

Analysis of a spectrum of subsolid pulmonary nodules by single-cell RNA sequencing provides insights into the immune regulation and cell–cell interactions in the tumor microenvironment during early lung tumor development.
Abstract
A greater understanding of molecular, cellular, and immunological changes during the early stages of lung adenocarcinoma development could improve diagnostic and therapeutic approaches in patients with pulmonary nodules at risk for lung cancer. To elucidate the immunopathogenesis of early lung tumorigenesis, we evaluated surgically resected pulmonary nodules representing the spectrum of early lung adenocarcinoma as well as associated normal lung tissues using single-cell RNA sequencing and validated the results by flow cytometry and multiplex immunofluorescence (MIF). Single-cell transcriptomics revealed a significant decrease in gene expression associated with cytolytic activities of tumor-infiltrating natural killer and natural killer T cells. This was accompanied by a reduction in effector T cells and an increase of CD4+ regulatory T cells (Treg) in subsolid nodules. An independent set of resected pulmonary nodules consisting of both adenocarcinomas and associated premalignant lesions corroborated the early increment of Tregs in premalignant lesions compared with the associated normal lung tissues by MIF. Gene expression analysis indicated that cancer-associated alveolar type 2 cells and fibroblasts may contribute to the deregulation of the extracellular matrix, potentially affecting immune infiltration in subsolid nodules through ligand–receptor interactions. These findings suggest that there is a suppression of immune surveillance across the spectrum of early-stage lung adenocarcinoma.
Significance:
Analysis of a spectrum of subsolid pulmonary nodules by single-cell RNA sequencing provides insights into the immune regulation and cell–cell interactions in the tumor microenvironment during early lung tumor development.
Introduction
Pulmonary nodules, identified as either solid or subsolid, are focal opacifications smaller than 3 cm and are commonly detected on CT of the chest (1). Radiographically, subsolid nodules are further categorized as either pure ground-glass (focal hazy opacifications in which underlying lung structures, such as vessels, remain visible), semi-consolidated (dense ground-glass), or part-solid nodules. Histologically, persistent or enlarging subsolid nodules often represent the early spectrum of lung adenocarcinoma (ADC), which includes premalignant atypical adenomatous hyperplasia (AAH), preinvasive ADC in situ (AIS), and minimally invasive ADC (MIA; refs. 2, 3). Although some subsolid nodules remain stable for many years, others progress to become invasive lung ADC. This unpredictable clinical trajectory of subsolid nodules combined with our poor understanding of the pathogenesis of disease at this stage lead to uncertainty in diagnostic and therapeutic approaches. A better understanding of the early molecular, cellular, and immunological properties that fuel progression would enable cancer interception at the earliest stages of disease (4).
Recent studies have begun interrogating the molecular and cellular characteristics of the early-spectrum lung cancer (5–8). Our recent study evaluating the lung cancer continuum, using whole-exome sequencing and IHC of formalin-fixed, paraffin-embedded (FFPE) specimens, demonstrated that persistent progression-associated putative tumor neoantigens correlated with T-cell infiltration and PD-L1 upregulation, suggesting potential immune recognition at the earliest stage of malignant progression (6). A recent single-cell RNA sequencing (scRNA-seq) study of early-stage non–small cell lung cancer (NSCLC) identified a cellular module, consisting of activated T cells, plasma cells and macrophages, which was associated with the responsiveness to immune checkpoint blockade therapy (9). Despite these advances, the immunopathogenesis of early-spectrum lung ADC remains obscure, due in part to the difficulty of obtaining tissue. Bronchoscopy and CT-guided core needle biopsies of pulmonary nodules are efficient for diagnostic purposes, but they yield low cell numbers and, more importantly, do not capture the complete cellular contexture of the nodule microenvironment.
Here, we report an atlas of early-spectrum ADC using a prospective cohort of patients undergoing surgical resection of radiologically detected high-risk pulmonary nodules. Fresh surgically resected tissue was obtained from nine primary and/or synchronous pulmonary nodules from six patients with evolving subsolid nodules and subjected to analysis with scRNA-seq, flow cytometry (FC), and multiplex immunofluorescence (MIF; Fig. 1A). ScRNA-seq and FC concomitantly provided evidence of immunosuppression mediated by immune and nonimmune cells within the tumor microenvironment (TME). Findings from single-cell studies were validated by MIF staining in an independent retrospective cohort of 16 patients with pulmonary nodules that included AAH, AIS, MIA lesions, and the associated invasive ADC. MIF staining revealed an incremental increase in regulatory T-cell (Treg) infiltration in premalignant and preinvasive lesions. These findings in the independent set of pulmonary nodules corroborate our scRNA-seq results, indicating gradual immune exhaustion and development of immunosuppressive phenotypes with disease progression.
Figure 1.

Single-cell transcriptional profiling of human immune cells in lung nodules and associated normal lung tissue. A, Schematic of cohorts and assays used in this study. B, UMAP plot of immune (red) and nonimmune cells (light blue). C and D, UMAP plot visualizing immune cell clusters colored by tissue types (C) and cell lineages (D), including NK, NKT, CD8, CD4, γδT, B, plasma, mast, DC, pDC, macrophages (MΦ), monocytes (Mono), and neutrophils (Neutro). E, Summary of fold change and their statistical P values between subsolid nodules and nLung for each cell lineage based on scRNA-seq and FC. P values were calculated by the LME model. The abundance of myeloid and B cells was not subjected to assessment (n.a.) by FC. F, Correlation of relative abundance of T (CD4+, CD8+, and NKT) and NK cells identified by scRNA-seq and FC. G, Difference between Stage I tumor (subsolid and solid nodules) and nLung in the UCLA (x-axis) and Leader and colleagues (9) studies (y-axis). Crosses represent mean ± SEM. Red boxes emphasize top lineages altered in both cohorts. (A, Created with BioRender.com.)
Materials and Methods
Human specimen collection
The study and its protocols were reviewed and conducted with the approval of UCLA Institutional Review Board (IRB # 10–001096). After obtaining written informed consent, fresh tissues were obtained from patients undergoing lung cancer surgery at UCLA Medical Centers (Santa Monica and Los Angeles, CA). Solid and subsolid lesions were identified by the operating thoracic surgeon, based on careful palpation and correlation with the imaging. In cases where the diagnosis had not already been verified by needle biopsy, review of the frozen section by a dedicated thoracic pathologist confirmed the early-spectrum lung ADC. A portion of the primary subsolid lesion, any associated solid tumor, as well as a sample of normal lung tissue from a grossly normal site of the lobectomy specimen, distant (>2 cm) from the tumor site(s), were then harvested, placed on ice in RPMI medium immediately, and delivered to the laboratory for tissue dissociation. Because of the nature of subsolid nodules, which are frequently small with ill-defined margins, we obtained our fresh samples from the center of the nodules to avoid contaminating the tumor specimens with peripheral normal tissue. The remaining nodules and adjacent tissue were preserved as FFPE blocks for pathology review of the nodules and assessment of the surrounding tissues for premalignancy. Mutation status was assessed by targeted DNA sequencing and FISH as per the clinical protocol. FFPE tissue blocks from a retrospective cohort with premalignant lesions and lung ADC were obtained from UCLA Lung Cancer Tissue Repository, and subjected to pathology review by two independent pathologists to identify specific histologic areas.
Sample dissociation for scRNA-seq
The single-cell dissociation protocol was adapted from Leelatian and colleagues (10). In brief, dissociation was performed in RPMI medium supplemented with 10% FBS. Tissues were sliced to approximately 1 mm3 pieces and dissociated in 200 U/mL collagenase type II (Sigma-Aldrich, #C6885) and 100 Kunitz U/mL DNAse I (Sigma-Aldrich, #DN25) at 37°C for approximately up to 1 hour until homogenous, followed by passing through a 40-μm strainer to remove cell aggregates and red blood cell lysis with 1 mL of ACK buffer (Sigma-Aldrich, #11814389001). Cells were resuspended in 5 mL DPBS + 0.4% BSA, counted, and immediately used to prepare the sequencing libraries.
scRNA-seq and read alignment
The 10X Genomics platform (10X Genomics) was used for assessing human scRNA-seq. Single-cell capture, library construction, and sequencing were performed at the University of California Los Angeles Technology Center for Genomics & Bioinformatics (RRID:SCR_012204) according to the manufacturer's protocols. The Chromium Single Cell 3′ Library & Gel Bead Kit v2 and v3 were used for library preparation. Libraries were sequenced using Illumina NovaSeq 6000 instrument. CellRanger 3.1.0 software (10X Genomics) was used to align and annotate reads using human genome assembly GRCh38p13 and gene annotation GENCODE34 and then generate count matrices.
Bioinformatics analysis
Single-cell data filtering, normalization, and batch adjustment
Count matrices were processed by following the Seurat pipeline. In brief, count matrices of individual samples were first combined and low-quality cells, which had >17% mitochondrial content and <475 detected genes, were filtered out. A total of 88,638 cells were retained for further analysis. The data were normalized and batch-adjusted following the Seurat Standard Workflow pipeline in which the top 2000 highest variance genes were used to find anchors and integrate data from different batches.
Single-cell cluster analysis and annotation
The batch-adjusted data were subjected to the clustering analysis to separate immune cells from nonimmune cells in silico by using the top 50 principal components to determine the k-nearest neighbors of each cell and visualize cells by Uniform Manifold Approximation and Projection (UMAP). The Louvain algorithm-based cluster identification was tested at a range of resolution from 0.2 to 1.5 to determine whether the increase of resolution value produced new clusters associated with a specific sample. The optimized resolution was 1.0 because it was high enough to obtain clusters associated with cell lineage identity and still minimized the number of clusters associated with a specific sample. Cell clusters were annotated into immune and nonimmune cell types based on the enrichment analysis and expression markers described below. PanglaoDB (11) was used in this step identifying immune cells (including natural killer, T, B, plasma, mast, dendritic, plasmacytoid dendritic cells (DC), macrophages, alveolar macrophages, monocytes, and neutrophils) and nonimmune cells [including pulmonary alveolar type I and II cells, airway epithelial cells, Clara cells, Ciliated cells, airway Goblet cells, ionocytes, fibroblasts, stromal cells, and endothelial cells (EC)]. The separated immune and nonimmune cells were then reanalyzed separately to determine the highest variance genes, recalculate principal components, and cluster and visualize cells by using the top 30 new calculated principal components. Cell clusters were re-annotated again for immune and nonimmune lineages/sublineages by using multiple databases, including PanglaoDB (11) and CIBERSORT (12), and previously published articles, in which single-cell transcriptomes were available for the particular immune lineages, such as Villani and colleagues (13) for DCs and monocytes, Leader and colleagues (9) for T cells.
The following 2-tier pipeline was performed to annotate cell type associated with each cluster:
-
1. In tier 1 of the pipeline, the systematic approach was used to determine whether the lineage markers of a cell type enriched as the positive marker genes in a cluster. The enrichment score of cluster C as cell type T, RC,T, was calculated as:
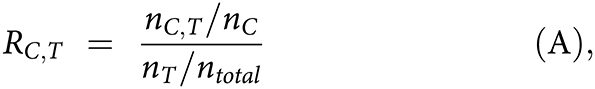
where nC,T is the number of genes that identified as positively expressed markers in both the cluster C and cell type T, whereas nc, nT, and ntotal are the total numbers of positively expressed markers in cluster C, cell type T, and the total number of genes detected by scRNA-seq, respectively. The positively expressed markers in each cluster were identified by FindAllMarkes using the MAST approach.
2. For each cluster, enrichment scores associated with different cell types were ranked from high to low. If the deviation between 1st and 2nd highest scores was >1, the cell type with the highest score was assigned to the cluster. If not, the expression of canonical markers was used in the next analysis step for cluster annotation.
3. In Tier 2, canonical expressions were used to either verify or clarify the ambiguity of the prediction in the previous step. The cluster was assigned to the cell lineage with the higher average z-transformed expression of canonical markers. Supplementary Table S3 lists markers utilized in each cluster.
Annotating functional subtypes by known gene modules
Some cell lineages composed of multiple clusters were associated with their subtypes or biological stages. Their functional subtypes were identified on the basis of their literature signature gene modules. In brief, the existing gene modules were modified to maintain only the mutually exclusive genes among modules. Each signature gene module has its score determined as the z-score average across all module genes for each cell. The module score at the cluster level was the average across all cells in the cluster.
Trajectory analysis
Monocle 2 (14) was used to establish linear trajectories for differentiating CD8 T cells. The reconstructed trajectories were built on functional gene sets (15, 16), markers for naïve and exhausted T cells detected in lung cancer.
Analysis of differentially expressed genes
Two different approaches, pseudo-bulk edgeR (17), and single-cell model MAST (18), were used to determine differentially expressed genes (DEG) between subsolid/solid and normal AT2 cells. The final DEGs were the intersection of both approaches. In brief, we used raw read counts of all cells identified as AT2 cells (regardless of their SFTPC expression level) and added a variable incorporating patient identity in addition to tissue histology when modeling expression changes in both pseudo-bulk and single-cell model approaches. Details of the pseudo-bulk approach can be found in the Bioconductor tutorial (https://bioconductor.org/books/release/OSCA/multi-sample-comparisons.html). A DEG was identified if its fold-change >2 and FDR < 0.1 in the pseudo-bulk edgeR approach, whereas it required the coefficient > 0.5 and FDR < 1e−50 in the MAST approach.
The DEGs between subsolid/solid lesions and normal tissue in cell types other than AT2 were assessed by using the single-cell model MAST approach because their transcriptome profiles were usually more homogeneous among samples than the AT2 population. MAST incoprorates patient identity as a variable in addition to tissue histology. Furthermore, cells in clusters associated with a specific sample (>70% cells in the cluster were from a single sample) were not used in analyzing the DEGs in these cell types.
Gene set enrichment analysis
Fisher exact test was used to determine the pathways or biological processes enriched by DEGs, and P values adjusted with the Benjamini–Hochberg method for multiple hypothesis testing. The statistically significant altered pathways (FDR <0.05) were then re-analyzed by the rank-based gene set enrichment analysis (GSEA) approach (19) using the Bioconductor fgsea package (bioRxiv 2016:060012) to assess the direction of deregulation in the subsolid/solid lesion samples. We used gene sets obtained from the Molecular Signature Database (MsigDB) version 7.1.
Ligand–receptor interaction among cell types
We defined the strength of each interaction of given ligand li in cell type S (source) to its related receptor ri in cell type R (receiver) as the product of average ligand expression across all cells of cell type S and average receptor expression across all cells of cell type R as suggested by Kumar and colleagues (20):
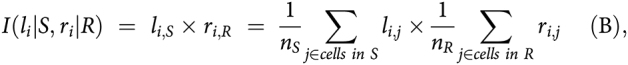 |
where nS and nR are the total number of cells in cell type S and R, respectively, and li,j (rij) is the expression (in log scale) of gene li (ri) in cell j. For a given cell type S and R, a ligand–receptor (LR) pair would have two different represented scores calculated by assuming the ligand in S interacting with the receptor in R, and vice versa. Furthermore, interaction scores were also computed for each lesion because lesions might have different microenvironments even though they are from the same subject. We used the LR database in the R-based iTALK package (21), which categorized literature LR pairs into four categories: Immune checkpoint, cytokine/chemokine, growth factors, and others, based on ligand functions. To calculate average gene expression of each cell type, we selected cells in clusters that were: (i) Composed of cells from multiple samples, and (ii) associated with disease status. For instance, we selected ECs in cluster 2 (normal), 14 and 15 (malignancy), and fibroblast cells in cluster 11 and 18 (normal), and 8 and 9 (malignancy).
LR scores were then dichotomized on the basis of two-sided Wilcoxon rank-sum tests (P < 0.05) comparing scores between malignant and normal samples for each specific LR interaction. A LR interaction was identified essential for the commutation among cell types in subsolid/solid lesion if: (i) Its Wilcoxon rank-sum P value was <0.05 (ii), fold change of their scores between malignant and normal samples > 2, and (iii) either ligand (li,A) or receptor (ri,B) expression was differentially expressed (FDR < 0.1) in malignant lesion compared with normal lung tissue.
Scripts to reproduce clustering, cluster annotation, differential expression, LR interaction analyses, as well as supporting data for gene modules are available on GitHub (https://github.com/LinhTranUCLA/scRNA_subsolid).
FC staining and analysis
Single-cell tumor suspensions were incubated with surface antibodies for 20 minutes in the dark, followed by washing with FACS buffer (PBS + 2% FBS). Intracellular staining was performed using the eBioscience intracellular fixation and permeabilization buffer set (Invitrogen, Cat# 88–8824–00) according to the manufacturer's protocol. mAbs against human were CD45 (BioLegend Cat# 368503, RRID:AB_2566351), CD3 (BioLegend Cat# 344822, RRID:AB_2563420), CD8a (BioLegend Cat# 301042, RRID:AB_2563505), CD56 (BioLegend Cat# 362509, RRID:AB_2563926), CD16 (BioLegend Cat# 302043, RRID:AB_11219184), CD25 (BioLegend Cat# 302640, RRID:AB_2629672), CD45RA (BioLegend Cat# 304048, RRID:AB_2563129), CCR7 (BioLegend Cat# 353214, RRID:AB_10917387), FOXP3 (BioLegend Cat# 320008, RRID:AB_492980), Ki67 (BioLegend Cat# 350505, RRID:AB_10896915), TCRγ/δ (BioLegend Cat# 331228, RRID:AB_2650627), CD19 (BioLegend Cat# 302205, RRID:AB_314235), CD14 (BioLegend Cat# 325603, RRID:AB_830676), CD4 (BD Biosciences Cat# 562658, RRID:AB_2744420), and CD69 (BD Biosciences Cat# 562617, RRID:AB_2737680). Data acquisition was performed on NovoCyte Quanteon Flow Cytometer (Agilent), and data analyzed by FlowJo software version 10.7.1 (RRID:SCR_008520) using the gating strategy described in Supplementary Fig. S3.
MIF staining and analysis
MIF staining was performed on 5-μm-thick FFPE slides using an automated staining system (Roche Ventana Discovery Ultra) with antibodies against pan cytokeratin (Agilent Cat# M351501–2, RRID:AB_2631307, dilution 1:100), granzyme B (Leica Biosystems Cat# NCL-L-GRAN-B, RRID:AB_563751, dilution 1:200), FOXP3 (Cell Signaling Technology Cat# 98377, RRID:AB_2747370, dilution 1:200), Ki67 (Ventana Medical Systems Cat# 790–4286, RRID:AB_2631262, prediluted), CD8 (Leica Biosystems Cat# CD8–4B11-L-CE, RRID:AB_10555292, dilution 1:200), CD3 (Ventana Medical Systems Cat# 790–4341, RRID:AB_2335978, prediluted) and DAPI (Akoya Biosciences, Cat# FP1490). Multispectral whole-slide images were acquired by scanning stained slides using the Vectra Polaris imaging system (PerkinElmer) at ×20 magnifications and unmixed to single-spectral components using InForm image analysis software (PerkinElmer) and single-color libraries. Unmixed multiple spectral whole-slide images were then imported into HALO v3.4 (Indica Labs) for image analysis, including stitching, selecting regions of interest, cell segmentation, and quantifying intensities into binary levels for individual cells. Single-cell data were then analyzed by R SPIAT (22). In brief, cells with negative DAPI expression were filtered out, before subjected to phenotype annotation based on marker colocalization to identify populations of CD8+ (CD8+) and CD4+ (CD3+ and CD8−) T cells, and epithelial cells (PanCK+). Cell density was calculated on the basis of tissue area (i.e., excluding non-cell/glass area).
Statistical analysis
Statistical testing was performed using R 3.6. We used the linear mixed-effects (LME) model (R lmerTest package) to incorporate individual patient variation in evaluating changes between groups. The statistical tests at the sample level did not include data from synchronous solid nodules unless specified in additional comparisons to eliminate the bias due to the small sample size. Cell-level statistical tests include the solid nodule data when specified. Appropriate rank-based statistical tests were applied according to the nature of variables. The tests used to determine statistical significance are quoted next to the P values and in the appropriate figure legends.
Data availability
The raw RNA sequence was deposited in dbGaP (Study Accession: phs002371.v3.p1), which manages the access control. The normalized gene expression (data level 3) and cell annotation (data level 4) are also available at Human Tumor Atlas Network—Data Coordinating Center. Detailed information of data access and data levels can be found at the Data Coordinating Center (https://humantumoratlas.org/data-access). Other raw data generated in the study are available upon request from the corresponding authors.
Results
Characteristics of prospective and retrospective cohorts with pulmonary nodules
The prospective cohort, evaluated by scRNA-seq, FC and MIF, consisted of six patients with evolving subsolid nodules that were initially identified incidentally or in the context of lung cancer screening. Four patients had a single subsolid nodule, and two had synchronous nodules (Supplementary Table S1; Supplementary Fig. S1A). Case 2 had a semiconsolidative nodule and a partsolid nodule in the same lobe. The latter was predominantly solid and classified as solid for downstream analysis. In Case 4, two additional subsolid nodules were present in a different ipsilateral lobe from the dominant solid nodule. In total, nine nodules (seven subsolid and two solid) with an average diameter of 15 mm were resected for subsequent analysis (Supplementary Table S1). The histological evaluation of nodules identified two cases of MIA and the remainder were identified as invasive or mixed with a noninvasive component ADC, including acinar, lepidic, and papillary subtypes (Supplementary Table S1; Supplementary Fig. S1B–S1J). No association between radiological and pathological features was observed (Supplementary Fig. S1A).
Single-cell findings were validated using an independent retrospective cohort from the UCLA lung cancer repository consisting of 16 patients that underwent surgical resection of high-risk pulmonary nodules (n = 18 subsolid and 8 solid nodules) that were pathologically classified as ADC (Fig. 1A; Supplementary Table S2). Nodules were identified incidentally or through the UCLA lung cancer screening program. Eighty-eight pathological regions representing early-spectrum ADC, including 57 AAH, 13 AIS, 2 MIA, and 16 ADC, along with 16 matched adjacent nLung were identified for downstream analysis by MIF.
scRNA-seq reveals differential immune infiltration in subsolid nodules compared with associated normal lung
ScRNA-seq was performed on nine nodules and matching nLung from the prospective cohort (Fig. 1A). Although all nodules were subjected to downstream studies, statistical analyses were performed comparing subsolid nodules with nLung. Solid nodules were used to determine whether alterations in subsolid nodules were consistent with those in solid nodules. 88,638 cells passed the quality control and were subjected to batch-effect correction before entering the analysis pipeline. Unsupervised graph-based clustering separated cells into the following cell types: (i) Immune clusters expressing PTPRC (CD45) and either CD3A, CD79A, or CD68, ii) nonimmune clusters expressing either COL1A1 (fibroblasts), PECAM1 (ECs), EPCAM (epithelial cells), or SCGB1A1 (secretory cells; Fig. 1B; Supplementary Fig. S2A and S2B). Immune and nonimmune cells were analyzed independently. Reclustering of 60,509 immune cells revealed 30 cell clusters, visualized with the UMAP approach (Supplementary Fig. S2C). Cells were aggregated according to radiographic sample types (Fig. 1C) rather than origin of samples (Supplementary Fig. S2D; Supplementary Table S3), illustrating that clusters were not associated with specific samples. The 13 major immune cell types (Fig. 1D; Supplementary Fig. S2E), including natural killer (NK), natural killer T (NKT), CD8+ T (CD8), CD4+ T (CD4), gamma delta T (γδT), B, plasma, mast, DC, plasmacytoid DC (pDC), macrophages, monocytes, and neutrophils were identified on the basis of the enrichment scores for the lineage gene sets and the expression of canonical lineage markers. We observed a significant increase in CD4+ T, B cells, and DCs, as well as reduced numbers of NK and NKT cells in subsolid nodules compared with the associated nLung (Fig. 1E; Supplementary Fig. S2F). The infiltration frequencies of adaptive immune cells, including CD4+ T and B cells, were highly correlated with DCs (Supplementary Fig. S2G). The abundance of T and NK cells in nodules and the associated nLung were independently evaluated using FC (Supplementary Fig. S3) and highly correlated with findings from scRNA-seq (Fig. 1F). We observed a high concordance (correlation coefficient = 0.64, P = 0.04) between our results and a recent study by Leader and colleagues (9) that included Stage I patients with ADC (n = 16), specifically in regards to an enrichment of B and CD4+ T cells and a decrement in NK and NKT cells in pulmonary nodules compared with nLung (Fig. 1G).
Reduction of cytolytic NKT and NK cells in subsolid nodules
A three-fold reduction in the frequency of NKT cells was observed in subsolid nodules compared with nLung (Figs. 1E and 2A). NKT cells formed two distinct clusters, clusters 11 (C11) and 20 (C20). Within the NKT cell population, we found a steep decrease in C11-NKT in the context of an increase in C20-NKT cells in subsolid nodules (Fig. 2B). Gene ontology analysis revealed that C11-NKT cells expressed signature genes associated with cytolytic activity, whereas C20-NKT cells expressed genes encoding cytokines that have the capacity to modulate immune cells, including DCs and T cells (Fig. 2C). The expression of FCGR3A (CD16), known for mediating antibody-dependent cytotoxic activity in NK cells (23), was limited to C11 but not C20 cells (Fig. 2D). Furthermore, the expression of transcription factors associated with type 1 (TBX21) and type 2 (ZBTB16) NKT cells (24) was mutually exclusive in C11 and C20 clusters, suggesting that C11 and C20 correspond to NKT1 and NKT2 cells, respectively (Fig. 2D). To further characterize these NKT clusters, we used gene modules derived from either human NK (25) or murine innate lymphoid cells (ILC; ref. 26) based on the premise that NKT subsets are similar to their NK counterparts (24). C11 cells highly expressed NK1 signatures, associated with cytolytic activity, and C20 expressed NK2 signatures, associated with cytokine responses and ILC type 2 (Fig. 2E). Moreover, C20-NKT2 cells from subsolid and solid nodules expressed significantly higher NK2 and ILC2 signatures than C20 cells from matching nLung (Wilcoxon's rank test P < 1e−10). Comparing the top DEGs between C20-NKT cells in subsolid nodules and nLung revealed an absence of cytolytic markers (GZMB and IFNG) and activation markers (FCGR3A/CD16) accompanied by a decrease in the expression of CCL5, a T lymphocyte chemoattractant, in C20-NKT2 cells from subsolid nodules (Fig. 2F; Supplementary Table S4). FC results confirmed decreased percentage of cytotoxic CD16+ cells within NKT cells in subsolid nodules compared with nLung (Fig. 2G).
Figure 2.

Reduction of infiltrating cytolytic NKT and NK cells in subsolid nodules. A, Relative abundance of NKT cells among CD45+ cells in each sample (dot). B, The percentage of NKT subtypes in each sample (dot). C, Immune-regulated pathways enriched by markers identified in NKT clusters, C11 (left) and C20 (right). D, NKT marker expression (Expr.) associated with cytolytic activity (FCGR3A/CD16) and NKT subtypes 1 (TBX21) and 2 (ZBTB16). E, Violin plots illustrating the distribution of the functional NKT gene module scores in various clusters and nodule types. Dashed lines represent the median scores of nLung cells in the selected clusters. ***, P < 1e−10 based on the rank-based Wilcoxon test. F, Top DEGs (P < 1e−5) between nLung and nodule-associated C20 NKT cells. G, Relative abundance of CD16+ NKT cells in each sample (dots) assessed by FC. H, Relative abundance of NK cells (cluster 4) to total immune cells by scRNA-seq. I, Relative abundance of CD16+ NK cells by FC. Each dot in spaghetti plots (A,B, and G–I) represents a sample, with colors representing individual patients. Line patterns indicate the nLung-subsolid (dashed) and nLung-solid (solid) relationships from the same patient. P values were calculated on the basis of the LME model to compare either subsolid nodules (A, B, and I) or both sub- and solid nodules (G and H) and nLung. Data points of solid nodules (A, B, and I) illustrate observations in subsolid were consistent with those in matched solid nodules.
NK cells formed a single cluster (C4). NK cell infiltration decreased significantly in subsolid nodules compared with nLung as demonstrated by both scRNA-seq and FC (Figs. 1E and 2H). Furthermore, infiltration of cytotoxic CD16+ NK cells was significantly lower in subsolid and solid nodules compared with nLung based on FC (Fig. 2I). These data indicate a decreased abundance of both total and cytotoxic NK and NKT cells in subsolid nodules compared with nLung. The significant decline in the cytotoxic signatures suggests suppression of the NK- and NKT-mediated antitumor responses with tumor progression.
Suppression of T-cell immune responses in subsolid nodules
ScRNA-seq captured transcriptomics of 20,445 CD4+, 5,790 CD8+, and 531 γδ T cells (Supplementary Figs. S2E and S4A). Among the infiltrating CD45+ cells, the proportion of CD4+ T cells was higher in subsolid nodules than in nLung (Fig. 1E). Analysis of infiltrating CD4+ T cells identified central-like memory T cells (Tcm, cluster C0), effector memory T cells (Tem, C1), Treg (C7), and activated T cells (Tact, C14, and C21; Supplementary Fig. S4A). The percentage of C1-Tem was decreased significantly [linear mixed-effects (LME) model P = 0.02], whereas that of C7-Treg was increased (LME model P = 0.01) in subsolid nodules compared with the matched nLung (Fig. 3A). As a result, Tem:Treg ratio was significantly decreased in the subsolid nodules, suggesting a suppression of T-cell–mediated immune activity (Fig. 3A). A similar pattern of CD4+ cells infiltration with relative enrichment of immunosuppressive Tregs and a significant reduction of Tem:Treg ratio in subsolid nodules was also noted by FC (Fig. 3B).
Figure 3.

Profiles of the CD4+ T-cell subsets in subsolid nodules. A and B, The percentage of CD4+ T cells identified as Tem and Treg cells and Tem:Treg ratio in each sample (dot) via scRNA-seq (A) and FC (B) in the perspective cohort. Line patterns indicate the nLung-subsolid (dashed) and nLung-solid (solid in A and B) relationships from the same patients. C and D, Densities of conventional CD4+ and Treg (C) and percentage of Treg to total CD4+ T cells (D) evaluated by MIF staining in tissue areas (dot) associated with histology (x-axis) in the retrospective cohort. E, Illustration of CD8+ T-cell differentiation pathways inferred by Monocle. F, Distribution of pseudotime scores (x-axis) in tissue types (y-axis) for each differentiation path. G, Density of GZMB+CD8+ T cells in tissue area (dots) associated with histology (x-axis) in the retrospective cohort. n.s., nonsignificant.
During sample preparation, approximately half of each lesion was dissociated for single-cell analysis and another half was FFPE. MIF staining of FFPE sections for CD3, CD8, GZMB, FOXP3, Ki67, PanCK, and DAPI was used to corroborate results from scRNA-seq and FC studies (Supplementary Fig. S5). Nineteen regions of interest, including those subjected to scRNA-seq and additional associated premalignant (AAH, n = 2) and preinvasive (AIS, n = 1) lesions, were stained and quantified (tissue area 0.5 -150.9 mm2). The density of both conventional CD4 (CD3+CD8−FOXP3−) and regulatory (CD3+CD8−FOXP3+) T-cell infiltration increased in premalignant and preinvasive lesions and reached the highest levels in their associated invasive lesions (Supplementary Fig. S4B). In accord with the scRNA-seq and FC results, MIF staining demonstrated that the percentage of Treg in the CD4+ T-cell population increased significantly with disease progression (Supplementary Fig. S4C). Similarly, MIF staining of pulmonary nodules from an independent cohort of 16 subjects from the UCLA lung cancer repository revealed an incremental increase in conventional and regulatory CD4+ T-cell infiltration as well as an enrichment of Tregs within the CD4+ compartment in the developing tumors (Fig. 3C and D).
Analysis of CD8+ T cells revealed two clusters. The C5 cluster was characterized by low GZMB expression and high expression of exhaustion markers (PDCD1/PD1 and TOX), whereas C6 cluster had high GZMB expression (Supplementary Fig. S4A). Analysis of DEGs among C6 CD8+ T cells in subsolid nodules and nLung revealed decreased expression (MAST model P < 1e−16) of genes associated with cytotoxic function (GZMB and GNLY) and the activation of T-cells (LMNA) in C6 cells from subsolid nodules (Supplementary Fig. S4D), implying that these cells have low cytotoxic potential. Trajectory inference analysis revealed early divergence of two pathways from naïve cells (C6 cells with low GZMB expression) leading to either an exhausted phenotype (C5 cells) or a terminally differentiated effector memory re-expressing CD45RA (TEMRA) phenotype characterized by high expression of GZMB (C6 cells with high GZMB expression; Fig. 3E). Moreover, the TEMRA differentiation signature was reduced in subsolid and solid nodules compared with nLung (Kruskal–Wallis χ2 test P = 4e−12), but the T-cell exhaustion signature was similar amongst all groups (Fig. 3F). In accord with these observations, MIF staining of an independent cohort of patients from the UCLA lung cancer repository confirmed decreased infiltration of cytotoxic GZMB+CD8+ T cells (Fig. 3G) and increased infiltration of GZMB−CD8+ T cells with disease progression (Supplementary Fig. S4E). Taken together, our analyses indicate an enrichment of regulatory CD4+ T cells and a decrease in cytotoxic CD8+ T lymphocytes in pulmonary nodules compared with nLung, consistent with suppression of cell-mediated immunity in the early spectrum of lung cancer.
Reduction of DC1 and enrichment of tumor-promoting monocytes in subsolid nodules
DC infiltration was increased in subsolid nodules compared with nLung (Fig. 1E) and positively correlated with CD4+ T-cell and B-cell infiltration (Supplementary Fig. S2G). Six clusters comprised of 5,363 DCs (C10, C12, C23, C26, and C27) and 185 pDCs (C28) were identified. The five DC clusters were annotated using existing gene modules (13): (i) C10 as conventional DC2, (ii) C12 and C23 as monocyte-derived DC3/4, (iii) C26 as conventional DC1, (iv) C27 as rare DC5 (Supplementary Figs. S2E and S6A). DC1 and DC2 activate CD8+ and CD4+ T cells through MHC class I and II antigen presentation, respectively. Within the DC population, the relative frequency of DC2 substantially increased (LME P = 0.09), whereas DC1 marginally decreased in most subsolid nodules compared with nLung (Supplementary Fig. S6B). Compared with the nLung, the DC1:DC2 ratio decreased (LME model P = 0.06) in subsolid nodules, with the exception of Case 4 nodule 2 and Case 5, both of which had mixed invasive and noninvasive components (Supplementary Fig. S6C). A decrement in tumor-infiltrating DC1s, which are critical for antigen cross-presentation and CD8+ T-cell activation, was previously observed in a cohort of patients with early-stage NSCLC (27).
Two monocyte clusters, C3 and C18, were identified by scRNA-seq and annotated on the basis of existing gene modules (13) as nonclassical type 2 and classical type 1 monocytes, respectively (Supplementary Fig. S6D). A statistically significant enrichment of C3-nonclassical monocytes was observed in subsolid nodules compared with nLung (Supplementary Fig. S6E). Enrichment of peripheral nonclassical monocytes has been shown in cancer patients compared with healthy individuals (28).
Decreased alveolar type 2 pneumocyte differentiation in subsolid nodules
Nonimmune cells (n = 28,129) aggregated into 34 clusters corresponding to seven cell types: Fibroblasts (COLA1A), endothelial (PECAM1), alveolar type 1 (AT1; PDPN), alveolar type 2 (AT2; EPCAM and NKX2–1), Clara (SCGB1A1 and SCGB3A1), bronchioalveolar stem (BASC; SFTPC and SCGB1A1), and ciliated (FOXJ1) cells (Supplementary Fig. S7A–S7D; Supplementary Table S3). Although nonimmune cells were shared among multiple samples and tissue types, many AT2 and BASC clusters were specific to individual nodule samples (Supplementary Fig. S7E–S7F), suggesting these clusters associated with tumor cells with heterogeneous expression patterns because of genetic variations. AT2 clusters detected only in nodules (nonimmune clusters niC4, niC17, niC21, niC25, niC26, and niC32) had lower expression of surfactant encoding genes (SFTPC and SFTPA1) compared with AT2 clusters (niC0, niC10, niC28, and niC29) that were present in nodules and nLung (Supplementary Fig. S7D; Supplementary Table S3). Categorizing AT2 clusters by SFTPC expression yielded two AT2 subtypes. SFTPC-lo AT2 cells were only present in nodules, whereas SFTPC-hi AT2 cells were enriched in nLung (Fig. 4A–C). Average expression of other AT2 markers, assigned as AT2 module score, was also lower in SFTP-lo nodule-specific AT2 cells compared with SFTPC-hi normal AT2 cells (Fig. 4D). These data suggest a possible loss of AT2-functionality in nodule-specific SFTPC-lo AT2 cells.
Figure 4.

Deregulation of AT2 cells in subsolid nodules. A, AT2 cells from sub- and solid nodules cluster separately from AT2 cells from matching nLung. Colors represent nodule types. B, Two major AT2 clusters identified on the basis of high (hi) or low (lo) SFTPC expression. C, Proportions of SFTPC-high and -low cells obtained from nodules and nLung. Values indicate cell numbers in the respective groups. Colors indicate tissue types. D, Violin plots of module scores representing AT2 lineage marker expression in various nonimmune cell populations. E, Volcano plot representing DEGs between nodule- and nLung-derived AT2 cells. P values (y-axis) and fold changes (x-axis) were calculated by the edgeR approach. The red dots indicate DEGs identified by both edgeR and MAST approaches. F, GSEA plots of the top deregulated pathways in AT2 cells from subsolid/solid nodules compared with nLung.
Single-cell and pseudo-bulk analyses of AT2 cells from nodules and nLung samples identified 372 DEGs (Fig. 4E; Supplementary Table S5). In addition to SFTPC, EDNRB, which encodes a nonselective endothelin B receptor that induces apoptosis upon activation, was one of the most downregulated genes in nodule-specific AT2 cells. Downregulation of EDNRB, possibly resulting from hypermethylation of the EDNRB promoter (29), has been associated with a poor prognosis in patients with ADC (30). Many of the other top upregulated genes in nodule-specific AT2 cells, including SPINK1, CEACAM6, IGFBP2, and ABCC3, have been implicated in tumorigenesis in various cancers, including NSCLC (31–33). For example, SPINK1 was shown to promote tumor progression by altering the TME (34) and correlated with poor prognosis in NSCLC (35). Pathway analysis indicated downregulation of the apoptosis and NFκB pathways and upregulation of genes regulating extracellular matrix (ECM) degradation and drug metabolism in nodule-specific AT2 cells (Fig. 4F). Collectively, these findings demonstrate upregulation of oncogenic pathways in AT2 cells in early-spectrum lung ADC, accompanied by loss of the characteristic AT2 gene expression, which is consistent with the hypothesis that AT2 cells could potentially foster a tumorigenic microenvironment.
Enrichment of dysregulated cancer-associated fibroblasts and proangiogenic ECs in subsolid nodules
Using known signature gene modules (36), we identified 8,280 ECs that formed nine clusters corresponding to capillary (niC2–3, niC6–7, and niC23), arterial (niC22), venous (niC14), immature (niC15), and lymphatic (niC31) ECs (Fig. 5A; Supplementary Fig. S7A–S7E; Supplementary Table S3). More than 50% of cells in the venous (C14) and immature (C15) clusters originated from subsolid and solid nodules (Fig. 5B). The top DEGs in venous and immature clusters were plasmalemmal vesicle-associated protein (37) and markers of endothelial stalk (ACKR1; ref. 38) and tip cells (INSR, LAMA4; refs. 36, 39), which exist in new blood vessels (Fig. 5C). Gene module analysis confirmed that nodule-associated ECs highly expressed immature stalk and tip cell gene programs (Fig. 5D), which are responsible for EC proliferation and migration during angiogenesis (36, 38). These findings indicate an increased angiogenic potential of subsolid nodules.
Figure 5.

Cancer-associated endothelial cells and fibroblasts enriched in subsolid nodules. A, UMAP plot visualizing the distribution of five EC subtypes in nLung and subsolid/solid nodules. B, Proportion of EC subtypes in nodules and normal lung. Cap, capillary; Imma, immature; Lymph, lymphatic EC. Color indicates tissue type. C, Expression of top DEGs in cancer-associated clusters C14 and C15 compared with other EC clusters. D, Violin plots of module scores of cancer-associated immature stalk and tip-like signatures in EC clusters. E, UMAP plot visualizing seven fibroblast clusters in nLung (left) and subsolid/solid nodules (right). F, Expression of CAF subtype 1 (CAF-S1) markers in fibroblast clusters. G, Fibroblast clusters characterized by tissue-based contribution (left) and gene modules associated with normal fibroblasts, immune regulating CAF-S1 (middle), and six CAF-S1 subgroups (right). The horizontal bar plot (left) indicates tissue type proportions in each cluster. Heat maps represent average scores of gene modules (columns) in clusters (rows). H, Violin plots illustrating the distribution of ecm-myCAF and IFNG-iCAF signature scores in clusters expressing markers associated with normal fibroblast (niC11 and niC18) and immune-regulating CAF-S1 (niC8, 9, and 27).
A total of 5,886 fibroblasts formed seven clusters (Fig. 5E; Supplementary Fig. S7A-S7D). Although most clusters were similarly distributed between nodules and nLung, niC9 cluster, expressing canonical cancer-associated fibroblast (CAF) markers FAP, S100A4, and PDGFRA (Fig. 5F), was predominantly present in nodules (Figs. 5E and G). Recent studies identified FAPhiSMAhiCD29med-hi CAF, named as CAF subtype 1 (CAF-S1), which mediated immunosuppression in breast cancer (40). CAFs were further stratified into six subtypes based on the expression of gene ontology signatures (41). Using these gene modules, niC11 and niC18 were identified as healthy fibroblasts and niC9 as CAF-S1 (Fig. 5G). Fibroblasts in niC9 also highly expressed gene signatures associated with ECM remodeling myofibroblasts (ecm-myCAF) and response to IFNγ and cytokine-mediated signaling pathways in inflammatory CAFs (INFG_iCAF; Fig. 5G and H).
LR interactions in the microenvironment of the developing tumor
Interactions among various cellular phenotypes were investigated by LR analysis. The LR assessment is based on the premise that the upregulation of ligand expression in cell S (source) can either induce expression of the related receptor in cell R (receiver) or enhance migration of cell R toward cell S (defined by upregulation of migratory genes in local infiltrating cell R). The LR interaction strength in each sample was defined as the product of ligand and receptor expressions in related cells, as suggested by Kumar and colleagues (20), and was dichotomized on the basis of the comparison between subsolid/solid nodules and nLung. A high number of LR interactions was observed among nonimmune cells from nodules, including SFTPC-lo AT2, proangiogenic ECs, and CAFs compared with their nLung-associated counterparts (Fig. 6A). Although the interaction between AT2 and ECs in nodules was found to be bidirectional, where each cell type acted equally as source and receiver, CAFs interacted with AT2 cells and ECs predominantly as source cells (expressing ligands rather than receptors). In contrast, the number of interactions among immune cells was sporadic (Supplementary Fig. S8). Evaluating interactions between nonimmune and immune cells revealed that B, NKT, and T cells, including CD8+ T and Tregs, frequently acted as receiver cells whereas myeloid cells, including DCs and monocytes, served as source and receiver cells (Fig. 6B–C).
Figure 6.

Analysis of ligand–receptor interactions between different cell types. A, Interactions among nonimmune cells. Values on lines indicate the number of activated LR interactions in nodules by comparing their scores to that of nLung. B and C, Heat maps illustrating the number of unidirectional interactions from nonimmune to immune cells (B) and vice versa (C) activated in nodules compared with nLung. Rows indicate the source cells expressing ligand, whereas columns represent receiver cells expressing receptor genes. D and E, Circos plot illustrating the unidirectional LR interactions among non- and immune cells (D) and among immune cells (E) due to the differentially expressed ligands and receptors between nodule and nLung in the indicated cell types.
Two stringent interactome networks were reconstructed, which required related ligands and receptors to be differentially expressed in subsolid/solid nodules compared with nLung (Fig. 6D–E). The first network showed that compared with nLung there was an upregulation of the CXCL12–CXCR4 axis in early-spectrum lung ADC in which CAFs expressing CXCL12 communicated with CXCR4-expressing AT2 and immune cells, including NKT, DC, macrophages, and neutrophils (Fig. 6D). In the second network, enhanced CD40 signaling was observed among immune populations in nodules. DC and monocytes expressing CD40 interacted with either CD40LG ligand in Tem and Tcm cells, an interaction that is critical for T-cell–mediated DC-licensing, or LTB ligand in NKT, CD8, Treg, Tem, and Tcm cells, which is instrumental in driving the formation of tertiary lymphoid structures (Fig. 6E). These results demonstrate a dynamic cross-talk among cells in the TME and highlight the profound role of CAFs and myeloid cells in modulating the T-cell responses in early-spectrum lung ADC.
Discussion
In the present study, we used prospective and retrospective cohorts of patients with pulmonary nodules to provide a comprehensive atlas of the cellular composition of early-spectrum/stage lung cancer by scRNA-seq, FC and MIF. The key findings of scRNA-seq are (i) a reduction of essential cell mediators of antitumor immunity, including NK, NKT, and cytotoxic CD8+ T cells accompanied by an increase in Treg/Tem CD4+ ratio and (ii) altered gene expression signatures in AT2 cells, ECs, and CAFs known to be associated with angiogenesis, inflammatory responses and ECM remodeling in nodules compared with nLung. LR analyses revealed specific cross-talk between immune and nonimmune cells within the TME of nodules that could potentially orchestrate the expansion of the suppressive immunophenotypes and immune escape. MIF staining confirmed progressive immunosuppression across the continuum of lung ADC characterized by enrichment of Tregs and decrease of GZMB+CD8+ cytotoxic T cells, consistent with recent studies (9, 42).
Dysfunction in both the innate and adaptive arms of immunity contributed to immunosuppression in subsolid nodules. Nodule-infiltrating NKT cells exhibited decreased expression of genes associated with cytolytic capacity and elevated expression of cytokines associated with NKT2, which have the capacity to mitigate effective immune recognition. One of these cytokines, amphiregulin (AREG), was significantly upregulated in subsolid nodules. AREG activates EGFR signaling leading to increased PD-L1 expression in tumor cells (43) and enhances Treg-mediated immunosuppression (44). AREG was recently found to circumvent tyrosine kinase inhibitor as well as immunotherapy effectiveness for lung cancer (45, 46). We found a significant enrichment of Tregs resulting in a reduced CD4+ Tem/Treg ratio as well as a reduction in the frequency of CD8+ T cells expressing GZMB, a critical mediator of cytotoxicity, in subsolid nodules. The effectiveness of immune checkpoint blockade therapy for NSCLC, together with our observation of the significantly increased Treg abundance in pulmonary premalignancy, suggests that immunotherapies may be effective for lung cancer interception (4, 47). Further research is required to define the patient populations at highest risk and most likely to benefit from immune-based interception.
Tumor-associated DC subsets encompass diversity in developmental origin, phenotype, gene expression, and function (48). DC1 and DC2 subsets play central roles in dictating the fate of the developing tumor. We observed increased DC2 infiltration in subsolid nodules, which correlated with increased CD4+ T and B cells. Recent studies suggest that MHC II neoantigens and DC2 can potentially shape an effective antitumor response (49, 50). The DC2 gene signature has been strongly associated with a positive prognosis in patients with lung ADC (51). DC1 and cytotoxic GZMB+ CD8+ T cells were diminished in most invasive subsolid nodules. Paucity of DC1s could be a result of decreased NK and NKT cells, which attract DC1 to the tumor site to facilitate cytotoxic immune responses (52). DC1 possess a heightened antigen cross-presentation capacity for activation of CD8+ T cells (48). Intratumoral DC1 facilitate infiltration of IFNγ-secreting type-1 CD4+ and CD8+ T effectors to the tumor site via the production of CXCL9 and CXCL10 (52). This feedback loop amplifies antitumor-adaptive immune responses. Consistent with these mechanistic studies, recent reports have identified decreased density of NK and DC1 within the TME as an indicator of poor prognosis and decreased responsiveness to ICI (53). Studies suggest that the intratumoral injection of autologous gene-modified DC may help to resolve this DC deficit and facilitate recruitment of effector T cells (51, 54, 55).
Alterations of gene signatures of nonimmune cells in subsolid nodules, including downregulation of epithelial AT2 cell lineage markers, are consistent with the observations in organoid models in the context of KRAS activation (56). Interrogation of subsolid-associated AT2, ECs and CAFs revealed increased gene expression signatures consistent with the promotion of angiogenesis and ECM remodeling. A CAF subtype (C9) was identified as exclusively present in nodules and highly expressed CXCL12, whose receptor CXCR4 was found to be upregulated in both AT2 and immune cells, including DCs, macrophages, and NKT cells (Fig. 6D). Via CXCR4, CXCL12 stimulates increased secretion of matrix metalloproteinases, mediating the degradation of the ECM (57). Targeting the CXCL12/CXCR4 axis was found to have synergistic effects with immune checkpoint blockade and targeted therapy in murine models (58, 59). Although tumor-intrinsic properties can define distinct immune infiltration in the TME (60, 61), the LR interaction analyses reveal potential roles of tumor–stroma elements, including CAFs and ECs, in shaping the infiltrating immune cell contexture. CAFs appear to be the predominant cell type modulating the interactions with other nonimmune and immune cells in subsolid nodules. Interrogation of CAFs may hold promise in identifying biomarkers as well as therapeutic targets for early detection and interception of lung cancer.
Our results should be evaluated in the context of the limitations of our study. Many nodules in the prospective cohorts had either KRAS or EGFR driver mutations. Although a retrospective cohort validated observations from this prospective cohort, our study was not powered to investigate changes associated with specific driver mutations. In addition, evaluation of adjacent nLung should be taken with caution given mounting evidence demonstrating field cancerization in histologically normal appearing lung tissue surrounding lung cancers (62).
In summary, our results provide insights into alterations in both immune and nonimmune cells in subsolid pulmonary nodules representing early-spectrum lung ADC. The creation of an early-spectrum lung ADC atlas will enhance our understanding of disease pathogenesis and provide opportunities for both the early diagnosis and interception of lung cancer.
Supplementary Material
DEGs between subsolid nodule and nLung C20 NKT cells
DEGs between subsolid and nLung AT2 cells
Figure S1 - S8, Table S1-S2
Summary of immune and non-immune cell clusters based on quality control metrics
Acknowledgments
This work was supported by funding from a Stand Up To Cancer-LUNGevity-American Lung Association Lung Cancer Interception Dream Team Translational Cancer Grant (grant number: SU2C-AACR-DT23–17 to S.M. Dubinett and A.E. Spira). Stand Up To Cancer is a division of the Entertainment Industry Foundation; NCI HTAN PCA 1U2CCA233238 (to S.M. Dubinett and A.E. Spira); Janssen Research and Development sponsored research agreement (PIs: J.E. Beane, S.A. Mazzilli, and J.D. Campbell); NIH/NCI MCL 1U01CA196408 (to S.M. Dubinett and A.E. Spira); NIH/NCI EDRN 1U01CA214182 (to S.M. Dubinett, M.E. Lenburg, and D.R. Aberle); Merit Review, Lung Precision Oncology Program and SDR Research funding from the Department of Veterans Affairs (to S.M. Dubinett); TRDRP 27IR-0036 (to K. Krysan); Thoracic Surgery Foundation research grant (to J. Yanagawa); STOP Cancer I.C.O.N./Natasha Girard Seed grant (to J. Yanagawa); Department of Veterans Affairs Merit Review I01BX005721 (to L.M. Tran); National Heart Lung and Blood Institute T32-HL072752 (to R. Salehi-Rad). This work used computational and storage services associated with the Hoffman2 Shared Cluster provided by UCLA Institute for Digital Research and Education's Research Technology Group. The indicated Stand Up To Cancer grant is administered by the American Association for Cancer Research, the scientific partner of SU2C.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
J. Yanagawa reports personal fees from OncLive, AstraZeneca, NCCN, Ideology, and Lewis-Brisbois-Biscard, and other support from ICONA outside the submitted work; as well as a patent for organoids and high-throughput drug screening for the treatment of sarcoma issued and image-based lung surgical intervention treatment planning issued. A.E. Prosper reports grants from American College of Radiology outside the submitted work. C. Shea reports grants from Janssen Pharmaceuticals during the conduct of the study. A.E. Spira reports personal fees from Johnson & Johnson outside the submitted work. D.R. Aberle reports grants from Boston University/Johnson & Johnson (Prime) and personal fees from 2021: Japanese Society of CT Screening outside the submitted work. S.M. Dubinett serves on the Scientific Advisory Boards for Early Diagnostics Inc. and LungLife AI, Inc. He has received research funding from Johnson & Johnson Lung Cancer Initiative and Novartis. No disclosures were reported by the other authors.
Authors' Contributions
J. Yanagawa: Conceptualization, curation, supervision, investigation, writing–original draft, writing–review and editing. L.M. Tran: Conceptualization, data curation, formal analysis, supervision, validation, visualization, writing–original draft. R. Salehi-Rad: Formal analysis, investigation, writing–review and editing. R.J. Lim: Formal analysis, investigation, methodology. C. Dumitras: Resources, investigation. E. Fung: Investigation. W.D. Wallace: Formal analysis, investigation. A.E. Prosper: Formal analysis, investigation. G. Fishbein: Formal analysis, investigation. C. Shea: Data curation, formal analysis. R. Hong: Data curation, formal analysis. B. Kahangi: Investigation. J.J. Deng: Investigation. A.C. Gower: Data curation. B. Liu: Conceptualization, writing–original draft. J.D. Campbell: Conceptualization, data curation, funding acquisition, methodology, writing–original draft. S.A. Mazzilli: Conceptualization, funding acquisition, project administration. J.E. Beane: Supervision, funding acquisition, methodology. H. Kadara: Conceptualization, data curation. M.E. Lenburg: Conceptualization, supervision, funding acquisition. A.E. Spira: Conceptualization, supervision, funding acquisition. D.R. Aberle: Conceptualization, supervision, funding acquisition, investigation, methodology, writing–original draft. K. Krysan: Conceptualization, investigation, visualization, methodology, writing–original draft. S.M. Dubinett: Conceptualization, supervision, funding acquisition, methodology, project administration, writing–review and editing.
References
- 1. Mazzone PJ, Lam L. Evaluating the patient with a pulmonary nodule: a review. JAMA 2022;327:264–73. [DOI] [PubMed] [Google Scholar]
- 2. Travis WD, Asamura H, Bankier AA, Beasley MB, Detterbeck F, Flieder DB, et al. The IASLC lung cancer staging project: proposals for coding t categories for subsolid nodules and assessment of tumor size in part-solid tumors in the forthcoming eighth edition of the TNM classification of lung cancer. J Thorac Oncol 2016;11:1204–23. [DOI] [PubMed] [Google Scholar]
- 3. McWilliams A, Tammemagi MC, Mayo JR, Roberts H, Liu G, Soghrati K, et al. Probability of cancer in pulmonary nodules detected on first screening CT. N Engl J Med 2013;369:910–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blackburn EH. Cancer interception. Cancer Prev Res 2011;4:787–92. [DOI] [PubMed] [Google Scholar]
- 5. Kadara H, Choi M, Zhang J, Parra ER, Rodriguez-Canales J, Gaffney SG, et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann Oncol 2017;28:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krysan K, Tran LM, Grimes BS, Fishbein GA, Seki A, Gardner BK, et al. The immune contexture associates with the genomic landscape in lung adenomatous premalignancy. Cancer Res 2019;79:5022–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Teixeira VH, Pipinikas CP, Pennycuick A, Lee-Six H, Chandrasekharan D, Beane J, et al. Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre-invasive lung cancer lesions. Nat Med 2019;25:517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beane JE, Mazzilli SA, Campbell JD, Duclos G, Krysan K, Moy C, et al. Molecular subtyping reveals immune alterations associated with progression of bronchial premalignant lesions. Nat Commun 2019;10:1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leader AM, Grout JA, Maier BB, Nabet BY, Park MD, Tabachnikova A, et al. Single-cell analysis of human non–small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 2021;39:1594–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leelatian N, Doxie DB, Greenplate AR, Mobley BC, Lehman JM, Sinnaeve J, et al. Single cell analysis of human tissues and solid tumors with mass cytometry. Cytometry B Clin Cytom 2017;92:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Franzen O, Gan LM, Bjorkegren JLM. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019;2019:baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017;356:eaah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 2017;14:979–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 2019;176:775–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non–small cell lung cancer by single-cell sequencing. Nat Med 2018;24:978–85. [DOI] [PubMed] [Google Scholar]
- 17. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 2015;16:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kumar MP, Du J, Lagoudas G, Jiao Y, Sawyer A, Drummond DC, et al. Analysis of single-cell RNA-seq identifies cell–cell communication associated with tumor characteristics. Cell Rep 2018;25:1458–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y, Wang R, Zhang S, Song S, Jiang C, Han G, et al. iTALK: an R package to characterize and illustrate intercellular communication. Biorxiv 2019:507871. [Google Scholar]
- 22. Feng Y, Yang T, Zhu J, Li M, Doyle M, Ozcoban V, et al. Spatial analysis with SPIAT and spaSim to characterize and simulate tissue microenvironments. Nat Commun 2023;14:2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsukerman P, Stern-Ginossar N, Yamin R, Ophir Y, Stanietsky AM, Mandelboim O. Expansion of CD16 positive and negative human NK cells in response to tumor stimulation. Eur J Immunol 2014;44:1517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bennstein SB. Unraveling natural killer T cells development. Front Immunol 2017;8:1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Crinier A, Milpied P, Escaliere B, Piperoglou C, Galluso J, Balsamo A, et al. High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity 2018;49:971–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol 2015;16:306–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 2017;169:750–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell 2019;35:588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Knight LJ, Burrage J, Bujac SR, Haggerty C, Graham A, Gibson NJ, et al. Epigenetic silencing of the endothelin-B receptor gene in non–small cell lung cancer. Int J Oncol 2009;34:465–71. [PubMed] [Google Scholar]
- 30. Wei F, Ge Y, Li W, Wang X, Chen B. Role of endothelin receptor type B (EDNRB) in lung adenocarcinoma. Thorac Cancer 2020;11:1885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blumenthal RD, Leon E, Hansen HJ, Goldenberg DM. Expression patterns of CEACAM5 and CEACAM6 in primary and metastatic cancers. BMC Cancer 2007;7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li T, Forbes ME, Fuller GN, Li J, Yang X, Zhang W. IGFBP2: integrative hub of developmental and oncogenic signaling network. Oncogene 2020;39:2243–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao Y, Lu H, Yan A, Yang Y, Meng Q, Sun L, et al. ABCC3 as a marker for multidrug resistance in non–small cell lung cancer. Sci Rep 2013;3:3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen F, Long Q, Fu D, Zhu D, Ji Y, Han L, et al. Targeting SPINK1 in the damaged tumour microenvironment alleviates therapeutic resistance. Nat Commun 2018;9:4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guo M, Zhou X, Han X, Zhang Y, Jiang L. SPINK1 is a prognosis predicting factor of non–small cell lung cancer and regulates redox homeostasis. Oncol Lett 2019;18:6899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goveia J, Rohlenova K, Taverna F, Treps L, Conradi LC, Pircher A, et al. An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell 2020;37:21–36. [DOI] [PubMed] [Google Scholar]
- 37. Wisniewska-Kruk J, van der Wijk AE, van Veen HA, Gorgels TG, Vogels IM, Versteeg D, et al. Plasmalemma vesicle-associated protein has a key role in blood-retinal barrier loss. Am J Pathol 2016;186:1044–54. [DOI] [PubMed] [Google Scholar]
- 38. Chen W, Xia P, Wang H, Tu J, Liang X, Zhang X, et al. The endothelial tip-stalk cell selection and shuffling during angiogenesis. J Cell Commun Signal 2019;13:291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stenzel D, Franco CA, Estrach S, Mettouchi A, Sauvaget D, Rosewell I, et al. Endothelial basement membrane limits tip cell formation by inducing Dll4/Notch signalling in vivo. EMBO Rep 2011;12:1135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018;33:463–79. [DOI] [PubMed] [Google Scholar]
- 41. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov 2020;10:1330–51. [DOI] [PubMed] [Google Scholar]
- 42. Xing X, Yang F, Huang Q, Guo H, Li J, Qiu M, et al. Decoding the multicellular ecosystem of lung adenocarcinoma manifested as pulmonary subsolid nodules by single-cell RNA sequencing. Sci Adv 2021;7:eabd9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol 2015;10:910–23. [DOI] [PubMed] [Google Scholar]
- 44. Zaiss DM, van Loosdregt J, Gorlani A, Bekker CP, Grone A, Sibilia M, et al. Amphiregulin enhances regulatory T-cell–suppressive function via the epidermal growth factor receptor. Immunity 2013;38:275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taniguchi H, Takeuchi S, Fukuda K, Nakagawa T, Arai S, Nanjo S, et al. Amphiregulin triggered epidermal growth factor receptor activation confers in vivo crizotinib-resistance of EML4–ALK lung cancer and circumvention by epidermal growth factor receptor inhibitors. Cancer Sci 2017;108:53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu Q, Long Q, Zhu D, Fu D, Zhang B, Han L, et al. Targeting amphiregulin (AREG) derived from senescent stromal cells diminishes cancer resistance and averts programmed cell death 1 ligand (PD-L1)-mediated immunosuppression. Aging Cell 2019;18:e13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Forde PM, Spicer J, Lu S, Provencio M, Mitsudomi T, Awad MM, et al. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N Engl J Med 2022;386:1973–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol 2020;20:7–24. [DOI] [PubMed] [Google Scholar]
- 49. Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019;574:696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4(+) T-cell immunity. Cell 2019;177:556–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang SC, Hillinger S, Riedl K, Zhang L, Zhu L, Huang M, et al. Intratumoral administration of dendritic cells overexpressing CCL21 generates systemic antitumor responses and confers tumor immunity. Clin Cancer Res 2004;10:2891–901. [DOI] [PubMed] [Google Scholar]
- 52. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 dendritic cells are required for effector T-cell trafficking and adoptive T-cell therapy. Cancer Cell 2017;31:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med 2018;24:1178–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee JM, Lee MH, Garon E, Goldman JW, Salehi-Rad R, Baratelli FE, et al. Phase I trial of intratumoral injection of CCL21 gene-modified dendritic cells in lung cancer elicits tumor-specific immune responses and CD8(+) T-cell infiltration. Clin Cancer Res 2017;23:4556–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Perez CR, De Palma M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat Commun 2019;10:5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dost AFM, Moye AL, Vedaie M, Tran LM, Fung E, Heinze D, et al. Organoids model transcriptional hallmarks of oncogenic KRAS activation in lung epithelial progenitor cells. Cell Stem Cell 2020;27:663–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ghosh MC, Makena PS, Gorantla V, Sinclair SE, Waters CM. CXCR4 regulates migration of lung alveolar epithelial cells through activation of Rac1 and matrix metalloproteinase-2. Am J Physiol Lung Cell Mol Physiol 2012;302:L846–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 2013;110:20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jung K, Heishi T, Incio J, Huang Y, Beech EY, Pinter M, et al. Targeting CXCR4-dependent immunosuppressive Ly6C(low) monocytes improves antiangiogenic therapy in colorectal cancer. Proc Natl Acad Sci U S A 2017;114:10455–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015;5:860–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity 2018;49:178–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kadara H, Wistuba II. Field cancerization in non–small cell lung cancer: implications in disease pathogenesis. Proc Am Thorac Soc 2012;9:38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DEGs between subsolid nodule and nLung C20 NKT cells
DEGs between subsolid and nLung AT2 cells
Figure S1 - S8, Table S1-S2
Summary of immune and non-immune cell clusters based on quality control metrics
Data Availability Statement
The raw RNA sequence was deposited in dbGaP (Study Accession: phs002371.v3.p1), which manages the access control. The normalized gene expression (data level 3) and cell annotation (data level 4) are also available at Human Tumor Atlas Network—Data Coordinating Center. Detailed information of data access and data levels can be found at the Data Coordinating Center (https://humantumoratlas.org/data-access). Other raw data generated in the study are available upon request from the corresponding authors.