

Abstract
Purpose of Review
This review aims to provide an overview of neuroinflammation in ischemic and hemorrhagic stroke, including recent findings on the mechanisms and cellular players involved in the inflammatory response to brain injury.
Recent Findings
Neuroinflammation is a crucial process following acute ischemic stroke (AIS) and hemorrhagic stroke (HS). In AIS, neuroinflammation is initiated within minutes of the ischemia onset and continues for several days. In HS, neuroinflammation is initiated by blood byproducts in the subarachnoid space and/or brain parenchyma. In both cases, neuroinflammation is characterized by the activation of resident immune cells, such as microglia and astrocytes, and infiltration of peripheral immune cells, leading to the release of pro-inflammatory cytokines, chemokines, and reactive oxygen species. These inflammatory mediators contribute to blood-brain barrier disruption, neuronal damage, and cerebral edema, promoting neuronal apoptosis and impairing neuroplasticity, ultimately exacerbating the neurologic deficit. However, neuroinflammation can also have beneficial effects by clearing cellular debris and promoting tissue repair. The role of neuroinflammation in AIS and ICH is complex and multifaceted, and further research is necessary to develop effective therapies that target this process. Intracerebral hemorrhage (ICH) will be the HS subtype addressed in this review.
Summary
Neuroinflammation is a significant contributor to brain tissue damage following AIS and HS. Understanding the mechanisms and cellular players involved in neuroinflammation is essential for developing effective therapies to reduce secondary injury and improve stroke outcomes. Recent findings have provided new insights into the pathophysiology of neuroinflammation, highlighting the potential for targeting specific cytokines, chemokines, and glial cells as therapeutic strategies.
Keywords: Neuroinflammation, Ischemic stroke, Hemorrhagic stroke, Intracerebral hemorrhage, Glial cells, Immune cells, Cytokines, Chemokines, Secondary injury, Microglia, Astrocytes, Inflammatory response, Therapeutic targets, Pathophysiology, Brain damage, Edema
Introduction
Stroke is an acute neurologic injury caused by ischemia or hemorrhage that stems from a wide range of pathologies. Ischemia is responsible for approximately 87% of strokes, making it the most common etiology among stroke types [1]. In either stroke type, the central nervous system’s (CNS) metabolic demands cannot be met, leading to brain tissue injury and cell death. In acute ischemic stroke (AIS), this metabolic insufficiency is due to impaired cerebral blood flow (CBF) due to blood vessel occlusion or systemic hypoperfusion. In contrast, metabolic insufficiency is secondary to vascular rupture and blood loss in hemorrhagic stroke (HS).
Regardless of stroke type, the role of inflammation cannot be overlooked. Neuroinflammation has been recognized as a critical component of stroke pathophysiology, affecting both the disease’s acute and chronic phases. The inflammatory process in stroke involves activating various cells, including microglia, astrocytes, endothelial cells, and leukocytes. It also causes the release of pro-inflammatory cytokines such as inflammatory chemokines (e.g., CXCL8, CCL2, and CCL3) and adhesion molecules. The inflammatory response can exacerbate brain damage leading to secondary injury, impeding tissue repair and recovery, and contributing to post-stroke complications, such as motor and cognitive impairment. Therefore, understanding the mechanisms and regulation of inflammation in stroke is essential for developing effective prevention and treatment strategies. This review provides an overview of the current knowledge on the role of inflammation in AIS and intracerebral hemorrhage (ICH), one of the HS subtypes, including its causes, mediators, and outcomes.
Neuroinflammation in Ischemic Stroke: General Concepts
Cerebral ischemia occurs due to obstructed CBF. The lack of oxygen and glucose supply required to meet the cerebral metabolic demand leads to the ischemic cascade, a complex series of events resulting in cell death and neurodegeneration as neurons and glial cells are unable to maintain their ionic homeostasis [2••]. Neuroinflammation is the activation of the brain’s innate immune system in response to an inflammatory challenge due to stimuli such as cerebral ischemia, and it involves immune cells, blood vessels, and molecular mediators [3]. This immune response aims to eliminate the initial cause of cellular injury [4], clear out damaged necrotic cells, and initiate brain tissue repair. The inflammatory cascade is initiated a few hours after stroke and can persist for days, weeks, or even months as a delayed tissue reaction to injury [5••]. The most effective approach to reducing cerebral ischemic damage is the emergent restoration of CBF. However, this can evoke secondary reperfusion injury caused by the inflammatory response [6]. Changes resulting from the loss of cerebral glucose and oxygen lead to injury to microvasculature and the blood-brain barrier (BBB), but the precise role of secondary inflammatory cells in the pathophysiology of cerebral ischemia remains unclear [7]. Inflammatory mediators can have detrimental effects on ischemic tissue, but they may also exert beneficial effects on stroke recovery. The disruption of the cellular ionic gradients results in an excess release of glutamate, causing an increase in intracellular calcium influx and excitotoxicity that initiates necrotic and apoptotic cell death pathways [8] . Cellular ionic imbalances can generate reactive oxygen species (ROS) during cell death processes, mediating membrane, mitochondrial, and DNA damage and resulting in protein misfolding and inflammation. Importantly, reperfusion after cerebral ischemia also increases the production of ROS and the inflammatory response, potentially worsening neuronal injury. However, outside of the reperfusion with thrombolytics and endovascular procedures limited by the studied time window, there is no known effective treatment for cerebral ischemia once it occurs [9] . Although numerous neuroprotective strategies have been developed, they have shown disappointing results in clinical trials due to a lack of efficacy or undesirable side effects. Interestingly, evidence suggests that cerebral ischemia can induce immune suppression in addition to the pro-inflammatory response [10]. This immune suppression may be a protective mechanism to limit excessive inflammation and secondary tissue damage, mediated by the release of anti-inflammatory cytokines and the activation of regulatory T-cells that can help dampen the immune response. However, while immune suppression may be beneficial in the acute phase of cerebral ischemia, it can also have long-term negative consequences. Studies have shown that immune suppression can increase the risk of infection and impair tissue repair and regeneration [11, 12••]. Table 1 provides a summary of essential factors related to neuroinflammation in stroke.
Table 1.
The pathophysiology of neuroinflammation in stroke
| Pathophysiology | Mechanism | Outcome |
|---|---|---|
| Endothelial cell activation | Release of von Willebrand factor (vWF) stored in Weibel-Palade bodies, interaction with leukocytes | Platelet activation, leukocyte recruitment, capillary no-reflow |
| ROS production | Acute hypoxia and glucose deprivation, cytosolic enzyme and mitochondrial activity | Upregulation of AMPA and glutamate receptors, excitotoxicity, neuronal apoptosis, pro-inflammatory cytokine production |
| Inflammatory cascade activation | P-selectin interaction with P-selectin glycoprotein ligand-1 on leukocytes, immune cell recruitment | Increased cerebral inflammation, progression of ischemic injury |
| Monocyte and macrophage function | Expression of anti- and pro-inflammatory cytokines | Dual effects on inflammation, phagocytosis of degraded cells |
| T-cell function | IL-17 secretion by CD4 and CD8 cells | Aggravation of ischemic injury |
| Platelet activation | Granule release, interaction with leukocytes | Increased platelet activation, pro-inflammatory cytokine release, formation of platelet-leukocyte complexes |
| Neutrophil infiltration | Expression of adhesion molecules, cytokines and chemokines | Increased inflammation, formation of neutrophil extracellular traps (NETs) |
| Microglial activation | Release of pro- and anti-inflammatory cytokines, phagocytosis of apoptotic cells | Dual effects on inflammation, modulation of neurodegeneration |
| Astrocyte activation | Release of pro- and anti-inflammatory cytokines, upregulation of GFAP | Dual effects on inflammation, modulation of neurodegeneration, formation of glial scars |
| Blood-brain barrier dysfunction | Disruption of tight junctions, infiltration of immune cells | Increased inflammation, neurodegeneration |
| Cytokine and chemokine release | Production by immune cells, astrocytes, endothelial cells, neurons | Amplification of inflammation, recruitment and activation of immune cells, modulation of neurodegeneration |
| Complement system activation | Release of complement proteins, interaction with immune cells | Amplification of inflammation, modulation of neurodegeneration, potential for therapeutic targeting |
This table is not exhaustive and only includes some of the key pathophysiological processes involved in neuroinflammation in stroke
Pathomechanisms of Inflammatory Recruitment
Blood-Brain Barrier Breakdown
The BBB is a neurovascular unit component, and consists of pericytes, astrocytes, neuronal processes, perivascular microglia, and the basal lamina [13, 14]. The BBB’s integrity is maintained via tight junctions, protein complexes that seal the paracellular route between opposing brain microvascular endothelial cells. The breakdown of the BBB occurs in multiple phases and is a dynamic process [15••]. In the hyperacute stage, sudden hypoxia initiates a loss of tight junction proteins, leading to cytotoxic edema, increased paracellular permeability, and infiltration of immune cells into the brain. The neuroinflammatory response worsens the BBB disruption in the acute stage, leading to a higher permeability. This is followed by a basement membrane breakdown, allowing for an influx of larger molecules and ultimately leading to vasogenic edema [3]. Repair mechanisms, particularly neo-angiogenesis, occur in the subacute stage during the first 1–3 weeks [16]. BBB dysfunction is one of the limiting factors for the narrow therapeutic time window of thrombolytic therapy. During an ischemic event, microglia and astrocytes become activated and produce cytokines, matrix metalloproteinases (MMPs), chemokines, and vascular endothelial growth factor (VEGF) [17]. This inflammatory response leads to the breakdown of the BBB and upregulation of cellular adhesion molecules, allowing for infiltration of immune cells, such as neutrophils, into the brain tissue. The influx of immune cells further activates the inflammatory cascade, specifically cytokines, leading to more activation of glial and astrocyte cells and ultimately resulting in neuronal cell death. Tumor necrosis factor-α (TNF-α) is one of the key cytokines involved in BBB breakdown. TNF-α disrupts the BBB by stimulating the activation and proliferation of astrocytes and microglia and regulating apoptosis factors, such as cysteine, leading to increased production of ROS and pro-inflammatory cytokines. Studies have shown that the activation of microglia and astrocytes during ischemia is harmful to the BBB and can also contribute to the development of neuroinflammatory and neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease [18]. The mechanism of neuroinflammation and BBB breakdown is demonstrated in Fig. 1.
Fig. 1.
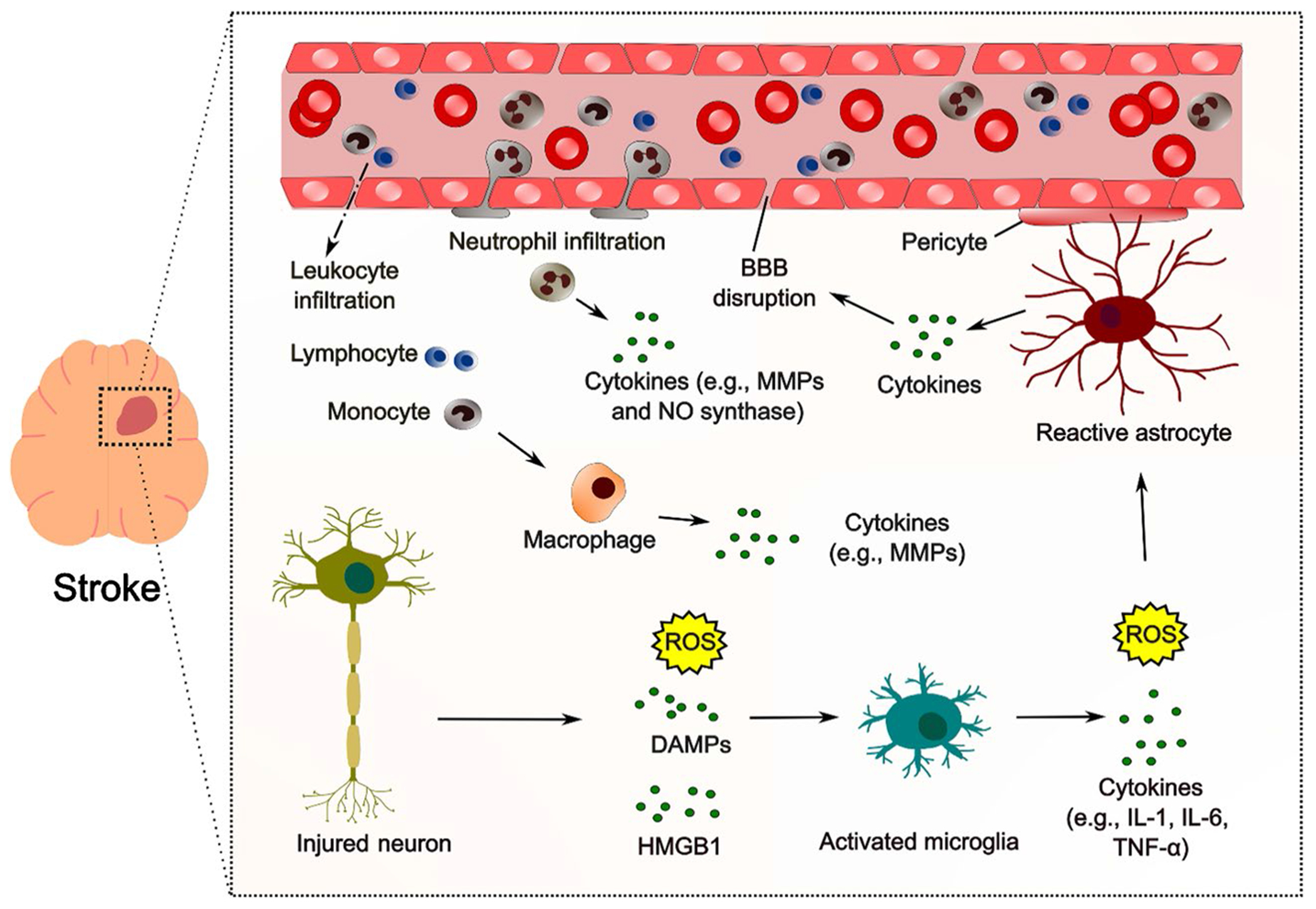
Schematic representation of neuroinflammation after stroke. Following stroke, activated microglia and astrocytes release proinflammatory cytokines and chemokines, leading to the recruitment and activation of immune cells such as monocytes and T cells from the periphery. This leads to further production of proinflammatory cytokines, perpetuating the inflammatory response. The inflammatory response contributes to the progression of brain damage, but also plays a role in tissue repair and neuroplasticity. Blood-brain barrier (BBB), matrix metalloproteinases (MMPs), nitric oxide (NO), danger-associated molecular patterns (DAMPs), high mobility group box-1 (HMGB1), interleukin (IL), and tumor necrosis factor (TNF). This figure is not exhaustive and only includes some of the main contributors and pathophysiological processes involved in neuroinflammation in stroke
The systemic inflammatory response triggered by lipopolysaccharides (LPS) can lead to the disruption of BBB. This response involves activating immune cells, including macrophages and microglia, which produce pro-inflammatory cytokines such as interleukin-1β (IL-1β). Pro-IL-1β, an inactive cytoplasmic precursor, is cleaved by caspase-1 to form IL-1β, which can lead to BBB breakdown [19]. Studies have also demonstrated that the mechanism of leukocyte infiltration into the CNS is induced by the relocation of stromal cell-derived factor-1 (SDF-1), also known as CXCL12 [20]. The SDF-1 chemokine plays a crucial role in leukocyte trafficking, and its expression is upregulated during neuroinflammation [21••]. Increased levels of IL-1β can lead to the activation of astrocytes, which in turn release SDF-1, causing the recruitment of leukocytes to the CNS [22].
Pericytes in Stroke Inflammatory Response
Following AIS, pericytes exert pro-inflammatory and immune-regulatory roles, while they also protect the brain from leukocyte invasion and have phagocytic and migratory properties following an ischemic attack [23••]. Pericytes differentiate into various cell types, including microglialike cells with phagocytic capacity in AIS [24], meaning their loss worsens leukocyte infiltration. Conversely, brain microvascular pericytes can also produce toll-like receptor 4 (TLR4) and a pro-inflammatory response [25].
Bloodborne Inflammatory Cells in Ischemic Stroke
After an AIS, various leukocyte subtypes infiltrate the brain parenchyma [26]. Neutrophils are among the first leukocytes to arrive and can cause inflammation and secondary injury by releasing pro-inflammatory proteins like nitric oxide synthase and MMPs. Activated leukocytes can also damage blood vessels by producing ROS and other proteases. Leukocytes can adhere to the endothelium, clogging the microvasculature and damaging blood vessels [3].
In addition to neutrophils, other leukocytes, such as monocytes and lymphocytes, can also infiltrate the ischemic brain. The expression of intracellular adhesion molecule (ICAM) and vascular cell adhesion molecule-1 (VCAM-1) on the endothelial cell and the interactions of the integrin proteins on the leukocyte cell surface further increases the aggregation along the endothelial cell on the vessel wall. Expression of platelet and endothelial cell adhesion molecule-1 (PECAM-1) and junctional proteins facilitate neutrophil diapedesis across the BBB [3]. Understanding the role of leukocytes in ischemic stroke is important for developing therapeutic strategies to minimize brain damage and promote recovery. Targeting the adhesion molecules and integrin proteins involved in leukocyte-endothelial cell interactions may provide a promising approach to reducing leukocyte infiltration and preventing BBB disruption. Additionally, targeting the inflammatory molecules released by leukocytes may help reduce neuroinflammation and potentially improve outcomes post-stroke.
Monocytes adhere to the vascular wall and move into the ischemic region 4–6 h after the neutrophil invasion [27]. This infiltration can lead to the differentiation of monocytes into macrophages, which can contribute to tissue repair by phagocytosing debris. However, macrophages can also exacerbate neuroinflammation by releasing pro-inflammatory cytokines and MMPs. Macrophages are subcategorized into two subtypes based on pro-inflammatory and anti-inflammatory properties. M1 macrophages produce pro-inflammatory cytokines such as IL-1β and TNF-α, which can enhance ischemic injury. M2 macrophages, also known as “healing macrophages,” promote the production of anti-inflammatory cytokines, including IL-10, which may help reduce neuroinflammation [28]. Peripheral blood monocytes can also migrate into ischemic brain tissue and differentiate into microglia and dendritic cells. Microglia are the resident immune cells in the brain, and dendritic cells are antigen-presenting cells which activate T-cells.
Lymphocytes can also release cytokines and other molecules that modulate the immune response. The role of lymphocytes in neuroinflammation is primarily related to T-cell functions. T-cells are infiltrated from BBB to the ischemic area by their attachment to endothelium with the help of adhesion molecules. IL-17 is secreted by infiltrated CD4 and CD8 cells into the brain parenchyma within hours after stroke onset, aggravating the ischemic injury [3]. However, other T-cells, such as regulatory T-cells (T-regs), play an important role in suppressing excessive immune responses and maintaining immune tolerance. Studies have shown that T-regs can be activated and recruited to the site of ischemic injury, where they can modulate the immune response and promote tissue repair [29]. T-regs can suppress the function of other immune cells, such as microglia and T-cells, by releasing anti-inflammatory cytokines, such as IL-10 and transforming growth factor beta (TGF-β) [30]. T-regs can also promote the production of neuroprotective factors, such as brain-derived neurotrophic factor (BDNF) and VEGF, which can promote angiogenesis and neuronal survival [31, 32••]. Understanding the role of T-regs in ischemic stroke may provide new therapeutic opportunities for modulating the immune response and promoting tissue repair.
The entry points of peripheral immune cells into the brain following an ischemic stroke are complex. The BBB is the primary entry site, and the first opening occurs within hours after cerebral ischemia due to increased transcytosis [16]. The second opening occurs 24 to 48 h later and is characterized by proteolytic degradation of tight junctions and basement membranes, resulting in the loss of vascular cells [33]. During this later phase, substantial infiltration of peripheral immune cells is observed.
The glia limitans, or glial limiting membrane, a thin barrier of astrocytic endfeet is associated with the parenchymal basal lamina that surrounds the brain and spinal cord, serves as a delaying mechanism for the activation of new transcriptional programs that enable entry into the brain parenchyma and can cause neutrophil accumulation around blood vessels [34]. The scavenger receptors CD36 and colony-stimulating factor 3 in cerebral endothelial cells allow peripheral immune cells to enter the brain parenchyma [35]. Neutrophils are also found on the abluminal surface of leptomeningeal vessels within hours after experimental stroke [36]. Accumulation of neutrophils in the meninges precedes their appearance in the brain parenchyma. Whether meningeal neutrophils enter the ischemic territory remains to be established. However, their entry is supported by the fact that they are observed on leptomeningeal vessels.
Additionally, it has been proposed that leukocytes, particularly monocytes and neutrophils, may enter the brain through vascular channels connecting the dura mater with the skull bone marrow [37, 38]. Due to its fenestrated endothelium, the choroid plexus is an entry site for patrolling lymphocytes, mostly CD4+ central memory T-cells, in the healthy brain [39]. The choroid plexus epithelium upregulates ICAM-1 and VCAM-1, together with the mucosal vascular adhesion molecule MAdCAM-1, in response to stroke to promote leukocyte trafficking [40, 41]. The choroid plexus epithelial cells also express the chemokine CCL20, which enables the entry of IL-17-secreting lymphocytes into the cerebrospinal fluid (CSF) through chemokine receptor 6 (CCR6). Monocytes enter the CSF through the choroid plexus after ischemic stroke. CD73, an ecto-ATPase expressed on choroid plexus epithelial cells and lymphocytes, mediates monocyte/macrophage trafficking across the choroid plexus. The choroid plexus stroma contains brain-resident macrophages and dendritic cells expressing MHC class II molecules, which may present antigens to T-cells.
Understanding the dual functions of monocytes and macrophages and the role of T-cells and other lymphocytes in neuroinflammation is crucial for developing effective therapeutic strategies to minimize brain damage and promote recovery after ischemic stroke. Targeting adhesion molecules and integrin proteins involved in leukocyte-endothelial cell interactions may be a promising approach to reducing leukocyte infiltration and preventing BBB disruption. Moreover, targeting the inflammatory molecules released by leukocytes, such as pro-inflammatory cytokines and MMPs, may help to reduce neuroinflammation and improve outcomes after ischemic stroke.
The Role of Platelets in Cerebral Inflammation
During an ischemic event, the endothelial cells release von Willebrand factor (vWF) stored in Weibel-Palade bodies. These bodies function to store two principal molecules, P-selectin and vWF, which are glycoproteins that play a crucial role in the coagulation process. Under normal conditions, circulating metalloprotease ADAMTS13 cleaves vWF into smaller fragments to prevent unwanted thrombus formation. However, during an ischemic event, the release of vWF from the endothelial cells can contribute to platelet activation and thrombus formation [42, 43•]. In addition to its role in coagulation, vWF is also involved in recruiting leukocytes to the site of ischemic injury.
ROS can form complexes with platelets, which are implicated in capillary no-reflow [44]. Capillary no-reflow is a phenomenon where erythrocytes clog the microvasculature due to the adherence of leukocytes, leading to obstruction of microvascular circulation and further tissue damage. Platelets also recruit leukocytes to the injury site by interacting with them through cell surface receptors, such as P-selectin and CD40 ligand, promoting leukocyte adhesion and activation. This interaction between platelets and leukocytes can contribute to the progression of neuroinflammation and parenchymal damage.
When activated, platelets release alpha and dense granules, further increasing platelet activation and acting as pro-inflammatory cytokines. Alpha granules are the most common granule in platelets and contain several growth factors [45]. They also contain proteins such as fibrinogen, vWF, and platelet factor 4. Upon platelet activation and subsequent aggregation, platelets release various factors, including several pro-inflammatory factors (these molecules include PF4, IL-8, PDGF, TGF-β, and VEGF) [46]. These pro-inflammatory factors are recruiters and activators of leukocytes, aiding in platelets’ immune regulation and inflammatory function, which can exacerbate tissue damage and contribute to the progression of ischemic injury [3].
In addition to their pro-inflammatory functions, platelets can release factors promoting tissue repair and regeneration. These factors include growth factors, such as platelet-derived growth factor (PDGF) and TGF-β, which can stimulate cell proliferation and extracellular matrix deposition [47, 48]. Therefore, targeting platelet activation and the release of pro-inflammatory factors while promoting the release of growth factors may provide a potential therapeutic strategy to improve outcomes after ischemic events [49].
Inflammatory Cascade Initiation
The inflammatory response triggered by ischemic stroke involves a complex interplay between various signaling pathways and cellular components. Several studies have shown that the activation of the inflammatory cascade occurs within minutes of ischemia onset and can continue for hours to days afterward [50••]. In addition, post-stroke inflammatory responses show sex differences and can affect infarct size and recovery. These responses are influenced by various intrinsic and extrinsic factors, such as differences in gonadal hormone exposure, sex chromosome complement, environmental, and social factors [51•].
P-selectin is released from Weibel-Palade bodies in endothelial cells and α-granules in platelets during an ischemic event. It appears on the cell surface and interacts with P-selectin glycoprotein ligand-1 on leukocytes, attracting them to the endothelial surface and promoting immune cell infiltration. This process contributes to the recruitment of immune cells and inflammation in the affected area.
Additionally, acute hypoxia within the cell and glucose deprivation leads to the production of ROS by cytosolic enzymes and mitochondria. The production of ROS leads to the generation of pro-inflammatory cytokines, further promoting cerebral inflammation. This cycle of inflammation and oxidative stress can contribute to the progression of ischemic injury and worsen outcomes. This oxidative stress can harm neurons, including the upregulation of 0α-amino-3-hydroxy-5-methyl-4 isoxazole-propionic acid (AMPA) and glutamate receptors. These receptors contribute to excitotoxicity, increasing intracellular calcium concentrations within neurons and ultimately leading to apoptosis.
Cerebral Cellular Involvement in Ischemic Stroke
Microglia
Microglia are the primary resident macrophage-like cells in the CNS and the first line of defense against stroke. When the brain experiences ischemia, microglia quickly become activated and release pro-inflammatory cytokines, such as TNF-α and IL-β, worsening the inflammatory response. In addition to their role in inflammation, microglia are also involved in maintaining a healthy neural network during development and repair and removing abnormal proteins [52].
Microglia have two distinct phenotypes that also play a role in neuroinflammation [46]. Similar to M1 and M2 macrophages, M1 microglia are pro-inflammatory, while M2 microglia are anti-inflammatory. Once activated, the microglia release TNF-α via the M1 phenotype, other pro-inflammatory cytokines, and ROS into circulation. M2 microglia produce anti-inflammatory cytokines, which help in remodeling and repair as well as angiogenesis. They also produce insulin-like growth factor, suppressing apoptosis and increasing neuronal cell growth and repair [53••, 54]. However, in ischemic stroke, microglia and macrophages switch from M2 to M1 phenotype in the acute stage, exacerbating neuronal death. Microglia can control their own polarization through autocrine and paracrine means, producing anti-inflammatory cytokines and promoting delayed microglia production (M2 phenotype) levels, which are neuroprotective. However, if this response is interrupted, chronic inflammation (M1 phenotype) can occur, exacerbating tissue damage [55–57]. Overall, the balance between M1 and M2 phenotypes plays a critical role in determining the outcome of ischemic stroke.
When in their resting state, multiple cells and receptors, including CD200, colony-stimulating factor 1 receptors, chemokines such as CX3CL1, and various neurotransmitters, are in constant motion to monitor the microenvironment of the CNS. However, when stimulated by injury or infection, microglia extend their processes and adopt an amoeboid shape. In response to ischemia, injured neurons release danger-associated molecular patterns (DAMPs), high mobility group box-1 (HMGB1) protein, and ROS, which activate the nuclear factor κβ (NF-κβ) pathway, resulting in the release of pro-inflammatory cytokines such as TNF-α [3].
Astrocytes
Astrocytes also play a crucial role in stroke pathophysiology. Increased glial fibrillary acidic protein (GFAP) expression leads to increased astrocyte production, which produces several pro-inflammatory mediators, such as IL-1β and TNF-α. However, they also have a dual function by means of activating anti-inflammatory cytokines by secreting co-stimulatory cells, such as CD200, that interact with microglia to promote an anti-inflammatory environment [58]. Moreover, astrocytes play a critical role in maintaining the homeostasis of neurotransmitters, particularly glutamate [59].
In ischemia, the impaired expression of excitatory amino acid transporter 2 (EAAT2), a major glutamate transporter expressed predominantly in astrocytes, interferes with the clearance of glutamate typically performed by astrocytes, leading to excitotoxicity and neuronal death. However, glutamate uptake by astrocytes needs a large amount of energy, which is challenging to provide in ischemic stroke [60]. In response to cytokines, astrocytes undergo hyperplasia and activation, releasing of vimentin, IL-1β, monocyte chemotactic protein-1, and MMPs, further weakening the BBB [3, 26].
Cytokine Involvement in AIS and ICH
Cytokines are small signaling polypeptides produced by cells within the body, particularly activated lymphocytes and macrophages. These small proteins include chemokines, interferons, lymphokines, and tumor necrosis factors. They play a significant role in the inflammatory process and are key to regulating the balance between cellular immunity and humoral immunity. They are divided into anti-inflammatory and pro-inflammatory types [61, 62]. IL-6 is special in that it has both pro- and anti-inflammatory properties [63••].
Chemokines primarily stimulate leukocyte exudation and regulate the distribution of leukocytes in lymph nodes and other tissues. Astrocytes and microglia are the major sources of chemokines in brain tissue. Various chemokine genes are upregulated, such as CXCL1, 2, 5, IL-8, and CCL20 [63††]. Overactivation of microglia can lead to a prolonged and excessive inflammatory response that causes further damage to the brain [64].
In ischemic stroke, cytokines such as IL-1, IL-6, and TNF-α are produced rapidly in response to the ischemic insult. They promote inflammation and BBB disruption, which can worsen stroke outcomes. IL-10 and TGF-β, on the other hand, have been shown to have neuroprotective effects and may help in reducing inflammation and promoting tissue repair. Overall, cytokines play a complex role in ischemic stroke pathology, and targeting their activity may have therapeutic potential in stroke treatment.
The role of inflammation in ICH is an evolving topic. After ICH, a type of sterile inflammation can occur, similar to that observed after ischemic stroke. This inflammation is triggered by molecules released from damaged or dead cells, such as HMGB1 protein. Hemoglobin, heme, and iron released after red blood cell lysis can also aggravate ICH-induced inflammatory injury [64]. This is because they promote oxidative stress and inflammation.
The tissue damage caused by the hemorrhage in ICH triggers an inflammatory response mediated by the release of DAMPs from damaged or dead cells. DAMPs are molecules normally found inside cells but released into the extracellular space when cells are damaged or die. Once released, they can activate immune cells and trigger an inflammatory response. In addition to DAMPs, other molecules released after ICH can also contribute to the inflammatory response. For example, when red blood cells lyse, they release hemoglobin, heme, and iron. These molecules can aggravate ICH-induced inflammatory injury by promoting oxidative stress and inflammation [64]. Microglia/macrophages are the first cells of the innate immune system that are activated rapidly post-HS. They are activated when toll-like receptors (TLRs), found on their membrane surface, bind to DAMPs. The key function of DAMPs is triggering the inflammatory response by communicating with microglia/macrophages that there is cellular damage. TLR4 is the predominant TLR found on microglial cells and is upregulated shortly after ICH, which leads to the upregulation of pro-inflammatory genes via the NF-κβ signaling pathway. Once activated, microglia phagocytose (engulf and digest) damaged cells, tissue debris, and hematoma. This helps to clear away damaged tissue and promote healing. However, if microglia become over-activated, they can release pro-inflammatory mediators that exacerbate ICH-induced cerebral injury. Pro-inflammatory mediators are molecules that promote inflammation and can include cytokines, chemokines, and ROS [62].
Understanding the mechanisms of neuroinflammation in AIS and ICH could lead to the development of new treatments to reduce secondary injury and improve outcomes.
Pro-inflammatory Cytokines in AIS and ICH
IL-1/IL-1β
The IL-1 family comprises eleven members [65]. The main sources in the CNS are microglia, astrocytes, and neurons. After binding to IL-1R1, both IL-1α and IL-1β induce further expression of IL-1. IL-1β is produced by microglia and monocytes, and its levels increase rapidly following an insult, typically within 15-30 minutes and are significantly elevated by 6 hours. Levels will only start to decline after 24–72 h. Following cerebral ischemia, IL-1β stimulates nuclear factor NF-κβ through TLRs and increases the production of chemokines and cytokines, leading to BBB disruption and edema formation, thereby exacerbating the ischemic injury. Microglia convert to the M1 type, which expresses IL-1β, causing neurotoxic effects. Studies in rats have shown that higher levels of IL-1β are associated with more significant cerebral injury [66]. Evidence also points towards both central and peripheral IL-1β contributing to cerebral injury. IL-1Ra is a naturally occurring protein that inhibits the activity of IL-1β by binding to its receptor. Preclinical studies have demonstrated that treatment with IL-1Ra reduces infarct size and BBB damage following AIS [67].
There is a positive correlation between IL-1β levels and disease severity and brain edema and a negative correlation with Glasgow Coma Scale (GCS) at 30 days in ICH [63••]. Rapid accumulation of DAMPs after ICH triggers an inflammatory response that involves interaction with TLRs on multiple cells. This interaction triggers massive production of IL-1β via the NF-κB signaling pathway, mainly by microglia and macrophages.
The NLRP3 inflammasome is a protein complex that plays an important role in inflammation following ICH. Following ICH, The NLRP3 inflammasome on microglia is activated via several mechanisms, including the complement system and construction of the MAC. Once activated, the NLRP3 inflammasome will activate caspase-1, which then converts pro-IL1-β and pro-IL-18 into mature IL-1β and IL-18. This process leads to a form of programmed cell death called pyroptosis, which leads to increases in pro-inflammatory cytokines [63••]. The NLRP3 inflammasome is an attractive therapeutic target in ICH due to its key role in the inflammatory response.
Clinical trials of subcutaneous IL-1Ra use in AIS and subarachnoid hemorrhage showed that it is safe and reduces plasma inflammatory markers such as IL-6 and C-reactive protein [68]. Further studies are needed to evaluate its use in everyday stroke practice and other hemorrhagic stroke types, such as ICH.
IL-6
IL-6 plays a complex and bivalent role in the pathogenesis of cerebral ischemia. Its production requires IL-1 and TNF-α to activate fibroblasts and endothelial cells. IL-6 enhances leukocyte recruitment by way of upregulating chemokines and adhesion molecule expression. It can also disrupt the BBB by decreasing the trans-endothelial electrical resistance value, thereby increasing BBB permeability.
However, IL-6 also demonstrates neuroprotective properties by promoting the repair of endothelial cells in the CNS, activating the PI3K/AKT and JAK-STAT pathways, facilitating angiogenesis and revascularization, decreasing NMDA-mediated neuronal cell injury, and protecting neurons against apoptosis. This bivalent action of IL-6 is dependent on the ischemic phase. While it is a pro-inflammatory cytokine in the acute phase, it exhibits neuroprotective properties in a subacute or chronic phase that may be exploited to develop novel therapeutic strategies for treating ischemic stroke [13, 69], and the timing of IL-6 targeting may be crucial for its therapeutic potential. IL-6 also produces acute-phase proteins such as hepatic cell fibrinogen, C-reactive protein (CRP), hepcidin, and serum amyloid A. These are now useful prognostic biomarkers in acute ischemic stroke [70].
IL-11
IL-11 is produced by multiple cells of mesenchymal origins, such as keratinocytes, synovial cells, bone cells, leukocytes, fibroblasts, and endothelial cells. In the CNS, IL-11 plays a role in multiple sclerosis (MS), promoting CD4 T-cells to differentiate into Th17 cells, thus increasing IL-17, among other cytokines. Oddly enough, activation of IL-11 reduces inflammatory demyelination, cleavage of caspase-9, and neuronal death. IL-11 levels are generally low in the CSF and healthy people’s serum, likely due to its short half-life. However, IL-11 levels increase after ICH, making it a potential marker of brain injury severity. Acute increases in IL-11 lead to endothelial cell proliferation and fibrosis around the cerebral aqueduct, which may contribute to brain edema and hydrocephalus. Most research focuses on using IL-11 as a marker to determine the severity and prognosis of ICH [63••].
IL-17
This pro-inflammatory cytokine is produced primarily by Th17 cells, and to a lesser extent, by γδT and NK cells. The primary subtype is IL-17A, but subtypes range to IL-17F. IL-17 binds to IL-17Rs found on epithelial cells, fibroblasts, endothelial cells, osteoblasts, and, importantly, hematopoietic cells. Once the IL-17Rs are bound by IL-17, downstream pathways are activated: NF-κβ, Mitogen-activated protein kinase (MAPK), and C/EBP’s. IL-17Rs recruit NF-κβ activator-1 via the SEF/IL-17R (SEFIR) domain, activating the NF-κβ and MAPK pathways. NF-κβ and MAPK pathways induce the production of inflammatory cytokines. The C/EBP pathway’s function is to act as a negative signal, inhibiting gene expression of pro-inflammatory cytokines. IL-17 receptors are expressed abundantly in the brain, and their signaling contributes directly or indirectly to damaging neurons and nerve function [63••].
After ICH, IL-17 levels increase via two pathways. Destruction of neurons leads to increased levels of peroxiredoxin. Peroxiredoxin and hemoglobin bind to TLR2/4 on macrophages, which stimulates the production of IL-23. IL-23 induces the differentiation of CD4 cells into Th17 cells, which greatly increases IL-17. This forms the IL-23/IL-17 inflammation axis. IL-23 stimulates γδT cells to a lesser degree, which also produces IL-17. Several studies suggest that IL-17 in the brain is mostly produced via γδT lymphocytes [71]. It was shown that γδT lymphocytes and IL-17 levels peak on the fourth day after ICH [72, 73•]. The significant rise in IL-17 after ICH promotes inflammatory reactions that aggravate brain injury [63••].
IL-23
IL-23 is a heterodimeric cytokine that belongs to the IL-12 family and is released by macrophages and activated dendritic cells. While IL-23 can induce the production of IFN-γ, the induction level is drastically lower than the induction by IL-12. The main role of IL-23 is to increase the levels of IL-17 by promoting CD4 T cell differentiation into Th17 cells. IL-23 can also contribute to increased levels of IL-17 through other pathways, such as the activation of IL-1β, which plays a significant role. If T-cell receptors are not activated, pathogen-associated molecular pattern molecules (PAMPs) activate IL-103B2; and IL-23. However, instead of T-cell differentiation to Th17, γδT cells are induced and produce IL-17 and IL-21. IL-1β and IL-23 activated γδT cells can also interact directly with CD4 T-cells and indirectly with dendritic cells to promote the production of IL-17. Current studies suggest that γδT cells are the primary producers of IL-17 following ICH [63••].
Interferon-γ
Interferon-γ (IFN-γ) is produced by various immune cells such as monocytes, natural killer cells, macrophages, B-lymphocytes, and T-cells. It has been implicated in atherogenesis. Following an ischemic insult, CD4+ T-cells can pass through the BBB and be activated by microglia to form either Th1 (pro-inflammatory) or Th2 (anti-inflammatory) cells. IFN-γ has a complex role in ischemic stroke. On the one hand, it can exacerbate the inflammatory response by inducing the expression of pro-inflammatory cytokines and chemokines, activating microglia (M1 type), and promoting neuroinflammation [74]. On the other hand, it can also have neuroprotective effects by promoting astrocyte activation and inducing neurotrophic factor expression [75]. Additionally, IFN-γ has been implicated in the pathogenesis of post-stroke depression (PSD) [76]. It has been shown that high levels of IFN-γ are associated with an increased risk of developing PSD [77••, 78]. Therefore, targeting IFN-γ signaling may be a potential therapeutic strategy for treating this common post-stroke complication.
TNF-α
TNF-α, a cell-signaling pro-inflammatory protein, binds to receptors TNF-R1 and TNF-R2. These are expressed in many different tissues throughout the body, but TNF-R2 is most implicated in the immune system. The primary pathways involved are NF-κB and MAPK pathways, which can result in apoptosis.
During cerebral ischemia, TNF-α mRNA is upregulated. In addition, multiple cell types, including mast cells, monocytes, neutrophils, macrophages, and fibroblasts, can upregulate its production. There are two types of TNF-α: the precursor transmembrane form, which produces local inflammation between cells, and the soluble form, which is the biologically active form. A TNF-converting enzyme produces the soluble form and has been shown to induce IL-1 and IL-6 production and initiate phagocytosis [17]. MMPs also produce soluble TNF-α, which contributes to BBB breakdown. It has been studied that there is an overexpression of TNF-α following both definitive and temporary occlusion of the middle cerebral artery.
TNF-α is one of the first cytokines to populate after ischemic stroke and has the first peak within 1–3 h and a successive peak around 24–36 h. Its levels decrease about 72–144 h post-stroke, associated with clinical improvement during the acute phase. Although TNF-α is a pro-inflammatory cytokine, it also has neuroprotective effects. Its exact mechanism is still being studied [13], but findings have shown that administering TNF-α before stroke induction significantly reduces infarct size and improves neurological function in rats [79, 80]. TNF-α can also induce the expression of neurotrophic factors such as BDNF and glial cell line-derived neurotrophic factor (GDNF), which promote neuroprotection and neuronal survival [81].
Following ICH, TNF-α is secreted by neurons and activates microglia and astrocytes. Their levels increase shortly after ICH. Once released, it decreases BBB stability by altering the cytoskeleton, tight junction protein expression, and serine protease production. It downregulates claudin-5 expression via the NF-κβ pathway and upregulates the expression of VCAM-1 via the MAPK pathway, thus contributing to leukocyte adhesion. It also causes neuronal cell death following ICH. Once TNF-α is released by activated microglia, it induces necroptosis of neurons and initiates neuronal apoptosis via the caspase-3 pathway. TNF-α also causes excitotoxicity by increasing the expression of AMPA receptors on synapses. TNF-α inhibitors have been shown to reduce the degree of brain edema, inflammation, and neurologic impairment; however, they do not alter hematoma volume. TNF-α inhibitors may have a role in ICH management in the future; however, more research is required in both animal and human models [63••].
Anti-inflammatory Cytokines
IL-4
IL-4 is important for humoral and adaptive immunity. It has several functions that contribute to its anti-inflammatory properties. It can induce the differentiation of naive Th0 lymphocytes into Th2 cells. It is produced by lymphoid cells and mast cells. It also promotes the activation of anti-inflammatory M2-type microglia and macrophages.
IL-4 is produced not only by immune-mediated cells, but it is also hypothesized that neurons produce IL-4 in response to cerebral ischemia. This cytokine is vital during the Th2 differentiation and can produce M2-type (“healing”) macrophages/microglia with anti-inflammatory properties. The neuroprotective effect of IL-4 is achieved through the STAT6 signal pathway, which belongs to the Spinal Transducer and Activator of Transcription family of proteins, leading to the inhibition of pro-inflammatory cytokines and stimulation of the PPARγ pathway [69]. These processes will help promote the phagocytosis of dead cells and the proteolysis of specific proteins that could support neuro-rehabilitation.
IL-4 treatment reduces infarct size and improves neurological deficits in animal models of ischemic stroke [82]. Additionally, IL-4 has been found to enhance neurogenesis in the subventricular zone and the dentate gyrus of the hippocampus, leading to improved functional recovery after stroke [83]. These findings suggest that IL-4 could be a potential therapeutic target for ischemic stroke, as it promotes anti-inflammatory responses and enhances neuroprotective and neuro-rehabilitative processes. Studies investigating the relationship between IL-4 and ICH have revealed that promoter polymorphisms of the IL-4 gene are closely related to ICH but not ischemic stroke. This suggests that IL-4 may be involved in the pathogenesis of ICH and that genetic variation in the IL-4 gene may be a potential indicator of disease severity. Further research is needed to understand the role of IL-4 in ICH fully and to explore its potential as a therapeutic target [63••].
IL-10
IL-10 is produced by monocytes, Th2 lymphocytes, CD4 regulatory T-cells, and mastocytes. Its expression in brain tissue promotes glial survival and inhibits the inflammatory pathway through different signaling pathways. IL-10 has potent and diverse effects on all hematopoietic cells infiltrating the brain after injury [84]. The neuroprotective effect is achieved by inhibiting the inflammatory cytokines through PI3K and STAT3 activation pathways. IL-10 also inhibits the effect of NF-κβ [85]. Increased IL-10 levels decrease CD11b cells related to infarct expansion via the nitric oxide pathway. It is important in the balance of Th1 and Th2 cell types. IL-10 is also an immensely powerful suppressor of lipopolysaccharide-induced TNF production. B7-H1 functions to suppress T-cell immune function through modulation of IL-10/IL-12 production and T-reg generation.
Several studies have investigated the potential therapeutic use of IL-10 in treating stroke. For example, treatment with IL-10 reduced brain injury and improved neurological function in rats with ischemic stroke [84]. Another study found that IL-10 gene therapy improved functional recovery and reduced inflammation in a mouse stroke model [86]. Overall, these findings suggest that IL-10 has potential as a therapeutic target for ischemic stroke by promoting anti-inflammatory responses and inhibiting neuroinflammation. However, elevated levels of IL-10 have also been found to contribute to post-stroke infection, particularly urinary tract infection in women, leading to poorer recovery [69]. Thus, the immunosuppressive effects of IL-10 may contribute to an increased risk of infections in stroke patients.
In ICH animal models, increasing the release of IL-10 by boosting T-reg cells reduced ICH-induced brain injury [72], and intraventricular injection of IL-10 reduced hematoma volume within 3 days after ICH [87]. Therefore, increasing the level of IL-10 is a potential target for neuroprotection in patients with ICH; however, learning the cellular mechanism by which it is regulated is necessary to help establish new therapeutic targets [63••].
IL-33
IL-33 has a trefoil structure that resembles other IL-1 cytokines. In the CNS, IL-33 is primarily produced by endothelial cells and astrocytes, with IL-33 receptors located mostly on astrocytes and microglia. While IL-33 has been shown to increase the production of pro-inflammatory cytokines, more evidence points to a larger role in anti-inflammatory processes. Primarily, it can facilitate macrophage differentiation into M2 macrophages, protecting the nervous system from further damage. It also plays a role in slowing Alzheimer’s disease (AD) progression by facilitating amyloid-β peptide uptake by activated microglia. This anti-inflammatory property also occurs after spinal cord injury.
After ICH, increases in pro-inflammatory cytokines and elevation of TNF-α induce the release of IL-33 by astrocytes. This essentially functions as a negative regulator by reducing the levels of pro-inflammatory cytokines and increasing anti-inflammatory cytokines and tissue repair by facilitating T-cell conversion to Th2 cells. Another neuroprotective feature of IL-33 is its role in increasing levels of the anti-apoptotic Bcl-2 and decreasing levels of cleaved caspase-3 expression, ultimately reducing neuronal apoptosis. The specific role of IL-33 in ICH is still under investigation, and its potential use in management is yet to be determined [63••].
TGF-β
TGF-β has been shown to promote the survival of neurons and facilitate the recovery of cerebral parenchyma following ischemic stroke. Among the five subtypes of TGF-β, subtypes β1 and β2 are particularly prominent after stroke [8]. TGF-β1 is produced by activated microglia and M2 macrophages, while TGF-β2 is produced by astrocytes. These subtypes of TGF-β can directly inhibit the pro-inflammatory cytokines, reducing cerebral damage in ischemic stroke [13].
TGF-β has anti-inflammatory roles in ICH primarily through its action in promoting the proliferation and differentiation of T-reg cells. In ICH, TGF-β levels decrease markedly at first but increase over the next 14 days, reaching normal levels between days 10 and 14. T-reg cells increase moderately during the first 3 days, increase markedly and peak on the 4th day, then decrease on the 7th day. TGF-β also facilitates brain repair by macrophages through the Smad2 and Smad3 pathways. T-reg cells promote the differentiation of microglia into the M2-like subtypes via the IL-10, GSK3-βPTEN axis.
Clinically, patients with early increases in plasma TGF-β1 levels had favorable outcomes 90 days after ICH, suggesting utility in recovery from ICH [88]. This finding is controversial as other data have shown that TGF-β level is elevated in the CSF of patients with severe traumatic brain injury and is associated with increased BBB permeability and development of brain injury after germinal matrix hemorrhage [89]. Another study revealed that chronically elevated TGF-β leads to microglia being less likely to differentiate from the M1 to the M2 phenotype [90]. Based on the mixed results of TGF-β’s effects during ICH, more research needs to be completed to delineate the mechanisms of this cytokine [63••].
Pro-inflammatory and Anti-inflammatory (Mixed Effect) Cytokines
IL-6
IL-6 is a special cytokine that has both pro-inflammatory and anti-inflammatory properties. It is a glycoprotein that balances the CD4 T-cells subsets and induces the production of B-cell antibodies, effectively acting as a connection between innate and acquired immunity. Its effects are achieved through membrane-bound receptors, and whether they are anti-inflammatory or pro-inflammatory depends on cytokine concentrations, site of injury, target cells, and stage of disease development.
In the CNS, IL-6 levels are very low unless an injury occurs. If the injury occurs, then IL-6 will be produced by neurons, microglia, and, most importantly, astrocytes. After ICH, IL-6 levels greatly increase, peaking 1 day following the inciting injury. IL-6’s role in secondary ICH is a topic of debate. It primarily exerts its detrimental CNS effects via the trans-signaling (pro-inflammatory) pathway. The anti-inflammatory effect via membrane-bound receptors is found mostly in microglia. IL-4, known to increase macrophage/microglial M2 (anti-inflammatory) phenotype, has been shown to induce the production of IL-6 along with other anti-inflammatory markers. IL-4 and IL-6 may act synergistically, playing a neuroprotective role in patients with ICH [63••].
Complement System Activation in AIS and ICH
The complement system, also known as the complement cascade, a part of the innate immune system, is also implicated in the pathophysiology of cerebral ischemia and stroke [91, 92]. The complement system consists of more than 30 plasma and membrane-bound proteins that interact with each other in a cascade-like manner. The complement system can be activated by three pathways: the classical, alternative, and lectin pathways, all of which converge on the formation of the C3 convertase, leading to the cleavage of C3 into C3a and C3b. C3a is a potent anaphylatoxin that activates immune cells, including microglia, while C3b plays a critical role in opsonization, phagocytosis, and clearance of pathogens and cellular debris [93]. The complement system also generates the membrane attack complex (MAC), which can lyse cells by creating pores in their membranes [93, 94].
In AIS, the complement system is locally and systemically activated [91, 92]. Studies have shown that cerebral ischemia leads to the activation of the classical and alternative complement pathways, resulting in the deposition of complement components, including C1q, C3, and MAC, in the ischemic brain tissue [95, 96]. The deposition of complement components is associated with BBB dysfunction, neuronal damage, and neuroinflammation [97]. In addition, the systemic activation of the complement system, as evidenced by elevated levels of complement components in the plasma, has been reported in patients with acute ischemic stroke and is associated with worse clinical outcomes [91, 98].
Endothelial cell activation and ROS production are key triggers of the complement system activation in AIS [97]. Endothelial cells, which line the cerebral vasculature, play a critical role in maintaining the integrity of the BBB and regulating cerebral blood flow. However, in response to ischemic injury, endothelial cells become activated, leading to the expression of adhesion molecules and the release of cytokines and chemokines, which recruit leukocytes and complement components to the site of injury. In addition, ROS, which is produced by activated endothelial cells, can directly activate the complement system, leading to the generation of C3a and C5a, which further amplify the inflammatory response [97].
The activation of the complement system in cerebral ischemia has numerous downstream effects on various immune and neuronal cells. Microglia express complement receptors and are activated by C3a and C5a, producing pro-inflammatory cytokines and chemokines [99]. In addition, microglia can phagocytose complement-opsonized cells and debris, leading to the generation of ROS and further complement activation [100, 101]. Astrocytes, another glial cell type, also express complement receptors and can be activated by complement components, producing pro-inflammatory cytokines and chemokines [100].
In addition to microglia and astrocytes, infiltrating neutrophils and peripheral macrophages also produce C1q and activate the complement cascade in the ischemic brain tissue. In the context of ischemic brain tissue, the presence of infiltrating neutrophils and peripheral macrophages can lead to the production of C1q, which can activate the complement cascade. This activation can result in the formation of the MAC, which can destroy cells, including neurons. The complement cascade can also be activated by DAMPs released by dying neurons, which can directly activate the alternative pathway of complement [102].
Activation of the complement system results in the formation of complement components, including C3a, C5a, and the MAC. The C3a and C5a fragments, known as anaphylatoxins, are potent inflammatory mediators that recruit and activate immune cells, including neutrophils, macrophages, and T-cells to the injury site. C5a also activates microglia and astrocytes, producing pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6. Furthermore, the MAC can directly damage neurons and glial cells, leading to cell death [97].
Several studies have suggested that complement activation exacerbates ischemic brain injury and worsens functional outcomes in stroke patients [91]. Inhibition of the complement system has been shown to reduce brain injury and improve outcomes in animal models of stroke [103]. For example, C5a receptor antagonists have been shown to reduce infarct volume, improve neurological deficits, and decrease neutrophil infiltration in the ischemic brain [104].
In addition, studies have shown that genetic deficiencies or pharmacological inhibition of complement components, such as C3, C5, and C6, can reduce brain injury and improve outcomes in animal models of stroke [96, 105–107]. Furthermore, administration of complement inhibitors, such as complement receptor 1-related protein-y, a soluble inhibitor of the classical and alternative complement pathways, or complement receptor 2-C3d fusion protein, which blocks the interaction between C3d and complement receptor 2, has been shown to reduce ischemic brain injury and improve functional outcomes in animal models of stroke [108].
In addition to promoting inflammation and neuronal damage, the complement system has also been implicated in promoting angiogenesis and tissue repair after AIS. For example, a study in a mouse model of stroke found that C3-deficient mice exhibited impaired angiogenesis and increased neuronal damage compared to wild-type mice [109]. In another study, inhibiting C5aR1 by PMX53 treatment reduced cell injury and inflammation and promote brain function recovery by inhibiting the TLR4 and NF-κB signaling pathway and reducing the production of TNF-α, IL-1β, and IL-6 in the middle cerebral artery occlusion/reperfusion rats [110].
In the context of ICH, several studies have shown that complement activation occurs rapidly following hemorrhage and is associated with significant tissue damage. For example, in a rat model of ICH, it was shown that complement activation occurs within minutes following hemorrhage and is associated with the infiltration of neutrophils and microglia into the brain tissue [111]. Similarly, it was shown that complement proteins are present in the hemorrhagic brain tissue and that the expression of complement proteins is increased in the perihematomal area [112].
The mechanisms by which complement activation contributes to the pathogenesis of ICH are complex and multifactorial. One of the main ways complement activation contributes to tissue damage in ICH is through the formation of the MAC, which can cause direct damage to the cell membrane and lead to cell lysis [113•]. In addition, the MAC can also activate microglia and astrocytes, leading to the production of pro-inflammatory cytokines and chemokines. These cytokines and chemokines can recruit additional immune cells to the injury site and exacerbate tissue damage.
Another way in which complement activation contributes to the pathogenesis of ICH is through the production of ROS. Several studies have shown that complement activation leads to the production of ROS, which can cause oxidative damage to cellular components. This oxidative damage can lead to further tissue damage and exacerbate the inflammatory response [114].
In addition to causing tissue damage, complement activation also plays an important role in the recruitment and activation of immune cells following ICH. For example, complement activation can produce of chemotactic factors that recruit neutrophils and monocytes to the site of injury [111]. Once recruited, these immune cells can exacerbate tissue damage through the production of pro-inflammatory cytokines and the phagocytosis of damaged cells.
In summary, complement activation plays a significant role in the pathophysiology of both AIS and ICH and is a potential therapeutic target for these conditions. However, the clinical efficacy of complement inhibitors in stroke has yet to be determined. There are concerns regarding the potential for increased risk of infection and impaired clearance of cellular debris. Further preclinical and clinical studies are needed to determine the safety and efficacy of complement inhibitors in stroke and to identify the optimal timing and dosage of these agents.
Transcription Factors in Post-ischemic Brain and ICH
Transcription factors are crucial in regulating neuroinflammation and can promote recovery and regeneration after injury by inducing endogenous stem cell proliferation. After ischemic stroke, different types of cells promote astrocyte activation by secreting various factors, including activated microglia, dead neurons, endothelial cells, and other cells [115]. These factors act by entering the cell via multiple pathways. One of these pathways is the NF-κβ pathway. NF-κβ regulates the transcription of a range of pro-inflammatory factors. After ischemic stroke, NF-κβ is activated in activated microglia and translocates from the cytoplasm into the nucleus, which induces the production of inflammatory cytokines and results in secondary brain injury after stroke [116••]. NF-κβ also induces microglia activation and polarization of the M1 phenotype [115]. Ischemia/reperfusion injury can activate transcription factors such as AP-1 and MAP kinase cascades, leading to cell death and inflammation [3]. However, there is also evidence that intravenous hedgehog agonist administration can induce endogenous stem cell proliferation after cerebral ischemia [117], suggesting the potential for transcription factors to play a role in promoting recovery and regeneration after injury [118]. AP-1 is a transcription factor that is activated by ischemia and reperfusion injury in the brain. It has been shown to promote neuronal cell death and inflammation by upregulating the expression of pro-inflammatory cytokines such as TNF-α and IL-6 [119]. Inhibition of AP-1 has been shown to reduce brain injury and improve functional recovery after ischemic stroke in animal models. MAP kinase cascades are also activated by ischemia/reperfusion injury in the brain. They play a role in promoting neuronal cell death and inflammation. Inhibition of MAP kinase cascades has been shown to reduce brain injury and improve functional recovery after ischemic stroke in animal models [120]. Hypoxia-inducible factor-1α (HIF-1α) is a transcription factor that is activated in response to hypoxia, a common occurrence after ischemic stroke [121]. HIF-1α regulates gene expression in cell death, inflammation, and angiogenesis. Peroxisome proliferator-activated receptor gamma (PPAR-γ) is another transcription factor that is involved in post-stroke injury [122]. PPAR-γ inhibits inflammation and oxidative stress and promotes neuroprotection. Cyclic adenosine monophosphate response elementbinding protein is a transcription factor that regulates neuronal survival and plasticity. Dysregulation of these transcription factors can lead to neuroinflammation and cell death. Nuclear factor erythroid 2-related factor 2 (Nrf2) and SRY-box 2 (Sox2) are transcription factors that have been shown to promote recovery and regeneration after stroke [123–125]. In conclusion, transcription factors play a significant role in the post-ischemic brain by regulating the transcription of pro-inflammatory factors and inducing microglia activation and polarization. Further research on these pathways may provide new ideas and directions for the diagnosis and clinical treatment of ischemic stroke.
Transcription factors have also been shown to play a role in the pathogenesis of ICH with neuroinflammation. NF-κB is a transcription factor that is activated by ICH [126]. It has been shown to promote neuroinflammation by upregulating the expression of pro-inflammatory cytokines such as TNF-α and IL-6. Inhibition of NF-κB has been shown to reduce brain injury and improve functional recovery after ICH in animal models. The hedgehog signaling pathway has been shown to promote recovery and regeneration after ICH restoring BBB function [127]. Nrf2 regulates gene expression in antioxidant defense and anti-inflammatory pathways [128, 129•]. Sox2 is a transcription factor involved in neural stem cell proliferation and differentiation [130]. Dysregulation of Nrf2 and Sox2 can lead to neuroinflammation and cell death in ICH [128, 131]. However, dysregulation of transcription factors can lead to neuroinflammation and cell death, highlighting the need for further research to develop effective therapies targeting transcription factors for stroke treatment and prevention.
Effect of Post-stroke Inflammation on Other Organs: Innate and Adaptive Immunity
Research is also focused on not only inflammation within the CNS post-stroke, but also on systemic inflammation and its effects on the rest of the body. The cytokines and inflammatory cells mentioned throughout this chapter can reach other organ systems through the breakdown of the BBB and through CSF and DAMPs that trigger the innate immune system. This causes an initial appearance of systemic inflammatory response in stroke patients that appears later in the course as depression of the immune system with leukopenia and post-stroke infections.
AIS and HS result in the release of DAMPs that can activate immunocompetent cells like microglia and astrocytes in the affected area [132–134]. Moreover, these DAMPs can also circulate in the bloodstream and promote the recruitment of circulating immune cells to the brain, resulting in a complex peripheral immune response to stroke [135]. Traditionally, the immune response to stroke was studied in the context of subacute immunosuppression, which is associated with increased susceptibility to bacterial infections in stroke patients [136]. However, more recent findings have highlighted a multiphasic immune response to stroke, with systemic inflammation occurring throughout all stages of the disease. During the hyperacute phase, the peripheral immune system is rapidly activated in response to stroke-induced brain injury [137, 138•]. This acute systemic response is followed by a state of immunosuppression characterized by the loss and unresponsiveness of immune cells [139, 140]. In the chronic phase after stroke, there is a low-grade sustained residual inflammation that could impact the long-term outcome of stroke patients. Understanding the different phases of the immune response to stroke is crucial for developing effective therapeutic strategies. For example, interventions aimed at reducing the acute systemic response could help prevent the onset of immunosuppression, while approaches targeting sustained residual inflammation could improve long-term outcomes.
Stroke also induces the mobilization of more leukocytes from the spleen and bone marrow and the activation of neurogenic pathways during the acute phase [12••]. In the subacute phase, which occurs within hours to days after stroke onset, a state of immunosuppression is triggered. The prolonged overactivation of neurogenic pathways, as well as the release of DAMPs and other pro-inflammatory mediators, causes lymphopenia due to massive cell death and a bias towards the monocyte differentiation pathway in bone marrow hematopoiesis [12••]. This leads to an imbalance between type 1 (Th1) and type 2 (Th2) helper T-cells, and circulating monocytes become less capable of providing costimulatory signals. Later, a long-term phase of compromised peripheral immunity is characterized by chronic and sustained high levels of DAMPs and pro-inflammatory cytokines. This sustained inflammation can potentially impact the long-term outcome of stroke patients, prevent immunosuppression, and reduce sustained residual inflammation. More research is needed to understand the mechanisms underlying the immune response to stroke fully and to develop effective therapeutic strategies.
A peripheral immune response to stroke is initiated within minutes after stroke onset by DAMPs that originate from dying or stressed cells within the ischemic brain or actively secreted by immune cells [141]. DAMP levels rapidly increase in blood within the first hours after stroke onset, including HMGB1, calprotectin, Prx-1 and Prx-5, heat shock proteins, and cell-free DNA. The exact mechanisms whereby the injured brain sends out these first DAMP signals to trigger acute systemic inflammation still remain unclear, but the opening of the BBB, as well as the glymphatic and meningeal lymphatic systems, have been identified as possible routes [142•].
Circulating DAMPs are recognized by pattern recognition receptors (PRRs) such as TLRs or the receptor for advanced glycation end products (RAGE), expressed by diverse immune cell subpopulations [143]. Signaling through these PRRs activates diverse downstream signaling pathways, including the NF-κB, MAPK, interferon regulatory factors (IRF), or the inflammasome signaling pathways [144]. These signaling pathways induce the production and release of pro-inflammatory cytokines by activated immune cells, which amplify and perpetuate the inflammatory response. The acute inflammatory response to stroke can contribute to the progression of brain damage by exacerbating BBB disruption, oxidative stress, and neuroinflammation. However, the acute inflammatory response can also play a beneficial role by promoting the clearance of cellular debris, the recruitment of immune cells to the site of injury, and the activation of tissue repair mechanisms. Therefore, therapeutic interventions that modulate the peripheral immune response to stroke are promising strategies for reducing the severity and extent of brain damage after stroke. For example, inhibition of DAMP signaling, cytokine production, and the modulation of immune cell polarization have shown neuroprotective effects in preclinical models of ischemic stroke. However, clinical translation of these approaches remains challenging, and further research is needed to identify safe and effective strategies for targeting the peripheral immune response to stroke.
The acute phase of stroke is characterized by a massive immune response that involves the release of pro-inflammatory cytokines, the mobilization of immune cells from peripheral reservoirs, and the dysbiosis of the gut microbiota. The spleen and bone marrow, two major reservoirs of immune cells, are rapidly exhausted within hours after stroke, activating neurogenic pathways that play a major role in releasing immune cells [102, 145]. Norepinephrine and epinephrine levels contribute to spleen shrinkage and massive exiting of immune cell populations from this organ. At the same time, the activation of sympathetic innervation is observed by an increase in the levels of tyrosine hydroxylase and norepinephrine within the first day after stroke [145, 146].
The gut has also been identified as an important reservoir of immune cells, from which leukocytes are mobilized to the circulation following stroke and even recruited to the ischemic brain [147]. The injured brain alters the gut microbiome, which can worsen the inflammatory response and cause secondary brain injury [148–150]. The intestinal microbiota also plays a role in modulating the phenotype of the immune cells within the acute inflammatory response to stroke. Stroke-induced alterations in gut microbiome composition have been associated with worse stroke outcomes, larger infarct volumes, poorer scores in functional tests, and even reduced intestinal motility and intestinal barrier dysfunction, leading to the translocation of intestinal bacteria into circulation and peripheral organs [151, 152]. In this regard, the dissemination of selective bacterial species from the gut microbiota after stroke has been proposed to be a plausible source of a post-stroke infection and could contribute further to the systemic pro-inflammatory immune activation after stroke. Moreover, activated endothelial and circulating innate immune cells after stroke could also promote immunothrombosis, the inflammation-dependent activation of coagulation that results in microvascular thrombosis and occlusion. Therefore, controlling the inflammatory response after stroke and modulating the gut microbiota may effectively reduce brain damage and improve stroke outcomes.
Given that the gut houses the largest portion of the human immune system, the bidirectional brain-gut axis is of interest in stroke and ICH as a possible target for future therapies that require further research to explore these possibilities [63••]. Microbiota diversity and abundance vary dynamically during the development of ischemic stroke [153]. Stroke gives rise to mucosal damage and bacterial translocation in the gut [154]. Brain injury made specific changes in the cecal microbiota through altered autonomic activity and mucoprotein production in mice [149]. Cerebral ischemia leads to gut-derived autonomic norepinephrine transmission, decreasing the number and function of intestinal epithelial cells and mucus secretion, giving rise to gut microbiota dysbiosis [155••]. Conventional lymphocytes and γδ T-cells can travel from the small intestine to the brain and meninges after stroke, exacerbating ischemic injury [156]. Additionally, neurohumoral signals generated by the ischemic brain can affect immune homeostasis, intestinal barrier function, and microbiota. Stroke patients often experience gastrointestinal complications, such as altered motility, microbial dysbiosis, and intestinal bleeding, which are associated with impaired functional recovery and increased mortality. In mice, β-adrenergic signaling disrupts intestinal mucin production, increases gut permeability, and changes microbial composition after stroke [157]. The gut barrier disruption is associated with increased bacterial translocation and the spread of intestinal bacteria to the blood, liver, and lungs. Aged mice are more susceptible to bacterial translocation and fail to clear bacteria due to more severe post-stroke immunosuppression. A recent study identified TREM1 upregulation in intestinal macrophages as a possible mediator of intestinal barrier disruption caused by β-adrenergic signaling [158••].
After the immune system’s early activation, systemic immunodepression follows, characterized by a reduction in circulating T, B, and NK cell counts and a shift from Th1 to Th2 helper T-cell ratio [157]. This immunosuppressive state is thought to be caused by the prolonged overactivation of the sympathetic nervous system (SNS) and the hypothalamic pituitary adrenal (HPA) axis [159]. Activating adrenergic receptors in splenocytes by circulating catecholamines triggers splenic atrophy and T-cell apoptosis. In contrast, glucocorticoids promote the production of anti-inflammatory cytokines, suppress the secretion of pro-inflammatory cytokines, and contribute to lymphocyte apoptosis in the spleen [157]. The SNS also results in a substantial increase in the myeloid lineage proliferation, a subsequent egress of myeloid cells to the circulation, and suppression of the lymphoid lineage progression [160, 161]. Experiments blocking the SNS and HPA pathways have reduced lymphocyte apoptosis and monocyte deactivation after experimental stroke. The SNS also plays a role in this response, as β-adrenergic receptor inhibition lowers bacteremia and lung colonization, increases survival rates, and preserves splenic and circulating lymphocytes [157]. Activation of α7-nicotinic acetylcholine receptor via vagus nerve stimulation (VNS) as a neuroprotective strategy in focal cerebral ischemia has recently attracted attention in the context of stroke treatment. It acts through various mechanisms such as its anti-inflammatory properties [162, 163]. The use of the surgical VNS paired with rehabilitation has been approved for use in moderate to severe upper extremity motor deficits associated with chronic stroke, and clinical trials for the utilization of non-invasive vagus nerve stimulation in acute stroke are being conducted [164, 165].
Post-stroke infection is one of the potentially life-threatening complications, affecting a large subset of stroke patients [166, 167]. The most common types of post-stroke infections are pneumonia and urinary tract infections [76, 167]. While current clinical strategies are based on broad-spectrum antibiotics, preventive antibiotic therapies are being evaluated to prevent the onset of these fatal complications, although beneficial effects have not been demonstrated. Factors that increase susceptibility to post-stroke infections include patients’ baseline characteristics, invasive devices and clinical procedures, and subacute immunosuppression after stroke. Alterations in the blood profile of several cytokines after stroke have been linked to an increased incidence of infections in stroke patients. The autonomic nervous system and the HPA pathway have been proposed as the main triggers of the cell-mediated loss of immunity. Mechanistically, an inflammasome-dependent mechanism of T-cell apoptosis has been found to have a crucial role in lymphopenia and the incidence of infections after stroke [12••, 168]. While the causal relationship between the post-stroke stress response, the subsequent immunosuppression, and the development of infections remains questionable, researchers are developing strategies to prevent post-stroke infections and improve the clinical management of stroke patients.
Chronic inflammation is increasingly recognized as a major factor in developing long-term stroke sequelae, including cognitive impairment, depression, and fatigue [76, 169••]. Inflammation contributes to ongoing damage to the brain and can lead to secondary injury in the days, weeks, and months after the initial stroke. Chronic inflammation after stroke is thought to be driven by several factors, including ongoing tissue damage, activation of resident immune cells, and infiltration of peripheral immune cells into the brain. In the chronic phase of stroke, peripheral tissues can become sensitized to the antigens they experience during the acute stroke phase [170]. Several inflammatory markers, including CRP, IL-6, and TNF-α have been associated with worse outcomes after stroke [171]. Resident immune cells, including microglia and astrocytes, play a key role in initiating and maintaining chronic neuroinflammation. These cells release pro-inflammatory cytokines and chemokines, activate complement, and phagocytose debris from damaged cells [172]. In addition, peripheral immune cells, including monocytes, lymphocytes, and neutrophils, can infiltrate the brain after a stroke and contribute to ongoing inflammation [173].
A better understanding of the mechanisms driving chronic inflammation after stroke may lead to new treatments to reduce the long-term sequelae of stroke. After a stroke, cellular stress and damage generate novel brain antigens that stimulate B- and T-lymphocytes in the spleen and lymph nodes [157, 174••]. Myeloid cells in the brain phagocytose dead cells and upregulate the MHC class II to become antigen-presenting cells, migrating to lymph nodes and the spleen. This antigen presentation process occurs both inside and outside the brain. T-cells migrate to the ischemic region or hematoma in an antigen-independent fashion within days after stroke and are present in the lesion. T-regs with a brain-specific signature accumulate during the chronic phase of stroke independently of antigen presentation [175•]. The T-regs expand locally and limit astrogliosis, suppress the neurotoxic phenotype of astrocytes, and promote functional recovery without affecting infarct volume. Similarly, although B-lymphocytes are not present in large numbers in the brain early after stroke, regulatory B-cells are protective and may also increase beneficial T-reg activity. Autoreactive CD4+ and CD8+ T-cells and B-cells increase four days after stroke, leading to antigen-dependent autoimmunity.
In stroke survivors, peripheral blood lymphocytes demonstrate more activity against myelin than those in controls or even people with MS [176, 177]. This autoimmunity, similar to antigen-independent responses, can be harmful or beneficial depending on T-lymphocytes polarization. Prior tolerization of T-regs specific to the brain antigen myelin oligodendrocyte glycoprotein leads to IL-10-dependent neuroprotection after stroke. Conversely, a pro-inflammatory stimulus after stroke will lead to a detrimental Th1 response against brain antigens and worse outcomes. Late B-lymphocyte responses also occur after stroke, and CNS antibody production may increase over time, with about half of the stroke survivors exhibiting intrathecal antibody synthesis after the first week. Chronic inflammation is neurotoxic, and the glial scar surrounding it lacks tight junctions in humans and rodents [178]. It is permeable to albumin and antibodies, possibly causing neuronal injury in the penumbra region.
Post-stroke atrophy has been attributed to the loss of network integrity that causes atrophy of the connected region. Brain atrophy can also occur from stroke risk factors in the absence of clinical stroke. Medial temporal atrophy, which may occur prior to stroke, is associated with cognitive decline after stroke [179]. The pro-inflammatory cytokine osteopontin (OPN) plays a role in neuroinflammatory diseases such as AD and mild cognitive impairment (MCI) [180]. In mice, atrophy after stroke partially depends on OPN [181]. Stroke doubles the risk of new dementia diagnosis, as stroke risk factors and recurrent stroke may contribute to increased dementia risk [180]. In animal models, inflammation can cause cognitive decline with or without stroke [180]. High-salt diets without stroke can increase peripheral Th17 cells, leading to cognitive decline. TLR4 signaling promotes aberrant neurogenesis causing cognitive decline after stroke [180]. B-lymphocytes can cause loss of long-term hippocampal potentiation and cognitive decline, perhaps as part of an autoimmune response triggered by a pro-inflammatory state [182]. An association between anti-MBP antibodies and cognitive decline has been shown in humans. A pro-inflammatory state in the blood 2 days after stroke is associated with subsequent cognitive decline [180, 183•].
Post-stroke fatigue (PSF) and post-stroke depression (PSD) are common and significant barriers to stroke recovery and rehabilitation. However, disentangling them from post-stroke cognitive impairment can be difficult [184]. Limited research exists on the links between neuroinflammation and fatigue, and depression after stroke, but some studies have suggested associations between PSD and inflammation, as well as certain immune cell activity [184]. There is a complex interplay between adaptive immunity, chronic inflammation, and long-term sequelae of stroke, including post-stroke dementia, atrophy, and PSF and PSD. Targeting the immune system and inflammation potentially improve outcomes in stroke survivors [184]. Research in this area is ongoing, and further investigation into the immune response to stroke and its impact on long-term outcomes may lead to new treatment strategies for stroke survivors [184–187].
Conclusion
This review highlights the importance of neuroinflammation in processes surrounding acute ischemic stroke and ICH. Neuroinflammation is a crucial aspect of stroke pathophysiology that has garnered increasing attention in recent years. After a stroke, the release of pro-inflammatory cytokines, chemokines, and other immune mediators leads to an infiltration of immune cells into the brain parenchyma. This neuroinflammatory response can have both beneficial and detrimental effects on the damaged tissue.
Neuroinflammation can help clear away dead cells and debris, promote tissue repair, and support neuroplasticity and neurogenesis. However, it can also exacerbate secondary injury by contributing to BBB disruption, edema formation, and neuronal death. Inflammatory cytokines can also impair the function of surviving neurons and promote the formation of glial scars, which can impede functional recovery.
The immune response after stroke is complex and involves both innate and adaptive immune cells, including microglia, macrophages, T-cells, B-cells, and NK cells. The phenotype of these immune cells can shift from pro-inflammatory to anti-inflammatory over time, with a temporal pattern that varies depending on the specific cell type and context of the injury. Therapeutic interventions that target neuroinflammation in stroke are currently an active area of research and hold promise for improving outcomes for stroke patients.
As discussed in this review, inflammation may start in the brain but certainly does not remain there. After a stroke, there is often a systemic inflammatory response that occurs throughout the body. This response is characterized by increased circulating cytokines, such as IL-6 and TNF-α, and other markers of inflammation, such as CRP. This systemic inflammatory response is thought to be triggered by releasing danger signals, DAMPs, from the damaged brain tissue. These signals can activate immune cells throughout the body, releasing pro-inflammatory cytokines and other mediators of inflammation. The systemic inflammatory response after stroke has been associated with a poorer prognosis and an increased risk of complications, such as infection and pneumonia. It is also thought to contribute to the development of secondary brain injury by exacerbating the inflammation and oxidative stress within the brain. As a result, there has been growing interest in targeting the systemic inflammatory response after stroke as a potential therapeutic strategy. The CNS and systemic inflammation should continue to be targeted for research and potential therapies, as ample evidence supports their significant role in the secondary injury that occurs after these neurologic emergencies.
Acknowledgements
The editors would like to thank Dr. John Brust for taking the time to review this manuscript.
Abbreviations
- AD
Alzheimer’s disease
- ADPKD
Autosomal dominant polycystic kidney disease
- AIS
Acute ischemic stroke
- AMPA
a-Amino-3-hydroxy-5-methyl-4 isoxazole-propionic acid
- ASCVD
Atherosclerotic cardiovascular disease
- AVMs
Arteriovenous malformations
- BBB
Blood-brain barrier
- BDNF
Brain-derived neurotrophic factor
- CAA
Cerebral amyloid angiopathy
- CBF
Cerebral blood flow
- CCR6
Chemokine receptor 6
- CNS
Central nervous system
- CRP
C-reactive protein
- CSF
Cerebrospinal fluid
- CVD
Cardiovascular disease
- DAMPs
Danger-associated molecular patterns
- EAAT2
Excitatory amino acid transporter 2
- GCS
Glasgow Coma Scale
- GDNF
Glial cell line-derived neurotrophic factor
- GFAP
Glial fibrillary acidic protein
- HIF-1α
Hypoxia-inducible factor-1alpha
- HMGB1
High mobility group box-1
- HPA
Hypothalamic pituitary adrenal
- HS
Hemorrhagic stroke
- ICAM
Intracellular adhesion molecule
- ICH
Intracerebral hemorrhage
- ICP
Intracranial pressure
- IFN-γ
Interferon-gamma
- IL-1β
Interleukin-1beta
- IRF
Interferon regulatory factor
- LAA
Large artery atherosclerosis
- LPS
Lipopolysaccharides
- MAC
Membrane attack complex
- MAdCAM-1
Mucosal vascular adhesion molecule-1
- MAPK
Mitogen-activated protein kinase
- MCI
Mild cognitive impairment
- MMPs
Matrix metalloproteinases
- MS
Multiple sclerosis
- NETs
Neutrophil extracellular traps
- NF-Kβ
Nuclear factor kappa beta
- Nrf2
Nuclear factor erythroid 2-related factor 2
- OPN
Osteopontin
- PAMPs
Pathogen-associated molecular pattern molecules
- PDGF
Platelet-derived growth factor
- PECAM-1
Platelet and endothelial cell adhesion molecule-1
- PPAR-γ
Peroxisome proliferator-activated receptor gamma
- PRRs
Pattern recognition receptors
- PSD
Post-stroke depression
- PSF
Post-stroke fatigue
- RAGE
Receptor for advanced glycation end products
- ROS
Reactive oxygen species
- SDF-1
Stromal cell-derived factor-1
- SEFIR
SEF/IL-17R
- SNS
Sympathetic nervous system
- Sox2
SRY-box 2
- SVD
Small vessel disease
- TGF-β
Transforming growth factor beta
- TLR
Toll-like receptor
- TNF-α
Tumor necrosis factor-alpha
- T-regs
Regulatory T-cells
- VCAM-1
Vascular cell adhesion molecule-1
- VEGF
Vascular endothelial growth factor
- VNS
Vagus nerve stimulation
- vWF
Von Willebrand factor
Footnotes
Compliance with Ethical Standards
Conflict of Interest Diana L. Alsbrook, Mario Di Napoli, Kunal Bhatia, Jose Biller, Sasan Andalib, Archana Hinduja, Roysten Rodrigues, Miguel Rodriguez; Sara Y. Sabbagh, Magdy Selim, Maryam Hosseini Farahabadi, Alibay Jafarli, and Afshin A. Divani each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Kleindorfer DO, Towfighi A, Chaturvedi S, Cockroft KM, Gutierrez J, Lombardi-Hill D, et al. 2021 Guideline for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline from the American Heart Association/American Stroke Association. Stroke. 2021;52(7):e364–467. 10.1161/STR.0000000000000375. [DOI] [PubMed] [Google Scholar]
- 2.••.Jung S, Gilgen M, Slotboom J, El-Koussy M, Zubler C, Kiefer C, et al. Factors that determine penumbral tissue loss in acute ischaemic stroke. Brain. 2013;136(Pt 12):3554–60. 10.1093/brain/awt246. [DOI] [PubMed] [Google Scholar]; This article examines the factors that contribute to the loss of penumbral tissue in cases of acute ischemic stroke. The study identifies variables such as time to treatment, collateral circulation, and infarct core size as critical determinants of penumbral tissue loss.
- 3.Jurcau A, Simion A. Neuroinflammation in cerebral ischemia and ischemia/reperfusion injuries: from pathophysiology to therapeutic strategies. Int J Mol Sci. 2021;23(1):14. 10.3390/ijms23010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. Journal of neurochemistry. 2016;139(Suppl 2):136–53. 10.1111/jnc.13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.••.Schaeffer S, Iadecola C. Revisiting the neurovascular unit. Nat Neurosci. 2021;24(9):1198–209. 10.1038/s41593-021-00904-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article revisits the concept of the neurovascular unit, emphasizing its crucial role in maintaining brain function and highlighting its relevance in neurological disorders. The review provides insights into the molecular and cellular mechanisms underlying neurovascular unit dysfunction and highlights its potential as a therapeutic target for various neurological conditions.
- 6.Jurcau A, Ardelean IA. Molecular pathophysiological mechanisms of ischemia/reperfusion injuries after recanalization therapy for acute ischemic stroke. J Integr Neurosci. 2021;20(3):727–44. 10.31083/jjin2003078. [DOI] [PubMed] [Google Scholar]
- 7.Khoshnam SE, Winlow W, Farzaneh M, Farbood Y, Moghaddam HF. Pathogenic mechanisms following ischemic stroke. Neurol Sci. 2017;38(7):1167–86. 10.1007/s10072-017-2938-1. [DOI] [PubMed] [Google Scholar]
- 8.Yang C, Hawkins KE, Dore S, Candelario-Jalil E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol. 2019;316(2):C135–C53. 10.1152/ajpcell.00136.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67(2):181–98. 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haeusler KG, Schmidt WU, Fohring F, Meisel C, Helms T, Jungehulsing GJ, et al. Cellular immunodepression preceding infectious complications after acute ischemic stroke in humans. Cerebrovasc Dis. 2008;25(1-2):50–8. 10.1159/000111499. [DOI] [PubMed] [Google Scholar]
- 11.Dropulic LK, Lederman HM. Overview of infections in the immunocompromised host. Microbiol Spectr. 2016;4(4):1–50. 10.1128/microbiolspec.DMIH2-0026-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.••.Simats A, Liesz A. Systemic inflammation after stroke:implications for post-stroke comorbidities. EMBO Mol Med. 2022;14(9):e16269. 10.15252/emmm.202216269. [DOI] [PMC free article] [PubMed] [Google Scholar]; The review highlights that stroke triggers an inflammatory response throughout the body, not just in the brain, which can have significant consequences for various organs and systems. Understanding the relationship between systemic inflammation and post-stroke comorbidities is crucial for the development of preventive and therapeutic strategies aimed at reducing the burden of these complications and improving the long-term outcomes of stroke patients.
- 13.Maida CD, Norrito RL, Daidone M, Tuttolomondo A, Pinto A. Neuroinflammatory mechanisms in ischemic stroke: focus on cardioembolic stroke, background, and therapeutic approaches. Int J Mol Sci. 2020;21(18):6454. 10.3390/ijms21186454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57(2):173–85. 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 15.••.Lochhead JJ, Yang J, Ronaldson PT, Davis TP. Structure, function, and regulation of the blood-brain barrier tight junction in central nervous system disorders. Front Physiol. 2020;11:914. 10.3389/fphys.2020.00914. [DOI] [PMC free article] [PubMed] [Google Scholar]; The review highlights the critical role of the BBB in maintaining brain homeostasis by tightly regulating the passage of molecules between the blood and the brain. Understanding the mechanisms underlying BBB tight junction disruption provides insights into potential therapeutic targets for the treatment of CNS disorders and the development of strategies to enhance drug delivery to the brain.
- 16.Bernardo-Castro S, Sousa JA, Bras A, Cecilia C, Rodrigues B, Almendra L, et al. Pathophysiology of blood-brain barrier permeability throughout the different stages of ischemic stroke and its implication on hemorrhagic transformation and recovery. Front Neurol. 2020;11:594672. 10.3389/fneur.2020.594672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nayak AR, Kashyap RS, Kabra D, Purohit HJ, Taori GM, Daginawala HF. Time course of inflammatory cytokines in acute ischemic stroke patients and their relation to inter-alfa trypsin inhibitor heavy chain 4 and outcome. Ann Indian Acad Neurol. 2012;15(3):181–5. 10.4103/0972-2327.99707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaur D, Sharma V, Deshmukh R. Activation of microglia and astrocytes: a roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology. 2019;27(4):663–77. 10.1007/s10787-019-00580-x. [DOI] [PubMed] [Google Scholar]
- 19.Mastronardi C, Whelan F, Yildiz OA, Hannestad J, Elashoff D, McCann SM, et al. Caspase 1 deficiency reduces inflammation-induced brain transcription. Proc Natl Acad Sci U S A. 2007;104(17):7205–10. 10.1073/pnas.0701366104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hill WD, Hess DC, Martin-Studdard A, Carothers JJ, Zheng J, Hale D, et al. SDF-1 (CXCL12) is upregulated in the ischemic penumbra following stroke: association with bone marrow cell homing to injury. J Neuropathol Exp Neurol. 2004;63(1):84–96. 10.1093/jnen/63.1.84. [DOI] [PubMed] [Google Scholar]
- 21.••.Sadri F, Rezaei Z, Fereidouni M. The significance of the SDF-1/CXCR4 signaling pathway in the normal development. Mol Biol Rep. 2022;49(4):3307–20. 10.1007/s11033-021-07069-3. [DOI] [PubMed] [Google Scholar]; This article highlights the significance of the SDF-1/CXCR4 signaling pathway in normal development processes. The review emphasizes the crucial role of this pathway in various biological events, including embryogenesis, organogenesis, tissue repair, and immune cell trafficking.
- 22.Linnerbauer M, Wheeler MA, Quintana FJ. Astrocyte crosstalk in CNS inflammation. Neuron. 2020;108(4):608–22. 10.1016/jneuron.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.••.Zhou SY, Guo ZN, Zhang DH, Qu Y, Jin H. The role of pericytes in ischemic stroke: fom cellular functions to therapeutic targets. Front Mol Neurosci. 2022;15:866700. 10.3389/fnmol.2022.866700. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article explores the role of pericytes in ischemic stroke, highlighting their diverse cellular functions and their potential as therapeutic targets. The review emphasizes the involvement of pericytes in maintaining blood-brain barrier integrity, regulating cerebral blood flow, modulating inflammation, and influencing neurovascular remodeling, thereby suggesting their importance in stroke pathophysiology and potential for targeted interventions.
- 24.Sakuma R, Kawahara M, Nakano-Doi A, Takahashi A, Tanaka Y, Narita A, et al. Brain pericytes serve as microglia-generating multipotent vascular stem cells following ischemic stroke. J Neuroinflammation. 2016;13(1):57. 10.1186/s12974-016-0523-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guijarro-Munoz I, Compte M, Alvarez-Cienfuegos A, Alvarez-Vallina L, Sanz L. Lipopolysaccharide activates Toll-like receptor 4 (TLR4)-mediated NF-kappaB signaling pathway and proinflammatory response in human pericytes. J Biol Chem. 2014;289(4):2457–68. 10.1074/jbc.M113.521161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin R, Liu L, Zhang S, Nanda A, Li G. Role of inflammation and its mediators in acute ischemic stroke. J Cardiovasc Transl Res. 2013;6(5):834–51. 10.1007/s12265-013-9508-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen R, Zhang X, Gu L, Zhu H, Zhong Y, Ye Y, et al. New insight into neutrophils: a potential therapeutic target for cerebral ischemia. Front Immunol. 2021;12:692061. 10.3389/fimmu.2021.692061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao X, Wang H, Sun G, Zhang J, Edwards NJ, Aronowski J. Neuronal interleukin-4 as a modulator of microglial pathways and ischemic brain damage. J Neurosci. 2015;35(32):11281–91. 10.1523/JNEUROSCI.1685-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liesz A, Hu X, Kleinschnitz C, Offner H. Functional role of regulatory lymphocytes in stroke: facts and controversies. Stroke. 2015;46(5):1422–30. 10.1161/STROKEAHA.114.008608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15(2):192–9. 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- 31.Lužnik Z, Anchouche S, Dana R, Yin J. Regulatory T cells in angiogenesis. J Immunol. 2020;205(10):2557–65. 10.4049/jimmunol.2000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.••.Li P, Gan Y, Sun BL, Zhang F, Lu B, Gao Y, et al. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann Neurol. 2013;74(3):458–71. 10.1002/ana.23815. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that adoptive regulatory T-cell (Treg) therapy has a protective effect against cerebral ischemia, offering potential therapeutic benefits. The findings highlight the role of Treg cells in modulating immune responses and reducing inflammation in the brain, suggesting their potential as a targeted therapy for the treatment of cerebral ischemia and related conditions.
- 33.Haley MJ, Lawrence CB. The blood-brain barrier after stroke: structural studies and the role of transcytotic vesicles. J Cereb Blood Flow Metab. 2017;37(2):456–70. 10.1177/0271678X16629976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hasel P, Cooper ML, Marchildon AE, Rufen-Blanchette UA, Kim RD, Ma TC et al. Defining the molecular identity and morphology of glia limitans superficialis astrocytes in mouse and human. 10.1101/2023.04.06.535893v1.abstract.2023 [DOI] [Google Scholar]
- 35.Ioghen O, Chitoiu L, Gherghiceanu M, Ceafalan LC, Hinescu ME. CD36 - A novel molecular target in the neurovascular unit. Eur J Neurosci. 2021;53(8):2500–10. 10.1111/ejn15147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otxoa-de-Amezaga A, Gallizioli M, Pedragosa J, Justicia C, Miro-Mur F, Salas-Perdomo A, et al. Location of neutrophils in different compartments of the damaged mouse brain after severe ischemia/reperfusion. Stroke. 2019;50(6):1548–57. 10.1161/STROKEAHA.118.023837. [DOI] [PubMed] [Google Scholar]
- 37.Cugurra A, Mamuladze T, Rustenhoven J, Dykstra T, Beroshvili G, Greenberg ZJ, et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science. 2021;373(6553):eabf7844. 10.1126/science.abf7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herisson F, Frodermann V, Courties G, Rohde D, Sun Y, Vandoorne K, et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci. 2018;21(9):1209–17. 10.1038/s41593-018-0213-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Angelini G, Bani A, Constantin G, Rossi B. The interplay between T helper cells and brain barriers in the pathogenesis of multiple sclerosis. Front Cell Neurosci. 2023;17:1101379. 10.3389/fncel.2023.1101379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steffen BJ, Breier G, Butcher EC, Schulz M, Engelhardt B. ICAM-1, VCAM-1, and MAdCAM-1 are expressed on choroid plexus epithelium but not endothelium and mediate binding of lymphocytes in vitro. The American journal of pathology. 1996;148(6):1819–38. [PMC free article] [PubMed] [Google Scholar]
- 41.Strominger I, Elyahu Y, Berner O, Reckhow J, Mittal K, Nemirovsky A, et al. The choroid plexus functions as a niche for t-cell stimulation within the central nervous system. Front Immunol. 2018;9:1066. 10.3389/fimmu.2018.01066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stegner D, Klaus V, Nieswandt B. Platelets as modulators of cerebral ischemia/reperfusion injury. Front Immunol. 2019;10:2505. 10.3389/fimmu.2019.02505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.•.Denorme F, De Meyer SF. The VWF-GPIb axis in ischaemic stroke: lessons from animal models. Thromb Haemost. 2016;116(4):597–604. 10.1160/TH16-01-0036. [DOI] [PubMed] [Google Scholar]; This article explores the role of the von Willebrand factor (VWF)-GPIb axis in ischemic stroke using animal models. It provides insights into the mechanisms underlying VWF-GPIb interactions and their implications in stroke pathophysiology, including thrombus formation, platelet activation, and endothelial dysfunction. The findings highlight the potential of targeting the VWF-GPIb axis as a therapeutic approach for preventing or treating ischemic stroke.
- 44.Rezkalla SH, Kloner RA. No-reflow phenomenon. Circulation. 2002;105(5):656–62. 10.1161/hc0502.102867. [DOI] [PubMed] [Google Scholar]
- 45.Blair P, Flaumenhaft R. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev. 2009;23(4):177–89. 10.1016/j.blre.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cognasse F, Duchez AC, Audoux E, Ebermeyer T, Arthaud CA, Prier A, et al. Platelets as key factors in inflammation: focus on CD40L/CD40. Front Immunol. 2022;13:825892. 10.3389/fimmu.2022.825892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen QL, Okuno N, Hamashima T, Dang ST, Fujikawa M, Ishii Y, et al. Vascular PDGFR-alpha protects against BBB dysfunction after stroke in mice. Angiogenesis. 2021;24(1):35–46. 10.1007/s10456-020-09742-w. [DOI] [PubMed] [Google Scholar]
- 48.Krupinski J, Issa R, Bujny T, Slevin M, Kumar P, Kumar S, et al. A putative role for platelet-derived growth factor in angiogenesis and neuroprotection after ischemic stroke in humans. Stroke. 1997;28(3):564–73. 10.1161/01.str.28.3.564. [DOI] [PubMed] [Google Scholar]
- 49.Rawish E, Nording H, Munte T, Langer HF. Platelets as mediators of neuroinflammation and thrombosis. Front Immunol. 2020;11:548631. 10.3389/fimmu.2020.548631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.••.Endres M, Moro MA, Nolte CH, Dames C, Buckwalter MS, Meisel A. Immune pathways in etiology, acute phase, and chronic sequelae of ischemic stroke. Circ Res. 2022;130(8):1167–86. 10.1161/CIRCRESAHA.121.319994. [DOI] [PubMed] [Google Scholar]; This article examines immune pathways involved in the etiology, acute phase, and chronic sequelae of ischemic stroke. It highlights the complex interplay between inflammatory responses, immune cell activation, and the neurovascular unit in different stages of stroke, providing insights into potential therapeutic targets for mitigating acute brain damage and preventing long-term complications.
- 51.•.Tariq MB, Lee J, LD MC. Sex differences in the inflammatory response to stroke. In: Seminars in immunopathology. Berlin/Heidelberg: Springer Berlin Heidelberg; 2022. p. 1–19. 10.1007/s00281-022-00969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article explores sex differences in the inflammatory response to stroke, highlighting the distinct immune responses observed between males and females. The review emphasizes the importance of considering sex as a biological variable in stroke research and suggests that understanding these sex-specific differences in inflammation could lead to tailored therapeutic strategies and improved outcomes for both male and female stroke patients.
- 52.Harry GJ, McPherson CA. Microglia: neuroprotective and neurodestructive properties. In: Kostrzewa RM, editor. Handbook of Neurotoxicity. New York, NY: Springer New York; 2014. p. 109–32. [Google Scholar]
- 53.••.Wang Y, Leak RK, Cao G. Microglia-mediated neuroinflammation and neuroplasticity after stroke. Front Cell Neurosci. 2022;16:980722. 10.3389/fncel.2022.980722. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review article focuses on microglia-mediated neuroinflammation and neuroplasticity following a stroke. It highlights the role of microglia, the resident immune cells in the brain, in the post-stroke inflammatory response and their influence on neuroplasticity, which refers to the brain’s ability to reorganize and form new connections. The article discusses the complex interactions between microglia, inflammatory signaling pathways, and neuronal plasticity processes, shedding light on the potential therapeutic targets for promoting post-stroke recovery and rehabilitation.
- 54.Zhao SC, Ma LS, Chu ZH, Xu H, Wu WQ, Liu F. Regulation of microglial activation in stroke. Acta Pharmacol Sin. 2017;38(4):445–58. 10.1038/aps.2016.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee SS, Pang L, Cheng Y, Liu JX, Ng ACK, Leung GKK. A previous hemorrhagic stroke protects against a subsequent stroke via microglia alternative polarization. Commun Biol. 2022;5(1):654. 10.1038/s42003-022-03621-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43(11):3063–70. 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- 57.Anttila JE, Whitaker KW, Wires ES, Harvey BK, Airavaara M. Role of microglia in ischemic focal stroke and recovery: focus on Toll-like receptors. Prog Neuropsychopharmacol Biol Psychiatry. 2017;79(Pt A):3–14. 10.1016/j.pnpbp.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Varnum MM, Kiyota T, Ingraham KL, Ikezu S, Ikezu T. The anti-inflammatory glycoprotein, CD200, restores neurogenesis and enhances amyloid phagocytosis in a mouse model of Alzheimer’s disease. Neuro of aging. 2015;36(11):2995–3007. 10.1016/jneurobiolaging.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mahmoud S, Gharagozloo M, Simard C, Gris D. Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells. 2019;8(2):184. 10.3390/cells8020184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen XY, Gao ZK, Han Y, Yuan M, Guo YS, Bi X. Activation and role of astrocytes in ischemic stroke. Front Cell Neurosci. 2021;15:755955. 10.3389/fncel.2021.755955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marshall JS, Warrington R, Watson W, Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol. 2018;14(Suppl 2):49. 10.1186/s13223-018-0278-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tschoe C, Bushnell CD, Duncan PW, Alexander-Miller MA, Wolfe SQ. Neuroinflammation after intracerebral hemorrhage and potential therapeutic targets. J Stroke. 2020;22(1):29–46. 10.5853/jos.2019.02236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.••.Zhu H, Wang Z, Yu J, Yang X, He F, Liu Z, et al. Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. Prog Neurobiol. 2019;178:101610. 10.1016/jpneurobio.2019.03.003. [DOI] [PubMed] [Google Scholar]; This review article examines the role and mechanisms of cytokines in the secondary brain injury following intracerebral hemorrhage (ICH). It discusses how cytokines, as key mediators of the inflammatory response, contribute to the pathogenesis of secondary brain injury after ICH. The article explores the various cytokines involved, their sources, signaling pathways, and the impact they have on processes such as blood-brain barrier disruption, neuroinflammation, oxidative stress, and neuronal cell death. Understanding the role of cytokines in secondary brain injury can provide insights into potential therapeutic targets for mitigating the harmful effects of ICH and improving patient outcomes.
- 64.Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. 2014;115:25–44. 10.1016/j.pneurobio.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 65.Palomo J, Dietrich D, Martin P, Palmer G, Gabay C. The interleukin (IL)-1 cytokine family--Balance between agonists and antagonists in inflammatory diseases. Cytokine. 2015;76(1):25–37. 10.1016/j.cyto.2015.06.017. [DOI] [PubMed] [Google Scholar]
- 66.Garcia JH, Liu KF, Relton JK. Interleukin-1 receptor antagonist decreases the number of necrotic neurons in rats with middle cerebral artery occlusion. Am J Clin Pathol. 1995;147(5):1477–86. [PMC free article] [PubMed] [Google Scholar]
- 67.Betz AL, Yang GY, Davidson BL. Attenuation of stroke size in rats using an adenoviral vector to induce overexpression of interleukin-1 receptor antagonist in brain. J Cereb Blood Flow Metab. 1995;15(4):547–51. 10.1038/jcbfm.1995.68. [DOI] [PubMed] [Google Scholar]
- 68.Galea J, Ogungbenro K, Hulme S, Patel H, Scarth S, Hoadley M, et al. Reduction of inflammation after administration of interleukin-1 receptor antagonist following aneurysmal subarachnoid hemorrhage: results of the Subcutaneous Interleukin-1Ra in SAH (SCIL-SAH) study. J Neurosurg. 2018;128(2):515–23. 10.3171/2016.9.JNS16615. [DOI] [PubMed] [Google Scholar]
- 69.Zhu H, Hu S, Li Y, Sun Y, Xiong X, Hu X, et al. Interleukins and ischemic stroke. Front Immunol. 2022;13:828447. 10.3389/fimmu.2022.828447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Di Napoli M, Papa F, Villa Pini Stroke Data Bank I. Inflammation, hemostatic markers, and antithrombotic agents in relation to long-term risk of new cardiovascular events in first-ever ischemic stroke patients. Stroke. 2002;33(7):1763–71. 10.1161/01.str.0000019124.54361.08. [DOI] [PubMed] [Google Scholar]
- 71.Tabarkiewicz J, Pogoda K, Karczmarczyk A, Pozarowski P, Giannopoulos K. The role of IL-17 and Th17 lymphocytes in autoimmune diseases. Arch Immunol Ther Exp (Warsz). 2015;63(6):435–49. 10.1007/s00005-015-0344-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gao L, Lu Q, Huang LJ, Ruan LH, Yang JJ, Huang WL, et al. Transplanted neural stem cells modulate regulatory T, gammadelta T cells and corresponding cytokines after intracerebral hemorrhage in rats. Int J Mol Sci. 2014;15(3):4431–41. 10.3390/ijms15034431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.•.Jiang C, Wang Y, Hu Q, Shou J, Zhu L, Tian N, et al. Immune changes in peripheral blood and hematoma of patients with intracerebral hemorrhage. FASEB J. 2020;34(2):2774–91. 10.1096/fj.201902478R. [DOI] [PubMed] [Google Scholar]; This study investigates immune changes in the peripheral blood and hematoma of patients with intracerebral hemorrhage (ICH). The findings reveal alterations in immune profiles, including inflammatory and immune cell responses, in both peripheral blood and the hematoma of ICH patients. These findings provide valuable insights into the immune mechanisms involved in ICH pathophysiology and may have implications for developing immune-based therapeutic interventions for ICH patients.
- 74.Lively S, Schlichter LC. Microglia responses to pro-inflammatory stimuli (LPS, IFNgamma+TNFalpha) and reprogramming by resolving cytokines (IL-4, IL-10). Front Cell Neurosci. 2018;12:215. 10.3389/fncel.2018.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun L, Li Y, Jia X, Wang Q, Li Y, Hu M, et al. Neuroprotection by IFN-gamma via astrocyte-secreted IL-6 in acute neuroinflammation. Oncotarget. 2017;8(25):40065–78. 10.18632/oncotarget.16990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Divani AA, Majidi S, Barrett AM, Noorbaloochi S, Luft AR. Consequences of stroke in community-dwelling elderly: the health and retirement study, 1998 to 2008. Stroke. 2011;42(7):1821–5. 10.1161/STROKEAHA.110.607630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.••.Hu J, Wang L, Fan K, Ren W, Wang Q, Ruan Y, et al. The association between systemic inflammatory markers and poststroke depression: a prospective stroke cohort. Clin Interv Aging. 2021;16:1231–9. 10.2147/CIA.S314131. [DOI] [PMC free article] [PubMed] [Google Scholar]; This prospective stroke cohort study examines the association between systemic inflammatory markers and poststroke depression. The findings suggest that increased levels of systemic inflammatory markers are associated with a higher risk of developing post-stroke depression, highlighting the potential role of inflammation in the pathogenesis of post-stroke psychiatric complications. These results contribute to our understanding of the underlying mechanisms of post-stroke depression and may have implications for the development of targeted interventions for better management of psychiatric outcomes in stroke patients.
- 78.Chen H, Huang X, Zeng C, Sun D, Liu F, Zhang J, et al. The role of indoleamine 2,3-dioxygenase 1 in early-onset post-stroke depression. Front Immunol. 2023;14:1125634. 10.3389/fimmu.2023.1125634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, et al. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2(7):788–94. 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 80.Ginis I, Jaiswal R, Klimanis D, Liu J, Greenspon J, Hallenbeck JM. TNF-alpha-induced tolerance to ischemic injury involves differential control of NF-kappaB transactivation: the role of NF-kappaB association with p300 adaptor. J Cereb Blood Flow Metab. 2002;22(2):142–52. 10.1097/00004647-200202000-00002. [DOI] [PubMed] [Google Scholar]
- 81.Abd-El-Basset EM, Rao MS, Alshawaf SM, Ashkanani HK, Kabli AH. Tumor necrosis factor (TNF) induces astrogliosis, microgliosis and promotes survival of cortical neurons. AIMS Neurosci. 2021;8(4):558–84. 10.3934/Neuroscience.2021031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lively S, Hutchings S, Schlichter LC. Molecular and cellular responses to interleukin-4 treatment in a rat model of transient ischemia. J Neuropathol Exp Neurol. 2016;75(11):1058–71. 10.1093/jnen/nlw081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang J, Rong P, Zhang L, He H, Zhou T, Fan Y, et al. IL4-driven microglia modulate stress resilience through BDNF-dependent neurogenesis. Sci Adv. 2021;7(12):eabb9888. 10.1126/sciadv.abb9888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garcia JM, Stillings SA, Leclerc JL, Phillips H, Edwards NJ, Robicsek SA, et al. Role of interleukin-10 in acute brain injuries. Frontiers in neurology. 2017;8:244. 10.3389/fneur.2017.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang P, Wu P, Siegel MI, Egan RW, Billah MM. Interleukin (IL)-10 inhibits nuclear factor kappa B (NF kappa B) activation in human monocytes. IL-10 and IL-4 suppress cytokine synthesis by different mechanisms. J Biol Chem. 1995;270(16):9558–63. 10.1074/jbc.270.16.9558. [DOI] [PubMed] [Google Scholar]
- 86.Ooboshi H, Ibayashi S, Shichita T, Kumai Y, Takada J, Ago T, et al. Postischemic gene transfer of interleukin-10 protects against both focal and global brain ischemia. Circulation. 2005;111(7):913–9. 10.1161/01.CIR.0000155622.68580.DC. [DOI] [PubMed] [Google Scholar]
- 87.Li Q, Lan X, Han X, Durham F, Wan J, Weiland A, et al. Microglia-derived interleukin-10 accelerates post-intracerebral hemorrhage hematoma clearance by regulating CD36. Brain Behav Immun. 2021;94:437–57. 10.1016/jbbi.2021.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Taylor RA, Chang CF, Goods BA, Hammond MD, Mac Grory B, Ai Y, et al. TGF-beta1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J Clin Invest. 2017;127(1):280–92. 10.1172/JCI88647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Morganti-Kossmann MC, Hans VH, Lenzlinger PM, Dubs R, Ludwig E, Trentz O, et al. TGF-beta is elevated in the CSF of patients with severe traumatic brain injuries and parallels blood-brain barrier function. J Neurotrauma. 1999;16(7):617–28. 10.1089/neu.1999.16.617. [DOI] [PubMed] [Google Scholar]
- 90.Chang CF, Wan J, Li Q, Renfroe SC, Heller NM, Wang J. Alternative activation-skewed microglia/macrophages promote hematoma resolution in experimental intracerebral hemorrhage. Neuro dis. 2017;103:54–69. 10.1016/jnbd.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ma Y, Liu Y, Zhang Z, Yang GY. Significance of complement system in ischemic stroke: a comprehensive review. Aging Dis. 2019;10(2):429–62. 10.14336/AD.2019.0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pedersen ED, Waje-Andreassen U, Vedeler CA, Aamodt G, Mollnes TE. Systemic complement activation following human acute ischaemic stroke. Clin Exp Immunol. 2004;137(1):117–22. 10.111/j1365-2249.2004.02489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dalakas MC, Alexopoulos H, Spaeth PJ. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat Rev Neurol. 2020;16(11):601–17. 10.1038/s41582-020-0400-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ling M, Murali M. Analysis of the complement system in the clinical immunology laboratory. Clin Lab Med. 2019;39(4):579–90. 10.1016/jcll.2019.07.006. [DOI] [PubMed] [Google Scholar]
- 95.Alawieh A, Elvington A, Tomlinson S. Complement in the homeostatic and ischemic brain. Front Immunol. 2015;6:417. 10.3389/fimmu.2015.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mocco J, Mack WJ, Ducruet AF, Sosunov SA, Sughrue ME, Hassid BG, et al. Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circ Res. 2006;99(2):209–17. 10.1161/01.RES.0000232544.90675.42. [DOI] [PubMed] [Google Scholar]
- 97.Schartz ND, Tenner AJ. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J Neuroinflammation. 2020;17(1):354. 10.1186/s12974-020-02024-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang P, Zhu Z, Zang Y, Bu X, Xu T, Zhong C, et al. Increased serum complement C3 levels are associated with adverse clinical outcomes after ischemic stroke. Stroke. 2021;52(3):868–77. 10.1161/STROKEAHA.120.031715. [DOI] [PubMed] [Google Scholar]
- 99.Alawieh A, Langley EF, Tomlinson S. Targeted complement inhibition salvages stressed neurons and inhibits neuroinflammation after stroke in mice. Sci Transl Med. 2018;10(441):eaao6459. 10.1126/scitranslmedaao6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sierra A, Abiega O, Shahraz A, Neumann H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci. 2013;7:6. 10.3389/fncel.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu S, Lu J, Shao A, Zhang JH, Zhang J. Glial cells: role of the immune response in ischemic stroke. Front Immunol. 2020;11:294. 10.3389/fimmu.2020.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Monsour M, Borlongan CV. The central role of peripheral inflammation in ischemic stroke. J Cereb Blood Flow Metab. 2023;43(5):1. 10.1177/0271678X221149509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kumar P, Hair P, Cunnion K, Krishna N, Bass T. Classical complement pathway inhibition reduces brain damage in a hypoxic ischemic encephalopathy animal model. PloS one. 2021;16(9):e0257960. 10.1371/journal.pone0257960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim GH, Mocco J, Hahn DK, Kellner CP, Komotar RJ, Ducruet AF, et al. Protective effect of C5a receptor inhibition after murine reperfused stroke. Neurosurgery. 2008;63(1):122–5. 10.1227/01.NEU.0000335079.70222.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Carpanini SM, Torvell M, Morgan BP. Therapeutic inhibition of the complement system in diseases of the central nervous system. Front Immunol. 2019;10:362. 10.3389/fimmu.2019.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Arumugam TV, Woodruff TM, Lathia JD, Selvaraj PK, Mattson MP, Taylor SM. Neuroprotection in stroke by complement inhibition and immunoglobulin therapy. Neuroscience. 2009;158(3):1074–89. 10.1016/jneuroscience.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Elvington A, Atkinson C, Zhu H, Yu J, Takahashi K, Stahl GL, et al. The alternative complement pathway propagates inflammation and injury in murine ischemic stroke. J Immunol. 2012;189(9):4640–7. 10.4049/jimmunol.1201904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Atkinson C, Zhu H, Qiao F, Varela JC, Yu J, Song H, et al. Complement-dependent P-selectin expression and injury following ischemic stroke. J Immunol. 2006;177(10):7266–74. 10.4049/jimmunol.177.10.7266. [DOI] [PubMed] [Google Scholar]
- 109.Alawieh A, Elvington A, Zhu H, Yu J, Kindy MS, Atkinson C, et al. Modulation of post-stroke degenerative and regenerative processes and subacute protection by site-targeted inhibition of the alternative pathway of complement. J Neuroinflammation. 2015;12:247. 10.1186/s12974-015-0464-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shi Y, Jin Y, Li X, Chen C, Zhang Z, Liu X, et al. C5aR1 Mediates the progression of inflammatory responses in the brain of rats in the early stage after ischemia and reperfusion. ACS Chem Neurosci. 2021;12(21):3994–4006. 10.1021/acschemneuro.1c00244. [DOI] [PubMed] [Google Scholar]
- 111.Zheng Y, Fan L, Xia S, Yang Q, Zhang Z, Chen H, et al. Role of complement C1q/C3-CR3 signaling in brain injury after experimental intracerebral hemorrhage and the effect of minocycline treatment. Front Immunol. 2022;13:919444. 10.3389/fimmu.2022.919444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang M, Xia F, Wan S, Hua Y, Keep RF, Xi G. Role of complement component 3 in early erythrolysis in the hematoma after experimental intracerebral hemorrhage. Stroke. 2021;52(8):2649–60. 10.1161/STROKEAHA.121.034372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.•.Wang M, Hua Y, Keep RF, Wan S, Novakovic N, Xi G. Complement inhibition attenuates early erythrolysis in the hematoma and brain injury in aged rats. Stroke. 2019;50(7):1859–68. 10.1161/STROKEAHA.119.025170. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that complement inhibition can reduce early erythrolysis in the hematoma and brain injury in aged rats. The findings suggest that targeting the complement system has potential therapeutic benefits in attenuating the detrimental effects of hematoma-induced erythrolysis and brain injury, particularly in aged individuals.
- 114.Morris G, Gevezova M, Sarafian V, Maes M. Redox regulation of the immune response. Cell Mol Immunol. 2022; 19(10):1079–101. 10.1038/s41423-022-00902-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Qin C, Yang S, Chu YH, Zhang H, Pang XW, Chen L, et al. Signaling pathways involved in ischemic stroke: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther. 2022;7(1):215. 10.1038/s41392-022-01064-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.••.Dong R, Huang R, Wang J, Liu H, Xu Z. Effects of microglial activation and polarization on brain injury after stroke. Front neuro. 2021;12:620948. 10.3389/fneur.2021.620948. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article explores the effects of microglial activation and polarization on brain injury following stroke. The review highlights the dynamic and complex role of microglia, the resident immune cells in the brain, in the post-stroke inflammatory response. It discusses how microglial activation and polarization can have both beneficial and detrimental effects on brain injury, influencing tissue damage, neuroinflammation, and tissue repair processes. Understanding the intricacies of microglial activation and polarization can provide insights into potential therapeutic strategies aimed at modulating these processes to promote better outcomes and enhance stroke recovery.
- 117.Zhu SZ, Szeto V, Bao MH, Sun HS, Feng ZP. Pharmacological approaches promoting stem cell-based therapy following ischemic stroke insults. Acta Pharmacol Sin. 2018;39(5):695–712. 10.1038/aps.2018.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang R, Zhang Z, Chopp M. Function of neural stem cells in ischemic brain repair processes. J Cereb Blood Flow Metab. 2016;36(12):2034–43. 10.1177/0271678X16674487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Diaz-Canestro C, Reiner MF, Bonetti NR, Liberale L, Merlini M, Wust P, et al. AP-1 (activated protein-1) transcription factor JunD regulates ischemia/reperfusion brain damage via IL-1beta (interleukin-lbeta). Stroke. 2019;50(2):469–77. 10.1161/STROKEAHA.118.023739. [DOI] [PubMed] [Google Scholar]
- 120.Gugliandolo A, Silvestro S, Sindona C, Bramanti P, Mazzon E. MiRNA: Involvement of the MAPK pathway in ischemic stroke. A promising therapeutic target. Medicina (Kaunas). 2021;57(10):1053. 10.3390/medicina57101053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dong P, Li Q, Han H. HIF-1alpha in cerebral ischemia (review). Mol Med Rep. 2022;25(2):1–11. 10.3892/mmr.2021.12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cai W, Yang T, Liu H, Han L, Zhang K, Hu X, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma): a master gatekeeper in CNS injury and repair. Prog Neurobiol. 2018;163–164:27–58. 10.1016/jpneurobio.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kang TC. Nuclear factor-erythroid 2-related factor 2 (Nrf2) and mitochondrial dynamics/mitophagy in neurological diseases. Antioxidants (Basel). 2020;9(7):617. 10.3390/antiox9070617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Stevanovic M, Drakulic D, Lazic A, Ninkovic DS, Schwirtlich M, Mojsin M. SOX transcription factors as important regulators of neuronal and glial differentiation during nervous system development and adult neurogenesis. Front Mol Neurosci. 2021;14:654031. 10.3389/fnmol.2021.654031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Farina M, Vieira LE, Buttari B, Profumo E, Saso L. The Nrf2 pathway in ischemic stroke: a review. Molecules. 2021;26(16):5001. 10.3390/molecules26165001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu ZC, Meng LQ, Song JH, Gao J. Dynamic protein expression of NF-kappaB following rat intracerebral hemorrhage and its association with apoptosis. Exp Ther Med. 2018;16(5):3903–8. 10.3892/etm.2018.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xing G, Zhao T, Zhang X, Li H, Li X, Cui P, et al. Astrocytic sonic hedgehog alleviates intracerebral hemorrhagic brain injury via modulation of blood-brain barrier integrity. Front Cell Neurosci. 2020;14:575690. 10.3389/fncel.2020.575690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Loan JJM, Al-Shahi Salman R, McColl BW, Hardingham GE. Activation of Nrf2 to optimise immune responses to intracerebral haemorrhage. Biomolecules. 2022;12(10):1438. 10.3390/biom12101438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.•.Christopher E, JJM L, Samarasekera N, McDade K, Rose J, Barrington J, et al. Nrf2 activation in the human brain after stroke due to supratentorial intracerebral haemorrhage: a case-control study. BMJ Neurol Open. 2022;4(1):e000238. 10.1136/bmjno-2021-000238. [DOI] [PMC free article] [PubMed] [Google Scholar]; This case-control study investigates the activation of Nrf2 (nuclear factor erythroid 2-related factor 2) in the human brain following stroke caused by supratentorial intracerebral hemorrhage. The findings suggest that Nrf2 activation plays a role in the response to hemorrhagic stroke, potentially influencing oxidative stress and neuroinflammation, highlighting its potential as a therapeutic target for mitigating the damage caused by intracerebral hemorrhage.
- 130.Turnbull MT, Zubair AC, Meschia JF, Freeman WD. Mesenchymal stem cells for hemorrhagic stroke: status of preclinical and clinical research. NPJ Regen Med. 2019;4:10. 10.1038/s41536-019-0073-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li R, Liu Z, Wu X, Yu Z, Zhao S, Tang X. Lithium chloride promoted hematoma resolution after intracerebral hemorrhage through GSK-3beta-mediated pathways-dependent microglia phagocytosis and M2-phenotype differentiation, angiogenesis and neurogenesis in a rat model. Brain res bull. 2019;152:117–27. 10.1016/jbrainresbull.2019.07.019. [DOI] [PubMed] [Google Scholar]
- 132.Kumar V Toll-like receptors in the pathogenesis of neuroinflammation. J Neuroimmunol. 2019;332:16–30. 10.1016/jjneuroim.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 133.Maehara N, Taniguchi K, Okuno A, Ando H, Hirota A, Li Z, et al. AIM/CD5L attenuates DAMPs in the injured brain and thereby ameliorates ischemic stroke. Cell Rep. 2021;36(11):109693. 10.1016/jcelrep.2021.109693. [DOI] [PubMed] [Google Scholar]
- 134.Liang C, Liu L, Bao S, Yao Z, Bai Q, Fu P, et al. Neuroprotection by Nrf2 via modulating microglial phenotype and phagocytosis after intracerebral hemorrhage. Heliyon. 2023;9(2):e13777. 10.1016/jheliyon.2023.e13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–7. 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhu W, Tian L, Yue X, Liu J, Fu Y, Yan Y. LncRNA expression profiling of ischemic stroke during the transition from the acute to subacute stage. Front neuro. 2019;10:36. 10.3389/fneur.2019.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006;26(5):654–65. 10.1038/sjjcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- 138.•.Luo J, Chen Y, Tang G, Li Z, Yang X, Shang X, et al. Gut microbiota composition reflects disease progression, severity and outcome, and dysfunctional immune responses in patients with hypertensive intracerebral hemorrhage. Front Immunol. 2022;13:869846. 10.3389/fimmu.2022.869846. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals that the composition of gut microbiota in patients with hypertensive intracerebral hemorrhage (ICH) reflects disease progression, severity, and outcome, as well as dysfunctional immune responses. The findings highlight the potential role of gut microbiota in influencing the pathophysiology of ICH and suggest that targeting the gut microbiota may hold promise for developing novel therapeutic strategies for managing this condition.
- 139.Westendorp WF, Dames C, Nederkoorn PJ, Meisel A. Immunodepression, infections, and functional outcome in ischemic stroke. Stroke. 2022;53(5):1438–48. 10.1161/STROKEAHA.122.038867. [DOI] [PubMed] [Google Scholar]
- 140.Xue M, Yong VW. Neuroinflammation in intracerebral haemorrhage: immunotherapies with potential for translation. The Lancet Neuro. 2020;19(12):1023–32. 10.1016/S1474-4422(20)30364-1. [DOI] [PubMed] [Google Scholar]
- 141.Gulke E, Gelderblom M, Magnus T. Danger signals in stroke and their role on microglia activation after ischemia. Ther Adv Neurol Disord. 2018;11:1756286418774254. 10.1177/1756286418774254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.•.Yankova G, Bogomyakova O, Tulupov A. The glymphatic system and meningeal lymphatics of the brain: new understanding of brain clearance. Rev Neurosci. 2021;32(7):693–705. 10.1515/revneuro-2020-0106. [DOI] [PubMed] [Google Scholar]; This article presents new insights into the glymphatic system and meningeal lymphatics of the brain, shedding light on the mechanisms of brain clearance. The review highlights the importance of these systems in clearing waste products and metabolic byproducts from the brain, emphasizing their role in maintaining brain health and preventing the accumulation of toxic substances. Understanding the functions and regulation of the glymphatic system and meningeal lymphatics contributes to our knowledge of brain clearance processes and opens up potential avenues for therapeutic interventions targeting brain waste removal.
- 143.Balanca B, Desmurs L, Grelier J, Perret-Liaudet A, Lukaszewicz AC. DAMPs and RAGE pathophysiology at the acute phase of brain injury: an overview. Int J Mol Sci. 2021;22(5):2439. 10.3390/ijms22052439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Duran-Laforet V, Pena-Martinez C, Garcia-Culebras A, Cuartero MI, Lo EH, Moro MA, et al. Role of TLR4 in neutrophil dynamics and functions: contribution to stroke pathophysiology. Front Immunol. 2021;12:757872. 10.3389/fimmu.2021.757872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Courties G, Herisson F, Sager HB, Heidt T, Ye Y, Wei Y, et al. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ Res. 2015;116(3):407–17. 10.1161/CIRCRESAHA.116.305207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yan HQ, Ma X, Chen X, Li Y, Shao L, Dixon CE. Delayed increase of tyrosine hydroxylase expression in rat nigrostriatal system after traumatic brain injury. Brain res. 2007;1134(1):171–9. 10.1016/jbrainres.2006.11.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Panther EJ, Dodd W, Clark A, Lucke-Wold B. Gastrointestinal microbiome and neurologic injury. Biomedicines. 2022;10:2. 10.3390/biomedicines10020500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Anrather J, Iadecola C. Inflammation and stroke: an overview. Neurotherapeutics. 2016;13(4):661–70. 10.1007/s13311-016-0483-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Houlden A, Goldrick M, Brough D, Vizi ES, Lenart N, Martinecz B, et al. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav Immun. 2016;57:10–20. 10.1016/jbbi.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Singh V, Roth S, Llovera G, Sadler R, Garzetti D, Stecher B, et al. Microbiota dysbiosis controls the neuroinflammatory response after stroke. J Neurosci Res. 2016;36(28):7428–40. 10.1523/JNEUROSCI.1114-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chidambaram SB, Rathipriya AG, Mahalakshmi AM, Sharma S, Hediyal TA, Ray B, et al. The influence of gut dysbiosis in the pathogenesis and management of ischemic stroke. Cells. 2022;11(7):1239. 10.3390/cells11071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Peh A, O’Donnell JA, Broughton BRS, Marques FZ. Gut microbiota and their metabolites in stroke: a double-edged sword. Stroke. 2022;53(5):1788–801. 10.1161/STROKEAHA.121.036800. [DOI] [PubMed] [Google Scholar]
- 153.Zhang S, Jin M, Ren J, Sun X, Zhang Z, Luo Y, et al. New insight into gut microbiota and their metabolites in ischemic stroke: a promising therapeutic target. Biomed Pharmacother. 2023;162:114559. 10.1016/biopha.2023.114559. [DOI] [PubMed] [Google Scholar]
- 154.Tascilar N, Irkorucu O, Tascilar O, Comert F, Eroglu O, Bahadir B, et al. Bacterial translocation in experimental stroke: what happens to the gut barrier? Bratisl Lek Listy. 2010;111(4):194–9. [PubMed] [Google Scholar]
- 155.••.Lei C, Liu C, Chen E, Lin L, Liu T, Liu Z. Bidirectional microbiota-gut-brain axis after stroke and its implications for treating ischemic stroke. Explor Res Hypothesis Med. 2023;8(1):48–56. 10.14218/ERHM.2022.00040. [DOI] [Google Scholar]; The review discusses the influence of the gut microbiota on stroke outcomes and the impact of stroke on the gut microbiota composition. It proposes the modulation of the microbiota-gut-brain axis as a potential therapeutic approach for improving stroke recovery and preventing post-stroke complications.
- 156.Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat Med. 2016;22(5):516–23. 10.1038/nm.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Iadecola C, Buckwalter MS, Anrather J. Immune responses to stroke: mechanisms, modulation, and therapeutic potential. J Clin Invest. 2020;130(6):2777–88. 10.1172/JCI135530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.••.Liu Q, Johnson EM, Lam RK, Wang Q, Bo Ye H, Wilson EN, et al. Peripheral TREM1 responses to brain and intestinal immunogens amplify stroke severity. Nat Immunol. 2019;20(8):1023–34. 10.1038/s41590-019-0421-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals that peripheral triggering receptor expressed on myeloid cells 1 (TREM1) responses to immunogens in the brain and intestines can exacerbate stroke severity. The findings emphasize the role of peripheral immune activation in influencing stroke outcomes and suggest that targeting TREM1 signaling may hold therapeutic potential for mitigating the detrimental effects of immune amplification in stroke patients.
- 159.Datta A, Saha C, Godse P, Sharma M, Sarmah D, Bhattacharya P. Neuroendocrine regulation in stroke. Trends Endocrinol Metab. 2023;34(5):260–77. 10.1016/j.tem.2023.02.005. [DOI] [PubMed] [Google Scholar]
- 160.Prass K, Meisel C, Hoflich C, Braun J, Halle E, Wolf T, et al. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med. 2003;198(5):725–36. 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lunardi Baccetto S, Lehmann C. Microcirculatory changes in experimental models of stroke and CNS-injury induced immunodepression. Int J Mol Sci. 2019;20(20):5184. 10.3390/ijms20205184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wu H, Li L, Su X. Vagus nerve through alpha7 nAChR modulates lung infection and inflammation: models, cells, and signals. Biomed Res Int. 2014;2014:283525. 10.1155/2014/283525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tang H, Li J, Zhou Q, Li S, Xie C, Niu L, et al. Vagus nerve stimulation alleviated cerebral ischemia and reperfusion injury in rats by inhibiting pyroptosis via alpha7 nicotinic acetylcholine receptor. Cell Death Discov. 2022;8(1):54. 10.1038/s41420-022-00852-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Liu CY, Russin J, Adelson DP, Jenkins A, Hilmi O, Brown B, et al. Vagus nerve stimulation paired with rehabilitation for stroke: Implantation experience from the VNS-REHAB trial. J Clin Neurosci. 2022;105:122–8. 10.1016/j.jocn.2022.09.013. [DOI] [PubMed] [Google Scholar]
- 165.van der Meij A, van Walderveen MAA, Kruyt ND, van Zwet EW, Liebler EJ, Ferrari MD, et al. NOn-invasive Vagus nerve stimulation in acute Ischemic Stroke (NOVIS): a study protocol for a randomized clinical trial. Trials. 2020;21(1):878. 10.1186/s13063-020-04794-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Akimoto T, Hara M, Ishihara M, Ogawa K, Nakajima H. Post-stroke pneumonia in real-world practice: background, microbiological examination, and treatment. Neurol Int. 2023;15(1):69–77. 10.3390/neurolint15010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ghelani DP, Kim HA, Zhang SR, Drummond GR, Sobey CG, De Silva TM. Ischemic stroke and infection: a brief update on mechanisms and potential therapies. Biochem Pharmacol. 2021;193:114768. 10.1016/j.bcp.2021.114768. [DOI] [PubMed] [Google Scholar]
- 168.Garcia-Pupo L, Van San E, Delgado-Hernandez R, Vanden Berghe T, Vanden BW. Emerging immune and cell death mechanisms in stroke: saponins as therapeutic candidates. Brain Behav Immun Health. 2020;9:100152. 10.1016/j.bbih.2020.100152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.••.Parikh NS, Merkler AE, Iadecola C. Inflammation, autoimmunity, infection, and stroke: epidemiology and lessons from therapeutic intervention. Stroke. 2020;51(3):711–8. 10.1161/STROKEAHA119.024157. [DOI] [PMC free article] [PubMed] [Google Scholar]; The review highlights the complex interplay between these factors and their impact on stroke incidence, progression, and outcomes. Understanding these relationships can inform the development of effective therapeutic strategies for stroke prevention and treatment, taking into account the contributions of inflammation, autoimmunity, and infection.
- 170.Becker KJ, Kindrick DL, Lester MP, Shea C, Ye ZC. Sensitization to brain antigens after stroke is augmented by lipopolysaccharide. J Cereb Blood Flow Metab. 2005;25(12):1634–44. 10.1038/sjjcbfm.9600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tirandi A, Sgura C, Carbone F, Montecucco F, Liberale L. Inflammatory biomarkers of ischemic stroke. Intern Emerg Med. 2023;18(3):723–32. 10.1007/s11739-023-03201-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Perry VH, Teeling J. Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol. 2013;35(5):601–12. 10.1007/s00281-013-0382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li Y, Zhu ZY, Huang TT, Zhou YX, Wang X, Yang LQ, et al. The peripheral immune response after stroke-A double edge sword for blood-brain barrier integrity. CNS Neurosci Ther. 2018;24(12):1115–28. 10.1111/cns.13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.••.Wang Z, He D, Zeng YY, Zhu L, Yang C, Lu YJ, et al. The spleen may be an important target of stem cell therapy for stroke. J Neuroinflammation. 2019;16(1):20. 10.1186/s12974-019-1400-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; The spleen is identified as a potentially significant target for stem cell therapy in the treatment of stroke, according to a study published in the Journal of Neuroinflammation. The research highlights the role of the spleen in mediating inflammatory responses and suggests that stem cell therapy targeting the spleen may provide therapeutic benefits by modulating neuroinflammation in stroke patients.
- 175.•.Yamamoto S, Matsui A, Ohyagi M, Kikutake C, Harada Y, Iizuka-Koga M, et al. In vitro generation of brain regulatory T cells by co-culturing with astrocytes. Front Immunol. 2022;13:960036. 10.3389/fimmu.2022.960036. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents a method for generating brain regulatory T cells (Tregs) in vitro by co-culturing T cells with astrocytes. The findings suggest that the interaction between T cells and astrocytes plays a crucial role in promoting the differentiation and function of brain-specific Tregs, offering insights into potential strategies for modulating immune responses in neurological disorders through the manipulation of astrocyte-T cell interactions.
- 176.Becker KJ. Activation of immune responses to brain antigens after stroke. J Neurochem. 2012;123(2):148–55. 10.1111/j.1471-4159.2012.07953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Vogelgesang A, Dressel A. Immunological consequences of ischemic stroke: immunosuppression and autoimmunity. J Neuroimmunol. 2011;231(1-2):105–10. 10.1016/jneuroim.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 178.Stevenson R, Samokhina E, Rossetti I, Morley JW, Buskila Y. Neuromodulation of glial function during neurodegeneration. Front Cell Neurosci. 2020;14:278. 10.3389/fncel.2020.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Henon H, Pasquier F, Durieu I, Pruvo JP, Leys D. Medial temporal lobe atrophy in stroke patients: relation to pre-existing dementia. J Neurol Neurosurg Psychiatry. 1998;65(5):641–7. 10.1136/jnnp.65.5.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sun Y, Yin XS, Guo H, Han RK, He RD, Chi LJ. Elevated osteopontin levels in mild cognitive impairment and Alzheimer’s disease. Mediators Inflamm. 2013;2013:615745. 10.1155/2013/615745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kliper E, Bashat DB, Bornstein NM, Shenhar-Tsarfaty S, Hallevi H, Auriel E, et al. Cognitive decline after stroke: relation to inflammatory biomarkers and hippocampal volume. Stroke. 2013;44(5):1433–5. 10.1161/STROKEAHA111000536. [DOI] [PubMed] [Google Scholar]
- 182.Doyle KP, Quach LN, Sole M, Axtell RC, Nguyen TV, Soler-Llavina GJ, et al. B-lymphocyte-mediated delayed cognitive impairment following stroke. J Neurosci. 2015;35(5):2133–45. 10.1523/JNEUROSCI.4098-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.•.Lemaitre P, Tareen SH, Pasciuto E, Mascali L, Martirosyan A, Callaerts-Vegh Z, et al. Molecular and cognitive signatures of ageing partially restored through synthetic delivery of IL2 to the brain. EMBO Mol Med. 2023;15(5):e16805. 10.15252/emmm.202216805. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that synthetic delivery of interleukin-2 (IL2) to the brain partially restores molecular and cognitive signatures of aging. The findings suggest that IL2 treatment has the potential to mitigate age-related molecular changes and improve cognitive function, highlighting its therapeutic potential for combating age-related cognitive decline.
- 184.Wen H, Weymann KB, Wood L, Wang QM. Inflammatory signaling in post-stroke fatigue and depression. Eur Neurol. 2018;80(3-4):138–48. 10.1159/000494988. [DOI] [PubMed] [Google Scholar]
- 185.Davis HE, McCorkell L, Vogel JM, Topol EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol. 2023;21(3):133–46. 10.1038/s41579-022-00846-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mijajlovic MD, Pavlovic A, Brainin M, Heiss WD, Quinn TJ, Ihle-Hansen HB, et al. Post-stroke dementia - a comprehensive review. BMC Med. 2017;15(1):11. 10.1186/s12916-017-0779-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Liesz A, Kleinschnitz C. Regulatory T cells in post-stroke immune homeostasis. Transl Stroke Res. 2016;7(4):313–21. 10.1007/s12975-016-0465-7. [DOI] [PubMed] [Google Scholar]