

Abstract
Background:
NAFLD caused by abnormalities in hepatic lipid metabolism is associated with an increased risk of developing HCC. The molecular mechanisms underlying the progression of NAFLD-related HCC are not fully understood. We investigated the molecular mechanism and role of KDM6B downregulation in NAFLD-related HCC after the KDM6B gene was identified using microarray analysis as commonly downregulated in mouse NAFLD-related HCC and human nonhepatitis B and nonhepatitis C viral-HCC.
Methods:
The 5-hydroxymethylcytosine levels of KDM6B in HCC cells were determined using glycosylated hydroxymethyl-sensitive PCR. Microarray and chromatin immunoprecipitation analyses using KDM6B-knockout (KO) cells were used to identify KDM6B target genes. Lipotoxicity was assessed using a palmitate-treated cell proliferation assay. Immunohistochemistry was used to evaluate KDM6B expression in human HCC tissues.
Results:
KDM6B expression levels in HCC cells correlated with the 5-hydroxymethylcytosine levels in the KDM6B gene body region. Gene set enrichment analysis revealed that the lipid metabolism pathway was suppressed in KDM6B-KO cells. KDM6B-KO cells acquired resistance to lipotoxicity (p < 0.01) and downregulated the expression of G0S2, an adipose triglyceride lipase/patatin like phospholipase domain containing 2 (ATGL/PNPLA2) inhibitor, through increased histone H3 lysine-27 trimethylation levels. G0S2 knockdown in KDM6B-expressed HCC cells conferred lipotoxicity resistance, whereas ATGL/PNPLA2 inhibition in the KDM6B-KO cells reduced these effects. Immunohistochemistry revealed that KDM6B expression was decreased in human NAFLD-related HCC tissues (p < 0.001), which was significantly associated with decreased G0S2 expression (p = 0.032).
Conclusions:
KDM6B-disrupted HCC acquires resistance to lipotoxicity via ATGL/PNPLA2 activation caused by epigenetic downregulation of G0S2 expression. Reduced KDM6B and G0S2 expression levels are common in NAFLD-related HCC. Targeting the KDM6B-G0S2-ATGL/PNPLA2 pathway may be a useful therapeutic strategy for NAFLD-related HCC.

INTRODUCTION
NAFLD, which is caused by hepatic lipid metabolism abnormalities, is associated with metabolic risk factors, such as obesity, dyslipidemia, hypertension, and diabetes1. The prevalence of NAFLD is increasing with the rise in the obese population worldwide2. Although NAFLD has a better prognosis than alcohol-associated fatty liver disease, some cases have been found to progress to NASH with fibrosis, which can lead to the development of HCC3,4. HCC is the world’s fourth most common cancer2. In recent years, the increase in the number of HBV surface antigen‑negative and HCV antibody-negative HCC (NBNC-HCC) cases has exceeded 30%1. The rise in NBNC-HCC is thought to be due to an increase in the number of patients with NAFLD5. Nagaoki et al6 reported that 14.9% of HCC cases are associated with NASH/NAFLD in patients with NBNC-HCC.
Studies on NASH-related HCC have been conducted using high-fat diet-fed and genetically modified mice7. Our group previously reported a mouse model with a knockout (KO) of the melanocortin type 4 receptor (MC4R −/− ), which develops NASH-like lesions and multiple liver tumors when fed a high-fat diet, similar to patients with obesity8. In addition, the HCC gene expression pattern from MC4R −/− mice closely resembles that of human HCC with metabolic risk factors9, suggesting that the MC4R −/− mice are a useful model for HCC with metabolic syndrome.
In hepatic cells, epigenetic mechanisms, including DNA methylation and histone modification, are crucial for regulating lipid metabolism, insulin resistance, oxidative stress response, and inflammation10,11. NAFLD has recently been linked to dynamic changes in 5-hydroxymethylcytosine (5hmC) patterns throughout the genome12. Evidence suggests that histone modification changes cause transcriptional dysregulation of lipogenic-related and glycolytic-related genes involved in the development of NAFLD and NASH12–14. However, the molecular mechanism of NAFLD-related HCC progression via epigenetic dysregulation is not fully understood.
To investigate the role of epigenetic factors in NAFLD-related HCC in this study, we first reanalyzed microarray data from mouse MC4R −/− -HCC and human NBNC-HCC9. KDM6B (lysine K-specific demethylase 6B, also known as JMJD3) was found to be a commonly downregulated gene in both HCC types. KDM6B is an epigenetic regulator, which acts as a histone H3 lysine-27 (H3K27) demethylase that induces transcriptional gene activation15. Downregulation of Kdm6b expression in the liver causes intrinsic defects in β-oxidation and, thereby, induces hepatosteatosis and glucose-insulin intolerance, suggesting that Kdm6b regulates energy homeostasis in the liver16. Here, we demonstrate the molecular mechanism by which KDM6B loss epigenetically downregulates the expression of G0S2, a selective inhibitor of adipose triglyceride lipase/patatin like phospholipase domain containing 2 (ATGL/PNPLA2), in HCC cells, leading to increased resistance to lipotoxicity. We also show that the loss of KDM6B-G0S2 is common in human NAFLD-related HCC.
METHODS
Human tissue samples
A total of 87 patients who underwent curative hepatic resection for HCC between 2006 and 2012 at Tokyo Medical and Dental University Hospital with approval of the ethics committees (permission number; G2017-018) of the Faculty of Medicine in Tokyo Medical and Dental University were collected, and written informed consent was obtained from all patients. The patients were anonymously coded in accordance with ethical guidelines, as instructed by the Declaration of Helsinki and Istanbul.
Cell lines and reagents
Human HCC cell lines (HuH7, JHH5, HLE, and HLF) were purchased from the American Type Culture Collection (Manassas, VA) and the Human Science Research Resources Bank (Osaka, Japan). Cells were cultured in an appropriate culture medium (Supporting Information, http://links.lww.com/HC9/A541) under 5% CO2 at 37°C. These cell lines were authenticated by DNA fingerprinting of short tandem repeats (BEX Co. Ltd, Tokyo, Japan). Cells were tested with a mycoplasma contamination detection assay before the experiment. Palmitate (Merck KGaA, Darmstadt, Germany) stock solution was prepared by coupling palmitate to bovine serum albumin (BSA; Merck KGaA, Darmstadt, Germany) as previously described17. ATGL/PNPLA2 inhibitor, Atglistatin (Selleck Biotech, Houston, TX), was dissolved in DMSO and used at a concentration of 0, 10, or 50 µM18.
5hmC detection
5hmC was analyzed using the EpiMark 5hmC and 5-mC analysis kit (E333175; New England Biolabs, New England Biolabs, Ipswich, MA) according to the manufacturer’s instructions. Briefly, genomic DNA was incubated with the T4β-glucosyltransferase that protects against Msp1 digestion (CCGG) by adding a glucose moiety specifically to 5hmC19. The 5hmC was detected by glycosylated hydroxymethyl-sensitive PCR using primers designed around the analyzed Msp1 digestion sites. The primer sequences are shown in Supplemental Table S1 (http://links.lww.com/HC9/A539).
RNA interference
HuH7 and JHH5 cells were transfected with small interfering RNAs (siRNAs) for KDM6B (SASI_Hs02_00314716, SASI_Hs02_00314717; Merck KGaA), G0S2 (SASI_Hs01_00133110, SASI_Hs02_00348390), ACSL1 (SASI_Hs01_00202187, SASI_Hs01_00202188), ATGL/PNPLA2 (SASI_Hs01_00225605, SASI_Hs01_00225606), or with negative control (MISSION siRNA Universal Negative Control; Merck KGaA) using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific, Waltham, MA) at a final concentration of 50 nM.
Establishment of KDM6B-KO cell lines
The CRISPR-targeting sequences (5′-CACAGCGCCCTTCGATACGGAGG-3′) were designed based on the Optimized CRISPR Design web system (http://crispr.mit.edu/). Oligos were cloned into the gRNA Cloning Vector (plasmid #41824; Addgene Addgene, Watertown, MA) following CRISPR gRNA Synthesis Protocol20 and transfected with the hCas9 (Addgene; plasmid #41815). After transfection of the vectors into HuH7 and JHH5 cells and selection by G418, Geneticin (Thermo Fisher Scientific), several KDM6B-KO clones were obtained by limiting dilution.
Quantitative reverse transcription PCR
Total RNA from the cultured cells was extracted using the RNeasy Protect Mini Kit (Qiagen). First-strand cDNA was synthesized from total RNA using the SuperScriptIII cDNA Synthesis kit (Thermo Fisher Scientific). Quantitative reverse transcription PCR was performed using the TB Green Premix Ex Taq kit (Takara Bio, Shiga, Japan) and gene-specific primers in the StepOne real-time PCR System (Thermo Fisher Scientific). Sequences of primers are presented in Supplemental Table S1 (http://links.lww.com/HC9/A539). Relative expression of mRNA was calculated using the comparative C t method after normalization to glyceraldehyde 3-phosphate dehydrogenase expression. All experiments were conducted in triplicate and repeated at least twice.
Microarray analysis
The integrity of the obtained RNA was confirmed by using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Complementary RNA was prepared from 100 ng of total RNA from each sample with a 3′ IVT Express Kit (Affymetrix Santa Clara, CA). Hybridization and signal detection of the Gene Chip Human Genome U133 Plus 2.0 Array (Affymetrix) were performed in accordance with the manufacturer’s instructions. The microarray datasets of pairs of the KDM6B wild-type (WT) and KDM6B-KO cells were normalized using the robust multiarray average method in the R statistical software and Affy Bioconductor package.
Western blotting
Cells were washed with PBS for total cell lysis and dissolved in an RIPA buffer (Thermo Fisher Scientific) with a protease inhibitor cocktail (Merck KGaA). After SDS-PAGE, the proteins were transferred onto polyvinylidene difluoride membranes (Immobilon-P; Merck KGaA). The membranes were probed overnight with first primary antibodies for KDM6B (Cell Signaling Technology), G0S2 (Abcam, Cambridge, UK), ACSL1 (Cell Signaling Technology), ATGL/PNPLA2 (Cell Signaling Technology), α-tubulin (Santa Cruz Biotechnology, Dallas, TX), or glyceraldehyde 3-phosphate dehydrogenase (Cell Signaling Technology). Goat antirabbit or antimouse IgG (H/L) with horseradish peroxidase–labeled secondary antibodies were used for visualization using the Western ECL substrate kit (BioRad Laboratories, Hercules, CA). The antibodies used for western blotting are listed in Supplemental Table S2 (http://links.lww.com/HC9/A539).
Lipidome analysis
Lipidome analysis was performed according to the Lipidome Lab Nontargeted Lipidome Scan package (Lipidome Lab, Akita, Japan) using liquid chromatograph orbitrap mass spectrometry, as shown in Supporting Information (http://links.lww.com/HC9/A541). The raw data files were postprocessed using the Lipid Search 4.2 software (Mitsui Knowledge Industries Co. Ltd, Tokyo, Japan), which identified individual intact lipid molecules. The relative values were calculated using the ratio of the chromatographic peak area of each analyte to that of the total analyte. The annotation method corresponds to an equivalent of the “Fatty Acyl/Alkyl Level or Hydroxyl Group Level” that is defined by the Lipidomics Standard Initiative21.
Lipid quantification
Intracellular lipids were extracted using the Lipid Extraction Kit (Cell Biolabs Inc., San Diego, CA) and then solubilized in a 100 μL methanol/chloroform mixture. Triglycerides were quantified using the Lipid Quantification Kit (Fluorometric) (Cell Biolabs Inc.), according to the manufacturer’s protocol.
Lipid staining
For neutral lipid and cholesteryl ester staining, cells were fixed with 4% paraformaldehyde and stained with Oil red O (Merck KGaA) as described previously22. Lipids were quantified using the ImageJ software (National Institutes of Health).
Cell proliferation assay
Cell viability was evaluated by a WST-8 assay using the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). Briefly, cells were plated at a density of 5.0×103 cells per well in 96-well plates and cultured for 48 hours. Cells were incubated in a fresh culture medium containing 10% Cell Counting Kit-8 reagent for 2 hours under 5% CO2 at 37°C. Absorbance was evaluated at 450 nm using a spectrophotometer (iMark; BioRad Laboratories). To assess the lipotoxic effects, the cell reduction rate was determined as the ratio of viable cells of the palmitate-treated and palmitate-untreated (control) groups after 48 hours of incubation.
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed by using the ChIP-IT Express Kit (Active Motif, Carlsbad, CA) according to the manufacturer’s protocol. The antibodies used in this study were anti-H3K27me3 (Active Motif) and anti-KDM6B (Cell Signaling Technology). Anti-PanH3 (Merck KGaA) and input DNA samples were used as internal controls. The primer sequences of ChIP-PCR are listed in Supplemental Table S1 (http://links.lww.com/HC9/A539).
ATGL activity measurement
ATGL activity was assessed using the EnzChek lipase substrate (Thermo Fisher Scientific). ATGL cell extracts from 7.0×106 cells were prepared in 300 μL of enzyme reaction buffer (50 mM HEPES, pH 7.2, 100 mM NaCl, 5 mM CaCl2, 0.5 mM DTT, 2% DMSO, and 0.1% Triton X-100). The ATGL activity assay was conducted in a 96-well plate containing 100 μg of ATGL cell extracts. The EnzChek lipase substrate was added to each well to a final concentration of 1 µM to initiate the reaction at 37°C. Fluorescence (excitation: 485 nm and emission: 510 nm) was recorded every 30 seconds for 90 minutes using Varioskan LUX (Thermo Fisher Scientific).
Bioinformatics analysis
The human and mouse microarray datasets, previously deposited in Gene Expression Omnibus (GSE102083), were used for this study. For analyzing human microarray data, 32 pairs of HCC and adjacent liver tissues surgically resected from NBNC patients were extracted. The microarray data were normalized using the robust multiarray average method in R statistical software and the Affy Bioconductor package. Public transcriptome data of HCC tissues and adjacent liver tissues were obtained from The Cancer Genome Atlas (TCGA) Research Network and downloaded from the cBioPortal site. Gene set enrichment analysis was performed using MSigDB gene sets23.
Immunohistochemistry
As described previously, formalin-fixed and paraffin-embedded tissues of human and MC4R −/− mouse HCC samples were used for immunohistochemistry9. The tissue sections were incubated with primary antibodies against KDM6B (Abcam) and G0S2 (Abcam) overnight at 4°C (Supplemental Table S2, http://links.lww.com/HC9/A539). After reaction with the Histofine Simple Stain MAX PO system (Nichirei Biosciences, Tokyo, Japan) for 1 hour at 20°C, immunoreactivity was visualized with 3-3′-diaminobenzidine (Wako, Osaka, Japan), followed by counterstaining with Mayer hematoxylin (Wako). The KDM6B expression in HCC tissues was determined as high and low according to the percentage of positive cells and intensity of the nuclear staining14,24. The percentage of positive cells was scored as follows; 0, no positive cells; 1, 1%–25%; 2, 26%–50%; 3, 51%–75%; and 4, 76%–100%. Positive staining intensity was graded according to the mean density; 0, negative; 1, weak; 2, moderate; and 3, strong. KDM6B expression was divided into low (score 0–4) and high (score 5–12) based on the staining score calculated as the multiplication of 2 scores. According to a previous report, cytoplasmic G0S2 expression in HCC was also defined as high and low25. Since KDM6B and G0S2 were expressed in normal liver tissues, we also determined low expression when their expression levels in HCC tissues were lower than those in adjacent nontumor liver tissues.
Statistical analysis
Statistical analysis was performed using R statistical software and Microsoft Excel for Mac 2019. Welch t test was used to analyze the differences between the continuous values of the 2 independent groups. The χ2 test was used for the analysis of categorical variables. For all analyses, p values <0.05 were considered statistically significant.
RESULTS
KDM6B expression is downregulated in NBNC and MC4R −/− mouse HCCs
Using our previous dataset, we reanalyzed the expression levels of epigenetic regulator genes between human and MC4R −/− mouse HCC (Figure 1A, upper panels, https://www.ncbi.nlm.nih.gov/geo; accession number GSE102083). Microarray analysis detected 12 downregulated epigenetic regulator genes in human NBNC-HCC samples and 16 downregulated epigenetic regulator genes in MC4R −/− mouse HCC (Figure 1A, B). In contrast, 66 upregulated epigenetic regulator genes were found in human NBNC-HCC, and 16 upregulated epigenetic genes were found in MC4R −/− mouse HCC (Figure 1A, B). KDM6B was downregulated in human HCC tissues among the common genes detected using the microarray in this study (Figure 1A, lower panels). We used public data to confirm the findings of our microarray data. Public transcriptome data provided by TCGA Research Network showed a lower expression level of KDM6B in nonviral HCC than in adjacent liver tissues (Figure 1C, left panel, p = 7.00×10−15), which was consistent with our human nonviral (Figure 1C, middle panel, p = 4.46×10−5) and mouse MC4R −/− -HCC data (Figure 1C, right panel, p = 0.018). Immunohistochemical analysis confirmed the loss of KDM6B protein expression in human NAFLD-related HCC of the nonviral type and mouse HCC tissues (Figure 1D), both of which had low KDM6B expression in our microarray analysis.
FIGURE 1.
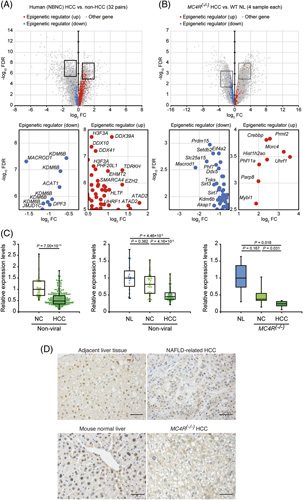
Identification of differentially expressed genes and KDM6B expression levels in human and mice. (A) Volcano plot of differentially expressed gene between NBNC-HCC and non-HCC in humans. (B) Volcano plot of differentially expressed gene between MC4R−/− HCC and MC4R(+/+) (wild-type) normal livers in mice. Representative upregulated and downregulated genes are shown in the lower panel. Blue dots indicated 21 downregulated (|log2FC| > 1.5 and FDR < 0.001) probes in humans and 19 downregulated (|log2FC| > 1 and FDR < 0.05) probes in mice, respectively. Red dots indicated 93 upregulated (|log2FC| < 1.5 and FDR > 0.001) probes in humans and 16 upregulated (|log2FC| < 2 and FDR > 0.05) probes in mice, respectively. (C) Box and beeswarm plots of KDM6B expression levels in humans and mice. The left, middle, and right panels show TCGA, human data, and mice data, respectively. Boxes represent the interquartile range (range from the 25th to 75th percentile), and horizontal lines show the median values. p values were calculated using the Mann-Whitney U test. (D) Immunohistochemical staining of KDM6B in human NAFLD-related HCC and MC4R−/− HCC tissues used in microarray analysis (Figure 2C, middle and right). NAFLD-HCC and MC4R−/− HCC exhibited low KDM6B expression compared to human liver tissue adjacent to NAFLD-HCC and normal mouse liver tissue, respectively. Magnification: ×200; scale bar: 50 μm. Abbreviations: FC, fold change; FDR, false discovery rate; MC4R, melanocortin type 4 receptor; NBNC-HCC, HBV surface antigen‑negative and HCV antibody‑negative HCC; NC, adjacent liver tissues; NL, normal liver tissues; TCGA, The Cancer Genome Atlas.
KDM6B expression correlates with 5hmC levels at the gene body region
In human HCC cell lines, KDM6B mRNA expression was high in HuH7 and JHH5 cells but low in HLE and HLF cells (Figure 2A). It has recently been reported that a gain of 5hmC at the CpG island located within exons 17 to 18 correlates with increased Kdm6b expression in mouse neural cells26 (Figure 2B). Since human genomic KDM6B contains a region with high sequence homology to the mouse putative Kdm6b regulatory region, we evaluated the 5hmC levels at the conserved region by glycosylated hydroxymethyl-sensitive PCR. HuH7 and JHH5 cells with high KDM6B expression exhibited detectable levels of 5hmC at 2 CpG sites, while HLE and HLF cells with low KDM6B expression did not, suggesting that the regulation of KDM6B expression may depend on the 5hmC levels at the region (Figure 2C).
FIGURE 2.
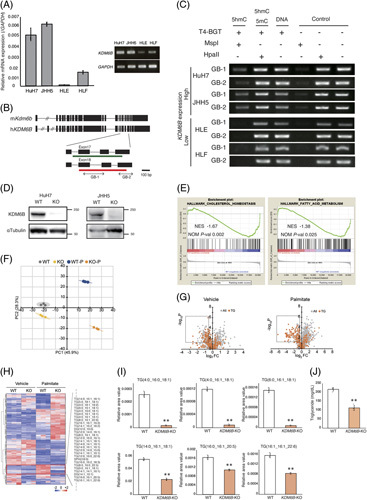
KDM6B knockout effects in HuH7 and JHH5 cells. (A) Endogenous relative expression of KDM6B in human liver cell lines. The relative mRNA expression levels were calculated using the comparative C t method and normalization to GAPDH. Data from repeated triplicate experiments were averaged and expressed as mean±SD. Each measurement is shown in a scatter plot. (B) Structure of the mouse and human KDM6B gene. Black squares indicate exons. The green bar indicates the mouse CG-rich region that is reported to show a positive correlation between KDM6B expression and 5hmC25. Red bars indicate CpG islands registered in the UCSC genome browser (https://genome.ucsc.edu/). PCR was performed at the gene body region indicated by bilateral arrows (GB-1 and GB-2). (C) Comparison of 5hmC levels in the CpG island in the gene body of KDM6B in HCC cell lines. KDM6B expression epigenetically correlates with 5hmC levels in the gene body. (D) Western blot analysis of KDM6B and α-tubulin in WT control cells and KDM6B-KO clones. (E) Gene expression analysis of KDM6B-KO HuH7 cells. Enrichment plots of gene sets associated with lipid metabolism. (F) Plot of principal component analysis of lipidome assay data using liquid chromatograph orbitrap mass spectrometry. Ellipses indicate groups of KDM6B-WT and KO cells with/without palmitate treatment and represent +3 SD. (G) Volcano plots of differentially detected metabolites between the KDM6B-WT and KDM6B-KO cells. Boxed areas contain the metabolites with significantly decreased levels in the KDM6B-KO cells (log2FC < −0.5, p < 0.05). Left panel: vehicle; right panel: treated with palmitate. (H) Heatmap of metabolites detected using lipidome assay. FC were calculated from the mean values of HuH7 KDM6B-WT and KDM6B-KO cells. p values were calculated using Welch t test. Z scores were calculated for each lipid, and principal component and clustering analyses were performed using Python with Matplotlib and Seaborn packages. (I) Candidate metabolites. Data are expressed as mean±SD. Each measurement is shown in a scatter plot (n = 3). (J) Quantification of intracellular triglyceride levels. Data are expressed as mean±SD. Each measurement is shown in a scatter plot (n = 3). ** p < 0.01. Abbreviations: FC, fold change; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GB, gene body; 5hmC, 5-hydroxymethylcytosine; hKDM6B, human KDM6B; KO, knockout; KO-P, KDM6B-knockout cells with palmitate treatment; mKdm6B, mouse Kdm6b; NES, normalized enrichment score; NOM P-val, nominal p value; PC, principal component; TG, triglyceride; WT, wild-type; WT-P, KDM6B wild-type cells with palmitate treatment.
KDM6B-KO reduces gene expression in lipid metabolism pathways and increases resistance to lipotoxicity in HCC cells
To clarify the role of KDM6B dysfunction, we established the KDM6B-KO subclone from HuH7 and JHH5 cells using the CRISPR-cas9 system (Figure 2D, Supplemental Figure S1, http://links.lww.com/HC9/A540), and then, genome-wide expression analysis between KDM6B-WT HuH7 cells and KDM6B-KO HuH7 cells by DNA microarrays was conducted. Gene set enrichment analysis demonstrated that Myc-targets, mTORC1-signaling, and metabolic pathways were suppressed when KDM6B was knocked out (Supplemental Table S3, http://links.lww.com/HC9/A539). Among them, the inhibition of gene signatures involved in the cholesterol and fatty acid metabolism pathways was noted in KDM6B-KO HuH7 cells (Figure 2E, Supplemental Table S3, http://links.lww.com/HC9/A539, normalized p values of 0.002 and 0.025, respectively). As hepatic lipid accumulation is an important feature of NAFLD1 and MC4R −/− mice8, we further studied the effects on cellular metabolites, especially lipid metabolism, due to the addition of fatty acids in KDM6B-KO cells by lipidome analysis. Principal component analysis of the 1059 molecular species revealed a clear difference in lipid profiles between KDM6B-WT and KDM6B-KO cells under vehicle and palmitate treatment (Figure 2F). Notably, metabolites that specifically reduced in KDM6B-KO cells (log2fold change < −0.5 and p < 0.05) were predominantly composed of triglyceride species (vehicle, p = 1.15×10−4; palmitate, p = 2.07×10−40, Figure 2G). In addition, a subset of 106 molecular species (including 55 molecular species corresponding to triglyceride) exhibited marked accumulation in KDM6B-WT cells compared to KDM6B-KO cells upon palmitate treatment (Figure 2H, Supplemental Table S4, http://links.lww.com/HC9/A539), with 6 representative molecular species of triglyceride exemplified in Figure 2I. We subsequently performed lipid quantification using the 2 cell lines and validated that the intracellular triglyceride level in KDM6B-KO cells was lower than that in KDM6B-WT cells under palmitate treatment (p < 0.05, Figure 2J).
Next, we investigated the biological changes in cells by adding palmitate. Oil red O staining showed lipid accumulation when KDM6B-WT HCC cells were treated with a 300 μM (HuH7) or 150 μM (JHH5) dose of palmitate27,28. Lipid accumulation in the KDM6B-KO cells was lower than that in KDM6B-WT cells (Figure 3A, p < 0.05). We then performed a cell proliferation assay to evaluate the lipotoxicity in KDM6B-KO cells treated with palmitate. Reduction rates of KDM6B-KO cells upon the addition of palmitate were significantly lower than those of WT cells (Figure 3B, p < 0.01), indicating that KDM6B-KO cells may increase resistance to lipotoxicity29. Similarly, decreased lipid accumulation and reduction rates were observed in HuH7 cells when KDM6B expression was inhibited by siRNA treatment (Supplemental Figure S2A–C, http://links.lww.com/HC9/A540).
FIGURE 3.
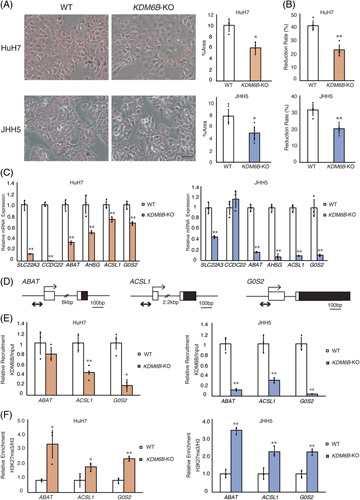
Knockout of KDM6B reduces lipid accumulation and increases lipotoxicity resistance. (A) Lipid accumulation in HuH7 and JHH5 followed by 48 hours of palmitate treatment (HuH7: 300 μM and JHH5: 150 μM). Visualization and quantification of lipids by Oil Red O staining. Scale bar: 100 μm. Five different fields were imaged by a microscope. The percentage of Oil-red-O-stained area in cells was calculated using the ImageJ software. (B) Cell proliferation evaluated by WST assay. The reduction rates of WT and KO cells were calculated by cell proliferation rate with and without palmitate treatment after 48 hours of incubation (n = 4). (C) Identification of target genes of KDM6B by epigenetic regulation. Quantitative reverse transcription PCR analysis of metabolism-related genes downregulated by KDM6B-KO in the HuH7 and JHH5 cells (n = 3). (D) The promoter regions of the 3 genes are shown. Transcriptional start sites are indicated by arrows, exons by squares, 5′ untranslated regions by white squares, and translated regions by black squares. ChIP-PCR was performed at the promoter regions indicated by bilateral arrows. (E) Quantitative ChIP analysis of candidate metabolism–related genes downregulated by KDM6B-KO in HuH7 and JHH5 cells. HuH7: left panel; JHH5: right panel. The enrichment levels of KDM6B at the promoter region were commonly decreased in AHSG, ACSL1, and G0S2 (n = 3). (F) Quantitative ChIP analysis of candidate genes regulated by KDM6B. HuH7: left panel; JHH5: right panel. The enrichment levels of H3K27me3 at the promoter region were commonly increased in ACSL1 and G0S2 (n = 3). Data from repeated experiments were performed in triplicate. All values are represented as the mean±SD. Each measurement is shown in a scatter plot. * p < 0.05; ** p < 0.01. Abbreviations: ChIP, chromatin immunoprecipitation; KO, knockout; WT, wild-type.
Increased H3K27 trimethylation at the promoter regions of G0S2 and ACSL1 in KDM6B-KO cells
Microarray analysis detected 30 downregulated (<2−2 fold) probes in the KDM6B-KO HuH7 cells when compared to that of KDM6B-WT cells (Supplemental Table S5, http://links.lww.com/HC9/A539, https://www.ncbi.nlm.nih.gov/geo; accession number GSE211848). Among them, 6 known lipid metabolism–related genes (SLC22A3, CCDC22, ABAT, AHSG, ACSL1, and G0S2) were selected, and then, mRNA expression levels were validated by quantitative reverse transcription PCR analysis. The HuH7 and JHH5 cells with KDM6B-KO showed significantly decreased expression levels of 6 genes, except CCDC22 in JHH5 cells (Figure 3C, p < 0.01). Moreover, mRNA levels of the 3 target genes (ABAT, ACSL1, and G0S2) were commonly downregulated by KDM6B knockdown in HuH7 and JHH5 cells (Supplemental Figure S2D, http://links.lww.com/HC9/A540, p < 0.05).
To clarify how KDM6B regulates downstream target gene expression, we quantified KDM6B occupancy and H3K27me3 levels at the promoter region of 3 genes (ABAT, ACSL1, and G0S2) between KDM6B-WT and KO cells. A ChIP assay using anti-KDM6B antibodies demonstrated that KDM6B was significantly recruited at the promoter region of ACSL1 and G0S2 in KDM6B-WT cells (Figure 3D, E, p < 0.01). The H3K27me3 levels at the promoter regions of ACSL1 and G0S2 were higher in KDM6B-KO cells than those in KDM6B-WT cells (Figure 3F, p < 0.05). However, KDM6B occupancy and H3K27me3 levels were changed at the ABAT promoter region in JHH5 cells but not in HuH7 cells after KDM6B-KO. These data indicate that KDM6B can bind the promoter regions of the lipid metabolism–related genes, ACSL1 and G0S2, and induce H3K27 demethylation, resulting in positive regulation of these gene expressions.
KDM6B-KO reduces lipid accumulation and gains lipotoxicity resistance by suppressing G0S2 expression
To investigate whether KDM6B-KO-mediated reduction of lipid accumulation is due to the decreased expression of G0S2 and ACSL1, we silenced the expression of G0S2 and ACSL1 by siRNA in HuH7 and JHH5 cells (Figure 4A, B, Supplemental Figure S3A, B, http://links.lww.com/HC9/A540). Lipid accumulation in HuH7 and JHH5 cells was significantly lower in cells transfected with G0S2 or ACSL1 siRNA than in cells transfected with negative control siRNA, followed by palmitate treatment (Figure 4C, Supplemental Figure S3C, http://links.lww.com/HC9/A540, p < 0.01). Suppression of G0S2 expression showed resistance to lipotoxicity (Figure 4D, p < 0.05), whereas suppression of ACSL1 expression did not (Supplemental Figure S3D, http://links.lww.com/HC9/A540). Rescued G0S2 expression in KDM6B-KO cells increased lipid accumulation and the cell reduction rate (Supporting Information, Supplemental Figure S4, http://links.lww.com/HC9/A540), suggesting that reduced lipid accumulation and enhanced resistance to lipotoxicity by KDM6B-KO may be due to the decreased expression of G0S2.
FIGURE 4.
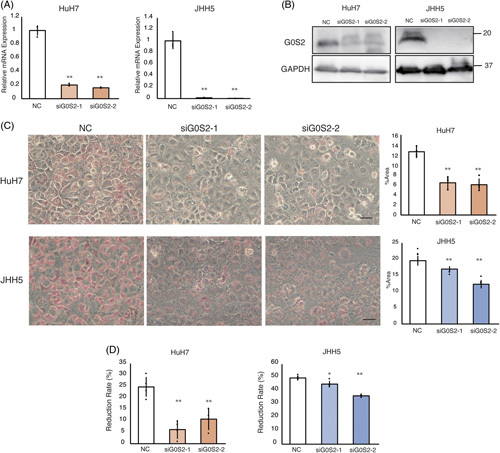
Knockdown of G0S2 reduces lipid accumulation and increases resistance to lipotoxicity. HuH7 and JHH5 cells were treated with control or G0S2 small interfering RNA at 50 nM concentration for 48 hours. (A) Changes in G0S2 mRNA levels (n = 3). (B) Western blot analysis for G0S2 and GAPDH in HuH7 and JHH5 cells. (C) Under the conditions described in (A) and (B), cells were stained with Oil Red O and evaluated for proliferation, followed by palmitate treatment for 48 hours (HuH7: 300 μM and JHH5: 150 μM). Scale bar: 100 μm. The percentage of Oil-red-O-stained area in cells was calculated from 3 representative locations using the ImageJ software. (D) Cell proliferation evaluated by WST assay. The reduction rate was calculated as described in Figure 3 (n = 5). All values are represented as the mean±SD. Each measurement is shown in a scatter plot. * p < 0.05; ** p < 0.01. Abbreviations: GAPDH, glyceraldehyde 3-phosphate dehydrogenase; NC, negative control.
Decreased lipid accumulation and acquired resistance to lipotoxicity by loss of KDM6B-G0S2 are mediated by ATGL/PNPLA2
G0S2 specifically interacts with the intracellular triglyceride hydrolyzing enzyme ATGL/PNPLA2 and inhibits the lipolytic enzyme activity of ATGL30. ATGL/PNPLA2 hydrolyzes intracellular triglycerides, stored in lipid droplets, to fatty acids31. To determine whether the reduction of lipid accumulation and acquisition of lipotoxicity by KDM6B-KO are due to enhanced ATGL/PNPLA2-dependent lipolysis caused by G0S2 downregulation, we examined the role of ATGL/PNPLA2 in HCC cells. A fluorescence-based ATGL activity assay demonstrated that ATGL activity in KDM6B-KO cells was higher than that in WT cells (Figure 5A). Similarly, G0S2-knockdown KDM6B-WT cells also increased ATGL activity compared with the negative control-transfected cells (Figure 5B).
FIGURE 5.
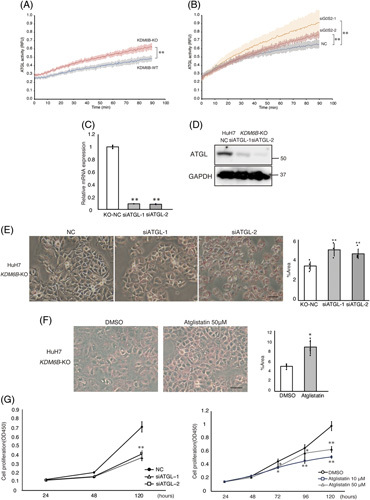
In HuH7 KDM6B-KO cells, suppression of ATGL/PNPLA2 expression, or pharmacological ATGL/PNPLA2 inhibition increases lipid accumulation and inhibits cell proliferation. (A) ATGL activity in HuH7 KDM6B-WT and KO cells. A fluorescence assay was performed to assess ATGL enzyme activity, which was recorded every 30 seconds for 90 minutes. Enzyme activity was measured twice or more for 1 sample and measured in 3 independent experiments. Solid lines indicate mean values; shaded areas indicate the 5%–95% interquartile range. Blue: HuH7 KDM6B WT and red: HuH7 KDM6B KO. ** p < 0.01. (B) ATGL activity in HuH7 KDM6B-WT cells with G0S2 knockdown. Gray: negative control small interfering RNA (NC); orange: siG0S2-1; and brown: siG0S2-2. ** p < 0.01. (C) Changes in ATGL/PNPLA2 mRNA levels. Each measurement is shown in a scatter plot (n = 3). (D) Western blot analysis of ATGL/PNPLA2 and GAPDH. (E) ATGL/PNPLA2 knockdown increases lipid accumulation analyzed by Oil Red O staining. (F) Representative images of HuH7 KDM6B-KO cells supplemented with Atglistatin, ATGL/PNPLA2 inhibitor, or DMSO, cultured for 5 days, fixed, and stained with Oil Red O are shown. Magnification: ×100; scale bar: 100 μm. The percentage of Oil-red-O-stained area in cells was calculated in representative locations using the ImageJ software. Each measurement is shown in a scatter plot (n = 5). * p < 0.05; ** p < 0.01. (G) Left panel: growth curve is plotted for HuH7 KDM6B-KO cells treated with or without small interfering RNA against ATGL/PNPLA2. Right panel: growth curve for HuH7 KDM6B-KO cells in the presence or absence of Atglistatin. Cell proliferation was inhibited in a concentration-dependent manner by Atglistatin (n = 5). All values are represented as the mean±SD. *p < 0.05; **p < 0.01. Abbreviations: ATGL, adipose triglyceride lipase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; KO, knockout; NC, Negative control; PNPLA2, patatin like phospholipase domain containing 2; WT, wild-type.
We then performed ATGL/PNPLA2 knockdown in HuH7 KDM6B-KO cells (Figure 5C, D), and the suppression of ATGL/PNPLA2 expression increased lipid accumulation in KDM6B-KO cells (Figure 5E, p < 0.01). Similarly, treatment with Atglistatin, a selective inhibitor of ATGL/PNPLA2, showed significantly higher lipid accumulation in KDM6B-KO cells compared to that in control cells (Figure 5F, p < 0.05). In contrast, ATGL/PNPLA2 knockdown or Atglistatin treatment showed significant inhibition of cell proliferation of KDM6B-KO cells compared to that in control cells (Figure 5G, p < 0.01). However, there was no apparent effect of the suppression of ATGL/PNPLA2 or Atglistatin on lipid accumulation or cell proliferation in KDM6B-WT cells (Supplemental Figure S5, http://links.lww.com/HC9/A540). These data suggest that the loss of KDM6B and resultant G0S2 downregulation decrease lipid accumulation and acquire resistance to lipotoxicity through the activation of ATGL/PNPLA2.
Low G0S2 expression in human HCC tissues is correlated with downregulated KDM6B expression
Finally, we evaluated the relationship between KDM6B and G0S2 expression in 87 human HCC specimens by immunohistochemistry. KDM6B expression was low in 25 of 87 HCC specimens (28.7%, Figure 6A, Table 1). The clinicopathological characters of the low KDM6B expression groups revealed the NBNC-HCC type (N = 20, 80%, p < 0.001). These findings were consistent with the data on our microarray and TCGA transcriptome analyses, which showed that KDM6B expression was downregulated in nonviral HCC (Figure 1C). Furthermore, the low KDM6B expression group was accompanied by NAFLD (N = 20, 80%, p < 0.001) and NASH (categorized as Matteoni type 3 and 4, N = 19, 76%, p < 0.001) that were pathologically diagnosed in the adjacent liver tissue. Moreover, adverse metabolic risk factors, such as obesity (N = 18, 72%, p < 0.001) and hypertension (N = 20, 80%, p = 0.003), were observed in the low KDM6B expression group.
FIGURE 6.
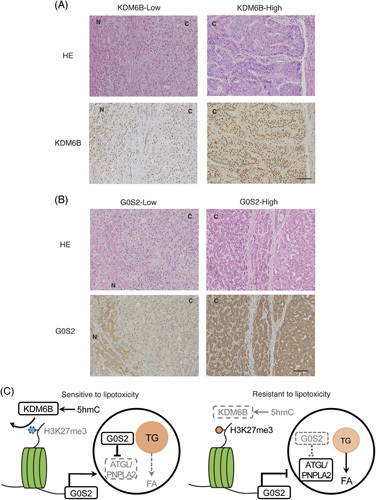
Immunohistochemistry evaluation of KDM6B and G0S2 expression. (A) HE staining and immunohistochemical analysis of KDM6B expression in HCC in representative KDM6B-high and KDM6B-low tissues. (B) Immunohistochemical staining of G0S2 in human HCC tissues. Low G0S2 expression was correlated with low KDM6B expression (p = 0.036). Magnification: ×100; scale bar: 100 μm. (C) Schematic illustration of the regulation mechanism of intracellular lipid by KDM6B-G0S2-ATGL/PNPLA2 pathway. Abbreviations: ATGL, adipose triglyceride lipase; C, cancer; FA, fatty acid; HE, hematoxylin-eosin; 5hmC, 5-hydroxymethylcytosine; N, noncancerous tissue; PNPLA2, patatin like phospholipase domain containing 2; TG, triglyceride.
TABLE 1.
Relationship between KDM6B expression and clinicopathological factors
| KDM6B expression | |||
|---|---|---|---|
| Clinicopathological factor | High (n=62) | Low (n=25) | p |
| Sex | 0.901 | ||
| Male | 43 | 17 | |
| Female | 19 | 8 | |
| Age (mean±SD) (y) | 69±11 | 69±8 | 1.000 |
| HBV | 18 | 2 | 0.035a |
| HCV | 21 | 3 | 0.038a |
| NBNC | 23 | 20 | <0.001a |
| NAFLD | 9 | 20 | <0.001a |
| NASH | 7 | 19 | <0.001a |
| Obesity | 17 | 18 | <0.001a |
| Hypertension | 28 | 20 | 0.003a |
| Diabetes | 21 | 9 | 0.553 |
| Dyslipidemia | 10 | 7 | 0.206 |
| Child-Pugh A/B | 58/4 | 25/0 | 0.190 |
| Plt (<10×104) | 10 | 2 | 0.320 |
| PT (<80%) | 14 | 9 | 0.199 |
| Alb (<3.5 g/dL) | 8 | 4 | 0.705 |
| AST (>40 IU/L) | 25 | 9 | 0.708 |
| ALT (>40 IU/L) | 20 | 9 | 0.738 |
| Tbil (>1.0 mg/dL) | 8 | 6 | 0.212 |
| ICG-15 (>15%) | 24 | 14 | 0.140 |
| AFP (>20 ng/mL) | 31 | 4 | 0.003a |
| PIVKA-II (>40 mAU/mL) | 35 | 12 | 0.474 |
| Tumor size (>5 cm) | 23 | 5 | 0.122 |
| Tumor number (>2) | 11 | 9 | 0.067 |
| Differentiation (well, moderate/poor) | 45/10 | 24/1 | 0.089 |
Statistical significance.
Abbreviations: Alb, albumin; ALT, alanine aminotransferase; AFP, alpha-fetoprotein; AST, aspartate aminotransferase; ICG-15, indocyanine green retention rate at 15 minutes; NBNC, nonhepatitis B and nonhepatitis C viral; PIVKA-II, protein induced by vitamin K absence II; Plt, platelet; PT, prothrombin time; Tbil, total bilirubin.
G0S2 expression was reduced in 26 of 85 HCC tissues (30.5%, Figure 6B), which is associated with a high tumor number (p = 0.022, Supplemental Table S6, http://links.lww.com/HC9/A539). Decreased G0S2 expression cases significantly exhibited low KDM6B expression (N = 11, p = 0.036, χ2 test, Supplemental Table S6, http://links.lww.com/HC9/A539). Moreover, the KDM6B/G0S2-double low expression group (N = 11) significantly correlated with NBNC (p = 0.020), NAFLD (p < 0.001), NASH (p < 0.001), obesity (p = 0.001), tumor size (p = 0.016), and tumor number (p = 0.009) in contrast to the KDM6B/G0S2-double high expression group (N = 47, Table 2).
TABLE 2.
Relationship between G0S2 and KDM6B expression and clinicopathological factors
| G0S2 and KDM6B | |||
|---|---|---|---|
| Clinicopathological factor | High (n=47) | Low (n=11) | p |
| Sex | 0.880 | ||
| Male | 31 | 7 | |
| Female | 16 | 4 | |
| Age (mean±SD) (y) | 70±10 | 68±8 | 1.000 |
| HBV | 12 | 1 | 0.239 |
| HCV | 17 | 1 | 0.080 |
| NBNC | 18 | 9 | 0.020a |
| NAFLD | 9 | 9 | <0.001a |
| NASH | 6 | 9 | <0.001a |
| Obesity | 13 | 9 | 0.001a |
| Hypertension | 23 | 8 | 0.154 |
| Diabetes | 17 | 5 | 0.568 |
| Dyslipidemia | 9 | 4 | 0.218 |
| Child-Pugh A/B | 34/13 | 8/3 | 0.979 |
| Plt (<10×104) | 10 | 1 | 0.353 |
| PT (<80%) | 12 | 5 | 0.191 |
| Alb (<3.5 g/dL) | 8 | 2 | 0.920 |
| AST (>40 IU/L) | 16 | 4 | 0.880 |
| ALT (>40 IU/L) | 13 | 3 | 0.980 |
| Tbil (>1.0 mg/dL) | 8 | 1 | 0.513 |
| ICG-15 (>15%) | 20 | 4 | 0.708 |
| AFP (>20 ng/mL) | 20 | 3 | 0.351 |
| PIVKA-II (>40 mAU/mL) | 29 | 9 | 0.206 |
| Tumor size (>5 cm) | 23 | 1 | 0.016a |
| Tumor number (>2) | 8 | 6 | 0.009a |
| Differentiation | 39/8 | 11/0 | 0.140 |
Statistical significance.
Abbreviations: Alb, albumin; ALT, alanine aminotransferase; AFP, alpha-fetoprotein; AST, aspartate aminotransferase; ICG-15, indocyanine green retention rate at 15 minutes; NBNC, nonhepatitis B and nonhepatitis C viral; PIVKA-II, protein induced by vitamin K absence II; Plt, platelet; PT, prothrombin time; Tbil, total bilirubin.
DISCUSSION
In this study, we observed that KDM6B expression frequently decreased in NAFLD-related HCC. We used microarray and public database analyses to demonstrate that KDM6B transcription was downregulated in nonviral type HCC. Moreover, in patients with nonviral type HCC, KDM6B expression was significantly lower than that in adjacent liver tissues, which is consistent with the results of the TCGA database. Several factors such as STAT3, HOXC9, and miRNA32,33 are known to activate KDM6B expression at the transcriptional level. We found that KDM6B expression corresponded to 5hmC levels at the KDM6B gene body region in HCC cells, suggesting that this region is important for the regulation of KDM6B expression26. A genome-wide analysis of liver 5hmC patterns in a mouse model of diet-induced obesity found that hepatic steatosis is associated with reversible 5hmC changes in numerous metabolic-related genes12. Moreover, global DNA hydroxy methylation levels in HCC are lower than those in non-neoplastic liver tissues34. Aberrant DNA demethylation may be one of the mechanisms underlying KDM6B downregulation in our HCC cases.
In this study, according to gene set enrichment analysis, the metabolic pathways of cholesterol and fatty acids were suppressed in KDM6B-KO cells. Furthermore, a subset of the lipid metabolism–related metabolites including triglyceride molecular species was remarkably different in KDM6B-WT and KDM6B-KO cells treated with palmitate by lipidome analysis, and intracellular triglyceride levels were lower in KDM6B-KO cells than in WT cells. Kdm6b expression is decreased in the livers of mice exposed to a high-fat diet35. Our results indicate that KDM6B is deeply involved in lipid metabolism, and the loss of KDM6B contributes to the development of NAFLD-related HCC. In addition, KDM6B transcriptionally activates global autophagy-network genes, such as Tfeb and Atg7, by decreasing H3K27me3 levels, thereby inducing autophagy-mediated lipid degradation in the livers of fasting mice36. Moreover, fasting-induced KDM6B epigenetically upregulates the expression of β-oxidation–promoting genes, including Fgf21, Cpt1a, and Mcad 16. Here, we identified 2 novel KDM6B downstream target genes, G0S2 and ACSL1, which are directly regulated by KDM6B-dependent H3K27me3 demethylation at their promoter regions. G0S2, identified initially as a G0/G1 switch protein 2 in lymphocytes, plays a role in terminal differentiation and cell cycle progression37. Recent studies indicate that the G0S2 protein content in the liver is increased in rats fed high-fat diets, promoting lipid accumulation by coupling with ATGL/PNPLA2 in the liver38,39; this suggests that G0S2 is a lipid metabolism regulator. ACSL1 facilitates the process of synthesizing triglycerides from fatty acids in hepatocytes40,41, especially in directing fatty acids toward mitochondrial β-oxidation42. Both G0S2 and ACSL1 knockdown inhibited lipid accumulation, but only the G0S2 knockdown demonstrated resistance to lipotoxicity. Our findings suggest that G0S2 downregulation in KDM6B-KO cells may explain the acquisition of lipotoxicity resistance.
G0S2 is a specific inhibitor of ATGL (also known as PNPLA2). ATGL/PNPLA2, a paralog of PNPLA3, is activated by binding to ABHD5, causing intracellular triglyceride hydrolysis43,44. PNPLA3 also binds to ABHD5 but functionally competes with ATGL/PNPLA2 to prevent its activation and promote intracellular triglyceride deposition44. A genome-wide association study analysis identified a genetic polymorphism of PNPLA3 as a major risk for fatty liver disease45. The PNPLA3 (I148M) variant has been shown to have a stronger functional competitive effect with ATGL/PNPLA2 than wild-type PNPLA3, resulting in the development of hepatic steatosis46,47. In our study, KDM6B-KO cells possessed a high ATGL/PNPLA2 activation level. Knockdown or pharmacological inhibition of ATGL/PNPLA2 increased lipid accumulation and decreased cell proliferation in KDM6B-KO cells. ATGL/PNPLA2 overexpression facilitates the growth of cancer cells, including HCC cells43,48. In NASH-related HCC, KDM6B loss may promote cell survival through activating ATGL (Figure 6C).
KDM6B expression patterns differ between cancer types, with downregulation in pancreatic and colon cancers but upregulation in prostate, kidney, gastric ovarian, and lung cancers, indicating that KDM6B functional roles are highly cell type–specific and pathologic context-dependent49. KDM6B expression is high in HCC with distant metastasis and has stem cell-like properties32. In our study, the high KDM6B expression group had viral infection and high alpha-fetoprotein levels (Table 1). In contrast, low KDM6B expression was correlated with NAFLD-related HCC and high metabolic risk factors, such as obesity and hypertension. Thus, the biological characteristics and mechanisms of low and high expressions of KDM6B in HCC carcinogenesis are distinct. Notably, KDM6B expression levels in primary HCC tissues were correlated with G0S2 expression levels, particularly in NAFLD-related HCC. These data indicate that KDM6B/G0S2-double low expression could be used as a biomarker for NAFLD-related HCC with resistance to lipotoxicity.
Understanding cancer metabolism, especially lipid metabolism, is critical for elucidating the mechanism of cancer development and progression and establishing new therapeutic strategies50. Our data suggest that KDM6B-deficient HCC cells confer resistance to lipotoxicity via epigenetic downregulation of G0S2 expression and subsequent ATGL/PNPLA2 activation. Therefore, targeting the KDM6B-G0S2-ATGL/PNPLA2 pathway may be a novel therapeutic strategy for NAFLD-related HCC.
Supplementary Material
Acknowledgments
DATA AVAILABILITY STATEMENT
The microarray data are available to the public using the specific Gene Expression Omnibus accession number (GSE211848). The other data available are within the article or its Supplemental Materials (http://links.lww.com/HC9/A541).
ACKNOWLEDGMENTS
The authors thank Hiromi Nagasaki and Dr. Norimichi Chiyonobu for their technical and clerical assistance. They also thank Editage (www.editage.com) for English language editing.
FUNDING INFORMATION
This work was supported by Grants-in-Aid for Scientific Research (A; 19H01055 and B; 20H03526, 23H02979), Challenging Research (Exploratory; 20K21627), and Young Scientists (23K15388) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; Research Grant from the Princess Takamatsu Cancer Research Fund; and P-CREATE (JP19cm0106540) and Program for Basic and Clinical Research on Hepatitis (JP21fk0210090, JP22fk0210102, and JP22fk0210106) from AMED (Japan Agency for Medical Research and Development).
CONFLICTS OF INTEREST
The authors have no conflicts to report.
Footnotes
Abbreviations: ATGL, adipose triglyceride lipase; ChIP, chromatin immunoprecipitation; G0S2, G0/G1 switch 2; H3K27 me3, histone H3 lysine-27 trimethylation; 5hmC, 5-hydroxymethylcytosine; KO, knockout; MC4R, melanocortin type 4 receptor; NBNC, nonhepatitis B and nonhepatitis C viral; PNPLA2, patatin like phospholipase domain containing 2; siRNA, small interfering RNA; TCGA, The Cancer Genome Atlas; WT, wild-type.
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal’s website, www.hepcommjournal.com.
Contributor Information
Megumi Hatano, Email: hatmem@tmd.ac.jp.
Yoshimitsu Akiyama, Email: yakiyama.monc@tmd.ac.jp.
Shu Shimada, Email: shimada.monc@tmd.ac.jp.
Kohei Yagi, Email: yagimsrg@tmd.ac.jp.
Keiichi Akahoshi, Email: akahmsrg@tmd.ac.jp.
Michiko Itoh, Email: mito.mem@tmd.ac.jp.
Minoru Tanabe, Email: tana.msrg@tmd.ac.jp.
Yoshihiro Ogawa, Email: yogawa@intmed3.med.kyushu-u.ac.jp.
Shinji Tanaka, Email: tanaka.monc@tmd.ac.jp.
REFERENCES
- 1.Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ, Karin M. Preclinical models for studying NASH-driven HCC: How useful are they? Cell Metab. 2019;29:18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2021;18:223–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calle E, Rodriguez C, Walker-Thurmond KTM. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N Engl J Med. 2003;348:1625–1638. [DOI] [PubMed] [Google Scholar]
- 4.Massoud O, Charlton M. Nonalcoholic fatty liver disease/nonalcoholic steatohepatitis and hepatocellular carcinoma. Clin Liver Dis. 2018;22:201–211. [DOI] [PubMed] [Google Scholar]
- 5.Oda K, Uto H, Mawatari S, Ido A. Clinical features of hepatocellular carcinoma associated with nonalcoholic fatty liver disease: A review of human studies. Clin J Gastroenterol. 2015;8:1–9. [DOI] [PubMed] [Google Scholar]
- 6.Nagaoki Y, Hyogo H, Ando Y, Kosaka Y, Uchikawa S, Nishida Y, et al. Increasing incidence of non-HBV- and non-HCV-related hepatocellular carcinoma: Single-institution 20-year study. BMC Gastroenterol. 2021;21:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farrell G, Schattenberg JM, Leclercq I, Yeh MM, Goldin R, Teoh N, et al. Mouse models of nonalcoholic steatohepatitis: Toward optimization of their relevance to human nonalcoholic steatohepatitis. Hepatology. 2019;69:2241–2257. [DOI] [PubMed] [Google Scholar]
- 8.Itoh M, Suganami T, Nakagawa N, Tanaka M, Yamamoto Y, Kamei Y, et al. Melanocortin 4 receptor-deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am J Pathol. 2011;179:2454–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiyonobu N, Shimada S, Akiyama Y, Mogushi K, Itoh M, Akahoshi K, et al. Fatty acid binding protein 4 (FABP4) overexpression in intratumoral hepatic stellate cells within hepatocellular carcinoma with metabolic risk factors. Am J Pathol. 2018;188:1213–1224. [DOI] [PubMed] [Google Scholar]
- 10.Hyun J, Jung Y. DNA methylation in nonalcoholic fatty liver disease. Int J Mol Sci. 2020;21:8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J, Kim Y, Friso S, Choi SW. Epigenetics in non-alcoholic fatty liver disease. Mol Aspects Med. 2017;54:78–88. [DOI] [PubMed] [Google Scholar]
- 12.Lyall MJ, Thomson JP, Cartier J, Ottaviano R, Kendall TJ, Meehan RR, et al. Non-alcoholic fatty liver disease (NAFLD) is associated with dynamic changes in DNA hydroxymethylation. Epigenetics. 2020;15:61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S, Woo DC, Kang J, Ra M, Kim KH, Lee SR, et al. The role of the histone methyltransferase EZH2 in liver inflammation and fibrosis in STAM NASH mice. Biology (Basel). 2020;9:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen Y, Guo X, Wang Y, Qiu W, Chang Y, Zhang A, et al. Expression and significance of histone H3K27 demethylases in renal cell carcinoma. BMC Cancer. 2012;12:470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park D, Hong S, Salinas R, Liu S, Sun S, Sgualdino J, et al. Activation of neuronal gene expression by the JMJD3 demethylase is required for postnatal and adult brain neurogenesis. Cell Rep. 2014;8:1290–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seok S, Kim YC, Byun S, Choi S, Xiao Z, Iwamori N, et al. Fasting-induced JMJD3 histone demethylase epigenetically activates mitochondrial fatty acid ß-oxidation. J Clin Invest. 2018;128:3144–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ichioka M, Suganami T, Tsuda N, Shirakawa I, Hirata Y, Satoh-Asahara N, et al. Increased expression of macrophage-inducible C-type lectin in adipose tissue of obese mice and humans. Diabetes. 2011;60:819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayer N, Schweiger M, Romauch M, Grabner GF, Eichmann TO, Fuchs E, et al. Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat Chem Biol. 2013;9:785–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nechin J, Tunstall E, Raymond N, Hamagami N, Pathmanabhan C, Forestier S, et al. Hemimethylation of CpG dyads is characteristic of secondary DMRs associated with imprinted loci and correlates with 5-hydroxymethylcytosine at paternally methylated sequences. Epigenetics Chromatin. 2019;12:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oba A, Shimada S, Akiyama Y, Nishikawaji T, Mogushi K, Ito H, et al. ARID2 modulates DNA damage response in human hepatocellular carcinoma cells. J Hepatol. 2017;66:942–951. [DOI] [PubMed] [Google Scholar]
- 21.Liebisch G, Ahrends R, Arita M, Arita M, Bowden J, Ejsing C, et al. Lipidomics needs more standardization. Nat Metab. 2019;1:745–747. [DOI] [PubMed] [Google Scholar]
- 22.Hatano M, Migita T, Ohishi T, Shima Y, Ogawa Y, Morohashi KI, et al. SF-1 deficiency causes lipid accumulation in Leydig cells via suppression of STAR and CYP11A1. Endocrine. 2016;54:484–496. [DOI] [PubMed] [Google Scholar]
- 23.Subramanian A, Tamayo P, Mootha V, Mukherjee S, Ebert B, Gillette M, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamamoto K, Tateishi K, Kudo Y, Sato T, Yamamoto S, Miyabayashi K, et al. Loss of histone demethylase KDM6B enhances aggressiveness of pancreatic cancer through downregulation of C/EBPα. Carcinogenesis. 2014;35:2404–2414. [DOI] [PubMed] [Google Scholar]
- 25.Xu X, Feng L, Liu Y, Zhou WX, Ma YC, Fei GJ, et al. Differential gene expression profiling of gastric intraepithelial neoplasia and early-stage adenocarcinoma. World J Gastroenterol. 2014;20:17883–17893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montibus B, Cercy J, Bouschet T, Charras A, Maupetit-Méhouas S, Nury D, et al. TET3 controls the expression of the H3K27me3 demethylase Kdm6b during neural commitment. Cell Mol Life Sci. 2020;78:757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao NQ, Li XY, Wang L, Feng ZL, Li XF, Wen YF, et al. Palmitate induces fat accumulation by activating C/EBPβmediated G0S2 expression in HepG2 cells. World J Gastroenterol. 2017;23:7705–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asakawa M, Itoh M, Suganami T, Sakai T, Kanai S, Shirakawa I, et al. Upregulation of cancer-associated gene expression in activated fibroblasts in a mouse model of non-alcoholic steatohepatitis. Sci Rep. 2019;9:19601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myoung SH, Sun YP, Shinzawa K, Kim S, Kun WC, Lee JH, et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J Lipid Res. 2008;49:84–97. [DOI] [PubMed] [Google Scholar]
- 30.Yang X, Lu X, Lombès M, Rha GB, Chi YI, Guerin TM, et al. The G0/G1 switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010;11:194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heckmann BL, Zhang X, Saarinen AM, Liu J. Regulation of G0/G1 switch gene 2 (G0S2) protein ubiquitination and stability by triglyceride accumulation and ATGL interaction. PLoS One. 2016;11:e0156742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sherry-Lynes MM, Sengupta S, Kulkarni S, Cochran BH. Regulation of the JMJD3 (KDM6B) histone demethylase in glioblastoma stem cells by STAT3. PLoS One. 2017;12:e0174775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang Y, Zhang L, Tu T, Li Y, Murray D, Tu Q, et al. MicroRNA-99a is a novel regulator of KDM6B-mediated osteogenic differentiation of BMSCs. J Cell Mol Med. 2018;22:2162–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Udali S, Guarini P, Moruzzi S, Ruzzenente A, Tammen SA, Guglielmi A, et al. Global DNA methylation and hydroxymethylation differ in hepatocellular carcinoma and cholangiocarcinoma and relate to survival rate. Hepatology. 2015;62:496–504. [DOI] [PubMed] [Google Scholar]
- 35.Zhao F, Ke J, Pan W, Pan H, Shen M. Synergistic effects of ISL1 and KDM6B on non-alcoholic fatty liver disease through the regulation of SNAI1. Mol Med. 2022;28:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Byun S, Seok S, Kim YC, Zhang Y, Yau P, Iwamori N, et al. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat Commun. 2020;11:807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heckmann BL, Zhang X, Xie X, Liu J. The G0/G1 switch gene 2 (G0S2): Regulating metabolism and beyond. Biochim Biophys Acta. 2013;1831:276–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Zhang Y, Qian H, Lu J, Zhang Z, Min X, et al. The G0/G1 Switch gene 2 is an important regulator of hepatic triglyceride metabolism. PLoS One. 2013;8:e72315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El-Assaad W, El-Kouhen K, Mohammad AH, Yang J, Morita M, Gamache I, et al. Deletion of the gene encoding G0/G1 switch protein 2 (G0S2) alleviates high-fat-diet-induced weight gain and insulin resistance and promotes browning of white adipose tissue in mice. Diabetologia. 2015;58:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan S, Yang XF, Liu HL, Fu N, Ouyang Y, Qing K. Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: An update. World J Gastroenterol. 2015;21:3492–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li T, Li X, Meng H, Chen L, Meng F. Acsl1 affects triglyceride levels through the pparγ pathway. Int J Med Sci. 2020;17:720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ellis JM, Li LO, Wu P-C, Koves TR, Ilkayeva O, Stevens RD, et al. Adipose acyl-CoA synthetase-1 (ACSL1) directs fatty acids towards β-oxidation and is required for cold thermogenesis. Cell Metab. 2010;12:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang R, Meng J, Yang S, Liu W, Shi L, Zeng J, et al. Recent advances on the role of ATGL in cancer. Front Oncol. 2022;12:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Romeo S, Savage DB. Lipase tug of war: PNPLA3 sequesters ABHD5 from ATGL. Nat Metab. 2019;1:505–506. [DOI] [PubMed] [Google Scholar]
- 45.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Kory N, BasuRay S, Cohen JC, Hobbs HH. PNPLA3, CGI-58, and inhibition of hepatic triglyceride hydrolysis in mice. Hepatology. 2019;69:2427–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang Y, He S, Li JZ, Seo YK, Osborne TF, Cohen JC, et al. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci USA. 2010;107:7892–7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu M, Yu X, Lin L, Deng J, Wang K, Xia Y, et al. ATGL promotes the proliferation of hepatocellular carcinoma cells via the p-AKT signaling pathway. J Biochem Mol Toxicol. 2019;33:e22391. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Liu L, Yuan X, Wei Y, Wei X. JMJD3 in the regulation of human diseases. Protein Cell. 2019;10:864–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond). 2018;38:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The microarray data are available to the public using the specific Gene Expression Omnibus accession number (GSE211848). The other data available are within the article or its Supplemental Materials (http://links.lww.com/HC9/A541).