

Abstract

D2O is commonly used as a solvent instead of H2O in spectroscopic studies of proteins, in particular, in infrared and nuclear-magnetic-resonance spectroscopy. D2O is chemically equivalent to H2O, and the differences, particularly in hydrogen-bond strength, are often ignored. However, replacing solvent water with D2O can affect not only the kinetics but also the structure and stability of biomolecules. Recent experiments have shown that even the mesoscopic structures and the elastic properties of biomolecular assemblies, such as amyloids and protein networks, can be very different in D2O and H2O. We discuss these findings, which probably are just the tip of the iceberg, and which seem to call for obtaining a better understanding of the H2O/D2O-isotope effect on water–water and water–protein interactions. Such improved understanding may change the differences between H2O and D2O as biomolecular solvents from an elephant in the room to an opportunity for protein research.
Introduction
D2O, or heavy water, is a stable isotopomer of H2O, containing deuterium instead of the most common hydrogen isotope protium. Deuterium was discovered in 1931 by H. Urey,1 who was awarded the Nobel Prize for this finding in 1934. The chemical and physical properties of D2O were first studied by G. Lewis and co-workers in the early 1930s2,3 and are very similar to those of H2O (Table 1). For this reason, D2O is often used as a solvent instead of H2O in experiments where the H atoms of water form a problem, such as in nuclear magnetic resonance, neutron scattering, and infrared spectroscopy and imaging. This holds in particular for studies of biomolecules: in both protein NMR and infrared spectroscopy and imaging,4−6 it is standard practice to use D2O as a solvent. In the case of infrared spectroscopy, this is done because the vibrational modes of the amide groups, which carry crucial information on the protein structure,7 have spectral overlap with the bending mode of H2O (both are in the 1600–1700 cm–1 frequency range). The D2O-bending frequency is 1250 cm–1, eliminating the overlap problem and making D2O the seemingly perfect replacement of H2O.
Table 1. Selected Physical and Chemical Properties of H2O and D2O8−11.
| property | H2O | D2O |
|---|---|---|
| molecular weight (g/mol) | 18.02 | 20.03 |
| melting point (°C) | 0 | 3.82 |
| boiling point (°C) | 100 | 101.4 |
| molar density (mol/L, 25.0 °C, 1 atm) | 55.35 | 55.14 |
| molecular polarizability (Å3) | 1.45 | 1.26 |
| viscosity (25 °C) | 0.891 | 1.095 |
| pH/pD (25 °C) | 6.9976 | 7.43 |
| dielectric constant (25 °C) | 78.37 | 78.06 |
The effect of H/D substitution on the kinetics of chemical reactions is well-known, and has been extensively studied and applied, for instance to study reaction mechanisms12 and to monitor protein folding.13 Interestingly, recent work shows that the kinetic effects induced by substituting D2O for H2O might also be useful for biomedical purposes:14 epithelial cells grown in a medium containing 45% D2O show significantly reduced migration and proliferation rates (Figure 1), and a similar slowdown in dynamics was observed in other cells,15−17 an effect that might find use for the storage of biological materials such as organs, or for anticancer treatment.15
Figure 1.

“Cells in slow motion.”14 (a) Epithelial cell proliferation is much slower in a D2O-rich medium. The duration of the cell cycle is roughly 17 h under normal (H2O) conditions and 44 h in water containing 45% D2O. (b) Independent of the culturing conditions, the actin cytoskeleton (orange) and the nucleus (blue) remain intact. Shown are typical pictures, which suggest that structures are comparable for the different H2O and D2O concentrations. Figure adapted from ref (14), Copyright 2021 Schnauß et al. (licensed under CC BY-NC 4.0).
While the effect of H/D substitution on kinetics is well established, it is often (implicitly) assumed that the effect of H2O/D2O substitution on the structure of biomolecules and biomolecular assemblies is small. However, although the use of isotopic substitution in spectroscopic experiments has been mostly successful, there is ample evidence that replacing H2O with D2O can alter the thermodynamic and structural properties of proteins18−35 and even the formation process and structure of protein assemblies.14,25,33,36−41 The fact that H2O/D2O replacement can change the structure of biopolymers and their assemblies should not come as a complete surprise: the hydrogen-bonda structures of H2O and D2O are known to be different,42 and hydration and the hydrophobic effect are essential for all biomacromolecules, ranging from polysaccharides to proteins. Hydrating water molecules create a water network around solutes that not only acts as structure stabilizer but also mediates intra- and intermolecular interactions. As stated recently by Fischer et al.,43 hydration represents “an additional evolutionary constraint upon protein sequence to maintain ligand binding and modulate the affinity of those interactions”, to which we might add that since evolution has optimized protein structure and dynamics in H2O rather than D2O, and since the hydrogen-bond structures of these two liquids are different, differences in structure and dynamics are to be expected when replacing one with the other.
The H2O/D2O-induced changes in biomolecular structure seem to call for more detailed studies of the difference between liquid D2O and H2O, but they also suggest fascinating new research opportunities. In this Perspective, we first briefly describe the differences between H2O and D2O; then we summarize and discuss the existing experimental evidence for isotope-induced structural changes in biomolecules and biomolecular assemblies; finally, we discuss the current challenges and perspectives, in particular the possibility of using D2O to investigate the role of hydration in protein stability and interactions.
H2O versus D2O
The interplay of nuclear quantum effects (NQEs) underlying the physical and chemical differences between liquid D2O and H2O is quite subtle. Simply put, the low mass of the hydrogen atom makes it behave more as a delocalized quantum particle than the heavier deuterium. This delocalization can have a substantial effect on the hydrogen bond strength.10 Specifically, for an O–H···O hydrogen bond, the hydrogen-bond strength is a function of the O···O distance (the shorter, the stronger) and the O–H···O bond angle (the straighter, the stronger). The larger distance spread for H vs D leads to a strengthening of the H-bond, while the larger angular spread leads to a weakening. Hence, these two nuclear quantum effects have contrary consequences for the H-bond strength. Depending on the details of the H-bond, one or the other effect may dominate, resulting in a weakening or strengthening of H-bonds upon isotopic substitution. Short hydrogen bonds are typically strengthened due to NQEs, whereas long ones are weakened.10 Here we summarize the most important differences that are generally agreed upon in the literature, focusing on the points that are relevant for understanding how replacing H2O with D2O can change the structures of biomolecules and biomolecular assemblies.
The structure of liquid D2O and water has been investigated using different methods, in particular X-ray, γ-ray, and neutron scattering. By combining X-ray measurements with molecular simulations, it was found that the covalent bond between oxygen and protium (O–H) is 3% longer with respect to the one between oxygen and deuterium (O–D), see Figure 2 (neutron scattering studies indicate a somewhat smaller isotope effect on the covalent bond length10). In D2O, the hydrogen-bond network is more tetrahedral than that in H2O and the hydrogen-bond coordination number is higher,42 both effects indicating stronger hydrogen bonds and a more structured hydrogen-bond network. The average hydrogen-bond distance (the O···O distance of two hydrogen-bonded water molecules) is 4% longer in D2O, as is also reflected in its lower molar density compared to H2O (cf. the situation in ice, where the hydrogen bonds are also stronger than in liquid water). In ab initio calculations on hydrogen-bonded oligomers, it was also found that the hydrogen-bond strength is 0.2–0.3 kcal/mol larger in D2O than in H2O.44 Finally, the macroscopic thermodynamical properties (such as the specific heat and the melting point) of H2O and D2O also indicate stronger hydrogen bonding between D2O molecules, with a difference in hydrogen-bond energy similar to that found in the ab initio calculations.10,45
Figure 2.

Average lengths of the covalent and hydrogen bonds in liquid H2O (a) and D2O (b).
Isotope-Induced Effects on Biomolecular Structure
We will now discuss examples of how the stronger hydrogen bonding in D2O can influence biomolecular structure and stability. First, we discuss the effects on individual biomolecules and then the more recently discovered D2O-induced effects on protein assemblies.
Effects of Replacing H2O with D2O on Protein Stability, Structure, and Hydration
D2O-induced changes in protein stability depend in a complicated manner on changes in the (local) hydration, with both enthalpic and entropic contributions. Yet, the simple argument that the stronger hydrogen bonding between D2O molecules suppresses protein unfolding, favoring compact, folded proteins with minimal hydration seems to be sound. In Table 2, we give an overview of experimental results demonstrating the effect of D2O on biomolecular stability, structure, and rigidity, based on (and somewhat extending) the excellent overview given in ref (26). Most studies focus on the conformational stability in H2O and D2O. This is motivated by the potential use of D2O as a way to slow down thermal degradation, especially in pharmaceutical applications. Several studies have shown that the native or folded states of globular proteins such as bovine serum albumin (BSA), lysozyme, and tubulin are more stable in D2O than in H2O.18−21 For instance, using differential scanning calorimetry (DSC), it was found that the denaturation temperature of lysozyme and BSA is 2–3 °C higher in heavy water than in water.18 Circular dichroism (CD) experiments, which are more structure-sensitive than DSC measurements, showed that the onset temperature of the irreversible thermal denaturation (i.e., the temperature of the irreversible change of the secondary structure) of BSA is 58 °C in D2O while it is 50 °C in H2O.19 Upon heat-treatment at 65 °C, BSA also retains a larger percentage of monomers in heavy water than in water (85% versus 75%, respectively), again indicating that the BSA monomeric form is more stable in D2O.20 Similar results have been found for other, nonglobular proteins, such as acyl carrier proteins,22 collagen,24 ribonuclease A27 and Drosophila signal-transduction protein Drk.26 Similar enhanced stability of the folded state was also observed for κ-carrageenan, which undergoes to a liquid-to-gel transition by forming double helices, that are stabilized significantly more in D2O.25
Table 2. Effects of D2O on the Properties of Proteins and Other Biomoleculesa.
| biomolecule | method | effect |
|---|---|---|
| bovine serum albumin18 | DSC | enhanced stability of the native state, TD2Od – TH2Od ≈ 2–3 °C |
| bovine serum albumin19 | CD | enhanced stability of the native state Irr. TD2Od – Irr. TH2Od ≈ 8 °C |
| bovine serum albumin20 | DLS, Fl, UV–vis, SE-HPLC | enhanced stability of the native state monomer % at 65 °C: 85% in D2O, 75% in H2O |
| lysozyme18 | DSC | enhanced stability of the native state, TD2Od – TH2Od ≈ 2–3 °C |
| tubulin21 | CD, DSC, Fl | enhanced stability of the native state, TD2Od – TH2Od ≈ 3 °C |
| acyl carrier proteins22 | NMR | enhanced stability of the native state, ΔGD2ON→U = 2.3 kcal/mol; ΔGH2ON→U = 1.8 kcal/mol |
| collagen peptides24 | CD, DSC | enhanced stability of the folded state, TD2Om – TH2Om ≈ 4 °C |
| ribonuclease A27 | DSC | enhanced stability of the native state, TD2Om – TH2Om ≈ 4 °C |
| Drosophila signal transduction protein26 | NMR | enhanced stability of the folded state, TD2Om – TH2Om ≈ 12 °C |
| κ-carragenean25 | DSC | enhanced stability of the folded state, TD2Ogel→liq – TH2Ogel→liq ≈ 3 °C |
| elastin-like peptides28 | DSC, CD, IR | enhanced stability of the collapsed state, Propensity to form β-turn/β-aggregate, LCSTH2O – LCSTD2O ≈ 2–5 °C |
| peptides containing alanine29 | CD | propensity for PPII structure: 5–200% higher PPII signal in D2O |
| plastocyanin32 | MD | altered solvent–protein interactions: 10–30% reduction of protein–water H-bonds |
| test polypeptides34 | MD | altered solvent–protein interactions |
| agarose (Ag2)33 | NMR | lower solvent–polysaccharide affinity, NH2Ow/ND2Ow ≈ 3.8 |
| ribonuclease T131 | luminescence | increased protein rigidity, IPLD2O = 36 ms, IPLH2O = 28 ms |
| β-lactoglobulin31 | luminescence | increased protein rigidity, IPLD2O = 44 ms, IPLH2O = 30 ms |
| liver alcohol dehydrogenase31 | luminescence | increased protein rigidity IPLD2O = 819 ms, IPLH2O = 630 ms |
| alkaline phosphatase31 | luminescence | increased protein rigidity, IPLD2O = 2142 ms, IPLH2O = 2060 ms |
| apo-azurin31 | luminescence | increased protein rigidity IPLD2O = 603 ms, IPLH2O = 564 ms |
| TAS1R2/TAS1R3 receptor30 | MD | smaller radius of gyration RD2Og is ≈3% smaller than RH2Og |
| azurin,35 lactoglobulin, ribonuclease | MD | smaller radius of gyration RD2Og is ≈1% smaller than RH2Og |
Abbreviations: Td = denaturation temperature; Irr. Td = irreversible denaturation temperature; Tm = melting temperature of the native state; T0 = transition temperature from folded-to-unfolded; Rg = radius of gyration; IPL = intrinsic Trp phosphorescence lifetime; ΔGN→U = Gibbs energy of unfolding; Tgel→liq = gel-to-liquid transition temperature; LCST = lower critical solution temperature; Nw = number of hydration waters per mass unit of agarose; DSC = differential scanning calorimetry; SE-HPLC = size exclusion high-performance liquid chromatography; CD = circular dichroism; DLS = dynamic light scattering; Fl = fluorescence measurements; NMR = nuclear magnetic resonance; MD = molecular dynamics simulations.
Part of this table is taken from ref (26).
The increased stability of folded and native structures in D2O indicates a stronger tendency to adopt a more compact, less solvent-exposed conformation in this solvent. For instance, a D2O-induced tightening of the helical structure has been proposed for actin, based on combined rheological and fluorescence experiments.14 Similarly, Cremer et al. have shown that elastin-like polypeptides (ELPs) undergo a hydrophobic collapse that is accompanied by the formation of β-turn structures, which are significantly more stable in D2O.28 Increased stability of intermolecular β-sheet structures in D2O has been suggested for insulin dimers because of the 2-fold slower assembly kinetics in heavy water with respect to water, as observed with infrared and two-dimensional infrared spectroscopy, and because of a larger fraction of dimer in D2O than H2O in the initial structures as revealed by molecular simulations based on solution-phase small-angle X-ray scattering experiments.39 This again suggests a general preference for a more compact conformation in D2O. Moreover, specific secondary structures can be enhanced when proteins are dissolved in D2O. Circular-dichroism studies by Chellgren et al. have demonstrated that peptides containing alanine have a stronger propensity to form polyproline II (PP II) structure in D2O than in H2O.29 Since it is believed that the PP II conformation perturbs the bulk hydrogen-bond network of the surrounding water less strongly than does an α-helical conformation, this effect was attributed to the increased energetic cost of protein solvation in D2O.
The difference in protein stability and the preference for PP II structure suggest that interactions between solvent and protein might be modified in D2O compared to H2O, leading to changes in the intraprotein hydrogen-bond network. This possibility has been investigated mostly by means of molecular dynamics simulations of various biomolecules, such as plastocyanin,32 RNA hairpins,23 and peptides.34 Interestingly, in ref (32), it was observed that a reduction of the number of hydrogen bonds between solvent and protein occurs mostly when polar and positively charged side groups are involved, while the opposite is observed for negatively charged side groups. Overall, however, a 10–30% reduction in the number of water molecules engaged in hydrogen bonds with the protein was observed in D2O compared to H2O, which was correlated to the enhancement of intramolecular interactions in this solvent.32 A lower affinity between D2O and solute was also observed in NMR studies on agarose.33 The increased rigidity which Cioni et al. have observed for different proteins (see Table 2) also supports the idea that protein–solvent interactions are altered in D2O:31 using luminescence methods it was found for 5 proteins out of the 7 analyzed that D2O increases protein rigidity, with a protein-dependent rigidity enhancement. In this respect it is interesting to note that some proteins crystallize more efficiently in D2O than in H2O,46 a phenomenon that in the case of ref (46) was even accompanied by a difference in crystal symmetry and structure (whereas in general protein crystal structures seem to be independent of whether H2O or D2O is used47−49). The D2O-induced damping of conformational fluctuations can be attributed to stronger solvent–solvent interactions,31 which reduce protein hydration and promote intramolecular interactions (as was observed in ref (32)). The reduction in structural fluctuations in D2O may thus be explained by the fact that water–protein interactions can destabilize proteins by lowering the free-energy barriers between different conformations.
We conclude our list of proteins with the well-known and intriguing fact that D2O tastes sweet. A recent molecular-dynamics study of this isotope effect by the Jungwirth group30 has shown that the transmembrane part of the human sweet-taste sensor protein is more compact, stiffer, and subject to less structural fluctuations in D2O than in H2O (Figure 3). This study again supports the idea of a reduction in protein hydration in D2O compared to H2O. Indeed, in a more recent study the same group has found that in D2O, water has a stronger propensity to form water/water hydrogen bonds than water/amino-acid hydrogen bonds (interestingly, this behavior does not follow the hydrophobicity scale of the amino acids).35 It was also found that globular proteins (azurin, lactoglobulin, and ribonuclease) are significantly more compact in D2O than in H2O. Jungwirth et al. conclude that “D2O is a somewhat worse solvent for biomolecules than H2O. This also implies that association between proteins or between a protein and a biomembrane may be positively affected by water deuteration”. In the next section, we will see experimental results that support this idea.
Figure 3.

Differences between the behavior of the transmembrane part of the human sweet taste receptor in H2O vs D2O. (a) Structure of the TMD of the TAS1R2/TAS1R3 receptor with the probability density (volumetric map) of H2O (blue) or D2O (red) molecules within 10 Å of the protein. The conserved water molecules in the X-ray templates are shown in cyan. Water molecules predicted with the software OpenEye52 are shown in licorice representation. (b) Time evolution of the radii of gyration in H2O (blue) and D2O (red) from three microsecond time scale simulations (separated by vertical dashed lines) with total mean values as dashed lines, showing that the protein is more compact in D2O. (c) Snapshot of the transmembrane part of the human sweet taste receptor color-coded that red/blue represents parts more/less rigid in D2O vs H2O. The embedding lipid membrane is represented in gray. (d) Difference in root-mean-square fluctuations in MD trajectories. Negative/positive values mean that structures are more/less rigid in D2O than in H2O. The red line represents the sum of all residues. INT, intracellular; EXT, extracellular. Adapted from ref (30), copyright 2021 Ben Abu et al. (licensed under CC BY).
D2O-Induced Changes in Protein Assemblies and Networks
We have seen that D2O increases the stability of the folded state of proteins, in particular promoting the formation of secondary structures that least disrupt the hydrogen-bond network of water, and that protein hydration is reduced in D2O. More recently, it has become clear that these changes at the molecular level can affect the propensity and mechanisms of aggregation/assembly of biopolymers into larger supramolecular structures, leading to different mechanical and thermodynamic properties of the final aggregate/assembly (Table 3). In particular, Salvatella et al. have found that androgen receptors have a stronger tendency to form biomolecular condensates by liquid–liquid phase separation (LLPS) in D2O than in H2O.36 Interestingly, in this study, it was shown that replacing less than 10% water (as is common in NMR) with D2O can already significantly affect the phase equilibrium of the condensation, with a decrease of the cloud point by 0.5 °C for each added percent of D2O, and that the size of the condensates becomes larger with increasing amount of added D2O. These changes were attributed to the enhancement in D2O of the intermolecular interactions that drive the initial oligomerization. Similarly, an elegant study by Beckett et al. has shown that the dimerization of the Escherichia coli protein BirA is more favorable in D2O than in H2O, with a dimer dissociation constant that is 10 times smaller in the former.50 A similar D2O-induced alteration of the aggregation propensity (and possibly the final aggregate size) has been proposed for BSA aggregates, based on thioflavin fluorescence, turbidity, and circular dichroism experiments.19,20,51
Table 3. Effects of D2O on Biomolecular Self-Assemblya.
| protein | method | effect |
|---|---|---|
| Escherichia coli protein BirA50 | SE | increased binding energy, KH2Odim/KD2Odim ≈ 10 |
| androgen receptor36 | NMR, DLS, microscopy | enhanced condensation, larger condensates 25 °C shift of cloud point at a H2O/D2O fraction of 1:1 |
| κ-carrageenan25 | rheology | faster assembly, higher elastic modulus, G′D2O/G′H2O ≈ 1.1–1.2 |
| gelatin37 | U-tube, rheology | faster assembly, higher shear modulus, rD2O/rH2O ≈ 2.5, GD2O/GH2O ≈ 3 |
| casein38b | rheology | faster assembly, higher elastic modulus: Gel.On.D2ORG = 9.1 ± 0.1 min; Gel.On.H2ORG = 14.6 ± 0.1 min; Gel.On.D2OTG = 1.3 ± 0.4 min; Gel.On.H2OTG = 11.3 ± 1.1 min; G′D2ORG = 1636.7 ± 75.7 Pa; G′H2ORG = 1183 ± 55.1 Pa; G′D2OTG = 504 ± 27.7 Pa; G′H2OTG = 210 ± 26 Pa |
| insulin39 | 2DIR, IR, Fl | slower assembly, τH2Olag ≈ 16 h; τD2Olag ≈ 20 h |
| α-synuclein40 | Fl, NMR, SANS | faster assembly, τH2Olag ≈ 34 h; τD2Olag ≈ 23 h (0.150 M NaCl) |
| actin52 | static light scattering | formation of multifilament bundles in D2O, DCRD2O(70%)/DCRH2O ≈ 2.5 |
| agarose33 | turbidity | change in the network, τD2O/τH2O ≈ 1.1–1.3 |
| pectin41 | SAXS | change in network fractal dimension |
Abbreviations: Kdim = equilibrium dissociation constant for dimerization; τlag= lag time; G′ = elastic modulus at a frequency of 1 Hz; r = rate of initial gelation; G = shear modulus; DCR = derived count rate (light-scattering intensity); Gel.On. = gelation onset; τ = initial turbidity; SE = sedimentation equilibrium measurements; 2DIR = two-dimensional infrared spectroscopy; SAXS= small angle X-ray scattering; SANS = small-angle neutron scattering.
Two methods were used to induce gelation, referred to as RG and TG.
Several studies have shown a significant difference in protein assembly rates in water and D2O, with assembly occurring faster in the latter. For instance, the aggregation and simultaneous double-helix formation of κ-carrageenan occurs faster in D2O than in H2O.25 Faster aggregation in D2O was also observed for gelatin,37 casein,38 and bovine serum albumin.19 These examples all show faster assembly in D2O, but self-assembly processes can also become slower in D2O. Recently, a ground-breaking study by Cho et al. has shown that amyloid formation of insulin occurs slower in D2O than in H2O (Figure 4).39 This effect was attributed to the presence of intermediates that adopt intermolecular beta-sheet structures, which are more favored in D2O than in H2O. Using D2O as a solvent instead of H2O increases the free-energy barrier for unfolding these intermediates, which is a necessary step for the final fibril formation. A similar enhancement of oligomer stability in heavy water was suggested for transthyretin tetramer.49 Interestingly, it was recently found that the fibrillization of alpha-synuclein (the protein responsible for Parkinson’s disease) proceeds faster in D2O than in water.40 This acceleration was attributed to enhanced protein–protein interactions in D2O that facilitate the refolding of alpha-synuclein, which is required for initiating its fibrillization.
Figure 4.
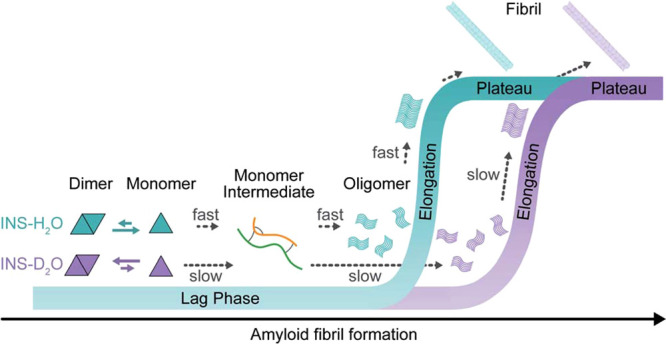
Insulin (INS) fibrillization kinetics in H2O and D2O and proposed fibrillization mechanism explaining the slower assembly in D2O. Reproduced from ref (39), Chun et al., published by the Royal Society of Chemistry (licensed under CC BY-NC 3.0).
Surprisingly, not only protein-assembly kinetics but even the viscoelastic properties of biopolymer networks can be different in D2O and H2O. The mechanical properties of reconstituted actin networks are affected by using D2O instead of H2O: in D2O the actin filaments behave as a transiently cross-linked network rather than the typical behavior of an entangled network (as is observed in H2O). This peculiar behavior in D2O was recently explained by the finding that D2O induces the formation of multifilament bundles, leading to a structural reorganization of the actin network and different mechanical properties.52 The difference in the network structure was attributed to a larger stickiness between actin filaments in D2O because of enhanced intermolecular interactions in this solvent.14,52 Similarly, the elastic modulus of gels formed by the aggregation of κ-carrageenan is ∼10–20% higher in D2O than in H2O because of the larger number of cross-links formed between the chains.25 Such increased network rigidity has also been observed in gelatin and casein gels.37,38 In contrast, Brenner et al. found that in agarose gel the mechanical properties are the same in D2O and H2O, even though D2O does enhance the stability of the helical structure and gives rise to gels with larger heterogeneity on the micrometer scale (and not the nanometer scale).33 Finally, an intriguing topological difference in biopolymer-network structure has been found in the case of pectin, for which recent experiments have shown that the fractal dimension of the gel network formed is higher in D2O than in H2O (indicating that in D2O the gel is more clustered),41 an observation that again “highlights the need to be mindful of changes induced when substituting D2O in systems with significant hydrogen bonding”.41
The Origin of D2O-Induced Changes in Stability and Structure
In D2O, biopolymers are exposed to a more strongly hydrogen-bonded water network,42 and therefore creating a solvation cavity to accommodate the protein (or increasing the solvent-exposed surface area of a protein) is energetically less favorable in D2O because of the additional enthalpic cost required to break the water hydrogen bonds. This energetic loss is enhanced when the solvent needs to reorganize around nonpolar groups, and hence hydrophobic patches have a stronger tendency to cluster in D2O than in H2O, an effect we may refer to as isotopically enhanced hydrophobic effect. However, a theoretical analysis by Graziano and Pica has shown that the H2O/D2O effect on the hydrogen-bond structure may not be sufficient to explain D2O-enhanced protein stability.11 Due to the lower molecular polarizability of D2O, van der Waals attractive interactions are less favorable in D2O, and thus fewer interactions take place between protein and water. Reduced van der Waals interactions affect the binding affinity of D2O to biomacromolecules, which may lead to changes in the hydration shell surrounding the biomolecules.23,35 The combination of reduced van der Waals interaction and the higher enthalpic cost of water–water hydrogen-bond breaking will likely change the hydration capability of D2O with respect to H2O in a synergistic way. Since contacts between water and protein can reduce the free energy barrier between the different protein conformations, the lower number of water–protein interactions in D2O will lead to structurally more stable and less fluctuating proteins, as reported in the literature (Table 2). This proposed stabilization mechanism is also suggested in a recent study by Haidar et al.53 From collision-induced unfolding and ion-mobility mass spectrometry, it was found that the stability of lysozyme, cytochrome c, and bovine ubiquitin in the gas phase is independent of whether the protein is hydrogenated or fully deuterated, in contrast with the increased stability of these proteins in D2O solution, again indicating that the changes in protein properties are due to solvent effects. This idea seems to be further confirmed by the general absence of significant differences between the crystal structures of hydrogenated and perdeuterated proteins.47,49 A decrease in water–protein interaction in D2O compared to H2O is also consistent with the enhanced rigidity observed, for instance, in collagen peptides, where intramolecular hydrophobic interactions are minimal and thus enhanced hydrophobic effect alone cannot explain the increased rigidity.24
We have seen that biomolecular assembly can occur at different rates in D2O and H2O (Table 3 and Figure 4). If the aggregation is driven by hydrophobic or hydrophilic interactions, the kinetics are expected to be different in D2O. As discussed before, D2O enhances the hydrophobic interactions (enhancing the aggregation) and has reduced protein hydration compared to H2O. This latter effect implies that the desolvation enthalpy, i.e., the energy required to break the hydrogen bonds between water and hydrophilic groups to allow the formation of bonds between hydrophilic groups, is lower in D2O than in H2O. This is consistent with the faster assembly rate reported for several systems.25,37,38 However, if the aggregation process involves the formation of intermediates stabilized by hydrophobic interactions, the assembly might be slower in D2O, as observed in the case of amyloid formation.39 To form fibrils, intermediates have to undergo partial unfolding, a process that is energetically more unfavorable in D2O since the intermediates are more stable due to the enhanced hydrophobic effect.
From Elephant in the Room to Opportunity for Protein Research
Although in general replacing H2O with D2O has a limited effect on protein structure (as is demonstrated by the large number of successful studies in which this procedure was used), the experiments and simulations discussed above show that replacing H2O with D2O can in some cases significantly change the structure and stability of proteins and protein assemblies. On the one hand, this means that experiments on proteins in which H2O has been replaced with D2O should be interpreted with caution. On the other hand, the possibility of “tuning” the hydration strength by varying the isotopic composition provides a unique tool to investigate protein hydration, and might be useful for gaining a better understanding of the role of water in defining protein structure. Water strongly influences the properties of proteins and is also believed to regulate and mediate protein–protein/ligand interactions in many biopolymers, such as collagen or silk fibroin, and water is also believed to play a crucial role in determining collagen interactions with minerals in bone tissue.54 Experiments designed to investigate protein hydration usually measure how the protein properties change upon varying the solvent, for instance, by replacing or mixing water with an organic solvent. This clearly changes the protein hydration but unfortunately also modifies many other solvent properties, such as the dielectric constant and the molecular size, which might affect protein intra- and intermolecular interactions. Replacing water with D2O is a unique method to specifically modify the water hydrogen bonding without changing the other solvent properties. Comparing protein behavior in H2O and D2O and their mixtures thus constitutes an elegant way to determine specifically the contribution of water hydrogen bonding to the physical and chemical properties of proteins without having to resort to changes in the solvent that alter more than the protein hydration. Such D2O vs H2O experiments may not always be easy to realize, but for instance two-dimensional infrared spectroscopy on proteins in H2O has already been reported.39,55−57 This recent advancement enables researchers to study proteins in more natural systems, such as in cells or in blood serum.58,59 Since the protein amide-I frequencies and line shapes may change upon H/D exchange, extracting structural information from such 2D-IR spectra in H2O will require adaptation of the currently existing theoretical and modeling framework, which was developed mainly for interpreting 2D-IR spectra of proteins in D2O; see ref. 58 for an excellent future perspective on this topic.
Since D2O enhances the hydrophobic effect, a comparison of protein secondary structure in H2O and D2O can reveal the role of hydrophobic interactions in the stabilization of the proteins or in promoting their collapse. Similarly, comparing self-assembly kinetics in water and D2O can be a valuable method to gain a better understanding of the aggregation process, in particular in the case of fibril formation. Fibril formation can occur spontaneously via a nucleation-and-growth mechanism (1-step-nucleation or 1SN) or in two steps via the formation of intermediate aggregates (2SN) stabilized by hydrophobic effects. Intermediates subsequently need to undergo structural transformations to attain the fibrillar conformation, representing the rate-limiting step for fibrillization. Since D2O stabilizes hydrophobic interactions, the aggregation rate in D2O with respect to H2O is reduced if the mechanism involves intermediates, because their unfolding is energetically more unfavorable in D2O. Comparing the fibrillization rate in water and D2O can therefore reveal whether intermediates are present and hence if the amyloid formation occurs by a 2SN or 1SN mechanism. On the same note, the ability of D2O to slow the aggregation and stabilize the intermediates can be used to study the intermediate species. Intermediates are transient and metastable aggregates, which are quite challenging to detect and characterize structurally. By using D2O, we can follow the protein self-assembly in “slow motion”.
Thus, we believe that the difference in biopolymer hydration in H2O and D2O can be exploited to gain a better understanding of biopolymers, in particular, of biopolymer–solvent interactions and their role in defining the structure and dynamics of proteins and protein assemblies. This constitutes an interesting next challenge for the scientific community working on proteins and protein assemblies.
Acknowledgments
We thank Ioana Ilie for fruitful discussions. This publication is part of the project (with Project Number VI.Veni.212.240) of the NWO Talent Programme Veni 2021, which is financed by the Dutch Research Council (NWO).
Biographies

Giulia Giubertoni is a research fellow at the University of Amsterdam in the group of Sander Woutersen, which she joined in 2020 after obtaining her Ph.D. at the FOM-Institute for Atomic and Molecular Physics in Amsterdam, some 20 years after her two co-authors obtained their Ph.D. there. She is also a guest researcher at the Max Planck Institute for Polymer Research under the supervision of Mischa Bonn. Giulia studies the role of hydration in the self-assembly of proteins that form the building blocks of biomaterials, using physical methods that range from multidimensional infrared spectroscopy to rheology. In 2021, she was granted an NWO-VENI grant to investigate the molecular origin of osteogenesis imperfecta.

Mischa Bonn is a Director at the Max Planck Institute for Polymer Research since 2011, heading the Molecular Spectroscopy Department. Mischa completed his M.Sc. degree in physical chemistry in 1993 at the University of Amsterdam and performed his Ph.D. research at the FOM-Institute for Atomic and Molecular Physics in Amsterdam, where he shared an office with (and had to listen to the music of) Sander Woutersen. After postdoctoral stays at the Fritz Haber Institute and Columbia University, he went to Leiden University and returned to the Institute for Atomic and Molecular Physics as group leader in 2004. His scientific interests focus on the development and application of ultrafast spectroscopies to study natural phenomena, specifically at interfaces and often involving Mischa’s favorite molecule: water.

Sander Woutersen obtained his M.Sc. in physical chemistry at the University of Amsterdam (1995) and did his Ph.D. research at the FOM-Institute for Atomic and Molecular Physics in Amsterdam, where he shared an office with (and was regularly made fun of by) Mischa Bonn. After a postdoctoral fellowship with Peter Hamm at the Max Born Institute in Berlin, he returned to Amsterdam, where he eventually became professor in physical chemistry. Sander’s research is at the interface between spectroscopy and soft matter.
The authors declare no competing financial interest.
Footnotes
We will use the term “hydrogen bond” to denote both hydrogen bonds and deuterium bonds.
References
- Urey H. C.; Brickwedde F. G.; Murphy G. M. A Hydrogen Isotope of Mass 2. Phys. Rev. 1932, 39, 164–165. 10.1103/PhysRev.39.164. [DOI] [Google Scholar]
- Lewis G. N.; Macdonald R. T. The viscosity of H2H2O. J. Am. Chem. Soc. 1933, 55, 4730–4731. 10.1021/ja01338a505. [DOI] [Google Scholar]
- Heavy Hydrogen and Heavy Water. Nature 1934, 134, 388–389. 10.1038/134388b0 [DOI] [Google Scholar]
- Yang H.; Yang S.; Kong J.; Dong A.; Yu S. Obtaining information about protein secondary structures in aqueous solution using Fourier transform IR spectroscopy. Nat. Protoc. 2015, 10, 382–396. 10.1038/nprot.2015.024. [DOI] [PubMed] [Google Scholar]
- Pleitez M. A.; Khan A. A.; Soldà A.; Chmyrov A.; Reber J.; Gasparin F.; Seeger M. R.; Schätz B.; Herzig S.; Scheideler M.; et al. Label-free metabolic imaging by mid-infrared optoacoustic microscopy in living cells. Nat. Biotechnol. 2020, 38, 293–296. 10.1038/s41587-019-0359-9. [DOI] [PubMed] [Google Scholar]
- Alperstein A. M.; Ostrander J. S.; Zhang T. O.; Zanni M. T. Amyloid found in human cataracts with two-dimensional infrared spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 6602–6607. 10.1073/pnas.1821534116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth A.; Zscherp C. What vibrations tell us about proteins. Q. Rev. Biophys. 2002, 35, 369–430. 10.1017/S0033583502003815. [DOI] [PubMed] [Google Scholar]
- Yang J.Deuterium: Discovery and Applications in Organic Chemistry; Elsevier, 2016. [Google Scholar]
- Li L.; Jakowski J.; Do C.; Hong K. Deuteration and Polymers: Rich History with Great Potential. Macromolecules 2021, 54, 3555–3584. 10.1021/acs.macromol.0c02284. [DOI] [Google Scholar]
- Ceriotti M.; Fang W.; Kusalik P. G.; McKenzie R. H.; Michaelides A.; Morales M. A.; Markland T. E. Nuclear Quantum Effects in Water and Aqueous Systems: Experiment, Theory, and Current Challenges. Chem. Rev. 2016, 116, 7529–7550. 10.1021/acs.chemrev.5b00674. [DOI] [PubMed] [Google Scholar]
- Pica A.; Graziano G. Effect of heavy water on the conformational stability of globular proteins. Biopolymers 2018, 109, e23076 10.1002/bip.23076. [DOI] [PubMed] [Google Scholar]
- Bell R. P.The proton in chemistry; Cornell University Press: Cornell, 1973. [Google Scholar]
- Kaltashov I. A.; Eyles S. J. Studies of biomolecular conformations and conformational dynamics by mass spectrometry. Mass Spectrom. Rev. 2002, 21, 37–71. 10.1002/mas.10017. [DOI] [PubMed] [Google Scholar]
- Schnauß J.; Kunschmann T.; Grosser S.; Mollenkopf P.; Zech T.; Freitag J. S.; Prascevic D.; Stange R.; Röttger L. S.; Rönicke S.; et al. Cells in Slow Motion: Apparent Undercooling Increases Glassy Behavior at Physiological Temperatures. Adv. Mater. 2021, 33, 2101840. 10.1002/adma.202170230. [DOI] [PubMed] [Google Scholar]
- Jandova J.; Hua A. B.; Fimbres J.; Wondrak G. T. Deuterium Oxide (D2O) Induces Early Stress Response Gene Expression and Impairs Growth and Metastasis of Experimental Malignant Melanoma. Cancers 2021, 13, 605. 10.3390/cancers13040605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant I.; Shashidhar R. Understanding the influence of heavy water stress on the physiology of Salmonella Typhimurium. Applied Radiation and Isotopes 2020, 159, 108990. 10.1016/j.apradiso.2019.108990. [DOI] [PubMed] [Google Scholar]
- Kampmeyer C.; Johansen J.; Holmberg C.; Karlson M.; Gersing S.; Bordallo H.; Kragelund B. B.; Lerche M.; Jourdain I.; Winther J.; et al. Mutations in a Single Signaling Pathway Allow Cell Growth in Heavy Water. ACS Synth. Biol. 2020, 9, 733–748. 10.1021/acssynbio.9b00376. [DOI] [PubMed] [Google Scholar]
- Efimova Y. M.; Haemers S.; Wierczinski B.; Norde W.; van Well A. A. Stability of globular proteins in H2O and D2O. Biopolymers 2007, 85, 264–273. 10.1002/bip.20645. [DOI] [PubMed] [Google Scholar]
- Fu L.; Villette S.; Petoud S.; Fernandez-Alonso F.; Saboungi M.-L. H/D Isotope Effects in Protein Thermal Denaturation: The Case of Bovine Serum Albumin. J. Phys. Chem. B 2011, 115, 1881–1888. 10.1021/jp104769v. [DOI] [PubMed] [Google Scholar]
- Reslan M.; Kayser V. The effect of deuterium oxide on the conformational stability and aggregation of bovine serum albumin. Pharm. Dev. Technol. 2018, 23, 1030–1036. 10.1080/10837450.2016.1268157. [DOI] [PubMed] [Google Scholar]
- Das A.; Sinha S.; Acharya B. R.; Paul P.; Bhattacharyya B.; Chakrabarti G. Deuterium oxide stabilizes conformation of tubulin: a biophysical and biochemical study. BMB reports 2008, 41, 62–67. 10.5483/BMBRep.2008.41.1.062. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Yang D. Equilibrium folding dynamics of meACP in water, heavy water, and low concentration of urea. Sci. Rep. 2017, 7, 16156. 10.1038/s41598-017-16449-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak A. K.; Bandyopadhyay T. Water isotope effect on the thermostability of a polio viral RNA hairpin: A metadynamics study. J. Chem. Phys. 2017, 146, 165104. 10.1063/1.4982049. [DOI] [PubMed] [Google Scholar]
- Mizuno K.; Bächinger H. P. The effect of deuterium oxide on the stability of the collagen model peptides H-(Pro-Pro-Gly)10-OH, H-(Gly-Pro-4(R)Hyp)9-OH, and Type I collagen. Biopolymers 2010, 93, 93–101. 10.1002/bip.21305. [DOI] [PubMed] [Google Scholar]
- Cardoso M. V. C.; Sabadini E. The gelling of κ-carrageenan in light and heavy water. Carbohydr. Res. 2010, 345, 2368–2373. 10.1016/j.carres.2010.08.015. [DOI] [PubMed] [Google Scholar]
- Stadmiller S. S.; Pielak G. J. Enthalpic stabilization of an SH3 domain by D2O. Protein Sci. 2018, 27, 1710–1716. 10.1002/pro.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasisanker P.; Oleinikova A.; Weingärtner H.; Ravindra R.; Winter R. Solvation properties and stability of ribonuclease A in normal and deuterated water studied by dielectric relaxation and differential scanning/pressure perturbation calorimetry. Phys. Chem. Chem. Phys. 2004, 6, 1899–1905. 10.1039/B314070A. [DOI] [Google Scholar]
- Cho Y.; Sagle L. B.; Iimura S.; Zhang Y.; Kherb J.; Chilkoti A.; Scholtz J. M.; Cremer P. S. Hydrogen Bonding of β-Turn Structure Is Stabilized in D2O. J. Am. Chem. Soc. 2009, 131, 15188–15193. 10.1021/ja9040785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellgren B. W.; Creamer T. P. Effects of H2O and D2O on Polyproline II Helical Structure. J. Am. Chem. Soc. 2004, 126, 14734–14735. 10.1021/ja045425q. [DOI] [PubMed] [Google Scholar]
- Ben Abu N.; Mason P. E.; Klein H.; Dubovski N.; Ben Shoshan-Galeczki Y.; Malach E.; Pražienková V.; Maletínská L.; Tempra C.; Chamorro V. C.; et al. Sweet taste of heavy water. Communications Biology 2021, 4, 440. 10.1038/s42003-021-01964-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioni P.; Strambini G. B. Effect of Heavy Water on Protein Flexibility. Biophys. J. 2002, 82, 3246–3253. 10.1016/S0006-3495(02)75666-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzzi R.; Arcangeli C.; Bizzarri A. R. A molecular dynamics simulation study of the solvent isotope effect on copper plastocyanin. Biophys. Chem. 1999, 82, 9–22. 10.1016/S0301-4622(99)00097-6. [DOI] [PubMed] [Google Scholar]
- Brenner T.; Tuvikene R.; Cao Y.; Fang Y.; Rikukawa M.; Price W. S.; Matsukawa S. Hydrogen isotope replacement changes hydration and large scale structure, but not small scale structure, of agarose hydrogel networks. Eur. Phys. J. E 2019, 42, 53. 10.1140/epje/i2019-11816-9. [DOI] [PubMed] [Google Scholar]
- Sheu S.-Y.; Schlag E. W.; Selzle H. L.; Yang D.-Y. Molecular Dynamics of Hydrogen Bonds in Protein-D2O: The Solvent Isotope Effect. J. Phys. Chem. A 2008, 112, 797–802. 10.1021/jp0771668. [DOI] [PubMed] [Google Scholar]
- Tempra C.; Chamorro V. C.; Jungwirth P. Effects of Water Deuteration on Thermodynamic and Structural Properties of Proteins and Biomembranes. J. Phys. Chem. B 2023, 127, 1138–1143. 10.1021/acs.jpcb.2c08270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielskutė S.; Garcia-Cabau C.; Frigolé-Vivas M.; Szulc E.; De Mol E.; Pesarrodona M.; García J.; Salvatella X. Low amounts of heavy water increase the phase separation propensity of a fragment of the androgen receptor activation domain. Protein Sci. 2021, 30, 1427–1437. 10.1002/pro.4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakenfull D.; Scott A. Gelatin gels in deuterium oxide. Food Hydrocolloids 2003, 17, 207–210. 10.1016/S0268-005X(02)00053-X. [DOI] [Google Scholar]
- Bayrak M.; Mata J.; Raynes J. K.; Greaves M.; White J.; Conn C. E.; Floury J.; Logan A. Investigating casein gel structure during gastric digestion using ultra-small and small-angle neutron scattering. J. Colloid Interface Sci. 2021, 594, 561–574. 10.1016/j.jcis.2021.03.087. [DOI] [PubMed] [Google Scholar]
- Chun S. Y.; Son M. K.; Park C. R.; Lim C.; Kim H. I.; Kwak K.; Cho M. Direct observation of protein structural transitions through entire amyloid aggregation processes in water using 2D-IR spectroscopy. Chemical Science 2022, 13, 4482–4489. 10.1039/D1SC06047C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens A. D.; Kölbel J.; Moons R.; Chung C. W.; Ruggiero M. T.; Mahmoudi N.; Shmool T. A.; McCoy T. M.; Nietlispach D.; Routh A. F.; et al. Decreased Water Mobility Contributes To Increased α-Synuclein Aggregation. Angew. Chem., Int. Ed. 2023, 62, e202212063. 10.1002/anie.202212063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansel B. W.; Chu C. Y.; Leis A.; Hemar Y.; Chen H. L.; Lundin L.; Williams M. A. Zooming in: Structural Investigations of Rheologically Characterized Hydrogen-Bonded Low-Methoxyl Pectin Networks. Biomacromolecules 2015, 16, 3209–3216. 10.1021/acs.biomac.5b00870. [DOI] [PubMed] [Google Scholar]
- Soper A. K.; Benmore C. J. Quantum Differences between Heavy and Light Water. Phys. Rev. Lett. 2008, 101, 065502. 10.1103/PhysRevLett.101.065502. [DOI] [PubMed] [Google Scholar]
- Darby J. F.; Hopkins A. P.; Shimizu S.; Roberts S. M.; Brannigan J. A.; Turkenburg J. P.; Thomas G. H.; Hubbard R. E.; Fischer M. Water Networks Can Determine the Affinity of Ligand Binding to Proteins. J. Am. Chem. Soc. 2019, 141, 15818–15826. 10.1021/jacs.9b06275. [DOI] [PubMed] [Google Scholar]
- Scheiner S.; Čuma M. Relative Stability of Hydrogen and Deuterium Bonds. J. Am. Chem. Soc. 1996, 118, 1511–1521. 10.1021/ja9530376. [DOI] [Google Scholar]
- Némethy G.; Scheraga H. A. Structure of Water and Hydrophobic Bonding in Proteins. IV. The Thermodynamic Properties of Liquid Deuterium Oxide. J. Chem. Phys. 1964, 41, 680–689. 10.1063/1.1725946. [DOI] [Google Scholar]
- Liu B.; Garnett J. A.; Lee W.-c.; Lin J.; Salgado P.; Taylor J.; Xu Y.; Lambert S.; Cota E.; Matthews S. Promoting crystallisation of the Salmonella enteritidis fimbriae 14 pilin SefD using deuterium oxide. Biochemical and biophysical research communications 2012, 421, 208–213. 10.1016/j.bbrc.2012.03.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artero J.-B.; et al. A comparison of refined X-ray structures of hydrogenated and perdeuterated rat γE-Crystallin in H2O and D2O. Acta Crystallographica Section D: Biological Crystallography 2005, 61, 1541–1549. 10.1107/S0907444905028532. [DOI] [PubMed] [Google Scholar]
- Chatake T.; Ishikawa T.; Yanagisawa Y.; Yamada T.; Tanaka I.; Fujiwara S.; Morimoro Y. High-resolution X-ray study of the effects of deuteration on crystal growth and the crystal structure of proteinase K. Acta Crystallographica Section F 2011, 67, 1334–1338. 10.1107/S1744309111031903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee A. W.; Moulin M.; Breteau N.; Haertlein M.; Mitchell E. P.; Cooper J. B.; Boeri Erba E.; Forsyth V. T. Impact of deuteration on the assembly kinetics of transthyretin monitored by native mass spectrometry and implications for amyloidoses. Angew. Chem., Int. Ed. 2016, 55, 9292–9296. 10.1002/anie.201602747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eginton C.; Beckett D. A. Large Solvent Isotope Effect on Protein Association Thermodynamics. Biochemistry 2013, 52, 6595–6600. 10.1021/bi400952m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun M. K.; Wolf M.; Matsarskaia O.; Da Vela S.; Roosen-Runge F.; Sztucki M.; Roth R.; Zhang F.; Schreiber F. Strong isotope effects on effective interactions and phase behavior in protein solutions in the presence of multivalent ions. J. Phys. Chem. B 2017, 121, 1731–1739. 10.1021/acs.jpcb.6b12814. [DOI] [PubMed] [Google Scholar]
- Mollenkopf P.; Prascevic D.; Bayerl T. M.; Käs J. A.; Schnauß J. Heavy water induces bundling in entangled actin networks. RSC Adv. 2023, 13, 24795–24800. 10.1039/D3RA03917J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haidar Y.; Konermann L. Effects of Hydrogen/Deuterium Exchange on Protein Stability in Solution and in the Gas Phase. J. Am. Soc. Mass Spectrom. 2023, 34, 1447. 10.1021/jasms.3c00130. [DOI] [PubMed] [Google Scholar]
- Nyman J. S.; Ni Q.; Nicolella D. P.; Wang X. Measurements of mobile and bound water by nuclear magnetic resonance correlate with mechanical properties of bone. Bone 2008, 42, 193–199. 10.1016/j.bone.2007.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume S.; Greetham G. M.; Donaldson P. M.; Towrie M.; Parker A. W.; Baker M. J.; Hunt N. T. 2D-IR Spectroscopy of Proteins in Water: Using the Solvent Thermal Response as an Internal Standard. Anal. Chem. 2020, 92, 3463–3469. 10.1021/acs.analchem.9b05601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford S. H.; Greetham G. M.; Parker A. W.; Nordon A.; Baker M. J.; Hunt N. T. Measuring proteins in H2O using 2D-IR spectroscopy: pre-processing steps and applications toward a protein library. J. Chem. Phys. 2022, 157, 205102. 10.1063/5.0127680. [DOI] [PubMed] [Google Scholar]
- Hume S.; Hithell G.; Greetham G. M.; Donaldson P. M.; Towrie M.; Parker A. W.; Baker M. J.; Hunt N. T. Measuring proteins in H2O with 2D-IR spectroscopy. Chemical science 2019, 10, 6448–6456. 10.1039/C9SC01590F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford S. H.; Baker M. J.; Hunt N. T. 2D-IR spectroscopy of proteins in H2O—A Perspective. J. Chem. Phys. 2023, 030901. 10.1063/5.0129480. [DOI] [PubMed] [Google Scholar]
- Rutherford S. H.; Greetham G. M.; Donaldson P. M.; Towrie M.; Parker A. W.; Baker M. J.; Hunt N. T. Detection of glycine as a model protein in blood serum using 2D-IR spectroscopy. Anal. Chem. 2021, 93, 920–927. 10.1021/acs.analchem.0c03567. [DOI] [PubMed] [Google Scholar]