

Abstract
Air pollution is the leading cause of lung cancer after tobacco smoking, contributing to 20% of all lung cancer deaths. Increased risk associated with living near trafficked roads, occupational exposure to diesel exhaust, indoor coal combustion and cigarette smoking, suggest that combustion components in ambient fine particulate matter (PM2.5), such as polycyclic aromatic hydrocarbons (PAHs), may be central drivers of lung cancer. Activation of the aryl hydrocarbon receptor (AhR) induces expression of xenobiotic-metabolizing enzymes (XMEs) and increase PAH metabolism, formation of reactive metabolites, oxidative stress, DNA damage and mutagenesis. Lung cancer tissues from smokers and workers exposed to high combustion PM levels contain mutagenic signatures derived from PAHs. However, recent findings suggest that ambient air PM2.5 exposure primarily induces lung cancer development through tumor promotion of cells harboring naturally acquired oncogenic mutations, thus lacking typical PAH-induced mutations. On this background, we discuss the role of AhR and PAHs in lung cancer development caused by air pollution focusing on the tumor promoting properties including metabolism, immune system, cell proliferation and survival, tumor microenvironment, cell-to-cell communication, tumor growth and metastasis. We suggest that the dichotomy in lung cancer patterns observed between smoking and outdoor air PM2.5 represent the two ends of a dose–response continuum of combustion PM exposure, where tumor promotion in the peripheral lung appears to be the driving factor at the relatively low-dose exposures from ambient air PM2.5, whereas genotoxicity in the central airways becomes increasingly more important at the higher combustion PM levels encountered through smoking and occupational exposure.
Keywords: Air pollution, Diesel exhaust, Smoking, Occupational exposure, Carcinogenesis, Genotoxicity, Inflammation, Tumor promotion, Tumor microenvironment, Tumor metastasis
1. Introduction
Lung cancer has long been recognized as one of the leading causes of cancer‑associated mortality [1–3]. It is a complex process which develops slowly over time, and consequently, most people diagnosed with lung cancer are 65 or older [4]. Central steps in the development include tumor initiation, tumor formation and progression, matrix remodeling, intravasation, extravasation and metastasis [5]. Each step is determined by genetic predispositions and mutations acquired over an individual’s lifetime due to endogenous processes, lifestyle factors and/or environmental exposures.
Although smoking remains the biggest risk factor for lung cancer, about 25% of the cases are not attributable to tobacco [6]. The Global Burden of Disease (GBD) Project has estimated that 19% of lung cancer deaths are associated with exposure to air pollution making it the second largest risk factor [7]. The majority of this is mainly attributed to fine particulate matter, PM2.5 (with particle aerodynamic diameter of less than 2.5 μm), derived from combustion sources such as traffic exhaust, coal and biomass burning, and industrial activities [7]. Outdoor air PM and diesel exhaust particles (DEP) have been classified as Group 1 known human carcinogens by the International Agency for Research on Cancer [8,9]. Other combustion PM sources such as cigarette smoke [10,11] and indoor combustion of coal [12] have also been classified as Group 1 human carcinogens, while emissions from the burning of biomass/wood have been classified as a Group 2A (probable) human carcinogen [12]. Epidemiological studies indicate that PM2.5 exposure may increase both the incidence and mortality rates associated with lung cancer [13], and also decrease the survival time of patients with lung cancer [14]. Several studies have also reported an increased association between living near busy roadways and lung cancer incidence and mortality in Asia, Europe and North-America, pointing towards a central role of direct exposure to combustion emissions from road vehicles such as ultrafine particles and/or volatile/semi-volatile organic compounds [15–19].
The causal links between combustion PM exposure and lung cancer development are further supported by both in vitro and in vivo studies [8,9,20,21]. Combined epidemiological and experimental studies have provided essential information on cancer acquisition hallmarks including genetic instability, sustained proliferative signaling, insensitivity to antigrowth signals, resistance to cell death, replicative immortality, replicative immortality, dysregulated metabolism, tumor promoting inflammation, angiogenesis, tissue invasion and metastasis [5,22]. Thus, modifications of a variety of biological processes seem to contribute to the carcinogenic effects of PM2.5.
Combustion-derived PM typically consists of aggregates of smaller carbon particles with mixtures of organic chemicals adhered to their surface [23]. Their carcinogenic properties have largely been attributed to extractable organic material (EOM) and the content of polycyclic aromatic hydrocarbons (PAHs) [24]. PAHs are a highly diverse group of chemicals originating from combustion of organic materials. Numerous PAHs are considered important air pollutants and particle toxicants. Some of them are classified either as carcinogenic or probably carcinogenic to human respiratory organ [8,9,25–27]. Other effects that have been linked to PAHs exposure via PM2.5 inhalation are impairment of respiratory functions, exacerbation of asthma and increased morbidity/mortality of obstructive lung diseases [28].
Several PAHs are considered complete carcinogens contributing to both tumor initiation and promotion [5,25,29]. Nevertheless, the carcinogenicity of PAHs is most often linked to their metabolism and genotoxicity: the formation of reactive electrophilic metabolites forming covalent DNA adducts leading to mutations in oncogenes and tumor suppressor genes [30]. Importantly, the mutagenic signatures of EOM of cigarette smoke, combustion PM and air pollution PM resemble the mutation pattern of benzo[a]pyrene (B[a]P), and the same mutations are also found in lung cancers from smokers and people exposed to high levels of combustion aerosols from indoor use of smoky coal or in occupational settings [24].
The metabolism and genotoxicity of PAHs are largely regulated by the aryl hydrocarbon receptor (AhR) through transcriptional control of xenobiotic metabolizing enzymes [31]. The AhR, which is the main cellular sensor of PAHs and other aromatic compounds, is a basic helix-loop-helix PAS transcription factor, expressed in almost all tissues including a number of lung cell types such as bronchial epithelial cells, alveolar type II cells, club (Clara) cells, endothelial cells and macrophages [32,33]. The prototypic genes regulated by AhR are the cytochrome P450 (CYP) family 1 members CYP1A1 and CYP1B1. While the CYP enzymes are generally considered to be important detoxification enzymes, CYP1A1 and CYP1B1are also involved in the metabolic activation of chemicals such as B[a]P into its ultimate carcinogen B[a]P-7,8-dihydrodiol-9,10-epoxide (BPDE) [34]. In line with this, the AhR appears to be essential for the carcinogenic effects of B[a]P [35,36].
While PAH-induced genotoxicity may be central to lung cancer development in smokers, it has become increasingly clear that the pattern of mutations and lung cancer subtypes in never-smokers are distinctly different [6,24,37]. Lung cancer in never-smokers rather appears to derive from naturally occurring mutations [37]. As PM2.5 is regarded as the main cause of lung cancer in never-smokers, it is possible that the carcinogenic effects of air pollution differ from those of smoking and that PAH-induced genotoxicity is of lesser importance. In support of this, a recent study suggests that tumor promotion is the main driver of air pollution-induced lung cancers [38]. However, this does not exclude other roles of PAHs and AhR in cancer development, which extends far beyond metabolic activation and genotoxic effects of PAHs. One of the best described roles of AhR is the tumor promoting action of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) [39–42]. In fact, AhR appears to be involved in all of the major stages in cancer development, including cancer initiation, promotion, progression, invasion, and metastasis. It has thus emerged as a regulator of malignant cell progression and immune evasion associated with poor cancer outcomes [43,44].
In light of the emerging evidence suggesting that lung cancer development from air pollution differs from what is seen in smokers [6,37,38], this review aims to address the many-faceted roles of PAHs and AhR in cancer development associated with combustion particle exposure (Fig. 1). We will discuss their potential involvement in all stages of carcinogenesis, from DNA damage to promotion, progression, invasion, and metastasis, and whether some of the differences observed between smoking and urban air PM2.5 may rather be a matter of the dose.
Fig. 1.
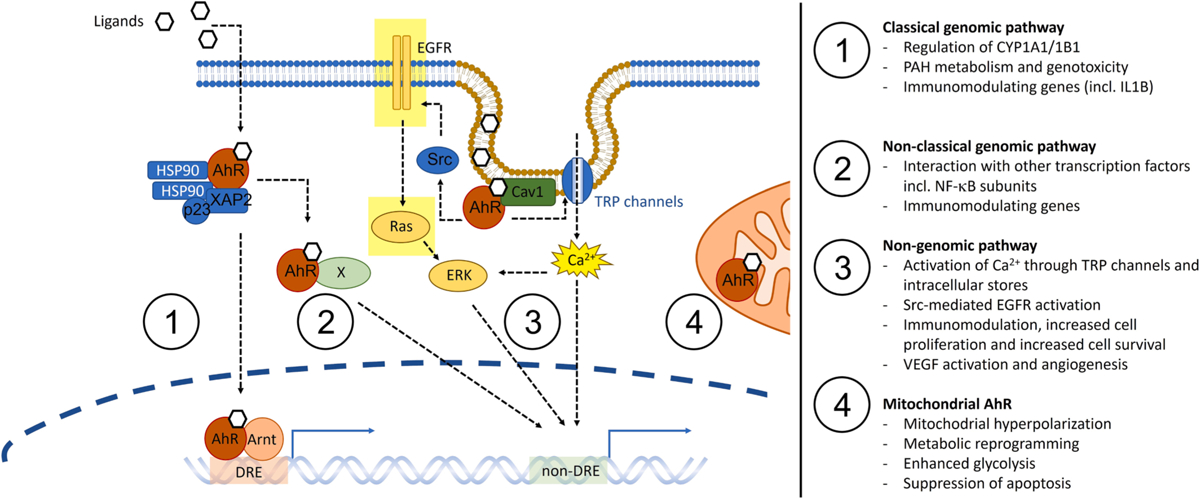
Overview of the main signaling pathways of AhR and the cancer-related responses regulated by these. AhR may induce effects through at least four different signaling modes. In the classical genomic pathway (1), inactive AhR resides in the cytosol bound to heat-shock protein 90 (HSP90), XAP2, and p23 proteins. Upon ligand activation, AhR translocates to the nucleus, dimerizes with its binding partner Arnt, and the AhR:Arnt dimer binds to dioxin response elements (DREs) in the regulatory region of target genes. The prototypical genes activated are the CYP1A1/−1B1 enzymes, which may metabolize PAHs into genotoxic metabolites. However, a number of genes express DRE sites and are affected by classical AhR signaling, including IL1B and other proinflammatory cytokines. AhR may also dimerize with other binding partners (X) such as NF-κB subunits through non-classical genomic signaling (2), activating alternative binding sites and regulate other genes including various immunomodulating factors. A subfraction of AhR appears to be localized in close connection interacting with caveolin-1 (Cav1) in caveolae, acting as a cytosolic signaling molecule in the so-called non-genomic pathway (3). Non-genomic AhR signaling regulates rapid activation of Ca2 + signaling from transient receptor potential (TRP) channels and intracellular stores, and Src-mediated activation of EGFR-RAS-ERK signaling which may regulate cell proliferation, cell survival, angiogenesis and immunomodulating responses. Importantly mutations in the KRAS (Ras) and EGFR genes are characteristic of lung cancers in smokers and never-smokers, respectively, underscoring the potential importance of the non-genomic pathway. Another subfraction of AhR has been localized in the inter-membrane space of mitochondria, mitochondrial AhR (4), and may regulate mitochondrial polarization, metabolic reprogramming, glycolysis and apoptosis, which is also associated with lung cancer development.
2. PM2.5, sources, and PAH characteristics
Potential mediators/modulators of the carcinogenic effects of PM2.5 and combustion-derived PM include the particle shape and size, surface reactivity (charge and presence of reactive groups including redox-active transition metals) and adherence of various organic components (PAHs, PAH-quinones and bacterial endotoxins) [45]. While the levels of organic chemicals are often found to be in the range 20–30% of total particle mass, it may reach as much as 90% [46]. The specific composition and the relative amount of chemicals attached to PM2.5 are highly dependent on sources, including combustion technology and fuel burned. Traditionally, diesel engine particles (DEP) have received most attention, and DEP emissions can be distinguished from gasoline emissions and wood smoke particles (WSP) by a high level of unresolved alkanes [47,48]. DEP also contained higher levels of alkylated and nitrated PAHs (alkyl-PAHs and nitro-PAHs) compared to other combustion PM [25,47]. By contrast, WSP may contain somewhat higher levels of oxygenated and hydroxylated PAHs (oxy-PAHs and hydroxy-PAHs), as compared to traffic emissions [47,49].
The relative contribution of different sources to PAHs measured on PM2.5 is changing as combustion technologies develop. The introduction of ever improved emission aftertreatment, such as EURO-classified diesel particulate filter (DPF) has considerably reduced both PM and PAH emissions from modern diesel vehicles, and today light-duty gasoline vehicles represent the dominating PAH source from traffic [50]. Notably, exhaust from modern gasoline vehicles contains very low levels of PM, and the majority of organic chemicals emitted occur in the gas phase, and then condenses to form secondary aerosols in the atmosphere [51,52]. Nevertheless, traffic emissions remain a major source of increased urban air PAH levels. Recent studies of road tunnels PM suggested that PAHs on traffic PM2.5 were primarily attached to aggregates of ultrafine PM originating from the combustion of transportation fuel [53,54]. More US EPA PAHs have been found in the ultrafine and fine PM (PM2.5) samples than in the coarse PM, which to a large degree seem to originate from non-combustion sources such as bitumen and tires [54]. Phenanthrene > pyrene > fluoranthrene were the most abundant species. However, high amounts of PAHs with 4 rings (benz[a]anthracene, chrysene) and 5 rings (B[a]P, benzo[e]pyrene, benzo[k]fluoranthene, benzo[j]fluoranthene, dibenz[a,h]anthracene), as well as the strong mutagen cyclopenta[c,d]pyrene were also found in these combustion PM samples [54]. In addition to PAHs, oxygenated (oxy-PAHs; 9H-fluoren-9-one and anthracene-9,10-dione) and nitrated (nitro-PAHs; 1-nitronaphtalene, 9-nitroanthracene and 1-nitropyrene) PAH derivatives from diesel engine emissions are found both in ultrafine and fine PM [9,54,55].
In general, specific profiles of PAHs associated with PM of various origin can lead to distinct toxic and carcinogenic potencies being linked with PM exposure. These may include both genotoxic and non-genotoxic modes of action, as discussed further in sections to follow. Airborne PM usually contain relatively high levels of carcinogenic priority PAHs (chrysene, benzo[b]fluoranthene, benzo[k[fluoranthene, B[a]P and indeno[1,2,3-cd]pyrene). Mixtures of PAHs associated with DEP have significantly higher total sum of PAHs in comparison to airborne PM samples; specifically, they contain higher levels of fluoranthene, pyrene, chrysene, benzo[j]fluoranthene, benzochrysenes and monomethylated anthracenes, phenanthrenes, pyrenes and benz[a]anthracenes [56]. DEP also contains high concentrations of nitro-PAHs formed through electrophilic substitution in the presence of NO2 [57]. Some nitro-PAHs such as 1-nitropyrene (1-NP) are formed mainly during the combustion process and have been suggested as a marker of DEP exposure, while others are formed through atmospheric processes between NO2 and gas-phase PAHs [57,58].
The PAHs composition in urban air PM2.5 does not depend only on the combustion sources, but it is largely affected by the environmental conditions. Volatility is reduced by size; therefore, smaller PAHs (four or fewer aromatic rings) are to a greater extent found in the gas phase, while high-molecular weight PAHs (five or more aromatic rings) are mainly detected on the particle [25]. However, as low-molecular weight PAHs are usually formed to a much greater extent than the larger PAHs, they also tend to be the dominating PAHs bound to PM. Accordingly, levels of e.g., phenanthrene and pyrene on DEP and urban air PM2.5 exceed the level of B[a]P [25,59]. The amount and type of PAHs being present on PM2.5 are further modified by ambient air temperature and photooxidation processes. As condensation and evaporation processes are directly regulated by temperature, higher levels of PAHs condense onto ambient particulates at low temperatures. The total PAH content in urban air PM2.5 can therefore be an order of magnitude higher in winter as compared to summer, and the relative amount of different PAH species may also change due to seasonal variation in sources, such as residential heating and forest fires [60–63]. Furthermore, photooxidation leads to formation of oxy-PAHs which contributes to SOA formation by reducing the vapor pressure compared to their parent PAHs and increasing the condensation process [64,65]. Importantly, while photooxidation of PAHs may increase their redox and direct mutagenic activities, it also leads to a reduced affinity towards AhR [66–68]. However, photo-oxidation also increases water solubility, which has been suggested to limit the bioavailability of oxy-PAHs [69].
3. Lung cancer
There are two main histopathological lung cancer groups: non-small cell lung cancer (NSCLC) [70] and small-cell lung cancer (SCLC) [71]. NSCLC accounts for 80% of the lung cancer in humans [72]. The majority of NSCLC are adenocarcinomas (ADC), the other histopathological NSCLC subtypes are squamous cell carcinoma (SCC) and large cell carcinoma. Although the cellular origin(s) of lung cancer remain largely unknown it has been speculated that different histopathological subtypes arise from distinct cells localized in defined microenvironments [73]. Due to their proximal-to-distal distribution pattern, SCC is often thought to arise from the proximal airway and ADC from more distal locations [74].
Lung cancers develop through a process involving multiple genetic and epigenetic alterations in the cells of origin(s). Examples of genes that have been linked to lung carcinogenesis are oncogenes/growth promoting proteins (e.g., v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog [KRAS], epidermal growth factor receptor [EGFR], tyrosine protein kinase c-Src, B-Raf proto-oncogene [BRAF], mitogen activated protein/extracellular regulated kinase [MEK-1], human epidermal growth factor receptor 2 [HER2], hepatocyte growth factor receptor [MET], anaplastic lymphoma kinase [ALK], and rearranged during transfection [RET]). Lung carcinogenesis also typically involves inactivation of tumor suppressor genes/proteins (e.g., TP53/p53, phosphatase with tensin homology [PTEN], and liver kinase B1 [LKB-1]) [30,75]. Mutations in the TP53 gene are frequent in almost all types of cancers [76], and they are present in approximately 50% of all NSCLC cases [77]. A frequent transversion, G:C to T:A, is correlated with exposure to carcinogens found in tobacco [78]. At several TP53 mutational hotspots, such as codons 248 and 273, a large fraction of the mutations is G to T events in overall lung cancers, while almost exclusively G to A transitions are found in non-tobacco-related cancers [6]. There seems to be a strong coincidence of G to T transversion hotspots in lung cancers and sites of preferential formation of PAH adducts along the TP53 gene [24,78].
EGFR and KRAS are two other frequently mutated genes in lung cancer. The EGFR receptor regulates cell survival and proliferation, and it is overexpressed in 50% of lung cancers. KRAS belongs to the Ras family of small GTPases which regulates downstream signaling of EGFR to the extracellular regulated kinases (ERK1/2), which is central for the cell growth and proliferation [6]. The EGFR-Ras-ERK1/2 pathway also regulates several proinflammatory genes which may affect the tumor microenvironment as discussed later. KRAS mutations are frequent in smokers but occur in only 5 to 10% of lung cancers in never- or light-smokers [79–81]. The KRAS mutations are often generated by G to T transversions associated with tobacco use and PAH exposure, and they lead to loss of the GTPase activity which is necessary for the inactivation of Ras in the GDP-bound form leaving the protein constitutively active [6]. EGFR mutations, on the other hand, are present in 15 to 50% of NSCLC patients from never-smokers, and the mutational pattern seems to be dominated by transition mutations (G to A) [80–82]. Deletion in exon 19 and the single amin acid substitution L858R in exon 21 (replacing leucin with arginine in codon 858) of the EGFR gene account for about 85% of observed EGFR mutations in NSCLC. This destabilizes the inactive form of the receptor leading to increased dimerization and activation compared to wildtype EGFR [83]. As EGFR and Ras are part of the same signaling pathway, both mutations target the peripheral airways and give rise to ADC [6]. However, while lung cancer in never smokers with EGFR driver mutations may be sensitive to EGFR tyrosine kinase inhibitor (EGFR TKI) treatment, lung cancers in smokers with KRAS mutations are often resistant to EGFR TKI treatment underscoring that upstream activation of EGFR is not necessary for the Ras activity in these patients [6]. Furthermore, while smoking tends to induce SCLC and SCC in the central airways, ADC in the peripheral regions is the most prevalent lung cancer type in never-smokers [6]. Thus, both the mutation pattern and lung cancer subtypes seem to be distinctly different in smokers and never-smokers.
Tissue stem cells are attractive candidates for cellular origin of cancer, as their long lifespan allows them to accumulate genetic mutations essential for cancer development [84]. A subtype of lung adenocarcinoma with KRAS mutations has been suggested to evolve from airway epithelium, having a distinct differentiation pattern with suppression of ciliated and exocrine bronchiolar cell (Clara cell)-related genes [85]. Based on histological observations and studies with genetically engineered mouse models, alveolar type 2 (AT2) cells have been hypothesized to be the cells of origin of another subpopulation of lung adenocarcinoma [86].
More recently, high-resolution mutational profiles of lung epithelial cells exposed to individual tobacco smoke chemicals support a role for PAHs like B[a]P [87]. Such studies have revealed that lung cancer with metastasis is a process not only linked to lung cancer stem cells transformation and epithelial-mesenchymal transition (EMT), but also to modifications of the tumor microenvironment of lung cancer [88,89] and mechanisms linked to angiogenesis and lymph angiogenesis [90]. Central influencing factors of lung cancer also include many noncoding RNAs (ncRNAs, miRNA) [91].
4. Lung cancer induced by combustion PM/PAHs
PM2.5 exposure from polluted air is the main risk factor for lung cancer in never-smokers, which predominately develops as ADC with EGFR driver-mutations in the peripheral lung [6]. A genomic analysis found that most of these tumors appeared to originate from natural mutations accumulating with age [37]. This implies that mutagens and genotoxic effects may not be the main drivers of air pollution induced lung cancer. While the frequency of EGFR-driven lung cancers seems to increase with increasing PM2.5 exposure, there are no changes in the accompanying EGFR mutation pattern, indicating that PM2.5 primarily induces ADC through promotion [38]. Studies in mouse models and in vitro support and extend this hypothesis by suggesting that macrophages exposed to PM2.5 induced a progenitor-like state in AT2 cells containing natural acquired mutated EGFR (L858R). Furthermore, interleukin (IL)-1β seems to be required for the promotion phase [38]. This aligns with earlier findings by Riva et al (2020) reporting that only 3 out of 20 tested suspected human carcinogens induced carcinogen-specific mutations in mice [92]. These authors therefore hypothesized that “key driver mutations are likely to be acquired through endogenous mutagenic processes rather than by the direct action of chemical exposures on the genome” and further speculated that inflammation could be a driving factor for tumor promotion [92].
Notably, IL-1β release and inflammation are also considered the driving force in silica- and asbestos-induced lung cancer [93,94], but EGFR mutations appear to be less frequent in never-smokers occupationally exposed to such mineral particles [95]. By contrast, never-smokers occupationally exposed to diesel exhaust particles and PAHs had equal or higher frequency of EGFR mutations compared to controls [95]. This indicates that additional mechanisms and properties associated with combustion particle exposure such as PAHs, may be necessary to promote EGFR-driven lung cancers. In line with this, an important role of IL-1β has been identified in inflammation-induced and AhR-dependent tumor promotion of lymphoma in mice [96].
Based on the differences in lung cancer subtypes and mutation spectra found in smokers versus never-smokers, it has been proposed that lung cancer in never-smokers is “a different disease” than lung cancer in smokers [6]. However, 8% of lung cancers in smokers lack evidence of smoking-induced mutagenesis, suggesting that also smoking may promote cancer through non-genotoxic mechanisms [97]. The marked reduction in risk of lung cancer following smoke cessation further points to a major role for tumor promotion also in smoking-induced cancers [24]. Moreover, lung cancer development from secondhand smoke (SHS) resembles never smokers in that ADC also seem to be the predominant cancer subtype and tobacco-induced mutations are lacking [37,98].The differences observed between smoking versus air pollution and SHS may rather be a matter of exposure dose. In further support of this, a meta-analysis of 16,000 lung cancer cases concluded that occupational exposure to diesel exhaust were associated with all lung cancer types, but the dose-dependency were much stronger for SCC than for ADC [99]. In other words, the ratio of SCC:ADC increased at higher DEP exposure levels. In line with this, the SCC:ADC ratio has been reported to be almost 3:1 in smokers who are exposed to very high PM doses, but inversed (more than 1:3) in never-smokers only exposed to low PM concentrations through ambient air [6]. Indoor exposure to smoky coal, which is considered to be 100-fold more carcinogenic than cigarette smoke and represents a high-dose exposure to combustion particles compared to outdoor air PM2.5 levels, has also been reported to cause an overrepresentation of G to T transversions in the TP53 gene similar to what is found in smokers and PAH exposed workers [6,12,24]. A systematic review of indoor exposure to coal and biomass smoke also concluded that the odds ratio (OR) of developing SCC was higher than the OR for developing ADC (3.58 vs. 2.33), again pointing towards a pattern of lung cancer subtypes more in the direction of smoking [100].
Occupational exposure to diesel exhaust and indoor exposure to solid fuel smoke represents much higher combustion PM exposures levels than those that are normally encountered in outdoor environments. Thus, low dose exposure to combustion particles appears mainly to induce ADC in the peripheral lung regions, but as concentrations increase, the risk of SCC development in the central airways increases much more than ADC, and becomes the dominant cancer type [99]. This apparent dose-dependent shift in lung cancer subtypes associated with various combustion PM exposures could likely be related to a dose-dependent increase in cilia dysfunction and impairment of particle clearance, as observed with tobacco smoking [101,102]. Thus, increased inhalation of combustion PM may exponentially increase the effective PM dose on bronchial epithelial cells by impairing the mucociliary clearance of deposited particles. This could explain the increased risk of SCC development from smoking, occupational diesel exposure and indoor air solid fuel smoke, compared to ADC [99].
At higher exposure doses, combustion PM-induced genotoxicity also appears to become more important. A number of experimental studies in rodents have proven the carcinogenic potency of PM and/or extractable organic matter (EOM) from a variety of combustion and urban air PM (primarily PM2.5) [8,9,12,103,104]. In a recent review, the carcinogenic potency of EOM on Sencar mouse skin from a variety of combustion emissions, coal tar, and B[a]P were presented [24]. B[a]P was found to have the highest carcinogenic potential. Most interestingly, the carcinogenic potency of EOM of urban air pollution as well as diesel and gasoline exhaust could be at least two orders of magnitude higher than EOM for tobacco smoke. EOM of ambient air PM, various combustion particles and cigarette smoke predominately induced G to T transversion in the Salmonella (Ames) mutagenicity assay [24]. The mutation spectra observed in experimental studies therefore provide further support for the suggestion that air pollution and tobacco smoking could lead to comparable patterns of lung cancer development given exposure to comparable dose levels.
Based on the above, we hypothesize that the discrepancies in mutation patterns and cancer subtypes induced by smoking and air pollution (never smokers) reflect the two ends of a combustion PM dose–response continuum. We further suggest that the tumor promoting effects of combustion PM are most important for lung cancer development in the lower dose-range, but that their mutagenic effects become increasingly more important as the exposure dose increases. Accordingly, a series of in vitro studies performed in rat liver epithelial cells showed that only a few environmental PAHs and methylated PAHs elicit major genotoxic effects, determined as formation of stable DNA adduct production and/or p53 activation [105–109]. Dibenzo[a,l]pyrene (dibenzo[def,p]chrysene) has been observed to be the most potent genotoxin, while several PAHs, including benzo[g]chrysene, B[a]P, 5-methylchrysene, 1- and 3-methylbenzo[a]pyrene exhibited significant genotoxic potencies. Other PAHs and methyl-PAHs, including benz[a] anthracene, chrysene, benzo[b]- and benzo[k]fluoranthene and dibenzo [a,h]anthracene, induced only a moderate DNA adduct production in rat liver epithelial cells, and numerous other PAHs or monomethylated PAHs showed only a minimal or no genotoxicity potencies.
In line with this, the AhR-dependent proliferation of rat liver epithelial cells (WB-F344) exposed to EOM of urban dust PM (SRM1649a) has been reported to occur at an order of magnitude lower doses than DNA damage [110]. It was therefore suggested that non-genotoxic effects of AhR activation could be an important determinant of the effects of complex PAH mixtures from PM [110]. Transcriptional activation of AhR appears to be among the most sensitive, if not the most sensitive, endpoint induced in vitro by combustion PM in airway epithelial cells [111,112]. As discussed in this review, the role of AhR in lung cancer development extends far beyond the regulation of PAH metabolism, adduct formation, and genotoxicity. AhR plays a central role in cancer promotion pointing towards the non-genotoxic properties of PAHs. For instance, the AhR may directly regulate inflammatory responses and immune cells in the tumor microenvironment [113]. Moreover, nuclear AhR translocation, a hallmark of AhR activation, appears to be more common in female non-smokers with ADC, and it is associated with EGFR mutations [114–116]. At the same time, it seems that AhR may suppress KRAS-driven ADC [117]. These observations are in coherence with the suggested role of inflammation and tumor promotion in air pollution-induced lung cancer, as well as the long-recognized role of genotoxicity and mutagenesis in tobacco smoke-induced lung cancer.
It is pertinent to emphasize that the differences discussed here represent the main trends and patterns seen in lung cancer development. Some never-smokers also develop SCC and express KRAS-mutations and G to T transversion, while some smokers develop ADC and express EGFR mutations and G to A transversion [6,97]. Indeed, exposure to ambient air pollution and traffic emissions appear to be consistently associated with elevated urinary excreted PAH metabolites and biomarkers of genotoxicity, and also smoking may promote cancer development by increasing selection of cells with naturally acquired mutations [97,118,119]. In the following sections, we will discuss the potential involvement of AhR and PAHs at different stages of cancer development and progression.
5. Canonical AhR signaling and PAH metabolism
In the absence of a ligand, AhR resides in the cytosol as part of a multiprotein complex consisting of AhR-interacting protein (ARA9 or XAP2), a heat shock protein 90 dimer (Hsp90) and co-chaperone p23. In its major signaling route, the so-called canonical or classical AhR pathway, ligand-activated AhR dissociates from the multiprotein complex and translocates to the nucleus, where it dimerizes with the AhR nuclear translocator (Arnt). The AhR/Arnt heterodimer then binds to the so-called xenobiotic response elements (XREs), also known as dioxin response elements (DREs), in regulatory regions of target phase I and phase II genes (Fig. 1).
Several studies have revealed that PM2.5, more specifically the organic fractions of PM2.5/DEP may, through cell specific mechanism, form reactive metabolites and display CYP1A1 activation [21,22,120,121]. The AhR-dependent induction of CYP1A1 expression seems to represent a particular sensitive biomarker of DEP-exposure [111]. PAHs are among the most likely candidates contributing to such effects on combustion PM. Due to their lipophilic nature, PAHs may detach from the particle and diffuse across the plasma membrane into the cell. Highly depending on cell type, the PAHs may be metabolized to reactive electrophilic metabolites and/or give rise to a canonical AhR-response modifying PAH-metabolism. In the following we briefly summarize the main metabolic steps of PAHs using B[a]P as an example.
There are three major pathways for PAH/B[a]P metabolism, which are characterized by specific sets of enzymes: i) the cytochrome P450 (CYP)1A1/CYP1B1 and epoxide hydrolase, ii) aldo–keto reductase and iii) the CYP peroxidase enzyme [31,122]. i) In the CYP1A1/CYP1B1 and epoxide hydrolase pathway, PAHs/ B[a]P are first oxidized by the CYP1 enzymes to epoxides, which next are hydrolyzed by epoxide hydrolase to PAH dihydrodiols/B[a]P-7,8-dihydrodiol. A second CYP1-catalyzed oxidation at the double bond adjacent to the diol forming stereospecific PAH dihydrodiol-epoxides/B[a]P-7,8-dihydroxy-9,10-epoxide. Some of these are highly reactive electrophilic metabolites which can form stable DNA adducts or promote depurination at damaged nucleotide sites [123]. ii) In the aldo–keto reductase pathway, the PAHs are first metabolized by CYP1A1/CYP1B1 followed by epoxide hydrolase. However, here the PAH dihydrodiols/B[a]P-7,8-dihydrodiol can be further oxidized by aldo–keto reductases to a PAH dione/B[a]P-7,8-dione. Several human aldo–keto reductases have been implicated in this pathway, which may generate ROS and oxidative DNA damage via redox cycling of PAH o-quinones. iii) PAHs can also be metabolized by peroxidase reactions to reactive radical cations, which in the case of B[a] P will occur in the C6 position. The one-electron oxidations mediated by peroxidases or other enzymes resulting in PAH radical cations and ROS mainly result in unstable DNA adducts subjected to depurinations [124].
The AhR regulates the induction of CYP1-enzymes including CYP1A1, CYP1A2, CYP1B1 and phase II enzymes NADPH:quinone oxidoreductase (NQO1), glutathione S-transferase (GST) A2, and UDP-glucuronosyltransferase (UGT)1A1 and UGT1A6 [31]. The AhR can also directly or indirectly regulate expression of several aldo–keto reductases, together with Nrf2 [27,125]. Many of these AhR-regulated enzymes are central to the total metabolism of PAHs and directly participate in production of reactive PAH metabolites. NSCLC samples are found to express increased levels of AhR mRNA wich correlates positively with CYP1A1 expression in cases of ADC [126]. Also, polymorphisms in CYP1A1 and CYP1B1 have been linked to increased lung cancer risk [127–129]. Notably, most studies on the molecular mechanisms illustrating various steps to be involved in the carcinogenicity of PAHs were based on studies of a single compound, typically B[a]P. In real life, we are exposed to mixtures which may contain hundreds of different PAHs and other compounds likely to interfere with the metabolic activation/detoxication processes [27]. Although many factors are important determinators for the toxicity, the central role of AhR-induced upregulation of CYP1 enzymes in the bioactivation of PAHs is further illustrated by other associations found between tissue specific AhR-dependent aryl hydrocarbon hydroxylase induction/CYP1 isoforms and rates of cancer, mutagenesis, DNA adducts and toxicity of PAHs [130].
Apart from regulation of enzymes associated with PAH metabolism, the AhR also acts as a “master regulator” of numerous other genes that are linked with the process of carcinogenesis. Therefore, in addition to the regulation of formation of genotoxic PAH metabolites, activation of the AhR by PAHs can be associated with further non-genotoxic mechanisms of action of PAHs, including e.g.: perturbation of cell cycle progression, cell proliferation and programmed cell death [27], deregulation of action of hormones and/or their metabolism (including e.g. increased catabolism of steroid hormones) [131], as well as deregulation of numerous genes linked with cancer development [132]. Therefore, estimation of the AhR-activating relative potencies (REPs) (calculated relative to TCDD or to B[a]P as model AhR agonists) can provide an important information about toxicity/carcinogenicity of PAHs and their mixtures that are associated with PM.
A comprehensive evaluation of AhR REPs of individual PAHs, monomethylated and oxygenated PAHs has been carried out using rat hepatoma H4IIE cell line, stably transfected with a luciferase reporter gene under the control of dioxin-responsive enhancers designed as DR-CALUX assay [133]. REP values calculated relative to the TCDD-induced AhR activity, were developed for thirty abundant environmental PAHs [134], dibenzoanthracenes and benzochrysenes [106], and monomethylated chrysenes, benz[a]anthracenes and B[a]P [107–109]. Additional data have been developed also for other PAH compounds using either DR-CALUX or its variant, PAH-CALUX assays [135,136]. In general, AhR REP values expressed relative to TCDD ranging from 1 × 10−3 (for benzo [k]fluoranthene, dibenzo[a,h]anthracene and dibenzo[a,k]fluoranthene) to 1 × 10−8 for fluoranthene. Since various classes of AhR ligands may differentially activate human and rodent AhR, human AhR-inducing REPs have also been developed [137], using the gene reporter AZ-AhR cell line [138]. The order for REPs of individual PAHs in human cells largely corresponded with the available data from rodent-based DR-CALUX assay, although some differences up to one order of magnitude in REP values of PAHs between human and rodent cells have been observed. Higher REP values were found in human cells for some important AhR ligands among PAHs, such as indeno[1,2,3-cd]pyrene, benz[a]anthracene or benzo[b]fluoranthene, while lower REP values have been determined for methyl-substituted PAHs. The same experimental models have also been used for estimation of AhR-mediated activities of PM extracts and chromatographic fractions (non-polar and polar) of these extracts. Taken together, the AhR-mediated activity of PAHs is an important parameter for hazard/risk assessment of both PM mixtures and individual environmental PAHs, as this mode of action is highly relevant for both genotoxic and non-genotoxic effects of PAHs, as well as PAH-containing mixtures, as further discussed below.
6. AhR - Reactive metabolites and genotoxicity
DNA damage, mutations and genomic instability is considered a universal hallmark of all cancers including lung cancer [139]. Exogenous DNA damage may arise from cellular exposure to radiation and environmental carcinogenic compounds including PAHs from combustion PM. As the AhR regulates the induction of phase I and phase II enzymes, AhR strongly influence the formation of DNA-reactive PAH-metabolites as well as the biological stability of the parent compounds which have implication for the duration of AhR signaling. However, most mutations in human tissues are of endogenous origin. DNA damage is naturally occurring due to chemical DNA instability (e.g. depurination). It can be induced by various cellular processes including somatic recombination, endogenous reactive chemicals (e.g. aldehydes and S-adenosylmethionine), ROS and products generated as a consequence of oxidative stress (e.g. lipid peroxides) [140,141]. Because of the low contribution of exogenous agents to the mutation rate of normal cells, initiation and mutations increasing DNA instability are expected to be chiefly due to endogenous causes [140]. In fact, oxidative DNA damage is often considered to be a driver of carcinogenesis [142]. Guanine is the most frequently oxidized base. Following oxidation, it will form 8-oxo-7, 8-dihydro-guanine (8-oxoG) [143]. Due to mispairing, such lesions may result in G:C to A:T transversions during replication, one of the most common mutagenic features seen in many cancers including lung [144].
Like many other cancer types, lung cancers often have a high level of mutations in the tumor suppressor gene TP53. The TP53 gene provides instructions for making the tumor protein p53 (or p53). p53 is central in the maintenance of genomic stability, responding to DNA damage by promoting cell cycle arrest and repair, balancing transcriptional regulation of DNA repair genes and induction of apoptosis. Cells with non-functional p53 will thus accumulate more DNA damage and be more resistant to cell death. Furthermore, as the presence of TP53 mutations are found in preneoplastic lesions in the lung, it is hypothesized to be an early marker of lung cancer development [145].
Mutagenic and genotoxic effects of PM/EOM from combustion PM are well known [8,11,12]. A number of studies have shown that people exposed to combustion PM have increased levels of genotoxicity biomarkers including chromosome aberrations, micronuclei, DNA damage measured by 32P-postlabeling or the comet assay.
There are several approaches suggested for a rapid assessment of the carcinogenic potencies of combustion PM from various sources. These are most often based on in vitro assays for genotoxic/mutagenic activity of PM or EOM [22,24,146]. The mutagenic potency of EOM from a variety of combustion emissions in the Salmonella test have been found to span two orders of magnitude [24]. Chemical analysis combined with mutagenicity studies of fractionated EOM have shown that the mutagenicity is most likely due to just a few chemical classes out of which PAHs are often found to play a central role [8,9,11]. This hypothesis is further supported by studies of EOM in vitro which have revealed mutagenic pattern similar to that seen following exposure to PAHs [24,147], as discussed in the section below. A similar approach has been used to derive mutagenic potencies of PAHs based on mutation assay in human B-lymphoblastoid cells [148,149]. As an alternative to genotoxicity testing of PM, the levels of PAHs in organic extracts from combustion PM can be also combined with information of the specific carcinogenic or AhR potencies of PAHs based on in vivo and/or in vitro data [134,137,150]. Other carcinogenicity-linked endpoints have been also proposed to quantify relative carcinogenic potencies of PAHs [26,151] Carcinogenicity risk assessment of PAHs is often based on toxic equivalency factors (TEFs) expressed relative to B[a]P, based on meta-analysis of animal carcinogenicity studies, as proposed by Nisbet and LaGoy [150], which serve to derive carcinogenicity of mixtures of PAHs, where an individual PAH concentration is multiplied by its respective TEF [152 153]. Such approach may serve to identify principal contributors of carcinogenicity or specific toxic action of PAH mixtures, and it has suggested that cyclopenta[c,d]pyrene, in addition to B[a]P, could be a prominent contributor to the estimated mutagenicity of the PAHs found in combustion PM samples [154,155]. Similarly, dibenzo[a,l] pyrene and to a lesser extent benzo[b]fluoranthene were found to be the major contributors to mutagenic potency in extracts of DEP collected from an industrial forklift [56]. Regarding AhR REPs, specific patterns of PAH contributors to the AhR-mediated activity were identified in extracts of standard reference materials (SRM) of urban air PM (SRM 1649a), diesel exhaust particles (DEP) from heavy duty diesel engine (SRM 1650b) and DEP collected from an industrial forklift (SRM 2975) [56]. Here, the following major AhR-active compounds were identified: benzo[k]fluoranthene and to a lesser extent indeno[1,2,3-cd]pyrene, benzo[j]fluoranthene, dibenzo[a,h]anthracene in SRM1649a; benzo[k] fluoranthene, indenopyrene, chrysene, benzo[b]chrysene and benzo[j] fluoranthene in SRM1650b; chrysene, indenopyrene, benzo[k]-, benzo [b]-, benzo[j]-, and dibenzo[b,k]-fluoranthene and 9-methylbenz[a] anthracene in SRM 2975. Generally, mutagenic, AhR-mediated and carcinogenic potencies of individual PAHs seem to be independent parameters. A number of non-priority PAHs such as cyclopenta[c,d]pyrene, benzo[j]fluoranthene, benzochrysenes and methylbenz[a] anthracenes belong among significant AhR agonists and/or genotoxic PAHs. For example, contribution of environmental six-ring PAHs with molecular weight 302 to overall AhR-mediated activity of airborne PM and DEP is even comparable with the overall contribution of carcinogenic US EPA PAHs [156]. It is of note that potent carcinogens, such as B [a]P and 5-methylchrysene, may combine multiple types of toxic activities, including genotoxicity, AhR-mediated activity and tumor promotion activities (see section 9), and they occur at relatively high concentrations in polluted air.
A central role for PAHs-induced mutagenesis in human lung cancer is further substantiated by analyses of mutation spectra in various types of lung cancers. As most hotspot codons are also for the most part mutated in non-lung cancers, the location of mutations seems to be mutagen independent [157]. However, both the TP53 and KRAS mutations found in lung cancer of smokers are predominantly G:C to T:A (G to T) transversions, while other types of cancers are generally dominated by G:C to A:T (G to A) transitions including the TP53 mutations in lung cancers of never-smokers [6,81,157]. B[a]P is metabolically activated into BPDE which reacts with DNA predominantly at the N2-position of guanine to produce primarily N2 -guanine lesions e.g. B[a]P 7,8-diol-9,10-epoxide-N2-deoxyguanosine (BPDE-N2-dG) adduct. As tobacco as well as the ultimate reactive B[a]P metabolite BPDE most often form G to T transversions, some have argued that B[a]P could be the carcinogen responsible for these mutations [24,158]. Importantly, production of pro-inflammatory mediators in target tissue that is associated with PM exposure may further increase production of genotoxic B[a]P metabolites, including BPDE [159,160].
Importantly, several other DNA lesions are also formed after tobacco/B[a]P exposure. Furthermore, there are studies that have failed to find significant differences in the spectrum of mutations between smokers and never-smokers although confirming the predominance of G to T transversions in lung cancer [161]. They proposed that spectra of TP53 mutations was due to an enhanced biological selection and that smoke exposure enhanced the effects of an endogenous mutagen. G to T transversions have also been suggested to be the predominant base substitution induced by PM from urban air [147] and smoky coal (Granville et al., 2003). Other PAHs, like the highly mutagenic cyclopenta[c,d]pyrene, induce similar types of mutations (guanine as well as adenine transversions) as observed for B[a]P [162]. Furthermore, this mutation pattern may not only be reflective of PAHs, but also aromatic amines [103,163]. G to T transversions are also formed via oxidative DNA damage, including PAH o-quinones under redox-cycling conditions [157].
Next-generation sequencing and computational analyses have revealed very complex high-resolution mutational profiles in cancers including changes in single base substitutions, doublet base substitutions, small insertion/deletions, and copy number mutations in human cancers [164,165]. The complexity of the new data reflects the fact that the mutations are due to various endogenous factors as well as a huge number of environmental exposures, each of them resulting in a spectrum of DNA damage. Despite this complexity, there still seems to be specific mutational signatures across the spectrum of human cancer types. Each mutational signature is hypothesized to correspond to specific mutagenic processes, thus considered to help further elucidating the etiology of cancer.
The large-scale analyses allowed to comprehensively evaluate mutational spectra in various lung cancer types [165,166] as well as those induced by cigarette smoke and individual components of cigarette smoke in experimental setting [87,164,166]. Such studies have confirmed the hypothesis suggesting a role for B[a]P-induced mutations in lung cancer from tobacco smokers [87,164]. More specifically, a study with human pluripotent stem cells exposed to various environmentally relevant chemicals and then clonally expanded suggests that the in vitro high-resolution mutational signatures from B[a]P, dibenzo[a, h]anthracene, 5-methylchrysene, and dibenz[a,j]-acridine are similar [167]. Similarly, mutational profiles of lung epithelial cells exposed to individual tobacco smoke chemicals have confirmed and extended the previously characterized B[a]P mutational signatures [87]. Here, the mutational signatures arising from B[a]P and norharmane were both found to be similar to human lung cancer signatures attributed to tobacco smoking [87].
There is, however, no strong mutational signature seen in populations exposed to outdoor air pollution. As this may be due to a lower dose when compared to cigarette smoke as previously discussed in section 4, a central issue for lung cancer development is to explore the rate limiting steps in the development. Several approaches have been taken, as to look for sensitive endpoints for toxic responses. The low contribution of exogenous agents to the mutation rate of normal cells suggests that carcinogens including combustion PM at low doses primarily act via other pathways. Furthermore, B[a]P-induced gene mutations and/or chromosomal aberrations appear to be less sensitive endpoints than the initial DNA damage induced, as BPDE-dGs most often are efficiently eliminated by nucleotide excision repair [168]. However, the induced DNA damage will modulate the transcription of many genes which are predominantly involved in cell cycle regulation, apoptosis, and DNA repair [169,170]. In addition, B[a]P and other PAHs/PAHs-derivatives may modulate gene transcription via interactions with AhR [169], as it is further discussed in sections below.
The exception to this scenario might be a situation of sustained excessive exposure to carcinogenic agents. This seems to be the case in cigarette smokers and persons occupationally exposed to high levels of other combustion PM based on the change in mutation spectra induced, which suggest PAHs-induced mutations. It may be that the higher concentration of combustion PM/PAHs/B[a]P simply increases the relative mutagenic probability from B[a]P over that of endogenous sources for DNA damage; possibly partly as a result of impaired detoxication pathways and/or DNA repair mechanisms at higher concentrations [171].
7. Reactive metabolites - Cell death, inflammation and compensatory cell proliferation
PM2.5, DEP, and some compounds attached to such particles may elicit formation of reactive molecules including ROS and electrophilic compounds reacting with macromolecules in various lung epithelial cells. Depending on their nature and half-life, the reactive metabolite will preferentially react with proteins or DNA giving rise to cell death, chromosomal aberrations or gene mutations [172,173]. Realizing that DNA damage from endogenous processes is probably far more prevalent than those resulting from exogenous agents [174,175], it becomes clear that processes changing the level of DNA damage by which cells will survive, enter S-phase or go into mitosis will increase the probability of accumulating gene mutation/chromosomal aberrations. Cell deaths may result in compensatory cell proliferation which is of great importance for fixation of DNA lesion, as well as an activation of ROS release in inflammatory cells which may further amplify epithelial tissue/DNA damage [176]. Accordingly, chronic tissue irritation with cell death is now regarded as an important part of lung cancer development. Importantly, the particles also contain compounds, including PAHs, which may change the level of DNA damage that the cell may tolerate and survive [177–179].
Silencing or mutation of TP53 tumor suppressor gene is considered the most prevalent oncogenic driver in lung cancer development. Genotoxic as well as various non-genotoxic mechanisms of p53 inactivation that are linked to PAHs have been reported. Repeated PM2.5 exposure has recently been reported to inhibit p53 expression via promoter hypermethylation [180], but p53 activity may also be more directly reduced. For a long time, it has been known that several PAHs may have so-called “stealth properties” [181,182], as they are able to covalently bind to DNA without being detected by the cells defense system. More specifically, several of the electrophilic PAH metabolites bind to DNA without triggering a proper G1-arrest. An increase in p53 can be seen, but often this p53 seems to be transcriptionally inactive as it does not lead to increased levels of p21waf1/cip, which are responsible for cell cycle control, blocking the transition from phase G1 to phase S. Furthermore, some PAHs are found to reduce an activation of p53 by induction of mouse double minute 2 (mdm2) protein which is a major negative regulator of p53 [183]. Reduced p53 nuclear translocation, stimulation of cell survival signals such as phosphorylation of Akt and Bad, and inhibition of DNA damage-induced apoptosis have been reported after exposure to certain PAH [177–179]. Cellular stress caused by DNA damage induces checkpoint kinase-2 (CHK2)-mediated phosphorylation and stabilization of the E2F1 transcription factor. The activation of a subset of pro-apoptotic E2F1 target genes, including apoptotic peptidase activating factor 1 (APAF1/Apaf1) and tumor protein 73 (TP73/p73) leading to apoptosis is attenuated by AhR-binding to E2F1 [184].
Importantly, B[a]P itself (as well as other PAHs) forms numerous metabolites with poorly characterized toxicological profiles, which might further modulate cellular responses to DNA damage [27]. Some of these PAHs have also been reported to have AhR-dependent activity linked to the regulation of cell proliferation, differentiation, senescence and programmed cell death [185]. The link between AhR-signaling and control of cell growth and proliferation is complex and may depend on cell phenotype as further discussed in section 9. Weak mitogenic activity which may also occur via increased intracellular calcium concentrations [Ca2+]i activation of EGFR and insulin receptor signaling, or estrogen receptors (ER) [186–188], elicited either by parent PAHs or their metabolites. Furthermore, an AhR-dependent disruption of contact inhibition induced by PAHs has been reported for a number of AhR-activating PAHs, probably linked to induction of JunD/cyclin A pathway [189].
Chemicals interfering with the cellular defense system, giving anti-apoptotic or mitotic signaling, would change the balance between cell death, cell survival and cell proliferation following endogenous DNA damaging events. If not compensated with increased DNA repair, it is likely that the result would increase the probability of permanent genetic damage. This hypothesis is supported by the fact that low doses of combustion PM/PM2.5 mostly result in cancers with “natural” mutations, in line with important roles also for the non-genotoxic properties of PAHs in lung cancer development.
8. Intracellular Ca2+-signaling, non-classical genomic and non-genomic AhR-pathways
While the classical or canonical genomic AhR-pathway leading to activation of CYP1A1/−1A2 and CYP1B1 through dimerization with Arnt is clearly essential for the formation of mutagenic metabolites and oxidative stress responses from PAHs in combustion PM, it cannot explain all effects observed from AhR ligands [190]. Non-classical or non-canonical effects involve alternative genomic pathways where AhR interacts with other transcription factors, such as the estrogen receptors (ERs) or the RelA and RelB subunits of the nuclear factor-κB (NF-κB), and which regulates a number of other genes, independently of a canonical XRE/DRE (xenobiotic or dioxin response elements) binding [191–193]. In addition, AhR may also function as signaling molecule in the cytosol controlling activation of c-Src and calcium (Ca2+) signaling through the so-called non-genomic pathway [190,192]. These non-classical pathways enable regulation of several processes relevant for carcinogenesis and tumor development, including inflammation, cell-to-cell communication, cell growth and proliferation, and cell migration which is discussed in more detail in the sections to follow (Fig. 1).
The NF-κB family of transcription factors are key regulators of inflammatory responses, including a number of cytokines, chemokines, and adhesion molecules which play central roles in cancer development [176,194]. Extensive crosstalk between AhR and NF-κB has been reported [195–197]. TCDD exposure and AhR overexpression increased NF-κB activity and IL-6 expression in lung cells [198]. TCDD also induced dimerization of AhR and RelB of the alternative NF-κB pathway and up-regulation of CXCL8 through a novel RelB/AhR response element (RelBAHRE) in macrophages and breast cancer cells [196,199]. Furthermore, B[a]P may induce CXCL8 expression in primary human lung macrophages through binding of AhR to consensus XRE sites in the CXCL8 promoter, and B[a]P administration increased pulmonary inflammation in mice [200]. AhR can also dimerize with the p65-submunit of NF-κB and activate κB-sites in the IL-6 and c-myc promoters [198,201]. However, AhR-deficient mice have been reported to display elevated NF-κB activity and inflammation in the lungs after inhalation of lipopolysaccharide (LPS), cigarette smoke, or crystalline silica [202,203]. AhR knockout has been also shown to increase inflammatory signaling in lung adenocarcinoma A549 cells [204]. Furthermore, AhR activation may suppress pulmonary inflammation induced by crystalline silica [203]. The receptor therefore seems to elicit both pro- and anti-inflammatory functions through enhancement and suppression of NF-κB activity in the lung and other tissues. A study in human bronchial BEAS-2B cells shows that this dual action may occur even within the same cell type. Both constitutive and ligand-activated AhR elicited a weak to moderate pro-inflammatory signal increasing CXCL8 and CCL5 release but seemed to suppress p65 activation and chemokine responses in combination with stronger activators of the classical NF-κB pathway, such as polyinosinic:polycytidylic acid (Poly I: C) or tumor necrosis factor (TNF)-α [205]. The interaction of AhR with members of the NF-kB family is an important aspect, as unresolved chronic inflammation is considered to be an important hallmark of cancer [194].
While non-activated AhR in its resting state is often depicted as “freely floating” in the cytosol, some studies suggest that at least a fraction of the AhR is anchored to the cell membrane, most likely in close connection with cholesterol rich regions such as the caveolae. AhR appears to bind directly to caveolin-1 (Cav1), and this binding is affected by exposure to AhR ligands [206,207]. A close connection between AhR and the cell membrane makes sense, as most AhR ligands are highly lipophilic and thereby distribute within the phospholipid bilayer, rather than dissolving into the aqueous cytosol [208,209]. Caveolae are believed to be central in the uptake of lipids and lipophilic compounds [210,211]. In line with this, polychlorinated biphenyls (PCBs) have been shown to accumulate in caveolae [212], suggesting that AhR is located at the regions where its ligand occur at the highest concentrations. This also places AhR in close contact with major cell signaling components, since a variety of different receptors and ion-channels cluster in cholesterol-rich micro domains. Studies in human microvascular endothelial cells suggest that pyrene and PAH-rich DEP-derived EOM trigger AhR-dependent Ca2+-signaling, possibly through activation of transient receptor potential canonical (TRPC) channels [213,214]. This response occurred rapidly after approximately two min of exposure, preceding transcriptional regulation. Similarly, DEP-EOM and phenanthrene were reported to stimulate Ca2+-influx and membrane depolarization in airway sensory nerve fibers from guinea pigs through AhR-dependent activation of TRPA ion channels [215]. AhR-mediated Ca2+-signaling through the so-called non-genomic pathway seems to be a central step in the regulation of TCDD induced cyclooxygenase 2 (COX-2) activation, prostaglandin release and inflammation [190]. Dysregulation of Ca2+-signaling is frequent in many cancer types and has been linked to tumor progression. Furthermore, aberrant expression of TRP-channel such as TRPC and TRPM has been reported in lung cancer and other cancer types and has been linked to EMT, cell proliferation, invasion and promotion of cell survival and suppression of apoptosis [216,217]. Importantly, these effects have been described for pyrene and phenanthrene, PAHs that traditionally have been considered weak AhR activators due to limited effects on classical AhR:Arnt signaling [150]. Although the potential role of AhR-induced Ca2+-responses in lung cancer development remains to be clarified, this underscores that models developed to assess AhR REP based on XRE/DRE driven reporters may not account for the non-classical effects of AhR ligands.
It should also be considered that PAHs may further activate intracellular Ca2+-signaling not only through AhR-driven responses. Beta-adrenergic receptors (β-ARs) have been detected in cancer cells of the breast, prostate, and skin as well as in lung cancer [218,219]. Numerous studies have linked this receptor to a variety of cellular phenomena such as cell proliferation and motility, cell apoptosis resistance, EMT, metastasis, and angiogenesis. Some constituents of tobacco smoke (e.g. 4-methylnitrosamino-1-(3-pyridyl)-1-butanone, a derivative of nicotine) are known agonists of β-ARs [220,221], and may regulate tumor cell proliferation and migration which are inhibited by beta-blockers (e.g. propranolol). Interestingly, β-AR, especially β2-AR, is also associated to the intracellular Ca2+ increase induced by B[a]P. Indeed, Mayati and coworkers demonstrated using an endothelial cell model that B[a]P induced intracellular calcium concentration through binding to β2-AR, and activation of G protein/adenylyl cyclase/cAMP/EPAC/phospholipase C pathway [222]. This effect was also inhibited by beta-blockers. Besides, β-AR pathway can modulate lung cancer cell resistance, and some works indicate that beta-blockers can slow down the onset of therapeutics resistance especially those associated or interacting with EGFR [223]. Although there is no consensus on the effects of betablocker treatment, it is interesting to note the role of β-ARs in lung cancer primarily have been linked to ADC- and EGFR-driven mutations, as reviewed elsewhere [219,224].
Another central part of AhR non-genomic signaling is the rapid c-Src-mediated activation of EGFR [225–227]. EGFR appears to regulate cytokine responses in DEP-exposed bronchial epithelial cells [228] and it may contribute to the AhR-induced inflammatory responses. The AhR-dependent activation of c-Src has also been found to be important in the TCDD-mediated regulation of COX-2 and prostaglandins [229]. COX-2 is known to be a key enzyme producing prostaglandins which may contribute to tumorigenesis including lung cancer [230–232]. Importantly, different ligands induce different responses upon AhR activation, also in the case of EGFR-mediated effects. A recent study revealed that in contrast to dioxin-like chemicals, the treatment of human epithelial cells with PAHs including B[a]P results in an auto-/paracrine activation of EGFR, which can be an important contributing factor in AhR-mediated tumor promotion [233]. AhR-induced activation of EGFR may also occur in concert with traditional genomic signaling and may induce cancer cell proliferation [116,234], and has also been reported to cause resistance towards EGFR tyrosin kinase inhibitor (EGFR-TKI) treatment of adenocarcinoma through Src-mediated non-genomic signaling [115]. Similar to AhR, EGFR may localize in the caveolae and interact with Cav1. Downregulation of Cav1 has been reported to enhance sensitivity towards EGFR-TKIs in lung adenocarcinoma cells (PC9) harboring EGFR mutations [235]. Both AhR overexpression and exposure to the AhR ligand PCB77 appear to increase Cav1 levels in caveolae [207,236]. Thus, the role of AhR in regulation EGFR activation and EGFR-TKI sensitivity, likely involves both c-Src and Cav1. As reviewed elsewhere, Cav1 has also been implicated in multiple stages of lung cancer development, including cell proliferation, migration, apoptosis and drug resistance [237]. Hence, the importance of AhR-Cav1 crosstalk likely extends beyond regulation of EGFR and warrants further studies into the role of non-genomic AhR signaling in ordered membrane microdomains for development of lung cancer. Collectively, these findings point towards a potential role of AhR in air pollution-mediated lung ADCs with EGFR driven mutations and lung cancer.
The pattern of AhR signaling with both genomic and non-genomic pathways and localization of at least a pool of cellular AhR at the caveolae interacting with Cav1, strongly resembles steroid receptor signaling pathways. Also, a pool of the estrogen, androgen, progesterone and glucocorticoid receptors (ER, AR, PR and GR) interact with Cav1 and signal through non-genomic pathways, in addition to their classical genomic pathways, in a pattern similar to AhR, involving both rapid c-Src and calcium responses. As reviewed elsewhere, these non-genomic steroid receptors signaling pathways appear important in cancer development, especially in estrogen and androgen sensitive cancers such as breast and prostate cancers. [238,239]. Due to the many shared features, it seems reasonable to expect that crosstalk between AhR and ER/AR non-genomic signaling may occur. More specifically, the interactions between AhR and the genomic signaling of steroid receptors are well known and include interference with ER, AR, PR and GR, although the crosstalk with ER is by far the best described. AhR can interfere with ER signaling through several mechanisms including induction of CYP1A1/1B1 which can metabolize estrogen, thereby reducing intracellular estrogen concentrations and ER activation, AhR: Arnt-mediated suppression of transcriptional activity of ER (“squelching”), and direct interactions leading to AhR:ER dimerization. However, AhR may both suppress and induce ER-regulated genes [191]. As reported for AhR, there also seems to be a crosstalk between ER and EGFR signaling in lung ADC. ERα (but not ERβ) appears to be highly correlated with presence of EGFR mutations in lung ADCs of female never-smokers [240]. The EGFR driver mutations observed in air-pollution associated ADC in never-smokers, were also far more frequent in women [38] which also appear more likely to develop ADC than SCC and to have a higher risk of developing lung cancer from smoking, compared to men [32]. However, while these observations are compatible with the involvement of sex steroid hormones, the interaction between PAHs and ER in lung cancer development remains elusive, and AhR-ER crosstalk has so far not been explored in lung cells with EGFR driver mutations.
Besides the presence of AhR at the plasma membrane, previous works have also pointed to the existence of a pool of AhR located in mitochondria, with possible consequences in terms of the metabolic reprogramming involved in tumor development. Thus, AhR has been shown to interact with one sub-unit of the mitochondrial F0F1-ATPase, namely the ATP5a1, in several cell lines (hepatic cells, lymphoma cells) [241]. Interestingly, the authors demonstrated that upon activation of AhR by TCDD, the AHR:ATP5α1 interaction was disrupted and a mitochondrial hyperpolarization occurred in an AhR-dependent and transcription-independent manner. It is noteworthy that under such conditions, a decrease in ATP production was also observed, although not significant. This led the authors to propose a role in the regulation of mitochondrial metabolism for this so-called «mito-AhR» which was shown to be located in the inter-membrane space of the organelle in Hepa1c1c7 cells [242]. Interestingly, Lagadic-Gossmann and coworkers previously showed in the epithelial hepatic cell line F258 that B[a]P was capable not only to induce a mitochondrial hyperpolarization [243], but also to trigger a glycolytic reprogramming [243], both being involved in survival signals supporting tumorigenesis [244]. Metabolic reprogramming is one of the hallmarks of development of lung cancer and other tumors [245,246] and recent data suggest that enhanced glycolysis may be central in PM2.5 induced NSCLC [247]. Intriguingly, DEP has also been reported to induce mitochondrial hyperpolarization in primary human T-cells [248] and PM2.5 has been reported to suppress mitochondrial-driven apoptosis through AhR dependent mechanisms [249]. Collectively this suggests that the role of the mitochondrial pool of AhR in lung cancer could be worth exploring. Furthermore, as cancer-related metabolic reprogramming can rely on changes in pH homeostasis [250] and as B[a]P is capable of eliciting changes in intracellular pH [251], it would also be interesting to test a role for such pH modifications. In line with this, note that calcineurin homologous protein isoform 2 (CHP2) was described to support tumor survival in non-small cell lung cancer, via the sodium/hydrogen exchanger (Na+/H+ exchanger, NHE) isoform 1 [252], i.e. an important transmembrane pH regulator that we showed to be activated by carcinogenic PAHs, including B[a]P [253]. Another important player worth investigating in this network would be the ATPase inhibitory factor 1 (IF1), that is, the physiological inhibitor of the F0F1-ATPase. Indeed, the activity of this peptide is sensitive to pH variations and has been linked to metabolic reprogramming and tumorigenesis [252,254]. Its gene expression seems to be modulated upon PAH exposure via AhR as well as β2-AR [255,256]. With respect to that, a previous paper has found IF1 as a target for PM2.5, possibly related to immune and inflammatory responses in pulmonary fibrosis [257].
9. Cancer promotion including cell-to-cell communication, EGFR activity, extracellular vesicles and miRNA
9.1. Disturbance of cell-to-cell junctions and contact inhibition
Disruption of intercellular communication mediated via various types of cell-to-cell junctions, including gap junctions (GJs), adherens junctions (AJs) or tight junctions (TJs), and associated deregulation of cell adhesion are important mechanisms linked with cancer development and cancer promotion. The GJs, which connect neighboring cells allow continuous exchange of small molecules, and thus contribute the maintenance of tissue homeostasis, proliferation control and regulation of epithelial cell polarity, which makes them important players also in lung tumorigenesis [258,259]. It has been reported that connexins have tumor suppressive roles in lung tissue [260,261]. Overall, both connexin proteins themselves and GJs (which they form) play a major role in cancer development and progression [262].
The down-regulation of gap junctional intercellular communication (GJIC) that is facilitated by GJs via the action of tumor promoting compounds, can contribute to the removal of an initiated cell from the growth suppression of neighboring cells, and it may thus serve as a marker of tumor promotion [263–265]. A number of carcinogenic chemicals have been observed to down-regulate GJIC and/or connexin expression in cell models derived from various tissues, including the lungs. The shortlist of potential tumor promoters acting via GJIC inhibition also includes PAHs, in particular those with low molecular weight that are associated with PM, but primarily are present in gas phase of polluted air. Several low molecular weight PAHs (including both parent PAH compounds and methylated PAH derivatives) have been demonstrated to inhibit GJIC in rat liver cell lines [266–268]. This toxic mode of action of PAHs might be inversely related with their ability to activate the AhR as illustrated for methylated benz[a]anthracenes [107]. Down-regulation of GJIC has also been observed for complex mixtures of PAHs, including cigarette smoke, cigarette smoke condensate or extracts of DEP [269–271].
Although PAHs and their impact on GJIC have been studied mostly in the context of liver tissue, several studies have also addressed their impact on cell models derived from lung epithelium. Lung alveolar epithelial cells express several connexin species, proteins, which couple cells via formation of GJs [272]. In murine C10 lung cells, a non-tumorigenic type II alveolar pneumocyte and progenitor cell type of lung adenocarcinoma, 1-methylanthracene, a well-known GJIC inhibitor, has been shown to block GJIC, activate ERK1/2 and to induce expression of pro-inflammatory regulators [273]. PAHs, such as fluoranthene and B[a]P may also interact to elicit genotoxic effects, GJIC inhibition and up-regulation of inflammatory mediators in this lung cell model [274]. In human bronchial epithelial HBE1 cells, low molecular weight PAHs have been reported to inhibit GJIC [275], again confirming that this mode of action is not limited to liver cells.
At present, most of the reported effects of PAHs on GJs and GJIC appear to be AhR-independent. Nevertheless, inhibition of GJIC seems to be connected also with AhR-regulated disruption of cell adhesion and cell proliferation control, which will be further outlined below. More-over, inflammation is known to modulate effects of PAHs on GJIC and related endpoints [276,277]. The exposure to PAHs is a part of complex effects of PM on lung tissue, which include induction of oxidative stress and inflammation. It is likely that a combination of these effects will lead to suppression of GJIC in alveolar and/or bronchial epithelium during PM exposure, thus contributing to promoting effects of PM and associated PAHs.
AhR activity has also been reported to contribute to alterations of AJs and cell adhesion [278,279]. Exposure to PAHs or their mixtures have been linked with down-regulation of E-cadherin, which is a principal constituent of AJs. The disruption of cell-to-cell junctions mediated by E-cadherin and their homeostatic functions may lead to deregulation of cell proliferation in target cells. Notably, AhR has been shown to play an active role in proliferation control in lung adenocarcinoma cells [280,281]. Furthermore, PAHs have been documented to exhibit tumor-promoting properties in cell transformation assay in vitro [282]. PAHs have been found to inhibit growth suppressive mechanisms such as contact inhibition, leading to an AhR-dependent enhanced cell proliferation [189,283]. In several liver cell models, activation of the AhR leads to disruption of contact inhibition, as well as to deregulation of proteins forming AJs and participating in intracellular signaling. PAHs acting as AhR ligands can alter cell proliferation control leading to disruption of contact inhibition and to down-regulate GJIC via enhanced Cx43 degradation in rat liver epithelial cells [284]. The AhR-mediated disruption of contact inhibition and increased cell proliferation are linked with disruption of Wnt/β-catenin signaling as well as down-regulation of E-cadherin [285,286]. Together, these data suggest a connection between disruption of growth suppression via deregulation of contact inhibition and removal of cells from the growth suppression of neighboring cells, which is paralleled by GJIC inhibition and down-regulation of other types of cell-to-cell junctions.
In addition to their impact on GJs and AJs, PM or PAH exposure can also affect tight junction proteins and disrupt the integrity of lung TJs, which are important for formation of epithelial barrier, preventing access of inhaled material to sub-epithelial layers [287]. Inflammation, which plays a key role in the development of lung diseases, leads to deregulation of TJ functions and their constituents, which can be also associated with induction of EMT [288,289], as discussed further on. Disruption of lung TJs may contribute to increased susceptibility to lung diseases and promote inflammatory responses within lung tissue. PM components have been shown to disrupt TJs and deregulate expression of TJ proteins within lung or bronchial epithelium [290]. Their effects could be linked to induction of pro-inflammatory cytokines, such as IL-6 and generation of oxidative stress [291]. Exposure to combustion particles may also result in disruption of epithelial barrier integrity, as evidenced e.g. for DEP exposure [292] or during exposure to wood smoke [293]. Regarding the effects of individual PAHs, B[a]P has been reported to disrupt barrier in endothelial cells, without directly affecting expression of TJ proteins [294]. These results again confirm that PAHs or their complex mixtures may affect multiple types of cell-to-cell junctions, and that at least some of these effects are dependent on the AhR activation.
Activation of the AhR has been reported to activate numerous signaling pathways that are associated with both the deregulation of inflammatory responses and simultaneous regulation of epithelial cell phenotype, including cell-to-cell junctions. Non-canonical genomic AhR-signaling involves crosstalk with several other transcription factors and signaling molecules independently of Arnt activation [193]. As previously discussed, AhR ligands may also act through non-genomic AhR-signaling where AhR functions as a signaling molecule in the cytosol, regulating c-Src non-receptor tyrosine kinase and Ca2+ signaling, and affecting ordered lipid domains within cell membranes [190,295], thus providing a direct link between cell junction protein complexes and membrane structure. Activation of c-Src, often linked also with an increased activity of MAP kinases, can indeed impact both structural and signaling functions of cell-to-cell junctions, including GJs, AJs or TJs, but it has been also implicated in TCDD-mediated upregulation of COX-2 [229], a key enzyme producing prostaglandins which may contribute to tumorigenesis including lung cancer [231,232]. As mentioned above, the study of Vogeley et al. [233] also revealed that the treatment with PAHs such as B[a]P results in an auto-/paracrine activation of EGFR, which could be another contributing factor in AhR-mediated tumor promotion.
9.2. EGFR-mediated tumor promotion
The receptor tyrosine kinase EGFR regulates the activity of pro-oncogenic pathways including the mitogen activated protein kinases (MAPKs) ERK1/2 and mammalian target of rapamycin (mTOR), which both promote cancer cell proliferation. Mutations such as the L858R mutations in exon 21, or deletions in exon 19 may lead to overactivation of the EGFR enhancing the stimulation of cell proliferation [296]. Furthermore, several studies suggest that AhR may regulate EGFR activation, through a non-genomic pathway involving c-Src [226,227,233,234]. Thus, modulation of the EGFR activity via PAHs and other AhR ligands could be a contributing factor to cancer cell proliferation and tumor promotion.
As previously discussed, ambient air PM2.5 appears to stimulate tumor promotion of cells harboring EGFR driver-mutations, such as the L858R mutation [38]. Several studies suggest that AhR could be involved in this process. Nuclear localization of AhR has been reported to be more common in lung cancer from women, non-smokers, adenocarcinoma and NSCLC patients with the EGFR exon 19 (E746–750A) deletion [114]. High AhR expression has also been reported from adenocarcinoma cell lines and in human ADC biopsies AhR immunostaining was higher than in normal bronchial tissue and SCC [297]. By contrast, AhR appears to suppress KRAS-driven lung tumor formation [117], which is more common in smokers than never-smokers [6]. As such, it appears that the role of AhR in lung tumor promotion may be more restricted to ADC with EGFR driver-mutations. AhR may also strengthen the resistance towards EGFR tyrosine kinase inhibitor (EGFR-TKI) in NSCLCs through non-genomic Src signaling [115,116]. Intriguingly, cancer cells appear to utilize this AhR-mediated pathway. Cancer-associated fibroblast (CAFs) have been reported to stimulate AhR-dependent proliferation and EGFR-TKI resistance in NSCLCs through production and release of the tryptophane metabolite and potent AhR ligand kynurenine [116]. The kynurenine-AhR axis is dysregulated in a number of cancers and has been associated not only with increased cell proliferation, but also immune evasion, neo-angiogenesis, metastasis, and chemoresistance [298]. Moreover, kynurenine from tumor-repopulating T-cells (TRCs) have been reported to drive AhR dependent upregulation of programmed cell death protein 1 (PD-1) in CD8+ T cells, with potential consequences for cancer immunotherapies [299]. PD-1 inhibits immune responses and promotes self-tolerance by modulating T-cell activity which may contribute to immune evasion [300]. This AhR-kynurenine-PD-1 pathway may also be activated in air pollution induced lung cancer. PM2.5, cigarette smoke and B[a]P has been shown to induce PD-1 ligand (PD-L1) in lung epithelial cells and macrophages, and the therapeutic effects of anti-PD-L1 antibody treatment (pembrolizumab) appear to be limited to lung cancers with high AhR expression levels, in both patients and mouse models [38,301].
In extension of the above, Wang et al [302] recently reported that long-term PM2.5 exposure [90 days) induced persistent activation of EGFR, cell proliferation, anchorage-independent growth, and tumor growth (xenograft mouse model) in human adenocarcinoma NCI-H1975 cells which harbors both the EGFR L858R and T790M mutations. Induction of proliferation and anchorage-independent growth was also observed in human lung cancer PC9 cells which carry a Glu746-Ala750 deletion mutation in exon 19 of the EGFR gene, while in human A549 lung cancer cells with KRAS-mutations but wild-type EGFR PM2.5 exposure only induced a transient EGFR activation and a nonsignificant increase in anchorage-independent growth [302]. In H1975 cells, the exposure to PM2.5 induced approximately 5-fold increase in colony formation ability, but in PC9 and A549 cells PM2.5 exposure caused a less than 2-fold increase [302]. These data suggest that PM2.5 stimulates EGFR activation and cell proliferation in a variety of lung cancer cell lines, but the responses were considerably enhanced in cells harboring both the L858R and T790M mutations [302], which both are common in ADC from never smokers [38].
PM2.5-exposure has also been shown to induce an AhR-dependent transcriptional activation of transmembrane serine protease 2 (TMPRSS2) and subsequent expression of the IL-1 family member IL-18 that may promote cancer progression. AhR nuclear expression also correlated with TMPRSS2 and IL18 expression and cancer stage in human lung cancer tissue [302]. Although the link between the AhR-TMPRSS2-IL18 pathway and EGFR activation was not specifically explored, the study provides a potential link between AhR activation and ADC with EGFR driver mutations. On the other hand, AhR expression has been reported to suppress lung cancer metastasis after orthotopic implantation of human adenocarcinoma cell lines (H1975, A549 and H1299) in SCID CB.17 mice, suggesting that AhR suppresses lung carcinogenesis irrespective of the dominant oncogenic driver [303]. Low AhR expression levels were also associated with faster cancer progression and reduced survival in lung ADC patients [303]. A likely explanation for this apparent contradiction could be thedifferences in effects of constitutive AhR activity versus PAH-induced AhR activation. There is a considerable diversity in AhR-regulated responses induced by different ligands [191], and the native AhR in unstimulated cells appears to affect the regulation of different gene clusters than those regulated upon ligand activation [304]. Importantly, while PAH exposure activated EGFR, this was not the case for dioxins, which underscores the variability in effects induced by different ligands [233].
Based on the above, we suggest that AhR may induce proliferation of lung cancer cells through mechanisms involving both non-genomic activation of EGFR, and genomic activation of NF-κB and its target genes such as TMPRSS2, leading to inflammatory responses regulated by members of the IL-1 cytokine family, such as IL-1β and IL-18. It seems that these effects may be enhanced in EGFR-driven adenocarcinoma, especially by the L858R mutation in exon 21, and that the involvement of AhR is restricted to PAH-mediated activation, since dioxins may not activate EGFR to a similar extent and unliganded constitutive AhR appears to suppress lung tumor progression independent of the driver mutation.
9.3. Extracellular vesicles and miRNA
Extracellular vesicles (EVs) are nanostructures produced by all cells, mediating cell-to-cell communication by exchanging proteins, nucleic acids and lipids or organelles (e.g. mitochondria) [305–307]. They constitute a heterogeneous population including exosomes (Exo; less than 200 nm), microvesicles (MV; 100–1000 nm) and apoptotic bodies. EVs are detected in various biological fluids [308], and suggested to participate in the maintenance of cellular homeostasis and intercellular communication including immune responses, cell proliferation, tissue repair and angiogenesis. EVs contribute to inflammation by containing cytokines, accordingly EVs containing high concentrations of biologically active TNF-α produced by alveolar macrophages was detected in bronchoalveolar lavage fluids (BALFs) during lung injury [309,310]. Damaged epithelial cells may also produce EVs that recruited pro-inflammatory M1 macrophages [311]. These nanostructures are also suggested to contribute to the growth and worsening of cancers. EVs produced by lung cancer cells are reported to stimulate the production of the pro-angiogenic factor vascular endothelial growth factor (VEGF) and increase vascular permeability and extracellular matrix remodeling [312]. Furthermore, an increase in EVs containing cell death protein ligand-1 (PD-L1) suggested to be involved in tumor immune evasion observed in patients with non-small cell lung cancers, who were non-responders to treatment [313]. Notably, PD-L1 is known to be under the control of AhR [301].
An increasing amount of evidence suggests that environmental pollutants can modify the production of EVs, and that they are involved in the appearance or progression of diseases linked to environmental exposures including lung cancer [314]. Tobacco-smoke, PM2.5 and PAHs have been shown to trigger EV release from different lung cell types (macrophages, bronchial epithelial cells, endothelial cells, platelets) [315]. PAHs such as B[a]P, dibenz[a,h]anthracene, or benz[a]anthracene, have been shown to increase EVs production by endothelial cells [316]. However, until now only limited data exist concerning the role of AhR in EVs production and content, especially upon exposure to air pollutants such as tobacco-smoke, PM2.5 or PAHs. Recently, it was demonstrated, using endothelial and hepatic cell models, that PAHs such as B[a]P may increase exosome production through AhR activation [316,317]. The inhibitory effect of naringenin (a flavonoid targeting AhR pathway) on EV production in BEAS-2B cells exposed to cigarette smoke extract could indicate a role for AhR also in lung epithelial cells [318]. Furthermore, pyrene, a weak agonist of canonical AhR signaling but potent inducer of AhR non-genomic Ca2+ signaling [214], increased exosome production using constitutive androstane receptor (CAR) pathway [317].
EVs may also contain miRNAs/ncRNA, a class of RNAs that regulate gene expression by interacting with their target mRNAs to induce their silencing, thereby influencing the cell response [319]. Via regulation of oncogenes or tumor suppressors, miRNAs can modulate tumor formation and contribute to lung cancer development [320–323]. Interestingly, several experimental and epidemiological studies report that exposure to various sources of combustion PM such as DEP, industrial/biomass combustion and cigarette smoking alter miRNA levels [324]. For example, DEP exposure in human lung cells upregulated miR-21 which has previously been identified as an ‘oncomir’ candidate by targeting cell proliferation and EMT through regulation of the PTEN/AKT signaling pathway [325]. Furthermore, loss of miR-29a is associated with cdc7 kinase accumulation and has been suggested as a mechanism to acquire resistance to cigarette smoke-induced DNA damage allowing the cells to proliferate [326].
The miRNAs have also been studied as biomarkers of interest in lung cancer [327] as diagnostic and/or prognostic tools [328,329]. More recently, EV-derived from biological fluids and their miRNAs have been proposed as a potential source of biomarkers for exposure and effects of environmental pollutants. Changes in extracellular miRNAs have been correlated to different sources of PM including DEP [330,331], traffic-related air pollution [332] and cigarette smoke [333–336]. Some miRNAs are commonly deregulated in lung cancers and as a result of exposure to air pollution, and they have been suggested as interesting biomarkers for the detection of sensitive human populations [337]. Furthermore, miRNAs following exposure to cigarette smoke are also suggested to contribute to a modification of the tumor microenvironment towards a pro-inflammatory response [338] and to be pro-angiogenic [339,340].
Finally, an increasing number of studies have shown that some miRNAs target AhR and vice versa, that AhR regulate miRNAs following oncogenic changes induced by PAHs [341,342]. In fact, AhR has been proposed as a key regulator in controlling miRNA levels in lung [343]. Accordingly, without activation, AhR suppressed the expression of the cancer-associated miR-96, whereas chronic cigarette smoke markedly increased its level by a mechanism independent of classic AhR activation by ligands [343]. Such ligand-independent regulation of miR-196a by AhR has been described by Hetch et al. [344] in lung fibroblasts controlling their apoptosis and potentially regulating the hallmarks of cancer as previously suggested [345]. By contrast, we and others recently reported the ligand-dependent AhR activation of miR-132 expression in blood cells [346,347]. This miRNA may possess pro- or anti-tumor functions depending on cancer [348]. Altogether, these elements reveal the interest and the complexity of miRNAs in air pollution-induced lung cancers and underline the need to further explore biological importance of the AhR in miRNA-induced processes, notably in link with EVs.
10. Role of PAHs and AhR in regulating the tumor microenvironment (TME)
10.1. Tumor microenvironment - Immune cells and stromal cells
Tumor cells are surrounded by non-malignant stromal cells which play a critical role for the survival, growth, progression, and metastasis of cancer cells. It is important to note that the development of metastasis is the cause of more than 90% of cancer mortality, and that the metastasis of tumor cells depends on the support of their microenvironment. Non-malignant stromal cells are a heterogeneous cell population forming the structure of the tumor microenvironment (TME) and include cancer associated fibroblasts (CAFs), endothelial cells, adipocytes and pericytes. Interestingly, a recent study showed that elevation of the protein fibroblast growth factor 2 (FGF-2) expression involves AhR signaling resulting in pericyte proliferation in the TME. Consequently, increased FGF-2 signaling and proliferation of pericytes leads to accumulation of tumor associated macrophages (TAMs) and metastasis [349].
Moreover, infiltrating adaptive and innate immune cells play a critical role in the TME and exert an anti- or pro-tumorigenic effect on the development of cancer. For instance, regulatory B cells producing IL-10 may contribute to immunosuppression in the tumor microenvironment. Regulatory B cells differentiation is promoted by the key tryptophan metabolite L-kynurenine (L-Kyn) in an indoleamine 2,3-dioxygenase (IDO) and AhR-dependent mechanism [350]. In addition to B cells, recent studies have shown that AhR activation by TCDD leads to accumulation of tumor associated myeloid cells (TAMCs) including myeloid derived suppressor cells (MDSCs) or TAMs [351]. The importance of immunosuppressive TAMCs and the central role of the TME has been demonstrated for the progression and metastasis of various malignancies including lung and breast cancer [352,353].
Furthermore, recent reports have shown a critical role of AhR in the recruitment of MDSCs and TAMs in adipose tissue of TCDD-treated mice [354] and during the development of glioblastoma [355,356]. The AhR has been found to induce the expression of immunoregulatory enzymes and factors such as arginase 1 (Arg1], IDO, IL-10 and the S100 calcium binding protein S100A9 which are important for the immunosuppressive function of TAMCs by creating a tumor-promoting microenvironment [357–360]. Additionally, cytokines, chemokines, and growth factors are soluble factors and important components of the TME since they regulate the recruitment and migration of immune cells as well as tumor cells [361]. The important role of IL-1β has also been demonstrated in AhR-mediated (TCDD-induced) development of lymphoma [96]. These studies indicate that IL-1β signaling creates a tumor-promoting microenvironment contributing to tumor growth and metastasis as reported previously [362,363]. Additionally, numerous studies confirmed the AhR-dependent upregulation of IL-1β in macrophages and other cell types after treatment with PM, PAHs and TCDD [352,353].
In summary, the literature supports the conclusion that activation of AhR generates a pro-tumorigenic microenvironment that tumors evolve to escape the immune response, enabling progressive tumor growth and metastasis. Consequently, the AhR may play a critical role in the TME of various cancer types by modulating the recruitment and function of infiltrating immune cells. Because AhR can be regulated by small molecules, the AhR has been suggested to be an attractive target for the tumor microenvironment and immunotherapy to treat cancer [113,364,365].
10.2. Angiogenesis and tumor growth
Formation of new blood vessels, neo-angiogenesis, is an essential part of tumor development in lung cancer and other cancers [366]. Development of different AhR knockout mouse models in the 1990 s revealed that AhR deficiency caused cardiac hypertrophy, vascular abnormalities in multiple organs and altered blood pressure [367]. These studies pointed towards a central role of AhR in angiogenesis. The central role of AhR cardiovascular development and homeostasis has been extensively reviewed elsewhere [367–370] and will therefore not be discussed in detail here. Among the angiogenic factors affected by AhR activation is the vascular endothelial growth factor (VEGF), which is a key regulator of angiogenesis.
In vitro exposure of a coculture of eosinophilic (EoL-1) cells and human umbilical vein endothelial cells (HUVECs) to B[a]P, was reported to promote HUVEC growth through ERK1/2 mediated VEGF expression and release from the EoL-1 cells [371]. Similarly, benzyl butyl phtalate induced VEGF release, stimulation angiogenesis in vitro and in vivo through AhR non-genomic activation of ERK1/2 in hepatocarcinoma (Huh7) cells [372]. AhR has been reported to induce VEGF expression in HepG2 cells through activating transcription factor 4 (ATF4), which may be under regulation of the ERK172 pathway [373]. Thus, the angiogenic VEGF-signal appears to arise from activation of AhR in both immune cells and cancer cells, which in the case of lung cancer would be bronchial and alveolar epithelial cells. However, AhR knockdown has also been shown to impair angiogenesis and compromise tumor xenograft growth in mice, by a mechanism involving AhR-dependent VEGF activation in endothelial cells [374]. VEGF is also regulated by the hypoxia-inducible factor-α (HIF-1 α), a PAS family member [375]. Angiogenesis as well as upregulation of the expression of HIF-1 α, ARNT, and VEGF induced by ischemia are enhanced in AhR knockout mice [376]. Indeed, HIF-1 α and AhR crosstalk has been shown to impact both hypoxia-driven gene expression and AhR target genes, presumably via competition for their common dimerization partner, Arnt, as well as by additional mechanisms relevant e.g. for immune cell regulation [377]. Moreover, the role of AhR in VEGF and angiogenesis regulation could be significantly affected by metabolism of PAHs. In fish cell models, both benzo[k]fluoranthene and B[a]P have been shown to alter expression of hypoxia reporter gene, presumably via their metabolites [378]. Interestingly, in human lung adenocarcinoma A549 cells, B[a]P has been found to promote induction of HIF-1 α target genes, including VEGF and carbonic anhydrase IX (CA IX) [379]. Another study indicated that a metabolite of B[a]P, B[a]P-3,6-dione, can induce HIF-1 α degradation in A549 cells [380]. By contrast BPDE and dihydrodiol epoxide metabolite of chrysene have both been reported to stimulate VEGF induction independently of HIF-1 α [381]. Thus, effects of PAHs on HIF-1a-driven angiogenesis in tumor cells could be regulated not only by their AhR activity but they could be directed also by a pattern of their metabolites being formed in target cells.
11. Role of PAHs and AhR in regulation of cancer cell stemness and metastasis
Acquisition of stem cell-like tumor phenotype (cancer stemness) and cancer stem cells are playing an important role in chemoresistance, tumor progression and metastasis. Cancer stem cells have been found to be multidrug-resistant (MDR) based on high expression of the multidrug transporter ATP-binding cassette super-family G member 2 (ABCG2) which is an efflux protein, also called the breast cancer resistance protein (BCRP) [382]. Interestingly, ABCG2 has been identified as a direct transcriptional target of AhR [383]. Consequently, the AhR has been implicated in cancer stemness serving as a sensor and molecular bridge between environmental exposure to PM and PAHs and an increased risk to develop metastases. In the lung, AhR has been shown to induce the expression of ABCG2 and other critical genes involved in cancer stemness [384] which has been found to be associated with an increase of stem population in osteosarcoma cells [385]. Further, the stabilization and activation of AhR has been associated with the expression of deubiquitinase UCHL3 promoting cancer stemness in non-small cell lung carcinoma [386]. The role of AhR in metastasis and cancer stemness seems to be rather complex and may involve various signaling pathways and cell types. Nonetheless, there is increasing evidence that chronic and sustained activation of AhR by environmental toxins (e.g. dioxins and PAHs) promotes carcinogenesis by supporting cancer stemness, chemoresistance and metastasis [364,387].
Atmospheric PM and associated pollutants have been also shown to alter EMT in lung epithelial and bronchial epithelial cell models. EMT plays a central role in various lung diseases, including pulmonary fibrosis and lung cancer. Effects of PM and other particles on EMT have been reviewed extensively in a recent work of Cochard and colleagues [388]. EMT is defined as a process by which cells lose their epithelial phenotype and acquire mesenchymal traits, which include increased ability to migrate and invade. As such, it plays a central role in cancer metastasis. This physiological process occurring during embryogenesis and organ development, which is usually defined by a loss of expression of E-cadherin and acquisition of expression of N-cadherin and vimentin, consists of numerous transition steps, which are only partially recapitulated in cancer cells [389–391]. Nevertheless, already partially executed EMT program may drive cancer metastasis and affects plasticity of tumor cells [389].
Regarding the impact of PM (and PAHs) on EMT in pulmonary cells, numerous studies have been carried out in vitro during recent years, and the cellular models used included both bronchial and alveolar epithelial cell models. The treatments included ambient PM2.5, DEP, PM derived from biomass burning and a number of standard reference materials (SRM), in both particulate forms and applied as their organic extracts [388]. There is a significant variability in dosing regimens or exposure times, but in general, a wide spectrum of PMs, or their extracts, have been shown to cause EMT in cell models derived from respiratory cells [388]. Studies using PM, DEP and/or individual PAHs as model PAH have indicated that these treatments may cause EMT-like phenotype in alveolar epithelial A549 or in human immortalized bronchial epithelial cells [281,392–395]. Their effects were mostly associated with the loss of E-cadherin expression and increased motility of target cells; nevertheless, the mechanisms underlying these effects remain only partially understood. Interestingly, a two-week exposure to B[a]P, but not TCDD, promoted mesenchymal-like phenotype in A549 cells. While TCDD increased the proliferative rate of A549 cells, exposure to B[a]P decreased cell proliferation and induced EMT-like phenotype, which was associated with enhanced cell migration, invasion, and altered cell morphology. These changes were mediated by the p21Cip1 -dependent delay in cell cycle progression [281]. Thus, activation of the AhR alone was not sufficient to elicit EMT in this cell model.
In human bronchial BEAS-2B cells, a short-term exposure to PM induced matrix metalloproteinase MMP1, extracellular matrix (ECM) remodeling genes, and several other genes related to EMT [392]. PM, cigarette smoke condensate and B[a]P have induced EMT in human bronchial epithelial cells (HBEC). However, twelve weeks of chronic exposure to these mixtures or to B[a]P were necessary to establish mesenchymal-like phenotype [396]. Deregulation of serpin family B member 2 (SERPINB2) expression is another mechanism that has been suggested to link EMT and PM exposure in human bronchial cells [397]. The upregulation of SERPINB2 via AhR-dependent mechanism [398] induced morphological alterations but it reduced cell migration after short-term exposure to PM2.5; in contrast, in transformed mesenchymal-like HBEC has been strongly SERPINB2 down-regulated. The overexpression of SERPINB2 in PM-exposed bronchial cells might be interpreted as an initial protective mechanism, helping to maintain the epithelial character of the cells [397].
Comparative HPLC-MS/MS analysis of parental HBEC-12KT and B [a]P-transformed HBEC-12KT-B1 (the cells with acquired mesenchymal-like phenotype) has revealed significant changes in sphingolipid (SL) and glycosphingolipid (GSL) profiles, favoring those SLs and GSLs which have been reported to act as positive modulators of EMT and other pro-carcinogenic processes [399]. Being both intracellular signaling molecules and important integral components of membrane lipid signaling domains, specific SLs and GSLs have been reported to be involved in cancer development, via playing multiple roles in promoting cancer cell growth and survival, as well as in EMT, cell migration and invasion [400–402]. Interestingly, exosomes isolated from mesenchymal-like HBEC-12KT-B1 cells contained similarly altered SL/GSL profiles indicating a possibility that exosomes derived from transformed mesenchymal-like cells might contribute to cancer progression also in recipient cells [399].
Taken together, multiple mechanisms leading to EMT in airway epithelial cells (both normal and cancer cells) have been reported after exposures to PM, DEP, their extracts or to individual PAHs. The AhR-dependent action of PAHs could also be modified by toxic effects of other PM components [388], leading to generation of oxidative stress, inflammatory responses or disruption of DNA integrity and cell proliferation. Together, these effects may lead to activation of transcription factors regulating EMT response. Overall, the mechanisms underlying induction of mesenchymal-like phenotype in lung epithelium will require further attention, as this mechanism may significantly contribute to dissemination of lung cancer cells and formation of metastases. Another line of evidence supporting this comes from the experiments with cigarette smoke, which contains large quantities of PAHs and AhR ligands, and which has been documented to induce EMT in lung adenocarcinoma A549 cells [403]. The cigarette smoke extract-induced intracellular ROS increased expression of runt-related transcription factor 2 (RUNX-2) and galectin-3, a novel mechanism likely to contribute to EMT induction [403]. The effects of PAHs and their mixtures on EMT are mostly non-genotoxic. They might be relevant for normal cells of respiratory epithelium, during early stages of cell transformation, as well as during cancer progression, where they promote cancer cell dissemination.
12. Framework for development of adverse outcome pathways (AOPs) for air pollution induced lung cancer
Recently, an adverse outcome pathway (AOP) was proposed for breast-cancer related cell death, with AhR as the molecular initiating event (MIE), decreased apoptosis and increased motility, inflammation, and endothelial migration as cellular key events (KE) [29]. As discussed in the present review, AhR and PAHs appear to affect many of the same responses in the lungs and a corresponding AOP could likely be developed for lung cancer development from PM2.5 and combustion particles. However, the AOP for AhR-induced breast cancer, which was based on an artificial intelligence tool, provides limited molecular insight into the KEs induced by AhR activation in breast cancer cells [29]. By contrast, the recent studies on air pollution induced lung cancer discussed in this review provide a more detailed mapping of the molecular and cellular events contributing to adenocarcinoma development from ambient air PM2.5 exposure. Air pollution induced lung cancer in never-smokers appear primarily to be due to promotion of AT2 cells harboring naturally acquired EGFR mutations. The collective evidence suggest that AhR plays a central role by regulating proinflammatory cytokines in various lung cells. Additional evidence for a central role of AhR in EGFR driven lung cancers from combustion particle exposure comes from the well-established link between AhR non-genomic signaling and activation of EGFR, and the observations that AhR nuclear translocation, a marker of AhR activation, is common in lung cancer from never-smokers. Based on this we suggest a framework for the role of AhR in lung cancer development from air pollution and other low concentrations of combustion PM, were AhR activation in macrophages and epithelial cells may represent the MIE leading release of IL-1 family cytokines such as IL-1 β and IL-18, and activation of EGFR which both contributes to induce proliferation of AT2 cells with EGFR driver mutations subsequently leading to tumor growth and lung ADC development (Fig. 2a). Although the role of AhR non-genomic signaling in EGFR activation is well established, it still remains unclear whether and how PM2.5 contribute to activation of EGFR with oncogenic mutations. Furthermore, additional AhR-regulated mechanisms clearly contribute to cancer progression through enhancing cell survival/suppression of apoptosis, altered tumor microenvironment, reduction of contact inhibition and increased angiogenesis (Fig. 2a). At higher combustion PM exposure doses, AhR-induced PAH metabolism and mutations in particular in TP53 and KRAS, become more important (Fig. 2b). Additional effects of AhR activation on inflammation, tumor microenvironment, cell-to-cell communication, cell proliferation and survival, are likely to occur also in these cases. However, it should be noted that proliferation and colony formation of lung cancer cells with KRAS mutations may be less affected by PM2.5 exposure than lung cancer cells harboring tEGFR mutations, and AhR has also been reported to suppress KRAS-driven NSCLC. It should also be considered that other combustion-derived mutagens not discussed in this review (e.g. aldehydes, nitrosamine, metals, and ultrafine-/nanoparticles as such) may contribute significantly to lung cancer development at high combustion PM exposure.
Fig. 2. A framework for development of Adverse Outcome Pathways (AOPs) for AhR in lung cancer from air pollution and combustion PM.
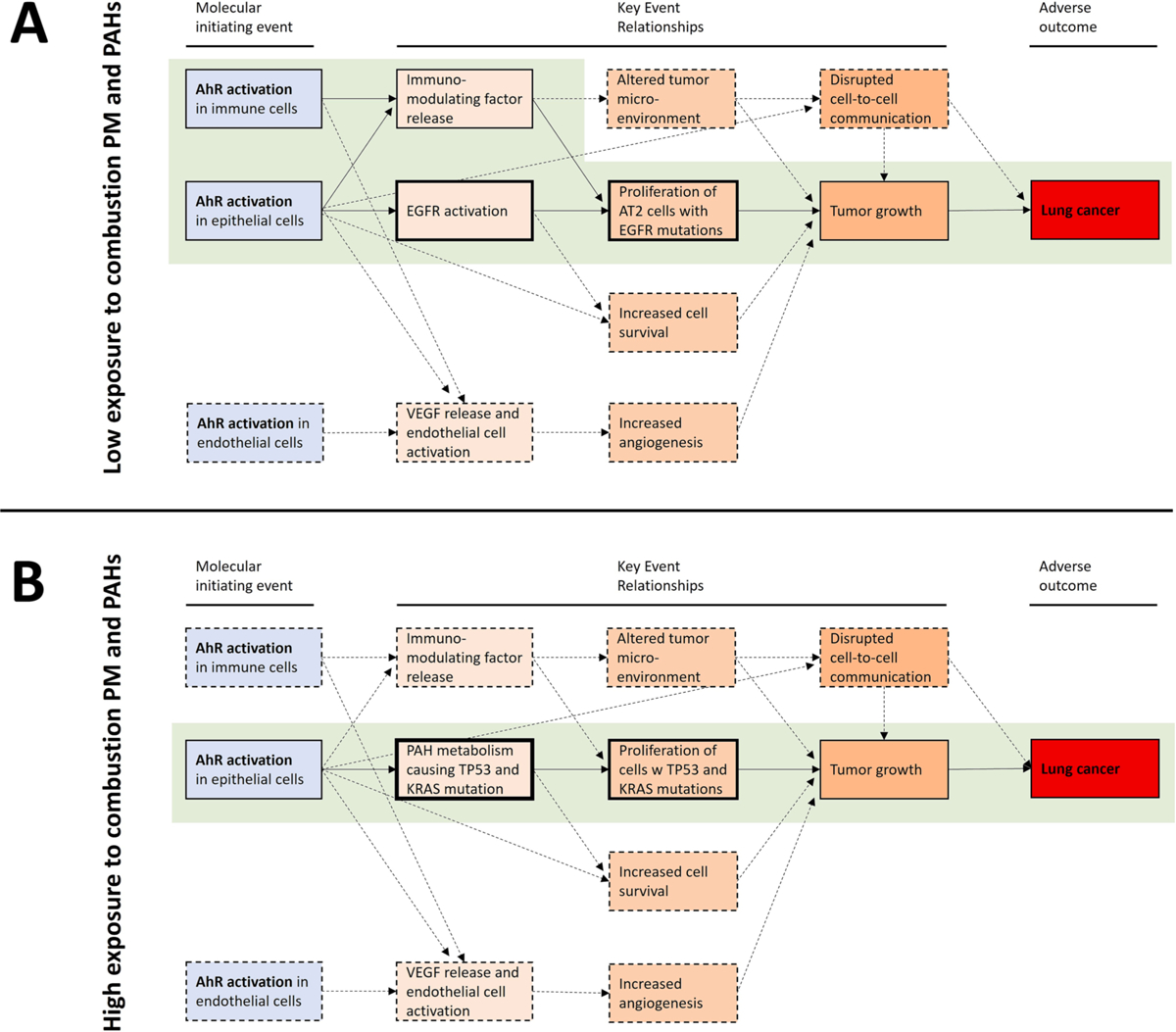
The figure presents a framework for development of AOPs for the link between AhR activation in different lung cell types and development of lung cancer after exposure of low levels of combustion PM and PAHs from outdoor air (A) and high-level exposures from smoking or occupational settings (B). At low-level exposure (A), AhR activation is primarily suggested to induce lung cancer by tumor promotion, through release of proinflammatory IL-1 family cytokines and nongenomic activation of EGFR. At high-level exposure (B) AhR induced CYP1 expression with subsequent PAH metabolism, formation of genotoxic metabolites and mutations in TP53 and KRAS is believed to be a central, early key events. AhR induced tumor promotion likely also affects cancer development in the high-level exposure scenario, but the role is less clear and suppressive effects of AhR on KRAS-driven cancers have been reported. It should be noted that some of MIE like VEGF release resulting in increased angiogenesis are first of importance in the later stage of cancer development; while key events like release of immunomodulating factors, DNA damage/mutations, increased cell survival, disrupted cell-to-cell-communication are of importance during a much longer period of cancer development than indicated in the figures. Well documented connections between AhR activation as the molecular initiating event, different key events, and the adverse outcome (lung cancer) are highlighted by solid lines on green background. Dotted lines represent connections that are indicated in the literature but where more uncertainty still exists.
Conclusive evidence for the role of AhR and PAHs in many of these processes is still lacking. However, this suggested framework for AhR signaling in lung cancer may provide a guidance for future studies and development of AOPs for AhR in lung cancer from exposure to ambient air PM2.5 and combustion PM. For instance, there is a need to explore how AhR knockdown or pharmacological inhibition would affect PM2.5 induced tumor promotion in lung AT2 cells with EGFR driver mutations and to compare the impact of PM2.5 with high or low PAH content on these responses. It is, however, important to consider that the role of AhR in lung cancer development is highly complex and, as is often the case in AhR research, that contrasting findings have been reported. The key to understanding the apparent multifaceted role of AhR in tumor development may lie in the diversity of responses regulated by unliganded constitutively active AhR, and upon distinct modes of AhR activation being elicited by its different ligands. Activation of AhR by PAHs and other ligands does not merely function as an on–off switch for transcription of target genes. AhR rather appears to bind and regulate a large number of gene clusters in unstimulated cells, and ligand-dependent activation causes considerable qualitative shifts in the genes regulated by the receptor [304]. A similar ligand promiscuity has also been described for the non-genomic effects of AhR [214,233]. These qualitative shifts in signaling and responses could likely explain some of the apparent contradictory results reported from studies on AhR in lung cancer based on knockout or overexpression models versus those based on exposure to different AhR ligands. Moreover, AhR could play specific roles in different types of lung cancers, where some express high AhR levels and others do not, and some are induced by AhR, while others are suppressed by AhR activity. Clarifying the underlying mechanisms for this “Janus-faced” role of AhR in lung cancer will be important. Another central question relates to the dose–response (or concentration-effect) relationship between PM or PAH exposure and different responses regulated by the AhR. The wide range of cellular processes regulated by AhR are presumably activated at somewhat different dose levels. Identifying the most sensitive biological responses induced by AhR may provide important information on the main mechanisms driving lung cancer development at relatively low PM-exposure levels encountered in outdoor air. After all, activation of AhR appears to be among the most sensitive endpoints reported from in vitro exposure of lung cell models to PM or DEP [111,112].
13. Conclusion
After more than half a century of research originating from studies on PAH metabolism, our understanding of the role of AhR in cancer development has expanded dramatically. For lung cancer, as for many other cancer types, AhR has been implicated at all stages of tumor development including initiation, promotion, progression, invasion, and metastasis.
We propose that lung cancer from smoking (and occupational and domestic exposure to high combustion PM levels) and lung cancer from air pollution (and secondhand smoke) in never-smokers represent the two ends of a dose–response continuum (Fig. 2a and b). In the case of lung adenocarcinomas (ADC) development in never-smokers from PM2.5 exposure from air pollution, tumor promotion appears to be a key mechanism acting on lung cells with EGFR driver-mutations acquired naturally through ageing. PAHs from combustion PM are likely candidate components contributing to these responses, through AhR-mediated activation of IL-1 family cytokines such as IL-1β and IL-18 induced through genomic pathways, and possibly also through non-genomic activation of EGFR. Moreover, AhR signaling upregulates immune-regulatory factors and can generate a pro-tumorigenic microenvironment enabling tumor promotion as discussed in this review. For lung squamous cell carcinoma (SCC) development in the central airways induced by higher exposure levels of combustion PM from smoking, occupational exposure, or indoor coal combustion, the initiation step appears to be a key mechanism driven by mutagenic PAH-metabolites through the classical AhR:Arnt-CYP pathway, acting in combination with other combustion-derived mutagens. The tumor promoting effects of AhR may also be involved in SCC, but they might be less prominent here. Accordingly, AhR has been reported to suppress some lung cancers, including those with KRAS-driver mutations characteristic of PAH-induced genotoxicity and smoking.
Clarifying the role of AhR in lung cancer development associated with air pollution and combustion PM may provide tools for detecting vulnerable populations and give a deeper understanding of essential risk factors. Hopefully this will lead to more efficient measures to reduce exposure to the most harmful air pollutants which can help to intervene and mitigate the development of cancer, especially for people at higher risk through environmental exposure to air pollution.
Funding
JØ has received funding from the European Union’s Horizon 2020 research and innovation program through the ULTRHAS project, Ultrafine Particles from Transportation – Health Assessment of Sources, under grant agreement No 955390. CV is supported in part by the Molecular Oncology Program at the UC Davis Comprehensive Cancer Center and the National Institute of Environmental Health Sciences of the United States National Institutes of Health under Award Number R01ES029126 and R01ES032827. JV and MM are supported by the Czech Science Foundation (project no. 21-00533S). JV acknowledges institutional support of the Institute of Biophysics of the Czech Academy of Sciences (RVO: 68081707).
Abbreviations:
- ATF4
Activating transcription factor 4
- ADC
adenocarcinomas
- AJs
adherens junctions
- AT2
alveolar type 2
- AOP
adverse outcome pathway
- ALK
anaplastic lymphoma kinase
- AR
androgen receptor
- APAF1/Apaf1
apoptotic peptidase activating factor 1
- Arg1
arginase 1
- AhR
aryl hydrocarbon receptor activities
- REPs
AhR-activating relative potencies
- ARA9 or XAP2
AhR-interacting protein
- Arnt
AhR nuclear translocator
- ABCG2
ATP-binding cassette super-family G member 2
- β-ARs
beta-adrenergic receptors
- B[a]P
benzo[a]pyrene
- BPDE
B[a]P-7,8-dihydrodiol-9,10-epoxide
- BPDE-N2-Dg
B[a]P 7,8-diol-9,10-epoxide-N2-deoxyguanosine
- BRAF
B-Raf proto-oncogene
- BCRP
breast cancer resistance protein
- BALFs
bronchoalveolar lavage fluids
- CHP2
calcineurin homologous protein isoform 2
- CA IX
carbonic anhydrase IX
- Cav1
caveolin-1
- KE
cellular key events
- CHK2
checkpoint kinase-2
- CXCL8
chemokine CXC-motif ligand 8
- COPD
chronic obstructive pulmonary disease
- CAR
constitutive androstane receptor
- COX-2
cyclooxygenase 2
- CYP
cytochrome P450
- DEP
diesel exhaust particles
- DREs
dioxin response elements
- DTT
dithiothreitol
- EC
elemental carbon
- (EoL-1) cells
eosinophilic
- EGFR
epidermal growth factor receptor
- EGFR-TKI
EGFR tyrosin kinase inhibitor
- EMT
epithelial-mesenchymal transition
- EGFR TKI
EGFR tyrosine kinase inhibitor
- ER
estrogen receptors
- ERR1/2
extracellular regulated kinase
- ECM
extracellular matrix
- EVs
extracellular vesicles
- EOM
extractable organic material
- FGF-2
fibroblast growth factor 2
- GJs
gap junctions
- GJIC
gap junctional intercellular communication
- GBD
Global Burden of Disease
- GR
glucocorticoid receptor
- GST
glutathione S-transferase
- GSL
glycosphingolipid
- Hsp90
heat shock protein 90 dimer
- MET
hepatocyte growth factor receptor
- HBEC3-KT
human bronchial epithelial cells
- BEAS-2B
human bronchial epithelial cell
- HER2
human epidermal growth factor receptor 2
- HUVECs
human umbilical vein endothelial cells
- HIF-1a
hypoxia-inducible factor-a
- IDO
indoleamine 2,3-dioxygenase
- IL
interleukin
- LDH
intracellular calcium concentrations [Ca2+]i lactate dehydrogenase
- LKB-1
liver kinase B1
- L-Kyn
L-kynurenine
- mTOR
mammalian target of rapamycin
- MV
microvesicles
- MEK-1
mitogen activated protein/extracellular regulated kinase kinase
- MAPK
mitogen activated kinase
- mdm2
mouse double minute 2
- MDR
multidrug-resistant
- MDSCs
myeloid derived suppressor cells
- NQO1
NADPH:quinone oxidoreductase
- nitro-PAHs
nitrated PAHs
- 1-NP
1-nitropyrene
- ncRNAs
noncoding RNAs
- miRNA
microRNA
- NSCLC
non-small cell lung cancer
- NF-kB
nuclear factor-kB
- OR
odds ratio
- OC
organic carbon
- 8-oxoG
8-oxo-7,8-dihydro-guanine
- oxy-PAHs
oxygenated PAHs
- PM
particulate matter
- EOM
PM-extractable organic material
- PTEN
phosphatase with tensin homology
- PAHs
polycyclic aromatic hydrocarbons
- PCBs
polychlorinated biphenyls
- Poly I:C
polyinosinic:polycytidylic acid
- PR
progesterone receptor
- PD-1
programmed cell death protein 1
- PD-L1
PD-1 ligand
- WB-F344
rat liver epithelial cells
- ROS
reactive oxygen species
- RET
rearranged during transfection
- RelBAHRE
RelB/AhR response element
- PM-EOM
residual particles after the extractions
- RUNX-2
runt-related transcription factor 2
- SERPINB2
serpin family B member 2
- SCLC
small-cell lung cancer
- NHE
sodium hydrogen exchanger Na+/H+ exchanger
- SL
sphingolipid
- p65
RelA and RelB, subunits of NF-kB
- SRM
standard reference material
- cancer stemness
stem cell-like tumor phenotype
- SCC
squamous cell carcinoma
- SHS
secondhand smoke
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- TEFs
toxic equivalency factors
- TJs
tight junctions
- TMPRSS2
transmembrane serine protease 2
- TRPC
transient receptor potential canonical
- TAMs
channels,tumor associated macrophages
- TME
tumor microenvironment
- TNF
tumor necrosis factor
- TP53
-α,tumor protein 53
- TP73/p73
tumor protein 73
- TRCs
tumor-repopulating T-cells
- UGT
tyrosine protein kinase c-Src,UDP-glucuronosyltransferase
- VEGF
vascular endothelial growth factor
- KRAS
v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
- WSP
wood smoke particles
- XRE/DRE
xenobiotic or dioxin response elements
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Landis SH, Murray T, Bolden S, Wingo PA. Cancer statistics, 1999. CA Cancer J. Clin 1999;49(1):8–31, 1. [DOI] [PubMed] [Google Scholar]
- [2].Jemal A, Thun MJ, Ries LA, Howe HL, Weir HK, Center MM, et al. , Annual report to the nation on the status of cancer, 1975–2005, featuring trends in lung cancer, tobacco use, and tobacco control, J. Natl Cancer Inst 100 (23) (2008) 1672–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. , Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J. Clin 71 (3) (2021) 209–249. [DOI] [PubMed] [Google Scholar]
- [4].American Cancer SocietyTrusted Source A, Lung cancer and age: statistics, risk factors, and more, Healthline. (2021). https://www.healthline.com/health/lung-cancer/lung-cancer-age.
- [5].Goodson WH 3rd, Lowe L, Carpenter DO, Gilbertson M, Manaf Ali A, de Cerain L, Salsamendi A, et al. , Assessing the carcinogenic potential of low-dose exposures to chemical mixtures in the environment: the challenge ahead, Carcinogenesis 36 Suppl 1(Suppl 1) (2015) S254–S296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sun S, Schiller JH, Gazdar AF, Lung cancer in never smokers — a different disease, Nat. Rev. Cancer 7 (10) (2007) 778–790. [DOI] [PubMed] [Google Scholar]
- [7].H.E. Institute, H., State of global air 2020, Health Effects Institute, Boston, MA, US, 2020. [Google Scholar]
- [8].Iarc, Outdoor air pollution, IARC Monogr. Eval. Carcinog. Risks Hum 109 (2016) 33–444. [PMC free article] [PubMed] [Google Scholar]
- [9].Iarc, Diesel and gasoline engine exhausts and some nitroarenes, IARC Monogr. Eval. Carcinog. Risks Hum 105 (2014) 9–699. [PMC free article] [PubMed] [Google Scholar]
- [10].IARC. Tobacco smoking., IARC Monogr. Eval. Carcinog. Risk Chem. Hum 38 (1986) 35–394. [PubMed] [Google Scholar]
- [11].Iarc, Tobacco smoke and involuntary smoking, IARC Monogr. Eval. Carcinog. Risks Hum 83 (2004) 1–1438. [PMC free article] [PubMed] [Google Scholar]
- [12].Iarc, Household use of solid fuels and high-temperature frying, IARC Monogr. Eval. Carcinog. Risks Hum 95 (2010) 1–430. [PMC free article] [PubMed] [Google Scholar]
- [13].Vinikoor-Imler LC, Davis JA, Luben TJ, An ecologic analysis of county-level PM2.5 concentrations and lung cancer incidence and mortality, Int. J. Environ. Res. Public Health 8 (6) (2011) 1865–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Eckel SP, Cockburn M, Shu YH, Deng H, Lurmann FW, Liu L, et al. , Air pollution affects lung cancer survival, Thorax 71 (10) (2016) 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bidoli E, Pappagallo M, Birri S, Frova L, Zanier L, Serraino D. Residential proximity to major roadways and lung cancer mortality. Italy, 1990–2010: An observational study. Int J Environ Res Public Health. 2016;13(2):191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shao Y, Wang Y, Yu H, Zhang Y, Xiang F, Yang Y, et al. , Geographical variation in lung cancer risk associated with road traffics in Jiading District, Shanghai. Sci Total Environ. 652 (2019) 729–735. [DOI] [PubMed] [Google Scholar]
- [17].Cakmak S, Hebbern C, Vanos J, Crouse DL, Tjepkema M, Exposure to traffic and mortality risk in the 1991–2011 Canadian Census Health and Environment Cohort (CanCHEC), Environ. Int 124 (2019) 16–24. [DOI] [PubMed] [Google Scholar]
- [18].Vineis P, Hoek G, Krzyzanowski M, Vigna-Taglianti F, Veglia F, Airoldi L, et al. , Lung cancers attributable to environmental tobacco smoke and air pollution in non-smokers in different European countries: a prospective study, Environ. Health 6 (2007) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gowda SN, DeRoos AJ, Hunt RP, Gassett AJ, Mirabelli MC, Bird CE, et al. , Ambient air pollution and lung cancer risk among never-smokers in the Women’s Health Initiative, Environ Epidemiol. 3 (6) (2019) e076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Claxton LD, Woodall GM Jr., A review of the mutagenicity and rodent carcinogenicity of ambient air, Mutat. Res 636 (1–3) (2007) 36–94. [DOI] [PubMed] [Google Scholar]
- [21].Cho CC, Hsieh WY, Tsai CH, Chen CY, Chang HF, Lin CS, In vitro and in vivo experimental studies of PM(2.5) on disease progression, Int. J. Environ. Res. Public Health 15 (7) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nemmar A, Holme JA, Rosas I, Schwarze PE, Alfaro-Moreno E, Recent advances in particulate matter and nanoparticle toxicology: a review of the in vivo and in vitro studies, Biomed Res. Int 2013 (2013), 279371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cassee FR, Héroux ME, Gerlofs-Nijland ME, Kelly FJ, Particulate matter beyond mass: recent health evidence on the role of fractions, chemical constituents and sources of emission, Inhal. Toxicol 25 (14) (2013) 802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].DeMarini DM, Linak WP, Mutagenicity and carcinogenicity of combustion emissions are impacted more by combustor technology than by fuel composition: a brief review, Environ. Mol. Mutagen 63 (3) (2022) 135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Boström CE, Gerde P, Hanberg A, Jernström B, Johansson C, Kyrklund T, et al. , Cancer risk assessment, indicators, and guidelines for polycyclic aromatic hydrocarbons in the ambient air, Environ. Health Perspect 110 Suppl 3(Suppl 3) (2002) 451–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dreij K, Mattsson Å, Jarvis IWH, Lim H, Hurkmans J, Gustafsson J, et al. , Cancer risk assessment of airborne PAHs based on in vitro mixture potency factors, Environ. Sci. Tech 51 (15) (2017) 8805–8814. [DOI] [PubMed] [Google Scholar]
- [27].Vondracek J, Machala M, The role of metabolism in toxicity of polycyclic aromatic hydrocarbons and their non-genotoxic modes of action, Curr. Drug Metab 22 (8) (2021) 584–595. [DOI] [PubMed] [Google Scholar]
- [28].Lag M, Ovrevik J, Refsnes M, Holme JA, Potential role of polycyclic aromatic hydrocarbons in air pollution-induced non-malignant respiratory diseases, Respir. Res 21 (1) (2020) 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Benoit L, Jornod F, Zgheib E, Tomkiewicz C, Koual M, Coustillet T, et al. , Adverse outcome pathway from activation of the AhR to breast cancer-related death, Environ. Int 165 (2022), 107323. [DOI] [PubMed] [Google Scholar]
- [30].Cooper WA, Lam DC, O’Toole SA, Minna JD, Molecular biology of lung cancer, J. Thorac. Dis 5 Suppl 5(Suppl 5) (2013) S479–S490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nebert DW, Dalton TP, The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis, Nat. Rev. Cancer 6 (12) (2006) 947–960. [DOI] [PubMed] [Google Scholar]
- [32].Tsay JJ, Tchou-Wong KM, Greenberg AK, Pass H, Rom WN, Aryl hydrocarbon receptor and lung cancer, Anticancer Res 33 (4) (2013) 1247–1256. [PMC free article] [PubMed] [Google Scholar]
- [33].Shivanna B, Chu C, Moorthy B, The aryl hydrocarbon receptor (AHR): A novel therapeutic target for pulmonary diseases? Int. J. Mol. Sci 23 (3) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Moserová M, Kotrbová V, Aimová D, Sulc M, Frei E, Stiborová M, Analysis of´ benzo[a]pyrene metabolites formed by rat hepatic microsomes using high pressure liquid chromatography: optimization of the method, Interdiscip. Toxicol. 2 (4) (2009) 239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nebert DW, Dalton TP, Okey AB, Gonzalez FJ, Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer, J. Biol. Chem 279 (23) (2004) 23847–23850. [DOI] [PubMed] [Google Scholar]
- [36].Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, et al. , Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor, PNAS 97 (2) (2000) 779–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang T, Joubert P, Ansari-Pour N, Zhao W, Hoang PH, Lokanga R, et al. , Genomic and evolutionary classification of lung cancer in never smokers, Nat. Genet 53 (9) (2021) 1348–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hill W, Lim EL, Weeden CE, Lee C, Augustine M, Chen K, et al. , Lung adenocarcinoma promotion by air pollutants, Nature 616 (7955) (2023) 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pitot HC, Goldsworthy T, Campbell HA, Poland A, Quantitative evaluation of the promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin of hepatocarcinogenesis from diethylnitrosamine, Cancer Res. 40 (10) (1980) 3616–3620. [PubMed] [Google Scholar]
- [40].Poland A, Palen D, Glover E, Tumour promotion by TCDD in skin of HRS/J hairless mice, Nature 300 (5889) (1982) 271–273. [DOI] [PubMed] [Google Scholar]
- [41].Shu HP, Paustenbach DJ, Murray FJ, A critical evaluation of the use of mutagenesis, carcinogenesis, and tumor promotion data in a cancer risk assessment of 2,3,7,8-tetrachlorodibenzo-p-dioxin, Regul. Toxicol. Pharm 7 (1) (1987) 57–88. [DOI] [PubMed] [Google Scholar]
- [42].Beebe LE, Anver MR, Riggs CW, Fornwald LW, Anderson LM, Promotion of N-nitrosodimethylamine-initiated mouse lung tumors following single or multiple low dose exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin, Carcinogenesis 16 (6) (1995) 1345–1349. [DOI] [PubMed] [Google Scholar]
- [43].Wang Z, Snyder M, Kenison JE, Yang K, Lara B, Lydell E, et al. , How the AHR became important in cancer: The role of chronically active AHR in cancer aggression, Int. J. Mol. Sci 22 (1) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xue P, Fu J, Zhou Y, The aryl hydrocarbon receptor and tumor immunity, Front. Immunol 9 (2018) 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Øvrevik J, Refsnes M, Låg M, Holme JA, Schwarze PE, Activation of proinflammatory responses in cells of the airway mucosa by particulate matter: oxidant- and non-oxidant-mediated triggering mechanisms, Biomolecules 5 (3) (2015) 1399–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].NTP. Diesel exhaust particulates. 2011.
- [47].Totlandsdal AI, Øvrevik J, Cochran RE, Herseth JI, Bølling AK, Låg M, et al. , The occurrence of polycyclic aromatic hydrocarbons and their derivatives and the proinflammatory potential of fractionated extracts of diesel exhaust and wood smoke particles, J. Environ. Sci. Health A Tox. Hazard. Subst. Environ. Eng 49 (4) (2014) 383–396. [DOI] [PubMed] [Google Scholar]
- [48].Li Y.-y., Liang D, Shen H, An analysis of background interference on fire debris, Procedia Eng. 52 (2013) 664–670. [Google Scholar]
- [49].Avagyan R, Nyström R, Lindgren R, Boman C, Westerholm R, Particulate hydroxy-PAH emissions from a residential wood log stove using different fuels and burning conditions, Atmos. Environ 140 (2016) 1–9. [Google Scholar]
- [50].Karavalakis GM, Robledo T, Miguel A. A review of polycyclic aromatic hydrocarbon and polycyclic aromatic hydrocarbon derivative emissions from off-road, light-duty, heavy-duty, and stationary sources. University of California, Riverside, and Bourns College of Engineering-Center for Environmental Research and Technology (CE-CERT); 2020. [Google Scholar]
- [51].Hartikainen AH, Ihalainen M, Yli-Pirilä P, Hao L, Kortelainen M, Pieber SM, et al. , Photochemical transformation and secondary aerosol formation potential of Euro6 gasoline and diesel passenger car exhaust emissions, J. Aerosol Sci 171 (2023), 106159. [Google Scholar]
- [52].Platt SM, El Haddad I, Pieber SM, Zardini AA, Suarez-Bertoa R, Clairotte M, et al. , Gasoline cars produce more carbonaceous particulate matter than modern filter-equipped diesel cars, Sci. Rep 7 (1) (2017) 4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Skuland T, Grytting VS, Låg M, Jørgensen RB, Snilsberg B, Leseman D, et al. , Road tunnel-derived coarse, fine and ultrafine particulate matter: physical and chemical characterization and pro-inflammatory responses in human bronchial epithelial cells, Part. Fibre Toxicol 19 (1) (2022) 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Holme JA, Låg M, Skuland T, Parenicova M, Ciganek M, Penciková K, et al. , Characterization of elements, PAHs, AhR-activity and pro-inflammatory responses of road tunnel-derived particulate matter in human hepatocyte-like and bronchial epithelial cells, Toxicol. In Vitro 90 (2023), 105611. [DOI] [PubMed] [Google Scholar]
- [55].Feilberg A, Poulsen MWB, Nielsen T, Skov H, Occurrence and sources of particulate nitro-polycyclic aromatic hydrocarbons in ambient air in Denmark, Atmos. Environ 35 (2001) 353–366. [Google Scholar]
- [56].Pěnčíková K, Ciganek M, Neča J, Illés P, Dvořák Z, Vondráček J, et al. , Modulation of endocrine nuclear receptor activities by polyaromatic compounds present in fractionated extracts of diesel exhaust particles, Sci. Total Environ 677 (2019) 626–636. [DOI] [PubMed] [Google Scholar]
- [57].Lee Y-Y, Hsieh Y-K, Huang B-W, Mutuku JK, Chang-Chien G-P, Huang S, An overview: PAH and nitro-PAH emission from the stationary sources and their transformations in the atmosphere, Aerosol Air Qual. Res 22 (7) (2022), 220164. [Google Scholar]
- [58].Scheepers PT, Martens MH, Velders DD, Fijneman P, van Kerkhoven M, Noordhoek J, et al. , 1-Nitropyrene as a marker for the mutagenicity of diesel exhaust-derived particulate matter in workplace atmospheres, Environ. Mol. Mutagen 25 (2) (1995) 134–147. [DOI] [PubMed] [Google Scholar]
- [59].Pálková L, Vondráček J, Trilecová L, Ciganek M, Pěnčíková K, Neča J, et al. , The aryl hydrocarbon receptor-mediated and genotoxic effects of fractionated extract of standard reference diesel exhaust particle material in pulmonary, liver and prostate cells, Toxicol. In Vitro 29 (3) (2015) 438–448. [DOI] [PubMed] [Google Scholar]
- [60].Gualtieri M, Øvrevik J, Holme JA, Perrone MG, Bolzacchini E, Schwarze PE, et al. , Differences in cytotoxicity versus pro-inflammatory potency of different PM fractions in human epithelial lung cells, Toxicol. In Vitro 24 (1) (2010) 29–39. [DOI] [PubMed] [Google Scholar]
- [61].Hu T, Zhang J, Xing X, Zhan C, Zhang L, Liu H, et al. , Seasonal variation and health risk assessment of atmospheric PM2.5-bound polycyclic aromatic hydrocarbons in a classic agglomeration industrial city, central China, Air Qual. Atmos. Health 11 (6) (2018) 683–694. [Google Scholar]
- [62].Ishihara Y, Kado SY, Bein KJ, He Y, Pouraryan AA, Urban A, et al. , Aryl hydrocarbon receptor signaling synergizes with TLR/NF-κB-signaling for induction of IL-22 through canonical and non-canonical AhR pathways, Front Toxicol. 3 (2021), 787360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Young TM, Black GP, Wong L, Bloszies CS, Fiehn O, He G, et al. , Identifying toxicologically significant compounds in urban wildfire ash using In vitro bioassays and high-resolution mass spectrometry, Environ. Sci. Tech 55 (6) (2021) 3657–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Goldfarb JL, Suuberg EM, Vapor pressures and thermodynamics of oxygen-containing polycyclic aromatic hydrocarbons measured using Knudsen effusion, Environ. Toxicol. Chem 27 (6) (2008) 1244–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kramer AL, Suski KJ, Bell DM, Zelenyuk A, Massey Simonich SL, Formation of polycyclic aromatic hydrocarbon oxidation products in α-pinene secondary organic aerosol particles formed through ozonolysis, Environ. Sci. Tech 53 (12) (2019) 6669–6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Machala M, Ciganek M, Bláha L, Minksová K, Vondráck J, Aryl hydrocarbon receptor-mediated and estrogenic activities of oxygenated polycyclic aromatic hydrocarbons and azaarenes originally identified in extracts of river sediments, Environ. Toxicol. Chem 20 (12) (2001) 2736–2743. [PubMed] [Google Scholar]
- [67].Larsson M, Hagberg J, Giesy JP, Engwall M, Time-dependent relative potency factors for polycyclic aromatic hydrocarbons and their derivatives in the H4IIE-luc bioassay, Environ. Toxicol. Chem 33 (4) (2014) 943–953. [DOI] [PubMed] [Google Scholar]
- [68].McCarrick S, Cunha V, Zapletal O, Vondráček J, Dreij K, In vitro and in vivo genotoxicity of oxygenated polycyclic aromatic hydrocarbons, Environ. Pollut 246 (2019) 678–687. [DOI] [PubMed] [Google Scholar]
- [69].Lammel G, Kitanovski Z, Kukučka P, Novák J, Arangio AM, Codling GP, et al. Oxygenated and nitrated polycyclic aromatic hydrocarbons in ambient air-levels, phase partitioning, mass size distributions, and inhalation bioaccessibility, Environ. Sci. Tech 54 (5) (2020) 2615–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].van Zandwijk N, Mooi WJ, Rodenhuis S, Prognostic factors in NSCLC, Recent experiences. Lung Cancer. 12 (Suppl 1) (1995) S27–S33. [DOI] [PubMed] [Google Scholar]
- [71].Schiller JH, Current standards of care in small-cell and non-small-cell lung cancer, Oncology 61 (Suppl 1) (2001) 3–13. [DOI] [PubMed] [Google Scholar]
- [72].Ramalingam S, Pawlish K, Gadgeel S, Demers R, Kalemkerian GP, Lung cancer in young patients: analysis of a surveillance, epidemiology, and end results database, J. Clin. Oncol 16 (2) (1998) 651–657. [DOI] [PubMed] [Google Scholar]
- [73].Hanna JM, Onaitis MW, Cell of origin of lung cancer, J Carcinog. 12 (2013) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Giangreco A, Groot KR, Janes SM, Lung cancer and lung stem cells: strange bedfellows? Am. J. Respir. Crit. Care Med 175 (6) (2007) 547–553. [DOI] [PubMed] [Google Scholar]
- [75].Larsen JE, Minna JD, Molecular biology of lung cancer: clinical implications, Clin. Chest Med 32 (4) (2011) 703–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hollstein M, Sidransky D, Vogelstein B, Harris CC, p53 mutations in human cancers, Science 253 (5015) (1991) 49–53. [DOI] [PubMed] [Google Scholar]
- [77].Toyooka S, Tsuda T, Gazdar AF, The TP53 gene, tobacco exposure, and lung cancer, Hum. Mutat 21 (3) (2003) 229–239. [DOI] [PubMed] [Google Scholar]
- [78].Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P, Tobacco smoke carcinogens DNA damage and p53 mutations in smoking-associated cancers, Oncogene 21 (48) (2002) 7435–7451. [DOI] [PubMed] [Google Scholar]
- [79].Ferrer I, Zugazagoitia J, Herbertz S, John W, Paz-Ares L, Schmid-Bindert G, KRAS-Mutant non-small cell lung cancer: from biology to therapy, Lung Cancer 124 (2018) 53–64. [DOI] [PubMed] [Google Scholar]
- [80].Pinheiro G, Pereira T, Dias C, Freitas C, Hespanhol V, Costa JL, et al. , Identifying relationships between imaging phenotypes and lung cancer-related mutation status: EGFR and KRAS, Sci. Rep 10 (1) (2020) 3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Mack PC, Klein MI, Ayers KL, Zhou X, Guin S, Fink M, et al. , Targeted next-generation sequencing reveals exceptionally high rates of molecular driver mutations in never-smokers with lung adenocarcinoma, Oncologist 27 (6) (2022) 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Jorge SE, Kobayashi SS, Costa DB, Epidermal growth factor receptor (EGFR) mutations in lung cancer: preclinical and clinical data, Braz. J. Med. Biol. Res 47 (11) (2014) 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Harrison PT, Vyse S, Huang PH, Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer, Semin. Cancer Biol 61 (2020) 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Smalley M, Ashworth A, Stem cells and breast cancer: a field in transit, Nat. Rev. Cancer 3 (11) (2003) 832–844. [DOI] [PubMed] [Google Scholar]
- [85].Fukui T, Shaykhiev R, Agosto-Perez F, Mezey JG, Downey RJ, Travis WD, et al. , Lung adenocarcinoma subtypes based on expression of human airway basal cell genes, Eur. Respir. J 42 (5) (2013) 1332–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Sainz de Aja J, Dost AFM, Kim CF, Alveolar progenitor cells and the origin of lung cancer, J. Intern. Med 289 (5) (2021) 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Mingard C, Battey JND, Takhaveev V, Blatter K, Hürlimann V, Sierro N, et al. , Dissection of cancer mutational signatures with individual components of cigarette smoking, Chem. Res. Toxicol 36 (4) (2023) 714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Heng WS, Gosens R, Kruyt FAE, Lung cancer stem cells: origin, features, maintenance mechanisms and therapeutic targeting, Biochem. Pharmacol 160 (2019) 121–133. [DOI] [PubMed] [Google Scholar]
- [89].Canever H, Carollo PS, Fleurisson R, Girard PP, Borghi N, Molecular tension microscopy of e-cadherin during epithelial-mesenchymal transition, Methods Mol. Biol 2179 (2021) 289–299. [DOI] [PubMed] [Google Scholar]
- [90].Xie S, Wu Z, Qi Y, Wu B, Zhu X, The metastasizing mechanisms of lung cancer: Recent advances and therapeutic challenges, Biomed. Pharmacother 138 (2021), 111450. [DOI] [PubMed] [Google Scholar]
- [91].Liu J, Li D, Luo H, Zhu X, Circular RNAs: The star molecules in cancer, Mol. Aspects Med 70 (2019) 141–152. [DOI] [PubMed] [Google Scholar]
- [92].Riva L, Pandiri AR, Li YR, Droop A, Hewinson J, Quail MA, et al. , The mutational signature profile of known and suspected human carcinogens in mice, Nat. Genet 52 (11) (2020) 1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Sayan M, Mossman BT, The NLRP3 inflammasome in pathogenic particle and fibre-associated lung inflammation and diseases, Part. Fibre Toxicol 13 (1) (2016) 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Borm PJ, Tran L, Donaldson K, The carcinogenic action of crystalline silica: a review of the evidence supporting secondary inflammation-driven genotoxicity as a principal mechanism, Crit. Rev. Toxicol 41 (9) (2011) 756–770. [DOI] [PubMed] [Google Scholar]
- [95].Paris C, Do P, Mastroianni B, Dixmier A, Dumont P, Pichon E, et al. , Association between lung cancer somatic mutations and occupational exposure in never-smokers, Eur. Respir. J 50 (4) (2017). [DOI] [PubMed] [Google Scholar]
- [96].Vogel CFA, Ishihara Y, Campbell CE, Kado SY, Nguyen-Chi A, Sweeney C, et al. , A protective role of aryl hydrocarbon receptor repressor in inflammation and tumor growth, Cancers (Basel) 11 (5) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Frankell AM, Dietzen M, Al Bakir M, Lim EL, Karasaki T, Ward S, et al. , The evolution of lung cancer and impact of subclonal selection in TRACERx, Nature (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Kim CH, Lee YC, Hung RJ, McNallan SR, Cote ML, Lim WY, et al. , Exposure to secondhand tobacco smoke and lung cancer by histological type: a pooled analysis of the International Lung Cancer Consortium (ILCCO), Int. J. Cancer 135 (8) (2014) 1918–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Ge C, Peters S, Olsson A, Portengen L, Schuz J, Almansa J, et al. , Diesel engine exhaust exposure, smoking, and lung cancer subtype risks. a pooled exposure-response analysis of 14 case-control studies, Am. J. Respir. Crit. Care Med 202 (3) (2020) 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kurmi OP, Arya PH, Lam KB, Sorahan T, Ayres JG, Lung cancer risk and solid fuel smoke exposure: a systematic review and meta-analysis, Eur. Respir. J 40 (5) (2012) 1228–1237. [DOI] [PubMed] [Google Scholar]
- [101].Xavier RF, Ramos D, Ito JT, Rodrigues FM, Bertolini GN, Macchione M, et al. , Effects of cigarette smoking intensity on the mucociliary clearance of active smokers, Respiration 86 (6) (2013) 479–485. [DOI] [PubMed] [Google Scholar]
- [102].Tilley AE, Walters MS, Shaykhiev R, Crystal RG, Cilia dysfunction in lung disease, Annu. Rev. Physiol 77 (2015) 379–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Iarc, Some aromatic amines, organic dyes, and related exposures, France International Agency for Research on Cancer, Lyon, 2010. [Google Scholar]
- [104].Loomis D, Grosse Y, Lauby-Secretan B, El Ghissassi F, Bouvard V, Benbrahim-Tallaa L, et al. , The carcinogenicity of outdoor air pollution, Lancet Oncol. 14 (13) (2013) 1262–1263. [DOI] [PubMed] [Google Scholar]
- [105].Topinka J, Marvanová S, Vondrácek J, Sevastyanova O, Nováková Z, Krcmár P, et al. , DNA adducts formation and induction of apoptosis in rat liver epithelial ‘stem-like’ cells exposed to carcinogenic polycyclic aromatic hydrocarbons, Mutat. Res 638 (1–2) (2008) 122–132. [DOI] [PubMed] [Google Scholar]
- [106].Svihálko L, Machala M, Pencíková K, Marvanová S, Neca J, Topinka J, et al. , Dibenzanthracenes and benzochrysenes elicit both genotoxic and nongenotoxic events in rat liver ‘stem-like’ cells, Toxicology 232 (1–2) (2007) 147–159. [DOI] [PubMed] [Google Scholar]
- [107].Marvanová S, Vondrácek J, Penccíková K, Trilecová L, Krcmárr P, Topinka J, et al. , Toxic effects of methylated benz[a]anthracenes in liver cells, Chem. Res. Toxicol 21 (2) (2008) 503–512. [DOI] [PubMed] [Google Scholar]
- [108].Machala M, Svihálková-Sindlerová L, Pencíková K, Krcmár P, Topinka J, Milcová A, et al. , Effects of methylated chrysenes on AhR-dependent and-independent toxic events in rat liver epithelial cells, Toxicology 247 (2–3) (2008) 93–101. [DOI] [PubMed] [Google Scholar]
- [109].Trilecová L, Krčková S, Marvanová S, Pěnčíkova K, Krčmář P, Neča J, et al. , Toxic effects of methylated benzo[a]pyrenes in rat liver stem-like cells, Chem. Res. Toxicol 24 (6) (2011) 866–876. [DOI] [PubMed] [Google Scholar]
- [110].Andrysík Z, Vondráček J, Marvanová S, Ciganek M, Neča J, Pěnčíková K, et al. , Activation of the aryl hydrocarbon receptor is the major toxic mode of action of an organic extract of a reference urban dust particulate matter mixture: the role of polycyclic aromatic hydrocarbons, Mutat. Res 714 (1–2) (2011) 53–62. [DOI] [PubMed] [Google Scholar]
- [111].Totlandsdal AI, Cassee FR, Schwarze P, Refsnes M, Låg M, Diesel exhaust particles induce CYP1A1 and pro-inflammatory responses via differential pathways in human bronchial epithelial cells, Part. Fibre Toxicol 7 (2010) 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Fizeșan I, Chary A, Cambier S, Moschini E, Serchi T, Nelissen I, et al. , Responsiveness assessment of a 3D tetra-culture alveolar model exposed to diesel exhaust particulate matter, Toxicol. In Vitro 53 (2018) 67–79. [DOI] [PubMed] [Google Scholar]
- [113].Sweeney C, Lazennec G, Vogel CFA, Environmental exposure and the role of AhR in the tumor microenvironment of breast cancer, Front. Pharmacol 13 (2022) 1095289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Su JM, Lin P, Chang H, Prognostic value of nuclear translocation of aryl hydrocarbon receptor for non-small cell lung cancer, Anticancer Res 33 (9) (2013) 3953–3961. [PubMed] [Google Scholar]
- [115].Ye M, Zhang Y, Gao H, Xu Y, Jing P, Wu J, et al. , Activation of the aryl hydrocarbon receptor leads to resistance to EGFR TKIs in non-small cell lung cancer by activating Src-mediated bypass signaling, Clin. Cancer Res 24 (5) (2018) 1227–1239. [DOI] [PubMed] [Google Scholar]
- [116].Feng H, Cao B, Peng X, Wei Q, Cancer-associated fibroblasts strengthen cell proliferation and EGFR TKIs resistance through aryl hydrocarbon receptor dependent signals in non-small cell lung cancer, BMC Cancer 22 (1) (2022) 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Nacarino-Palma A, Rejano-Gordillo CM, González-Rico FJ, Ordiales-Talavero A, Román ÁC, Cuadrado M, et al. , Loss of aryl hydrocarbon receptor favors K-Ras(G12D)-driven non-small cell lung cancer, Cancers (Basel) 13 (16) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Demetriou CA, Raaschou-Nielsen O, Loft S, Møller P, Vermeulen R, Palli D, et al. , Biomarkers of ambient air pollution and lung cancer: a systematic review, Occup. Environ. Med 69 (9) (2012) 619–627. [DOI] [PubMed] [Google Scholar]
- [119].DeMarini DM, Genotoxicity biomarkers associated with exposure to traffic and near-road atmospheres: a review, Mutagenesis 28 (5) (2013) 485–505. [DOI] [PubMed] [Google Scholar]
- [120].Bonvallot V, Baeza-Squiban A, Baulig A, Brulant S, Boland S, Muzeau F, et al. , Organic compounds from diesel exhaust particles elicit a proinflammatory response in human airway epithelial cells and induce cytochrome p450 1A1 expression, Am. J. Respir. Cell Mol. Biol 25 (4) (2001) 515–521. [DOI] [PubMed] [Google Scholar]
- [121].Cavanagh JA, Trought K, Brown L, Duggan S, Exploratory investigation of the chemical characteristics and relative toxicity of ambient air particulates from two New Zealand cities, Sci. Total Environ 407 (18) (2009) 5007–5018. [DOI] [PubMed] [Google Scholar]
- [122].Moorthy B, Chu C, Carlin DJ, Polycyclic aromatic hydrocarbons: from metabolism to lung cancer, Toxicol. Sci 145 (1) (2015) 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Xue W, Warshawsky D, Metabolic activation of polycyclic and heterocyclic aromatic hydrocarbons and DNA damage: a review, Toxicol. Appl. Pharmacol 206 (1) (2005) 73–93. [DOI] [PubMed] [Google Scholar]
- [124].Henkler F, Stolpmann K, Luch A, Exposure to polycyclic aromatic hydrocarbons: bulky DNA adducts and cellular responses, Exp Suppl. 101 (2012) 107–131. [DOI] [PubMed] [Google Scholar]
- [125].Penning TM, Human aldo-keto reductases and the metabolic activation of polycyclic aromatic hydrocarbons, Chem. Res. Toxicol 27 (11) (2014) 1901–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Oyama T, Sugio K, Uramoto H, Kawamoto T, Kagawa N, Nadaf S, et al. , Cytochrome P450 expression (CYP) in non-small cell lung cancer, Front Biosci 12 (2007) 2299–2308. [DOI] [PubMed] [Google Scholar]
- [127].Zhan P, Wang Q, Qian Q, Wei SZ, Yu LK, CYP1A1 MspI and exon7 gene polymorphisms and lung cancer risk: an updated meta-analysis and review, J. Exp. Clin. Cancer Res 30 (1) (2011) 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Chen Z, Li Z, Niu X, Ye X, Yu Y, Lu S, et al. , The effect of CYP1A1 polymorphisms on the risk of lung cancer: a global meta-analysis based on 71 case-control studies, Mutagenesis 26 (3) (2011) 437–446. [DOI] [PubMed] [Google Scholar]
- [129].Liu C, Cui H, Gu D, Zhang M, Fang Y, Chen S, et al. , Genetic polymorphisms and lung cancer risk: Evidence from meta-analyses and genome-wide association studies, Lung Cancer 113 (2017) 18–29. [DOI] [PubMed] [Google Scholar]
- [130].Nebert DW, Shi Z, Gálvez-Peralta M, Uno S, Dragin N, Oral benzo[a]pyrene: understanding pharmacokinetics, detoxication, and consequences–Cyp1 knockout mouse lines as a paradigm, Mol. Pharmacol 84 (3) (2013) 304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Vondráček J, Pivnička J, Machala M, Polycyclic aromatic hydrocarbons and disruption of steroid signaling, Current Opinion in Toxicology. 11–12 (2018) 27–34. [Google Scholar]
- [132].Kolluri SK, Jin UH, Safe S, Role of the aryl hydrocarbon receptor in carcinogenesis and potential as an anti-cancer drug target, Arch. Toxicol 91 (7) (2017) 2497–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Murk AJ, Legler J, Denison MS, Giesy JP, van de Guchte C, Brouwer A, Chemical-activated luciferase gene expression (CALUX): a novel in vitro bioassay for Ah receptor active compounds in sediments and pore water, Fundam. Appl. Toxicol 33 (1) (1996) 149–160. [DOI] [PubMed] [Google Scholar]
- [134].Machala M, Vondrácek J, Bláha L, Ciganek M, Neca JV, Aryl hydrocarbon receptor-mediated activity of mutagenic polycyclic aromatic hydrocarbons determined using in vitro reporter gene assay, Mutat. Res 497 (1–2) (2001) 49–62. [DOI] [PubMed] [Google Scholar]
- [135].Larsson M, Orbe D, Engwall M, Exposure time-dependent effects on the relative potencies and additivity of PAHs in the Ah receptor-based H4IIE-luc bioassay, Environ. Toxicol. Chem 31 (5) (2012) 1149–1157. [DOI] [PubMed] [Google Scholar]
- [136].Pieterse B, Felzel E, Winter R, van der Burg B, Brouwer A, PAH-CALUX, an optimized bioassay for AhR-mediated hazard identification of polycyclic aromatic hydrocarbons (PAHs) as individual compounds and in complex mixtures, Environ. Sci. Tech 47 (20) (2013) 11651–11659. [DOI] [PubMed] [Google Scholar]
- [137].Vondráček J, Pěnčíková K, Neča J, Ciganek M, Grycová A, Dvořák Z, et al. , Assessment of the aryl hydrocarbon receptor-mediated activities of polycyclic aromatic hydrocarbons in a human cell-based reporter gene assay, Environ. Pollut 220 (Pt A) (2017) 307–316. [DOI] [PubMed] [Google Scholar]
- [138].Novotna A, Pavek P, Dvorak Z, Novel stably transfected gene reporter human hepatoma cell line for assessment of aryl hydrocarbon receptor transcriptional activity: construction and characterization, Environ. Sci. Tech 45 (23) (2011) 10133–10139. [DOI] [PubMed] [Google Scholar]
- [139].Burgess JT, Rose M, Boucher D, Plowman J, Molloy C, Fisher M, et al. , The therapeutic potential of DNA damage repair pathways and genomic stability in lung cancer, Front. Oncol 10 (2020) 1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].De Bont R, van Larebeke N, Endogenous DNA damage in humans: a review of quantitative data, Mutagenesis 19 (3) (2004) 169–185. [DOI] [PubMed] [Google Scholar]
- [141].Melis JP, van Steeg H, Luijten M, Oxidative DNA damage and nucleotide excision repair, Antioxid. Redox Signal 18 (18) (2013) 2409–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Nikolaev A, Yang ES, The impact of DNA repair pathways in cancer biology and therapy, Cancers (Basel) 9 (9) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Lindahl T, Instability and decay of the primary structure of DNA, Nature 362 (6422) (1993) 709–715. [DOI] [PubMed] [Google Scholar]
- [144].Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. , Patterns of somatic mutation in human cancer genomes, Nature 446 (7132) (2007) 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Tammemagi MC, McLaughlin JR, Bull SB, Meta-analyses of p53 tumor suppressor gene alterations and clinicopathological features in resected lung cancers, Cancer Epidemiol. Biomarkers Prev 8 (7) (1999) 625–634. [PubMed] [Google Scholar]
- [146].Nahta R, Al-Mulla F, Al-Temaimi R, Amedei A, Andrade-Vieira R, Bay SN, et al. , Mechanisms of environmental chemicals that enable the cancer hallmark of evasion of growth suppression, Carcinogenesis (2015;36 Suppl 1(Suppl 1),) S2–S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].DeMarini DM, Shelton ML, Bell DA, Mutation spectra in Salmonella of complex mixtures: comparison of urban air to benzo[a]pyrene, Environ. Mol. Mutagen 24 (4) (1994) 262–275. [DOI] [PubMed] [Google Scholar]
- [148].Durant JL, Busby WF Jr., Lafleur AL, Penman BW, Crespi CL, Human cell mutagenicity of oxygenated, nitrated and unsubstituted polycyclic aromatic hydrocarbons associated with urban aerosols, Mutat. Res 371 (3–4) (1996) 123–157. [DOI] [PubMed] [Google Scholar]
- [149].Durant JL, Lafleur AL, Busby WF Jr., Donhoffner LL, Penman BW, Crespi CL, Mutagenicity of C24H14 PAH in human cells expressing CYP1A1, Mutat. Res 446 (1) (1999) 1–14. [DOI] [PubMed] [Google Scholar]
- [150].Nisbet IC, LaGoy PK, Toxic equivalency factors (TEFs) for polycyclic aromatic hydrocarbons (PAHs), Regul. Toxicol. Pharm 16 (3) (1992) 290–300. [DOI] [PubMed] [Google Scholar]
- [151].de Oliveira Galvão MF, Sadiktsis I, Marques Pedro T, Dreij K, Determination of whole mixture-based potency factors for cancer risk assessment of complex environmental mixtures by in vitro testing of standard reference materials, Environ. Int 166 (2022), 107345. [DOI] [PubMed] [Google Scholar]
- [152].Chen SC, Liao CM, Health risk assessment on human exposed to environmental polycyclic aromatic hydrocarbons pollution sources, Sci. Total Environ 366 (1) (2006) 112–123. [DOI] [PubMed] [Google Scholar]
- [153].Roy R, Jan R, Gunjal G, Bhor R, Pai K, Satsangi PG, Particulate matter bound polycyclic aromatic hydrocarbons: Toxicity and health risk assessment of exposed inhabitants, Atmos. Environ 210 (2019) 47–57. [Google Scholar]
- [154].Tong HY, Karasek FW, Quantitation of polycyclic aromatic hydrocarbons in diesel exhaust particulate matter by high-performance liquid chromatography fractionation and high-resolution gas chromatography, Anal. Chem 56 (12) (1984) 2129–2134. [DOI] [PubMed] [Google Scholar]
- [155].Nesnow S, Ross JA, Nelson G, Wilson K, Roop BC, Jeffers AJ, et al. , Cyclopenta[cd]pyrene-induced tumorigenicity, Ki-ras codon 12 mutations and DNA adducts in strain A/J mouse lung, Carcinogenesis 15 (4) (1994) 601–606. [DOI] [PubMed] [Google Scholar]
- [156].Vondráček J, Pěnčíková K, Ciganek M, Pivnička J, Karasová M, Hýžd’alová M, et al. , Environmental six-ring polycyclic aromatic hydrocarbons are potent inducers of the AhR-dependent signaling in human cells, Environ. Pollut 266 (Pt 2) (2020), 115125. [DOI] [PubMed] [Google Scholar]
- [157].Park JH, Gelhaus S, Vedantam S, Oliva AL, Batra A, Blair IA, et al. , The pattern of p53 mutations caused by PAH o-quinones is driven by 8-oxo-dGuo formation while the spectrum of mutations is determined by biological selection for dominance, Chem. Res. Toxicol 21 (5) (2008) 1039–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Hussain SP, Amstad P, Raja K, Sawyer M, Hofseth L, Shields PG, et al. , Mutability of p53 hotspot codons to benzo(a)pyrene diol epoxide (BPDE) and the frequency of p53 mutations in nontumorous human lung, Cancer Res. 61 (17) (2001) 6350–6355. [PubMed] [Google Scholar]
- [159].Umannova L, Machala M, Topinka J, Schmuczerová J, Krčmář P, Neča J, et al. , Benzo[a]pyrene and tumor necrosis factor-α coordinately increase genotoxic damage and the production of proinflammatory mediators in alveolar epithelial type II cells, Toxicol. Lett 206 (2) (2011) 121–129. [DOI] [PubMed] [Google Scholar]
- [160].Smerdová L, Šmerdová J, Kabátková M, Kohoutek J, Blažek D, Machala M, et al. , Upregulation of CYP1B1 expression by inflammatory cytokines is mediated by the p38 MAP kinase signal transduction pathway, Carcinogenesis 35 (11) (2014) 2534–2543. [DOI] [PubMed] [Google Scholar]
- [161].Rodin SN, Rodin AS, Human lung cancer and p53: the interplay between mutagenesis and selection, PNAS 97 (22) (2000) 12244–12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Keohavong P, Melacrinos A, Shukla R, In vitro mutational spectrum of cyclopenta[cd]pyrene in the human HPRT gene, Carcinogenesis 16 (4) (1995) 855–860. [DOI] [PubMed] [Google Scholar]
- [163].DeMarini DM, Influence of DNA repair on mutation spectra in Salmonella, Mutat. Res 450 (1–2) (2000) 5–17. [DOI] [PubMed] [Google Scholar]
- [164].Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. , Signatures of mutational processes in human cancer, Nature 500 (7463) (2013) 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. , The repertoire of mutational signatures in human cancer, Nature 578 (7793) (2020) 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Nakagawa H, Fujita M, Whole genome sequencing analysis for cancer genomics and precision medicine, Cancer Sci. 109 (3) (2018) 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Kucab JE, Zou X, Morganella S, Joel M, Nanda AS, Nagy E, et al. A compendium of mutational signatures of environmental agents. Cell. 2019;177 (4):821–36.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Seeberg E, Steinum AL, Nordenskjöld M, Söderhäll S, Jernström B, Strand-break formation in DNA modified by benzo[alpha]pyrene diolepoxide. Quantitative cleavage by Escherichia coli uvrABC endonuclease, Mutat. Res 112 (3) (1983) 139–145. [DOI] [PubMed] [Google Scholar]
- [169].Hockley SL, Arlt VM, Brewer D, Te Poele R, Workman P, Giddings I, et al. , AHR- and DNA-damage-mediated gene expression responses induced by benzo(a) pyrene in human cell lines, Chem. Res. Toxicol 20 (12) (2007) 1797–1810. [DOI] [PubMed] [Google Scholar]
- [170].Clewell RA, Thompson CM, Clewell HJ 3rd., Dose-dependence of chemical carcinogenicity: Biological mechanisms for thresholds and implications for risk assessment, Chem. Biol. Interact 301 (2019) 112–127. [DOI] [PubMed] [Google Scholar]
- [171].Hartwig A, Arand M, Epe B, Guth S, Jahnke G, Lampen A, et al. , Mode of action-based risk assessment of genotoxic carcinogens, Arch. Toxicol 94 (6) (2020) 1787–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Holme JA, Trygg B, Søderlund E, Species differences in the metabolism of 2-acetylaminofluorene by hepatocytes in primary monolayer culture, Cancer Res. 46 (4 Pt 1) (1986) 1627–1632. [PubMed] [Google Scholar]
- [173].Rannug U, Holme JA, Hongslo JK, Srám R, International commission for protection against environmental mutagens and carcinogens. an evaluation of the genetic toxicity of paracetamol, Mutat. Res 327 (1–2) (1995) 179–200. [DOI] [PubMed] [Google Scholar]
- [174].Barnes JL, Zubair M, John K, Poirier MC, Martin FL, Carcinogens and DNA damage, Biochem. Soc. Trans 46 (5) (2018) 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Lu H, Yang M, Zhou Q, Reprogramming transcription after DNA damage: recognition, response, repair, and restart, Trends Cell Biol. (2022). [DOI] [PubMed] [Google Scholar]
- [176].Øvrevik J, Refsnes M, Lag M, Brinchmann BC, Schwarze PE, Holme JA, Triggering mechanisms and inflammatory effects of combustion exhaust particles with implication for carcinogenesis, Basic Clin. Paharmacol. Toxicol 121 (Suppl 3) (2017) 55–62. [DOI] [PubMed] [Google Scholar]
- [177].Solhaug A, Refsnes M, Låg M, Schwarze PE, Husøy T, Holme JA, Polycyclic aromatic hydrocarbons induce both apoptotic and anti-apoptotic signals in Hepa1c1c7 cells, Carcinogenesis 25 (5) (2004) 809–819. [DOI] [PubMed] [Google Scholar]
- [178].Landvik NE, Gorria M, Arlt VM, Asare N, Solhaug A, Lagadic-Gossmann D, et al. , Effects of nitrated-polycyclic aromatic hydrocarbons and diesel exhaust particle extracts on cell signalling related to apoptosis: possible implications for their mutagenic and carcinogenic effects, Toxicology 231 (2–3) (2007) 159–174. [DOI] [PubMed] [Google Scholar]
- [179].Liamin M, Boutet-Robinet E, Jamin EL, Fernier M, Khoury L, Kopp B, et al. , Benzo[a]pyrene-induced DNA damage associated with mutagenesis in primary human activated T lymphocytes, Biochem. Pharmacol 137 (2017) 113–124. [DOI] [PubMed] [Google Scholar]
- [180].Zhou W, Tian D, He J, Wang Y, Zhang L, Cui L, et al. , Repeated PM2.5 exposure inhibits BEAS-2B cell P53 expression through ROS-Akt-DNMT3B pathway-mediated promoter hypermethylation, Oncotarget 7 (15) (2016) 20691–20703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Khan QA, Vousden KH, Dipple A, Cellular response to DNA damage from a potent carcinogen involves stabilization of p53 without induction of p21(waf1/cip1), Carcinogenesis 18 (12) (1997) 2313–2318. [DOI] [PubMed] [Google Scholar]
- [182].Dipple A, Khan QA, Page JE, Pontén I, Szeliga J, DNA reactions, mutagenic action and stealth properties of polycyclic aromatic hydrocarbon carcinogens (review), Int. J. Oncol 14 (1) (1999) 103–111. [DOI] [PubMed] [Google Scholar]
- [183].Malmlöf M, Pääjarvi G, Högberg J, Stenius U, Mdm2 as a sensitive and mechanistically informative marker for genotoxicity induced by benzo[a]pyrene and dibenzo[a, l]pyrene, Toxicol. Sci 102 (2) (2008) 232–240. [DOI] [PubMed] [Google Scholar]
- [184].Marlowe JL, Fan Y, Chang X, Peng L, Knudsen ES, Xia Y, et al. , The aryl hydrocarbon receptor binds to E2F1 and inhibits E2F1-induced apoptosis, Mol. Biol. Cell 19 (8) (2008) 3263–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Barouki R, Coumoul X, Fernandez-Salguero PM, The aryl hydrocarbon receptor, more than a xenobiotic-interacting protein, FEBS Lett. 581 (19) (2007) 3608–3615. [DOI] [PubMed] [Google Scholar]
- [186].Tannheimer SL, Barton SL, Ethier SP, Burchiel SW, Carcinogenic polycyclic aromatic hydrocarbons increase intracellular Ca2+ and cell proliferation in primary human mammary epithelial cells, Carcinogenesis 18 (6) (1997) 1177–1182. [DOI] [PubMed] [Google Scholar]
- [187].Tannheimer SL, Ethier SP, Caldwell KK, Burchiel SW, Benzo[a]pyrene- and TCDD-induced alterations in tyrosine phosphorylation and insulin-like growth factor signaling pathways in the MCF-10A human mammary epithelial cell line, Carcinogenesis 19 (7) (1998) 1291–1297. [DOI] [PubMed] [Google Scholar]
- [188].Plísková M, Vondrácek J, Vojtesek B, Kozubík A, Machala M, Deregulation of cell proliferation by polycyclic aromatic hydrocarbons in human breast carcinoma MCF-7 cells reflects both genotoxic and nongenotoxic events, Toxicol. Sci 83 (2) (2005) 246–256. [DOI] [PubMed] [Google Scholar]
- [189].Andrysík Z, Vondrácek J, Machala M, Krcmár P, Svihálková-Sindlerová L, Kranz A, et al. , The aryl hydrocarbon receptor-dependent deregulation of cell cycle control induced by polycyclic aromatic hydrocarbons in rat liver epithelial cells, Mutat. Res 615 (1–2) (2007) 87–97. [DOI] [PubMed] [Google Scholar]
- [190].Matsumura F, The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin-activated Ah receptor to cause toxic effects, Biochem. Pharmacol 77 (4) (2009) 608–626. [DOI] [PubMed] [Google Scholar]
- [191].Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B, Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor, Toxicol. Sci 124 (1) (2011) 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Sondermann NC, Faßbender S, Hartung F, Hätälä AM, Rolfes KM, Vogel CFA, et al. , Functions of the aryl hydrocarbon receptor (AHR) beyond the canonical AHR/ARNT signaling pathway, Biochem. Pharmacol 208 (2023), 115371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Guyot E, Chevallier A, Barouki R, Coumoul X, The AhR twist: ligand-dependent AhR signaling and pharmaco-toxicological implications, Drug Discov. Today 18 (9–10) (2013) 479–486. [DOI] [PubMed] [Google Scholar]
- [194].Taniguchi K, Karin M, NF-κB, inflammation, immunity and cancer: coming of age, Nat. Rev. Immunol 18 (5) (2018) 309–324. [DOI] [PubMed] [Google Scholar]
- [195].Tian Y, Rabson AB, Gallo MA, Ah receptor and NF-kappaB interactions: mechanisms and physiological implications, Chem. Biol. Interact 141 (1–2) (2002) 97–115. [DOI] [PubMed] [Google Scholar]
- [196].Vogel CF, Sciullo E, Li W, Wong P, Lazennec G, Matsumura F, RelB, a new partner of aryl hydrocarbon receptor-mediated transcription, Mol. Endocrinol 21 (12) (2007) 2941–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Vogel CF, Matsumura F, A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-kappaB family, Biochem. Pharmacol 77 (4) (2009) 734–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Chen PH, Chang H, Chang JT, Lin P, Aryl hydrocarbon receptor in association with RelA modulates IL-6 expression in non-smoking lung cancer, Oncogene 31 (20) (2012) 2555–2565. [DOI] [PubMed] [Google Scholar]
- [199].Kobayashi S, Okamoto H, Iwamoto T, Toyama Y, Tomatsu T, Yamanaka H, et al. , A role for the aryl hydrocarbon receptor and the dioxin TCDD in rheumatoid arthritis, Rheumatology (Oxford) 47 (9) (2008) 1317–1322. [DOI] [PubMed] [Google Scholar]
- [200].Podechard N, Lecureur V, Le Ferrec E, Guenon I, Sparfel L, Gilot D, et al. , Interleukin-8 induction by the environmental contaminant benzo(a)pyrene is aryl hydrocarbon receptor-dependent and leads to lung inflammation, Toxicol. Lett 177 (2) (2008) 130–137. [DOI] [PubMed] [Google Scholar]
- [201].Kim DW, Gazourian L, Quadri SA, Romieu-Mourez R, Sherr DH, Sonenshein GE, The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells, Oncogene 19 (48) (2000) 5498–5506. [DOI] [PubMed] [Google Scholar]
- [202].Thatcher TH, Maggirwar SB, Baglole CJ, Lakatos HF, Gasiewicz TA, Phipps RP, et al. , Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB, Am. J. Pathol 170 (3) (2007) 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Beamer CA, Seaver BP, Shepherd DM, Aryl hydrocarbon receptor (AhR) regulates silica-induced inflammation but not fibrosis, Toxicol. Sci 126 (2) (2012) 554–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Vázquez-Gómez G, Karasová M, Tylichová Z, Kabátková M, Hampl A, Matthews J, et al. , Aryl hydrocarbon receptor (AhR) limits the inflammatory responses in human lung adenocarcinoma A549 cells via interference with NF-κB signaling, Cells. 11 (4) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Øvrevik J, Låg M, Lecureur V, Gilot D, Lagadic-Gossmann D, Refsnes M, et al. , AhR and Arnt differentially regulate NF-κB signaling and chemokine responses in human bronchial epithelial cells, Cell Commun. Signal 12 (2014) 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Oesterling E, Toborek M, Hennig B, Benzo[a]pyrene induces intercellular adhesion molecule-1 through a caveolae and aryl hydrocarbon receptor mediated pathway, Toxicol. Appl. Pharmacol 232 (2) (2008) 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Rey-Barroso J, Alvarez-Barrientos A, Rico-Leo E, Contador-Troca M, Carvajal-Gonzalez JM, Echarri A, et al. , The Dioxin receptor modulates Caveolin-1 mobilization during directional migration: role of cholesterol, Cell Commun. Signal 12 (2014) 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Plant AL, Knapp RD, Smith LC, Mechanism and rate of permeation of cells by polycyclic aromatic hydrocarbons, J. Biol. Chem 262 (6) (1987) 2514–2519. [PubMed] [Google Scholar]
- [209].Barhoumi R, Mouneimne Y, Ramos KS, Safe SH, Phillips TD, Centonze VE, et al. , Analysis of benzo[a]pyrene partitioning and cellular homeostasis in a rat liver cell line, Toxicol. Sci 53 (2) (2000) 264–270. [DOI] [PubMed] [Google Scholar]
- [210].Abumrad NA, Cabodevilla AG, Samovski D, Pietka T, Basu D, Goldberg IJ, Endothelial cell receptors in tissue lipid uptake and metabolism, Circ. Res 128 (3) (2021) 433–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Luo S, Yang M, Zhao H, Han Y, Jiang N, Yang J, et al. , Caveolin-1 regulates cellular metabolism: A potential therapeutic target in kidney disease, Front. Pharmacol 12 (2021), 768100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Penn A, Murphy G, Barker S, Henk W, Penn L, Combustion-derived ultrafine particles transport organic toxicants to target respiratory cells, Environ. Health Perspect 113 (8) (2005) 956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Brinchmann BC, Le Ferrec E, Podechard N, Lagadic-Gossmann D, Shoji KF, Penna A, et al. , Lipophilic chemicals from diesel exhaust particles trigger calcium response in human endothelial cells via aryl hydrocarbon receptor non-genomic signalling, Int. J. Mol. Sci 19 (5) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Brinchmann BC, Le Ferrec E, Bisson WH, Podechard N, Huitfeldt HS, Gallais I, et al. , Evidence of selective activation of aryl hydrocarbon receptor nongenomic calcium signaling by pyrene, Biochem. Pharmacol 158 (2018) 1–12. [DOI] [PubMed] [Google Scholar]
- [215].Robinson RK, Birrell MA, Adcock JJ, Wortley MA, Dubuis ED, Chen S, et al. Mechanistic link between diesel exhaust particles and respiratory reflexes. J Allergy Clin Immunol. 2018;141(3):1074–84.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Tran MT, Overview of Ca(2+) signaling in lung cancer progression and metastatic lung cancer with bone metastasis, Explor Target Antitumor Ther. 2 (3) (2021) 249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Zhong T, Zhang W, Guo H, Pan X, Chen X, He Q, et al. , The regulatory and modulatory roles of TRP family channels in malignant tumors and relevant therapeutic strategies, Acta Pharm. Sin. B 12 (4) (2022) 1761–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Coelho M, Soares-Silva C, Brandão D, Marino F, Cosentino M, Ribeiro L, β-Adrenergic modulation of cancer cell proliferation: available evidence and clinical perspectives, J. Cancer Res. Clin. Oncol 143 (2) (2017) 275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Nilsson MB, Le X, Heymach JV , β-adrenergic signaling in lung cancer: A potential role for beta-blockers, J. Neuroimmune Pharmacol 15 (1) (2020) 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Schuller HM, Tithof PK, Williams M, Plummer H 3rd., The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid, Cancer Res. 59 (18) (1999) 4510–4515. [PubMed] [Google Scholar]
- [221].Askari MDF, Tsao M-S, Schuller HM, The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through β-adrenergic transactivation of EGF receptors, J. Cancer Res. Clin. Oncol 131 (10) (2005) 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Mayati A, Levoin N, Paris H, N’Diaye M, Courtois A, Uriac P, et al. , Induction of intracellular calcium concentration by environmental benzo(a)pyrene involves a β2-adrenergic receptor/adenylyl cyclase/Epac-1/inositol 1,4,5-trisphosphate pathway in endothelial cells, J. Biol. Chem 287 (6) (2012) 4041–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Rosell R, Cardona AF, Arrieta O, Aguilar A, Ito M, Pedraz C, et al. , Coregulation of pathways in lung cancer patients with EGFR mutation: therapeutic opportunities, Br. J. Cancer 125 (12) (2021) 1602–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Lei Z, Yang W, Zuo Y, Beta-blocker and survival in patients with lung cancer: A meta-analysis, PLoS One 16 (2) (2021) e0245773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Madhukar BV, Brewster DW, Matsumura F, Effects of in vivo-administered 2,3,7,8-tetrachlorodibenzo-p-dioxin on receptor binding of epidermal growth factor in the hepatic plasma membrane of rat, guinea pig, mouse, and hamster, PNAS 81 (23) (1984) 7407–7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Kohle C, Gschaidmeier H, Lauth D, Topell S, Zitzer H, Bock KW, 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-mediated membrane translocation of c-Src protein kinase in liver WB-F344 cells, Arch. Toxicol 73 (3) (1999) 152–158. [DOI] [PubMed] [Google Scholar]
- [227].Cheon H, Woo YS, Lee JY, Kim HS, Kim HJ, Cho S, et al. , Signaling pathway for 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced TNF-alpha production in differentiated THP-1 human macrophages, Exp. Mol. Med 39 (4) (2007) 524–534. [DOI] [PubMed] [Google Scholar]
- [228].Øvrevik J, Refsnes M, Totlandsdal AI, Holme JA, Schwarze PE, Låg M, TACE/TGF-α/EGFR regulates CXCL8 in bronchial epithelial cells exposed to particulate matter components, Eur. Respir. J 38 (5) (2011) 1189–1199. [DOI] [PubMed] [Google Scholar]
- [229].Vogel C, Boerboom AM, Baechle C, El-Bahay C, Kahl R, Degen GH, et al. , Regulation of prostaglandin endoperoxide H synthase-2 induction by dioxin in rat hepatocytes: possible c-Src-mediated pathway, Carcinogenesis 21 (12) (2000) 2267–2274. [DOI] [PubMed] [Google Scholar]
- [230].Vogel CF, Li W, Sciullo E, Newman J, Hammock B, Reader JR, et al. , Pathogenesis of aryl hydrocarbon receptor-mediated development of lymphoma is associated with increased cyclooxygenase-2 expression, Am. J. Pathol 171 (5) (2007) 1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Sandler AB, Dubinett SM, COX-2 inhibition and lung cancer, Semin. Oncol 31 (2 Suppl 7) (2004) 45–52. [DOI] [PubMed] [Google Scholar]
- [232].Wölfle D, Marotzki S, Dartsch D, Sc W ¨ hafer, H. Marquardt, Induction of cyclooxygenase expression and enhancement of malignant cell transformation by 2,3,7,8-tetrachlorodibenzo-p-dioxin, Carcinogenesis 21 (1) (2000) 15–21. [DOI] [PubMed] [Google Scholar]
- [233].Vogeley C, Sondermann NC, Woeste S, Momin AA, Gilardino V, Hartung F, et al. , Unraveling the differential impact of PAHs and dioxin-like compounds on AKR1C3 reveals the EGFR extracellular domain as a critical determinant of the AHR response, Environ. Int 158 (2022), 106989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Xie G, Peng Z, Raufman JP, Src-mediated aryl hydrocarbon and epidermal growth factor receptor cross talk stimulates colon cancer cell proliferation, Am. J. Physiol. Gastrointest. Liver Physiol 302 (9) (2012) G1006–G1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Cui Y, Zhu T, Song X, Liu J, Liu S, Zhao R, Downregulation of caveolin-1 increased EGFR-TKIs sensitivity in lung adenocarcinoma cell line with EGFR mutation, Biochem. Biophys. Res. Commun 495 (1) (2018) 733–739. [DOI] [PubMed] [Google Scholar]
- [236].Lim EJ, Májková Z, Xu S, Bachas L, Arzuaga X, Smart E, et al. , Coplanar polychlorinated biphenyl-induced CYP1A1 is regulated through caveolae signaling in vascular endothelial cells, Chem. Biol. Interact 176 (2–3) (2008) 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Shi YB, Li J, Lai XN, Jiang R, Zhao RC, Xiong LX, Multifaceted roles of caveolin-1 in lung cancer: A new investigation focused on tumor occurrence, development and therapy, Cancers (Basel) 12 (2) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Thiebaut C, Vlaeminck-Guillem V, Tredan O, Poulard C, Le Romancer M, Non-genomic signaling of steroid receptors in cancer, Mol. Cell. Endocrinol 538 (2021), 111453. [DOI] [PubMed] [Google Scholar]
- [239].Mauvais-Jarvis F, Lange CA, Levin ER, Membrane-initiated estrogen, androgen, and progesterone receptor signaling in health and disease, Endocr. Rev 43 (4) (2022) 720–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Mazières J, Rouquette I, Lepage B, Milia J, Brouchet L, Guibert N, et al. , Specificities of lung adenocarcinoma in women who have never smoked, J. Thorac. Oncol 8 (7) (2013) 923–929. [DOI] [PubMed] [Google Scholar]
- [241].Tappenden DM, Lynn SG, Crawford RB, Lee K, Vengellur A, Kaminski NE, et al. , The aryl hydrocarbon receptor interacts with ATP5alpha1, a subunit of the ATP synthase complex, and modulates mitochondrial function, Toxicol. Appl. Pharmacol 254 (3) (2011) 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Hwang HJ, Dornbos P, Steidemann M, Dunivin TK, Rizzo M, LaPres JJ, Mitochondrial-targeted aryl hydrocarbon receptor and the impact of 2,3,7,8-rachlorodibenzo-p-dioxin on cellular respiration and the mitochondrial proteome, Toxicol. Appl. Pharmacol 304 (2016) 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Hardonniere K, Saunier E, Lemarie A, Fernier M, Gallais I, Helies-Toussaint C, et al. , The environmental carcinogen benzo[a]pyrene induces a Warburg-like metabolic reprogramming dependent on NHE1 and associated with cell survival, Sci. Rep 6 (2016) 30776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Hardonniere K, Huc L, Sergent O, Holme JA, Lagadic-Gossmann D, Environmental carcinogenesis and pH homeostasis: Not only a matter of dysregulated metabolism, Semin. Cancer Biol 43 (2017) 49–65. [DOI] [PubMed] [Google Scholar]
- [245].Li X, Liu M, Liu H, Chen J, Tumor metabolic reprogramming in lung cancer progression, Oncol. Lett 24 (2) (2022) 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Vanhove K, Graulus GJ, Mesotten L, Thomeer M, Derveaux E, Noben JP, et al. , The metabolic landscape of lung cancer: New insights in a disturbed glucose metabolism, Front. Oncol 9 (2019) 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Chen Q, Wang Y, Yang L, Sun L, Wen Y, Huang Y, et al. , PM2.5 promotes NSCLC carcinogenesis through translationally and transcriptionally activating DLAT-mediated glycolysis reprograming, J. Exp. Clin. Cancer Res 41(1):229 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Pierdominici MM, Diesel exhaust particle exposure in vitro impacts T lymphocyte phenotype and function, Part. Fibre Toxicol 11 (74) (2014) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Ferecatu IB, Polycyclic aromatic hydrocarbon components contribute to the mitochondria-antiapoptotic effect of fine particulate matter on human bronchial epithelial cells via the aryl hydrocarbon receptor, Part. Fibre Toxicol 7 (18) (2010) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Andersen AP, Moreira JM, Pedersen SF, Interactions of ion transporters and channels with cancer cell metabolism and the tumour microenvironment, Philos. Trans. R. Soc. Lond. B Biol. Sci 369 (1638) (2014) 20130098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Lagadic-Gossmann D, Hardonniere K, Mograbi B, Sergent O, Huc L, Disturbances in H(+) dynamics during environmental carcinogenesis, Biochimie 163 (2019) 171–183. [DOI] [PubMed] [Google Scholar]
- [252].Cottle WT, Wallert CH, Anderson KK, Tran MF, Bakker CL, Wallert MA, et al. Calcineurin homologous protein isoform 2 supports tumor survival via the sodium hydrogen exchanger isoform 1 in non-small cell lung cancer. Tumour Biol. 2020;42(7):1010428320937863. [DOI] [PubMed] [Google Scholar]
- [253].Huc L, Sparfel L, Rissel M, Dimanche-Boitrel M-T, Guillouzo A, Fardel O, et al. , Identification of Na+/H+ exchange as a new target for toxic polycyclic aromatic hydrocarbons in liver cells, FASEB J. 18 (2) (2004) 1–26. [DOI] [PubMed] [Google Scholar]
- [254].Sánchez-Cenizo L, Formentini L, Aldea M, Ortega AD, García-Huerta P, Sánchez-Aragó M, et al. , Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype, J. Biol. Chem 285 (33) (2010) 25308–25313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Paul D, Sinha AN, Ray A, Lal M, Nayak S, Sharma A, et al. , A-to-I editing in human miRNAs is enriched in seed sequence, influenced by sequence contexts and significantly hypoedited in glioblastoma multiforme, Sci. Rep 7 (1) (2017) 2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Hardonnière K, Lagadic-Gossmann D, ATPase inhibitory factor 1 (IF1): a novel player in pollutant-related diseases? Current Opinion in Toxicology. 8 (2018) 42–47. [Google Scholar]
- [257].Han X, Liu H, Zhang Z, Yang W, Wu C, Liu X, et al. , Epitranscriptomic 5-methylcytosine profile in PM(2.5)-induced mouse pulmonary fibrosis, Genomics Proteomics Bioinformatics 18 (1) (2020) 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Mesnil M, Crespin S, Avanzo JL, Zaidan-Dagli ML, Defective gap junctional intercellular communication in the carcinogenic process, BBA 1719 (1–2) (2005) 125–145. [DOI] [PubMed] [Google Scholar]
- [259].Ruch RJ, Porter S, Koffler LD, Dwyer-Nield LD, Malkinson AM, Defective gap junctional intercellular communication in lung cancer: loss of an important mediator of tissue homeostasis and phenotypic regulation, Exp. Lung Res 27 (3) (2001) 231–243. [DOI] [PubMed] [Google Scholar]
- [260].Avanzo JL, Mesnil M, Hernandez-Blazquez FJ, Mackowiak II, Mori CM, da Silva TC, et al. , Increased susceptibility to urethane-induced lung tumors in mice with decreased expression of connexin43, Carcinogenesis 25 (10) (2004) 1973–1982. [DOI] [PubMed] [Google Scholar]
- [261].King TJ, Lampe PD, The gap junction protein connexin32 is a mouse lung tumor suppressor, Cancer Res. 64 (20) (2004) 7191–7196. [DOI] [PubMed] [Google Scholar]
- [262].Zhou M, Zheng M, Zhou X, Tian S, Yang X, Ning Y, et al. , The roles of connexins and gap junctions in the progression of cancer, Cell Commun. Signal 21 (1) (2023) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Ruch RJ, Klaunig JE, Effects of tumor promoters, genotoxic carcinogens and hepatocytotoxins on mouse hepatocyte intercellular communication, Cell Biol. Toxicol 2 (4) (1986) 469–483. [DOI] [PubMed] [Google Scholar]
- [264].Yamasaki H, Role of disrupted gap junctional intercellular communication in detection and characterization of carcinogens, Mutat. Res 365 (1–3) (1996) 91–105. [DOI] [PubMed] [Google Scholar]
- [265].Rosenkranz HS, Pollack N, Cunningham AR, Exploring the relationship between the inhibition of gap junctional intercellular communication and other biological phenomena, Carcinogenesis 21 (5) (2000) 1007–1011. [DOI] [PubMed] [Google Scholar]
- [266].Bláha L, Kapplová P, Vondrácek J, Upham B, Machala M, Inhibition of gap-junctional intercellular communication by environmentally occurring polycyclic aromatic hydrocarbons, Toxicol. Sci 65 (1) (2002) 43–51. [DOI] [PubMed] [Google Scholar]
- [267].Upham BL, Bláha L, Babica P, Park JS, Sovadinova I, Pudrith C, et al. , Tumor promoting properties of a cigarette smoke prevalent polycyclic aromatic hydrocarbon as indicated by the inhibition of gap junctional intercellular communication via phosphatidylcholine-specific phospholipase C, Cancer Sci. 99 (4) (2008) 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Vondrácek J, Svihálková-Sindlerová L, Pencíková K, Marvanová S, Krcmár P, Ciganek M, et al. , Concentrations of methylated naphthalenes, anthracenes, and phenanthrenes occurring in Czech river sediments and their effects on toxic events associated with carcinogenesis in rat liver cell lines, Environ. Toxicol. Chem 26 (11) (2007) 2308–2316. [DOI] [PubMed] [Google Scholar]
- [269].Rivedal E, Myhre O, Sanner T, Eide I, Supplemental role of the Ames mutation assay and gap junction intercellular communication in studies of possible carcinogenic compounds from diesel exhaust particles, Arch. Toxicol 77 (9) (2003) 533–542. [DOI] [PubMed] [Google Scholar]
- [270].Rutten AA, Jongen WM, de Haan LH, Hendriksen EG, Koeman JH, Effect of retinol and cigarette-smoke condensate on dye-coupled intercellular communication between hamster tracheal epithelial cells, Carcinogenesis 9 (2) (1988) 315–320. [DOI] [PubMed] [Google Scholar]
- [271].Roemer E, Lammerich HP, Conroy LL, Weisensee D, Characterization of a gap-junctional intercellular communication (GJIC) assay using cigarette smoke, Toxicol. Lett 219 (3) (2013) 248–253. [DOI] [PubMed] [Google Scholar]
- [272].Koval M, Sharing signals: connecting lung epithelial cells with gap junction channels, Am. J. Physiol. Lung Cell. Mol. Physiol 283 (5) (2002) L875–L893. [DOI] [PubMed] [Google Scholar]
- [273].Osgood RS, Upham BL, Hill T 3rd, Helms KL, Velmurugan K, Babica P, et al. , Polycyclic aromatic hydrocarbon-induced signaling events relevant to inflammation and tumorigenesis in lung cells are dependent on molecular structure, PLoS One 8 (6) (2014) e65150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Bauer AK, Velmurugan K, Plottner S, Siegrist KJ, Romo D, Welge P, et al. ,¨ Environmentally prevalent polycyclic aromatic hydrocarbons can elicit co-carcinogenic properties in an in vitro murine lung epithelial cell model, Arch. Toxicol 92 (3) (2018) 1311–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Brózman O, Novák J, Bauer AK, Babica P, Airborne PAHs inhibit gap junctional intercellular communication and activate MAPKs in human bronchial epithelial cell line, Environ. Toxicol. Pharmacol 79 (2020), 103422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Kabátková M, Svobodová J, Pěnčíková K, Mohatad DS, Šmerdová L, Kozubík A, et al. , Interactive effects of inflammatory cytokine and abundant low-molecular-weight PAHs on inhibition of gap junctional intercellular communication, disruption of cell proliferation control, and the AhR-dependent transcription, Toxicol. Lett 232 (1) (2015) 113–121. [DOI] [PubMed] [Google Scholar]
- [277].Romo D, Velmurugan K, Upham BL, Dwyer-Nield LD, Bauer AK, Dysregulation of gap junction function and cytokine production in response to non-genotoxic polycyclic aromatic hydrocarbons in an in vitro lung cell model, Cancers (Basel) 11 (4) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Dietrich C, Kaina B, The aryl hydrocarbon receptor (AhR) in the regulation of cell-cell contact and tumor growth, Carcinogenesis 31 (8) (2010) 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Kung T, Murphy KA, White LA, The aryl hydrocarbon receptor (AhR) pathway as a regulatory pathway for cell adhesion and matrix metabolism, Biochem. Pharmacol 77 (4) (2009) 536–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Shimba S, Komiyama K, Moro I, Tezuka M, Overexpression of the aryl hydrocarbon receptor (AhR) accelerates the cell proliferation of A549 cells, J. Biochem 132 (5) (2002) 795–802. [DOI] [PubMed] [Google Scholar]
- [281].Hýžďalová M, Procházková J, Strapáčová S, Svržková L, Vacek O, Fedr R, et al. , A prolonged exposure of human lung carcinoma epithelial cells to benzo[a] pyrene induces p21-dependent epithelial-to-mesenchymal transition (EMT)-like phenotype, Chemosphere 263 (2021), 128126. [DOI] [PubMed] [Google Scholar]
- [282].Misaki K, Takamura-Enya T, Ogawa H, Takamori K, Yanagida M, Tumour-promoting activity of polycyclic aromatic hydrocarbons and their oxygenated or nitrated derivatives, Mutagenesis 31 (2) (2016) 205–213. [DOI] [PubMed] [Google Scholar]
- [283].Chramostová K, Vondrácek J, Sindlerová L, Vojtesek B, Kozubík A, Machala M, Polycyclic aromatic hydrocarbons modulate cell proliferation in rat hepatic epithelial stem-like WB-F344 cells, Toxicol. Appl. Pharmacol 196 (1) (2004) 136–148. [DOI] [PubMed] [Google Scholar]
- [284].Andrysík Z, Procházková J, Kabátková M, Umannová L, Simečková P, Kohoutek J, et al. , Aryl hydrocarbon receptor-mediated disruption of contact inhibition is associated with connexin43 downregulation and inhibition of gap junctional intercellular communication, Arch. Toxicol 87 (3) (2013) 491–503. [DOI] [PubMed] [Google Scholar]
- [285].Procházková J, Kabátková M, Bryja V, Umannová L, Bernatík O, Kozubík A, et al. , The interplay of the aryl hydrocarbon receptor and β-catenin alters both AhR-dependent transcription and Wnt/β-catenin signaling in liver progenitors, Toxicol. Sci 122 (2) (2011) 349–360. [DOI] [PubMed] [Google Scholar]
- [286].Svobodová J, Procházková J, Kabátková M, Krkoška M, Šmerdová L, Líbalová H, et al. , 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) disrupts control of cell proliferation and apoptosis in a human model of adult liver progenitors, Toxicol. Sci 172 (2) (2019) 368–384. [DOI] [PubMed] [Google Scholar]
- [287].Brune K, Frank J, Schwingshackl A, Finigan J, Sidhaye VK, Pulmonary epithelial barrier function: some new players and mechanisms, Am. J. Physiol. Lung Cell. Mol. Physiol 308 (8) (2015) L731–L745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Wittekindt OH, Tight junctions in pulmonary epithelia during lung inflammation, Pflugers Arch. 469 (1) (2017) 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Kyuno D, Takasawa A, Kikuchi S, Takemasa I, Osanai M, Kojima T, Role of tight junctions in the epithelial-to-mesenchymal transition of cancer cells, Biochim. Biophys. Acta Biomembr 1863 (3) (2021), 183503. [DOI] [PubMed] [Google Scholar]
- [290].Liu J, Chen X, Dou M, He H, Ju M, Ji S, et al. , Particulate matter disrupts airway epithelial barrier via oxidative stress to promote Pseudomonas aeruginosa infection, J. Thorac. Dis 11 (6) (2019) 2617–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Kim SS, Kim CH, Kim JW, Kung HC, Park TW, Shin YS, et al. , Airborne particulate matter increases MUC5AC expression by downregulating Claudin-1 expression in human airway cells, BMB Rep 50 (10) (2017) 516–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [292].Smyth T, Veazey J, Eliseeva S, Chalupa D, Elder A, Georas SN, Diesel exhaust particle exposure reduces expression of the epithelial tight junction protein Tricellulin, Part. Fibre Toxicol 17 (1) (2020) 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Zeglinski MR, Turner CT, Zeng R, Schwartz C, Santacruz S, Pawluk MA, et al. , Soluble wood smoke extract promotes barrier dysfunction in alveolar epithelial cells through a MAPK signaling pathway, Sci. Rep 9 (1) (2019) 10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Ho DH, Burggren WW, Blood-brain barrier function, cell viability, and gene expression of tight junction-associated proteins in the mouse are disrupted by crude oil, benzo[a]pyrene, and the dispersant COREXIT, Comp. Biochem. Physiol. C: Toxicol. Pharmacol 223 (2019) 96–105. [DOI] [PubMed] [Google Scholar]
- [295].Tomkiewicz C, Herry L, Bui LC, Métayer C, Bourdeloux M, Barouki R, et al. , The aryl hydrocarbon receptor regulates focal adhesion sites through a non-genomic FAK/Src pathway, Oncogene 32 (14) (2013) 1811–1820. [DOI] [PubMed] [Google Scholar]
- [296].Wee P, Wang Z, Epidermal growth factor receptor cell proliferation signaling pathways, Cancers (Basel) 9 (5) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Lin P, Chang H, Tsai WT, Wu MH, Liao YS, Chen JT, et al. , Overexpression of aryl hydrocarbon receptor in human lung carcinomas, Toxicol. Pathol 31 (1) (2003) 22–30. [DOI] [PubMed] [Google Scholar]
- [298].Gouasmi R, Ferraro-Peyret C, Nancey S, Coste I, Renno T, Chaveroux C, et al. , The kynurenine pathway and cancer: Why keep It simple when you can make it complicated, Cancers (Basel) 14 (11) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, et al. Tumor-repopulating cells induce PD-1 expression in CD8(+) T cells by transferring kynurenine and AhR activation. Cancer Cell. 2018;33(3):480–94.e7. [DOI] [PubMed] [Google Scholar]
- [300].Han Y, Liu D, Li L, PD-1/PD-L1 pathway: current researches in cancer, Am. J. Cancer Res 10 (3) (2020) 727–742. [PMC free article] [PubMed] [Google Scholar]
- [301].Wang GZ, Zhang L, Zhao XC, Gao SH, Qu LW, Yu H, et al. , The Aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy, Nat. Commun 10 (1) (2019) 1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Wang TH, Huang KY, Chen CC, Chang YH, Chen HY, Hsueh C, et al. , PM2.5 promotes lung cancer progression through activation of the AhR-TMPRSS2-IL18 pathway, EMBO Mol. Med 15 (6) (2023) e17014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [303].Nothdurft S, Thumser-Henner C, Breitenbücher F, Okimoto RA, Dorsch M, Opitz CA, et al. , Functional screening identifies aryl hydrocarbon receptor as suppressor of lung cancer metastasis, Oncogenesis. 9 (11) (2020) 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [304].Sartor MA, Schnekenburger M, Marlowe JL, Reichard JF, Wang Y, Fan Y, et al. , Genomewide analysis of aryl hydrocarbon receptor binding targets reveals an extensive array of gene clusters that control morphogenetic and developmental programs, Environ. Health Perspect 117 (7) (2009) 1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].van Niel G, D’Angelo G, Raposo G, Shedding light on the cell biology of extracellular vesicles, Nat. Rev. Mol. Cell Biol 19 (4) (2018) 213–228. [DOI] [PubMed] [Google Scholar]
- [306].Urabe F, Kosaka N, Ito K, Kimura T, Egawa S, Ochiya T, Extracellular vesicles as biomarkers and therapeutic targets for cancer, Am. J. Phys. Cell Phys 318 (1) (2020) C29–C39. [DOI] [PubMed] [Google Scholar]
- [307].Al Amir Dache Z, Otandault A, Tanos R, Pastor B, Meddeb R, Sanchez C, et al. , Blood contains circulating cell-free respiratory competent mitochondria, FASEB J. 34 (3) (2020) 3616–3630. [DOI] [PubMed] [Google Scholar]
- [308].Mohan A, Agarwal S, Clauss M, Britt NS, Dhillon NK, Extracellular vesicles: novel communicators in lung diseases, Respir. Res 21 (1) (2020) 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Mahida RY, Matsumoto S, Matthay MA, Extracellular vesicles in ARDS: New insights into pathogenesis with novel clinical applications, in: Vincent J--L (Ed.), Annual Update in Intensive Care and Emergency Medicine 2020, Springer International Publishing, Cham, 2020, pp. 53–65. [Google Scholar]
- [310].Soni S, Wilson MR, O’Dea KP, Yoshida M, Katbeh U, Woods SJ, et al. , Alveolar macrophage-derived microvesicles mediate acute lung injury, Thorax 71 (11) (2016) 1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Lee H, Zhang D, Zhu Z, Dela Cruz CS, Jin Y, Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs, Sci. Rep 6 (1) (2016) 35250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Liu Y, Gu Y, Han Y, Zhang Q, Jiang Z, Zhang X, et al. , Tumor exosomal RNAs promote lung pre-metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils, Cancer Cell 30 (2) (2016) 243–256. [DOI] [PubMed] [Google Scholar]
- [313].de Miguel-Perez D, Russo A, Arrieta O, Ak M, Barron F, Gunasekaran M, et al. , Extracellular vesicle PD-L1 dynamics predict durable response to immune-checkpoint inhibitors and survival in patients with non-small cell lung cancer, J. Exp. Clin. Cancer Res 41 (1) (2022) 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [314].Carberry CK, Rager JE, The impact of environmental contaminants on extracellular vesicles and their key molecular regulators: A literature and database-driven review, Environ. Mol. Mutagen 64 (1) (2023) 50–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [315].Eckhardt CM, Baccarelli AA, Wu H, Environmental Exposures and Extracellular Vesicles: Indicators of Systemic Effects and Human Disease, Current Environmental Health Reports. 9 (3) (2022) 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Le Goff M, Lagadic-Gossmann D, Latour R, Podechard N, Grova N, Gauffre F, et al. , PAHs increase the production of extracellular vesicles both in vitro in endothelial cells and in vivo in urines from rats, Environ. Pollut 255 (Pt 1) (2019), 113171. [DOI] [PubMed] [Google Scholar]
- [317].van Meteren N, Lagadic-Gossmann D, Chevanne M, Gallais I, Gobart D, Burel A, et al. , Polycyclic aromatic hydrocarbons can trigger hepatocyte release of extracellular vesicles by various mechanisms of action depending on their affinity for the aryl hydrocarbon receptor, Toxicol. Sci 171 (2) (2019) 443–462. [DOI] [PubMed] [Google Scholar]
- [318].Chen Z, Wu H, Fan W, Zhang J, Yao Y, Su W, et al. , Naringenin suppresses BEAS-2B-derived extracellular vesicular cargoes disorder caused by cigarette smoke extract thereby inhibiting M1 macrophage polarization, Front. Immunol 13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [319].Ghildiyal M, Zamore PD, Small silencing RNAs: an expanding universe, Nat. Rev. Genet 10 (2) (2009) 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Osada H, Takahashi T, let-7 and miR-17–92: Small-sized major players in lung cancer development, Cancer Sci. 102 (1) (2011) 9–17. [DOI] [PubMed] [Google Scholar]
- [321].Lin PY, Yu SL, Yang PC, MicroRNA in lung cancer, Br. J. Cancer 103 (8) (2010) 1144–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T, Impaired microRNA processing enhances cellular transformation and tumorigenesis, Nat. Genet 39 (5) (2007) 673–677. [DOI] [PubMed] [Google Scholar]
- [323].Iqbal MA, Arora S, Prakasam G, Calin GA, Syed MA, MicroRNA in lung cancer: role, mechanisms, pathways and therapeutic relevance, Mol. Aspects Med 70 (2019) 3–20. [DOI] [PubMed] [Google Scholar]
- [324].Cheng M, Wang B, Yang M, Ma J, Ye Z, Xie L, et al. , microRNAs expression in relation to particulate matter exposure: A systematic review, Environ. Pollut 260 (2020), 113961. [DOI] [PubMed] [Google Scholar]
- [325].Dai L, Chen F, Zheng Y, Zhang D, Qian B, Ji H, et al. , miR-21 regulates growth and EMT in lung cancer cells via PTEN/Akt/GSK3β signaling, FBL. 24 (8) (2019) 1426–1439. [DOI] [PubMed] [Google Scholar]
- [326].Barkley LR, Santocanale C, MicroRNA-29a regulates the benzo[a]pyrene dihydrodiol epoxide-induced DNA damage response through Cdc7 kinase in lung cancer cells, Oncogenesis. 2(7):e57–e (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].Yu S-L, Chen H-Y, Chang G-C, Chen C-Y, Chen H-W, Singh S, et al. , MicroRNA signature predicts survival and relapse in lung cancer, Cancer Cell 13 (1) (2008) 48–57. [DOI] [PubMed] [Google Scholar]
- [328].Fujita Y, Kuwano K, Ochiya T, Takeshita F, The impact of extracellular vesicle-encapsulated circulating microRNAs in lung cancer research, Biomed Res. Int 2014 (2014), 486413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [329].Izzotti A, Balansky R, Ganchev G, Iltcheva M, Longobardi M, Pulliero A, et al. , Blood and lung microRNAs as biomarkers of pulmonary tumorigenesis in cigarette smoke-exposed mice, Oncotarget 7 (51) (2016) 84758–84774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].Bollati V, Marinelli B, Apostoli P, Bonzini M, Nordio F, Hoxha M, et al. , Exposure to metal-Rrch particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes, Environ. Health Perspect 118 (6) (2010) 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [331].Rynning I, Arlt VM, Vrbova K, Neca J, Rossner P Jr., Klema J, et al. , Bulky DNA adducts, microRNA profiles, and lipid biomarkers in Norwegian tunnel finishing workers occupationally exposed to diesel exhaust, Occup. Environ. Med 76 (1) (2019) 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [332].Rodosthenous RS, Coull BA, Lu Q, Vokonas PS, Schwartz JD, Baccarelli AA, Ambient particulate matter and microRNAs in extracellular vesicles: a pilot study of older individuals, Part. Fibre Toxicol 13 (1) (2016) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [333].Banerjee A, Waters D, Camacho OM, Minet E, Quantification of plasma microRNAs in a group of healthy smokers, ex-smokers and non-smokers and correlation to biomarkers of tobacco exposure, Biomarkers 20 (2) (2015) 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [334].Ruiz-Vera T, Ochoa-Martínez ÁC, Pruneda-Álvarez LG, Zarazúa S, Pérez-Maldonado IN, Exposure to biomass smoke is associated with an increased expression of circulating miRNA-126 and miRNA-155 in Mexican women: a pilot study, Drug Chem. Toxicol 42 (3) (2019) 335–342. [DOI] [PubMed] [Google Scholar]
- [335].Jardim MJ, Fry RC, Jaspers I, Dailey L, Diaz-Sanchez D, Disruption of microRNA expression in human airway cells by diesel exhaust particles is linked to tumorigenesis-associated pathways, Environ. Health Perspect 117 (11) (2009) 1745–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [336].Xu Z, Wang N, Xu Y, Hua L, Zhou D, Zheng M, et al. , Effects of chronic PM2.5 exposure on pulmonary epithelia: transcriptome analysis of mRNA-exosomal miRNA interactions, Toxicol. Lett 316 (2019) 49–59. [DOI] [PubMed] [Google Scholar]
- [337].Sima M, Rossnerova A, Simova Z, Rossner P Jr., The impact of air pollution exposure on the microRNA machinery and lung cancer development, J Pers Med 11 (1) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [338].Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, et al. , MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response, PNAS 109 (31) (2012) E2110–E2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [339].Liu Y, Luo F, Wang B, Li H, Xu Y, Liu X, et al. , STAT3-regulated exosomal miR-21 promotes angiogenesis and is involved in neoplastic processes of transformed human bronchial epithelial cells, Cancer Lett. 370 (1) (2016) 125–135. [DOI] [PubMed] [Google Scholar]
- [340].Pontis F, Roz L, Mensah M, Segale M, Moro M, Bertolini G, et al. , Circulating extracellular vesicles from individuals at high-risk of lung cancer induce pro-tumorigenic conversion of stromal cells through transfer of miR-126 and miR-320, J. Exp. Clin. Cancer Res 40 (1) (2021) 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [341].Li Y, Wei Y, Guo J, Cheng Y, He W, Interactional role of microRNAs and bHLH-PAS proteins in cancer (Review), Int. J. Oncol 47 (1) (2015) 25–34. [DOI] [PubMed] [Google Scholar]
- [342].Stading R, Gastelum G, Chu C, Jiang W, Moorthy B, Molecular mechanisms of pulmonary carcinogenesis by polycyclic aromatic hydrocarbons (PAHs): Implications for human lung cancer, Semin. Cancer Biol 76 (2021) 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [343].Rogers S, de Souza AR, Zago M, Iu M, Guerrina N, Gomez A, et al. , Aryl hydrocarbon receptor (AhR)-dependent regulation of pulmonary miRNA by chronic cigarette smoke exposure, Sci. Rep 7 (1) (2017) 40539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [344].Hecht E, Zago M, Sarill M, Rico de Souza A, Gomez A, Matthews J, et al. , Aryl hydrocarbon receptor-dependent regulation of miR-196a expression controls lung fibroblast apoptosis but not proliferation, Toxicol. Appl. Pharmacol 280 (3) (2014) 511–525. [DOI] [PubMed] [Google Scholar]
- [345].Lee SS, Cheah YK, The interplay between micrornas and cellular components of tumour microenvironment (TME) on non-small-cell lung cancer (NSCLC) progression, J. Immunol. Res 2019 (2019) 3046379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [346].Abdullah A, Maged M, Hairul-Islam MI, Osama IA, Maha H, Manal A, et al. , Activation of aryl hydrocarbon receptor signaling by a novel agonist ameliorates autoimmune encephalomyelitis, PLoS One 14 (4) (2019) e0215981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [347].Souki R, Amosse J, Genêt V, Le Gall M, Saintpierre B, Letourneur F, et al. , Small RNA-sequencing reveals the involvement of microRNA-132 in benzo[a] pyrene-induced toxicity in primary human blood cells, Environ. Pollut 328 (2023), 121653. [DOI] [PubMed] [Google Scholar]
- [348].Wanet A, Tacheny A, Arnould T, Renard P, miR-212/132 expression and functions: within and beyond the neuronal compartment, Nucleic Acids Res. 40 (11) (2012) 4742–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [349].Wang Y, Sun Q, Ye Y, Sun X, Xie S, Zhan Y, et al. , FGF-2 signaling in nasopharyngeal carcinoma modulates pericyte-macrophage crosstalk and metastasis, JCI Insight 7 (10) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [350].Tousif S, Wang Y, Jackson J, Hough KP, Strenkowski JG, Athar M, et al. , Indoleamine 2, 3-dioxygenase promotes aryl hydrocarbon receptor-dependent differentiation of regulatory B cells in lung cancer, Front. Immunol 12 (2021), 747780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [351].Neamah WH, Busbee PB, Alghetaa H, Abdulla OA, Nagarkatti M, Nagarkatti P, AhR activation leads to alterations in the gut microbiome with consequent effect on induction of myeloid derived suppressor cells in a CXCR2-dependent manner, Int. J. Mol. Sci 21 (24) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [352].Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, et al. , The lung microenvironment: an important regulator of tumour growth and metastasis, Nat. Rev. Cancer 19 (1) (2019) 9–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [353].Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ, Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy, Cancer Immunol. Immunother 58 (1) (2009) 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [354].Vogel CF, Chang WL, Kado S, McCulloh K, Vogel H, Wu D, et al. , Transgenic overexpression of aryl hydrocarbon receptor repressor (AhRR) and AhR-mediated induction of CYP1A1, cytokines, and acute toxicity, Environ. Health Perspect 124 (7) (2016) 1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [355].Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao C-C, et al. , Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39, Nat. Neurosci 22 (5) (2019) 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [356].Gabriely G, Quintana FJ, Role of AHR in the control of GBM-associated myeloid cells, Semin. Cancer Biol 64 (2020) 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [357].Neamah WH, Singh NP, Alghetaa H, Abdulla OA, Chatterjee S, Busbee PB, et al. , AhR activation leads to massive mobilization of myeloid-derived suppressor cells with immunosuppressive activity through regulation of CXCR2 and microRNA miR-150–5p and miR-543–3p that target anti-inflammatory genes, J. Immunol 203 (7) (2019) 1830–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [358].Vogel CF, Goth SR, Dong B, Pessah IN, Matsumura F, Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase, Biochem. Biophys. Res. Commun 375 (3) (2008) 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [359].Bankoti J, Rase B, Simones T, Shepherd DM, Functional and phenotypic effects of AhR activation in inflammatory dendritic cells, Toxicol. Appl. Pharmacol 246 (1–2) (2010) 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [360].Dahlem C, Kado SY, He Y, Bein K, Wu D, Haarmann-Stemmann T, et al. , AHR signaling interacting with nutritional factors regulating the expression of markers in vascular inflammation and atherogenesis, Int. J. Mol. Sci 21 (21) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [361].Haarmann-Stemmann T, Bothe H, Abel J, Growth factors, cytokines and their receptors as downstream targets of arylhydrocarbon receptor (AhR) signaling pathways, Biochem. Pharmacol 77 (4) (2009) 508–520. [DOI] [PubMed] [Google Scholar]
- [362].Jain A, Kaczanowska S, Davila E, IL-1 receptor-associated kinase signaling and its role in inflammation, cancer progression, and therapy resistance, Front. Immunol 5 (2014) 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [363].Krelin Y, Voronov E, Dotan S, Elkabets M, Reich E, Fogel M, et al. , Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors, Cancer Res. 67 (3) (2007) 1062–1071. [DOI] [PubMed] [Google Scholar]
- [364].Lagunas-Rangel FA, Liu W, Schiöth HB, Can exposure to environmental pollutants be associated with less effective chemotherapy in cancer patients? Int. J. Environ. Res. Public Health 19 (4) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [365].Salemme V, Centonze G, Cavallo F, Defilippi P, Conti L, The crosstalk between tumor cells and the immune microenvironment in breast cancer: Implications for immunotherapy, Front. Oncol 11 (2021), 610303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [366].Hall RD, Le TM, Haggstrom DE, Gentzler RD, Angiogenesis inhibition as a therapeutic strategy in non-small cell lung cancer (NSCLC), Transl Lung Cancer Res 4 (5) (2015) 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [367].Zhang N, The role of endogenous aryl hydrocarbon receptor signaling in cardiovascular physiology, J Cardiovasc Dis Res. 2 (2) (2011) 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [368].Yi T, Wang J, Zhu K, Tang Y, Huang S, Shui X, et al. , Aryl hydrocarbon receptor: a new player of pathogenesis and therapy in cardiovascular diseases, Biomed Res. Int 2018 (2018) 6058784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [369].Lee W-J, Lin K-H, Wang J-S, Sheu WH-H, Shen C-C, Yang C-N, et al. Aryl hydrocarbon receptor deficiency augments dysregulated microangiogenesis and diabetic retinopathy. Biomedicine & Pharmacotherapy. 2022;155:113725. [DOI] [PubMed] [Google Scholar]
- [370].Li Y, Zhou C, Lei W, Wang K, Zheng J, Roles of aryl hydrocarbon receptor in endothelial angiogenic responses†, Biol. Reprod 103 (5) (2020) 927–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [371].Gu J, Chan LS, Wong CK, Wong NS, Wong CK, Leung KN, et al. , Effect of benzo[a]pyrene on the production of vascular endothelial growth factor by human eosinophilic leukemia EoL-1 cells, J. Environ. Pathol. Toxicol. Oncol 30 (3) (2011) 241–249. [DOI] [PubMed] [Google Scholar]
- [372].Tsai CF, Hsieh TH, Lee JN, Hsu CY, Wang YC, Lai FJ, et al. , Benzyl butyl phthalate induces migration, invasion, and angiogenesis of Huh7 hepatocellular carcinoma cells through nongenomic AhR/G-protein signaling, BMC Cancer 14 (2014) 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [373].Terashima J, Tachikawa C, Kudo K, Habano W, Ozawa S, An aryl hydrocarbon receptor induces VEGF expression through ATF4 under glucose deprivation in HepG2, BMC Mol. Biol 14 (2013) 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [374].Roman AC, Carvajal-Gonzalez JM, Rico-Leo EM, Fernandez-Salguero PM, Dioxin receptor deficiency impairs angiogenesis by a mechanism involving VEGF-A depletion in the endothelium and transforming growth factor-beta overexpression in the stroma, J. Biol. Chem 284 (37) (2009) 25135–25148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [375].Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. , Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1, Mol. Cell Biol 16 (9) (1996) 4604–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [376].Ichihara S, Yamada Y, Ichihara G, Nakajima T, Li P, Kondo T, et al. , A role for the aryl hydrocarbon receptor in regulation of ischemia-induced angiogenesis, Arterioscler. Thromb. Vasc. Biol 27 (6) (2007) 1297–1304. [DOI] [PubMed] [Google Scholar]
- [377].Button EL, Bersten DC, Whitelaw ML, HIF has Biff - Crosstalk between HIF1a and the family of bHLH/PAS proteins, Exp. Cell Res 356 (2) (2017) 141–145. [DOI] [PubMed] [Google Scholar]
- [378].Fleming CR, Billiard SM, Di Giulio RT, Hypoxia inhibits induction of aryl hydrocarbon receptor activity in topminnow hepatocarcinoma cells in an ARNT-dependent manner, Comp. Biochem. Physiol. C: Toxicol. Pharmacol 150 (3) (2009) 383–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [379].Zhang M, Hu Y, Yang F, Zhang J, Zhang J, Yu W, et al. , Interaction between AhR and HIF-1 signaling pathways mediated by ARNT/HIF-1β, BMC Pharmacol. Toxicol 23 (1) (2022) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [380].Li ZD, Liu LZ, Shi X, Fang J, Jiang BH, Benzo[a]pyrene-3,6-dione inhibited VEGF expression through inducing HIF-1alpha degradation, Biochem. Biophys. Res. Commun 357 (2) (2007) 517–523. [DOI] [PubMed] [Google Scholar]
- [381].Ding J, Li J, Chen J, Chen H, Ouyang W, Zhang R, et al. , Effects of polycyclic aromatic hydrocarbons (PAHs) on vascular endothelial growth factor induction through phosphatidylinositol 3-kinase/AP-1-dependent, HIF-1alpha-independent pathway, J. Biol. Chem 281 (14) (2006) 9093–9100. [DOI] [PubMed] [Google Scholar]
- [382].Zattoni IF, Delabio LC, Dutra JP, Kita DH, Scheiffer G, Hembecker M, et al. , Targeting breast cancer resistance protein (BCRP/ABCG2): functional inhibitors and expression modulators, Eur. J. Med. Chem 237 (2022), 114346. [DOI] [PubMed] [Google Scholar]
- [383].Tan KP, Wang B, Yang M, Boutros PC, Macaulay J, Xu H, et al. , Aryl hydrocarbon receptor is a transcriptional activator of the human breast cancer resistance protein (BCRP/ABCG2), Mol. Pharmacol 78 (2) (2010) 175–185. [DOI] [PubMed] [Google Scholar]
- [384].Yan B, Liu S, Shi Y, Liu N, Chen L, Wang X, et al. , Activation of AhR with nuclear IKKalpha regulates cancer stem-like properties in the occurrence of radioresistance, Cell Death Dis. 9 (5) (2018) 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [385].Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, et al. , The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype, Nat. Med 7 (9) (2001) 1028–1034. [DOI] [PubMed] [Google Scholar]
- [386].Ouyang L, Yan B, Liu Y, Mao C, Wang M, Liu N, et al. , The deubiquitylase UCHL3 maintains cancer stem-like properties by stabilizing the aryl hydrocarbon receptor, Signal Transduct. Target. Ther 5 (1) (2020) 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [387].Therachiyil L, Hussein OJ, Uddin S, Korashy HM, Regulation of the aryl hydrocarbon receptor in cancer and cancer stem cells of gynecological malignancies: an update on signaling pathways, Semin. Cancer Biol 86 (Pt 3) (2022) 1186–1202. [DOI] [PubMed] [Google Scholar]
- [388].Cochard M, Ledoux F, Landkocz Y, Atmospheric fine particulate matter and epithelial mesenchymal transition in pulmonary cells: state of the art and critical review of the in vitro studies, J. Toxicol. Environ. Health B Crit. Rev 23 (7) (2020) 293–318. [DOI] [PubMed] [Google Scholar]
- [389].Bakir B, Chiarella AM, Pitarresi JR, Rustgi AK, EMT, MET, plasticity, and tumor metastasis, Trends Cell Biol. 30 (10) (2020) 764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [390].Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. , Guidelines and definitions for research on epithelial-mesenchymal transition, Nat. Rev. Mol. Cell Biol 21 (6) (2020) 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [391].Lambert AW, Weinberg RA, Linking EMT programmes to normal and neoplastic epithelial stem cells, Nat. Rev. Cancer 21 (5) (2021) 325–338. [DOI] [PubMed] [Google Scholar]
- [392].Longhin E, Capasso L, Battaglia C, Proverbio MC, Cosentino C, Cifola I, et al. , Integrative transcriptomic and protein analysis of human bronchial BEAS-2B exposed to seasonal urban particulate matter, Environ. Pollut 209 (2016) 87–98. [DOI] [PubMed] [Google Scholar]
- [393].Yang D, Ma M, Zhou W, Yang B, Xiao C, Inhibition of miR-32 activity promoted EMT induced by PM2.5 exposure through the modulation of the Smad1-mediated signaling pathways in lung cancer cells, Chemosphere 184 (2017) 289–298. [DOI] [PubMed] [Google Scholar]
- [394].Li D, Yun Y, Gao R, Oxygenated polycyclic aromatic hydrocarbons (Oxy-PAHs) facilitate lung cancer metastasis by epigenetically regulating the epithelial-to-mesenchymal transition (EMT), Environ. Pollut 255 (Pt 2) (2019), 113261. [DOI] [PubMed] [Google Scholar]
- [395].Yue H, Yun Y, Gao R, Li G, Sang N, Winter polycyclic aromatic hydrocarbon-bound particulate matter from peri-urban North China promotes lung cancer cell metastasis, Environ. Sci. Tech 49 (24) (2015) 14484–14493. [DOI] [PubMed] [Google Scholar]
- [396].Bersaas A, Arnoldussen YJ, Sjøberg M, Haugen A, Mollerup S, Epithelial-mesenchymal transition and FOXA genes during tobacco smoke carcinogen induced transformation of human bronchial epithelial cells, Toxicol. In Vitro 35 (2016) 55–65. [DOI] [PubMed] [Google Scholar]
- [397].Longhin E, Camatini M, Bersaas A, Mantecca P, Mollerup S, The role of SerpinB2 in human bronchial epithelial cells responses to particulate matter exposure, Arch. Toxicol 92 (9) (2018) 2923–2933. [DOI] [PubMed] [Google Scholar]
- [398].Brauze D, SERPINB2-its regulation and interplay with aryl hydrocarbon receptor, J. Appl. Genet 62 (1) (2021) 99–105. [DOI] [PubMed] [Google Scholar]
- [399].Machala M, Slavík J, Kovác O, Procházková J, Pěnčíková K, Pařenicová M, et al. , Changes in sphingolipid profile of benzo[a]pyrene-transformed human bronchial epithelial cells are reflected in the altered composition of sphingolipids in their exosomes, Int. J. Mol. Sci 22 (17) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [400].Hannun YA, Obeid LM, Sphingolipids and their metabolism in physiology and disease, Nat. Rev. Mol. Cell Biol 19 (3) (2018) 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [401].Furukawa K, Ohmi Y, Ohkawa Y, Bhuiyan RH, Zhang P, Tajima O, et al. , New era of research on cancer-associated glycosphingolipids, Cancer Sci. 110 (5) (2019) 1544–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [402].Cumin C, Huang YL, Everest-Dass A, Jacob F, Deciphering the importance of glycosphingolipids on cellular and molecular mechanisms associated with epithelial-to-mesenchymal transition in cancer, Biomolecules 11 (1) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [403].Sharma JR, Agraval H, Yadav UCS, Cigarette smoke induces epithelial-to-mesenchymal transition, stemness, and metastasis in lung adenocarcinoma cells via upregulated RUNX-2/galectin-3 pathway, Life Sci. 318 (2023), 121480. [DOI] [PubMed] [Google Scholar]