soldan A, et al. CSF Alzheimer Disease Biomarkers: Time-Varying Relationships With MCI Symptom Onset and Associations With Age, Sex, and ApoE4. Neurology 2022;99:e1640–e1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vos SJ, Xiong C, Visser PJ, et al. Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study. Lancet Neurol 2013;12:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ossenkoppele R, Pichet Binette A, Groot C, et al. Amyloid and tau PET-positive cognitively unimpaired individuals are at high risk for future cognitive decline. Nat Med 2022;28:2381–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991;30:572–580. [DOI] [PubMed] [Google Scholar]
- 8.Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 2006;27:1372–1384. [DOI] [PubMed] [Google Scholar]
- 9.Masliah E, Mallory M, Alford M, et al. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 2001;56:127–129. [DOI] [PubMed] [Google Scholar]
- 10.Camporesi E, Nilsson J, Brinkmalm A, et al. Fluid Biomarkers for Synaptic Dysfunction and Loss. Biomark Insights 2020;15:1177271920950319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomez de San Jose N, Massa F, Halbgebauer S, Oeckl P, Steinacker P, Otto M Neuronal pentraxins as biomarkers of synaptic activity: from physiological functions to pathological changes in neurodegeneration. J Neural Transm (Vienna) 2022;129:207–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chapman G, Shanmugalingam U, Smith PD. The Role of Neuronal Pentraxin 2 (NP2) in Regulating Glutamatergic Signaling and Neuropathology. Front Cell Neurosci 2019;13:575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang MC, Park JM, Pelkey KA, et al. Narp regulates homeostatic scaling of excitatory synapses on parvalbumin-expressing interneurons. Nat Neurosci 2010;13:1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galasko D, Xiao M, Xu D, et al. Synaptic biomarkers in CSF aid in diagnosis, correlate with cognition and predict progression in MCI and Alzheimer’s disease. Alzheimers Dement (N Y) 2019;5:871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soldan A, Moghekar A, Walker KA, et al. Resting-State Functional Connectivity Is Associated With Cerebrospinal Fluid Levels of the Synaptic Protein NPTX2 in Non-demented Older Adults. Front Aging Neurosci 2019;11:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swanson A, Willette AA, Alzheimer’s Disease Neuroimaging I. Neuronal Pentraxin 2 predicts medial temporal atrophy and memory decline across the Alzheimer’s disease spectrum. Brain Behav Immun 2016;58:201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao MF, Xu D, Craig MT, et al. NPTX2 and cognitive dysfunction in Alzheimer’s Disease. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nilsson J, Gobom J, Sjodin S, et al. Cerebrospinal fluid biomarker panel for synaptic dysfunction in Alzheimer’s disease. Alzheimers Dement (Amst) 2021;13:e12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spellman DS, Wildsmith KR, Honigberg LA, et al. Development and evaluation of a multiplexed mass spectrometry based assay for measuring candidate peptide biomarkers in Alzheimer’s Disease Neuroimaging Initiative (ADNI) CSF. Proteomics Clin Appl 2015;9:715–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Libiger O, Shaw LM, Watson MH, et al. Longitudinal CSF proteomics identifies NPTX2 as a prognostic biomarker of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 2021;17:1976–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pletnikova O, Kageyama Y, Rudow G, et al. The spectrum of preclinical Alzheimer’s disease pathology and its modulation by ApoE genotype. Neurobiol Aging 2018;71:72–80. [DOI] [PubMed] [Google Scholar]
- 22.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 23.Albert M, Soldan A, Gottesman R, et al. Cognitive changes preceding clinical symptom onset of mild cognitive impairment and relationship to ApoE genotype. Curr Alzheimer Res 2014;11:773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albert M, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gervaise-Henry C, Watfa G, Albuisson E, et al. Cerebrospinal Fluid Abeta42/Abeta40 as a Means to Limiting Tube- and Storage-Dependent Pre-Analytical Variability in Clinical Setting. J Alzheimers Dis 2017;57:437–445. [DOI] [PubMed] [Google Scholar]
- 27.Toombs J, Foiani MS, Wellington H, et al. Amyloid beta peptides are differentially vulnerable to preanalytical surface exposure, an effect incompletely mitigated by the use of ratios. Alzheimers Dement (Amst) 2018;10:311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willemse E, van Uffelen K, Brix B, Engelborghs S, Vanderstichele H, Teunissen C. How to handle adsorption of cerebrospinal fluid amyloid beta (1–42) in laboratory practice? Identifying problematic handlings and resolving the issue by use of the Abeta(42)/Abeta(40) ratio. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 2017;13:885–892. [DOI] [PubMed] [Google Scholar]
- 29.Pettigrew C, Soldan A, Wang J, et al. Longitudinal CSF Alzheimer’s disease biomarker changes from middle age to late adulthood. Alzheimers Dement (Amst) 2022;14:e12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Na CH, Sathe G, Rosenthal LS, et al. Development of a novel method for the quantification of tyrosine 39 phosphorylated alpha- and beta-synuclein in human cerebrospinal fluid. Clin Proteomics 2020;17:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacLean B, Tomazela DM, Shulman N, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010;26:966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pettigrew C, Soldan A, Zhu Y, et al. Cortical thickness in relation to clinical symptom onset in preclinical AD. Neuroimage Clin 2016;12:116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akaike H A new look at the statistical model identification. IEEE Transactions on Automatic Control 1974;19:716–723. [Google Scholar]
- 34.Hilbe JM. Negative binomial regression. Cambridge, UK: Cambridge University Press, 2011. [Google Scholar]
- 35.Dakterzada F, Lopez-Ortega R, Arias A, et al. Assessment of the Concordance and Diagnostic Accuracy Between Elecsys and Lumipulse Fully Automated Platforms and Innotest. Front Aging Neurosci 2021;13:604119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y) 2018;4:575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shao K, Shan S, Ru W, Ma C. Association between serum NPTX2 and cognitive function in patients with vascular dementia. Brain Behav 2020;10:e01779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boiten WA, van Steenoven I, Xiao MF, et al. Pathologically Decreased CSF Levels of Synaptic Marker NPTX2 in DLB Are Correlated with Levels of Alpha-Synuclein and VGF. Cells 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao MF, Roh SE, Zhou J, et al. A biomarker-authenticated model of schizophrenia implicating NPTX2 loss of function. Sci Adv 2021;7:eabf6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pelkey KA, Barksdale E, Craig MT, et al. Pentraxins coordinate excitatory synapse maturation and circuit integration of parvalbumin interneurons. Neuron 2015;85:1257–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Ende EL, Xiao M, Xu D, et al. Neuronal pentraxin 2: a synapse-derived CSF biomarker in genetic frontotemporal dementia. J Neurol Neurosurg Psychiatry 2020;91:612–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data used in these analyses are available through standard application procedures described on the BIOCARD website (biocard-se.org).