

Abstract
Background:
Epigenetic regulation of vascular remodeling in pulmonary hypertension (PH) is poorly understood. Transcription regulating, histone acetylation code alters chromatin accessibility to promote transcriptional activation. Our goal was to identify upstream mechanisms that disrupt epigenetic equilibrium in PH.
Methods:
Human pulmonary artery smooth muscle cells (hPASMC), human idiopathic pulmonary arterial hypertension (iPAH):hPASMC cells, iPAH lung tissue, failed donor lung tissue (FDL), human pulmonary microvascular endothelial cells (hPMVECs), iPAH:PASMC and non-iPAH:PASMC RNA-seq databases, NanoString nCounter, and CUT&RUN were utilized to investigate histone acetylation, hyperacetylation targets, protein and gene expression, sphingolipid activation, cell proliferation, and gene target identification. SPHK2 KO were compared to control C57BL/6NJ mice after 3 weeks of hypoxia and assessed for indices of pulmonary hypertension.
Results:
We identified that hPASMC are vulnerable to the transcription promoting epigenetic mediator histone acetylation resulting in alterations in transcription machinery and confirmed its pathological existence in PH:PASMC cells. We report that Sphingosine Kinase 2 (SPHK2) is elevated as much as 20-fold in iPAH lung tissue and is elevated in iPAH:PASMC cells. During PH pathogenesis, nuclear SPHK2 activates nuclear bioactive lipid Sphingosine 1-phosphate (S1P) catalyzing enzyme and mediates transcription regulating histone H3K9 acetylation (Ac-H3K9) through Endothelial Monocyte Activating Polypeptide (EMAP) II. In iPAH lungs we identified a 4-fold elevation of the reversible epigenetic transcription modulator Ac-H3K9 : H3 ratio. Loss of SPHK2 inhibited hypoxic induced PH and Ac-H3K9 in mice. We discovered that pulmonary vascular endothelial cells are a priming factor of the EMAP II/SPHK2/S1P axis that alters the acetylome with a specificity for PASMC, through hyperacetylation of histone H3K9. Using CUT&RUN we further show that EMAP II mediated SPHK2 has the potential to modify the local transcription machinery of pluripotency factor Krὓppel-like factor (KLF4) by hyperacetylating KLF4 Cis-Regulatory Elements while deletion and targeted inhibition of SPHK2 rescues transcription altering Ac-H3K9.
Conclusion:
SPHK2 expression and its activation of the reversible histone H3K9 acetylation in hPASMC, represent new therapeutic targets that could mitigate PH vascular remodeling.
Keywords: Acetylation, epigenetic, Pulmonary Hypertension, Basic Science Research, Pulmonary Biology
Graphical Abstract
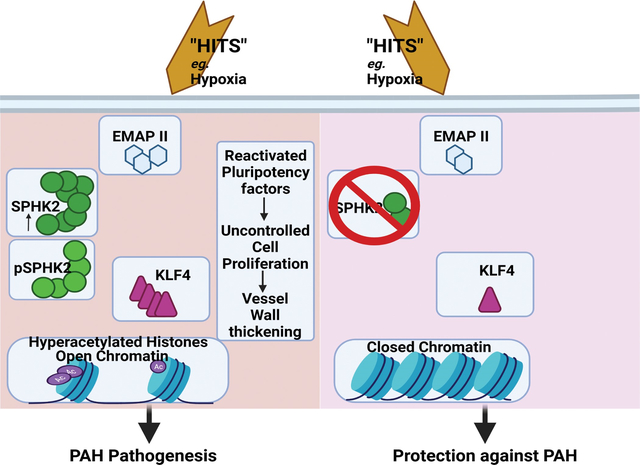
Introduction
Pulmonary Hypertension (PH), is a progressive, incurable and devastating vascular disease with pathophysiological differences1. Risk factors and sources of vascular injury in PH vascular remodeling are diverse and include factors such as genetic mutations, epigenetic modifications, and environmental triggers such as hypoxia, immune/inflammatory response, pathogens, and drugs1. Accumulation of enough “hits” to the pulmonary vasculature can lead to uncontrolled vascular cell proliferation that mirrors cancer cell phenotypes.
Pulmonary vascular remodeling in part is regulated by epigenetic machinery that alters gene expression2,3 and has already demonstrated potential clinical value in PH4,5. Reversible modification of chromatin availability by epigenetic regulating elements includes histone modifications and DNA methylation. Although recent studies suggest that histone acetylation impacts vascular endothelial cell (ECs) metabolism in PH6, targeting histone acetylation modifications through inhibition of downstream enzymatic regulator histone deacetylases (HDACs) were not encouraging7. Histone acetylation marks such as histone H3K9 acetylation play key roles in aging and vascular tone8,9 suggesting an indispensable epigenetic role in PH pathobiology10,11. Therefore, deeper molecular insight into the histone acetylation code in PH and identification of targets upstream of the histone acetylation process is necessary.
Nuclear Sphingosine Kinase 2 (SPHK2) and its product Sphingosine 1-Phosphate (S1P) are known to be involved in histone acetylation in different cell types and disease pathogenesis including uncontrolled cancer cells proliferation12. Previous studies demonstrate that as a predominant lipid constituent of all cellular membranes, sphingolipid homeostasis mediates cellular adaptation. Prior PH studies focus on the inflammatory role that isovariant Sphingosine Kinase 1 (SPHK1) activation of S1P inside-out cell surface-receptor signaling has in pulmonary vascular smooth muscle cells (SMCs) occlusive arteriopathy13,14. Differing in subcellular location with distinct kinetic properties, SPHK1 mainly localizes to the cytosol where it mediates inflammatory responses via S1P and its cell surface receptors via inside out signaling, while Sphingosine Kinase 2 (SPHK2) is cytoplasmic and nuclear, with nuclear localization and export signals15,16. A previous study reported that isovariant SPHK2 expression was not altered in PH, and thus a role for SPHK2 activation of nuclear S1P was minimized and the epigenetic role of nuclear isovariant SPHK2 / S1P axis in PH was not explored17. However, Gairhe et al.18 reported that SPHK2 was elevated in perivascular tissue of PH patients, but did not explore its significance, role, or epigenetic impact of SPHK2. Therefore, re-evaluation of the epigenetic role that the SPHK2/S1P axis has in PH is warranted and could provide greater insight into the contribution that epigenetic regulators have in the progression of PH.
Here, we report that the active chromatin mark, H3K9 acetylation and the SPHK2/S1P axis have a role in PH vascular remodeling. Moreover, SPHK2 phosphorylation, nuclear expression, and mediation of gene transcription is activated by a known pro-inflammatory mediator Endothelial Monocyte Activating Polypeptide II (EMAPII)1, activating histone H3K9 acetylation specifically in pulmonary vascular SMCs and transcriptional activation of known vascular remodeling gene Krὓppel-like factor (KLF4)19. These findings suggest that a second look at the role of SPHK2 isovariant in PH is warranted.
Methods
For expanded methods, please see the online supplement.
Human idiopathic (i)PAH (Group 1 PH) and FDL (Failed Donor Lungs) lung tissues and iPAH:PASMCs
All studies involving human lung tissues and cells were conducted according to the Institutional review board. Deidentified archived tissues and pulmonary arterial smooth muscle cells (PASMCs) obtained from the Pulmonary Hypertension Breakthrough Initiative (PHBI) biorepository from patients with Idiopathic Pulmonary Arterial Hypertension (iPAH: Group 1 PH) at time of lung transplantation and patients without PH that were failed donor lungs (FDL) (See Table S1). According to the PHBI biorepository (https://ipahresearch.org/phbi-research/), tissues were obtained in the operating room and immediately snap frozen at the collection site, cataloged, and stored to maintain tissue integrity. According to the PHBI biorepository (https://ipahresearch.org/phbi-research/), cells were isolated from the left lower lung, phenotypically characterized, and frozen in aliquots for distribution to PHBI biorepository requests. Tissue for histology were ethanol fixed paraffin embedded (EFPE) slides as per the PHBI biorepository(https://ipahresearch.org/phbi-research/).
Experimental PH Mouse model
Animal experimental procedure was performed in accordance with the guidelines issued by the University of Notre Dame/Indiana University Institutional Animal and Use Committee. Mice were maintained in a controlled pathogen free animal facility at all times with 12h light/dark cycles at a temperature of 22–24° Celsius with routine check on animal status and were provided standard irradiated mouse chow ad lib. In the experimental rodent PH model, 12–14 wk old male mice of SPHK2-deficient [SPHK2 KO] in C57BL/6NJ background20 and age-matched C57BL/6NJ21 [Wild type: WT] male mice were exposed to hypoxia (10% O2) in a ventilated chamber or normoxia for 21 days. Randomization on 12–14 week littermate cohorts was performed by using a coin flip methodology and distribution of littermates across all experimental groups and none of these animals were excluded. All procedures and analysis were performed in a blinded manner.
Cell culture and treatments
Primary human PASMCs (hPASMCs), pulmonary microvascular endothelial cells (hPMVECs) purchased from Lonza (Walkersville, MD) and iPAH: PASMCs were cultured in complete growth medium or conditioned media in a humidified atmosphere or 1% O2 with 5% CO2 at 37° C. For all studies, passages 5–10 were used for hPASMCs and hPMVECs.
Statistical analysis
The data are presented as means ±1 standard error of mean (SEM) or as median and inter-quartile range from at least three independent experiments, if not mentioned otherwise. Statistical significance was determined with unpaired Student’s t-test or one-way or two-way ANOVA for experiments with n≥6 and normality confirmed data sets by Shapiro-Wilk test after log transformation or otherwise Kruskal-Wallis or Kolmogorov-Smirnov non-parametric test for experiments with n<6 using GraphPad Prism software.
Results
Pulmonary expression of an active chromatin mark, acetylated H3K9, and an epigenetic modulator SPHK2 are elevated in iPAH patients.
Studies focused on the identification and role of a key active chromatin mark acetyl-H3K9 (Ac-H3K9) in vascular remodeling. Ac-H3K9, an active chromatin mark of gene promoter regions and enhancers, is an enzymatically regulated reversible modification of acetylation of lysine residues to facilitate the opening of chromatin spatial structures and the activation of transcription during proliferation22, inflammation23 and biological aging8. Using human tissue from 40 different individual patients lungs, 20 failed donor (FDL) and 20 PH lungs classified as idiopathic (iPAH) with unknown cause or etiology obtained from the PHBI biorepository, western blot analysis of the Ac-H3K9: H3 ratios (equal protein loading conditions, confirmed by Ponceau S staining) determined that H3K9 acetylation levels are significantly elevated in iPAH patient samples compared to FDL controls (Fig 1A,B). Utilization of Ac-H3K9: tubulin as a secondary validation of equal protein loading for challenging human tissues collected under variable circumstances confirmed that Ac-H3K9 levels are significantly elevated in iPAH (Fig 1C). Increased H3K9 was seen in both male and female iPAH patient cohort lung tissues (See Fig S1A). Cancer and inflammatory studies recently identified nuclear SPHK2 activated S1P as an important epigenetic modulator complex of Ac-H3K924,25. In contrast to known isovariant SPHK1 regulation of inside-out cell signaling in PH, isovariant SPHK2 has largely been ignored17. Transcript levels of SPHK2 were significantly elevated in human iPAH lung tissue specimens (Fig 1D, see Fig S1B). Analysis of SPHK2 expression using a C-terminal antibody that recognizes all SPHK2 isoforms determined that SPHK2 protein was significantly elevated (Fig 1E,F, see Fig S1C) compared to FDL tissues (SPHK2 antibodies 17096-1-AP, Proteintech and 32346, CST yielded similar results).
Figure 1. H3K9 acetylation and SPHK2 expression show a potential correlation in PAH patients’ lungs.
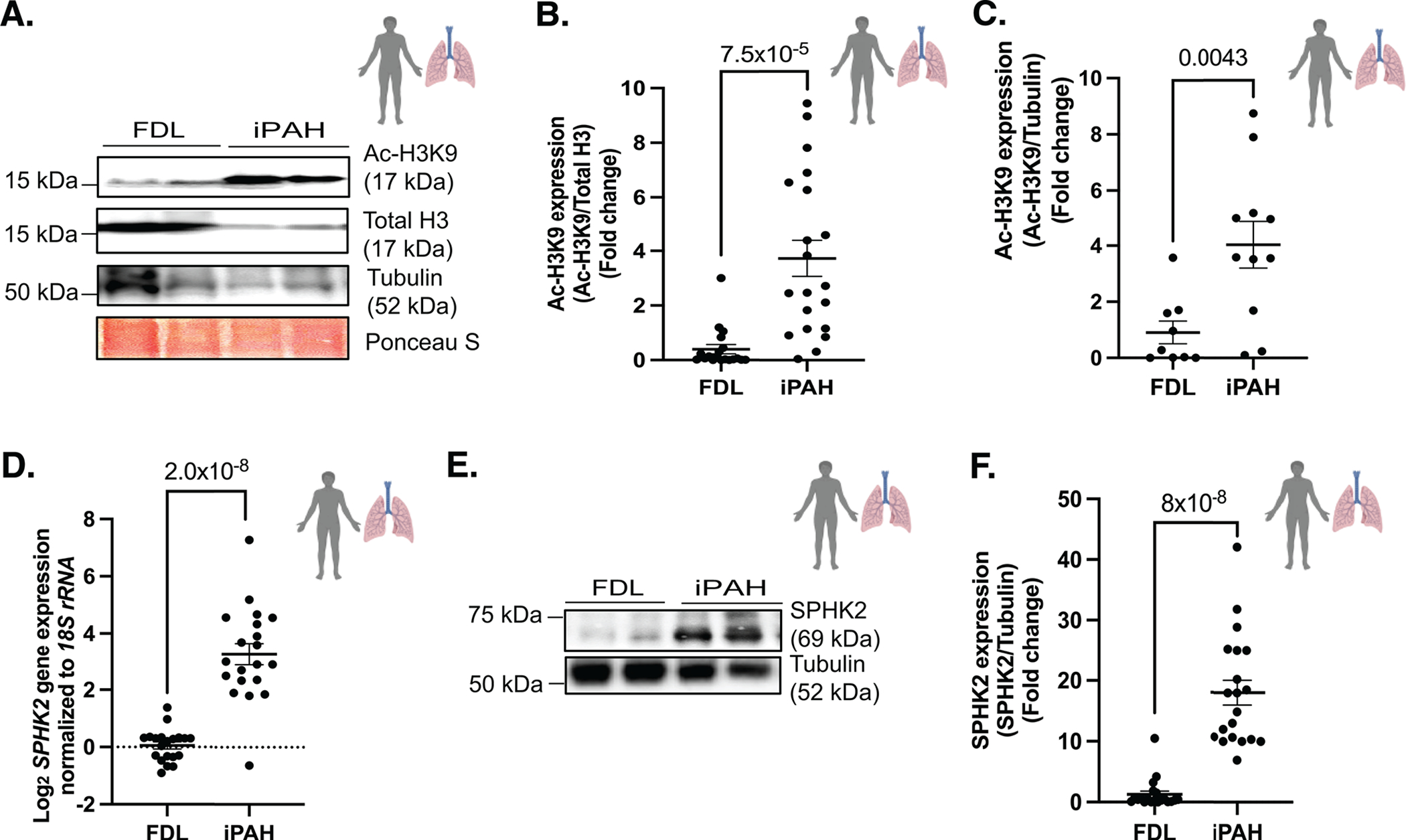
(A) Representative immunoblot probed for Ac-H3K9, total H3, tubulin and Ponceau S staining in protein lysates of human idiopathic pulmonary arterial hypertension (iPAH: type of Group 1 PH) lung or failed donor lung (FDL) tissue specimens and (B) quantitation of Ac-H3K9/Total H3, n=19–20 (C) quantitation of Ac-H3K9/Tubulin in protein lysates of human iPAH (n=11) or FDL (n=9). (D) SPHK2 expression levels normalized against 18S rRNA in iPAH lung and FDL tissues. n=20 (E) Representative immunoblot probed for SPHK2 and Tubulin in protein lysates of human iPAH (type of Group 1 PH) lung or FDL tissue specimens and (F) quantitation of SPHK2/Tubulin in protein lysates of human iPAH lung or FDL, n=20. P values are calculated using unpaired t-test and results are shown as means ± SEM.
SPHK2 deficiency confers protection against hypoxia induced-PH pulmonary vascular resistance, right ventricle hypertrophy and vascular remodeling in experimental PH mouse model.
A hypoxia induced-PH26 mouse model using 10% oxygen for 21 days was characterized by evaluating PH parameters: pulmonary vascular resistance (PVR) and pulmonary acceleration time [(PAT: which inversely and linearly correlates with mean pulmonary arterial pressure (MPAP)27) (PVR and PAT were assessed by non-invasive echocardiography)], right ventricle hypertrophy (RVH) (assessed by Fulton’s index), and distal vascular remodeling (assessed by histology studies). In this study male mice were used, as previous studies indicate that female mice have minimal chronic hypoxia-induced pulmonary hypertensive responses and experience possible fluctuation and interference from ovarian hormones9. C57BL/6NJ (WT) mice exposed to 10% O2 (WT hypoxia) showed elevated PVR, RVH and peripheral pulmonary artery muscularization and decreased PAT compared to the C57BL/6NJ mice exposed to 21% O2 (WT normoxia) (Fig 2A,B,C,D, see Fig S2A). In addition, cardiac output (assessed by non-invasive echocardiography) showed no significant difference among the groups (See Fig S2B). SPHK2 KO mice in hypoxia (SPHK2 KO hypoxia) demonstrated no change in cardio-pulmonary parameters and were similar to SPHK2 KO normoxia mice, while also demonstrating a reduced PVR and RVH, with reduction in pulmonary artery muscularization and PAT compared to its background WT hypoxia mice (Fig 2A,B,C,D). Moreover, hypoxic WT mice showed increased expression of Ac-H3K9: H3 while ablation of SPHK2 (SPHK2 KO) minimized the susceptibility for hypoxia induced hyperacetylation in mice (Fig 2E) suggesting that SPHK2 deficiency may offer protection against PH through rescuing disrupted epigenetic equilibrium of H3K9 acetylation.
Figure 2. SPHK2 ablation confers protection against experimental PH and hyperacetylation of H3K9 in hypoxia-induced experimental PH mouse model.
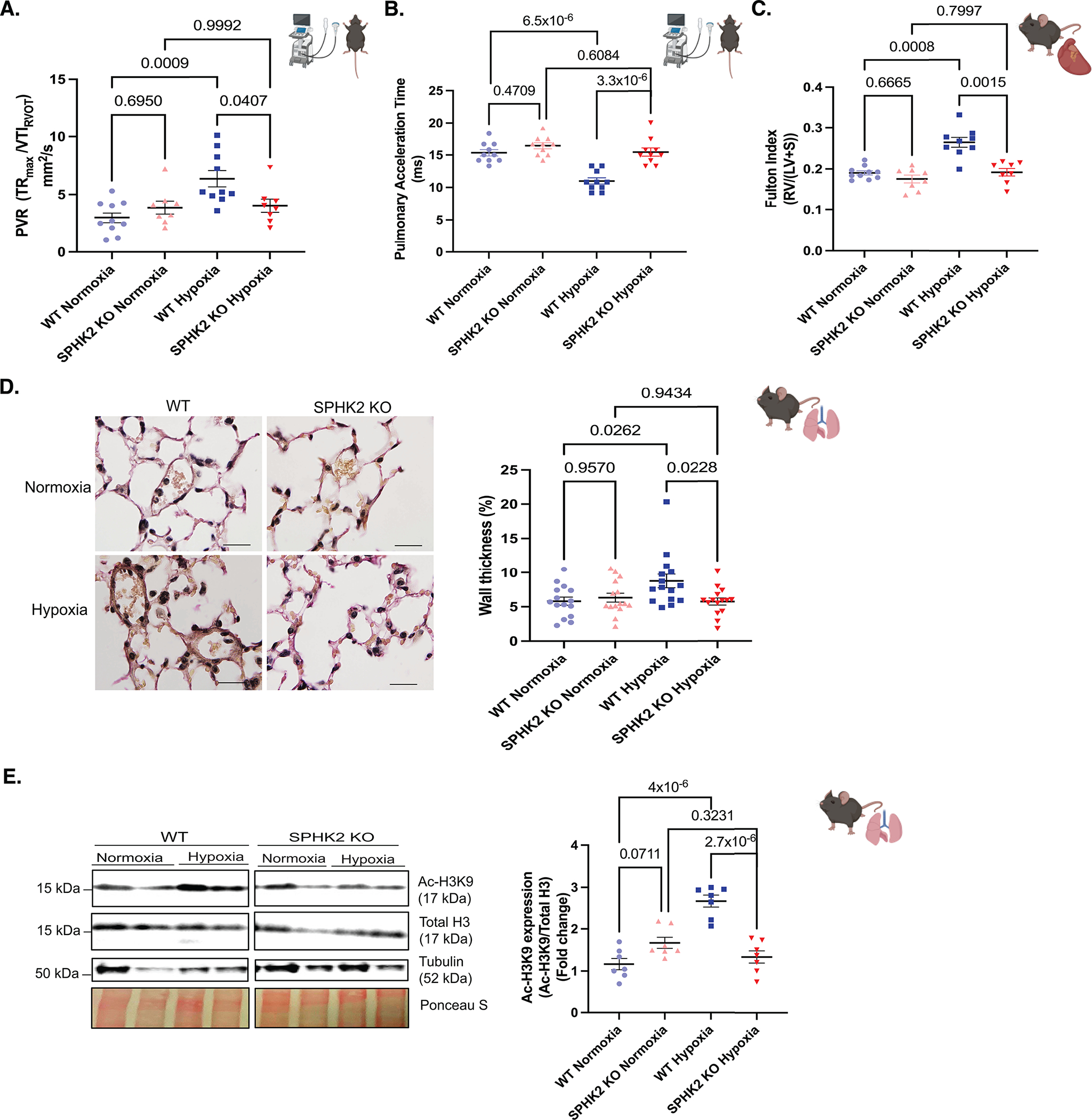
SPHK2 KO or wild type (WT) control mice (C57BL/6NJ) were subjected to 3 wks of hypoxia (10% O2) or normoxia (room air). (A) Pulmonary vascular resistance (the maximum velocity of tricuspid regurgitation/the velocity time integral of the right ventricular outflow tract, TRmax velocity/VTIRVOT) n=8–10/group. (B) Pulmonary acceleration time (PAT), n=8–10/group. (C) RV hypertrophy/Fulton Index (the weight ratio of the right ventricle divided by the sum of left ventricle and septum, RV/(LV + S)) n=8–10/group. (D) Representative images of elastin-stained distal pulmonary vessels and, wall thickness of distal pulmonary vessels in elastin-stained lung tissue sections (wall thickness (%) = (2 × medial wall thickness / external diameter) × 100) n= 3/group randomly selected mice per each group and n=15 images of distal pulmonary vessels, scale bar is 10 μm, n=5–6/group. (E) Representative immunoblot probed for Ac-H3K9, total H3, Tubulin and ponceau S staining in whole tissue lysates from WT or SPHK2 KO in normoxia or hypoxia on same blot n=5–7/group and, quantitation of Ac-H3K9/Total H3. P values are calculated using one-way ANOVA following Tukey’s multiple comparisons test, and results are shown as means ± SEM.
Pro-inflammatory mediator EMAPII is upregulated in human iPAH lung tissue and promotes histone H3K9 hyperacetylation in vascular smooth muscle cells but not in vascular endothelial cells.
Endothelial monocyte activating polypeptide II (EMAP II), proteolytically cleaved from the Aminoacyl tRNA Synthetase Complex Interacting Multifunctional Protein 1 (AIMP1, also known as SCYE-1 and p43), is a known pro-inflammatory mediator of vascular growth and promotes signs of pulmonary hypertension in a murine model of Bronchopulmonary dysplasia (BPD)28. In vitro studies determined that EMAPII activation of the SPHK1/S1P inside out signaling axis regulated by cell membrane S1P receptors modulates vascular SMC proliferation13. Our studies examined if EMAPII can initiate nuclear activation of the SPHK2/S1P axis. AIMP1 expression was increased in iPAH lung tissue (Fig 3A,B and See Fig S3A). In contrast to low basilar perivascular expression29, immunohistochemical analysis determined that AIMP1 expression was elevated in thickened remodeled vessels of iPAH lungs (See Fig S3B). In mechanistic studies, the potential of EMAPII in histone H3K9 acetylation was examined in two vessel wall cell populations, endothelial cells (human pulmonary microvascular endothelial cells: hPMVECs) and smooth muscle cells (human pulmonary artery smooth muscle cells: hPASMCs). In hPASMCs, recombinant (r) EMAPII treatment induced a dynamic time dependent H3K9 acetylation (Fig 3C,D) but not H4K5 acetylation (as nuclear SPHK2 has previously been reported to increase multiple acetylation patterns, our studies explored the more robustly impacted acetylation patterns of histone H3K9 and H4K524) (See Fig S3C). Hyperacetylation patterns were not detected in hPMVECs (Fig 3E,F).
Figure 3. EMAP II has the potential to be a key pathogenic mediator in PH through modulating epigenetic equilibrium via histone H3K9 acetylation uniquely in vascular SMCs.
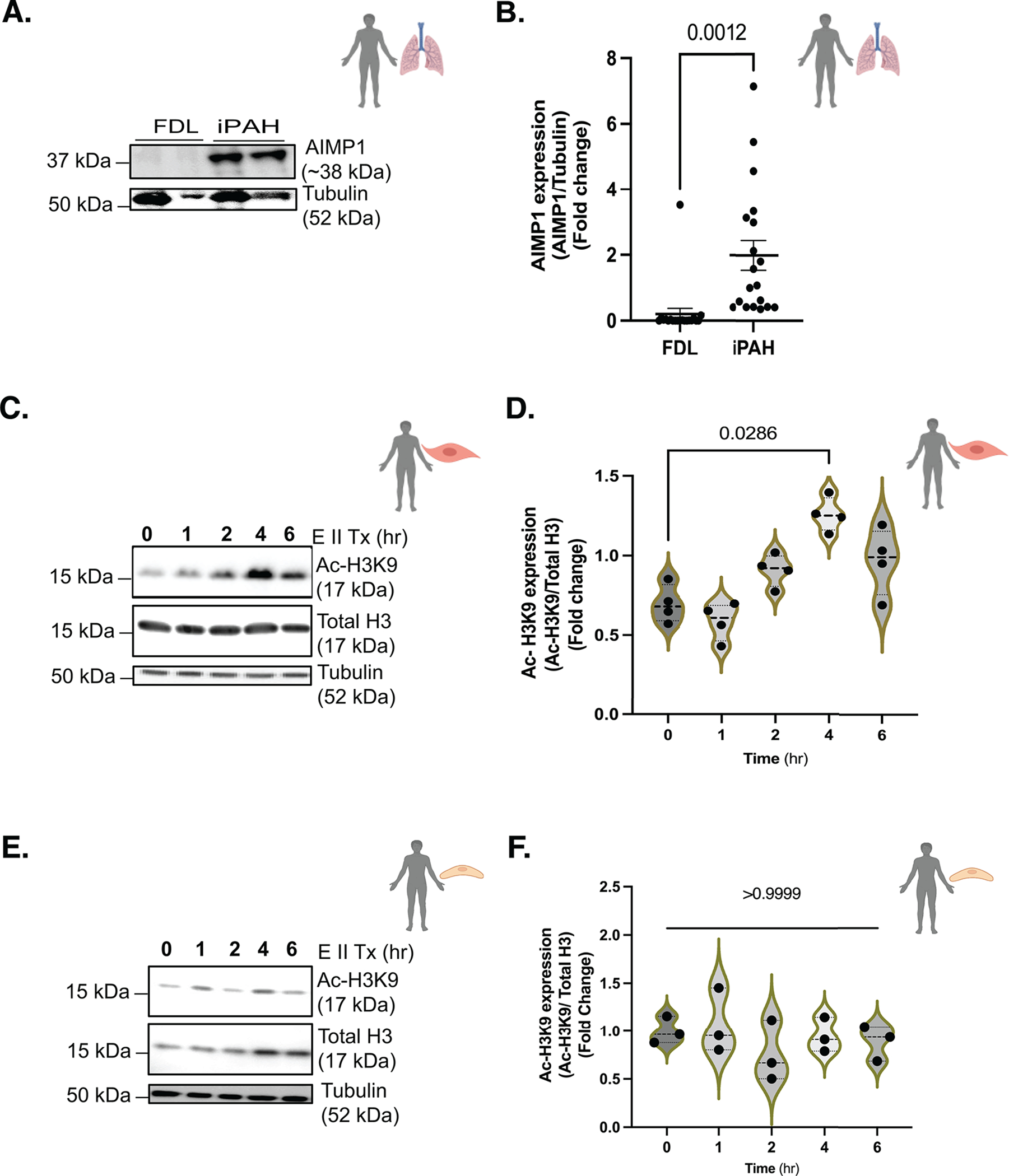
(A) Representative immunoblot probed for AIMP1 (precursor form of EMAP II) and Tubulin in protein lysates of human iPAH or FDL, n=19–20/group and, (B) quantitation of AIMP1 (AIMP1/Tubulin) in protein lysates of human iPAH or FDL, n=19–20/group. (C) Representative immunoblot probed for Ac-H3K9, total H3 or tubulin in hPASMCs following EMAP II treatment for 0, 1, 2, 4 and 6 hours and (D) quantitation of Ac-H3K9 expression levels normalized against total H3 in hPASMCs, n=4. (E) Representative immunoblot probed for Ac-H3K9, total H3 or tubulin in hPMVECs following EMAP II treatment for 0, 1, 2, 4 and 6 hours and (F) quantitation of Ac-H3K9 expression levels normalized against total H3 in hPMVECs, n=3. P values are calculated using unpaired t-test or Kolmogorov-Smirnov non-parametric testing and results are shown as means ± SEM or median and inter-quartile range.
EMAPII activates nuclear SPHK2 and nuclear lipid S1P to promote histone H3K9 acetylation and proliferative SMC phenotype via SPHK2.
Previous studies link acetylation of histone H3K9 with nuclear pSPHK2 and S1P24. In WT hypoxia mice, an increased expression of nuclear pSPHK2 and Ac-H3K9 compared to WT normoxia mice (See Fig S3D and Fig 2E) was observed, while SPHK2 ablation inhibited hypoxia induced changes in Ac-H3K9 (Fig 2E). Furthermore, the prominent co-expression of EMAPII and pSPHK2, suggests a potential interaction between EMAPII and phosphorylation of SPHK2 (See Fig S3B). rEMAPII treated hPASMCs induced the nuclear localization of SPHK2/pSPHK2 after 2 hours (Fig 4A,B,C,D); but not in hPASMCs pretreated with an EMAPII neutralizing antibody (see Fig S3E). Immunofluorescence in hPASMCs confirmed EMAPII induction of nuclear pSPHK2 (purple), superimposed with DAPI stained nuclei (blue), with minimal cytoplasmic expression (stained for F-actin-green) (Fig 4A). In contrast, in rEMAPII treated hPMVECs, SPHK2 activation was not significant compared to non-treated hPMVECs, where basal expression of pSPHK2, in both hPMVEC nuclear and cytoplasmic subcellular fractions of treated and non-treated cells was unchanged (See Fig S3F,G) suggesting a cell type-specific differential response and a more austere role for EMAPII activated SPHK2 in hPASMCs.
Figure 4. EMAP II promotes nuclear activation of SPHK2 that in turn generates nuclear lipid, S1P in vascular SMCs.
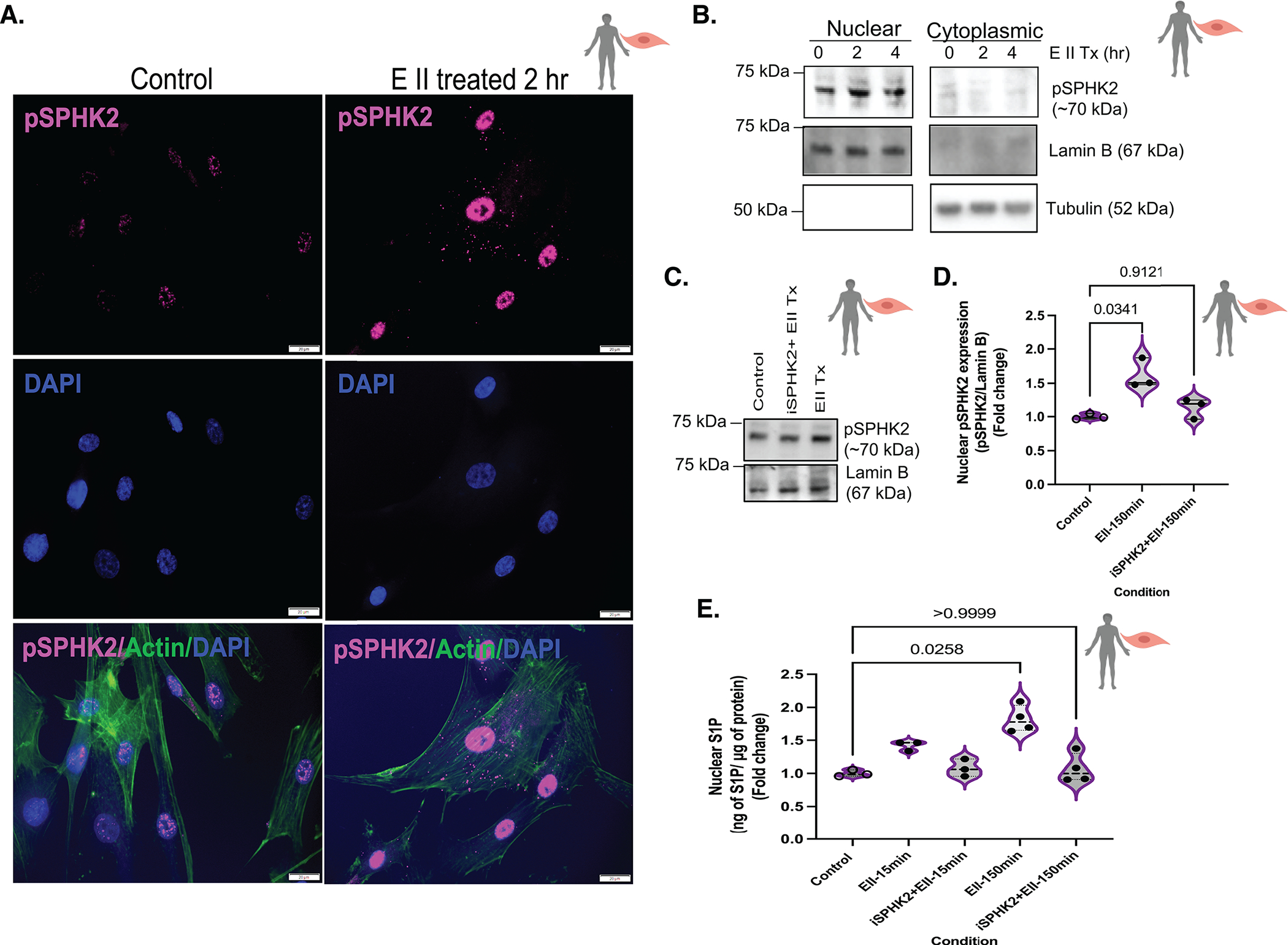
(A) Representative immunocytochemistry images of pSPHK2 (pink), actin (green, cytoplasmic marker) and DAPI (blue, nuclear) coimmunostaining in EMAP II treated (2 hr) or vehicle treated fixed hPASMCs, scale bar is 20 μm, n=3. (B) Representative immunoblot probed for pSPHK2, tubulin and lamin B in cytoplasmic and nuclear fractions of hPASMCs following EMAP II treatment for 0, 2 and 4 hours, n=3. (C) Representative immunoblot probed for pSPHK2 and lamin B in nuclear fractions of hPASMCs following EMAP II treatment (150 minutes) with or without SPHK2 inhibitor (D) quantification of nuclear pSPHK2/lamin B, n=3. (E) ELISA-nuclear C18-S1P levels normalized against 1 μg of nuclear proteins in the nuclear fractions of hPASMCs following EMAP II for 15 or 150 minutes with or without SPHK2 inhibitor, n=3 or 4/group. P values are calculated using Kruskal-Wallis against control or Kolmogorov-Smirnov non-parametric test and results are shown as median and inter-quartile range.
Historically, hyper-proliferation of PASMCs is a vital characteristic in PH pathogenesis. With the significant impact that EMAPII had on nuclear predominant activation of SPHK2 and histone H3K9 hyperacetylation in hPASMCs, these studies focused on EMAPII/SPHK2 mediated epigenetic activity.
Immunoblotting confirmed the EMAPII induced nuclear expression of pSPHK2 while pretreatment with SPHK2 inhibitor [iSPHK2: ABC294640 (Echelon Biosciences Inc., Salt Lake City, UT)] of 10 μM for 1 hour prior to EMAPII treatment ablated the EMAPII induced nuclear activation of SPHK2 (Fig 4C,D). Nuclear induction of pSPHK2 is associated with phosphorylation of sphingosine resulting in nuclear S1P. Following rEMAPII stimulation, there was an elevation of nuclear S1P (Fig 4E) in hPASMCs, whereas pretreating with iSPHK2 suppressed EMAPII generated nuclear S1P expression (Fig 4E). Furthermore, following rEMAPII treatment there was a significant decrease in the HDAC activity as compared to control while iSPHK2 rescued the loss of HDAC activity (See Fig S4A), demonstrating a cell specific impact of SPHK2 epigenetic activation in PASMC. Unchanged expression of HDAC1/2 (See Fig S4B,C) supports the previously reported function of nuclear S1P generated by nuclear pSPHK2 as an inhibitor of HDAC activity and promoter of H3K9 acetylation24.
EMAPII induction of the nuclear SPHK2/S1P axis promotes global acetylation of histone H3K9 in human vascular SMCs.
Knockdown of SPHK2 in hPASMCs using specific siRNA significantly attenuated rEMAPII induced Ac-H3K9 upregulation at 4 hours and successful knockdown of SPHK2 was confirmed by immunoblotting (Fig 5A,C). This observation further was confirmed and determined EMAPII mediated H3K9 hyperacetylation targets in hPASMCs by CUT&RUN experiments using an acetylated H3K9 antibody. In chromatin mapping by CUT&RUN, two independent biological replicates were used according to the modENCODE repository guidelines30. CUT&RUN analysis of rEMAP II treated hPASMCs demonstrated that rEMAPII induced a 2.8-fold hyperacetylated histone H3K9 as compared to the control (Fig 5D,F). Pretreatment with iSPHK2 partially diminished EMAPII promoted hyperacetylation of H3K9 (Fig 5F). Importantly, 10951 of 11578 (94.58%) of the loci were successfully associated with genes (NIH PAVIS, https://manticore.niehs.nih.gov/pavis2/) (Fig 5E) and gene ontology studies identified growth, cellular response to stress and protein modification processes were among the pathways altered by EMAPII mediated hyperacetylation in hPASMCs (ShinyGO v0.75, http://bioinformatics.sdstate.edu/go/) (Fig 5G). The role of EMAPII/SPHK2 in cell growth is supported by increased cell proliferation rate in EMAPII treated hPASMCs that is rescued by pretreatment with iSPHK2 (Fig 5H).
Figure 5. EMAP II mediated SPHK2 signaling promotes global hyperacetylation of histone H3K9 in vascular SMCs.
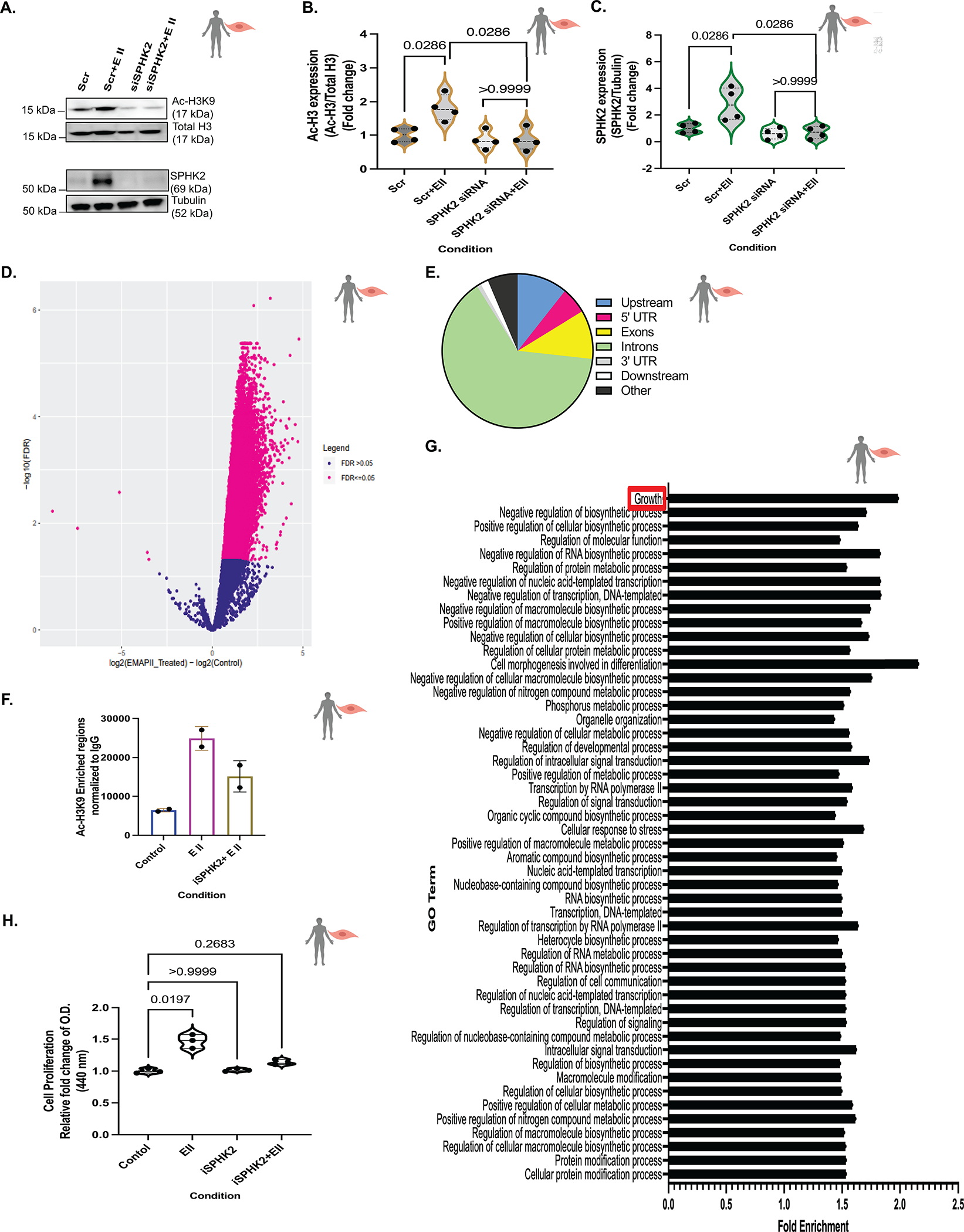
(A) Representative immunoblot probed for Ac-H3K9, total H3, SPHK2 and tubulin in whole cell lysates of hPASMCs following siRNA mediated SPHK2 silencing and post-transfection EMAP II treatment for 4 hours and (B) quantitation of Ac-H3K9/total H3 and (C) quantification of SPHK2/tubulin, n=4. (D) Volcano plot showed the log2-fold changes and statistical significance of hyperacetylated H3K9 regions calculated after differential binding analysis of EMAP II treated vs control hPASMCs. Pink points indicate significantly hyperacetylated H3K9 regions in EMAP II (right to 0) or in control (left to 0). FDR=0.05, n=2 (E) Genome wide distribution of differentially enriched hyperacetylated H3K9 peaks (log2-fold change > 1, p value < 0.05) n=2. (F) Number of peaks of Ac-H3K9 normalized to IgG in with or without SPHK2 inhibitor and EMAP II treated (2–3 hours) hPASMCs, n=2. (G) Gene Ontology results using differentially enriched Ac-H3K9 peaks in EMAP II treated hPASMCs, n=2. (H) Cell proliferation rate in hPASMCs treated with vehicle or EMAP II following SPHK2 inhibitor treatment for 24 hours, n=3. P values are calculated using Kruskal-Wallis against control or Kolmogorov-Smirnov non-parametric test and results are shown as means ± SEM or median and inter-quartile range.
KLF4 is a potential downstream epigenetic target of nuclear SPHK2/histone H3K9 acetylation pathway in PH patients and hypoxia-induced experimental PH mouse model.
Peak annotation and visualization identified 1186 upstream sites out of differentially acetylated sites by EMAPII compared to control (See Supplementary Data Set File) that can be responsible for transcription factors binding sites/regulatory elements. Moreover, 5554 differentially acetylated sites by EMAPII compared to control are altered by iSPHK2 pretreatment. The common 755 sites out of differentially acetylated upstream sites by EMAPII compared to control, that is also altered by iSPHK2 pretreatment, suggest the genes that have the highest potential to be transcriptionally regulated by EMAPII/SPHK2 axis through cistrome/epicistrome (Fig 6A, See Supplementary Table File). The local vascular remodeling gene, and a pluripotent/Yamanaka factor19, KLF4 frequently implicated in enhancer-dependent transcriptional regulation31, was among these 755 sites (See Supplementary Table File). Out of these 755 sites, the stem cell markers that possess H3K9 acetylation that lie on ENCODE candidate Cis-Regulatory Elements and other Yamanaka factors (SOX2 and OCT4) were transcriptionally profiled in human iPAH tissues using a custom Nanostring code set. Several stem cell markers showed an increasing trend in iPAH tissues compared to the control tissues (See Fig S5A) despite the heterogenous nature of lung tissue.
Figure 6. EMAP II mediated SPHK2 signaling promotes local hyperacetylation of histone H3K9 of KLF4 enhancers and alters the local transcription machinery of KLF4 in vascular SMCs.
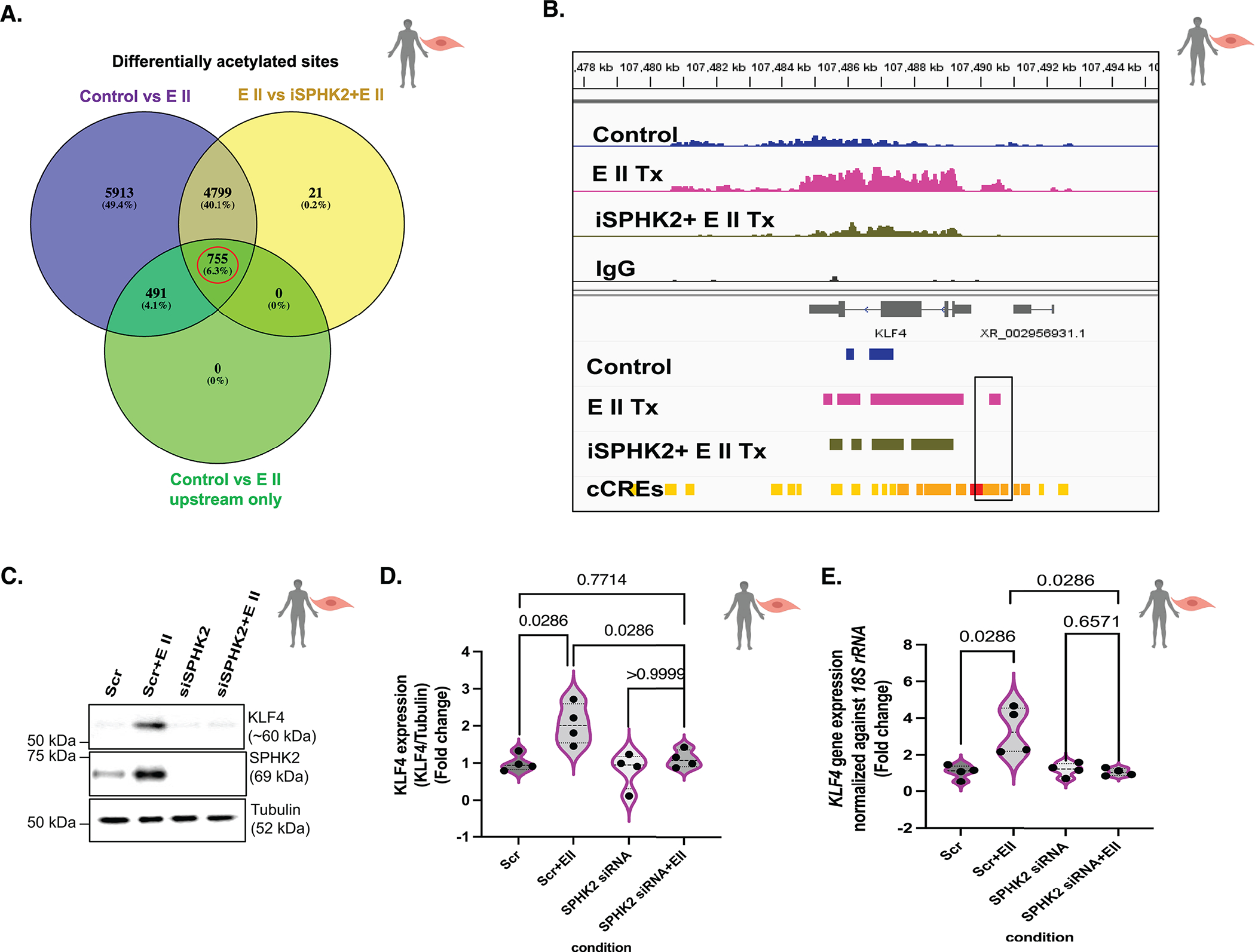
(A) The Venn’s diagram of differential acetylated sites in control vs EMAP II (total) (purple), EMAP II vs iSPHK2+EMAP II (yellow) and control vs EMAPII only in 5’UTR and upstream with fold enrichment greater than 2 (green). The red circle indicates the potential gene set with potential upstream candidate regulatory elements that would be differentially acetylated by EMAP II through SPHK2 in hPASMCs. Venn diagram is created using Venny 2.1 (an online interactive tool), n=2/group (B) Snapshot of IGV view of KLF4 gene in Ac-H3K9 CUT&RUN data of with or without SPHK2 inhibitor and EMAP II treated (2–3 hours) hPASMCs. (cCRE= candidate Cis-Regulatory Elements) n=2/group (C) Representative immunoblot probed for KLF4, SPHK2 and tubulin in whole cell lysates of hPASMCs following siRNA mediated SPHK2 silencing and post-transfection EMAP II treatment for 6–8 hours, and (D) quantitation of KLF4/tubulin and (E) KLF4 expression levels normalized against 18S rRNA in hPASMC cells following siRNA mediated SPHK2 silencing and EMAP II treatment for 6 hours, n=4. P values are calculated using Kolmogorov-Smirnov non-parametric testing and results are shown as median and inter-quartile range.
As KLF4 is a transcriptional regulator of cellular proliferation, differentiation, apoptosis and somatic cell reprogramming32,33, its activation in hPASMCs was explored. rEMAPII treatment increased differentially enriched Ac-H3K9 regions in KLF4 as compared to control (Fig 6B). Notably, pretreatment of hPASMCs with iSPHK2 rescued the level of Ac-H3K9 regions in EMAPII treated cells, suggesting that EMAPII activates local KLF4 gene transcription machinery through an SPHK2 mediated hyperacetylation mechanism (Fig 6). Gene annotation using NIH PAVIS showed 5’UTR of KLF4 as an Ac-H3K9 enriched region and moreover, ENCODE candidate Cis-Regulatory Elements overlapped with the enriched regions unique to EMAPII treated CUT&RUN peaks (Fig 6B). Knockdown of SPHK2 ablated EMAPII induced H3K9 acetylation and induction of KLF4 transcription and translation (Fig 6C,D,E) in PASMCs, suggesting EMAPII/SPHK2/S1P/epigenetics may play a critical role in KLF4 transcription machinery. The transcription and protein levels of KLF4 were increased in both human iPAH lung tissue extracts and experimental hypoxia induced-PH mouse lung tissues compared to their controls (See Fig S5B,C,D,E,F,G). Moreover, in contrast to hypoxia induced KLF4 expression in WT hypoxia mice as compared to WT normoxia mice, SPHK2 deficient mice were resistant to hypoxia induced KLF4 expression (See Fig S5B,C,D).
Vascular EC population is a priming factor of EMAPII/SPHK2/Ac-H3K9 mediated KLF4 pathway in vascular SMCs.
The potential of ECs to be the endogenous source of EMAPII to initiate SPHK2/Ac-H3K9/KLF4 pathway in SMCs was explored. Under chemical hypoxia, ECs showed an increase in AIMP1 expression (Fig S3H) but not PASMCs (Fig S3H). hPMVEC cultured in normoxia or hypoxia (1% O2) and cell culture supernatants (endothelial cells conditioned media: ECM) were collected and utilized to treat SMCs (Fig 7A). Hypoxia ECM showed an increased expression of EMAPII compared to the normoxia ECM (Fig 7B). hPASMCs treated with hypoxia derived ECM demonstrated increased KLF4 expression and Ac-H3K9 levels while preincubation with EMAPII neutralizing antibody restored levels consistent with normoxia ECM treated cells (control) suggesting a vital role of EC secreted EMAPII in SMC’s KLF4 expression (Fig 7B,C,D,E). Moreover, hPASMCs pretreated with SPHK2 siRNA showed a significant inhibition of hypoxia derived ECM induction of KLF4 and Ac-H3K9 expression (Fig 8C,D,E,F) and restored levels consistent with the control. Thus, suggesting a prominent role for endothelial cell secreted factors in activation of the histone acetylome and a contributor to reactivation of the pluripotency factor such as KLF4 and importantly, demonstrating the significance of SPHK2 inhibition in Ac-H3K9 mediated KLF4 pathway (Fig 7G).
Figure 7. EMAP II/SPHK2/Ac-H3K9 mediated KLF4 signaling is a novel pathway that exists in PH disease pathogenesis.
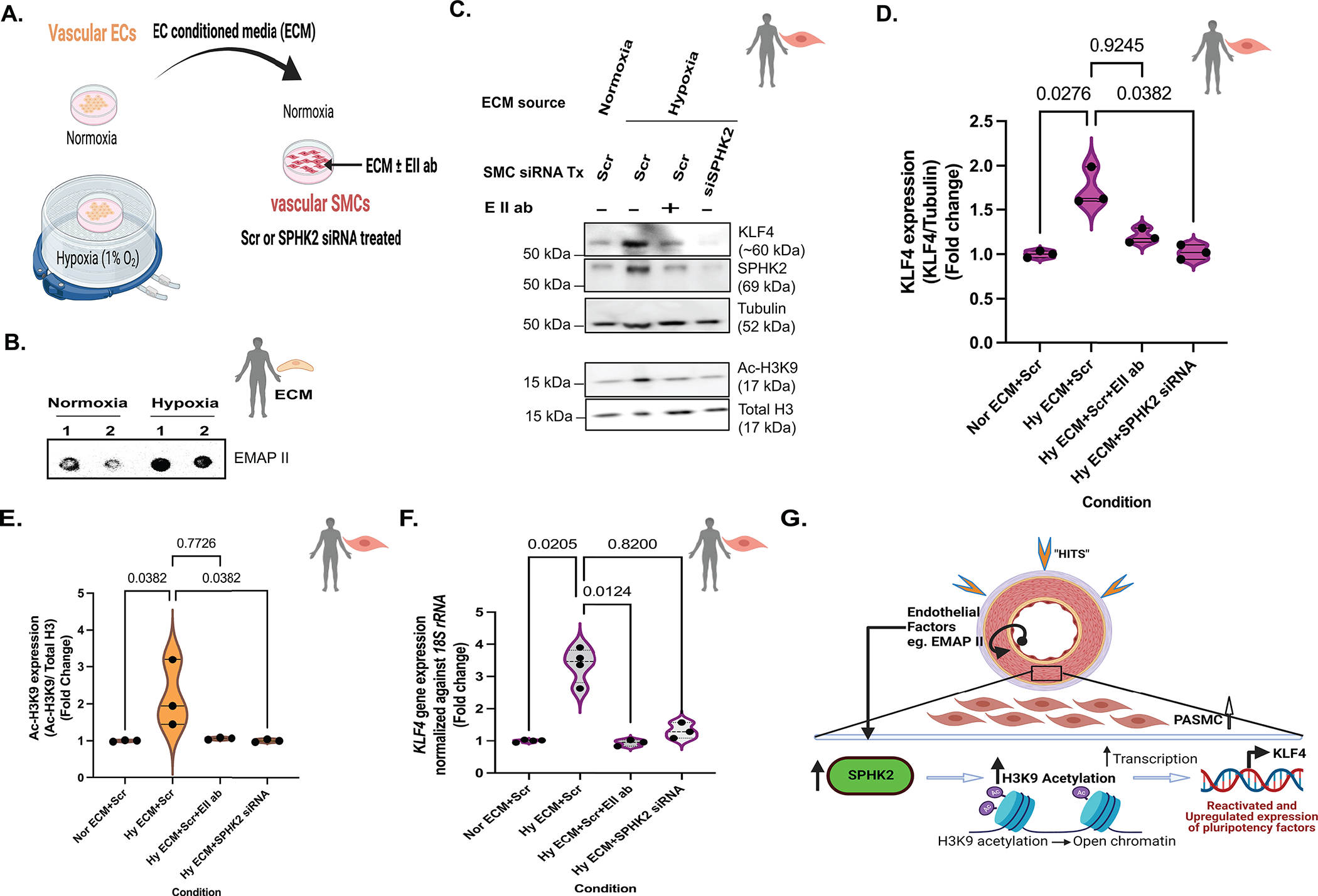
(A) Schematic diagram representing the collection of vascular endothelial cells (ECs) conditioned media (ECM) from ECs grown in 1%O2 or room air to treat vascular smooth muscle cells (SMCs) and, (B) representative dot blot probed for secreted EMAP II expression in ECM. (C) Representative immunoblot probed for KLF4, SPHK2, tubulin, Ac-H3K9 and total histone H3 in whole cell lysates of normoxia or hypoxia ECM with or without EMAP II neutralizing antibody treated hPASMCs pre-transfected with siRNA mediated SPHK2 or scramble silencing and, (D) quantification of KLF4/Tubulin, n=3 and (E) quantification of Ac-H3K9/total histone H3, n=3. (F) KLF4 expression levels normalized against 18S rRNA in normoxia or hypoxia ECM with or without EMAP II neutralizing antibody treated hPASMCs pre-transfected with siRNA mediated SPHK2 or scramble silencing, n=3–4. (G) EMAP II secreted by vascular ECs promote SPHK2/Ac-H3K9/KLF4 signaling in vascular SMCs that may promote PASMCs proliferation. P values are calculated using Kruskal-Wallis against Hy ECM+Scr or Kolmogorov-Smirnov non-parametric test if not mentioned otherwise, and results are shown as median and inter-quartile range.
Figure 8. EMAP II/SPHK2/Ac-H3K9 mediated KLF4 signaling is a novel pathway that exists in PH disease pathogenesis.
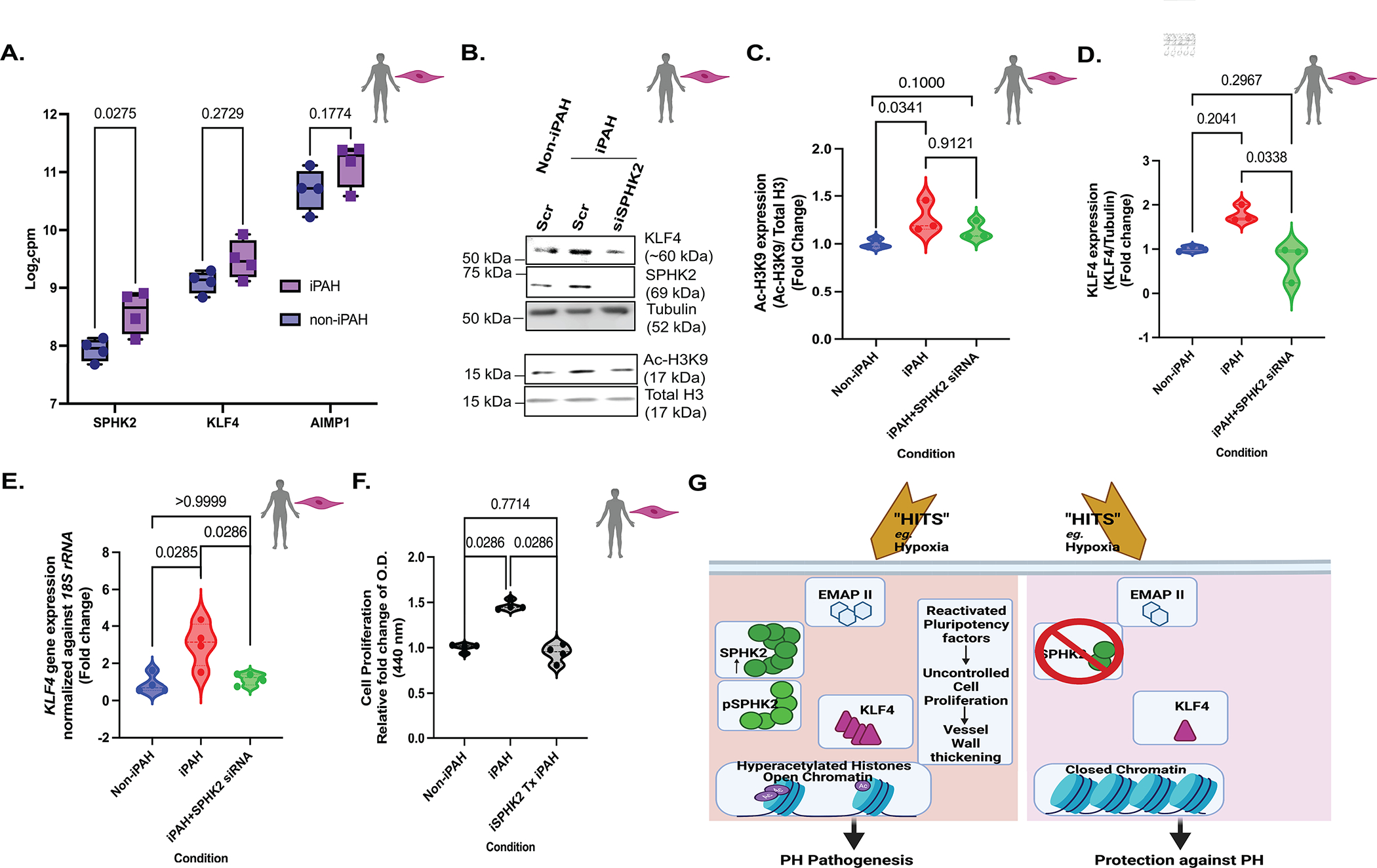
(A) RNA-seq data of SPHK2, KLF4 and AIMP1 in iPAH: PASMCs and non-iPAH:PASMCs in log2-fold of count per million (cpm). Following two-way ANOVA, Sidak’s multiple comparisons test for logarithmic values, n=4. (B) Representative immunoblot probed for KLF4, SPHK2, tubulin, Ac-H3K9 and total histone H3 in whole cell lysates of non: iPAH or iPAH PASMCs with scramble or SPHK2 siRNA transfection and, quantification of (C) Ac-H3K9/total histone H3 and, (D) KLF4/Tubulin, n=3 (E) KLF4 expression levels normalized against 18S rRNA in non: iPAH or iPAH PASMCs with scramble or SPHK2 siRNA transfection, n=4. (F) Cell proliferation rate of non: iPAH or iPAH PASMC with or without iSPHK2 pretreatment for 24 hours, n=4. (G) The proposed model: Endothelial monocyte activating polypeptide II (EMAP II) plays a key role in reawakening pluripotency factor, KLF4 in human pulmonary artery smooth muscle cells (PASMCs) through stimulation of the nuclear SPHK2/S1P epigenetic modulating axis, suggesting that cooperation between SPHK2 and EMAP II could be a major driving force for epigenetic-mediated vascular PASMCs reprogramming and remodeling in PH. Ablation of SPHK2 expression confers protection against PH by rescuing the global and local transcription machinery from histone acetylation and activation of the pluripotency factor, KLF4. P values are calculated using Kruskal-Wallis against iPAH or Kolmogorov-Smirnov non-parametric test if not mentioned otherwise, and results are shown as median and inter-quartile range.
EMAPII/SPHK2/Ac-H3K9 mediated KLF4 reprogramming in PH: vascular SMCs.
Cooperatively, the analysis of RNA sequencing of iPAH:PASMC dataset with accession number GSE144274 in NCBI Gene Expression Omnibus (GEO)34 and qPCR data (See Fig S6A) showed a significant increase in SPHK2 transcripts in iPAH:PASMC emphasizing the important role of SPHK2 in PH that has been previously overlooked17 (Fig 8A). In contrast to elevation of SPHK2 transcripts, SPHK1 transcripts were decreased in iPAH:PASMC compared to the non-iPAH:PASMCs (Fig S6C). Moreover, it showed an increasing trend for both KLF4 and AIMP1 expressions in iPAH:PASMC compared to the non-iPAH:PASMCs (Fig 8A). The elevated histone H3K9 acetylation (Fig 8B,C) was partially inhibited by successful knockdown of SPHK2 (Fig 8B and see Fig S6B) resulting in Ac-H3K9 levels similar to non-iPAH:PASMCs. However, the partial inhibition by SPHK2 siRNA might be due to the complex and dynamic nature of histone acetylation. The increased trend of transcription and expression of KLF4 (Fig 8 B,D,E) in iPAH:PASMCs was significantly inhibited by SPHK2 knockdown. Moreover, the significantly elevated proliferation rate of iPAH:PASMCs compared to non-iPAH:PASMCs, was resorted to levels consistent with non-iPAH: PASMCs following iSPHK2 treatment (Fig 8F). Altogether, these results validate the potential role of EMAPII/SPHK2/Ac-H3K9/KLF4 signaling in vascular SMCs in PH pathogenesis.
Discussion
The initiation and progression of PH is multi-factorial, with genetic and environmental factors functioning as major drivers of disease progression. Here, a key epigenetic role was identified for the SPHK2/S1P axis as a coherent upstream mechanism that cogently explains when, what, and how a disruption of epigenetic equilibrium can occur in PH. These studies show that activation of the nuclear SPHK2/S1P axis results in histone H3K9 epigenetic modifications that reawaken latent gene transcription of KLF4 in vascular SMCs, but not vascular ECs. Moreover, the pro-inflammatory mediator EMAPII was identified as an upstream mediator of the SPHK2/S1P axis that regulates the epigenetic equilibrium through H3K9 acetylation via modulating S1P homeostasis in vascular SMCs and PH (Fig 8G).
Accumulating studies show the importance and mechanistic regulation of global and local histone acetylation code6,9. However, a complete picture of the global histone acetylation code in PH pathogenesis is not available in part because in contrast to stable histone methylation, histone acetylation is very dynamic and transient. Ac-H3K9, an active chromatin mark of promoter regions and enhancers, is an enzymatically regulated reversible modification of acetylation of lysine residues to facilitate the opening of chromatin spatial structures and the activation of transcription during proliferation22, inflammation23 and biological aging8. Recently, Mendoza et al. identified the low-abundant Ac-H3K9 increase as a result of enzymatic acetate transfer to activating lysine sites upon gene transcription and proliferation22. A recent study showed that histone H3K9 acetylation regulates the iron-sulfur biogenesis protein, BolA Family Member 3 to promote pulmonary artery endothelial metabolic reprogramming and dysfunction in PH6. Here, studies identified how histone acetylation mediated reactivation of its downstream reprogramming gene targets unique to vascular SMCs could impact PH pathogenesis. The broad host of HDAC’s downstream targets and multiple isoforms could partially account for the off-targets and conflicting role7 of targeting HDACs in PH. SPHK2 is an integral part of the multi-protein co-repressor complex comprised of the core catalytic components HDAC1/224 within the nucleus. Phosphorylation of SPHK2 increases nuclear S1P that subsequently enhances gene transcription by functioning as an endogenous inhibitor of HDAC without affecting the activity of histone acetyltransferase24. With the discovery of decreased nuclear HDAC activity in human PH lung tissues by Mumby el al.35, expanding the understanding of upstream nuclear SPHK2/S1P/Ac-H3K9 epigenetic targets is an important next step in developing effective PH therapy.
Nuclear SPHK2/S1P signaling plays a critical role in epigenetic regulation of bacterial-mediated inflammatory lung injury23, cardiovascular diseases, and cancer progression12,24. A recent study alludes to SPHK2 signaling in TGFβ1/Smad signaling pathway36 in PH while SPHK2 phosphorylation and nuclear localization to airway epithelium suggests a critical role in epigenetic regulation of bacterial-mediated inflammatory lung injury through histone acetylation23. However, neither TGFβ1 nor SMAD7 were detected as a differentially acetylated region that overlaps with cCREs in CUT&RUN (data not shown). In breast cancer, SPHK2 is associated with HIF and hypoxia mediated cell proliferation25. Since nuclear SPHK2 is seen in both inflammation and cell proliferation, SPHK2 has the potential to act as an alarming molecule of tissue injury and to be associated with tissue repair processes. Here, the functional significance as an epigenetic modulator in human PH builds on the study that showed increased pulmonary arterial SPHK2 expression in human iPAH lung tissue sections37. Previously, Chen et al.17 reported conflicting results with us as they claimed that only SPHK1 is associated with PH, but not SPHK2. It is important to note that SPHK2 has two isoforms, SPHK2a and SPHK2b, that differ in subcellular location and function38 with the SPHK2b isoform of 654aa demonstrating a unique 1–37 aa N-terminal sequence, while the SPHK2a isoform is 618aa and lacks this N-terminal portion39. However, the Chen et al.17 study utilized an antibody generated to a peptide region from the 1–30 aa N-terminus of SPHK2b (long isoform) resulting in the SPHK2a (short isoform) being overlooked. Unfortunately, the tools are not available to dissect out the two isoforms, as the isoform SPHK2b specific antibody utilized by Chen et al.17 is no longer available for many years and is a limitation of this study. However, unbiased RNA sequencing data of iPAH:PASMCs available at the GEO database under GSE14427434 showed an increased expression of SPHK2 (Fig 8A) and no significant change in SPHK1 expression compared to the non-iPAH cells (see Fig S6C). In line with Gairhe et al.18 observations, we report that SPHK2 is elevated in PH patients, and for the first time, we explored its significance, role, or epigenetic impact of SPHK2 in PH. The SPHK2/S1P axis provides greater insight into the contribution that epigenetic regulators have in the progression of PH.
An experimental chronic hypoxia-PH mouse model was sufficient for the main investigational focus of mechanisms of pulmonary arteriolar muscularization26. The SPHK2 KO mice is generated in C57BL/6NJ background. Chen et al.17 mentioned control mice in their studies as C57BL6 mice. Importantly, there are marked contrasting differences in C57BL6 linage mice of inflammatory, genetic, and metabolic responses and signatures between C57BL/6J and C57BL/6NJ mice40,41. In addition, SPHK2a isoform is species-specific, expressed in humans but not in mice42, suggesting possible differences between human and rodent model findings. Previously, a protective role by SPHK2 against cardiac fibrosis43 was reported. However, no changes in cardiac function of SPHK2 KO mice (See Fig S2B) were identified. Defining the epigenetic role of the SPHK2/S1P axis is important in PH.
Little is known about the upstream initiation and modulation of SPHK2 expression and epigenetic activity. Previously the pro-inflammatory mediator EMAPII was reported to regulate SPHK1/S1P/S1PR homeostasis through ERK activated SPHK113 to promote hPASMC pro-proliferative factor IL644 but not proinflammatory cytokine TNFα expression. Previous studies indicated that ERK phosphorylates SPHK2 at Ser387 and Thr614 with the Ser387 phosphorylation site sequence is identical to SPHK145. Here our studies focused on the unique phosphorylation of SPHK2 via Thr614 via EMAPII. Prior studies show that AIMP1/EMAPII expression remains low in postnatal and adult lungs, confined to the sub-endothelium of large vessels in quiescent vessels where it functions as a mediator of endothelial cell growth, while upon tissue insults EMAPII expression can be significantly increased28,29,46–48 in line with the observed overexpression in vitro of AIMP1/EMAPII in hypoxic EC lysate and conditioned media (Fig S3H and Fig 7B) and not observed in PASMCs. Interestingly, an earlier study hints at the possibility that EMAPII promotes signs of PH secondarily in a murine BPD model49. This is consistent with observed increased protein expression and localization of EMAPII in cells within the distal vessels that expressed activated pSPHK2 in human PH lung sections was noted suggesting a potential interaction between EMAPII and SPHK2 in a PH setting.
This study is a sentinel step in the PH research field where epigenetic equilibrium is disrupted in part due to increased H3K9 acetylation in human Group 1 PH lung tissues. With the discovery of EMAPII altered acetylome in vascular SMCs, the potential targets that could modulate hyperproliferation of SMCs were tempting. Mechanistic investigations determined that the ectopic expression of OCT4, SOX2 and KLF4 can initiate the reprogramming of somatic cells to induce pluripotent stem cells (iPSCs) that closely resemble embryonic stem cells through epigenetic remodeling mechanisms50. rEMAPII treatment induced a significant increase in expression of SOX2 and KLF4 in vascular SMCs (See Fig 6C,D,E and Fig S5I). Importantly, only enhancers of KLF4 were under local hyperacetylation of histone H3K9 in CUT&RUN studies. Numerous studies emphasize the role of KLF4 in vascular SMC proliferation, phenotypic switching of PASMC from contractile to synthetic51 and KLF4 involvement in vascular gene transcription thorough histone acetylation19. However, CUT&RUN data showed that SPHK2 inhibition only partially diminishes EMAP II induced hyperacetylated regions suggesting the involvement of other signaling molecules. Consistent with our findings, a recent study showed that intra-nuclear SPHK2-S1P axis inhibits the HDAC activity and deacetylation of KLF4 promoter regions in M1-to-M2 microglia transition52. While this study does not define the exact role of KLF4 in PASMCs proliferation and is such a limitation, the utilization of chromatin mapping studies and the role in KLF4 expression, a pluripotency marker, is novel and a significant addition to the histone acetylation field in PH. Taken together, these findings challenge current dogma and support the critical need to identify mediators of PH epigenetic equilibrium.
In conclusion, this study establishes an epigenetic role for the EMAPII/SPHK2/S1P/histone H3K9 acetylation axis in the progression of PH through reprogramming the histone acetylome in vascular SMCs.
Supplementary Material
Novelty and Significance.
What Is Known?
Histone acetylation marks such as histone H3K9 acetylation play key roles in aging and vascular tone suggesting an indispensable epigenetic role in PH pathobiology.
Histone H3K9 acetylation is previously reported to be regulated by nuclear epigenetic regulator, SPHK2.
What New Information does this article contribute?
We show that SPHK2 can drive PH pathogenesis via histone H3K9 hyperacetylation, contributing to PASMC vascular remodeling.
SPHK2 deficiency confers reduced pulmonary vascular resistance, right ventricle hypertension and distal vessel wall thickness.
Employing CUT&RUN assay, we show that EMAP II has a key role in the stimulation of nuclear SPHK2/S1P epigenetic modulating axis suggesting that cooperation between SPHK2 and EMAPII could be a major driving force for epigenetic mediated vascular PASMC reprogramming and remodeling in PH.
We discovered that pulmonary vascular endothelial cells are a priming factor of the EMAPII/SPHK2/S1P axis that alters the acetylome with a specificity for PASMC, through hyperacetylation of histone H3K9.
Our findings indicate that SPHK2/S1P mediates upregulation of epigenetic mediator H3K9 acetylation in PASMC, contributing to epigenetic mediated rekindling of transcriptional regulators of cellular proliferation, differentiation, and somatic cell reprogramming. Targeting elevated SPHK2 expression could mitigate the pathological phenotype of PASMC that contributes to progression of PH.
Acknowledgement
Lung tissue and Ethanol-Fixed Paraffin-Embedded (EFPE) tissue sections from patients with idiopathic pulmonary arterial hypertension and from failed donor lungs (controls) were obtained from the Pulmonary Hypertension Breakthrough Initiative, funded by the National Institutes of Health (R24 HL 123767) and the Cardiovascular Medical Research and Education Fund (CMREF). Schematic diagrams are created using BioRender.com.
Sources of funding:
This publication was made possible in part by 5R01HL114977 (MAS) from NIH, the Lilly Endowment, Inc. Physician Scientist Initiative (MAS), the O’Brien Family Endowment for Excellence (a fellowship administered by the Institute for Precision Health, on behalf of Notre Dame Research) (ADCUR), National Science Foundation Award 1659556 (KMB), the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number T35HL110854 (MG) and Buckner Family Scholarship (MH).
Non-Abbreviations and Acronyms:
- PAH
Pulmonary Arterial Hypertension
- PH
Pulmonary Hypertension
- IPAH
Idiopathic Pulmonary Arterial Hypertension
- S1P
Sphingosine 1-Phosphate
- SPHK2
Sphingosine Kinase 2
- pSPHK2
Phosphorylated Sphingosine Kinase 2
- EMAP II
Endothelial Monocyte Activating Polypeptide II
- AIMP1
Aminoacyl tRNA synthetase complex Interacting Multifunctional Protein 1
- EC
Endothelial Cells
- hPASMC
human pulmonary artery smooth muscle cells
- hPMVECs
human pulmonary microvascular endothelial cells
- SMCs
Smooth muscle cells
- siRNA
Small interfering RNA
- H3K9
Histone H3 lysine 9
- Ac-H3K9
Acetyl histone H3 lysine 9
- CUT&RUN
Cleavage Under Targets & Release Using Nuclease
- KO
Knockout
- KLF4
Krüppel-like factor 4
- GO
Gene ontology
- cCRE
candidate Cis-Regulatory Elements
- Tx
Treatment
- FDL
Failed donor lung
- ELISA
Enzyme-linked immunosorbent assay
- RV
Right Ventricle
- ENCODE
The Encyclopedia of DNA Elements
Footnotes
Conflict of Interests The authors declare that they have no known competing financial interests or personal relationships or conflicts of interests that could have appeared to influence the work reported in this paper.
References:
- 1.Rabinovitch M Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 2012;122:4306–4313. doi: 10.1172/JCI60658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L, Moonen JR, Cao A, Isobe S, Li CG, Tojais NF, Taylor S, Marciano DP, Chen PI, Gu M, Li D, Harper RL, El-Bizri N, Kim YM, Stankunas K, Rabinovitch M. Dysregulated Smooth Muscle Cell BMPR2–ARRB2 Axis Causes Pulmonary Hypertension. Circ Res 2023;132:545–564. doi: 10.1161/CIRCRESAHA.121.320541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bisserier M, Mathiyalagan P, Zhang S, Elmastour F, Dorfmüller P, Humbert M, David G, Tarzami S, Weber T, Perros F, Sassi Y, Sahoo S, Hadri L. Regulation of the Methylation and Expression Levels of the BMPR2 Gene by SIN3a as a Novel Therapeutic Mechanism in Pulmonary Arterial Hypertension. Circulation 2021;144:52–73. doi: 10.1161/CIRCULATIONAHA.120.047978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benincasa G, Maron BA, Affinito O, D’Alto M, Franzese M, Argiento P, Schiano C, Romeo E, Bontempo P, Golino P, Berrino L, Loscalzo J, Napoli C. Association Between Circulating CD4+ T Cell Methylation Signatures of Network-Oriented SOCS3 Gene and Hemodynamics in Patients Suffering Pulmonary Arterial Hypertension. J Cardiovasc Transl Res 2023;16:17–30. doi: 10.1007/S12265-022-10294-1/FIGURES/6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benincasa G, Napoli C, Loscalzo J, Maron BA. Pursuing functional biomarkers in complex disease: Focus on pulmonary arterial hypertension. Am Heart J 2023;258:96–113. doi: 10.1016/J.AHJ.2022.12.009. [DOI] [PubMed] [Google Scholar]
- 6.Yu Q, Tai YY, Tang Y, Zhao J, Negi V, Culley MK, Pilli J, Sun W, Brugger K, Mayr J, Saggar R, et al. BOLA (BolA Family Member 3) Deficiency Controls Endothelial Metabolism and Glycine Homeostasis in Pulmonary Hypertension. Circulation 2019;139:2238–2255. doi: 10.1161/CIRCULATIONAHA.118.035889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chelladurai P, Boucherat O, Stenmark K, Kracht M, Seeger W, Bauer UM, Bonnet S, Pullamsetti SS. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br J Pharmacol 2021;178:54–71. doi: 10.1111/BPH.14932. [DOI] [PubMed] [Google Scholar]
- 8.Yi SJ, Kim K. New Insights into the Role of Histone Changes in Aging. Int J Mol Sci 2020;21:1–20. doi: 10.3390/IJMS21218241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li D, Shao NY, Moonen JRMD, Zhao Z, Shi M, Otsuki S, Wang L, Elaine Yan TN, Marciano DP, Contrepois K, Li CG, Wu JC, Snyder MP, Rabinovitch M. ALDH1A3 Coordinates Metabolism with Gene Regulation in Pulmonary Arterial Hypertension. Circulation 2021;143:2074–2090. doi: 10.1161/CIRCULATIONAHA.120.048845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ranasinghe ADCU Schwarz MA. Integrating epigenetics and metabolomics to advance treatments for pulmonary arterial hypertension. Biochem Pharmacol 2022;204:115245. doi: 10.1016/J.BCP.2022.115245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chakraborty R, Ostriker AC, Xie Y, Dave JM, Gamez-Mendez A, Chatterjee P, Abu Y, Valentine J, Lezon-Geyda K, Greif DM, Schulz VP, Gallagher PG, Sessa WC, Hwa J, Martin KA. Histone Acetyltransferases p300 and CBP Coordinate Distinct Chromatin Remodeling Programs in Vascular Smooth Muscle Plasticity. Circulation 2022;145:101161CIRCULATIONAHA121057599. doi: 10.1161/CIRCULATIONAHA.121.057599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.W S, D M, Y C, L H, S L, D Y, S Z, G Z, W Z, J W, J C. SphK2/S1P Promotes Metastasis of Triple-Negative Breast Cancer Through the PAK1/LIMK1/Cofilin1 Signaling Pathway. Front Mol Biosci 2021;8. doi: 10.3389/FMOLB.2021.598218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ranasinghe ADCU, Lee DD, Schwarz MA. Mechanistic regulation of SPHK1 expression and translocation by EMAP II in pulmonary smooth muscle cells. Biochim Biophys Acta - Mol Cell Biol Lipids 2020;1865:158789. doi: 10.1016/J.BBALIP.2020.158789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W, Toran PT, Sabol R, Brown TJ, Barth BM. Epigenetics and Sphingolipid Metabolism in Health and Disease. Int J Biopharm Sci 2018;1. doi: 10.31021/IJBS.20181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.R A, CJ van K, K D, M TB, D MZH. Regulation and functional roles of sphingosine kinases. Naunyn Schmiedebergs Arch Pharmacol 2007;374:413–428. doi: 10.1007/S00210-007-0132-3. [DOI] [PubMed] [Google Scholar]
- 16.Venkataraman K, Thangada S, Michaud J, Oo ML, Ai Y, Lee Y-M, Wu M, Parikh NS, Khan F, Proia RL, Hla T. Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem J 2006;397:461. doi: 10.1042/BJ20060251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Tang H, Sysol JR, Moreno-Vinasco L, Shioura KM, Chen T, Gorshkova I, Wang L, Huang LS, Usatyuk PV, et al. The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension. Am J Respir Crit Care Med 2014;190:1032–1043. doi: 10.1164/rccm.201401-0121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gairhe S, Joshi SR, Bastola MM, McLendon JM, Oka M, Fagan KA, McMurtry IF. Sphingosine-1-Phosphate is Involved in the Occlusive Arteriopathy of Pulmonary Arterial Hypertension: Http://DxDoiOrg/101086/687766 2017;6:369–380. doi: 10.1086/687766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moonen JR, Chappell J, Shi M, Shinohara T, Li D, Mumbach MR, Zhang F, Nair RV., Nasser J, Mai DH, Taylor S, Wang L, Metzger RJ, Chang HY, Engreitz JM, Snyder MP, Rabinovitch M. KLF4 recruits SWI/SNF to increase chromatin accessibility and reprogram the endothelial enhancer landscape under laminar shear stress. Nat Commun 2022 131 2022;13:1–16. doi: 10.1038/s41467-022-32566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.019140 - B6N.129S6-Sphk2<tm1Rlp>/J n.d. https://www.jax.org/strain/019140 (accessed September 1, 2021).Http://DxDoiOrg/101086/687766 2017;6:369–380. doi: 10.1086/687766 [DOI] [Google Scholar]
- 21.005304 - C57BL/6NJ n.d. https://www.jax.org/strain/005304 (accessed September 30, 2021). [Google Scholar]
- 22.Mendoza M, Egervari G, Sidoli S, Donahue G, Alexander DC, Sen P, Garcia BA, Berger SL. Enzymatic transfer of acetate on histones from lysine reservoir sites to lysine activating sites. Sci Adv 2022;8:5688. doi: 10.1126/SCIADV.ABJ5688/SUPPL_FILE/SCIADV.ABJ5688_SM.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DL E, EV B, IA B, Y L, C T, Y K, EV B, V S, AW H, A H, RM T, V N, P F. Pseudomonas aeruginosa stimulates nuclear sphingosine-1-phosphate generation and epigenetic regulation of lung inflammatory injury. Thorax 2019;74:579–591. doi: 10.1136/THORAXJNL-2018-212378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, Spiegel S. Regulation of Histone Acetylation in the Nucleus by Sphingosine-1-Phosphate. Science (80-) 2009;325:1254–1257. doi: 10.1126/SCIENCE.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hait NC, Maiti A, Xu P, Qi Q, Kawaguchi T, Okano M, Takabe K, Yan L, Luo C. Regulation of hypoxia-inducible factor functions in the nucleus by sphingosine-1-phosphate. FASEB J 2020;34:4293–4310. doi: 10.1096/FJ.201901734RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomez-Arroyo J, Saleem SJ, Mizuno S, Syed AA, Bogaard HJ, Abbate A, Taraseviciene-Stewart L, Sung Y, Kraskauskas D, Farkas D, Conrad DH, Nicolls MR, Voelkel NF. A brief overview of mouse models of pulmonary arterial hypertension: Problems and prospects. Am J Physiol - Lung Cell Mol Physiol 2012;302:977–991. doi: 10.1152/AJPLUNG.00362.2011/SUPPL_FILE/VIDEO1.MP4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah M, Phillips MR, Quintana M, Stupp G, McLean SE. Echocardiography allows for analysis of pulmonary arterial flow in mice with congenital diaphragmatic hernia. J Surg Res 2018;221:35. doi: 10.1016/J.JSS.2017.06.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee DD, Lal CV, Persad EA, Lowe C-W, Schwarz AM, Awasthi N, Schwarz RE, Schwarz MA. Endothelial Monocyte-Activating Polypeptide II Mediates Macrophage Migration in the Development of Hyperoxia-Induced Lung Disease of Prematurity. Am J Respir Cell Mol Biol 2016;55:602–612. doi: 10.1165/rcmb.2016-0091OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwarz M, Lee M, Zhang F, Zhao J, Jin Y, Smith S, Bhuva J, Stern D, Warburton D, Starnes V. EMAP II: A modulator of neovascularization in the developing lung. Am J Physiol - Lung Cell Mol Physiol 1999;276:365–375. doi: 10.1152/AJPLUNG.1999.276.2.L365/ASSET/IMAGES/LARGE/ALUNL0318706Z.JPEG. [DOI] [PubMed] [Google Scholar]
- 30.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 2012;22:1813. doi: 10.1101/GR.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.J Ji J, Wang J, Yang J, Wang X-P, Huang J-J, Xue T-F, Sun X-L. The Intra-nuclear SphK2-S1P Axis Facilitates M1-to-M2 Shift of Microglia via Suppressing HDAC1-Mediated KLF4 Deacetylation. Front Immunol 2019;10:1241. doi: 10.3389/FIMMU.2019.01241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheikh AQ, Saddouk FZ, Ntokou A, Mazurek R, Greif DM. Cell Autonomous and Non-cell Autonomous Regulation of SMC Progenitors in Pulmonary Hypertension. Cell Rep 2018;23:1152. doi: 10.1016/J.CELREP.2018.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheikh AQ, Misra A, Rosas IO, Adams RH, Greif DM. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci Transl Med 2015;7:308ra159. doi: 10.1126/SCITRANSLMED.AAA9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorr MW, Sriram K, Muthusamy A, Insel PA. Transcriptomic analysis of pulmonary artery smooth muscle cells identifies new potential therapeutic targets for idiopathic pulmonary arterial hypertension. Br J Pharmacol 2020;177:3505–3518. doi: 10.1111/BPH.15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mumby S, Gambaryan N, Meng C, Perros F, Humbert M, Wort SJ, Adcock IM. Bromodomain and extra-terminal protein mimic JQ1 decreases inflammation in human vascular endothelial cells: Implications for pulmonary arterial hypertension. Respirology 2017;22:157. doi: 10.1111/RESP.12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng J, Jiang B, Xiao X, Zhang R. Inhibition of sphingosine kinase 2 attenuates hypertrophic scar formation via upregulation of Smad7 in human hypertrophic scar fibroblasts. Mol Med Rep 2020;22:2573–2582. doi: 10.3892/MMR.2020.11313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.S G, SR J, MM B, JM M, M O, KA F, IF M. Sphingosine-1-phosphate is involved in the occlusive arteriopathy of pulmonary arterial hypertension. Pulm Circ 2016;6:369–380. doi: 10.1086/687766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hait NC, Bellamy A, Milstien S, Kordula T, Spiegel S. Sphingosine Kinase Type 2 Activation by ERK-mediated Phosphorylation. J Biol Chem 2007;282:12058–12065. doi: 10.1074/JBC.M609559200. [DOI] [PubMed] [Google Scholar]
- 39.Hatoum D, Haddadi N, Lin Y, Nassif NT, McGowan EM. Mammalian sphingosine kinase (SphK) isoenzymes and isoform expression: challenges for SphK as an oncotarget. Oncotarget 2017;8:36898. doi: 10.18632/ONCOTARGET.16370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MQ G, SC Y, JCW M, MSM I. Differential metabolic and inflammatory responses to intermittent hypoxia in substrains of lean and obese C57BL/6 mice. Life Sci 2019;238. doi: 10.1016/J.LFS.2019.116959. [DOI] [PubMed] [Google Scholar]
- 41.K M, A Y . Substrains matter in phenotyping of C57BL/6 mice. Exp Anim 2021;70:145–160. doi: 10.1538/EXPANIM.20-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hatoum D, Haddadi N, Lin Y, Nassif NT, McGowan EM, Hatoum D, Haddadi N, Lin Y, Nassif NT, McGowan EM. Mammalian sphingosine kinase (SphK) isoenzymes and isoform expression: challenges for SphK as an oncotarget. Oncotarget 2017;8:36898–36929. doi: 10.18632/ONCOTARGET.16370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan H, Zhao H, Yi S wei, Zhuang H, Wang D wen, Jiang J gang, Shen G fen. Sphingosine-1-Phosphate Protects Against the Development of Cardiac Remodeling via Sphingosine Kinase 2 and the S1PR2/ERK Pathway. Curr Med Sci 2022 424 2022;42:702–710. doi: 10.1007/S11596-022-2600-X. [DOI] [PubMed] [Google Scholar]
- 44.Wang AP, Yang F, Tian Y, Su JH, Gu Q, Chen W, Gong SX, Ma XF, Qin XP, Jiang ZS. Pulmonary Artery Smooth Muscle Cell Senescence Promotes the Proliferation of PASMCs by Paracrine IL-6 in Hypoxia-Induced Pulmonary Hypertension. Front Physiol 2021;12:307. doi: 10.3389/FPHYS.2021.656139/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hait NC, Bellamy A, Milstien S, Kordula T, Spiegel S. Sphingosine kinase type 2 activation by ERK-mediated phosphorylation. J Biol Chem 2007;282:12058–12065. doi: 10.1074/jbc.M609559200. [DOI] [PubMed] [Google Scholar]
- 46.Quintos-Alagheband ML, White CW, Schwarz MA. Potential Role for Antiangiogenic Proteins in the Evolution of Bronchopulmonary Dysplasia. Https://HomeLiebertpubCom/Ars 2004;6:137–145. doi: 10.1089/152308604771978444. [DOI] [PubMed] [Google Scholar]
- 47.Yuan C, Yan L, Solanki P, Vatner SF, Vatner DE, Schwarz MA. Blockade of EMAP II Protects Cardiac Function after Chronic Myocardial Infarction by Inducing Angiogenesis. J Mol Cell Cardiol 2015;79:224. doi: 10.1016/J.YJMCC.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matschurat S, Knies UE, Person V, Fink L, Stoelcker B, Ebenebe C, Behrensdorf HA, Schaper J, Clauss M. Regulation of EMAP II by Hypoxia. Am J Pathol 2003;162:93. doi: 10.1016/S0002-9440(10)63801-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee DD, Lal CV., Persad EA, Lowe C-W, Schwarz AM, Awasthi N, Schwarz RE, Schwarz MA. Endothelial Monocyte-Activating Polypeptide II Mediates Macrophage Migration in the Development of Hyperoxia-Induced Lung Disease of Prematurity. Am J Respir Cell Mol Biol 2016;55:602–612. doi: 10.1165/rcmb.2016-0091OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cherepanova OA, Gomez D, Shankman LS, Swiatlowska P, Williams J, Sarmento OF, Alencar GF, Hess DL, Bevard MH, Greene ES, et al. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat Med 2016;22:657–665. doi: 10.1038/nm.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yap C, Mieremet A, De Vries CJM, Micha D, De Waard V. Six Shades of Vascular Smooth Muscle Cells Illuminated by KLF4 (Krüppel-Like Factor 4). Arterioscler Thromb Vasc Biol 2021;41:2693–2707. doi: 10.1161/ATVBAHA.121.316600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.J Ji J, Wang J, Yang J, Wang XP, Huang JJ, Xue TF, Sun XL. The intra-nuclear SphK2-S1P axis facilitates M1-to-M2 shift of microglia via suppressing HDAC1-Mediated KLF4 deacetylation. Front Immunol 2019;10:1241. doi: 10.3389/FIMMU.2019.01241/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.