

Abstract
Type 1 diabetes (T1D) develops due to autoimmune targeting of the pancreatic islet β-cells. Clinical symptoms arise from reduced insulin in circulation. The molecular events and interactions between discrete immune cell populations, infiltration of such leukocytes into pancreatic and islet tissue, and selective targeting of the islet β-cells during autoimmunity and graft rejection are not entirely understood. One protein central to antigen presentation, priming of immune cells, trafficking of leukocytes, and vital for leukocyte effector function is the intercellular adhesion molecule-1 (ICAM-1). The gene encoding ICAM-1 is transcriptionally regulated and rapidly responsive (i.e., within hours) to pro-inflammatory cytokines. ICAM-1 is a transmembrane protein that can be glycosylated; its presence on the cell surface provides co-stimulatory functions for immune cell activation and stabilization of cell-cell contacts. ICAM-1 interacts with the β2-integrins, CD11a/CD18 (LFA-1) and CD11b/CD18 (Mac-1), which are present on discrete immune cell populations. A whole-body ICAM-1 deletion protects NOD mice from diabetes onset, strongly implicating this protein in autoimmune responses. Since several different cell types express ICAM-1, its biology is fundamentally essential for various physiological and pathological outcomes. Herein, we review the role of ICAM-1 during both autoimmunity and islet graft rejection to understand the mechanism(s) leading to islet β-cell death and dysfunction that results in insufficient circulating quantities of insulin to control glucose homeostasis.
Keywords: autoimmunity, ICAM-1, inflammation, pancreatic islet, Type 1 diabetes
1. Introduction
Pancreatic β-cells are housed within the islets of Langerhans and produce the protein hormone insulin [1]. Islet β–cells are capable of remarkable adaptive responses, such as increased insulin secretion and enhanced proliferation in response to pathophysiological situations, such as insulin resistance [2–4]. Consequently, when the islet β-cells are rendered dysfunctional or undergo cell death, this leads to insufficient quantities of insulin and triggers clinical symptoms of diabetes [2, 5–7]. Type 1 diabetes (T1D) is generally referred to as an autoimmune disease and the presence of auto-antibodies is one strategy to distinguish this form of diabetes from Type 2 diabetes (T2D).
Because autoimmune diseases in general feature the presence of immune cells that target a self-tissue, leading to dysfunction of the host tissue, there is major interest in understanding the mechanistic underpinnings that lead to initiation and progression of such disease(s). From the T1D perspective, there has never been a single trigger identified to explain onset in susceptible individuals. What is generally agreed upon is that the number of auto-antibodies present is an indicator of greater disease risk [8], that insulin in circulation is reduced to a level that requires exogenous sources (e.g., from injections of recombinant insulin, secretion from transplanted islets, etc.), and that immune cell infiltration (often referred to as insulitis) into pancreatic tissue is present [9]. The extent to which insulitis occurs is variable and the number of leukocytes required for its conclusive presence has been recommended [10].
Cytokines, secreted predominately from immune cells, are also likely to be involved in both physiology and pathophysiology of the autoimmune disease process. From the standpoint of leukocyte recruitment and activation, there are specialized proteins, termed chemotactic cytokines (aka chemokines), which promote immune cell recruitment towards a site of inflammation [11, 12]. In addition, there are various adhesion molecules present on antigen presenting cells, endothelial cells, and target tissues that normally facilitate physiological immune cell function but become dysregulated in conditions of autoimmunity [13]. One such adhesion molecule is the intercellular adhesion molecule-1 (ICAM-1), which is the focus of this review. We outline its importance in various models of T1D and in rejection of transplanted islets to gain greater insights into mechanisms that are relevant to T1D onset, progression, and eventual development of effective therapeutics.
ICAM-1 is a seven-exon gene that is strongly regulated at the expression level by cytokine signals [14–16]. The gene encodes a glycoprotein with multiple splice variants that can express on the cell surface as well as in secreted form [17–19]. ICAM-2, encoded by a separate gene is typically not inducible, but is also an LFA-1 ligand [20]. The function of the soluble version of the ICAM-1 protein (sICAM-1) is not completely understood. However, sICAM-1 levels are elevated in the circulation of Type 1 diabetic patients and people with genetic risk for T1D [21, 22]. Stimulation of T cells in vitro was greatly inhibited by sICAM-1 or chimeric immunoglobulin fused to sICAM-1 [23]. These functions of membrane-bound ICAM-1 versus sICAM-1 may be competitive to limit overactive signaling through LFA-1.
LFA-1 is a heterodimer of CD11a and CD18 proteins that function together as a β2 integrin. It was first discovered in mice [24, 25] and subsequently in humans [26]. LFA-1 is present on most, if not all, T-cells and facilitates interaction with antigen-presenting cells (APCs) [27]. Modulation of LFA-1 components regulates both inside-out signaling and cell spreading ability [28]. In addition, the LFA-1/ICAM-1 interaction controls the ability of T-cells to expand in response to T-cell receptor (TCR) activation, such as during exposure to viral infection. Thus, LFA-1/ICAM-1 interactions provide increased stability for cell-cell contacts (adhesion effects) and convey additional (co-stimulatory) signals that help modulate T-cell effector responses. Collectively, these interactions assist with appropriate refining of the physiological immune response.
Chemokines promote recruitment of immune cells to a site of inflammation [11, 12] while adhesion molecules such as ICAM-1 facilitate cell-cell contact as well as providing a secondary (co-stimulatory) signal for immune cell activation. With this in mind, it is perhaps not surprising that ICAM-1 is a central component of both healthy immunity as well as a promising target to control autoimmunity. Below we discuss several different scenarios by which ICAM-1 function is disrupted either by systemic interventions or by genetic approaches. Each of these model systems provides insights into the function of the ICAM-1 protein and its roles in the autoimmunity associated with onset of T1D.
2. Monoclonal Antibody and Peptide-based Strategies targeting the ICAM-1/LFA-1 interaction regulate diabetes development
The NOD mouse is the gold standard rodent model for pre-clinical studies designed to understand various aspects of autoimmunity relevant to T1D [29]. Similar to human disease, the onset of T1D is spontaneous, proceeds with the presence of specific immune populations (e.g., macrophages, T-cells, etc.), and requires insulin as a life-saving measure after diabetes onset. CD4 and CD8 T-lymphocytes, as well as macrophages, are important for the overall development of disease [30–33]. In addition, diabetes is accelerated in NOD mice upon administration of the drug cyclophosphamide [34], leading to reductions in total mass of the insulin-producing islet β-cell.
Insulin is a major auto-antigen in both mice and humans; priming of effector cells by antigen presenting cells allows selective targeting of β-cells within the islets of Langerhans [35–37]. The presentation of antigens occurs via the major histocompatibility complex (MHC), which is an important component of the autoimmune disease process. This is illustrated elegantly in the NOD mouse model of T1D, where preventing MHC action by deletion of the beta-2-microglobulin (B2M) gene prevents insulitis and diabetes [38]. Moreover, allogeneic grafts survived for an openended period when islets from B2M−/− mice were the donors [39]. Notably, antigen presentation through the MHC proteins to the T-cell receptor (TCR) also requires a secondary activation signal. Because ICAM-1 is an important component of antigen presentation by virtue of providing such a secondary signal (shown schematically in Figure 1), we predict that modulating expression of the Icam1 gene regulates the autoimmune process. The Icam1 gene is transcriptionally regulated [15] and is thus positioned to be a central mediator of the autoimmune mechanisms that are detrimental for islet β-cells (Figure 2). Early studies have examined this notion using a variety of pre-clinical model systems, which are discussed below.
Figure 1. ICAM-1 is involved at multiple levels and in cell types in autoimmunity.
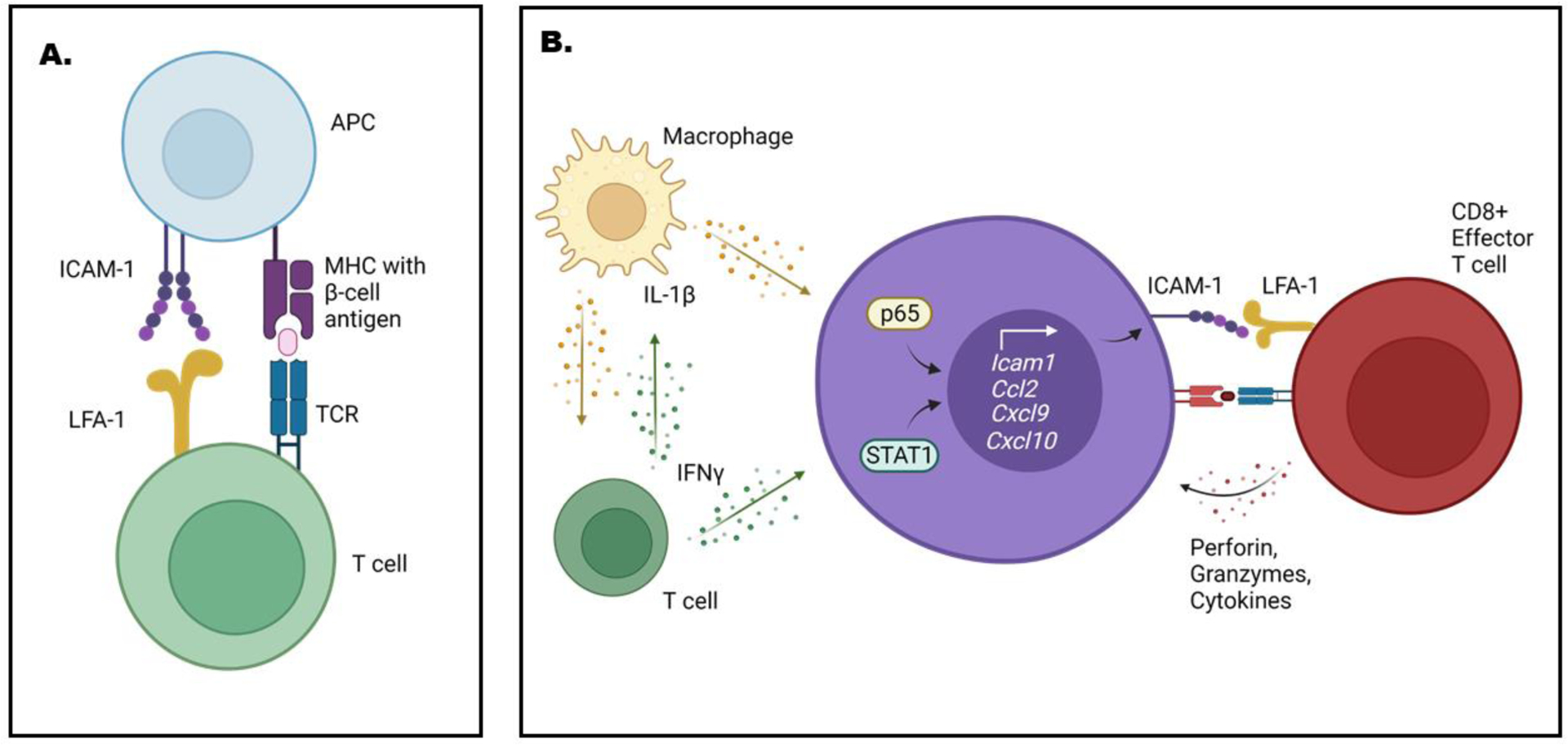
A. Antigen presenting cells (APC) use ICAM-1 interactions with LFA-1 on T-cells as a secondary signal to facilitate priming and activation responses. B. The islet beta-cell (purple) responds to variety of cytokines, including interleukin-1β (IL-1β; e.g., from macrophages) and interferon-gamma (IFNγ; e.g., from T-cells). IL-1β signals through the transcriptional factor p65 while IFN-γ activates STAT1 to control gene expression in islet β-cells. As examples, Icam1 and selected genes that encode chemokines are responsive to cytokines via p65/STAT1 activatsion. Chemokines are secreted proteins that recruit immune cells to a site of inflammation. Islet beta-cells increase ICAM-1 abundance to promote interaction with LFA-1. This ICAM-1/LFA-1 interaction could facilitate direct contact with effector T-cells (shown in red) in the islet. Collectively, these events are help explain some of the events driving the autoimmune process that ultimately leads to T1D. This figure was generated using BioRender.
Figure 2. Three major compartments where ICAM-1 influences immune cell activity.

Three possiblities for ICAM-1 to influence autoimmunity include: 1) priming of T-cells (left panel), 2) trafficking of immune cells (middle panel), and 3) effector function (right panel). While ICAM-1 likely performs these roles as part of healthy immune cell activity, each of these processes may contribute to ICAM-1 dependent events that lead to islet beta-cell loss during autoimmunity and islet graft rejection. This figure was generated using BioRender.
Monoclonal antibodies targeting ICAM-1 or LFA-1 reduced diabetes onset when injected into five week old NOD mice. This age was chosen because insulitis was either not present at all or very limited in terms of detectable immune cell infiltration. Onset of hyperglycemia was monitored until the mice were 30 weeks of age [40]. There was a noticeable reduction in immune cell accumulation near and within islets as a partial explanation for the reduced onset of disease. Remarkably, this strategy did not block cyclophosphamide-induced onset of hyperglycemia [40]. The use of both ICAM-1 and LFA-1 antibodies in combination provided complete protection against diabetes onset compared to each intervention individually. Similar to the above study, injections into young (2–4 weeks of age) female NOD mice afforded stable therapeutic benefit [41]. In addition, adoptive transfer of splenocytes from 22-week-old NOD that had been injected with antibodies against LFA-1 and ICAM-1 were not able to produce diabetes in the NOD-SCID mice recipients [41]. Furthermore, administration of cyclophosphamide did not promote diabetes onset in mice that were previously exposed to the monoclonal antibodies targeting LFA-1/ICAM-1.
Intravenous administration of an adenovirus encoding soluble ICAM-1/ Ig fusion proteins to NOD mice provided long-term protection against invasive insulitis and onset of diabetes without suppressing immune function [42]. This intervention also induced lasting remission (>6 months) in 50% of mice with overt diabetes. However, the exact mechanism of how this protection was achieved is unknown. In contrast to previous studies showing the requirement for LFA-1/ICAM-1 interaction to prime effector cells, this strategy may be effective by blocking T-cell adhesion and effector function to prevent or limit ongoing islet β-cell destruction (e.g., as shown schematically on the right hand side of Figure 2). There is no doubt that administration of sICAM-1 had a protective effect against diabetes onset in vivo in NOD mice [43]. In addition to reducing islet destruction, the sICAM-1 and sICAM-1-Ig therapy reduced the expression of Th1 type cytokines within the islet. NOD mice given a plasmid that encodes sICAM-1 increased the ratio of Th2/Th1 cytokine production, reduced islet leukocytic infiltration, and resulted in less islet destruction [44]. This outcome points to an immunosuppressive effect of sICAM-1, leading to a possible scenario whereby sICAM-1 induction is a protective mechanism against autoimmunity. However, more work is required to understand the induction mechanisms, source tissue(s), and consequences of sICAM-1 at various states of disease progression to gain a greater understanding about the autoimmunity mechanisms relevant to T1D.
Administration of soluble LFA-1 and ICAM-1 peptides, which block the membrane-associated ICAM-1 protein from binding its cognate receptor, reduced immune cell infiltration and preserved islet integrity in NOD mice. This intervention began in mice at 13 weeks of age; the caveat is that only 25% of the peptide-treated mice examined for infiltration were diabetic (blood glucose > 250mg/dL) compared to 60% of the saline treated animals. [45]. The effect of the soluble LFA-1 and ICAM-1 peptides is also present during transfer of NOD splenocytes to nondiabetic mice. This model provides rapid (1–2 weeks) infiltration of the islets by untreated T cells and macrophages, leading to rapid loss of glycemic control via islet destruction [46]. However, administration of ICAM-1 and LFA-1 antibodies to non-diabetic NOD mice is sufficient to completely protect the nondiabetic recipients from onset of diabetes induced by transfer of islet-derived mononuclear cells [41] or splenocyte transfer from diabetic NOD donors [47]. This protection correlated with mild insulitis or no detectable immune cell infiltration in recipient mice [41].
Blockade of ICAM-1 and LFA-1 protected against cyclophosphamide-induced diabetes in NOD mice, but this effect was dependent on the order of administration. For example, when 10-week old NOD mice were given cyclophosphamide and then subsequently anti-LFA-1 and ICAM-1 antibodies, there was no protection from diabetes. By contrast, NOD mice injected with a combination of LFA-1 and ICAM-1 monoclonal antibodies at 2 weeks of age were completely protected from diabetes when treated with cyclophosphamide at 12 weeks old [41]. Whether this indicates an effect of disrupting ICAM-1/LFA-1 interactions to limit immune cell trafficking or by enhancing the Treg ability to suppress pro-inflammatory responses is not clear [40]. Additionally, disruption of the Icam1 gene on the NOD background reduced insulitis severity in cyclophosphamide-injected mice [48]. However, C57BL/6 mice deficient for the Icam1 gene are not protected from immune cell infiltration or diabetes induced by multiple low doses of streptozotocin (STZ) [49]. Alternatively, antibody-mediated blockade of LFA-1 and ICAM-1 in combination, but not when administered separately, [50, 51] reduced hyperglycemia in STZ-treated mice [50, 52]. We note that chemically-induced hyperglycemia (e.g., by STZ delivery) vs. spontaneous autoimmunity leading to diabetes (as observed in NOD mice) are very different mechanisms, which clearly respond differently to interventions modulating LFA-ICAM-1 interactions.
3. Genetic deletion of ICAM-1 in NOD mice prevents diabetes
With the systemic antibody- and peptide-based strategies targeting ICAM-1 proving effective in reducing diabetes onset in NOD mice, studying ICAM-1 in NOD mice with genetic deletion of the protein gave further insights into autoimmune mechanisms. As noted earlier, the NOD mouse is the most often used rodent model to study aspects of Type 1 Diabetes [53]. T1D in humans and NOD mice each clearly require interactions between discrete populations of immune cells [54]. Chemokine proteins control the recruitment of specific immune cell types into the pancreatic islet and expression of chemokines as well as leukocyte recruitment occurs prior to diabetes onset [55, 56]. Since dendritic cells, macrophages, and T lymphocytes are critical for disease progression [57, 58], understanding how ICAM-1 integrates immune cell infiltration with effector function was undertaken from a genetic manipulation approach. Transgenic mice with whole-body deletion of the Icam1 gene were generated to understand how leukocyte invasion and islet β-cell mass were impacted in a genetic background prone to autoimmune-mediated T1D.
Similar to several studies using peptide-targeting or antibody approaches, NOD mice with genetic deletion of the Icam1 gene were completely protected from diabetes development [48]. In addition, NOD mice with whole body Icam1 deletion only display mild insulitis when compared with wild-type NOD mice. Mice heterozygous for ICAM-1 deletion exhibited an intermediate phenotype (delayed onset relative to ICAM+/+ mice, p = 0.09) suggesting gene dosage is critical for determining disease onset. Remarkably, this reduction in insulitis also extends to NOD mice injected with cyclophosphamide, a drug that accelerates autoimmune-mediated diabetes onset. One possible explanation for the reductions in diabetes frequency is decreased IFNγ expression in pancreatic tissue of the NOD ICAM−/−mice compared to ICAM+/+ mice. This decrease in IFNγ is congruent with reductions in immune cells that produce pro-inflammatory cytokines (e.g., CD4+, CD8+ cells, etc.).
4. ICAM-1 influences T-cell function
T-lymphocytes are primed by antigen presenting cells [59, 60]. This involves major histocompatibility complex (MHC):peptide interaction with the T-cell receptor. Additional co-regulatory signals strengthen the activation potential of an individual T-cell population [61]. The ICAM-1/LFA-1 interaction can provide such a co-stimulatory role to assist with full priming and activation of a particular lymphocytic population [62]. In addition, there is a T-cell:T-cell interaction that also requires ICAM-1/LFA-1 [63]. These interactions are likely to be essential for normal immune function as well as participate in the development of autoimmune disease. Data in support of ICAM-1 as a major contributor to autoimmunity is illustrated by the studies discussed below.
Disrupting the ICAM-1/LFA-1 interaction with a combined anti-ICAM-1 and anti-LFA-1 antibody strategy early in life (2–4 weeks old) can render the splenocytes from 22 week old NOD mice unable to transfer diabetes to NOD-SCID mice [41, 47]. These observations point to a possible modifiable phase in the early development of autoimmunity, wherein the ICAM-1/LFA-1 pathway is an important contributor to possible priming of specific T-cell subsets that influence onset of disease. Indeed, preventing the ICAM-1/LFA-1 interaction early in the mouse life span appears sufficient to spare the pancreatic islets from the full effect of the cytotoxic lymphocytic response and thus could be a critical signaling point in the early phases of autoimmunity leading to T1D.
For example, during the presentation of islet autoantigen(s) to naïve autoreactive T cells, a sustained ICAM-1 / LFA-1 interaction is likely to be one critical factor that tips the balance toward loss of self-tolerance. Many of the early studies into the role of the ICAM-1/LFA-1 pathway attempt to determine the specific mechanism of tolerance induced upon blockade. One possibility is the deletion or inactivation of autoreactive T cells. However, splenic T cells isolated from nondiabetic NOD mice (who had received protection due to treatment with ICAM1/LFA-1 mAbs early in life) were just as proliferative in response to the islet autoantigen GAD65 as controls. [41]. In addition, T cell numbers in the spleen or ex vivo proliferative responses from mice who were treated with the mAbs versus control mice remained similar [52]. On the other hand, splenocytes from normo- and hyperglycemic NOD mice treated with ICAM1/LFA-1 peptides at 13 weeks of age [45] or with an Icam1 genetic deletion [64] did not proliferate in response to islet antigen ex vivo.
It is possible that a population of suppressor T cells induced during the tolerizing phase by anti-ICAM-1/LFA-1 intervention helps control autoimmunity. Supporting this notion, systemic administration of the TREG and immunomodulatory cytokine IL-10 reduces insulitis and hyperglycemia in the NOD mouse [65]. In addition, the compartment of exposure may be an important factor, as transgenic production of IL-10 by beta-cells accelerated diabetes unless paired with Icam1 gene deficiency [exon 4] [64]. Moriyama et al. argue against an important role of suppressor T cells, since mixing the splenocytes of anti-ICAM-1/LFA-1 treated mice with splenocytes of acutely diabetic NODs and transferring them into nondiabetic recipients only slightly delayed diabetes onset [41]. Attempts to expand the TREG population with IL-2 while simultaneously reducing effector T cell and NK cell proliferation with anti-LFA-1 antibody were considered. Although the TREG population was indeed expanded and even reduced islet destruction by autoreactive T cells in transfer experiments, this treatment was ultimately not successful because of the robust expansion of effector cells in the pancreatic lymph node [66]. Thus, a balance between Treg control of inflammation and expansion of self-reactive effector T-cells is constantly in flux and is likely why onset of T1D is heterogeneous in both mice and humans.
5. ICAM-1 influences Islet Graft Rejection
Islet transplantation or transplantation of stem-cell derived β-cells are promising potential therapies for Type 1 Diabetes [67, 68]. However, several barriers prevent widespread use of the procedure. These include: 1) islets from multiple donors needed to achieve insulin-independence in one recipient patient. 2) The broad immunosuppressive drug regimen required to protect the transplanted tissue and the resulting side effects from such pharmacological interventions. 3) The transplanted tissue will be subjected to autoimmune attack by the host immune system. 4) Exactly how to make stable, differentiated, fully functional β-cells from stem cells is not entirely understood.
Efforts to understand how ICAM-1 is involved in survival of transplanted tissue has been a focus of past studies. Mouse allogeneic islet transplant survival improved when recipients of transplanted tissue received antibodies against ICAM-1 or LFA-1 [69–73]. The most common intervention strategy typically involved islet transplant immediately followed by mAb targeting either one or both proteins. LFA-1 blockade improves graft survival when treatment was started 7 days after transplant [71].
Blocking the ICAM-1 to LFA-1 interaction is effective in prolonging survival of xenografts. Combining treatments of monoclonal antibodies to both LFA-1 and ICAM-1 prolonged survival time of rat islet grafts when transplanted into mice [74–76]. Pre-treatment of human islets with anti-ICAM1 antibody prior to transplantation in an STZ-induced mouse model showed a 40% survival rate of the xenograft beyond 100 days compared to non-treated islets or islets treated with anti-ICAM2 and anti-LFA-3 antibodies [77]. Similar protection against graft rejection was obtained when islets are transplanted under the renal capsule or injected into the portal vein [72]. However, in other studies, islet graft survival was not found to be improved with ICAM-1 gene deficiency when either the donor or the recipient was ICAM-1 −/− [78]. Graft survival also did not improve in a BL6 to NOD transplant when the donor was ICAM-1 −/− [79]. However, experimental variations revealed that pretreatment of donor and recipient with ICAM-1 or LFA-1 blockade (using either Abs or oligonucleotide delivery) can improve graft survival compared with postoperative treatment [80]. In addition, antisense oligonucleotides against ICAM-1 were also effective [72]. We postulate that many of the discordant findings arise from differences in experimental design, genetics (e.g., mouse strain used), and/or timing of specific interventions.
At present, the precise mechanisms of tolerance and the fate of graft-reactive T cells is not completely understood. Mice that have achieved indefinite islet graft survival via ICAM-1/LFA-1 blockade will still reject third party skin grafts, so the beneficial effect is not attained through general immunosuppression or a permanent defect in antigen presentation [81]. The incongruities in the literature start with reactivity to donor-type tissue: some reports indicate that recipients with long-term transplanted islets reject donor-type skin or islet grafts [77, 81], while others report tolerance [71]. Some clues into the mechanism(s) come from work in xenogeneic transplantation. For example, in a specific case of rat to mouse xenograft, the use of anti-rat ICAM-1 mAb in combination with anti-mouse LFA-1 mAb was the most effective method of prolonging graft survival [75]. It is possible that anti-donor ICAM-1 blockade and anti-recipient LFA-1 blockade are the most effective combination because ICAM-1 on the surface of donor cells may be more critical in generating the autoimmune response. This finding is congruent with the work of Zeng et al, wherein anti-human ICAM-1 was the most potent antibody for inhibiting murine splenocyte proliferation in vitro in response to human islets, and it was effective at prolonging human to mouse xenotransplantation [77].
Another possibility is that the antigen presenting cell(s) are of donor origin. If true, the critical antigen presentation event in islet transplant rejection would be donor APCs arriving along with the transplanted tissue, who then present donor-type antigens to recipient host T cells, providing a direct method of rejection [82]. This would explain results showing that despite achieving over 100 days of tolerance in the ICAM-1-treated human xenograft, mice who were given a secondary untreated graft from the same donor still rejected the new graft [77]. If the main APCs in graft rejection are donor type APCs, then the secondary transplant would theoretically bring in a new batch of donor APCs, and the lack of inhibition would result in these new APCs presenting antigens to the host. This notion is supported by evidence that xenogeneic and allogeneic islet transplants survive significantly longer when the islets are highly purified (i.e., most if not all of the immune cells removed before transplant) [83]. In addition, when mice were simultaneously given anti-ICAM-1-treated islet grafts in the right kidney and untreated grafts in the left kidney, only the untreated graft was rejected [77]. If donor APCs had been brought in the untreated islets, it would be expected that presentation of antigens to the host, which are equally immunogenic against the islets in each kidney, unless blockade of ICAM-1 disrupted antigen presentation. Another possibility is that the transplanted tissue was not being integrated into the existing blood supply, but was rather attempting to recruit new blood vessels to supply the graft. It is possible, even likely, that multiple mechanisms of rejection (and tolerance) exist to explain the disparate outcomes within the various studies and the role ICAM-1 plays depends on the context.
The ICAM-1/LFA-1 pathway is not the only costimulatory pathway investigated as a therapeutic approach to prolong islet transplantation. Indeed, the blockade of several costimulatory receptors such as CD28, ICOS, CD40, and others have been reviewed [61, 84]. In mice, anti-LFA-1 treatment worked in conjunction with an antibody against CD2. Though each antibody prolonged islet graft survival, no additive effect was observed, and LFA-1 blockade was more effective than CD2 blockade [81]. Interventions targeting CD154 (CD40L) in a BL6 to NOD transplant context improved islet graft survival, and this augmentation was further enhanced by LFA-1 blockade [79]. Based on these data, it is perhaps not surprising that therapies targeting costimulatory blockade have reached clinical practice as part of the islet transplantation procedure in individuals with T1D in order to restore islet function and achieve maintenance of normoglycemia, and insulin independence. Along these lines, an LFA-1 antibody, efalizumab, showed better islet graft survival and improved glucose control in humans compared to the Edmonton Protocol, in addition to being a safer alternative [85, 86].
For example, in one trial, all eight subjects receiving islet transplantation with maintenance immunosuppression by efalizumab in combination with thymoglobulin and sirolimus, achieved insulin independence for varying lengths of time and lowering of HbA1c [85]. In addition, efalizumab was a safer alternative with regard to islet and renal function compared to other immunosuppressants such as corticosteroids and calcineurin inhibitors (e.g. tacrolimus) used in the Edmonton protocol. In a separate study, an efalizumab-based regimen was compared to the Edmonton protocol [86]. All four patients on the efalizumab-based intervention achieved insulin independence with a single islet cell infusion, whereas 6 of 8 islet graft recipients on the Edmonton protocol required multiple infusions. Individuals on the efalizumab-based intervention also experienced less adverse events and no allosensitization compared to study participants on the Edmonton protocol. Unfortunately, this antibody was withdrawn from the market after three patients out of roughly 40,000 subjects presented with progressive multifocal leukoencephalopathy [87]. Long-term follow up studies were not possible because efalizumab was withdrawn from the market and many patients on this regimen were switched to abatacept. Abatacept is an approved medication in the treatment of rheumatoid arthritis and is a CTLA4-Ig fusion protein that attenuates T-cell activation by blocking the CD28 co-stimulatory pathway. Two of the three subjects that remained on immunosuppressive therapy following the withdrawal of efalizumab rejected their islet grafts suggesting that, despite lower risk for toxicity compared to tacrolimus and sirolimus, blockade of the CD28 costimulatory pathway by abatacept may not be an effective strategy for preventing islet rejection [86]. Further studies are required to demonstrate this directly.
6. ICAM-1 in individual cell types: influencing autoimmunity and graft rejection.
Mechanisms driving autoimmune responses and those promoting graft rejection are both linked to immune cell recruitment and function. However, a key difference between the two is that during the rejection of grafted tissue, the immunogenic element is most likely donor-type MHC molecules. This is in contrast to autoimmunity of diabetes, wherein the antigens are β-cell specific autoantigens such as insulin and GAD65 [88]. For example, Bertry-Coussot et al., used a soluble ICAM-1 intervention, which reduced insulitis and diabetes in the NOD mouse, but did not prevent skin allograft rejection [42]. Although the antigens tolerized in the situations of autoimmunity and islet transplantation may be different, blockade of the ICAM-1/LFA-1 pathway clearly shows its relevance for the immune-mediated destruction of the islets. In both cases, the uninterrupted pathway performs the functions of leukocytic extravasation, antigen presentation to autoreactive or graft-reactive T cells, and enhancing the effector functions of cytotoxic lymphocytes (Figure 2). Below is an evaluation of the ICAM-1 role in individual cell types, and its putative contribution to autoimmunity and islet graft rejection.
6.1. Endothelial cells
Due to the role of ICAM-1 in extravasation of immune cells from the blood vessels, it is possible that protein expression in endothelial cells is a critical element of autoimmune-mediated loss of tissue function. Indeed, ICAM-1 is easily visible on islet endothelium during autoimmune diabetes progression [40, 89]. However, in a number of diabetes models, the inhibition of ICAM-1 does not actually prevent extravasation of immune cells to the pancreatic islets. Mice with a whole-body genetic deficiency in the Icam1 gene treated with STZ show no reduction in islet immune infiltration compared to wild-type mice [49]. The sICAM-1/Ig treatment used by Bertry-Coussot et al also demonstrated no inhibition of extravasation in their NODscid transfer experiments [42]. One group found slowed, but not inhibited, entrance of 3A9 T cells into the islets of IP-HEL mice, and they observed a similar result with BDC2.5 T cells injected into irradiated NOD mice with Icam1 genetic disruption [90]. Leukocyte migration into the islets was also not inhibited by LFA-1 blockade [73] or ICAM-1 gene deletion in islet transplantation contexts [78]. Redundancy within the range of pathways involved in extravasation means that other membrane receptors could mediate the trafficking process in the absence of ICAM-1. For example, treatment of NOD mice at two weeks of age with antibodies against VLA-4 and VCAM-1 effectively prevents autoimmune diabetes development by 30 weeks of age; however, this therapy required continuous administration over the entire experimental timeframe [91]. By contrast, blockade of the ICAM-1/LFA-1 pathway during a critical early tolerizing phase is sufficient, as tolerance remains after removal of the intervention. The study of Fabien et al showed that interference with L-selectin more effectively prevented islet infiltration than LFA-1 mAbs, despite the latter having a more beneficial effect in preserving islet function [92]. Thus, even if ICAM-1 contributes to autoimmune diabetes or islet graft rejection through its role in extravasation, the redundancy of transendothelial migration pathways may render the LFA-1/ICAM-1 binding event less important or even dispensable for leukocyte trafficking into the islet.
6.2. Stimulation of Naïve T Cells
Immune invasion into pancreatic tissue of the autoimmune-prone NOD mouse involves increased presence of macrophages and dendritic cells with ICAM-1 readily detectable on the surface of these cells [46, 89, 93]. Due to the role of ICAM-1 in providing a second signal for T cell activation, it is conceivable that professional APCs within the islet present β–cell specific antigens to activate naïve T cells. In addition, another possibility is that T-cells recognize antigen on the β-cell surface and use ICAM-1 on β-cells as the secondary signal for activation during autoimmunity. Prior to hyperglycemia, the islets of NOD mice are enriched with CD103+ dendritic cells, which are highly efficient at cross-presenting MHC-I bound by peptide to CD8+ T cells; the presence of a dendritic cell that presents antigen efficiently to CD4+ and CD8+ T cells enables islet destruction to proceed [94]. Thus, the presence or absence of ICAM-1 on that APC could be a critical element in the induction of autoimmunity. In addition, specific polymorphisms in T1D risk among humans are related to other genes involved in co-stimulation and the immune synapse, including the genes encoding CTLA4, PD-1, IL-2RA, and PTPN22 [95]. The mechanistic work probing a role for ICAM-1 during antigen presentation during islet autoimmunity and islet transplantation rejection makes a strong case for its role in each of these processes.
The work of Moriyama et al. using NOD mice showed that a two-week treatment of ICAM-1/LFA-1 mAbs early in life protected mice from disease [41]. It is plausible that interactions blocked by these antibodies include the initial antigen presentation events that precede the immune infiltration of the islets. Furthermore, donor T cells that could only recognize transgenic β-cell specific antigens when these were presented by donor APCs revealed that antigen presentation from APC only to T cell was sufficient to cause disease [93]. Moreover, anti-LFA-1 treatment reduced proliferation of transferred autoreactive T cells within the pancreatic lymph nodes of NOD mice [66]. In addition, CD4+ T cells specific for OVA peptide were primed in vitro by Drosophila APCs either in the presence or absence of ICAM-1 on the APC surface and then transferred into mice who expressed the OVA peptide under the control of the rat insulin promoter. Although both groups of T cells (those primed by APCs with or without ICAM-1) infiltrated the islets of their respective hosts, only the cells stimulated in the presence of ICAM-1 led to islet destruction [96]. In a mixed lymphocyte reaction (MLR) experiment with splenocytes from mice that had received islet transplants, APCs from ICAM-1 knockout mice caused a decreased proliferation response [79]. All of this evidence points to ICAM-1 on the surface of the APC as a vital element in both autoimmunity and transplant rejection.
6.3. Modulating Effector T-cell Function
The ICAM-1/LFA-1 pathway strengthens the immune interaction between the cytotoxic lymphocyte and the somatic cell targeted for killing. The immunological synapse that forms between these two cells is shorter-lived than APC stimulation of naïve T cells, but it is nonetheless important for effector cell function [27]. Treatment with a mAb against CD8 was sufficient to prevent insulitis and diabetes in NOD mice, pointing to the necessity of CD8+ CTLs in the disease process [47]. Hyperexpression of MHC I has been observed on the surface of endocrine cells of the diabetic NOD pancreas [46] and expression of MHC was increased by cytokines in vitro [97]. Pancreatic tissue, isolated islets, and beta cells in culture have little to no expression of ICAM-1 in the basal state, but they readily express the gene when exposed to IL-1β, IFN-γ, and TNF-α [15, 89, 97–99]. Whether the ICAM-1 protein is present on beta cells in the disease state in vivo was debated: some labs have observed its presence [100], while others have not [40]. Yagi et al. demonstrated that ICAM-1 is indeed expressed on the beta cells of diabetic NOD mice when examined under immunoelectron microscopy [98]. Their images also indicate LFA-1 on mononuclear cells, which contact the β-cells. With the more advanced imaging techniques available at present, in combination with co-localization antibody/antigen detection methods, it is clear that beta cells of diabetic mice, and more specifically within inflamed islets, express ICAM-1 [15, 99].
Due to several variables, including the transient nature of the ICAM-1/LFA-1 interactions and the lack of tissue-specific knockout models to directly address the role of ICAM-1, it has been difficult to impossible to investigate ICAM-1 within distinct tissue compartments to understand autoimmune disease. However, strategies using ex vivo approaches, which include isolation and expansion of CTLs that had invaded NOD islets, give some insights. These CTLs were incubated with NOD-derived beta cells and demonstrated significant cytotoxic activity; this cytotoxic activity was amplified by pretreatment of the beta cells with cytokines, and it was reduced in a concentration-dependent manner by mAbs against LFA-1 and ICAM-1 [98]. Monoclonal antibodies against LFA-1 improved graft survival even when administered 7 days after transplantation, when islets were already heavily infiltrated. Reduced islet rejection at such a late stage suggests that LFA-1 blockade may function, at least in part, by inhibiting effector T cell functions in the islet [70]. Thus, it is reasonable to speculate that ICAM-1 is normally induced on the β-cell in response to an inflammatory signal, such as viral infection; this surface expression would serve to alert the immune system that a virus is present and needs to be cleared. Consistent with this premise, infection by SV40 promoted ICAM-1 expression in the absence of cytokines [97]. On the other hand, reovirus infection did not promote ICAM-1 abundance on islet β-cells, indicating that ICAM-1 induction could be signal-specific [99].
7. Concluding Remarks and Future Directions
It is clear that ICAM-1 is required for progression of autoimmunity in NOD mice [48], while its role in islet graft rejection is still unresolved [75, 77, 78]. Nevertheless, there is solid evidence to show that ICAM-1 is critical for antigen presentation to naïve T cells, which likely contributes to autoimmune disease processes [79, 96]. However, less work has been done to investigate whether ICAM-1 directly on cells within the islet, especially β-cells, are critical for autoimmune and graft rejection processes. Effector T cell activity within the islet is indeed enhanced by the presence of ICAM-1 [98]. Thus, a possible mechanism for protection when ICAM-1 interaction with LFA-1 is inhibited or reduced is the reduction of immune activity in the islet overall, and an increase in the relative quantity of Th2 cytokines over Th1 cytokines in autoimmune diabetes [48] and graft rejection [69, 80]. Future studies will require tissue-targeted investigations into the role of ICAM-1 in each cellular compartment to provide new insights into the mechanisms underlying autoimmune disease onset and progression as well as leukocyte-mediated rejection of transplanted islets.
Acknowledgements
Funding for the authors’ laboratories was through the National Institutes of Health R03 AI151920 (J.J.C.), R01 DK123183 (J.J.C.), and P20 GM135002 (S.J.B.). Dr. Thomas Martin is supported by NIH T32 training grant AT004094-14.
Declaration of interests
Jason Collier reports financial support was provided by National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Campbell JE and Newgard CB, Mechanisms controlling pancreatic islet cell function in insulin secretion, Nat Rev Mol Cell Biol 22 (2021) 142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Burke SJ, Karlstad MD and Collier JJ, Pancreatic Islet Responses to Metabolic Trauma, Shock 46 (2016) 230–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sharma RB, O’Donnell AC, Stamateris RE, Ha B, McCloskey KM, Reynolds PR, Arvan P and Alonso LC, Insulin demand regulates β cell number via the unfolded protein response, J Clin Invest 125 (2015) 3831–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kahn BB, Type 2 diabetes: when insulin secretion fails to compensate for insulin resistance, Cell 92 (1998) 593–6. [DOI] [PubMed] [Google Scholar]
- [5].Esser N, Utzschneider KM and Kahn SE, Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia, Diabetologia 63 (2020) 2007–2021. [DOI] [PubMed] [Google Scholar]
- [6].Johnson JD, On the causal relationships between hyperinsulinaemia, insulin resistance, obesity and dysglycaemia in type 2 diabetes, Diabetologia 64 (2021) 2138–2146. [DOI] [PubMed] [Google Scholar]
- [7].Eizirik DL, Szymczak F and Mallone R, Why does the immune system destroy pancreatic beta-cells but not alpha-cells in type 1 diabetes?, Nat Rev Endocrinol (2023). [DOI] [PubMed] [Google Scholar]
- [8].Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, Winkler C, Ilonen J, Veijola R, Knip M, Bonifacio E and Eisenbarth GS, Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children, JAMA 309 (2013) 2473–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Atkinson MA, Eisenbarth GS and Michels AW, Type 1 diabetes, Lancet (London, England) 383 (2014) 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Campbell-Thompson ML, Atkinson MA, Butler AE, Chapman NM, Frisk G, Gianani R, Giepmans BN, von Herrath MG, Hyoty H, Kay TW, Korsgren O, Morgan NG, Powers AC, Pugliese A, Richardson SJ, Rowe PA, Tracy S and In’t Veld PA, The diagnosis of insulitis in human type 1 diabetes, Diabetologia 56 (2013) 2541–3. [DOI] [PubMed] [Google Scholar]
- [11].Baggiolini M, Chemokines and leukocyte traffic, Nature 392 (1998) 565–8. [DOI] [PubMed] [Google Scholar]
- [12].Collier JJ, Sparer TE, Karlstad MD and Burke SJ, Pancreatic islet inflammation: an emerging role for chemokines, J Mol Endocrinol 59 (2017) R33–R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bullard DC, Adhesion molecules in inflammatory diseases: insights from knockout mice, Immunol Res 26 (2002) 27–33. [DOI] [PubMed] [Google Scholar]
- [14].Dustin ML, Rothlein R, Bhan AK, Dinarello CA and Springer TA, Induction by IL 1 and interferon-gamma: tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1), J Immunol 137 (1986) 245–54. [PubMed] [Google Scholar]
- [15].Martin TM, Burke SJ, Batdorf HM, Burk DH, Ghosh S, Dupuy SD, Karlstad MD and Collier JJ, ICAM-1 Abundance Is Increased in Pancreatic Islets of Hyperglycemic Female NOD Mice and Is Rapidly Upregulated by NF-κB in Pancreatic β-Cells, J Immunol (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mizgerd JP, Spieker MR and Lupa MM, Exon truncation by alternative splicing of murine ICAM-1, Physiol Genomics 12 (2002) 47–51. [DOI] [PubMed] [Google Scholar]
- [17].Rothlein R, Dustin ML, Marlin SD and Springer TA, A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1, J Immunol 137 (1986) 1270–4. [PubMed] [Google Scholar]
- [18].Marlin SD and Springer TA, Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1), Cell 51 (1987) 813–9. [DOI] [PubMed] [Google Scholar]
- [19].Makgoba MW, Sanders ME, Ginther Luce GE, Dustin ML, Springer TA, Clark EA, Mannoni P and Shaw S, ICAM-1 a ligand for LFA-1-dependent adhesion of B, T and myeloid cells, Nature 331 (1988) 86–8. [DOI] [PubMed] [Google Scholar]
- [20].Staunton DE, Dustin ML and Springer TA, Functional cloning of ICAM-2, a cell adhesion ligand for LFA-1 homologous to ICAM-1, Nature 339 (1989) 61–4. [DOI] [PubMed] [Google Scholar]
- [21].Lampeter ER, Kishimoto TK, Rothlein R, Mainolfi EA, Bertrams J, Kolb H and Martin S, Elevated Levels of Circulating Adhesion Molecules in IDDM Patients and in Subjects at Risk for IDDM, Diabetes 41 (1992) 1668–1671. [DOI] [PubMed] [Google Scholar]
- [22].Fathollahi A, Massoud A, Amirzargar AA, Aghili B, Nasli Esfahani E and Rezaei N, sICAM-1, sVCAM-1 and sE-Selectin Levels in Type 1 Diabetes, Fetal and Pediatric Pathology 37 (2018) 69–73. [DOI] [PubMed] [Google Scholar]
- [23].Roep BO, Heidenthal E, de Vries RR, Kolb H and Martin S, Soluble forms of intercellular adhesion molecule-1 in insulin-dependent diabetes mellitus, Lancet 343 (1994) 1590–3. [DOI] [PubMed] [Google Scholar]
- [24].Davignon D, Martz E, Reynolds T, Kurzinger K and Springer TA, Lymphocyte function-associated antigen 1 (LFA-1): a surface antigen distinct from Lyt-2,3 that participates in T lymphocyte-mediated killing, Proc Natl Acad Sci U S A 78 (1981) 4535–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kurzinger K, Reynolds T, Germain RN, Davignon D, Martz E and Springer TA, A novel lymphocyte function-associated antigen (LFA-1): cellular distribution, quantitative expression, and structure, J Immunol 127 (1981) 596–602. [PubMed] [Google Scholar]
- [26].Sanchez-Madrid F, Krensky AM, Ware CF, Robbins E, Strominger JL, Burakoff SJ and Springer TA, Three distinct antigens associated with human T-lymphocyte-mediated cytolysis: LFA-1, LFA-2, and LFA-3, Proc Natl Acad Sci U S A 79 (1982) 7489–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gérard A, Cope AP, Kemper C, Alon R and Köchl R, LFA-1 in T cell priming, differentiation, and effector functions, Trends Immunol 42 (2021) 706–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pyszniak AM, Carpenito C and Takei F, The role of LFA-1 (CD11a/CD18) cytoplasmic domains in binding to intercellular adhesion molecule-1 (CD54) and in postreceptor cell spreading, Exp Cell Res 233 (1997) 78–87. [DOI] [PubMed] [Google Scholar]
- [29].Chen YG, Mathews CE and Driver JP, The Role of NOD Mice in Type 1 Diabetes Research: Lessons from the Past and Recommendations for the Future, Frontiers in endocrinology 9 (2018) 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shimizu J, Kanagawa O and Unanue ER, Presentation of beta-cell antigens to CD4+ and CD8+ T cells of non-obese diabetic mice, J Immunol 151 (1993) 1723–30. [PubMed] [Google Scholar]
- [31].Zakharov PN, Hu H, Wan X and Unanue ER, Single-cell RNA sequencing of murine islets shows high cellular complexity at all stages of autoimmune diabetes, J Exp Med 217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Haskins K and Cooke A, CD4 T cells and their antigens in the pathogenesis of autoimmune diabetes, Curr Opin Immunol 23 (2011) 739–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Toren E, Burnette KS, Banerjee RR, Hunter CS and Tse HM, Partners in Crime: Beta-Cells and Autoimmune Responses Complicit in Type 1 Diabetes Pathogenesis, Front Immunol 12 (2021) 756548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Brode S, Raine T, Zaccone P and Cooke A, Cyclophosphamide-induced type-1 diabetes in the NOD mouse is associated with a reduction of CD4+CD25+Foxp3+ regulatory T cells, J Immunol 177 (2006) 6603–12. [DOI] [PubMed] [Google Scholar]
- [35].Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF and Eisenbarth GS, Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice, Nature 435 (2005) 220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Roep BO and Peakman M, Antigen targets of type 1 diabetes autoimmunity, Cold Spring Harbor perspectives in medicine 2 (2012) a007781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nakayama M, Insulin as a key autoantigen in the development of type 1 diabetes, Diabetes Metab Res Rev 27 (2011) 773–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Serreze DV, Leiter EH, Christianson GJ, Greiner D and Roopenian DC, Major histocompatibility complex class I-deficient NOD-B2mnull mice are diabetes and insulitis resistant, Diabetes 43 (1994) 505–9. [DOI] [PubMed] [Google Scholar]
- [39].Markmann JF, Bassiri H, Desai NM, Odorico JS, Kim JI, Koller BH, Smithies O and Barker CF, Indefinite survival of MHC class I-deficient murine pancreatic islet allografts, Transplantation 54 (1992) 1085–9. [DOI] [PubMed] [Google Scholar]
- [40].Hasegawa Y, Yokono K, Taki T, Amano K, Tominaga Y, Yoneda R, Yagi N, Maeda S, Yagita H, Okumura K and et al. , Prevention of autoimmune insulin-dependent diabetes in non-obese diabetic mice by anti-LFA-1 and anti-ICAM-1 mAb, Int Immunol 6 (1994) 831–8. [DOI] [PubMed] [Google Scholar]
- [41].Moriyama H, Yokono K, Amano K, Nagata M, Hasegawa Y, Okamoto N, Tsukamoto K, Miki M, Yoneda R, Yagi N, Tominaga Y, Kikutani H, Hioki K, Okumura K, Yagita H and Kasuga M, Induction of tolerance in murine autoimmune diabetes by transient blockade of leukocyte function-associated antigen-1/intercellular adhesion molecule-1 pathway, J Immunol 157 (1996) 3737–43. [PubMed] [Google Scholar]
- [42].Bertry-Coussot L, Lucas B, Danel C, Halbwachs-Mecarelli L, Bach JF, Chatenoud L and Lemarchand P, Long-term reversal of established autoimmunity upon transient blockade of the LFA-1/intercellular adhesion molecule-1 pathway, J Immunol 168 (2002) 3641–8. [DOI] [PubMed] [Google Scholar]
- [43].Martin S, Heidenthal E, Schulte B, Rothe H and Kolb H, Soluble forms of intercellular adhesion molecule-1 inhibit insulitis and onset of autoimmune diabetes, Diabetologia 41 (1998) 1298–303. [DOI] [PubMed] [Google Scholar]
- [44].van den Engel NK, an Haack M, Martin S and Kolb H, Oral DNA vaccination with a plasmid encoding soluble ICAM-1 modulates cytokine expression profiles in nonobese diabetic mice, J Mol Med (Berl) 80 (2002) 301–8. [DOI] [PubMed] [Google Scholar]
- [45].Dotson AL, Novikova L, Stehno-Bittel L and Benedict SH, Elimination of T cell reactivity to pancreatic β cells and partial preservation of β cell activity by peptide blockade of LFA-1:ICAM-1 interaction in the NOD mouse model, Clin Immunol 148 (2013) 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].O’Reilly LA, Hutchings PR, Crocker PR, Simpson E, Lund T, Kioussis D, Takei F, Baird J and Cooke A, Characterization of pancreatic islet cell infiltrates in NOD mice: effect of cell transfer and transgene expression, Eur J Immunol 21 (1991) 1171–80. [DOI] [PubMed] [Google Scholar]
- [47].Chowdhury SA, Nagata M, Yamada K, Nakayama M, Chakrabarty S, Jin Z, Kotani R and Yokono K, Tolerance mechanisms in murine autoimmune diabetes induced by anti-ICAM-1/LFA-1 mAb and anti-CD8 mAb, Kobe J Med Sci 48 (2002) 167–75. [PubMed] [Google Scholar]
- [48].Martin S, van den Engel NK, Vinke A, Heidenthal E, Schulte B and Kolb H, Dominant role of intercellular adhesion molecule-1 in the pathogenesis of autoimmune diabetes in non-obese diabetic mice, J Autoimmun 17 (2001) 109–17. [DOI] [PubMed] [Google Scholar]
- [49].Martin S, Vinke A, Heidenthal E, Schulte B and van den Engel N, Development of low-dose streptozotocin-induced diabetes in ICAM-1-deficient mice, Horm Metab Res 31 (1999) 636–40. [DOI] [PubMed] [Google Scholar]
- [50].Hayashi T, Hashimoto S and Kameyama Y, Reduced streptozotocin-induced insulitis in CD-1 mice by treatment with anti-intercellular adhesion molecule-1 and anti-lymphocyte function associated antigen-1 monoclonal antibodies together with lactic dehydrogenase virus infection, Int J Exp Pathol 75 (1994) 117–121. [PMC free article] [PubMed] [Google Scholar]
- [51].Makino S, Harada M, Kishimoto Y and Hayashi Y, Absence of insulitis and overt diabetes in athymic nude mice with NOD genetic background, Jikken Dobutsu 35 (1986) 495–8. [DOI] [PubMed] [Google Scholar]
- [52].Herold KC, Vezys V, Gage A and Montag AG, Prevention of autoimmune diabetes by treatment with anti-LFA-1 and anti-ICAM-1 monoclonal antibodies, Cell Immunol 157 (1994) 489–500. [DOI] [PubMed] [Google Scholar]
- [53].Driver JP, Chen YG and Mathews CE, Comparative genetics: synergizing human and NOD mouse studies for identifying genetic causation of type 1 diabetes, Rev Diabet Stud 9 (2012) 169–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wallberg M and Cooke A, Immune mechanisms in type 1 diabetes, Trends Immunol 34 (2013) 583–91. [DOI] [PubMed] [Google Scholar]
- [55].Burke SJ, Karlstad MD, Eder AE, Regal KM, Lu D, Burk DH and Collier JJ, Pancreatic β-Cell production of CXCR3 ligands precedes diabetes onset, Biofactors 42 (2016) 703–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Burke SJ and Collier JJ, Transcriptional Regulation of Chemokine Genes: A Link to Pancreatic Islet Inflammation?, Biomolecules 5 (2015) 1020–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Unanue ER, Antigen presentation in the autoimmune diabetes of the NOD mouse, Annu Rev Immunol 32 (2014) 579–608. [DOI] [PubMed] [Google Scholar]
- [58].Carrero JA, McCarthy DP, Ferris ST, Wan X, Hu H, Zinselmeyer BH, Vomund AN and Unanue ER, Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of NOD mice, Proc Natl Acad Sci U S A 114 (2017) E10418–E10427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Janeway CA Jr., The priming of helper T cells, Semin Immunol 1 (1989) 13–20. [PubMed] [Google Scholar]
- [60].Pishesha N, Harmand TJ and Ploegh HL, A guide to antigen processing and presentation, Nat Rev Immunol 22 (2022) 751–764. [DOI] [PubMed] [Google Scholar]
- [61].Chen L and Flies DB, Molecular mechanisms of T cell co-stimulation and co-inhibition, Nat Rev Immunol 13 (2013) 227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chirathaworn C, Kohlmeier JE, Tibbetts SA, Rumsey LM, Chan MA and Benedict SH, Stimulation through intercellular adhesion molecule-1 provides a second signal for T cell activation, J Immunol 168 (2002) 5530–7. [DOI] [PubMed] [Google Scholar]
- [63].Gerard A, Khan O, Beemiller P, Oswald E, Hu J, Matloubian M and Krummel MF, Secondary T cell-T cell synaptic interactions drive the differentiation of protective CD8+ T cells, Nat Immunol 14 (2013) 356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Balasa B, La Cava A, Van Gunst K, Mocnik L, Balakrishna D, Nguyen N, Tucker L and Sarvetnick N, A mechanism for IL-10-mediated diabetes in the nonobese diabetic (NOD) mouse: ICAM-1 deficiency blocks accelerated diabetes, J Immunol 165 (2000) 7330–7. [DOI] [PubMed] [Google Scholar]
- [65].Pennline KJ, Roque-Gaffney E and Monahan M, Recombinant human IL-10 prevents the onset of diabetes in the nonobese diabetic mouse, Clin Immunol Immunopathol 71 (1994) 169–75. [DOI] [PubMed] [Google Scholar]
- [66].Brenu EW, Bartley TJ, Wright CM and Hamilton-Williams EE, CD11a/ICAM-1 blockade combined with IL-2 targeting therapy causes a paradoxical acceleration of type 1 diabetes, Immunol Cell Biol 95 (2017) 803–813. [DOI] [PubMed] [Google Scholar]
- [67].Rickels MR and Robertson RP, Pancreatic Islet Transplantation in Humans: Recent Progress and Future Directions, Endocr Rev 40 (2019) 631–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Helman A and Melton DA, A Stem Cell Approach to Cure Type 1 Diabetes, Cold Spring Harb Perspect Biol 13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Arai K, Sunamura M, Wada Y, Takahashi M, Kobari M, Kato K, Yagita H, Okumura K and Matsuno S, Preventing effect of anti-ICAM-1 and anti-LFA-1 monoclonal antibodies on murine islet allograft rejection, Int J Pancreatol 26 (1999) 23–31. [DOI] [PubMed] [Google Scholar]
- [70].Nishihara M, Gotoh M, Fukuzaki T, Ohta Y, Monden M, Yagita H, Okumura K, Miyasaka M and Mori T, Potent immunosuppressive effect of anti-LFA-1 monoclonal antibody on islet allograft rejection, Transplant Proc 27 (1995) 372. [PubMed] [Google Scholar]
- [71].Nishihara M, Gotoh M, Ohzato H, Ohta Y, Luo Z, Dono K, Umeshita K, Sakon M, Monden M, Yagita H, Okumura K and Miyasaka M, Awareness of donor alloantigens in antiadhesion therapy induces antigen-specific unresponsiveness to islet allografts, Transplantation 64 (1997) 965–70. [DOI] [PubMed] [Google Scholar]
- [72].Katz SM, Bennett F, Stecker K, Clark JH, Pham T, Wang ME, Kahan BD and Stepkowski SM, ICAM-1 antisense oligodeoxynucleotide improves islet allograft survival and function, Cell Transplant 9 (2000) 817–28. [DOI] [PubMed] [Google Scholar]
- [73].Nicolls MR, Coulombe M, Yang H, Bolwerk A and Gill RG, Anti-LFA-1 Therapy Induces Long-Term Islet Allograft Acceptance in the Absence of IFN-γ or IL-4, The Journal of Immunology 164 (2000) 3627–3634. [DOI] [PubMed] [Google Scholar]
- [74].Fukuzaki T, Gotoh M, Monden M, Ohta Y, Dono K, Kanai T, Yagita H, Okumura K, Miyasaka M and Mori T, Role of adhesion molecules in islet xenograft rejection, Transplant Proc 26 (1994) 1113. [PubMed] [Google Scholar]
- [75].Ohta Y, Gotoh M, Ohzato H, Fukuzaki T, Nishihara M, Dono K, Umeshita K, Sakon M, Yagita H, Okumura K, Tanaka T, Kawashima H, Miyasaka M and Monden M, Direct antigen presentation through binding of donor intercellular adhesion molecule-1 to recipient lymphocyte function-associated antigen-1 molecules in xenograft rejection, Transplantation 65 (1998) 1094–100. [DOI] [PubMed] [Google Scholar]
- [76].Grochowiecki T, Gotoh M, Dono K, Takeda Y, Sakon M, Yagita H, Okumura K, Miyasaka M and Monden M, Induction of unresponsiveness to islet xenograft by MMC treatment of graft and blockage of LFA-1/ICAM-1 pathway, Transplantation 69 (2000) 1567–71. [DOI] [PubMed] [Google Scholar]
- [77].Zeng Y, Gage A, Montag A, Rothlein R, Thistlethwaite JR and Bluestone JA, Inhibition of transplant rejection by pretreatment of xenogeneic pancreatic islet cells with anti-ICAM-1 antibodies, Transplantation 58 (1994) 681–9. [PubMed] [Google Scholar]
- [78].Sandberg JO and Korsgren O, Islet allo- and xenotransplantation in normal, intercellular adhesion molecule-1-deficient, and P selectin-deficient mice, Transplantation 64 (1997) 584–9. [DOI] [PubMed] [Google Scholar]
- [79].Berney T, Pileggi A, Molano RD, Poggioli R, Zahr E, Ricordi C and Inverardi L, The effect of simultaneous CD154 and LFA-1 blockade on the survival of allogeneic islet grafts in nonobese diabetic mice, Transplantation 76 (2003) 1669–74. [DOI] [PubMed] [Google Scholar]
- [80].Katz SM, Tian L, Stepkowski SM, Phan T, Bennett CF and Kahan BD, Effect of ICAM-1/LFA-1 blockade on pancreatic islet allograft survival, function, and early cytokine production, Transplant Proc 29 (1997) 748–9. [DOI] [PubMed] [Google Scholar]
- [81].Gotoh M, Fukuzaki T, Monden M, Dono K, Kanai T, Yagita H, Okumura K and Mori T, A potential immunosuppressive effect of anti-lymphocyte function-associated antigen-1 monoclonal antibody on islet transplantation, Transplantation 57 (1994) 123–6. [PubMed] [Google Scholar]
- [82].Lin CM and Gill RG, Direct and indirect allograft recognition: pathways dictating graft rejection mechanisms, Curr Opin Organ Transplant 21 (2016) 40–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Gotoh M, Maki T, Porter J and Monaco AP, Augmented survival of purified islet xeno- and allografts with reduced numbers, Transplant Proc 19 (1987) 984. [PubMed] [Google Scholar]
- [84].Ford ML, Adams AB and Pearson TC, Targeting co-stimulatory pathways: transplantation and autoimmunity, Nat Rev Nephrol 10 (2014) 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Posselt AM, Bellin MD, Tavakol M, Szot GL, Frassetto LA, Masharani U, Kerlan RK, Fong L, Vincenti FG, Hering BJ, Bluestone JA and Stock PG, Islet transplantation in type 1 diabetics using an immunosuppressive protocol based on the anti-LFA-1 antibody efalizumab, Am J Transplant 10 (2010) 1870–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Turgeon NA, Avila JG, Cano JA, Hutchinson JJ, Badell IR, Page AJ, Adams AB, Sears MH, Bowen PH, Kirk AD, Pearson TC and Larsen CP, Experience with a novel efalizumab-based immunosuppressive regimen to facilitate single donor islet cell transplantation, Am J Transplant 10 (2010) 2082–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Carson KR, Focosi D, Major EO, Petrini M, Richey EA, West DP and Bennett CL, Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a Review from the Research on Adverse Drug Events and Reports (RADAR) Project, Lancet Oncol 10 (2009) 816–24. [DOI] [PubMed] [Google Scholar]
- [88].Thayer TC, Wilson SB and Mathews CE, Use of nonobese diabetic mice to understand human type 1 diabetes, Endocrinology and metabolism clinics of North America 39 (2010) 541–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Prieto J, Kaaya EE, Juntti-Berggren L, Berggren PO, Sandler S, Biberfeld P and Patarroyo M, Induction of intercellular adhesion molecule-1 (CD54) on isolated mouse pancreatic beta cells by inflammatory cytokines, Clin Immunol Immunopathol 65 (1992) 247–53. [DOI] [PubMed] [Google Scholar]
- [90].Calderon B, Carrero JA, Miller MJ and Unanue ER, Cellular and molecular events in the localization of diabetogenic T cells to islets of Langerhans, Proc Natl Acad Sci U S A 108 (2011) 1561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Tsukamoto K, Yokono K, Amano K, Nagata M, Yagi N, Tominaga Y, Moriyama H, Miki M, Okamoto N, Yoneda R and et al. , Administration of monoclonal antibodies against vascular cell adhesion molecule-1/very late antigen-4 abrogates predisposing autoimmune diabetes in NOD mice, Cell Immunol 165 (1995) 193–201. [DOI] [PubMed] [Google Scholar]
- [92].Fabien N, Bergerot I, Orgiazzi J and Thivolet C, Lymphocyte function associated antigen-1, integrin alpha 4, and L-selectin mediate T-cell homing to the pancreas in the model of adoptive transfer of diabetes in NOD mice, Diabetes 45 (1996) 1181–6. [DOI] [PubMed] [Google Scholar]
- [93].Lo D, Reilly CR, Scott B, Liblau R, McDevitt HO and Burkly LC, Antigen-presenting cells in adoptively transferred and spontaneous autoimmune diabetes, Eur J Immunol 23 (1993) 1693–8. [DOI] [PubMed] [Google Scholar]
- [94].Unanue ER, Ferris ST and Carrero JA, The role of islet antigen presenting cells and the presentation of insulin in the initiation of autoimmune diabetes in the NOD mouse, Immunol Rev 272 (2016) 183–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Bluestone JA, Herold K and Eisenbarth G, Genetics, pathogenesis and clinical interventions in type 1 diabetes, Nature 464 (2010) 1293–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Camacho SA, Heath WR, Carbone FR, Sarvetnick N, LeBon A, Karlsson L, Peterson PA and Webb SR, A key role for ICAM-1 in generating effector cells mediating inflammatory responses, Nat Immunol 2 (2001) 523–9. [DOI] [PubMed] [Google Scholar]
- [97].Vives M, Soldevila G, Alcalde L, Lorenzo C, Somoza N and Pujol-Borrell R, Adhesion molecules in human islet beta-cells. De novo induction of ICAM-1 but not LFA-3, Diabetes 40 (1991) 1382–90. [DOI] [PubMed] [Google Scholar]
- [98].Yagi N, Yokono K, Amano K, Nagata M, Tsukamoto K, Hasegawa Y, Yoneda R, Okamoto N, Moriyama H, Miki M and et al. , Expression of intercellular adhesion molecule 1 on pancreatic beta-cells accelerates beta-cell destruction by cytotoxic T-cells in murine autoimmune diabetes, Diabetes 44 (1995) 744–52. [DOI] [PubMed] [Google Scholar]
- [99].Campbell IL, Cutri A, Wilkinson D, Boyd AW and Harrison LC, Intercellular adhesion molecule 1 is induced on isolated endocrine islet cells by cytokines but not by reovirus infection, Proc Natl Acad Sci U S A 86 (1989) 4282–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Calderon B, Carrero JA, Miller MJ and Unanue ER, Entry of diabetogenic T cells into islets induces changes that lead to amplification of the cellular response, Proc Natl Acad Sci U S A 108 (2011) 1567–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]