

Abstract
The expanding applications of non-viral genomic medicines in the lung remain restricted by delivery challenges. Leveraging a high-throughput platform, we here synthesize and screen a combinatorial library of biodegradable ionizable lipids to build inhalable delivery vehicles for mRNA and CRISPR-Cas9 gene editors. Lead lipid nanoparticles are amenable for repeated intratracheal dosing and could achieve efficient gene editing in lung epithelium, providing avenues for gene therapy of congenital lung diseases.
Congenital lung diseases such as surfactant protein (SP) deficiency disorders, cystic fibrosis (CF), and alpha-1 antitrypsin (AAT) deficiency can lead to lifelong morbidity and mortality1, 2. Although the genetic origins for these ailments have been identified, an effective treatment option remains elusive3,4. The intratracheal delivery of gene-editing tools, particularly CRISPR-Cas9, to the airway epithelium or other pulmonary cells presents a promising corrective approach to provide life-long health and quality of life benefits for patients5,6.
In vivo gene editing has been achieved using viral vectors such as an AAV, but these stable DNA-based vectors lead to the long-term expression of Cas9 ribonuclease and sgRNA in cells7. While the extended exposure to editing machinery may favour gene correction rates, it can also lead to the accumulation of off-target genetic alterations8, 9. Moreover, the immunogenicity of AAV capsids triggers neutralizing antibodies and T-cell responses that limit repeated dosing of AAV-based treatments10; however, gene editing in the lung benefits from repeated dosing due to higher cell turnover rate11. Additionally, size limitations pose challenges for integrating effective S. pyogenes CRISPR-Cas9 (SpCas9) constructs into AAVs12. These limitations can be overcome by non-viral, mRNA-based delivery platforms which enable transient expression and repeated dosing13. LNPs are the most clinically advanced non-viral vector, as seen with the widely accepted mRNA vaccine technologies developed by both Moderna and Pfizer/BioNTech and show great promise in Cas9 hepatic gene editing platforms14–16. However, an LNP-based Cas9 delivery system for efficient pulmonary gene modification has yet to be reported. Compared with the liver, the lung poses unique challenges for delivery due to its specialized cell types, mucus barrier, and mucociliary clearance. Therefore, there remains a need for efficient approaches as airway epithelia remain poorly transduced by most viral and non-viral approaches17.
Here we synthesized and screened a combinatorial library of biodegradable ionizable lipids to build nanoparticles for pulmonary mRNA delivery. Lead lipid formulations are amenable to multiple intratracheal dosing and can efficiently deliver Cre or Cas9 mRNA to mouse airway epithelium.
In this work, we designed a three-component reaction (3-CR) system in which a nitro ricinoleic acrylate (NRA) linker was coupled with aliphatic alcohols (lipid tails), followed by connecting with head groups containing primary, secondary, or tertiary amines (Fig. 1a–b). Compared to the conventional two-component reaction that conjugates amine headgroups directly with lipid tails, this 3-CR system simplifies the tedious synthesis process, increases lipid structural diversity, and rapidly generates a structurally diverse combinatorial library of biodegradable ionizable lipids. We synthesized a library of 720 new lipids containing the NRA linker with one of 10 distinct tails (variable in length, saturation, location of unsaturated bonds) and 72 headgroups (variable in cyclic structure, aromaticity, functional groups) (Fig. 1c). All the ionizable lipids in this library contain numerous ester and carbonate groups to improve the physiological biodegradability of ionizable lipids. This reduces the potential for adverse reactions and enhances the compatibility with multi-dosing regimens18.
Fig. 1 |. Overview of the lipid nanoparticle synthesis and screening.
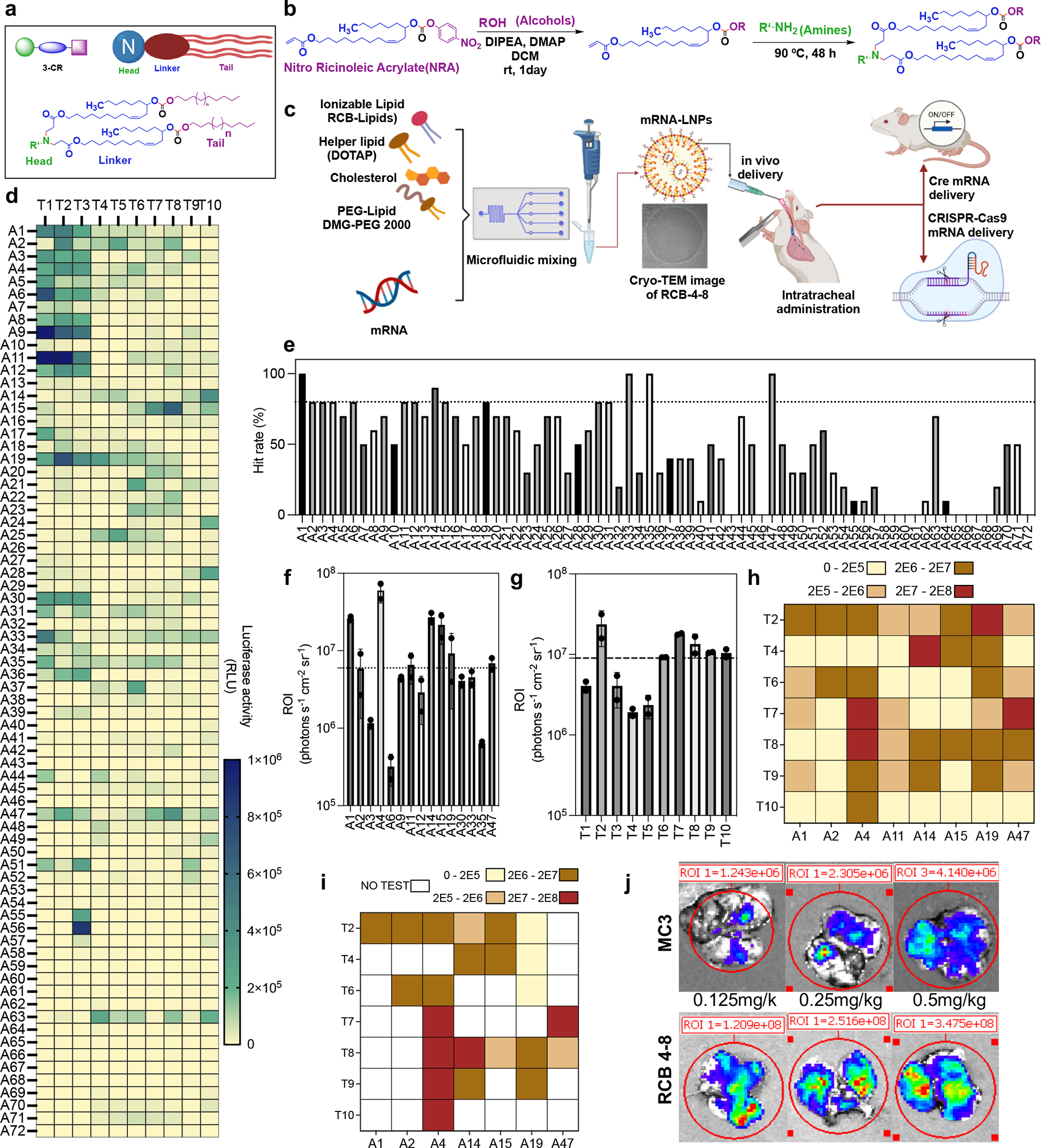
a, A illustration shows the three-component reaction (3-CR) of amine (headgroup), ricinoleic acrylate (linker), and alcohols (tail) for the high-throughput synthesis of lipids. b, The synthesis route of carbonate-based ionizable biodegradable lipids from Nitro Ricinoleic Acrylate (NRA). c, Structures of amine headgroups (72) and alcohol tails (10) in a combinatorial library of 720 biodegradable ionizable lipids. d, A schematic to illustrate the formulation of a newly designed RCB-4-8 LNP for pulmonary delivery of Cre-mRNA and CRISPR-Cas9 gene-editing tools. e, A549 cells were treated with mLuc-loaded LNPs. The relative luciferase expression after incubating with mLuc LNPs overnight is shown in a heat map. f, The hit rate of each amine headgroup was calculated by the percentage of LNPs with luciferase expression > 50,000. g-j, Results of the in vivo batch-based testing, where batch 1 analysis (g) determined the top-performing amine headgroup structures (intramuscular injection, 0.5 mg/kg mLuc, 10 LNP mixtures per mouse); batch 2 analysis (h) identified the top-performing lipid tail structures (intramuscular injection, 0.4 mg/kg, 8 LNP mixtures per mouse); batch 3 analysis (i) identified the individual lipids with top-performing mRNA delivery efficiency in the muscle (intramuscular injection, 0.25 mg/kg mLuc per mouse). Finally, batch 4 analysis (j) identified the individual lipids with top-performing mRNA delivery efficiency in the lung (intratracheal administration, 0.25 mg/kg mLuc per mouse). For all in vivo screening studies in c–f, results were obtained from two mice per group and presented as mean ± SD. k, Representative IVIS images of mouse lungs at 6 hr following intratracheal administration of MC3 or RCB-4-8 mLuc LNPs (0.125, 0.25, 0.5 mg/kg mLuc per mouse).
A visual overview of biological evaluations is provided in Fig. 1d. To identify lead candidates, LNPs composed of the new ionizable lipids, DOPE, C14-PEG2000 and cholesterol were prepared following a classical formulation ratio19 and loaded with firefly luciferase mRNA (mLuc). A549 cells were treated with mLuc-LNPs, and luciferase expression was measured after overnight incubation via Bright-Glo Assay (Fig. 1e). To select promising ones for in vivo tests, these new LNPs were categorized by 72 different amine headgroups (A1-A72) of ionizable lipids; ionizable lipids in each headgroup class contain the same amine headgroup but different lipid tails (T1-T10). The percentage of hit rate is calculated by the number of lipids with relative luminescence unit [RLU] >50,000 (1–10) divided by the total number of lipids in each group (10). Headgroups with a hit rate of at least 80% across lipid variants were chosen for further animal screening (Fig. 1f). To expedite screening, we adopted an orthogonal batched-based testing strategy, which dramatically reduces the number of animals required and the time demanded to identify lead LNPs. First, mLuc-LNPs carrying identical headgroups were pooled into 15 batches (10 LNPs per batch) and intramuscularly injected into mice. After six hours, the transfection potency of each cluster was measured via the luminescence signal at the injection site with IVIS imaging (Fig. 1g, Fig. S1), identifying eight outstanding amine headgroups (ROI > 6×106 p/sec/cm2/sr). Similarly, lipids carrying identical lipid tails were pooled into ten batches (8 LNPs per batch) and intramuscularly injected into mice (Fig. 1h, Fig. S2), identifying seven outstanding lipid tails (ROI > 9×106 p/sec/cm2/sr). We then examined the transfection potency of the ionizable lipid containing top-performing components (8 amine headgroups x 7 lipid tails = 56 promising lipids) by intramuscular injection in mice (Fig. 1i, Fig. S3). Those with an ROI > 2×106 p/sec/cm2/sr were further investigated in the lung via intratracheal administration (Fig. 1j, Fig. S4) and the top nine (ROI > 1×107 p/sec/cm2/sr) were further investigated for gene editing. While the strategy of batch-based testing can improve the efficiency of LNP screening, the potential for false negatives increases, in particular for those formulations that are only efficacious above the tested dose. To investigate the potential for the top nine LNPs to deliver CRISPR-Cas9 complexes, we tested their capacity to deliver SpCas9 mRNA and sgRNA to HEK-GFP (HEK 293T cell line that expresses the green fluorescent protein). LNPs formulated with SpCas9 mRNA/GFP-sgRNA were added to the HEK-GFP cells (Fig. S5). Six of the nine LNPs knocked out GFP in over 80% of cells, outperforming the positive controls of Lipofectamine MessengerMAX (~64%) and RNAiMAX (~35%). Notably, the RCB-4-8 LNP, which displayed the most substantial potency in mLuc lung transfection in vivo, showed the highest GFP knock-out efficiency in vitro (~95%).
Previous work has shown that replacing DOPE with DOTAP, a cationic lipid, could effectively increase the transfection efficacy of mLuc LNPs in the lung after systemic administration20. Here, we reformulated the lead LNP RCB-4-8 following the as-reported DOTAP formulation and found that the luciferase expression in the lung after intratracheal dosing was also further improved (Fig. S6a), probably because the composition of cationic lipids could enhance the pulmonary cell internalization and mucus retention of LNPs. This DOTAP formulation of RCB-4-8 LNPs was characterized by dynamic light scattering and cryo-transmission electron microscope (Fig. S6b–d) and adopted for the rest of studies in this work. While LNPs made in microfluidics have a smaller size, lower PDI, and slightly higher encapsulation efficiency than those made by direct pipette mixing (Fig. S6b). In a dose-response assay, RCB-4-8 mLuc-LNPs improved lung transfection efficiency approximately 100-fold compared to LNPs formulated with DLin-MC3-DMA (MC3), an ionizable lipid approved by the U.S. FDA for RNA delivery (Fig. 1k, Fig. S7). The potency of RCB-4-8 LNPs in mediating mRNA transfection in the lung is more than 10-fold higher than that of another LNP formulation recently developed for pulmonary mRNA delivery21. Moreover, less than 30% of the biodegradable RCB-4-8 remained in the lung 48 hrs post-administration, compared to over 90% retention of MC3 at the same time point (Fig. S8), suggesting lower toxicity.
Efficient gene editing in the lung provides a means to target several genetic disorders22. As a proxy for elaborate gene editing approaches, we examined Cre-recombinase mRNA (Cre-mRNA)-mediated editing through intratracheal delivery of the RCB-4-8 LNP. To visually detect gene editing in vivo, we intratracheally administered the LNPs loaded with Cre-mRNA (LNP-Cre) to Lox-3xSTOP-Lox (LSL)-tdTomato (Ai9) reporter mice23. The Cre recombinase translated from mRNA can catalyze genetic recombination to remove the LSL cassette, resulting in the expression of tdTomato, a red fluorescent protein gain-of-signal in genetically modified cells (Fig.2a). Mice were dosed either once (LNP-Cre x1) or three times (LNP-Cre x3) over four days with LNPs-Cre. We collected the lungs three days after the last dose and quantified tdTomato expression by flow cytometry. Results show that LNP-Cre x1 and LNP-Cre x3 could result in tdTomato-positive (tdTomato+) expression in 42.0±3.0% and 53.0±6.0% of total lung cells, respectively (Fig. 2b, Fig. S9), which is more than two-fold higher than Cre mRNA delivered by polymer polyplexes (~20%)24. Furthermore, to investigate the cellular distribution of edited cells, tdTomato and nuclei (DAPI) were visualized in frozen lung sections. LNP-Cre led to a well-distributed tdTomato signal in the airway, while multiple dosing could remarkably increase the number of tdTomato+ cells (Fig. 2c). To further validate this result, we also performed immunohistochemistry staining on lung tissue sections, which manifested an average of 17% and 39% tdTomato+ cells for LNP-Cre x1 and LNP-Cre x3, respectively (Fig. 2d–e). Importantly, both large (35%) and small (22%) airways can be edited after triple doses of LNP-Cre (Fig. S10a–c). Also, PCR analysis of genomic DNA (gDNA) from lung cells revealed that edited Ai9 alleles were present after single- and triple-doses of LNP-Cre (Fig. 2f); additionally, compared to LNP-Cre x1, LNP-Cre x3 led to increased PCR band intensity at 300bp, manifesting that multiple dosing improves gene editing. This also demonstrates that our LNPs are amenable to repeated dosing, which is a significant advantage compared to viral vectors.
Fig. 2 |. Gene editing in mouse lung with RCB-4-8 LNP-mRNA.
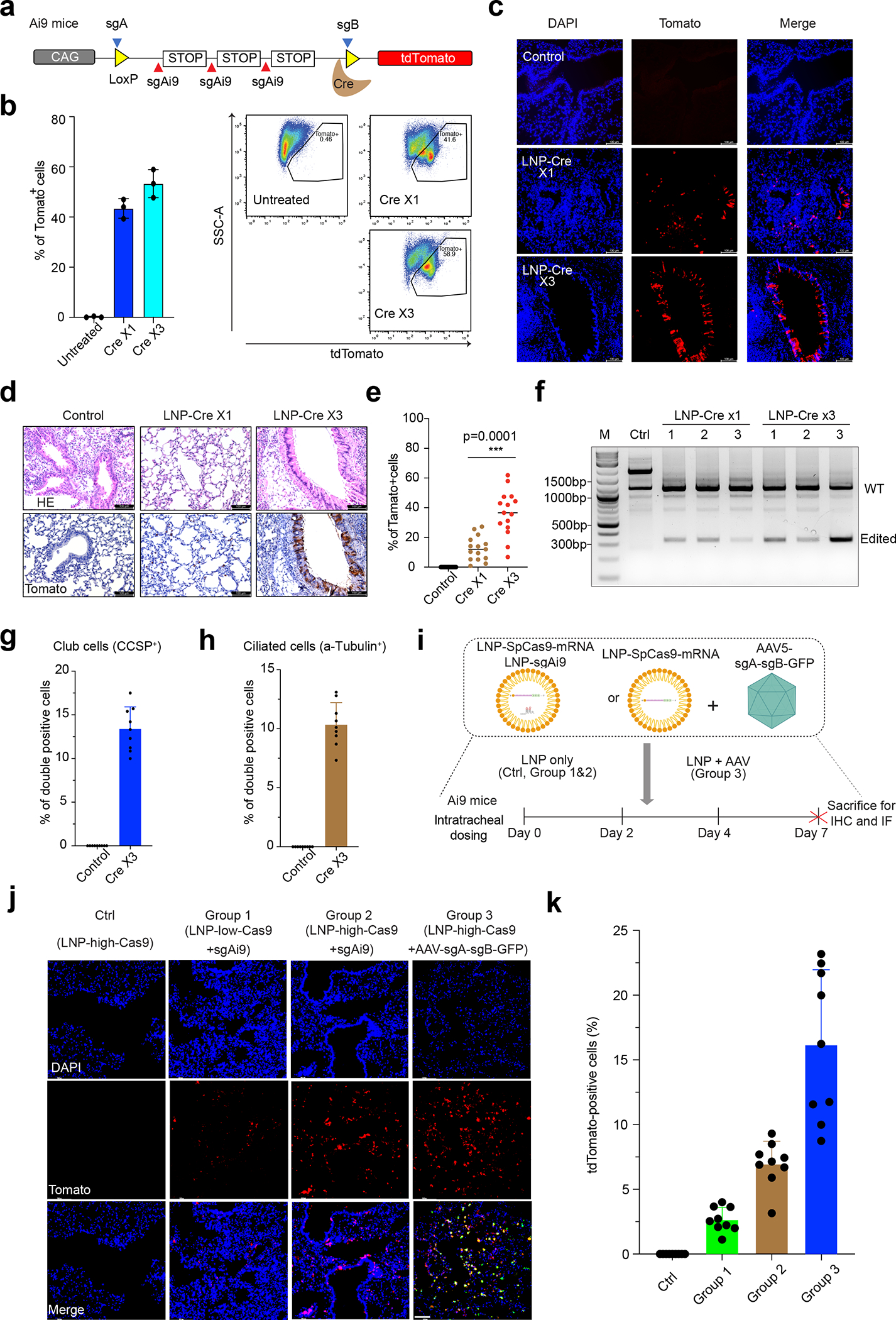
a, Measuring RCB-4-8 LNP-Cre-mRNA mediated editing using Lox-STOP-Lox tdTomato (Ai9) reporter mice. The Cre recombinase will delete the STOP cassettes and activate the tdTomato reporter. b, Ai9 mice were intratracheally administered with either one or three doses (over four days) of Cre-mRNA. To study the impact of AAV5 immunogenicity on the LNP, we delivered 6×1010 AAV5 with a dummy sequence in the mouse lung one week before dosing LNP-mRNA. Three days after the last dose, the lungs were collected and analyzed by flow cytometry (n=3 mice for each group). Results were obtained from three mice and presented as mean ± SD. c, Representative native fluorescence images of lung sections are shown. Results were obtained from 3 independent experiments. Saline serves as a negative control. Scale bar: 100μm. d, Paraffin-embedded lung sections were stained with antibodies for tdTomato. e, Quantification of IHC in d. Results were obtained from three mice (n=5 sections per mouse) and presented as mean ± SD. ***P<0.001 by one-way ANOVA with Tukey’s multiple comparisons test. f. Genomic DNA was collected from mice (n=3/group) treated with LNP-Cre mRNA or controls. PCR was performed with primers flanking the Ai9 locus. WT: wide type. M, marker. g, Quantification of tdTomato+ club cells. Results were obtained from three mice (3 airways per mouse) and presented as mean ± SD. h, Quantification of tdTomato+ ciliated cells. Results were obtained from three mice (3 airways per mouse) and presented as mean ± SD. i, Measuring RCB-4-8 LNP-SpCas9-mRNA/sgRNA mediated NHEJ using Ai9 reporter mice. The sgAi9 or sgA and sgB will delete the STOP cassettes and activate the tdTomato reporter. Ai9 mice were intratracheally administered with low or high doses of SpCas9-mRNA/sgAi9. To develop a combined viral and non-viral delivery of CRISPR system components in vivo, we intratracheally delivered 6×1010 scAAV5-sgA-sgB-GFP one week before dosing LNP-SpCas9-mRNA. Three days after the last dose, the lungs were collected. j-k, Quantification of tdTomato positive cells (k) and representative native fluorescence images of lung sections (j) are shown. LNPhigh-SpCas9mRNA serves as a negative control. Scale bar: 100μm. n=9 sections from 3 mice. Error bars are S.D. Results were obtained from three mice (3 airways per mouse) and presented as mean ± SD.
Club and ciliated cells are two major subtypes of airway epithelial cells in the lung25. Ciliated cells move mucus along the bronchiolar epithelium and cannot self-renew or transdifferentiate in response to injury26. Club cells protect the epithelium by secreting club cell secretory proteins and, in response to injury, can differentiate into ciliated cells to regenerate the bronchiolar epithelium27. Clara cell secretory protein (CCSP) and acetylated-tubulins are markers for club cells (formerly known as Clara cells) and ciliated cells, respectively28. Antibodies against tdTomato and CCSP or acetylated-tubulin were used to detect edited cells (double-positive) in paraffin-fixed lung sections. We observed 13±3% and 11±2% double-positive club and ciliated cells, respectively, in the large airways (Fig. 2g–h, Fig. S10d–g) under both confocal and standard fluorescence microscope. Together, these data demonstrate that the RCB-4-8 LNP can deliver mRNA to various cell types in respiratory epithelium and induce potent expression of functional proteins in these cells.
Next, the feasibility of using RCB-4-8 LNP to achieve efficient pulmonary delivery of CRISPR-Cas9 complexes was explored in the Ai9 tdTomato reporter mouse. As illustrated in Fig. 2i, a single-guide RNA (sgRNA), sgAi9, previously developed to delete the tdTomato STOP cassettes and activate tdTomato expression in cells, was co-encapsulated with SpCas9 mRNA into the RCB-4-8 LNP29. Then, we intratracheally administered these RCB-4-8 LNPs at low (LNPlow-SpCas9 mRNA+sgAi9) or high (LNPhigh-SpCas9 mRNA+sgAi9) dose (0.5 or 1 mg•kg−1 total RNA) at 2-day intervals (three doses) and quantified gene editing on the 7th day. In the absence of sgRNA, the negative control, RCB-4-8 LNPs carrying SpCas9 mRNA only (LNPhigh-SpCas9 mRNA) did not display any tdTomato signal in the lung tissue; in contrast, LNPlow-SpCas9 mRNA+sgAi9 and LNPhigh-SpCas9 mRNA+sgAi9 could result in 3±1.0% or 7±3.0% tdTomato+ cells, respectively (Fig. 2j–k). Compared to Cre recombinase, which is highly efficient in LoxP deletion, tdTomato reporter activation triggered by the SpCas9/sgRNA system requires specific double or triple deletions of STOP cassettes; as a result, while the tdTomato mouse enables visualization of edited cells in vivo, the number of tdTomato+ cells in this experiment tends to under-report Cas9 editing. On the other hand, as numerous AAV serotypes have been identified with variable tropism30, the hybrid approach of combining AAV encoding sgRNA with LNP carrying Cas9 mRNA is promising to enable tissue-specific gene editing in vivo. Therefore, in addition to co-delivery of Cas9 mRNA and sgRNA solely using LNPs, we also explored the efficacy of gene editing by combined viral (i.e., AAV) and non-viral (i.e., LNP) delivery of CRISPR system components in the lung. A dual sgRNA (sgA and sgB) system that is highly efficient for LoxP editing was adopted in this LNP-AAV experiment31. In contrast to single guide sgAi9, which could only remove two out of three STOP cassettes, these dual sgRNAs tend to be more efficient as deletion mediated by sgA and sgB could remove all three STOP cassettes. A cohort of Ai9 mice were intratracheally administered with sgA and sgB packaged in AAV (AAV-sgA+sgB, 6×1010) seven days before receiving LNPhigh SpCas9 mRNA. Immunostaining assay of lung tissue sections on the 14th day shows that the LNPhigh-SpCas9mRNA+AAV-sgA-sgB could activate tdTomato fluorescent reporter in 17.0±5.0% of total lung cells (Fig. 2j–k). While this editing efficiency is comparable to that of a recently reported dual-AAV-mediated CRISPR-Cas9 gene editing approach (~16%–26%) 31, the transient nature of RNA from LNPs would avoid the safety concerns associated with persistent SpCas9 expression from AAV transduction13, 32. Because the entire Ai9 locus (994bp between sgA and sgB) is too large for short-reads amplicon deep sequencing, we used PCR to make two libraries of sgA (167bp) and sgB (103bp) target sites. Indels were detected at both target sites in gDNA collected from mouse lungs treated with LNPhigh-SpCas9mRNA+AAV-sgA-sgB (Fig. S11). Notably, this sequencing strategy did not measure large deletions induced by sgA and sgB in tdTomato+ cells. Although different sgRNA designs prevent direct comparison of LNPhigh-SpCas9mRNA/AAV-sgA+sgB to LNPhigh-SpCas9mRNA/sgAi9, our proof-of-concept work convincingly demonstrates delivery approaches based on RCB-4-8 LNP-based for efficient CRISPR-Cas9 gene editing in the lung. Previously, intravenous injection of lung-targeted LNP formulations encapsulating SpCas9 mRNA/sgRNA reported a gene editing efficiency of 15.1% in mouse lungs33. Nevertheless, lung cells edited by that approach are primarily epithelial cells instead of endothelial cells, which are the primary targets for most congenital lung diseases, including CF, AAT and SP. In addition, pulmonary delivery of gene editors is better suited to avoid gene editing in undesirable tissues/organs primarily associated with systemic delivery34. Together, these compelling results demonstrate the promising potential of RCB-4-8 LNPs for enabling CRISPR/Cas9 gene editing in the lung.
This work demonstrates the power of combining rational design and high throughput screening to improve the in vivo performance of ionizable LNPs for mRNA delivery. Previously we found that incorporating alkyne structures in the ionizable lipid tail could increase nanoparticle fusogenicity and thus facilitate the endosomal escape of mRNA cargos35. However, alkyne-containing ionizable lipids (e.g., A6) could only be effective when combined with a classical alkene-containing non-biodegradable ionizable lipid such as MC3 and cKK-E1235. In contrast, RCB-4-8 demonstrates that alkyne-containing ionizable lipids could also work solely to enable efficient mRNA delivery. Moreover, secondary carbonate groups in RCB-4-8 allow it to retain the advantages of branched tails for mRNA delivery while being more biodegradable than the lipids with secondary ester groups, which degrade relatively slowly in the physiological environment36. To make the intratracheal dosing of these LNP formulations more clinically relevant, we are currently working on improving the stability of LNP formulations for nebulization. We expect this will enable patients to receive gene therapy by simply inhaling the mRNA LNPs via a nebulizer. On the other hand, many lung diseases, such as CF, are characterized by the accumulation of thick, sticky mucus. This poses an additional challenge for pulmonary gene delivery. Future work will explore the performance of LNP formulations in diseased animal models with abnormal mucus.
In summary, we have synthesized and evaluated a library of 720 biodegradable ionizable lipids, among which RCB-4-8 was identified to enable highly efficient pulmonary mRNA delivery and gene editing in the lung, using an alkyne-containing ionizable lipid as the sole ionizable compound used for potent mRNA transfection. In contrast to DNA-based AAV delivery systems, which lead to lasting Cas9 expression, mRNA is relatively short-lived and limits the potential to accumulate off-target gene alterations. Furthermore, unlike AAV, LNPs offer the potential for repeat administration, which may be essential to achieving therapeutic editing levels in the lung. We have shown that RCB-4-8 can deliver a Cas9 system to both ciliated and club epithelial cells in the lung, suggesting its potential to enable gene editing for various airway diseases, including CF (ciliated epithelial cells).
Additionally, we have developed a combined viral and non-viral delivery system for Cas9-mediated gene editing in the lung, which provides a dual-delivery approach to limit off-tissue targeting editing while limiting the expression of SpCas9. The RCB-4-8 LNP is an efficient non-viral delivery system for gene editing in the lung epithelium and opens new avenues for treating congenital lung diseases. Further studies are underway to test whether RCB-4-8 LNP can transduce basal cells in the trachea of mice and non-human primates.
Methods
Materials and lipid library synthesis
All amines and other starting materials are purchased from Sigma-Aldrich, Enamine stores, TCI America, Alfa-Aesar, and Ambeed. High-throughput synthesis of lipids was carried out in 96 deep-well plates with glass inserts (molar ratio of amine/ricinoleic acrylate 1:1.1). The reactions were performed at 90 °C for two days, with conversions typically over 80%. These lipids were purified by flash column chromatography, and structures were confirmed by 1H and 13C 500 MHz NMR spectrometry. High-resolution mass spectra were obtained using a BioTOF High-Resolution Mass Spectrometry (HRMS) at the MIT Department of Chemistry Instrumentation Facility.
General Procedure A: synthesis of carbonates of ricinoleic acrylates (lipid tails# 1–10)
To a round bottom flask, (Z)-12-(((4-nitrophenoxy)carbonyl)oxy)octadic-9-en-1-yl acrylate (2.0 mmol, 1.0 equiv), DMAP (0.4 mmol, 0.2 equiv) and aliphatic alcohol (5.0 mmol, 2.5 equiv) in DCM (20 mL) was added. After dissolving all the starting materials, DIPEA (6.0 mmol, 3.0 equiv) was slowly, drop-wise added and stirred overnight at room temperature or until consumption of the starting material. The reaction mass was diluted with brine (20 mL) and extracted with dichloromethane (3 X 100 mL). The organic layer was dried over anhydrous MgSO4 and concentrated in vacuo. Crude products were purified via SiO2 gel flash column chromatography using hexanes and ethyl acetate (30:1) as eluent to afford a carbonate-derived ricinoleic acrylate as a colourless liquid.
General Procedure B: synthesis of ionizable lipids
Into a 1-dram scintillation vial equipped with a magnetic stir bar was placed a carbonate-derived ricinoleic acrylate (0.5 mmol, 2.5 equiv) and aliphatic amine (0.2 mmol, 1.0 equiv). Generally, the ionizable lipids were synthesized with a molar ratio of amine/ricinoleic acrylate carbonates in 1:2.5 and 1:1.25 equiv for primary and secondary amines, respectively. The vial was sealed with a silicone-lined screwcap and stirred at 90 °C for 2–3 days or until the complete conversion of the starting amine. Purification was achieved by flash column chromatography on an Isco Combiflash system eluting with a gradient (0–100%) Ultra (3% concentrated ammonium hydroxide, 22% methanol, 75% dichloromethane) in dichloromethane to afford carbonate-derived ricinoleic acrylate-based ionizable lipids (RCB- head group#-Tail group#).
Nanoparticle formulation
All lipids, except for the ionizable lipids, were purchased from Avanti. The benchmark ionizable lipid, DLin-MC3-DMA (MC3), was purchased from Cayman Chemical. LNPs were synthesized by mixing an aqueous phase containing the mRNA with an organic phase containing the lipids at a 3:1 volume ratio either by pipetting or in a microfluidic chip device19. The aqueous phase was prepared with mRNA in 10 mM citrate buffer (pH 3.0, Fisher). All mRNAs were stored at −80 °C and were allowed to thaw on ice before use. The organic phase was prepared by solubilizing with ethanol a mixture of the synthesized ionizable lipid, helper phospholipids (1,2-dioleoyl-sn-glycerol-3-phosphoethanolamine [DOPE] or 1,2-dioleoyl-3-trimethylammonium-propane [DOTAP]), cholesterol and C14-PEG2000 at predetermined molar ratios (35:16: 46.5: 2.5 for DOPE formulation19; 30:39:30:1 for DOTAP formulation20). LNPs for high-throughput in vitro and in vivo screening were prepared in a 96-well plate by directly adding the ethanol phase to the aqueous phase, followed by direct pipette mixing. RCB-4-8 LNPs for in vivo studies were all prepared using the microfluidic device. LNPs are compared with each other only when they are made by the same method. For in vitro studies, LNPs were directly incubated with cells without further dialysis. For in vivo studies, LNPs were mixed and dialyzed against 1X PBS in a 20,000 MWCO cassette (Thermo Fisher) at 4 °C for six hours before injection into mice.
LNP characterization
The size, polydispersity index, and zeta potentials of LNPs were measured using dynamic light scattering (ZetaPALS, Brookhaven Instruments). Diameters are reported as the intensity mean peak average. A modified Quant-iT RiboGreen RNA assay (Invitrogen) was used to calculate the mRNA encapsulation efficiency. Briefly, mRNA standard solutions with different concentrations were prepared in TE (Tris-EDTA) buffer. mRNA-LNP samples were eluted in TE to obtain a sample containing approximately 250 ng/mL mRNA. Similar standard mRNA solutions and mRNA-LNP samples were also made in TE buffer supplemented by Triton-X100 surfactant (10%). Into the microwells of a 96 well-plate, 100 μL of each sample (including standard mRNA solutions and mRNA-LNP samples) was added, followed by the addition of 100 μL Ribogreen reagent (1:200 diluted). After a 5-min incubation in the dark and at room temperature, fluorescence intensity was recorded using a Tecan Infinite M200 Pro plate reader, applying the excitation and emission at 485 and 528 nm, respectively. The fluorescence obtained from mRNA-LNP samples dispersed in TE and TE/Triton-X100 was theoretically attributed to free (unencapsulated) and total mRNAs, respectively. Standard curves plotted using standard solutions in TE and TE/Triton-X100 were utilized to convert fluorescence intensity to concentration (Conc). Finally, the encapsulation efficiency (EE%) was calculated according to the following formula: EE% = Conc total RNA−Conc free RNA / Conc total RNA.
In vitro high-throughput screening
LNPs loaded with 50 ng firefly luciferase mRNA (mLuc) were added to 96-well plates pre-seeded with A549 cells. After overnight incubation, the mLuc transfection efficiency was measured using Bright-Glo Luciferase Assay System (Promega) according to the manufacturer’s instructions. The fluorescence and luminescence were quantified using the Tecan Infinite M200 Pro plate reader.
In vitro gene editing study
293T cells stably expressing GFP were seeded into 48-well polystyrene tissue culture plates (Corning) at 50,000 cells per well in DMEM medium with 10% FBS. After 24h incubation, cells were transfected with 150ng of SpCas9 mRNA and 50ng of sgGFP (20nt sequence: GCTGAAGCACTGCACGCCGT) encapsulated in LNPs, Lipofectamine™ MessengerMax Transfection Reagent (Invitrogen, Cat# LMRNA001), or Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen, Cat# 13778100) according to manufacturer instructions. 72h after transfection, GFP+/− cells were analyzed on an LSR HTS-II flow cytometer.
Animal studies
All animal experiments were authorized by the Division of Comparative Medicine at the Massachusetts Institute of Technology or the Institutional Animal Care and Use Committee (IACUC) at UMass medical school. Six-week-old female C57BL/6J mice and eight-week-old Ai9 (strain #007909) mice from Jackson laboratory were used. A 14-hour light/10-hour dark cycle and a temperature of 65–75°F (~18–23°C) with 40–60% humidity were used.
In vivo batch-based testing
LNP mixtures within one classification group were mixed and dialyzed before injection into mice. Then, 0.5mg/kg mLuc RNA for batch analysis 1 (10 LNP mixtures in each group, intramuscular injection), 0.4mg/kg mLuc RNA per mouse for batch analysis 2 (8 LNP mixtures in each group, intramuscular injection), 0.25mg/kg for batch analysis 3 (intramuscular injection) and batch analysis 4 (intratracheal injection) were and injected for each lipid group. At 6h after LNP dosing, mice were subjected to the bioluminescence analysis.
Bioluminescence analysis
At 6h after the intramuscular or intratracheal administration of the mRNA LNPs, mice were injected intraperitoneally with 0.2 ml d-luciferin (10 mg/ml in PBS). The mice were anesthetized in a ventilated anesthesia chamber with 1.5% isofluorane in oxygen and imaged 10 min after the injection with an in vivo imaging system (IVIS, PerkinElmer). Luminescence was quantified using the Living Image software (PerkinElmer).
Generation of plasmid
AAV-sgA-sgB was generated through Gibson assembly, by combining the following three DNA fragments: (1) gBlock sgAi9L driven by U6, (2) gBlock sgAi9R driven by U6, (3) a MluI/EagI-digested AAV backbone. SpCas9 is expressed from our published single-stranded AAV vector with U1A promoter.31 Target sequences of the gRNAs are:
sgRNA.A: aaagaattgatttgataccg
sgRNA.B: gtatgctatacgaagttatt
AAV vector production
AAV vectors (AAV5 or scAAV5 capsids) were packaged at the Viral Vector Core of the Horae Gene Therapy Center at the University of Massachusetts Medical School. 31 In brief, AAV vector plasmid carrying an expression cassette for the gene of interest flanked by AAV2 ITRs is co-transfected into HEK293 cells with a packaging plasmid and adenovirus helper plasmid. The packaging plasmid expresses regulatory proteins of AAV2 and capsid proteins of the AAV5 serotype, which will excise the recombinant gene from the AAV vector plasmid, replicate the gene, and package the gene into AAV virions. Adenovirus serotype 5 E1, E2a, and E4 proteins, and VA I and II RNAs expressed from the adenovirus helper plasmid provide helper functions essential for rAAV rescue, replication, and packaging. The recombinant viruses are purified by standard CsCl gradient sedimentation method and desalted by dialysis. The vectors are quality control tested by ddPCR titration for DNase-resistant vector gene (vg) concentration using probe and primers targeting the poly(A) region of the vg and silver-stained SDS-polyacrylamide gel analysis to establish the purity of each lot.
Isolation of lung single cells
The single cells were generated with the lung dissociation kit (Miltenyi Kit: Catalog #: 130-095-927). Briefly, the mouse lung was perfused with PBS and placed in a 10cm cell culture dish. The lung tissue was minced using scalpel blades and scissors and dissociated on a MACS tissue dissociator. GentleMACS program 37C_m_LDK_1 was run for 10mins, followed by another gentleMACS program m_lung_02. After the program’s termination, tubes were detached from the MACS tissue dissociator. Samples were centrifuged at 300g for 10min, after which the supernatant was removed. The cell pellets were resuspended in 10ml BSA/PBS, +2.5ml buffer, and then filtered with 70 um strainers. The cell suspension was centrifuged at 500g for another 10 min and then resuspended in 2ml RBC lysis buffer after 10 min. Subsequently, the cell suspension was centrifuged at 300g for 10 min at 4°C. The final cell pellets were resuspended in 1ml BSA buffer for cell number counting and FACS analysis.
In vivo gene editing study
For gene editing in mouse lungs with RCB-4-8 LNPs-Cre mRNA, eight-week-old Ai9 mice were randomly allocated into three groups. The mice were injected with 60 μl RCB-4-8 LNPs either once (day 0) or three times (day 0, 2, 4). For gene editing in mouse lungs with RCB-4-8 LNPs-SpCas9 mRNA/sgRNA, eight-week-old Ai9 mice were randomly allocated into four groups. The mice were injected with 60 μl RCB-4-8 LNPs at two types of doses (low: 0.5 mg/kg or high dose: 1 mg/kg; total RNA [4/1 mRNA/sgAi9, wt/wt]). To study the immunogenicity, 60 μl PBS containing 6×1010 vector genes (vg) of scAAV5 virus (dummy sequence) or scAAV5-sgA-sgB virus were delivered to mouse lung through intratracheal intubation one week before the LNP injection. Animals were sacrificed at the end of each experiment (3 days after the last injection). Lungs were fixed with formalin or stored at −80°C with OTC (TissueTek) freezing compound until further analyses. No sample size calculation was performed, and each group consisted of at least three mice for statistical analysis.
Immunohistochemistry (IHC) and immunofluorescence
For IHC studies, formalin-fixed, paraffin-embedded (FFPE) mouse lung samples were sectioned at 4 μm, deparaffinized, and the antigen was retrieved with ten mM citrate buffer for 9 minutes at 95°C. Then the slides were incubated overnight at 4 °C with anti-GFP (CST, Cat. #2956, 1:200) or anti-tdTomato (Rockland, Cat. #600-401-379, 1:300). Visualization was performed using the DAB Quanto kit (Fisher Scientific, Cat. # TA-125-QHDX) as instructed by the manufacturer. 20 airways from 5 IHC images for each mouse were used to generate the average percentage.
For CCSP double staining, the slides were incubated overnight at 4 °C with an anti-CCSP antibody (Santa Cruz, Cat. # SC-25555, 1:2000) and an anti-tdTomato antibody (ThermoFisher, Cat. #MA5-15257, 1:300). The sections were then incubated for one hour at room temperature with Alexa Fluor 488 Donkey anti-Rabbit IgG (CCSP) and Alexa Fluor 647 Donkey anti-mouse IgG (tdTomato). For acetylated tubulin double staining, Alexa Fluor 647 anti-acetylated tubulin antibody (Santa Cruz, Cat. # SC-23950, 1:200) and anti-tdTomato antibody (Rockland, Cat. #600-401-379) were used. Nuclei were counterstained with DAPI. Images were acquired on a ZEISS confocal microscope (40X oil lens) and a Leica DMi8 imaging microscope.
For direct fluorescence imaging, lungs were fixed in 4% buffered paraformaldehyde overnight at 4°C, then equilibrated in 30% sucrose overnight at 4°C before freezing in OCT. Three non-consecutive sections from each organ sample were mounted with DAPI to visualize nuclei and imaged for DAPI, tdTomato, and GFP. A Leica DMi8 scanner at 20X magnification was used to obtain and process the images. For quantification of editing, airways were identified in lung sections by morphology (thick ring of DAPI-stained nuclei), and particular areas were measured for classification into small (< 50,000 μm2) or large (> 50,000 μm2). The total number of tdTomato+ cells and DAPI-stained nuclei were counted in each airway. Editing efficiency was calculated as tdTomato+ cells/DAPI-stained nuclei and expressed as a percentage.
Targeted amplicon deep sequencing
Code and primers used for deep sequencing data analysis are provided in the supplementary information. Genomic DNA (gDNA) was isolated for indel analysis from the frozen lung of Ai9 mice injected with AAV-sgA-sgB and LNP-SpCas9mRNA. Genomic loci spanning the target sites were PCR amplified with locus-specific primers carrying tails complementary to the Truseq adapters. Targeted amplicon deep sequencing libraries were prepared by two PCR steps. 200ng of gDNA was used for the 1st PCR using a Phusion master mix (Thermo) with locus-specific primers that contain tails. PCR products from the 1st PCR were used for the 2nd PCR with i5 primers and i7 primers to complete the adaptors and include the i5 and i7 indices. All primers used for the amplicon sequence are listed in the Supplementary sequence below. PCR products were purified with Ampure beads (0.9X reaction volume), eluted with 25ul of TE buffer, and quantified by Tapestation and Qubit. An equal mole of each amplicon was pooled and sequenced using Illumina Miniseq. Amplicon sequencing data were analyzed with CRISPResso (https://crispresso.pinellolab.partners.org/). Briefly, demultiplexing and base calling were performed using bcl2fastq Conversion Software v2.18 (Illumina, Inc.), allowing 0 barcode mismatches with a minimum trimmed read length of 75. Alignment of sequencing reads to the Amplicon sequence (Supplementary Data 1) was performed using CRISPResso2 in standard mode using the parameters “−q 30”. For all experiments, indel frequency was calculated as the number of discarded reads divided by the total number of reads ((number of indel-containing reads)/(number of reference-aligned reads)).
Statistical analysis
Statistical analyses for plotted data were performed using GraphPad Prism 9. The sample size was not pre-determined by statistical methods but rather based on preliminary data. Group allocation was performed randomly. In all studies, data represent biological replicates (n) and are depicted as mean ± s.d. as indicated in the figure legends. A two-tailed Student’s t-test or a one-way analysis of variance (ANOVA) was performed when comparing two groups or more than two groups, respectively, as indicated in the figure legends. In all analyses, P values < 0.05 were considered statistically significant.
Display items
The cartoons of lipid components, lipid nanoparticles, microfluidic chip, pipettes, mice, CRISPR-Cas9 and AAV in Fig.1d and Fig. 2i were created with BioRender.com.
Supplementary Material
Acknowledgments
This work was supported by Translate Bio (Lexington, MA) and the National Institutes of Health (grant nos. UG3HL147367). B.L., R.S.M., A.G., and A. J. were supported by Translate Bio (Lexington, MA). B.L. was supported by the Leslie Dan Faculty of Pharmacy startup fund. A.V. was supported by the PRiME Postdoctoral Fellowship from the University of Toronto. S.L., G.G., W.X. and D.G.A were supported by grants from the National Institutes of Health (grant nos. UG3HL147367 and UH3HL147367). W.X. was supported by grants from the National Institutes of Health (grant nos. DP2HL137167 and P01HL131471), the American Cancer Society (grant nos. 129056-RSG-16-093), and the Cystic Fibrosis Foundation. The authors thank the Koch Institute Swanson Biotechnology Center for technical support, specifically the Animal Imaging & Preclinical Testing, Histology, Nanotechnology Materials, and Microscopy core facilities.
Footnotes
Competing Interests
B.L., R.S.M., A.G., and D.G.A. have filed a patent for developing the described lipids. D.G.A. receives research funding from Translate Bio and is a Founder of Orna Therapeutics. R.L. is a co-founder of Moderna; he also serves on the board and has equity in Particles for Humanity. For a list of entities with which R.L is, or has been recently involved, compensated or uncompensated, see https://www.dropbox.com/s/yc3xqb5s8s94v7x/Rev%20Langer%20COI.pdf?dl=0. The remaining authors declare no competing interests.
Data availability
Illumina Sequencing data have been submitted to the Sequence Read Archive. These datasets are available under BioProject Accession number PRJNA918691. The authors declare that all other data supporting the findings of this study are available within the paper and its Supplementary Information files or upon reasonable request. Plasmids are available from Addgene.
Reference
- 1.Rudnick DA & Perlmutter DH Alpha-1-antitrypsin deficiency: a new paradigm for hepatocellular carcinoma in genetic liver disease. Hepatology 42, 514–521 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Riordan JR et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073 (1989). [DOI] [PubMed] [Google Scholar]
- 3.Kwok AJ, Mentzer A & Knight JC Host genetics and infectious disease: new tools, insights and translational opportunities. Nature Reviews Genetics 22, 137–153 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bisserier M et al. Novel Insights into the Therapeutic Potential of Lung-Targeted Gene Transfer in the Most Common Respiratory Diseases. Cells 11, 984 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wan T & Ping Y Delivery of genome-editing biomacromolecules for treatment of lung genetic disorders. Advanced Drug Delivery Reviews 168, 196–216 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Treating Cystic Fibrosis with mRNA and CRISPR. Human Gene Therapy 31, 940–955 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lau C & Suh Y In vivo genome editing in animals using AAV-CRISPR system: applications to translational research of human disease [version 1; peer review: 2 approved]. F1000Research 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uddin F, Rudin CM & Sen T CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Frontiers in Oncology 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han HA, Pang JKS & Soh B-S Mitigating off-target effects in CRISPR/Cas9-mediated in vivo gene editing. Journal of Molecular Medicine 98, 615–632 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mingozzi F & High KA Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 122, 23–36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berical A, Lee RE, Randell SH & Hawkins F Challenges Facing Airway Epithelial Cell-Based Therapy for Cystic Fibrosis. Frontiers in Pharmacology 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swiech L et al. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol 33, 102–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yin H, Kauffman KJ & Anderson DG Delivery technologies for genome editing. Nat Rev Drug Discov 16, 387–399 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Yin H et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat Biotechnol 35, 1179–1187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu M et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of <i>Angptl3</i>. Proceedings of the National Academy of Sciences 118, e2020401118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gillmore JD, Maitland ML & Lebwohl D CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. Reply. N Engl J Med 385, 1722–1723 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Alapati D & Morrisey EE Gene Editing and Genetic Lung Disease. Basic Research Meets Therapeutic Application. American Journal of Respiratory Cell and Molecular Biology 56, 283–290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maier MA et al. Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics. Molecular Therapy 21, 1570–1578 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kauffman KJ et al. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett 15, 7300–7306 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Kauffman KJ et al. Rapid, Single-Cell Analysis and Discovery of Vectored mRNA Transfection In Vivo with a loxP-Flanked tdTomato Reporter Mouse. Mol Ther Nucleic Acids 10, 55–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lokugamage MP et al. Optimization of lipid nanoparticles for the delivery of nebulized therapeutic mRNA to the lungs. Nat Biomed Eng 5, 1059–1068 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doudna JA The promise and challenge of therapeutic genome editing. Nature 578, 229–236 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madisen L et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel AK et al. Inhaled Nanoformulated mRNA Polyplexes for Protein Production in Lung Epithelium. Adv Mater 31, e1805116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hogan BL et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 15, 123–138 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thebaud B Angiogenesis in lung development, injury and repair: implications for chronic lung disease of prematurity. Neonatology 91, 291–297 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Kotton DN & Morrisey EE Lung regeneration: mechanisms, applications and emerging stem cell populations. Nat Med 20, 822–832 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herriges M & Morrisey EE Lung development: orchestrating the generation and regeneration of a complex organ. Development 141, 502–513 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Staahl BT et al. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat Biotechnol 35, 431–434 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown D et al. Deep Parallel Characterization of AAV Tropism and AAV-Mediated Transcriptional Changes via Single-Cell RNA Sequencing. Front Immunol 12, 730825 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang SQ et al. AAV5 delivery of CRISPR-Cas9 supports effective genome editing in mouse lung airway. Mol Ther 30, 238–243 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yin H et al. Non-viral vectors for gene-based therapy. Nat Rev Genet 15, 541–555 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Cheng Q et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat Nanotechnol 15, 313–320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yin H et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol 34, 328–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miao L et al. Synergistic lipid compositions for albumin receptor mediated delivery of mRNA to the liver. Nature Communications 11, 2424 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X et al. Functionalized lipid-like nanoparticles for in vivo mRNA delivery and base editing. Sci Adv 6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Illumina Sequencing data have been submitted to the Sequence Read Archive. These datasets are available under BioProject Accession number PRJNA918691. The authors declare that all other data supporting the findings of this study are available within the paper and its Supplementary Information files or upon reasonable request. Plasmids are available from Addgene.