

Abstract
Engineered T cell therapies have shown significant clinical success. However, current manufacturing capabilities present a challenge in bringing these therapies to patients. Furthermore, the cost of development and manufacturing is still extremely high due to complexity of the manufacturing process. Increased automation can improve quality and reproducibility while reducing costs through minimizing hands-on operator time, allowing parallel manufacture of multiple products, and reducing the complexity of technology transfer. In this article, we describe the results of a strategic alliance between GSK and Miltenyi Biotec to develop a closed, automated manufacturing process using the CliniMACS Prodigy for autologous T cell therapy products that can deliver a high number of cells suitable for treating solid tumor indications and compatible with cryopreserved apheresis and drug product. We demonstrate the ability of the T cell Transduction – Large Scale process to deliver a significantly higher cell number than the existing process, achieving 1.5 × 1010 cells after 12 days of expansion, without affecting other product attributes. We demonstrate successful technology transfer of this robust process into three manufacturing facilities.
Keywords: GMP, manufacturing, T cell therapy, oncology, cell therapy, gene therapy, cell and gene therapy, automation, CliniMACS Prodigy, technology transfer
Graphical abstract
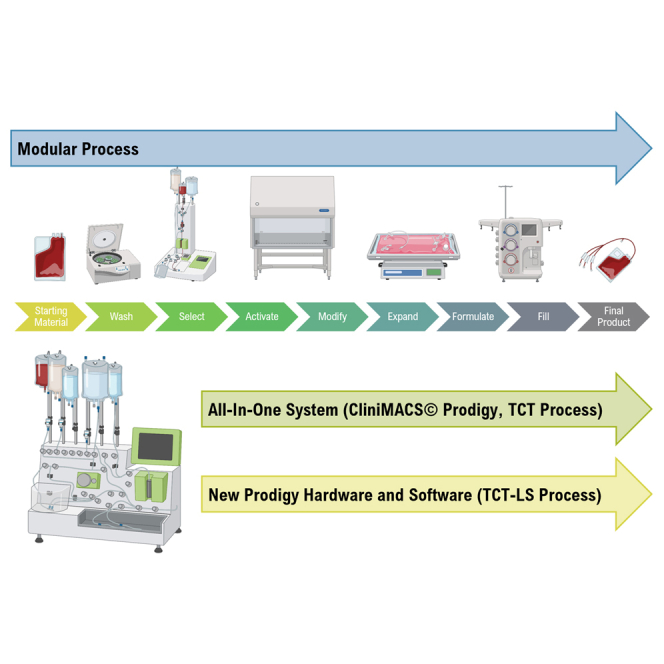
Francis and colleagues describe development of a novel manufacturing process using the CliniMACS Prodigy for production of large numbers of autologous T cells in a closed automated system, compatible with a cryopreserved starting material and drug product, and demonstrate successful technology transfer and clinical manufacturing for treating solid tumor indications.
Introduction
Adoptive T cell transfer, which involves ex vivo expansion of tumor-specific T cells followed by infusion into the patient, is a rapidly expanding approach that has demonstrated significant clinical success, particularly in the area of hematological malignancies, where several therapies are now commercially approved.1 However, application to the treatment of solid tumors, which account for more than 75% of cancer-related deaths, has been much more challenging.2 Early clinical trials demonstrated limited efficacy and high toxicity,3,4,5 although some successes have been shown.6,7,8,9,10 The challenges associated with translating the therapeutic success in hematological malignancies into solid tumors has been widely reviewed.2,11,12,13,14,15,16,17,18 However, one factor contributing to lack of efficacy may be the dose used: one modeling study showed that trafficking of chimeric antigen receptor (CAR)-T cells to solid tumors is less effective in humans than in mice, suggesting that the dose defined using animal models may be insufficient to achieve a similar clinical response in humans.19,20 Ongoing studies of CAR- and T cell receptor (TCR)-T cell therapies in solid tumors suggest that a higher dose of TCR-T cells is required compared to CAR-T cells.21 Therefore, manufacturing processes need to be able to expand a relatively small starting cell number into a much higher cell number over a period of days, without negatively affecting the function of the cells.
The cost of developing a lentiviral vector-modified autologous gene therapy from the start of clinical trials to commercialization was calculated to be $550 million, with cell process development and technology transfer accounting for a significant proportion of this. The costs are directly proportional to the complexity and degree of automation of the manufacturing process.22 The manufacturing process for autologous T cells therapies and the different technologies available have been extensively reviewed elsewhere.23,24,25 In many cases, manufacturing is performed using a modular platform (Figure 1), with different unit operations performed on different devices. Although this approach is more flexible, it is also more complex and requires a higher level of operator intervention, as well as implementation of additional controls to ensure no product cross-contamination occurs when manufacturing multiple products in parallel.
Figure 1.

Modular vs. all-in-one manufacturing process for T cell therapies
Copyright © 2023 Miltenyi Biotec B.V. & Co. KG. All rights reserved.
An all-in-one manufacturing platform (Figure 1) capable of performing multiple unit operations in an automated way within a closed system can reduce costs through reducing hands-on operator time from over 24 h with a modular manufacturing process based on a rocking-motion bioreactor to around 6 h,26 which in turn increases manufacturing throughput, reduces the complexity of technology transfer, and improves the quality and consistency of the manufacturing process to reduce product failure and subsequent re-manufacturing rates. Automation can also enable compliance with regulatory requirements for clinical and commercial manufacturing by allowing precise control of process parameters to demonstrate reproducibility and repeatability of the manufacturing process, including across multiple sites if required. Manufacturing within a closed system contributes to both cost reduction and quality of the product, as it enables parallel manufacturing of multiple products, allowing scale-out and reducing the requirement for the rigorous change-over processes needed to prevent cross-contamination; it also enables the majority of processing steps to be carried out within a lower-classification cleanroom while minimizing the risk of microbial, particulate, or cross-product contamination. The disadvantage of an all-in-one system include the high initial cost, a lack of flexibility once committed to a particular device, and a requirement for large numbers of devices to meet clinical or commercial manufacturing demands.
The CliniMACS Prodigy (Figure 1) has been used for clinical manufacturing of multiple different types of cell and gene therapies, including CAR-T cells,27,28,29 macrophages,30 and regulatory T cells,31 including in point-of-care settings.32,33 However, cell expansion in this system is limited by the size of the culture chamber, which has a maximum capacity of 250 mL and 5 × 109 total cells. In 2016, GSK and Miltenyi Biotec formed a strategic alliance to enable integration of greater automation and high-end processing technology into GSK manufacturing processes for cell and gene therapies. One of the primary aims of this alliance was the development of new hardware and software for the CliniMACS Prodigy to deliver a highly automated and closed process termed T cell Transduction – Large Scale (TCT-LS) compatible with a cryopreserved starting material and a cryopreserved drug product and capable of producing the higher cell doses required for TCR-T drug products. A full comparison of the already available TCT and the new TCT-LS processes is shown in Table 1. Furthermore, we could demonstrate that the new process does not negatively affect other product attributes, is very robust, and shows consistent high-quality clinical products after successful transfer to three different manufacturing sites.
Table 1.
Summary of differences between TCT and TCT-LS processes
| Process parameter | TCT process | TCT-LS process |
|---|---|---|
| Maximum chamber capacity (mL) | 250 | 600 |
| Maximum chamber capacity (cell number) | 5 × 109 | ≥ 1.5 × 1010a |
| Maximum number of target cells for selection | 3 × 109 | 3 × 109 |
| Selection reagent | CD4/CD8 microbeads (1 vial each) | CD4/CD8 microbeads (1 vial each) |
| Number of CD4+/CD8+ cells to start culture | 1 × 108 | 4 × 108 |
| Activation reagent | MACS GMP T cell TransAct (1 vial) | MACS GMP T cell TransAct – Large Scale (1 vial) |
| Cell culture conditions | Static culture until day 3 | Shaking from day 0 |
Greater cell numbers may be possible with further optimization of culture conditions.
Results
The TCT-LS process produces large numbers of transduced T cells without negatively affecting other product attributes
Healthy donor apheresis was used to manufacture genetically modified T cells transduced with lentiviral vector using either the standard (TCT) or large-scale (TCT-LS) process on the CliniMACS Prodigy. Cell count and viability were analyzed throughout the 12-day process. Figure 2A shows that the TCT-LS process is able to produce significantly higher cell numbers than the TCT process (mean at day 12 for TCT-LS, 1.70 × 1010; mean for TCT, 5.22 × 109, p < 0.0001), meeting the target of ≥1.5 × 1010 viable cells (shown by dotted line) in the majority of cases. Figure 2B shows viability throughout the manufacturing process. Although there is a decrease in viability on day 1, and this is slightly lower for TCT-LS than for TCT in this study, this difference is not statistically significant (mean for TCT, 81.9%; mean for TCT-LS%, 72.9%; p = 0.0637); for both processes the viability increases by the end of the process and is not significantly different at day 12 (mean for TCT-LS, 92.6%; mean for TCT, 94.2%; p = 0.0928). The proportion of CD3+ (mean for TCT-LS, 98.3%; mean for TCT, 92.6%; p = 0.5906), CD4+ (mean for TCT-LS, 58.6%; mean for TCT, 64.3%; p = 0.6033), and CD8+ (mean for TCT-LS, 39.5%; mean for TCT, 31.7%; p = 0.3313) T cells in the final product is comparable for both processes (Figure 2C). Performance during the selection step is also similar for both processes (Figure 2D), with no significant difference in recovery (calculated as the number of CD3+ T cells post selection divided by the number of CD3+ cells pre-selection; mean for TCT-LS, 49.9%; mean for TCT, 53.6%; p = 0.7892), purity as defined by the %CD3+ cells (mean for TCT-LS, 86.2%; mean for TCT, 87.3%; p = 0.9832), or viability (mean for TCT-LS, 84.9%; mean for TCT, 89.3%; p = 0.4324). This is as expected, as there are minor differences in the design of the selection column or the process itself at the selection stage. Analysis of different immune cell sub-types was performed pre and post selection. Figure 2E shows enrichment of T cells post-selection, with reduction in other immune cell types.
Figure 2.

Comparison of standard (TCT) and large-scale (TCT-LS) manufacturing processes
(A) Comparison of cell expansion over 12 days between TCT and TCT-LS processes (n = 10 for TCT and 13 for TCT-LS, individual data points shown, unpaired t test of day 12 values). (B) Comparison of viability over 12 days between TCT and TCT-LS processes (n = 10 for TCT and 13 for TCT-LS, individual data points shown, unpaired t test of day 12 values). (C) Comparison of %CD3+, %CD4+, or %CD8+ T cell subsets in drug product manufactured using either TCT or TCT-LS process (n = 10 for TCT and 13 for TCT-LS; graph shows individual data points with mean + SD; two-way ANOVA with Sidak’s multiple comparisons test). (D) Comparison of viability, purity (%CD3+ cells), and cell recovery during selection in TCT and TCT-LS processes (n = 7 for TCT and 13 for TCT-LS, graph shows individual data points with mean + SD; two-way ANOVA with Sidak’s multiple comparisons test). (E) Cellular composition pre and post selection for TCT and TCT-LS processes (n = 6 for TCT and 10 for TCT-LS, graph shows mean). (F) Comparison of memory T cell subsets in product manufactured using either TCT or TCT-LS in matched donors (n = 3, graph shows individual data points with mean + SD; two-way ANOVA with Sidak’s multiple comparisons test). (G) Comparison of CD4:CD8 ratio on day 0 and day 12 in product manufactured using either TCT or TCT-LS in matched donors (n = 3; graph shows individual data points with mean + SD; two-way ANOVA with Sidak’s multiple comparisons test).
To account for the high variability seen between different donors, extended characterization of memory T cell populations, activation markers, exhaustion markers, and CD4:CD8 ratio was analyzed in three runs with matched donors. Healthy donor apheresis was cryopreserved using the day -X process. Cells were thawed for selection, after which the CD4+/CD8+ cells were divided and expanded in either the TCT or TCT-LS process. Analysis of memory subsets was performed at days 0, 7, and 12. There is no significant difference in the proportion of different memory subsets in the final product manufactured using either the TCT or TCT-LS process (Figures 2F and S8; p > 0.8779). For both the TCT and TCT-LS processes, there is a significant decrease in naive T cells from day 0 to days 7 and 12, and an increase in stem cell memory and effector T cells, while proportions of central memory and effector memory T cells are variable throughout the process and between donors (Figure S8). Analysis of memory phenotypes in the total, transduced, and non-transduced cell populations was also performed at day 12 and shows no significant differences in any memory sub-population (Figures S8A–S8E).
Expression of the exhaustion markers LAG-3, PD-1, and TIM-3 was evaluated at days 0, 7, and 12 of the process. Expression of all exhaustion markers remains below 15% at time points during the process, with no differences seen between the TCT and TCT-LS processes (Figures S10A–S10C; p > 0.7588). Expression of the activation markers CD69 and CD25 was evaluated at days 0, 1, 7, and 12 of the process. Expression of the early activation marker CD69 peaks at day 1 and the late activation marker CD25 at day 7, as expected. There is no significant difference in marker expression between the TCT and TCT-LS processes at any time point (Figures S9D and S9E; p > 0.1041).
CD4:CD8 ratio was calculated based on the percentage of CD4+ and CD8+ T cells at day 0 and day 12 (Figure 2G). Although there is variability between donors and between time points, with one donor showing an increased CD4:CD8 ratio and two donors showing a decreased CD4:CD8 ratio at day 12 compared to day 0, there is no significant difference between the TCT and TCT-LS manufacturing processes (mean at day 12 for TCT-LS, 1.57; mean for TCT, 1.24; p = 0.2386). Analysis of CD4:CD8 ratio was also performed for the total, transduced, and non-transduced cell populations at day 12 and shows no significant difference (Figure S9F).
Accurate and consistent formulation and filling of apheresis using the CliniMACS Prodigy
Development work for this part of the manufacturing process (termed day -X) focused on development of a new custom software for the CliniMACS Prodigy. This software performs washing, concentration adjustment, formulation, and filling operations in a fully closed and automated manner within existing commercially available single-use tubing sets and using the CliniMACS Formulation Unit accessory.
Development studies were completed in two stages. The first stage evaluated accuracy of filling, post-thaw viability, and %CD3+ cells. The second stage evaluated performance of the cells cryopreserved using the day -X process throughout a single proof-of-concept TCT-LS process to produce a transduced T cell product. Results and acceptance criteria are shown in Table 2. All acceptance criteria were met, with a mean filling accuracy of 94% across 42 bags in 13 studies and a mean DMSO concentration of 5.1%. It was noted that the post-thaw viability of the CD3+ population was lower than the viability of the overall CD45+ population (mean for CD3+, 68.7%; mean for CD45+, 80.9%), showing CD3+ cells are more susceptible to damage during freeze-thaw; however, subsequent data from the TCT-LS process using both healthy donor (Figure 2; Table 2) and patient apheresis (Figure 5) cryopreserved using the day -X process shows cells are able to recover and expand to reach a high viability and number, demonstrating an ability to deliver a consistent final product.
Table 2.
Summary of day -X development studies
| Study | Parameter investigated | Acceptance criteria | Number of runs | Result (range) |
|---|---|---|---|---|
| Apheresis cryopreservation | filling Accuracy (%) | ±15% of target volume | 13 (42 bags filled) | 94.0% (85.2%–105.6%) |
| DMSO concentration (%) | for information only (target 5%) | 6 | 5.1% (4.5%–5.3%) | |
| post-thaw viability (%) | CD45: ≥70% CD3: for information only |
9 | CD45+: 80.9% (74.3%–87.4%) CD3+: 68.7% (50.9%–83.9%) |
|
| Drug product manufacturing | %CD3+ Cells | ≥80% | 1 | 99.6% |
| cell number | ≥1 × 1010 | 1 | 1.58 × 1010 | |
| transduction efficiency (%) | ≥10% | 1 | 39.4% | |
| vector copy number (copies/cell) | ≤5 copies/cell | 1 | 1.4 | |
| potency | antigen-specific killing detected | 1 | detected | |
| post-thaw viability (%) | ≥70% | 1 | 91.8% |
Figure 5.

Consistent performance of TCT-LS process at site 1 (n = 12), site 2 (n = 4), and site 3 (n = 3) manufacturing facilities during technology transfer using healthy donor material
(A) Comparison of recovery (CD3+ cells after selection/CD3+ cells before selection), viability, and purity (%CD3+ cells) post selection. (B) Comparison of viability, purity (%CD3+ cells), and transduction efficiency in final product. (C) Comparison of fold expansion (number of cells at end of process/cells seeded after selection). (D) Comparison of vector copy number in final product; dotted line shows acceptance criterion of ≤5 copies/cell. Graphs show mean + SD.
Development of formulation and fill process
At the end of the TCT-LS process, cells are washed to reduce any residual impurities relating to the cell culture process, such as medium, cytokines, or vector. The cells are resuspended in formulation buffer in the chamber of the tubing set, where the volume of cell suspension is adjusted to 2× the target cell concentration for filling. A final concentration of 5% DMSO is achieved through a 1:1 dilution of cell suspension and CryoStor 10 to formulate the bulk drug product.
A two-step approach toward automated formulation and filling of the drug product was chosen. In the first step, clinical studies were initiated using manual distribution of an automatically formulated bulk drug product into individual drug product and quality control (QC) bags. The Prodigy completed washing, cell concentration adjustment, and DMSO addition steps in an automated way. While still working in a closed system, operators then ensured a homogeneous cell suspension via a sequence of inversion of the bag and incubation on a horizontal shaker and distributed the formulated cells into the individual bags via gravity flow. The operators used gravity to allow the cell suspension to flow into the bags to be filled, using a balance to monitor filling, and manually opening and closing the bag clamps to control the liquid flow. This semi-automated process ensured flexibility for dose escalation with different planned filling volumes and dosing strategies and allowed rapid implementation for initiation of the clinical study.
The accuracy of this semi-automated formulation and fill process was evaluated by determination of bag weight, expressed as a percentage of the target volume, in the clinical batches manufactured for GSK3377794 (Figure 3A). Of the 53 successfully manufactured batches, one batch was filled into three product bags, 51 were filled into two product bags, and two were filled into one product bag (the number of bags filled is dependent on the number of cells at the end of expansion). Filling accuracy was high, with a mean of 99.8% (range, 78.5%–104.7%) across all bags filled. Of the 105 bags filled, 95 were within the required 5% volume accuracy (shown by the dotted lines), demonstrating the consistency and accuracy of this process for manufacturing clinical products. Cell concentration was determined via post-thaw analysis of the drug product filled into the QC bag and expressed as the percentage of the expected cell concentration (Figure 3B). This was more variable, with an average of 92% and range of 62.0%–117.4%, reflecting the complexity and variability of manufacturing a product using patient starting material.
Figure 3.

Development of a partially automated formulation and fill process
(A) Filling accuracy in product bags filled for clinical product; n = 108 bags across 55 batches, solid line indicates target of 100%, dotted line indicates acceptable range of ±5%. (B) Cell concentration in product bags filled for clinical product expressed as percentage of expected; n = 55 batches. (C) Cell viability from post-thaw drug product, n = 55 batches. Graph shows individual data points with mean + SD.
While initially allowing filling flexibility with high accuracy, this filling process still consisted of a labor-intensive, two-operator process step. To further increase automation and reduce operator hands-on time at later clinical stages, the filling process was automated in a second step. While the bulk drug product was initially formulated into a bag that required manual mixing and incubation on a horizontal shaker before distribution, the automated filling process allowed the 1:1 dilution of the formulated 2× drug product with CryoStor 10 in the culture chamber of the Prodigy tubing set itself, in which automated mixing with an optimized resuspension strategy was then possible. Depending on the pre-defined dosing strategy, the appropriate volumes of formulated cells were automatically transferred into bags of different sizes, including product bags, retain bags to be stored for further analysis as required, and QC bags to be aliquoted into cryovials prior to cryopreservation for QC analysis of the T cell product.
The filling accuracy (Figure 4A) was evaluated through weighing the filled bags and expressed as a percentage of the target fill volume. The filling accuracy was lower than in the manual gravity filling process (mean for product bags, 89.8% [range 84.8%–97.2%]; retain bags, 90.9% [87.8%–97.0%]; QC bags, 89.0% [83.6%–94.5%]). Therefore, the specification of the ongoing clinical study of +5% volume accuracy was not met without further development work, such as including the device camera for volume accuracy factors, and the automated filling project was discontinued at this stage.
Figure 4.

Development of a fully automated formulation and fill process
(A) Filling accuracy in product, retain, and QC bags filled in buffer runs (expressed as percentage of expected value); n = 66 for product bags, six for retain bags, 17 for QC bags; solid/dotted lines indicate target, 5% and 15% variability. (B) DMSO concentration measured by osmolality against a standard curve with known DMSO concentrations in product, retain, and QC bags filled in buffer runs; n = 30 for product bags, 10 for retain bags, six for QC bags; dotted line indicates target of 5%. (C) Cell concentration in product bags from runs expressed as the percentage of expected concentration (solid/dotted lines indicate target, 5% and 15% variability), n = 15 bags across four runs, graph shows mean + SD, one-way ANOVA with multiple comparisons (Dunnett’s multiple comparisons test). (D) Viability in product bags from runs (dotted line shows acceptance criterion of ≥70%), n = 15 bags across four runs, one-way ANOVA with multiple comparisons (Dunnett’s multiple comparisons test). Graphs show individual data points with mean + SD.
However, the automated formulation of the bulk drug product within the chamber using automated mixing still consisted of a step toward higher automation and was investigated further. First, the mixing strategy was optimized in buffer runs without cells. The DMSO concentration (Figure 4B) was measured using osmolality as a surrogate measure. The DMSO concentration was shown to be slightly lower than the 5% target in all bag types (mean for product bags, 4.6%; retain bags, 4.6%; QC bags, 4.5%), but this had no negative impact on the post-thaw viability and cell number.
Finally, the suitability of this mixing strategy was analyzed in four independent runs using T cells frozen with the day -X process and then expanded with the TCT-LS process. At the end of the culture process, T cells were automatically formulated to the final bulk drug product and mixed in the chamber before distribution into three to six bags. The bags were then frozen and thawed using the clinical study protocols, and cell concentration and viability were assessed both directly after thawing (post thaw [0 h]), and after a 2-h hold time at room temperature without prior DMSO wash-out (post thaw [2 h]). While cell concentrations were significantly higher for the post-thaw 0-h analysis, the concentrations at pre-freeze and post-thaw 2 h were close to the target and not significantly different (mean for pre-freeze, 104.5%; mean for post-thaw 0 h, 112%; mean for post-thaw 2 h, 101.8%) The entire T cell manufacturing and fill process also showed highly promising data for cell viability (Figure 4D). Viability was high pre-freeze (mean 90.1%) and did not drop significantly after the freeze-thaw process at either 0 h (mean 87.5%, p = 0.1977) or 2 h (mean 87.6%, p = 0.1721) time points. Cells in all 15 bags from all four runs met the acceptance criterion of >70% viability.
Successful technology transfer of an automated manufacturing process to multiple manufacturing facilities
Technology transfer of a manufacturing process from research and development (R&D) labs to a Good Manufacturing Practice (GMP) manufacturing facility contributes significantly to the cost of developing an autologous T cell therapy product due to the length and complexity of the process. Following successful development of the automated manufacturing process described above, GSK transferred this process to three GMP manufacturing facilities in the US and Europe to support manufacturing of three T cell therapy products. Figure 5 shows the results of technology transfer runs performed for each facility using healthy donor material. All runs met the pre-defined acceptance criteria.
Figure 5A shows the performance of the selection process in terms of cell recovery (number of CD3+ cells post selection compared to number of CD3+ cells pre-selection), viability and purity (%CD3+ cells) post selection. Most runs met the targets for recovery ≥40% (mean for site 1, 39.4%; site 2, 51.3% and site 3, 48.8%), viability ≥70% (mean for site 1, 73.9%; site 2, 81.8%; and site 3, 75.9%), and purity ≥80% (mean for site 1, 94.3%; site 2, 95.0%; and site 3, 92.3%), in all runs at all three sites, with no significant difference in any attribute between the different sites (p = 0.08). Figure 5B shows the attributes of the T cell product after genetic modification and expansion in terms of viability, purity, and transduction efficiency (percentage of cells expressing transgene). All runs at all sites met the acceptance criteria of ≥70% post-thaw viability (mean for site 1, 83.0%; site 2, 84.5%; and site 3, 88.0%), ≥80% purity (mean for site 1, 99.5%; site 2, 100.0%; and site 3, 99.2%), and ≥10% transduction efficiency (mean for site 1, 45.5%; site 2, 51.3%; and site 3, 49.9%), with no significant difference in any attribute between the different sites (p = 0.4529). Figure 5C shows that there is no significant difference in fold expansion (from cells seeded post selection to cells harvested at the end of the process) between the manufacturing sites (mean for site 1, 32.7; site 2, 41.4; and site 3, 25.0; p = 0.3227). Figure 5D shows that all products had a vector copy number (VCN) below the requirement of five copies per cell, with no significant difference between sites (mean for site 1, 2.2; site 2, 3.0; and site 3, 1.7; p = 0.7466). All runs met the acceptance criteria for antigen-specific cell killing of ≥3 points on the serial dilution curve with ≥17% antigen-specific cell killing, demonstrating comparable potency of the final product (data not shown).
Consistent and robust manufacturing of autologous T cell therapy products for clinical studies
Figure 6 shows a summary of data from clinical batches manufactured for three T cell therapy products in clinical studies at a single manufacturing facility. Batches were manufactured between October 2020 and November 2022. Of 72 batches manufactured, 64 met all acceptance criteria and were released, representing a batch success rate of 89%, demonstrating the robustness and consistency of the manufacturing process even with patient starting material. The success rate of manufacturing improved over time, as further process and operational improvements were implemented: the success rate for the 42 batches manufactured in 2021 was 82%, and for the 29 batches manufactured in 2022 was 97%. Of the batches that failed, either during manufacturing or due to failure to meet acceptance criteria on the final product, it was frequently because of the quality of the starting material: for example, the presence of particulates, low viability, or failure of the cells to expand. In many cases, re-manufacturing, either from additional cryopreserved apheresis from the day -X process or from a second apheresis collection, was successful. No batches failed due to microbial contamination, demonstrating the suitability of the Prodigy platform for manufacturing within a closed system at a lower cleanroom classification (grade C).
Figure 6.

Summary of clinical manufacturing data for three T cell products (n = 55 for GSK3377794, five for GSK3845097, and seven for GSK3901961)
(A) Purity (%CD3+ cells); acceptance criterion of 80% shown by dotted line. (B) Post-thaw viability; acceptance criterion of 70% shown by dotted line; GSK3377794 data are the same as post-thaw viability shown in Figure 3C. (C) Transduction efficiency. (D) Vector copy number; acceptance criterion of >5 copies/cell shown by dotted line. (E) Fold expansion (cells in final product/cells seeded for expansion). (F) Total number of transduced cells manufactured. Graphs show individual data points with mean + SD.
Data shown are from 67 batches (55 batches for GSK3377794, five batches for GSK3845097, and seven batches for GSK3901961) that were successfully manufactured and issued with a certificate of analysis. Product attributes, including purity (%CD3+ cells), transduction efficiency (percentage of transduced cells), vector copy number, and fold expansion (number of cells at the end of the process compared to the number of cells seeded after selection) were evaluated in the final product prior to freezing, and post-thaw viability, cell number, and antigen-specific cell killing were evaluated from a sentinel vial filled from the QC bag and cryopreserved in parallel to the product bags. The purity of the final product (Figure 6A) was >90% for all batches (mean for GSK3377794 = 98.7%, GSK3845097 = 99.6%, and GSK3901961 = 99.7%) exceeding the acceptance criterion of ≥80% (shown by the dotted line). Post-thaw viability (Figure 6B) was also high (mean for GSK3377794 = 88.3%, GSK3845097 = 88.2%, GSK3901961 = 86.3%). Transduction efficiency was more variable across batches and across different products (Figure 6C), ranging from 23% to 81% (mean for GSK3377794 = 58.9%, GSK3845097 = 54.8%, GSK3901961 = 44.9%), which may reflect variability both in patient material but also in different vector batches used. Vector copy number (Figure 6D) showed a similar variability, with a range of 0.7–4.2 copies/cell (mean for GSK3377794 = 2.6, GSK3845097 = 2.6, GSK3901961 = 1.7), but all batches met the acceptance criterion of ≤5 copies/cell (shown by the dotted line). However, the high fold expansion (Figure 6E) achieved, although still variable (ranging from 10 to 50; mean for GSK3377794 = 29, GSK3845097 = 19, GSK3901961 = 33), meant that even batches with lower transduction produced enough transduced cells for patient treatment in most cases (Figure 6F). In addition, all batches met the acceptance criteria for in vitro antigen-specific cell killing, with ≥3 points on the serial dilution curve with ≥17% antigen-specific cell killing, demonstrating potency of the final product.
Discussion
Recent years have demonstrated the enormous potential of T cell immunotherapy, notably in treating cancer, but in other diseases as well. However, the manufacturing process for autologous cell therapies is complex and expensive, resulting in high prices for these medicines and therefore limiting patient access. These costs are directly related to the complexity and degree of automation of the manufacturing process. Introducing an automated and closed manufacturing process can therefore significantly increase product safety and quality while reducing costs and complexity.
The CliniMACS Prodigy has previously been used for GMP production of several different types of cell therapy, including T cells. However, the size of the chamber for culturing cells poses a limitation where high cell numbers are required, such as in solid-tumor oncology indications.21 In this paper, we describe the results of a newly developed manufacturing platform using the CliniMACS Prodigy for GSK’s pipeline of CAR- and TCR-T cell therapies to treat multiple oncology indications. Our results demonstrate the ability of the new TCT-LS process to deliver the required target doses without affecting any relevant product attributes.
Implementing a cryopreservation step for the apheresis starting material prior to manufacturing avoids rescheduling or canceling a manufacturing slot. Cryopreservation of multiple bags of apheresis for each patient also enables re-manufacture in case of manufacturing or product failure without needing to schedule a repeat apheresis collection. Due to the rapid progression of their disease, patients are often on bridging chemotherapy while awaiting treatment, and a wash-out period of several weeks may be required before apheresis collection, significantly increasing the time until patient treatment. The results of this study demonstrate the ability of the day -X process to formulate apheresis accurately and consistently.
Automated formulation and fill of the final product also enables supply chain flexibility, as the product can be stored while all release testing is completed and can be shipped to and stored at the clinical site to enable the physician to schedule treatment. This step of the process is particularly challenging to automate due to complexities in dosing, where a clinical trial design may require dose escalation and or split-dosing strategies, and this is likely to change during clinical development of a product. Different products may also require different doses, making a platform manufacturing approach challenging. In this case, we demonstrate successful development of a partially automated formulation and fill process, where formulation is performed in the chamber of the Prodigy tubing set to achieve a homogeneous cell suspension, which is then manually filled into individual product bags. This process resulted in a filling accuracy within 5% of target volume and allows for a more flexible filling approach, which may be beneficial to early-phase clinical studies, as it enables the filling process to be entirely closed while also providing flexibility to manage complex dosing strategies.
We also show preliminary data for a fully automated formulation and fill process, using the formulation unit to control filling of individual product bags. In this case, it was not possible to completely automate the fill process while meeting GSK’s requirement for a filling accuracy of +5% of the target volume, although we were able to achieve filling to within ±15% of the target volume. Further optimization of this process is currently being developed by Miltenyi Biotec. Instead, we chose to focus on the automated formulation of the drug product in the chamber, optimizing the mixing and resuspension steps to ensure homogeneous filling of the product without negatively affecting viability.
Technology transfer of a manufacturing process from R&D to a GMP manufacturing facility contributes significantly to the cost of developing an autologous T cell therapy product, due to the length and complexity of the process. Development of an automated process can simplify this process, reducing the risk of run failures and helping to comply with regulatory requirements to demonstrate that the manufacturing process at each site results in a comparable product. Following successful development of the end-to-end manufacturing process described above, GSK transferred this process to three GMP manufacturing facilities in the US and Europe to support clinical and commercial manufacturing of three T cell therapy products meeting all acceptance criteria.
Finally, we show that the manufacturing process still performs consistently even when using patient apheresis as starting material, which is frequently highly variable. We summarize the manufacturing data from 69 batches, showing a manufacturing success rate of 89%. Where batches did fail, they were re-manufactured from existing cryopreserved apheresis in most cases. All products had a purity of more than 80%, and only two had a post-thaw viability of less than 70%. While transduction efficiency was more variable, the high total cell numbers achieved in the process meant that even batches with lower transduction produced a sufficient dose of transduced cells. All batches met the acceptance criteria for antigen-specific cell killing, demonstrating potency of the final product in vitro. All three T cell products have been manufactured in a certified multi-product facility, where manufacturing processes are performed in a single cleanroom suite at grade C classification. The closed nature of the Prodigy platform formed a critical component of our contamination control strategy in justifying this approach to the regulatory authorities.
While the closed tubing set of the Prodigy provides a high degree of sterility assurance, consideration must also be given to the preparation of the raw materials required for the process, such as viral vector, media, and supplements. The ability to sterile weld the container closures for such raw materials is an area still requiring further development by suppliers to aid simplification/streamlining of the manufacturing processes.
A significant challenge facing advanced therapies is the ability to link manufacturing data to clinical outcomes, to be able to define the critical quality attributes responsible for the safety and efficacy of a product. Extended characterization of many product attributes is required at early stages of development when there may be limited resources and expertise available. This can contribute to limited understanding of the complex product, its mechanism of action, and how manufacturing process parameters can affect the final product.
There is currently limited understanding of the dose requirements for product efficacy for T cell therapies in solid-tumor indications, as their therapeutic effect is largely driven by expansion in vivo following infusion and their ability to traffic to and persist in the tumor site.2 It is likely that other factors apart from the absolute number of transduced T cells are important for efficacy, such as the proportions of different T cell memory subsets and levels of exhaustion.34 Early studies focused on effector T cells, due to their high secretion of effector cytokines and proficiency in killing tumor cells in vitro; however, more recent evidence suggests that infusion of products with a higher proportion of less differentiated T cells, including naive, central memory, and stem cell memory T cells, may be advantageous due to their high proliferative capacity.35,36 However, a previous post hoc analysis of GSK3377794 product characteristics and clinical outcome found that a higher dose was correlated with a better clinical outcome and found no link with different memory phenotypes; instead, a higher number of CD8+ T cells in apheresis material was linked to a better response.37
There is evidence that these attributes can be influenced by the manufacturing process, such as the use of different cytokines for T cell expansion in vitro. While interleukin (IL)-2 was reported to increase in vitro expansion but to result in higher exhaustion and therefore poorer anti-tumor function, a combination of IL-7 and IL-15 resulted in better expansion, cytotoxicity, and cytokine secretion in vitro, and IL-21 resulted in greater expansion of less differentiated T cells and showed prolonged persistence in vivo.38 Our own analysis shows that a manufacturing process using IL-2 for expansion over 12 days results in a reduction in naive T cells and increase in stem cell memory and effector T cells compared to cells on day 0. However, we found that expansion was similar with either IL-2 or a combination of IL-7 and IL-15 (data not shown). There is also some evidence that reducing the manufacturing duration to 1–3 days may improve the anti-tumor activity of CAR-T cells in hematological indications.39,40 Expansion over a reduced number of days may also skew populations toward less differentiated sub-types,36 resulting in a lower available dose but a potentially more efficacious product, as well as significantly reducing the cost of manufacturing. Further characterization of the correlation between clinical outcomes and memory phenotypes in the product infused will be necessary to determine the optimal product characteristics, and, even then, designing a manufacturing process to produce the T cell sub-populations of choice will pose new challenges.
Materials and Methods
Source of cells
Experiments were performed at GSK (Medicines Research Centre, Stevenage, UK, or Upper Providence, Collegeville, USA) and Miltenyi Biotec (Bergisch Gladbach, Germany). Leukapheresis was obtained from healthy donors, WSQQied by Hemacare (Los Angeles, CA), BioIVT (Cambridge, UK), or Medizinische Hochschule (Hannover, Germany). Buffy coats were obtained from Medizinische Hochschule (Hannover, Germany). The human biological samples were sourced ethically, and their research use was in accordance with the terms of the informed consents under an Institutional Review Board/Ethics Committee (IRB/EC)-approved protocol. Cells were formulated in a 1:1 mixture of CryoStor Buffer and CryoStor 10 (both STEMCELL Technologies, Vancouver, Canada), resulting in a final concentration of 5% DMSO. Cells were cryopreserved using a controlled-rate freezer (CRF) and stored in vapor-phase liquid nitrogen.
Source of vector
Third-generation self-inactivating lentiviral vectors based on human immunodeficiency virus type one (HIV-1) pseudotyped with the envelope glycoprotein from vesicular stomatitis virus G (VSV-G) and encoding a therapeutic engineered T cell receptor or chimeric antigen receptor construct were used in these studies. Vector was manufactured at AGC Biologics (Bresso, Italy) using transient transfection of suspension culture-adapted HEK293T cells. Following transfection, the supernatant containing the viral particles was purified, concentrated, and stored at −80°C. The infectious titer of each batch was determined by flow cytometry for the TCR- or CAR-specific peptide complex in transduced cell lines.
Large-scale automated T cell production using the CliniMACS Prodigy
The TCT and TCT-LS processes comprise T cell selection, activation, transduction, and expansion steps of the manufacturing process. Cells were thawed using either a Sahara-TSC (Sarstedt, Nümbrecht, Germany) or Plasmatherm (Barkey, Leopoldshöhe, Germany) thawing device, and connected to the CliniMACS Prodigy (Miltenyi Biotec, Bergisch Gladbach, Germany) via sterile welding. For the TCT process, the TS520 tubing set (with a centricult chamber with capacity up to 250 mL) and the TCT software was used. For the TCT-LS process, the TS620 tubing set (with a MXC50 centricult chamber with capacity up to 600 mL) and the custom software developed in collaboration with Miltenyi Biotec, which is now commercially available, was used. After thawing, the cells were immediately diluted and loaded into the chamber.
Magnetic selection of CD4+ and CD8+ cells was performed using CD4 and CD8 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). For the TCT process, 1 × 108 CD4+/CD8+ cells were seeded into the chamber. For the TCT-LS process, 4 × 108 CD4+/CD8+ cells were seeded into the chamber. Cells were activated by addition of MACS GMP T cell TransAct Large Scale (Miltenyi Biotec, Bergisch Gladbach, Germany). The following day, transduction was performed with lentiviral vector at a previously determined multiplicity of infection (MOI). The cells were then expanded for up to 12 days in TexMACS medium (Miltenyi Biotec, Bergisch Gladbach, Germany) supplemented with 100 IU/mL IL-2 (Miltenyi Biotec, Bergisch Gladbach, Germany) and 5% AB serum (Access Biologicals, Vista, CA) for the first 5 days, after which the medium was changed to serum free. At the end of the manufacturing process, cells were automatically formulated in CryoStor CS5 (5% DMSO) at a concentration of 10–100 × 106 cells/mL and filled into CryoMACS bags (Miltenyi Biotec, Bergisch Gladbach, Germany) using the custom formulation software as described below and cryopreserved using a CRF. Cells were transferred into cryovials for analytical testing and cryopreserved in parallel to the product bags. Samples for cell count/viability and immunophenotype were taken throughout the process. Samples for transduction efficiency, vector copy number, and cytotoxicity were taken at the end of the process.
Apheresis formulation and fill
The day -X process comprises washing, formulation, and cryopreservation of apheresis material to enable it to be stored until required for manufacturing. Cells were connected to the TS710 tubing set on the CliniMACS Prodigy via sterile welding. The automated day -X process was used to wash the cells twice in CliniMACS PBS/EDTA buffer (Miltenyi Biotec, Bergisch Gladbach, Germany) supplemented with 0.5% human serum albumin (HSA; Irvine Scientific, Santa Ana, CA) and twice in CryoStor buffer (CSB; STEMCELL Technologies, Vancouver, Canada). The cell concentration was then adjusted to 40 × 106 CD3+ cells/mL by addition of further CSB, and cells were then transferred from the Prodigy chamber into CryoMACS bags provided as part of the CliniMACS Formulation Set (Miltenyi Biotec, Bergisch Gladbach, Germany) in an automated way using the CliniMACS Formulation Unit (Miltenyi Biotec, Bergisch Gladbach, Germany). Cells were then formulated by adding a 1:1 dilution of CryoStor CS10 to each bag containing the cell suspension, resulting in a final 5% DMSO concentration and a final concentration of 20 × 106 CD3+ cells/mL. The number of bags filled was determined by the cell number post wash, as each bag contains a set number of CD3+ cells sufficient to start the next stage of manufacturing. Cells were cryopreserved using a CRF. Samples for cell count/viability and immunophenotype were taken prior to the start of processing, after completion of the wash steps, and after cryopreservation.
Initial runs were performed using buffers only to evaluate filling accuracy and consistency, with CliniMACS PBS/EDTA buffer used in place of leukapheresis. The volume of each filled bag was measured using a balance to measure weight as a surrogate for volume, and the weight-to-volume calculation was adjusted according to the solution density. The DMSO concentration was measured using an osmometer, with results extrapolated from a standard curve generated from osmometer readings of samples with known DMSO concentrations ranging from 2.5% to 10%. Subsequent runs were performed with healthy donor leukapheresis obtained as described above.
Drug product formulation and fill
The drug product formulation and fill process includes washing, formulation, and cryopreservation of transduced T cells to enable the final product to be stored until required for patient treatment. This process takes place in the existing TS620 tubing set used for the TCT-LS process together with the CliniMACS Formulation Set to fill multiple product bags.
In the version of the formulation process used for clinical manufacturing, washing, resuspension of the cells in formulation buffer (0.9% saline [Fresenius Kabi, Bad Homburg, Germany] supplemented with 4% HSA [Grifols, Los Angeles, CA]), and concentration adjustment of the cell suspension at the end of expansion were performed in the chamber of the TS620 tubing set. The cell suspension was then transferred to a bulk product bag, where CryoStor CS10 (STEMCELL Technologies, Vancouver, Canada) was added in a 1:1 ratio to achieve a final 5% DMSO concentration. Mixing of the bulk bag, and subsequent transfer of the formulation cell suspension into individual product bags, was performed manually. This process was evaluated using transduced T cells produced using the TCT-LS process. Consistency and accuracy of filling across several product bags was assessed by measuring the volume and % DMSO. Samples for cell count/viability were taken prior to harvest, after filling and prior to cryopreservation, and after cryopreservation.
Further optimization to develop a completely automated formulation and fill process was performed. In this process, cell washing, concentration adjustment, and formulation of the drug product in formulation buffer and CS10 occurs in the chamber of the TS620 tubing set, with automated shaking to ensure homogeneous mixing of cell suspension and formulation buffer. Cells are then transferred from the Prodigy chamber into CryoMACS bags provided as part of the CliniMACS Formulation Set in an automated way using the CliniMACS Formulation Unit. Initial tests of the software were performed with buffer only to evaluate chamber mixing efficiency and accuracy of volume transfer, including determining the dead volume remaining in the tubing set, the accuracy of dilution with CS10, and the accuracy and consistency of filling across multiple bags. Subsequent runs were then performed with transduced T cells produced using the TCT-LS process to evaluate the accuracy and consistency of cell concentration across multiple bags.
Flow cytometry
Flow cytometry was performed on the MACSQuant Analyzer 10. An overview of the panels and antibodies used for analysis at different time points is shown in Table S1. Cells were transferred into a 96-well round-bottom plate (Corning, Corning, NY) and resuspended in fluorescence-activated cell sorting (FACS) buffer (CliniMACS + 0.5% HSA). Cells were washed and resuspended in FACS buffer, and dextramer reagent for detection of the engineered TCR was added (transduction panel only). Cells were incubated for 25 min, washed, resuspended in FACS buffer, and all other antibodies added as a pre-prepared master mix. Cells were incubated for 30 min, washed, and resuspended in FACS buffer. The gating strategies used are shown in Figures S1–S6. Appropriate fluorescence minus one (FMO) and compensation controls were used. Data analysis was performed using MACSQuantify software (Miltenyi Biotec, Bergisch Gladbach, Germany).
Antigen-specific cell killing
Analysis of antigen-specific cell killing of transduced T cells was performed using a flow cytometry assay to detect carboxyfluorescein succinimidyl ester (CFSE) in a cell line presenting the target peptide recognized by the engineered TCR on transduced T cells. The difference in live cell count between peptide- and no-peptide conditions is used to calculate the percentage of peptide-specific cell killing, providing a measure of cytotoxicity activity in transduced T cells.
On day 0, target cells were seeded in T75 flasks (Thermo Fisher, Waltham, MA) in RPMI 1640 medium (Gibco, Billings, Montana) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, Billings, Montana), 1× GlutaMAX (Gibco, Billings, Montana), and 50 U/mL penicillin/streptomycin (Gibco, Billings, Montana) and incubated overnight at 37°C, 5% CO2. The following day, transduced T cells were thawed, resuspended in complete RPMI, and incubated for 4 h at 37°C, 5% CO2. Target cells were resuspended at a concentration of 1 × 107 cells/mL in a 1 μM CFSE (BD Bioscience Franklin Lakes, NJ) solution in Dulbecco's phosphate-buffered saline (DPBS) (Gibco, Billings, Montana). Cells were incubated for 10 min and resuspended in assay medium. A 2-fold serial dilution of transduced T cells was prepared in a 96-deep-well plate (Costar, Washington D.C.) and 4 × 104 CFSE-labeled target cells were added to each well. Then 50 μL of target peptide (Peptide Protein Research, Fareham, UK) or 50 μL of complete RPMI was added to the sample and control wells, respectively. Cells were incubated for 18 h at 37°C, 5% CO2.
On day 2, cells were resuspended in a 1:100 dilution of 7-Aminoactinomycin D (7-AAD) (BD Bioscience, Franklin Lakes, NJ) in FACS buffer. Data were acquired using the MACSQuant Analyzer 10. Appropriate compensation controls were used. Data were analyzed using MACSQuantify software. The gating strategy used is shown in Figure S7. Control wells must have an average of ≥70% viability and ≥200 cells/μL. Transduced T cells should have ≥3 points on the serial dilution curve with ≥17% antigen-specific cell killing.
Vector copy number
The average number of copies of integrated lentivirus (LV) per cell (vector copy number; VCN)was determined using a digital droplet PCR (ddPCR) method on the QX200 AutoDG Droplet Digital PCR system (Bio-Rad, Hercules, CA). The method is performed with two sets of primers and probes: one specific for quantification of LV and one specific for quantification of the cellular genome. VCN is determined by the ratio of LV to genome.
Cell pellets were frozen at −80°C and then thawed for genomic DNA extraction using the DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany). DNA digestion was performed using the Fast Digest MIuI kit (Thermo Fisher, Waltham, MA). The PCR reaction mixture was prepared using the ddPCR Supermix for probes and ddPCR copy number assay kit (both Bio-Rad, Hercules, CA). Primers/probe mix were supplied by Invitrogen (Waltham, MA) or Thermo Fisher. Droplet generation was performed using the automated droplet generator (Bio-Rad, Hercules, CA). PCR amplification was performed using the QX200 system thermocycler (Bio-Rad, Hercules, CA). Droplet reading was performed using the QX200 droplet reader (Bio-Rad, Hercules, CA). Data analysis was performed using the QX manager software (Bio-Rad, Hercules, CA).
Manufacturing data from clinical batches
Manufacturing data from 67 clinical batches for three different TCR-T cell products that were issued a certificate of analysis (55 batches for GSK3377794, five batches for GSK3845097, and seven batches for GSK3901961) manufactured between 2020 and 2022 at a single manufacturing facility under trials NCT04526509 and NCT03967223 were evaluated. Product attributes, including purity (%CD3+ cells), transduction efficiency (% transduced cells), fold expansion, vector copy number, antigen-specific cell killing, and post-thaw viability, were evaluated in the final product either prior to freezing or from a sentinel vial cryopreserved in parallel to the product bags.
Statistical analyses
GraphPad Prism v7.03 (GraphPad Software, San Diego, CA) was used for statistical analysis and generation of graphs.
TCT-LS: unpaired t test was used to compare cell number and viability between TCT-LS and TCT at day 12. Two-way ANOVA with multiple comparison (with Sidak’s correction applied) was used for all other comparisons.
Formulation: one-way ANOVA with multiple comparisons (Dunnett’s multiple comparisons test) was used to compare mean values for post-thaw at 0 and 2 h with pre-freeze values.
Site-to-site comparison: one-way ANOVA with multiple comparisons (with Tukey’s correction applied) was used to compare mean values for each site with each other site.
Statistical significance for each comparison is indicated on the graphs as follows: no significant difference (NSD), ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.005.
Data and code availability
The data that support the findings of this study are available from the corresponding author on reasonable request, subject to the relevant legal agreements being in place.
Acknowledgments
The authors would like to gratefully acknowledge the hard work of all the members, past and present, of the Cell Process Development team in the Cell and Gene Therapy department and the other teams within Cell and Gene Therapy at GSK, in particular analytical development. We are further thankful to past and present members of the internal TCT-LS system and software development, as well as engineering teams at Miltenyi Biotec, for their valuable contributions. We express our sincere acknowledgment to the manufacturing and technology transfer teams at each of our manufacturing facilities who worked so hard to deliver these medicines to patients. The authors would also like to express their most sincere appreciation to all enrolled patients and their families.
Author contributions
A.K., N.F., S.W., M.B., and S.P. contributed to this study’s conceptualization and methodology. A.B., A.R., T.O., K.I., S.N., and S.L. contributed to data curation. N.F., T.O., A.R., A.B., S.L., S.P., and M.B. performed formal data analysis. A.B., A.R., T.O., K.I., C.H., S.N., and N.F. contributed to investigation during this study. B.G., S.W., S.P., A.K., N.F., and M.B. contributed to project administration. N.F. was responsible for writing the manuscript.
Declaration of interests
N.F., S.N., and A.K. are current or past employees and/or shareholders of GSK. M.B., S.P., C.H., A.B., A.R., T.O., S.L., K.I., B.G., and S.W. are current employees of Miltenyi Biotec.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.09.002.
Supplemental information
References
- 1.Medicine A.f.R. 2022. Regenerative Medicine: The Pipeline Momentum Builds.https://alliancerm.org/sector-report/h1-2022-report/ [Google Scholar]
- 2.Guedan S., Ruella M., June C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2019;37:145–171. doi: 10.1146/annurev-immunol-042718-041407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamers C.H.J., Sleijfer S., Vulto A.G., Kruit W.H.J., Kliffen M., Debets R., Gratama J.W., Stoter G., Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J. Clin. Oncol. 2006;24:e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 4.Morgan R.A., Dudley M.E., Rosenberg S.A. Adoptive cell therapy: genetic modification to redirect effector cell specificity. Cancer J. 2010;16:336–341. doi: 10.1097/PPO.0b013e3181eb3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kershaw M.H., Westwood J.A., Parker L.L., Wang G., Eshhar Z., Mavroukakis S.A., White D.E., Wunderlich J.R., Canevari S., Rogers-Freezer L., et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006;12:6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Louis C.U., Savoldo B., Dotti G., Pule M., Yvon E., Myers G.D., Rossig C., Russell H.V., Diouf O., Liu E., et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown C.E., Badie B., Barish M.E., Weng L., Ostberg J.R., Chang W.C., Naranjo A., Starr R., Wagner J., Wright C., et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015;21:4062–4072. doi: 10.1158/1078-0432.CCR-15-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J.D., Martinez-Lage M., Brem S., Maloney E., Shen A., et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beatty G.L., O'Hara M.H., Lacey S.F., Torigian D.A., Nazimuddin F., Chen F., Kulikovskaya I.M., Soulen M.C., McGarvey M., Nelson A.M., et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology. 2018;155:29–32. doi: 10.1053/j.gastro.2018.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown C.E., Alizadeh D., Starr R., Weng L., Wagner J.R., Naranjo A., Ostberg J.R., Blanchard M.S., Kilpatrick J., Simpson J., et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finck A.V., Blanchard T., Roselle C.P., Golinelli G., June C.H. Engineered cellular immunotherapies in cancer and beyond. Nat. Med. 2022;28:678–689. doi: 10.1038/s41591-022-01765-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Junghans R.P. The challenges of solid tumor for designer CAR-T therapies: a 25-year perspective. Cancer Gene Ther. 2017;24:89–99. doi: 10.1038/cgt.2016.82. [DOI] [PubMed] [Google Scholar]
- 13.Morgan R.A., Dudley M.E., Wunderlich J.R., Hughes M.S., Yang J.C., Sherry R.M., Royal R.E., Topalian S.L., Kammula U.S., Restifo N.P., et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bos R., van Duikeren S., Morreau H., Franken K., Schumacher T.N.M., Haanen J.B., van der Burg S.H., Melief C.J.M., Offringa R. Balancing between antitumor efficacy and autoimmune pathology in T-cell-mediated targeting of carcinoembryonic antigen. Cancer Res. 2008;68:8446–8455. doi: 10.1158/0008-5472.CAN-08-1864. [DOI] [PubMed] [Google Scholar]
- 15.Johnson L.A., Morgan R.A., Dudley M.E., Cassard L., Yang J.C., Hughes M.S., Kammula U.S., Royal R.E., Sherry R.M., Wunderlich J.R., et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parkhurst M.R., Yang J.C., Langan R.C., Dudley M.E., Nathan D.A.N., Feldman S.A., Davis J.L., Morgan R.A., Merino M.J., Sherry R.M., et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011;19:620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan R.A., Chinnasamy N., Abate-Daga D., Gros A., Robbins P.F., Zheng Z., Dudley M.E., Feldman S.A., Yang J.C., Sherry R.M., et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linette G.P., Stadtmauer E.A., Maus M.V., Rapoport A.P., Levine B.L., Emery L., Litzky L., Bagg A., Carreno B.M., Cimino P.J., et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown M.P., Ebert L.M., Gargett T. Erratum: Clinical chimeric antigen receptor T-cell therapy: a new and promising treatment modality for glioblastoma. Clin. Transl. Immunology. 2021;10 doi: 10.1002/cti2.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majzner R.G., Theruvath J.L., Nellan A., Heitzeneder S., Cui Y., Mount C.W., Rietberg S.P., Linde M.H., Xu P., Rota C., et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019;25:2560–2574. doi: 10.1158/1078-0432.CCR-18-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishihori T., Hoffman J.E., Huff A., Kapoor G.S., Eleftheriadou I., Zajic S., Urbano A., Suchindran S., Chisamore M., D'Souza J.W., et al. Safety and efficacy of letetresgene autoleucel alone or with pembrolizumab for relapsed/refractory multiple myeloma. Blood Adv. 2023;7:1168–1177. doi: 10.1182/bloodadvances.2022008460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krishna D., Rittié L., Tran H., Zheng X., Chen-Rogers C.E., McGillivray A., Clay T., Ketkar A., Tarnowski J. Short Time to Market and Forward Planning Will Enable Cell Therapies to Deliver R&D Pipeline Value. Hum. Gene Ther. 2021;32:433–445. doi: 10.1089/hum.2020.212. [DOI] [PubMed] [Google Scholar]
- 23.Wang X., Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol. Ther. Oncolytics. 2016;3 doi: 10.1038/mto.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vormittag P., Gunn R., Ghorashian S., Veraitch F.S. A guide to manufacturing CAR T cell therapies. Curr. Opin. Biotechnol. 2018;53:164–181. doi: 10.1016/j.copbio.2018.01.025. [DOI] [PubMed] [Google Scholar]
- 25.Mizukami A., Swiech K. Platforms for Clinical-Grade CAR-T Cell Expansion. Methods Mol. Biol. 2020;2086:139–150. doi: 10.1007/978-1-0716-0146-4_10. [DOI] [PubMed] [Google Scholar]
- 26.Lock D., Monjezi R., Brandes C., Bates S., Lennartz S., Teppert K., Gehrke L., Karasakalidou-Seidt R., Lukic T., Schmeer M., et al. Automated, scaled, transposon-based production of CAR T cells. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2022-005189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mock U., Nickolay L., Philip B., Cheung G.W.K., Zhan H., Johnston I.C.D., Kaiser A.D., Peggs K., Pule M., Thrasher A.J., Qasim W. Automated manufacturing of chimeric antigen receptor T cells for adoptive immunotherapy using CliniMACS prodigy. Cytotherapy. 2016;18:1002–1011. doi: 10.1016/j.jcyt.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 28.Zhu F., Shah N., Xu H., Schneider D., Orentas R., Dropulic B., Hari P., Keever-Taylor C.A. Closed-system manufacturing of CD19 and dual-targeted CD20/19 chimeric antigen receptor T cells using the CliniMACS Prodigy device at an academic medical center. Cytotherapy. 2018;20:394–406. doi: 10.1016/j.jcyt.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 29.Oberschmidt O., Morgan M., Huppert V., Kessler J., Gardlowski T., Matthies N., Aleksandrova K., Arseniev L., Schambach A., Koehl U., Kloess S. Development of Automated Separation, Expansion, and Quality Control Protocols for Clinical-Scale Manufacturing of Primary Human NK Cells and Alpharetroviral Chimeric Antigen Receptor Engineering. Hum. Gene Ther. Methods. 2019;30:102–120. doi: 10.1089/hgtb.2019.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraser A.R., Pass C., Burgoyne P., Atkinson A., Bailey L., Laurie A., W A McGowan N., Hamid A., Moore J.K., Dwyer B.J., et al. Development, functional characterization and validation of methodology for GMP-compliant manufacture of phagocytic macrophages: A novel cellular therapeutic for liver cirrhosis. Cytotherapy. 2017;19:1113–1124. doi: 10.1016/j.jcyt.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marín Morales J.M., Münch N., Peter K., Freund D., Oelschlägel U., Hölig K., Böhm T., Flach A.C., Keßler J., Bonifacio E., et al. Automated Clinical Grade Expansion of Regulatory T Cells in a Fully Closed System. Front. Immunol. 2019;10:38. doi: 10.3389/fimmu.2019.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson Z., Roe A., Sharma A.A., Lopes F.B.T.P., Talla A., Kleinsorge-Block S., Zamborsky K., Schiavone J., Manjappa S., Schauner R., et al. Automated Manufacture of Autologous CD19 CAR-T Cells for Treatment of Non-hodgkin Lymphoma. Front. Immunol. 2020;11:1941. doi: 10.3389/fimmu.2020.01941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castella M., Caballero-Baños M., Ortiz-Maldonado V., González-Navarro E.A., Suñé G., Antoñana-Vidósola A., Boronat A., Marzal B., Millán L., Martín-Antonio B., et al. Point-Of-Care CAR T-Cell Production (ARI-0001) Using a Closed Semi-automatic Bioreactor: Experience From an Academic Phase I Clinical Trial. Front. Immunol. 2020;11:482. doi: 10.3389/fimmu.2020.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stock S., Übelhart R., Schubert M.L., Fan F., He B., Hoffmann J.M., Wang L., Wang S., Gong W., Neuber B., et al. Idelalisib for optimized CD19-specific chimeric antigen receptor T cells in chronic lymphocytic leukemia patients. Int. J. Cancer. 2019;145:1312–1324. doi: 10.1002/ijc.32201. [DOI] [PubMed] [Google Scholar]
- 35.Barrett D.M., Singh N., Porter D.L., Grupp S.A., June C.H. Chimeric antigen receptor therapy for cancer. Annu. Rev. Med. 2014;65:333–347. doi: 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schafer J.M., Xiao T., Kwon H., Collier K., Chang Y., Abdel-Hafiz H., Bolyard C., Chung D., Yang Y., Sundi D., et al. Sex-biased adaptive immune regulation in cancer development and therapy. iScience. 2022;25 doi: 10.1016/j.isci.2022.104717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gyurdieva A., Zajic S., Chang Y.F., Houseman E.A., Zhong S., Kim J., Nathenson M., Faitg T., Woessner M., Turner D.C., et al. Biomarker correlates with response to NY-ESO-1 TCR T cells in patients with synovial sarcoma. Nat. Commun. 2022;13:5296. doi: 10.1038/s41467-022-32491-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe N., Mo F., McKenna M.K. Impact of Manufacturing Procedures on CAR T Cell Functionality. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.876339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engels B., Zhu X., Yang J., Price A., Sohoni A., Stein A.M., Parent L., Greene M., Niederst M., Whalen J., et al. Preservation of T-Cell Stemness with a Novel Expansionless CAR-T Manufacturing Process, Which Reduces Manufacturing Time to Less Than Two Days, Drives Enhanced CAR-T Cell Efficacy. Blood. 2021;138:2848. doi: 10.1182/blood-2021-146246. [DOI] [Google Scholar]
- 40.Ghassemi S., Durgin J.S., Nunez-Cruz S., Patel J., Leferovich J., Pinzone M., Shen F., Cummins K.D., Plesa G., Cantu V.A., et al. Rapid manufacturing of non-activated potent CAR T cells. Nat. Biomed. Eng. 2022;6:118–128. doi: 10.1038/s41551-021-00842-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request, subject to the relevant legal agreements being in place.