

Abstract
Convergent synthetic pathways were devised for efficient synthesis of a series of uniformly 13C labeled polycyclic aromatic hydrocarbons de novo from U-13C-benzene and other simple commercially-available 13C-starting compounds. All target products were obtained in excellent yields, including the alternant PAH U-13C-naphthalene, U-13C-phenanthrene, U-13C-anthracene, U-13C-benz[a]anthracene, U-13C-pyrene and the nonalternant PAH U-13C-fluoranthene.
Introduction
The biological and spectroscopic properties of polycyclic aromatic hydrocarbons (PAH) are a focus of attention in physico-chemical, toxicological, analytical, and environmental studies. PAH labeled with 13C have been useful in investigations encompassing such diverse areas as NMR hyperfine shifts and coupling mechanisms,1a–d metabolism, biological activity2 and environmental distribution, such as transport and aging of PAH in sediments.3 PAH have been identified as mutagenic and carcinogenic pollutants in many of the National Priorities List sites in the US4 and bioremediation has gained attention as an approach to degrading PAH in the environment, especially those PAH with five or fewer fused rings.5a–e The use of stable isotope probing to identify microorganisms which deplete PAH in the environment through metabolic transformation6a–d relies on the availability of PAH substrates highly enriched in 13C. However, such substrates are not commonly available but require de novo synthesis from a very limited pool of 13C labeled starting materials, severely limiting application of this technique. Currently, naphthalene is the only PAH available commercially as the U-13C isotopomer.
We report synthesis of the series of alternant U-13C-labeled PAH: naphthalene, phenanthrene, anthracene, benz[a]anthracene and pyrene and the nonalternant U-13C-labeled PAH fluoranthene in high yield from U-13C-benzene. By employing appropriate 13C-labeled and natural abundance (NA) isotopomers as starting materials, PAH partially 13C-labeled at specific sites can also be synthesized using the schemes presented below for use in spectroscopic studies or investigations of metabolism or biosynthesis. Multiply 13C-labeled PAH and derivatives are also valuable as standards in developing highly sensitive and specific isotope dilution mass spectrometric quantification protocols in bioanalytical or environmental analyses.
Results and discussion
Although there are a large number of classical methods for building PAH ring systems,7 the limited number of commercially available U-13C starting chemicals severely restricts the freedom of design of synthetic pathways. The high cost of the labeled starting compounds also demands that synthetic routes be efficient with high overall yields. U-13C-Tetralone (1), accessible through classical Haworth synthesis from commercially available U-13C-succinic anhydride and U-13C-benzene (Scheme 1), was chosen as the most versatile intermediate for the preparation of all target PAH. The route to each target was optimized by synthesis on the appropriate scale using NA isotopomers. Optimization of yields is critical to identifying practical routes and thus, while a number of synthetic routes are based on classical reactions, the reaction conditions and choice of catalyst established in this study are specifically geared towards application to PAH and are thus a significant addition to the arsenal of techniques available.
Scheme 1.
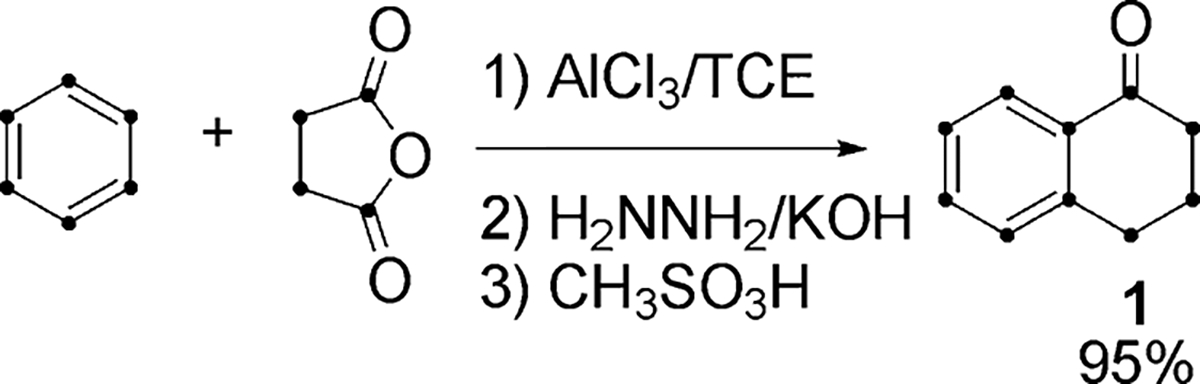
U-13C-anthracene (6)
For the synthesis of anthracene (Scheme 2), tetralone was first reduced to tetralin (2) for Friedel–Crafts acylation. Among many procedures for the reduction, Li–NH3 reduction was initially selected because of the mild reaction conditions and reported high yield;8 however, our yields were variable and inconsistent. This observation is in accord with a report of multiple products from the reduction8a involving partial reduction of the benzene ring, reduction of the carbonyl group and dimerization. In comparison, Wolff–Kishner reduction gave yields greater than 90% consistently.
Scheme 2.
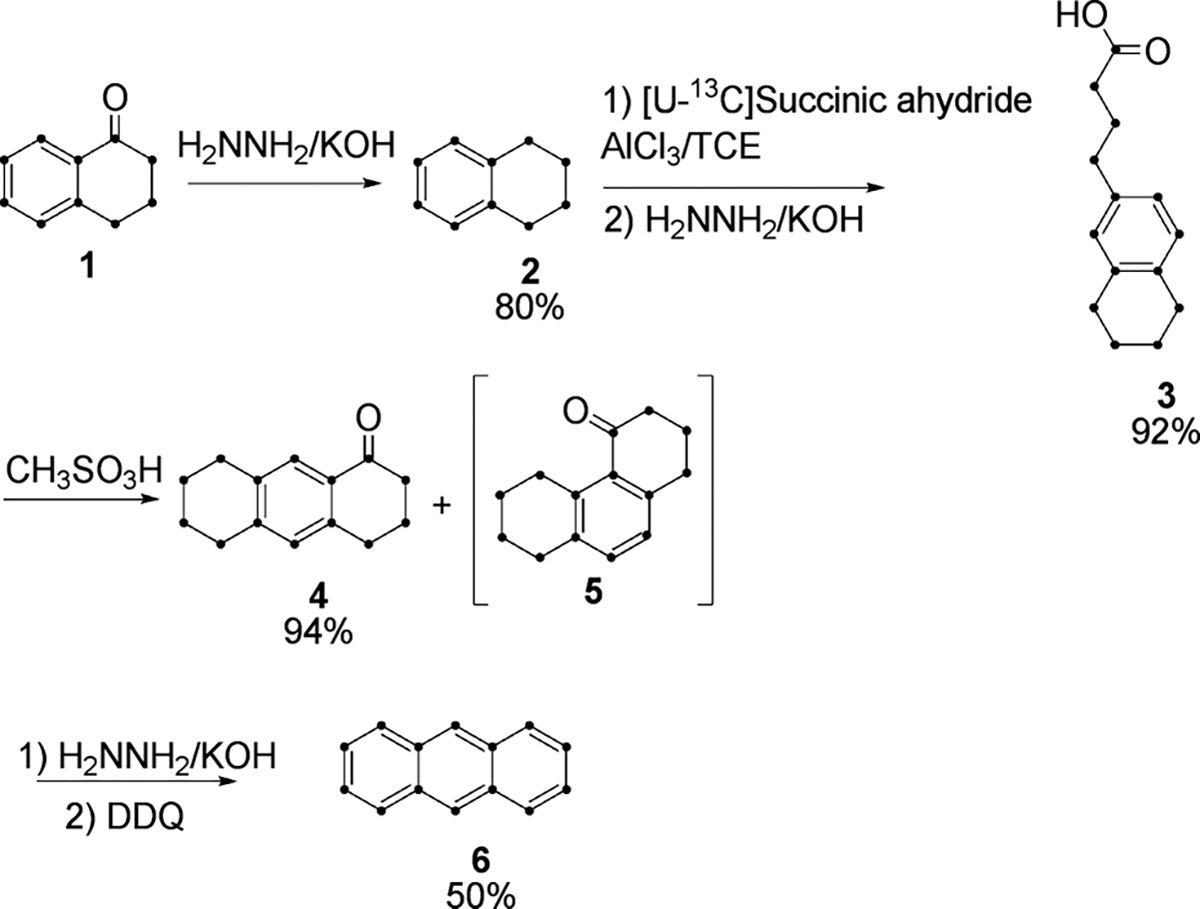
Friedel–Crafts acylation of 2 with succinic anhydride and a second Wolff–Kishner reduction yielded regiospecific substitution of a 4-carbon unit at C6 of tetralin (3).9 Cyclization of 3 by treatment with H2SO4, PPA, and/or combination of PCl5, SnCl4 and AlCl3 has been reported10a–d to give a mixture of 4 and its angular isomer 5 which are difficult to separate. However, cyclization with methanesulfonic acid (MSA)11 gave the desired linear isomer 4 in high yield when reaction time was less than 30 min. The proportion of angular isomer 5 increases with increasing reaction time, which is consistent with the reported reversibility of Friedel–Crafts acylation,12a–c and 4 being the kinetically favored product. Reduction of 4 followed by aromatization with DDQ gave 6. A small amount of contaminating phenanthrene was removed by recrystallization, for a final overall yield of 16% in 9 steps from U-13C-benzene.
U-13C-phenanthrene (11) and U-13C-pyrene (14)
Compounds 11 and 14 have been prepared6c,d from U-13C-naphthalene (7) via the Haworth synthesis. However, Friedel–Crafts succinoylation of 7 under the reported conditions leads to a mixture of 2,3-dihydrophenanthren-4(1H)-one (8) and 3,4-dihydrophenanthren-1(2H)-one in a 1 : 2 ratio. This route is thus inefficient when pyrene is the desired target, since only the minor product (8) can be utilized and separation of the ketones is difficult. Therefore, the alternative pathway described in Scheme 3 was devised.
Scheme 3.
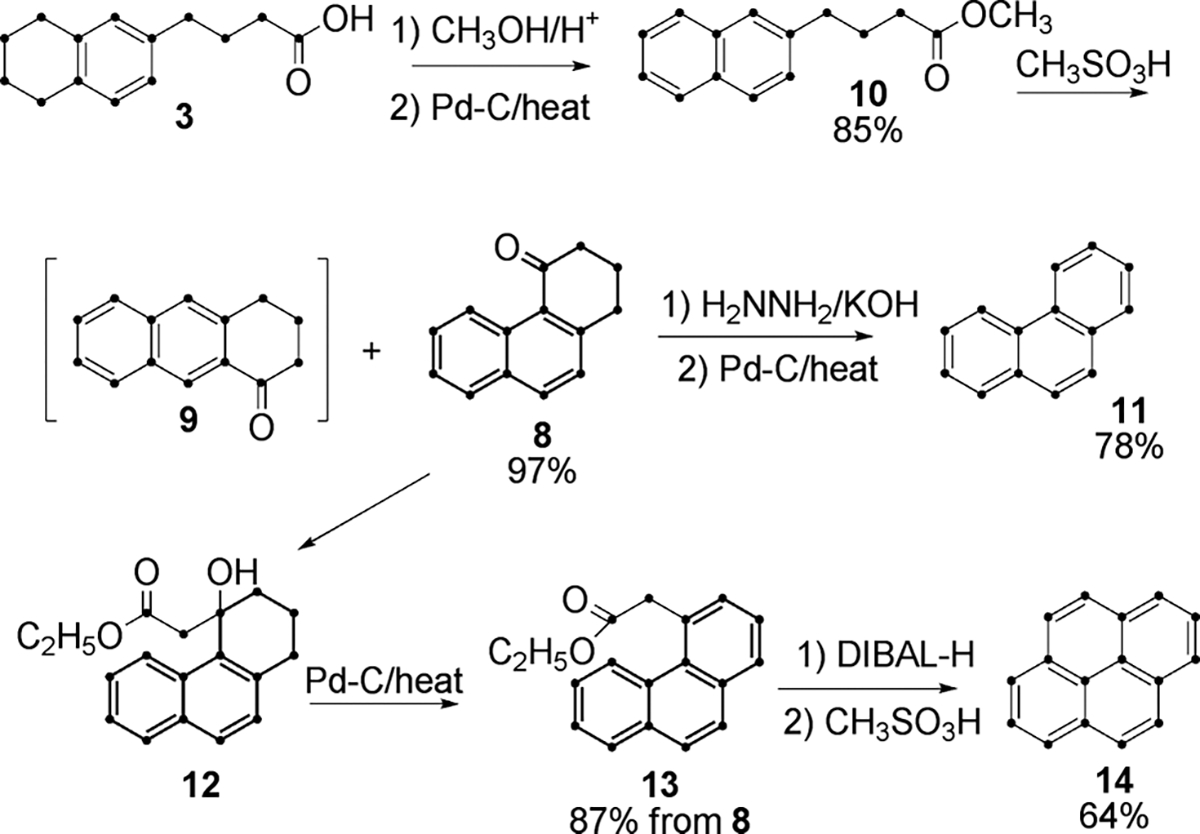
Esterification of 3 in methanol followed by aromatization with Pd–C yielded 10. Cyclization of fully aromatized naphthalene derivative 10 in MSA afforded 8 along with only ~ 3% of linear ketone 9 based on 1H NMR analysis. Transformation of 8 to 11 was straightforward, with anthracene side product removed by crystallization to give 11 in 67% overall yield from 8.
This convenient route to 8 also facilitated the synthesis of U-13C-pyrene. In the published route to pyrene from 6,6d the two-carbon K-region unit was added by a Reformatsky reaction with ethyl bromoacetate. While the Reformatsky reaction is robust, yields are usually moderate, particularly when stoichiometric quantities of reagents are employed; hence this approach is inefficient for use with labeled reagents. Instead, ketone 8 was treated with the lithium enolate of ethyl acetate13a–c at low temperature based on the modified procedure of Bodine.14 The resulting hydroxyl ester 12 was aromatized to 13, which was reduced to the corresponding aldehyde then cyclized to pyrene (14) in 4 steps in 58% yield from 8.
U-13C-naphthalene (7) and U-13C-benz[a]anthracene (17)
To take advantage of convergent synthetic pathways, we developed a route to the benz[a]anthracene skeleton based on the Friedel–Crafts acylation of naphthalene with phthalic anhydride, since phthalic anhydride may be prepared by the oxidation of tetralin, tetralone or naphthalene. Tetralone (1) from Scheme 1 served as the starting point for phthalic anhydride (Scheme 4). Tetralone can be oxidized to phthalic acid (15) in moderate yield either by KMnO415 or KO2/18-crown-6 ether.16 We selected oxidation by KMnO4 as a more straightforward procedure and were able to optimize conditions to achieve a yield of 72%.
Scheme 4.
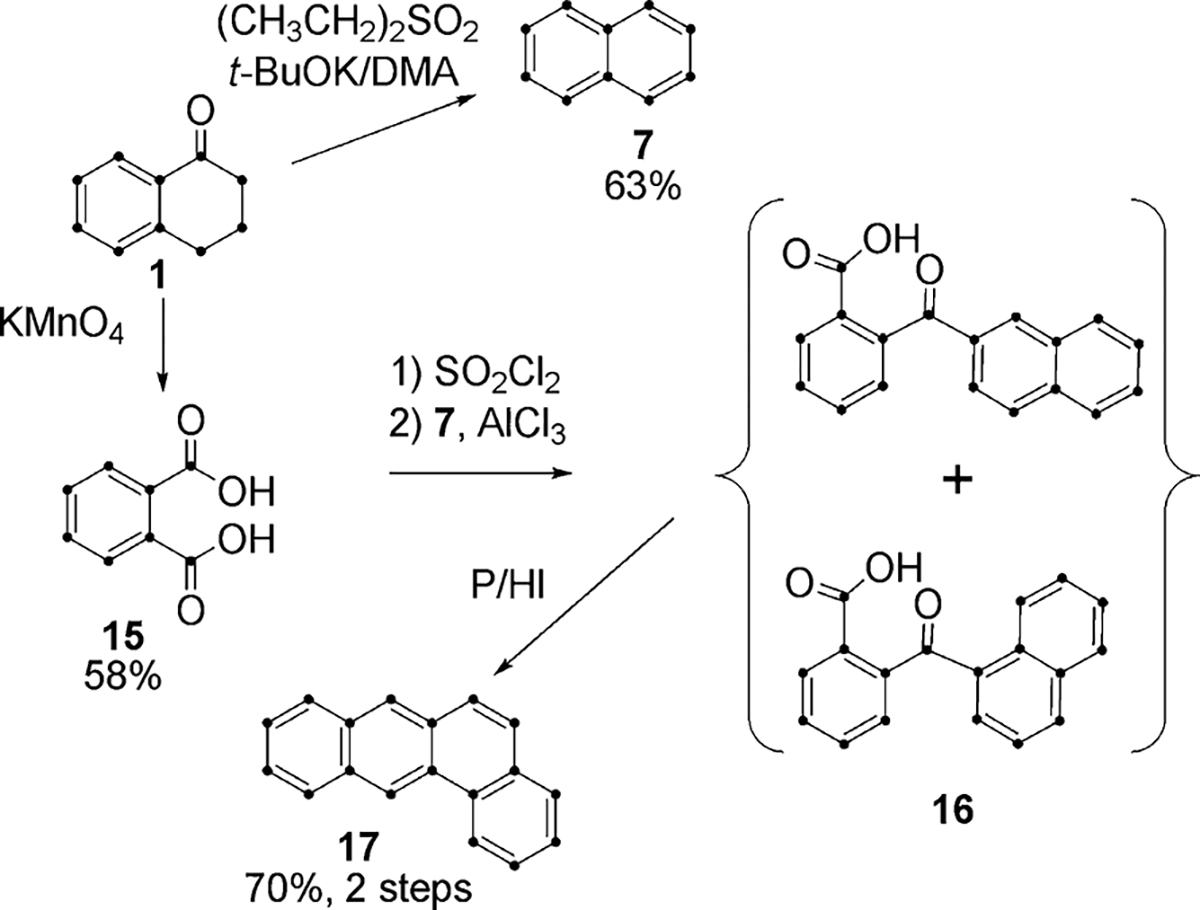
Tetralone was also the starting point for the synthesis of 7 (63%) in one step by treatment with diethylsulfone in dimethylacetamide/t-BuOK (Scheme 4).17 Friedel–Crafts condensation of phthalic anhydride with 7 provided the mixture of isomers 16.18a,b The mixture was then reduced by hydrogen iodide and phosphorus (Scheme 4) to benz[a]anthracene (17) in 70% overall yield.19
U-13C-fluoranthene (23)
Many of the published7 syntheses of the nonalternant PAH fluoranthene are based on fluorene or acenaphthylene, which are not available as U-13C isotopomers. Recently Larock20 and Meijere21 reported the direct coupling of substituted benzene and naphthalene to construct the unique 5-membered ring in nonalternant PAH. Pathways A and B in Scheme 5 apply this strategy starting with tetralone.
Scheme 5.
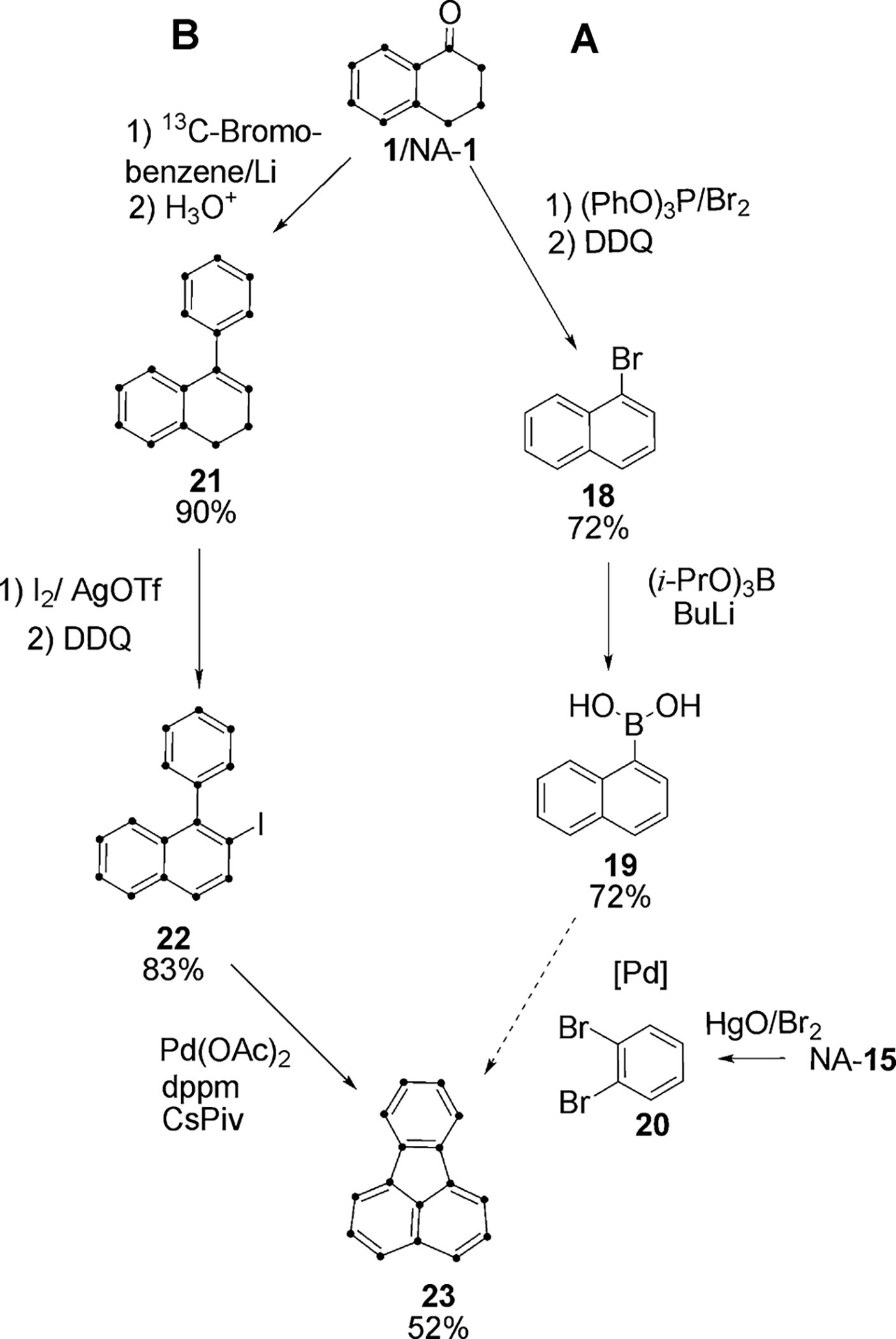
In pathway A, 1-naphthylboronic acid (19) for the coupling reaction could readily be obtained from 1-bromonaphthalene (18),22 which was prepared via bromination of NA tetralone23 followed by oxidation with DDQ. However, Hunsdiecker reactions with NA phthalic acid did not give consistent yields of o-dibromobenzene (20), the coupling partner.24a,b Consequently, we pursued pathway B, in which U-13C-bromobenzene was treated with Li to generate phenyllithium for coupling with 1, followed by dehydration to afford 4-phenyl-1,2-dihydronaphthalene (21).25a,b Iodination and dehydrogenation of 21 to 2-iodo-1-phenylnaphthalene (22) was followed by Pd-catalyzed migration and coupling to furnish fluoranthene (23) in 52% yield.
Conclusions
We have developed synthetic schemes to the U-13C alternant PAH: naphthalene, phenanthrene, anthracene, benz[a]anthracene and pyrene and the nonalternant PAH fluoranthene in quantities ranging from mg to >100 mg. By selection of appropriately labeled and NA starting compounds, the same routes can be used to synthesize compounds partially labeled with 13C at specific sites that will be useful in spectroscopic, toxicological, biological and analytical applications. Key features in the schemes are the optimization of reaction conditions to provide target compounds in high yield and the selection or modification of reaction conditions and catalysts to direct intramolecular cyclizations on the aromatic periphery to yield either angular or linear systems in order to avoid inefficient separation of mixtures, with consequent decrease in overall yield. Synthesis of the catacondensed pyrene and nonalternant fluoranthene structures represented a particular challenge. The pathways described here can be used as a basis for extending synthetic efforts to additional 13C-labeled aromatic ring systems.
Experimental section
General:
All reactions were first carried out with NA starting materials in order to optimize the reaction conditions. The NMR spectra of NA intermediates either matched those of authentic compounds or were consistent with assigned structures (see electronic supplementary information). Preparation of the target U-13C-PAH and key intermediates are presented in this section. All other procedures and characterization data are included as electronic supplementary information.
All labeled chemicals were purchased from Cambridge Isotope Laboratories, Inc. NMR spectra were recorded in chloroform-d unless otherwise noted. Chemical shift assignments for U-13C compounds are based on 1H and 13C NMR shifts reported for the NA compounds and resolved splittings.
U-13C-3,4,5,6,7,8-Hexahydroanthracen-1(2H)-one (4):
3 (575 mg, 2.4 mmol) was added to MSA (15 mL) and the mixture heated at 90 °C for 30 min under a nitrogen atmosphere. The mixture was then poured into ice water and extracted with ether (3 × 40 mL). The combined extracts were washed with dilute aqueous NaHCO3 (20 mL), water (2 × 15 mL) and brine (15 mL), then dried over anhydrous MgSO4. After filtration, the solvent was removed to yield a viscous liquid, which solidified upon standing (486 mg, 94% yield). Mp 44–46 °C. 1H NMR26 (300 MHz) δ 7.71 (bd, J = 153.9 Hz, 1H, H9), 6.91 (bd, J = 155.0 Hz, 1H, H10), 2.84 (bd, J = 128.0 Hz, 2H, C4H2), 2.74 (bd, J = 126.0 Hz, 4H, C5H2, C8H2), 2.58 (bd, J = 130.7 Hz, 2H, C2H2), 2.06 (bd, J = 130.5 Hz, 2H, C3H2), 1.75 (bd, J = 144.3 Hz, 4H, C6H2, C7H2). 13C NMR27 (75 MHz) δ 190.0 (dd, J = 47.9, 40.8 Hz, C1), 143.3–145.4 (m, C4a), 141.9 (dd, J = 95.2, 53.5 Hz, C10a), 136.2 (dd, J = 95.5, 54.8 Hz, C9a), 130.6–126.9 (m, C8a, C9, C10), 40.3–39.0 (m, C2H2), 30.8–28.8 (m, C4H2, C5H2, C8H2), 24.5–22.6 (m, C3H2, C6H2, C7H2).
U-13C-Anthracene (6):
U-13C-1,2,3,4,5,6,7,8-octahydroanthracene (112 mg, 0.56 mmol) was dissolved in benzene (10 mL); DDQ (520 mg, 2.3 mmol) was added and the mixture was refluxed for 1 h. After cooling to rt, the mixture was filtered and washed with benzene. The combined filtrate was concentrated and purified by chromatography (Al2O3, benzene) to give a white solid. (58 mg, 50%). Mp 203–205 °C. GC-EIMS m/z 192 (M+, 100). UV-Vis λmax(ethanol) 251.5, 340.5, 357.0, 375.0 nm. 1H NMR27 (300 MHz) δ 8.44 (bd, J = 154.6 Hz, 2H, H9, H10), 8.00 (bd, J = 155.1 Hz, 4H, H1, H4, H5, H8), 7.48 (bd, J = 161.1 Hz, 4H, H2, H3, H6, H7). 13C NMR (100 MHz)1c δ 133.1–130.8 (C4a, C8a, C9a, C10a), 129.5–127.0 (m, C1, C4, C5, C8), 127.0–125.0 (m, C2, C3, C6, C7, C9, C10).
U-13C-Phenanthrene (11):
U-13C-1,2,3,4-tetrahydrophenanthrene (224 mg, 1.1 mmol) was dissolved in triethylene glycol dimethyl ether (5 mL), Pd–C (60 mg, 10% on charcoal) was added. The mixture was heated to 240 °C under an argon atmosphere for 4 h. After cooling to rt, the reaction mixture was diluted with benzene (50 mL) and washed with water (10 × 15 mL) then brine (15 mL), and dried over anhydrous Na2SO4. The solvent was removed and the residue crystallized from ethanol to give 11 (172 mg, 78% yield). Mp 98–99 °C. GC-EIMS m/z 192 (M+, 100). UV-Vis. λmax (ethanol) 250.0, 274.0, 292.0 nm. 1H NMR1c (500 Hz) 8.70 (bd, J = 160.2 Hz, 2H, H4, H5), 7.89 (bd, J = 147.8 Hz, 2H, H1, H8), 7.74 (bd, J = 162.7 Hz, 2H, H9, H10), 7.66 (bd, J = 160.2 Hz, 2H, H2, H7), 7.60 (bd, J = 162.8 Hz, 2H, H3, H6) 13C NMR1c (100 MHz) δ 132.0–131.4 (m, C8a, C10a), 131.1–129.9 (m, C4a, C4b), 129.2–127.7 (m, C1, C8), 127.3–126.1 (m, C2, C3, C6, C7, C9, C10), 123.4–122.2 (m, C4, C5).
Ethyl U-13C-2-(phenanthren-4-yl)acetate (13):
To a mixture of N-isopropylcyclohexylamine (0.75 mL, 4.5 mmol) in anhydrous THF (5 mL) cooled at −78 °C under Ar, butyllithium (1.6 M in hexane, 2.5 mL, 4.0 mmol) was added. After stirring at −78 °C for 10 min, (1,2)-13C2-ethyl acetate (500 mg, 5.5 mmol) in anhydrous THF (2 mL) was added dropwise. After addition was complete, stirring was continued for 15 min. Compound 8 (500 mg, 2.45 mmol) in THF (5 mL) was added. The mixture was stirred for 1 h. Concd HC1 (1 mL) in 10 mL of THF was added at −78 °C to quench the reaction. The mixture was allowed to warm to room temperature and partitioned into water (50 mL) and ether (50 mL). The aqueous layer was extracted with ether (2 × 25 mL) and the combined extracts were washed with 5% HC1 (2 × 15 mL), water and brine, then dried over anhydrous MgSO4. Removal of the ether afforded 12 as a yellowish oil. The crude product was dehydrogenated as described above for 11 to give 13 (580 mg, 87% in 2 steps). 1H NMR28 (400 MHz) δ 8.56 (bd, J = 152.6 Hz, 1H, H5′), 7.87 (bd, J = 157.0 Hz, 1H, H8′), 7.81 (bd, J = 159.4 Hz, 1H, H1′), 7.68 (bd, J = 157.8 Hz, 2H, H9′, H10′), 7.52 (bd, J = 160.6 Hz, 4H, H2′, H3′, H6′, H7′), 4.40 (dd, J = 128.6, 6.8 Hz, 2H, ArCH2), 4.19 (dq, J = 7.2, 3.5 Hz, 2H, OCH2), 1.26 (t, J = 7.1 Hz, 3H, CH3). 13C NMR28 (100 MHz) δ 171.9 (d, J = 59.8 Hz, CO), 135.0–129.6 (m, C4′, C4a′, C4b′, C8a′, C10a′), 129.5–125.6 (m, C1′–C3′, C5′–C8′, C9′, C10′), 44.3 (dd, J = 59.6, 40.2 Hz, ArCH2).
U-13C-Pyrene (14):
To a solution of 13 (560 mg, 2 mmol) in dry toluene (30 mL), diisobutylaluminium hydride (4 mL, 1 M in hexane) was added at −78 °C under Ar. The reaction mixture was stirred for 1 h at −78 °C and the yellowish complex was hydrolyzed by the addition of conc HC1 (1 mL) in THF (9 mL). After warming to rt, the reaction mixture was washed with dilute HCl and the organic layer dried over anhydrous MgSO4. Removal of the toluene gave the aldehyde as a pale yellow oil. The crude product was dissolved in anhydrous CH2Cl2 (8 mL) and cooled in an ice bath, MSA (5 mL) was added slowly and the mixture was stirred at rt for 30 min. The mixture was then poured into ice and extracted with CH2Cl2 (3 × 25 mL). The extracts were washed with water and brine, then dried over anhydrous Na2SO4. After removal of solvent, the residue was further purified by chromatography (SiO2, hexane) to give pyrene (290 mg, 64% in two steps). Mp 139–140 °C. GC-EIMS m/z 218 (M+, 100). UV-Vis λmax(ethanol) 231.0, 240.0, 262.0, 272.0, 306.0, 319.0, 334.0 nm. 1H NMR1c,d (400 MHz) δ 8.16 (bd, J = 157.5 Hz, 4H, H1, H3, H6, H8), 8.05 (bd, J = 158.8 Hz, 4H, H4, H5, H9, H10), 7.98 (bd, J = 161.5 Hz, 2H, H2, H7). 13C NMR1c,d (100 MHz) δ 132.3–130.5 (m, C3a, C5a, C8a, C10a), 128.3–126.7 (m, C4, C5, C9, C10), 126.3–125.2 (m, C1–C3, C6–C8, C3a1, C5a1).
U-13C-Naphthalene (7):
U-13C-Tetralone (243 mg, 1.6 mmol) and diethyl sulfone (1.02 g, 8 mmol) were mixed in DMA (5 mL), t-BuOK (1.8 g, 16 mmol) was added and the mixture heated under argon at 150 °C over night. The reaction mixture was cooled and partitioned into water (25 mL) and ether (50 mL). The organic layer was washed extensively with water (5 × 15 mL) and brine (10 mL) and dried over anhydrous MgSO4. After evaporation of solvent, the residue was purified by chromatography (SiO2, hexane) to afford 7 (135 mg, 63% yield). Mp. 80–82 °C. GC-EIMS m/z 138 (M+, 100). UV-Vis λmax(ethanol) 220.0, 266.0, 275.0. 1H NMR29,30a,b (300 MHz) δ 7.84 (bd, J = 160.9 Hz, 4H, H1, H4, H5, H8), 7.49 (bd, J = 158.9 Hz, 4H, H2, H3, H6, H7) 13C NMR30a,b (75 MHz) δ 134.4–133.3 (m, C5a, C8a), 129.2–127.4 (m, C1, C4, C5, C8) 127.8–125.7 (m, C2, C3, C6, C7).
U-13C-Phthalic acid (15):
Tetralone (255 mg, 1.64 mmol) was mixed with water (20 mL), KMnO4 (1.44 g, 9.11 mmol) and KOH (510 mg, 9.1 mmol) and the mixture refluxed overnight. After cooling, methanol (2 mL) was added, followed by concd HCl (10 mL). The clear solution was extracted with ether and the extract was evaporated to afford a solid, which was washed with methylene chloride to give 15 (164 mg, 58%). 1H NMR (300 MHz, DMSO) δ 7.76 (bd, J = 158.4 Hz, 2H, H3, H6), 7.63 (bd, J = 162.6 Hz, 2H, H4, H5).
U-13C-Benz[a]anthracene (17):
A mixture of 16 and its α-isomer (160 mg, 0.92 mmol), was dissolved in HI (5 mL) and HOAc (10mL) and refluxed for 2 d. After cooling, the mixture was poured into water (100 mL) and extracted with CH2Cl2 (3 × 40 mL). The extract was washed with aqueous sodium sulfite (2 × 10 mL), brine and dried over anhydrous Na2SO4. After evaporation of solvent, the residue was purified by chromatography (SiO2, hexane) to give 17 (70% in two steps). Mp 151–152 °C. GC-EIMS m/z 246 (M+, 100)UV-Vis. λmax (ethanol)220.0,256.0,268.0,277.0,287.0,299.5, 327.5, 341.5, 357.0 nm. 1H NMR31 (500 MHz) δ 9.16 (bd, J = 161.3 Hz, 1H, H12), 8.83 (bd, J = 158.3 Hz, 1H, H1), 8.36 (bd, J = 159.1 Hz, 1H, H7), 8.11 (bd, J = 152.9 Hz, 1H, H8), 8.05–8.03 (bd, J = 167.5 Hz, 1H, H11), 7.91–7.34 (m, 7H, H2–H6, H9, H10). 13C NMR32 (75 MHz) δ 133.4–131.5 (m, C4a, C6a, C7a), 131.5–130.1 (m, C12a, C12b), 130.1–128.2 (m, C4, C8, C11a), 128.2–126.7 (m, C2, C3, C5–C7, C11), 126.7–124.8 (m, C9, C10), 123.8–122.5 (m, C1), 122.5–121.2 (m, C12).
U-13C-Fluoranthene (23):
To a solution of 22 (346 mg, 1 mmol) in DMF (8 mL), Pd(OAc)2 (23 mg, 0.1 mmol), 1,1-bis(diphenylphosphino)methane (dppm) (40 mg, 0.2 mmol) and CsO2CCMe3 (468mg,0.2mmol) were added. The reaction mixture was stirred under argon at 110 °C for 12 h. After cooling, the reaction mixture was diluted with ether (100 mL) and partitioned into water. The aqueous layer was extracted with ether (25 mL) and the combined extracts were washed with HCl (0.5 M, 15 mL), water (10 mL) and brine (15 mL) and dried over anhydrous MgSO4. After evaporation of solvent, the residue was purified by chromatography (SiO2, hexane) to give 23 (112 mg, 52% yield). Mp 103–105 °C GC-EIMS m/z 218 (M+, 100). UV-Vis λmax (ethanol) 235.0, 276.5, 281.5, 286.5, 322.5, 342.0, 358.0 nm. 1H NMR29 (300 MHz) δ 7.94 (bd, J = 160.8 Hz, 4H, H1, H6, H7, H10), 7.85 (bd, J = 156.2 Hz, 2H, H3, H4), 7.65 (bd, J = 158.3 Hz, 2H, H2, H5), 7.40 (bd, J = 159.9 Hz, 2H, H8, H9). 13C NMR33 (75 MHz) δ 140.1–138.7 (m, C6b, C10a), 137.9–136.0 (m, C6a, C10b), 133.3–131.4 (m, C10c), 130.1 (q, J = 54.6 Hz, C3a), 128.7–126.0 (m, C2–C5, C8, C9), 122.1–120.9 (C7, C10), 120.1 (t, J = 55.6 Hz, C1, C6).
Supplementary Material
Acknowledgements
This work was supported by NIEHS (Grant Nos. 5P30ES010126 and 5P42ES005948).
Footnotes
Electronic supplementary information (ESI) available: Reaction procedures for all intermediates not given in the text; complete 1H and 13C NMR spectra for all intermediates and target compounds.
Notes and References
- 1.(a) Janes DW and Shaw JD, Magn. Reson. Chem, 1985, 23, 787; [Google Scholar]; (b) Berger S, Bernd W and Diehl K, Magn. Reson. Chem, 1989, 27, 201; [Google Scholar]; (c) Lutnaes BF, Luthe G, Brinkman UAT, Johansen JE and Krane J, Magn. Reson. Chem, 2005, 43, 588; [DOI] [PubMed] [Google Scholar]; (d) Hanson PE, Poulsen OK and Berg A, Org. Magn. Reson, 1975, 7, 23. [Google Scholar]
- 2.Smith CJ, Charisse JW, Huang W, Maggio V, Grainger J and Patterson DG, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2002, 778, 157. [DOI] [PubMed] [Google Scholar]
- 3.Guthrie-Nichols E, Grasham A, Kazunga C, Sangaiah R, Gold A, Bortiatynski J, Salloum M and Hatcher P, Environ. Toxicol. Chem, 2003, 22, 40. [PubMed] [Google Scholar]
- 4.www.atsdr.cdc.gov.
- 5.(a) Andreoni V and Gianfreda L, Appl. Microbiol. Biotechnol, 2007, 76, 287; [DOI] [PubMed] [Google Scholar]; (b) Haritash AK and Kaushik CP, J. Hazard. Mater, 2009, 169, 1; [DOI] [PubMed] [Google Scholar]; (c) Doyle E, Muckian L, Hickey AM and Clipson N, Adv. Appl. Microbiol, 2008, 65, 27; [DOI] [PubMed] [Google Scholar]; (d) Peng RH, Xiong AS, Xue Y, Fu XY, Gao F, Zhao W, Tian YS and Yao QH, FEMS Microbiol. Rev, 2008, 32, 927; [DOI] [PubMed] [Google Scholar]; (e) Samanta SK, Singh OV and Jain RK, Trends Biotechnol, 2002, 20, 243. [DOI] [PubMed] [Google Scholar]
- 6.(a) Radajewski S, Ineson P, Parekh NR and Murrell JC, Nature, 2000, 403, 646; [DOI] [PubMed] [Google Scholar]; (b) Dumont MG and Murrell JC, Nat. Rev. Microbiol, 2005, 3, 499; [DOI] [PubMed] [Google Scholar]; (c) Singleton DR, Powell SN, Sangaiah R, Gold A, Ball LM and Aitken MD, Appl. Environ. Microbiol, 2005, 71, 1202; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Singleton DR, Sangaiah R, Gold A, Ball LM and Aitken MD, Environ. Microbiol, 2006, 8, 1736. [DOI] [PubMed] [Google Scholar]
- 7.Harvey RG, Polycyclic aromatic hydrocarbons: chemistry and carcinogenicity, Cambridge, New York: Cambridge University Press, 1991. [Google Scholar]
- 8.(a) Marcinow Z and Rabideau PW, J. Org. Chem, 1988, 53, 2117; [DOI] [PubMed] [Google Scholar]; (b) Hall SS, Lipsky SD, McEnroe FJ and Bartels AP, J. Org. Chem, 1971, 36, 2588. [Google Scholar]
- 9.(a) Newman MS and Zahm HV, J. Am. Chem. Soc, 1943, 65, 1097; [Google Scholar]; (b) Daly CM, Iddon B and Suschitzky H, J. Chem. Soc., Perkin Trans. 1, 1988, 1933. [Google Scholar]
- 10.(a) Rahman A, Vazquez AT and Khan AA, J. Org. Chem, 1963, 28, 3571; [Google Scholar]; (b) Boykin DW, Dewprashad B and Eisenbraun E, J. Org. Chem, 1990, 55, 425; [Google Scholar]; (c) Crawford M and Supanekar VR, J. Chem. Soc. C, 1966, 2252; [Google Scholar]; (d) Barton DHR, Sammes PG and Weingarten GG, J. Chem. Soc. C, 1971, 729. [Google Scholar]
- 11.(a) Premasagar V, Palaniswamy VA and Eisenbraun EJ, J. Org. Chem, 1981, 46, 2974; [Google Scholar]; (b) Isabelle ME, Wightman RH, Avdovich HW and Laycock DE, Can. J. Chem, 1980, 58, 1344. [Google Scholar]
- 12.(a) Agranat I and Shih Y-S, Synthesis, 1974, 865; [Google Scholar]; (b) Agranat I, Bentor Y and Shih Y-S, J. Am. Chem. Soc, 1977, 99, 7068; [Google Scholar]; (c) Mala’bi T, Pogodin S and Agranat I, Lett. Org. Chem, 2009, 6, 237. [Google Scholar]
- 13.(a) Rathke MW and Sullivan DF, J. Am. Chem. Soc, 1973, 95, 3050; [Google Scholar]; (b) Rathke MW, J. Am. Chem. Soc, 1970, 92, 3222; [Google Scholar]; (c) Rathke MW and Lindert A, J. Am. Chem. Soc, 1971, 93, 2318. [Google Scholar]
- 14.Bodine RS, Hylarides M, Daub GH and VanderJagt DL, J. Org. Chem, 1978, 43, 4025. [Google Scholar]
- 15.Susan AB, Ebert DA and Duncan WP, J. Labelled Compd. Radiopharm, 1979, 16, 579. [Google Scholar]
- 16.Sotiriou C, Lee W and Giese RW, J. Org. Chem, 1990, 55, 2159. [Google Scholar]
- 17.Garst ME, Dolby LJ, Esfandiari S, Okrent RA and Avey AA, J. Org. Chem, 2006, 71, 553. [DOI] [PubMed] [Google Scholar]
- 18.(a) Mahmoodi S, Russ. J. Org. Chem, 2003, 39, 1762; [Google Scholar]; (b) Fujisawa S, Oonishi I, Aoki J, Ohashi Y and Sasada Y, Bull. Chem. Soc. Jpn, 1985, 58, 3356. [Google Scholar]
- 19.Platt KL and Oesch F, J. Org. Chem, 1981, 46, 2601. [Google Scholar]
- 20.Huang Q, Campo MA, Yao T, Tian Q and Larock RL, J. Org. Chem, 2004, 69, 8251. [DOI] [PubMed] [Google Scholar]
- 21.Wegner HA, Scott LT and de Meijere A, J. Org. Chem, 2003, 68, 883. [DOI] [PubMed] [Google Scholar]
- 22.Rice JE and Cai ZW, J. Org. Chem, 1993, 58, 1415. [Google Scholar]
- 23.Spaggiari A, Vaccari D, Davoli P, Torre G and Prati F, J. Org. Chem, 2007, 72, 2216. [DOI] [PubMed] [Google Scholar]
- 24.(a) Davis JA, Herynk J, Carroll S, Bunds J and Johnson D, J. Org. Chem, 1965, 30, 415; [Google Scholar]; (b) Meyers AI and Fleming MP, J. Org. Chem, 1979, 44, 3405. [Google Scholar]
- 25.(a) Jamison TF, Lubell WD, Dener JM, Krishe MJ and Rapoport H, Organic Synthesis, 1993, 71, 220; [Google Scholar]; (b) Parhan WE and Piccirilli RM, J. Org. Chem, 1976, 41, 1268. [Google Scholar]
- 26.Boykin DW, J. Org. Chem, 1990, 55, 425. [Google Scholar]
- 27.Gobert F, Combrisson S, Platzer N and Ricard M, Org. Magn. Reson, 1976, 8, 293. [Google Scholar]
- 28.Assignments based on 4-Me-phenanthrene: Ibrom K, Kohn GW, Boeckmann KU, Kraft R, Holba-Schulz P and Ernst L, Org. Lett, 2000, 2, 4111. [DOI] [PubMed] [Google Scholar]
- 29.Pouchert CJ and Behnke J (eds), The Aldrich Library of 13C and 1H FTNMR Spectra, Aldrich Chemical: Milwaukee, WI, 1993. [Google Scholar]
- 30.(a) Kitching W, Bullpitt M, Doddrell D and Adcock W, Org. Magn. Reson, 1974, 6, 289; [Google Scholar]; (b) Seita J, Sandstrom J and Drakenberg T, Org. Magn. Reson, 1978, 11, 239. [Google Scholar]
- 31.Moody JD, Freeman JP and Cerniglia CE, Biodegradation, 2005, 16, 513. [DOI] [PubMed] [Google Scholar]
- 32.Cox RH and Levy LA, Org. Magn. Reson, 1983, 21, 173. [Google Scholar]
- 33.Ernst L, Org. Magn. Reson, 1976, 8, 161. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.