

SUMMARY
Lifelong immunosuppression is required for allograft survival after kidney transplantation but may not ultimately prevent allograft loss resulting from chronic rejection. We developed an approach that attempts to abrogate immune rejection and the need for post-transplantation immunosuppression in three patients with Schimke immuno-osseous dysplasia who had both T-cell immunodeficiency and renal failure. Each patient received sequential transplants of αβ T-cell–depleted and CD19 B-cell–depleted haploidentical hematopoietic stem cells and a kidney from the same donor. Full donor hematopoietic chimerism and functional ex vivo T-cell tolerance was achieved, and the patients continued to have normal renal function without immunosuppression at 22 to 34 months after kidney transplantation. (Funded by the Kruzn for a Kure Foundation.)
On the basis of recent estimates, the median survival of trans-planted kidney grafts is 11.7 years for organs from deceased donors and 19 years for organs from living donors; therefore, most children who receive a kidney transplant will receive more than one in their lifetime.1 The lifelong immunosuppression received by kidney-transplant recipients exposes them to risks of infections, cancer, and other complications and, even with full adherence to immunosuppression, does not eliminate the risk of chronic rejection and eventual allograft loss. Initial attempts to achieve immune tolerance by establishing transient mixed chimerism through allogeneic hematopoietic stem-cell transplantation (HSCT) have been successful 60 to 70% of the time.2,3 The establishment of stable mixed chimerism has been associated with immune tolerance in HLA-matched patients, whereas in HLA-mismatched kidney-transplant recipients, mixed chimerism has permitted a reduction in the number of immunosuppressive drugs needed but not their discontinuation.4 Alternative approaches to establish 95 to 100% donor engraftment after HSCT have been complicated by the risk of fatal graft-versushost disease (GVHD), which has occurred in some patients.5 The use of HSCT to eliminate the need for immunosuppression after kidney transplantation in children has not been evaluated as it has been in adults. We developed an approach that abrogates immune-mediated rejection and the need for long-term post-transplantation immunosuppression and tested it in three patients with Schimke immuno-osseous dysplasia (SIOD) who had been determined to need a kidney transplant.
SIOD is a rare autosomal recessive multisystem disease that characteristically includes glucocorticoid-resistant nephrotic syndrome, spondyloepiphyseal dysplasia, short stature, and variable T-cell immunodeficiency.6 The disease is due to biallelic mutations in the gene encoding SMARCAL1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin, subfamily A-like 1),7 which is implicated in replication-coupled DNA repair and telomere maintenance.8 The nephrotic syndrome in SIOD is a podocytopathy that usually progresses to focal segmental glomerulosclerosis and end-stage kidney disease.9,10
Previously, four of five patients with SIOD who underwent HSCT after myeloablative conditioning died from HSCT-related causes, including infections and GVHD11; the one long-term survivor has had full immune and hematopoietic reconstitution.12 The high post-HSCT mortality among the patients with SIOD may reflect, at least in part, the increased sensitivity of SMARCAL1-deficient cells to DNA-damaging agents,11,13 which are used in myeloablative conditioning regimens.
Success with αβ T-cell–depleted and CD19 B-cell–depleted haploidentical (αβ–haploidentical) HSCT has been achieved in patients undergoing transplantation who have nonmalignant diseases, with overall survival of 90%.14 The depletion of alloreactive T cells and B cells results in low rates of both acute and chronic GVHD and of transplant-related death.14 No deaths due to GVHD have occurred in a cohort of 70 patients who were treated in this manner, including patients with disorders such as Fanconi’s anemia and dyskeratosis congenita, in which radiosensitivity is a symptom, which necessitated the use of reduced-intensity conditioning to avoid toxic effects.15–18 We hypothesized that αβ–haploidentical HSCT after reduced-intensity conditioning could result in successful engraftment in patients with SIOD, whose cells are also radiosensitive. A predicted advantage of sequential HSCT and kidney transplantation has been suggested by outcomes among patients who first underwent HSCT for an unrelated disease and then subsequently received a kidney transplant from the same HSCT donor, which resulted in long-term kidney-graft survival without longterm immunosuppression.18–20
We report on the treatment and clinical courses of three children with SIOD and T-cell immunodeficiency who underwent sequential αβ–haploidentical HSCT and kidney transplantation from the same parental donor in order to establish functional tolerance and abrogate the need for post-transplantation immunosuppression.
METHODS
PATIENTS
Three patients with SIOD who had documented biallelic mutations in SMARCAL1 (Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org) underwent αβ–haploidentical HSCT after enrollment in studies that had been approved by the Stanford University institutional review board. Informed consent for the HSCT and kidney donation were obtained from the parents who served as donors. This research was conducted in accordance with the International Council for Harmonisation and the Declaration of Helsinki.
TREATMENT
The αβ T-cell and CD19 B-cell depletion was performed as previously reported.14 All patients underwent peripheral blood lymphocyte immunophenotyping by flow cytometry, karyotype analysis, determination of circulating leukocyte telomere length (see the Supplementary Methods section and Fig. S1), and chimerism studies conducted with the use of short-tandem-repeat polymerase chain reaction.21 The pre-HSCT reduced-intensity conditioning consisted of antithymocyte globulin (7.5 mg per kilogram of body weight), fludarabine (starting dose of 1 mg per kilogram daily for 4 days), cyclophosphamide (1200 mg per square meter of body-surface area), total-body irradiation (200 cGy), and rituximab (200 mg per square meter) (Fig. 1). Adjustment of fludarabine doses for all patients was based on levels in plasma and calculations of the area under the concentration–time curve (see the Supplementary Methods section and Fig. S2).
Figure 1. Sequential αβ T-Cell–Depleted and CD19 B-Cell–Depleted Haploidentical HSCT and Kidney Transplantation in Three Patients with SIOD.
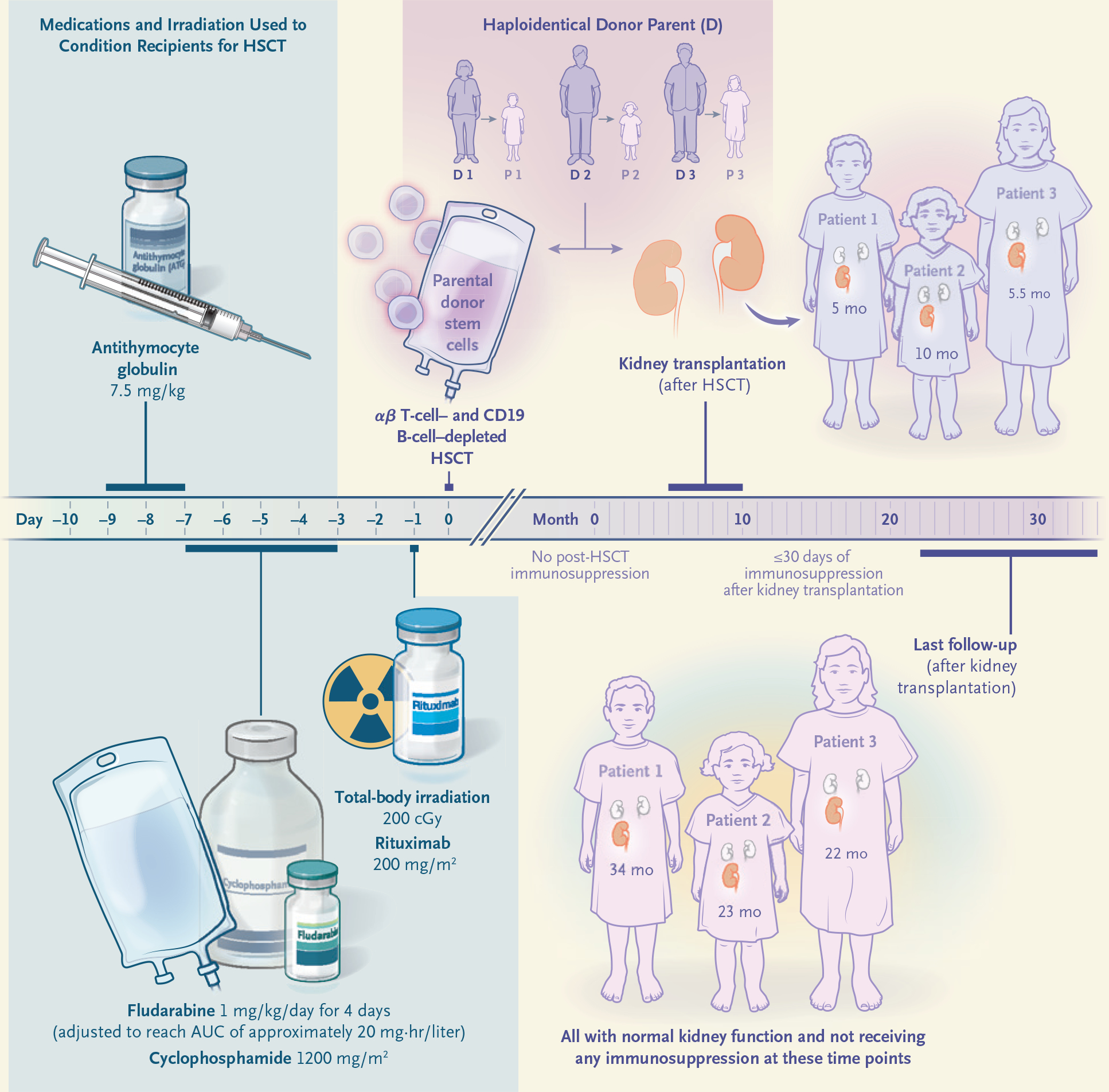
Three patients with Schimke immuno-osseous dysplasia (SIOD) received a reduced-intensity conditioning regimen consisting of antithymocyte globulin, fludarabine (the dose of which was adjusted because of the patients’ renal insufficiency), cyclophosphamide, totalbody irradiation, and rituximab. The graft used for hematopoietic stem-cell transplantation (HSCT), which was obtained from one of the parents, was depleted of αβ T cells and CD19 B cells, and no immunosuppression was administered after the graft infusion. Each patient received a kidney transplant 5 to 10 months later from the parent who served as the donor for HSCT. A short course of immunosuppression was stopped by 30 days after kidney transplantation. All three patients have had normal kidney function without any immunosuppression for a period of follow-up ranging from 22 to 34 months. AUC denotes area under the curve.
In the first 30 days after HSCT, Patients 1 and 2 received 1×106 donor-derived αβ T cells per kilogram; the cells had been engineered ex vivo to express an inducible caspase 9 (iC9) suicide gene,22 but the patients did not receive dimerization therapy to activate the iC9 transgene product. (At the time of the treatment of Patient 3, iC9 genetically modified T cells were no longer being manufactured.) When full (i.e., >95%) donor myeloid and lymphoid chimerism after HSCT had been confirmed, each patient received a living-donor kidney transplant from the parental HSCT donor. The patients received intraoperative methylprednisolone and postoperative low-dose oral prednisone (0.5 mg per kilogram per day with taper) and tacrolimus (target serum level of 3 to 5 ng per milliliter) to reduce potential reperfusion-related inflammation. All peri-transplantation immunosuppressive drugs were tapered off by day 30 after transplantation, and no subsequent immunosuppressive therapy was administered. One-way mixed-lymphocyte culture was performed to evaluate the functional tolerance of recipient T cells to the alloantigens of the kidney-transplant donor. Peripheral blood mononuclear cells (PBMCs) were used as a source of responder T cells, and irradiated Epstein–Barr virus–transformed lymphoblastoid cell lines derived from both parents of the patients and an unrelated healthy third-party donor were used as stimulators (see the Supplementary Methods section).
RESULTS
OUTCOMES
Because all three patients with SIOD had both moderate adaptive immunodeficiency (e.g., diminished antibody responses to vaccines and diminished T-cell responses to mitogens, vaccine antigens, or both) (Table S1) and advanced renal insufficiency, we performed parental αβ–haploidentical HSCT to establish a competent donor-derived immune system that would be tolerant to a subsequent kidney allograft from the same donor. No post-HSCT pharmacologic immunosuppression was used (Table 1). All three patients had successful engraftment, with full donor lymphoid and myeloid chimerism at the time of the neutrophil engraftment for Patients 1 and 3 and by day 150 after transplantation for Patient 2, and normal T-cell immune reconstitution, including normal mitogen responsiveness (Table 1). When adequate nutritional status (i.e., a body-mass index [the weight in kilograms divided by the square of the height in meters] of >18.5) had been reached through the use of enteral feedings and an ambulatory level of activity had been attained, kidney transplantation from the HSCT parental donor was performed (at 5, 10, and 5.5 months after HSCT in Patients 1, 2, and 3, respectively) (Fig. 1). Immunosuppression was withdrawn by day 30 after transplantation, and at the time of this report, 34, 23, and 22 months had passed since transplantation; renal function remained normal, without any clinical signs of rejection (Table 1).
Table 1.
Characteristics of the Patients and the Hematopoietic Stem-Cell Transplantations.*
| Characteristic | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| Age at HSCT (yr) | 6.0 | 4.8 | 8.6 |
| HLA match of HSCT graft (no. of alleles matched/total no.) | 8/10 | 5/10 | 7/10 |
| Donor-specific antibodies | Negative | Negative | Negative |
| CKD stage at HSCT† | 5 | 3 | 5 |
| Renal dialysis at HSCT | Yes | No | Yes |
| CMV IgG serostatus | |||
| Donor | Positive | Negative | Positive |
| Recipient | Negative | Positive | Negative |
| ABO and Rh type | |||
| Donor | O+ | A+ | O+ |
| Recipient | A+ | A+ | O+ |
| Graft composition (cells/kg) | |||
| CD34+ HSCs | 15×106 | 20×106 | 19.9×106 |
| αβ+ T cells | 0.69×105 | 0.67×105 | 0.07×105 |
| γδ+ T cells | 3.75×106 | 5.76×106 | 4.33×106 |
| NK cells | 16.4×106 | 4.07×106 | 66.6×106 |
| B cells | 3.75×105 | 50.2×105 | 0.9×105 |
| Post-HSCT day of neutrophil engraftment‡ | 12 | 14 | 12 |
| Post-HSCT day of platelet engraftment‡ | 16 | 14 | 15 |
| Treatment-related toxic effects | Mucositis grade II | Mucositis grade II | Mucositis grade I |
| CMV reactivation after HSCT or KT | None | None | None |
| Opportunistic infections after HSCT or KT | None | None | None |
| Chimerism at hematopoietic engraftment | |||
| Whole blood (% donor) | 100 | 71§ | 100 |
| Day after HSCT | 12 | 14 | 12 |
| Pre-KT chimerism | |||
| Lymphoid (% donor CD3+) | 100 | 96 | 97 |
| Day after HSCT | 148 | 150 | 61 |
| Myeloid (% donor CD15+) | 100 | 99 | 99 |
| Day after HSCT | 148 | 150 | 61 |
| Post-KT chimerism | |||
| Lymphoid (% donor CD3+) | 100 | 97 | 100 |
| Day after KT | 219 | 313 | 201 |
| Myeloid (% donor CD15+) | 100 | 96 | 100 |
| Day after KT | 219 | 313 | 201 |
| PHA response before KT (counts/min) | 135,082 | 161,795 | 104,191 |
| Day of immune reconstitution of naive T cells after HSCT¶ | |||
| CD4+ | 180 | 180 | 180 |
| CD8+ | 365 | 365 | 270 |
| Graft-versus-host disease | |||
| Acute | Grade II skin only | None | None |
| Chronic | None | None | None |
| Virus reactivated after HSCT | HHV-6 | None | None |
| iC9 genetically modified donor T cells | Yes | Yes | No |
| Day after HSCT | 27 | 24 | — |
| Creatinine level at last follow-up (mg/dl) | 0.25 | 0.29 | 0.32 |
| Response to vaccination after HSCT and KT | |||
| Streptococcus pneumoniae IgG titers, 11 serotypes | Protective titers | Protective titers | Patient vaccinated but titers not yet available |
| Tetanus antibody level (lU/ml) | >5.33 | >5.33 | Patient vaccinated but titers not yet available |
The HSCT conditioning regimen received by all three patients consisted of fludarabine, cyclophosphamide, total-body irradiation (200 cGy), antithymocyte globulin, and rituximab. To convert values for creatinine to micromoles per liter, multiply by 88.4. CMV denotes cytomegalovirus, DSA donor-specific antibody, HHV-6 human herpesvirus 6, HLA human leukocyte antigen, HSC hematopoietic stem cell, HSCT hematopoietic stem-cell transplantation, KT kidney transplant, and PHA phytohemagglutinin.
Chronic kidney disease (CKD) stages range from 1 to 5, with stage 5 indicating end-stage kidney disease. Stage 3 CKD is defined as a glomerular filtration rate (GFR) of 30 to 59 ml per minute per 1.73 m2, and stage 5 CKD as a GFR of less than 15 ml per minute per 1.73 m2.
A definition of engraftment is provided by Kharfan-Dabaja et al.23
Full chimerism was present in Patient 2 by day 150 after HSCT.
Immune reconstitution of naive CD4+ and CD8+ T cells has been defined as the presence of more than 50 cells per microliter. The gating strategy for naive CD4+ T cells was CD45+/singlets/CD3+/CD4+CD8−/CD27+CD45RO−, and the gating strategy for naive CD8+ T cells was CD45+/singlets/CD3+/CD4−CD8+/CD27+CD45RO−.
To evaluate the recipients for functional T-cell tolerance after kidney transplantation, PBMCs were isolated from the patients and evaluated in a one-way mixed-lymphocyte culture assay (see the Supplementary Methods section). After HSCT and kidney transplantation, the T cells of all three recipients did not proliferate in response to stimulation by the irradiated cells from their parental donors, whereas they did proliferate after stimulation by cells from the nondonor parent or third-party control cells (Fig. 2). These results indicated that after sequential αβ–haploidentical HSCT and transplantation of a kidney from the same donor, the circulating donorderived T cells were functionally tolerant to the alloantigens of the transplanted kidney and, therefore, potentially unable to mediate graft rejection even in the absence of immune suppression.
Figure 2. One-Way Mixed-Lymphocyte Cellular Assay of T-Cell Alloreactivity at 1 Year after Kidney Transplantation.
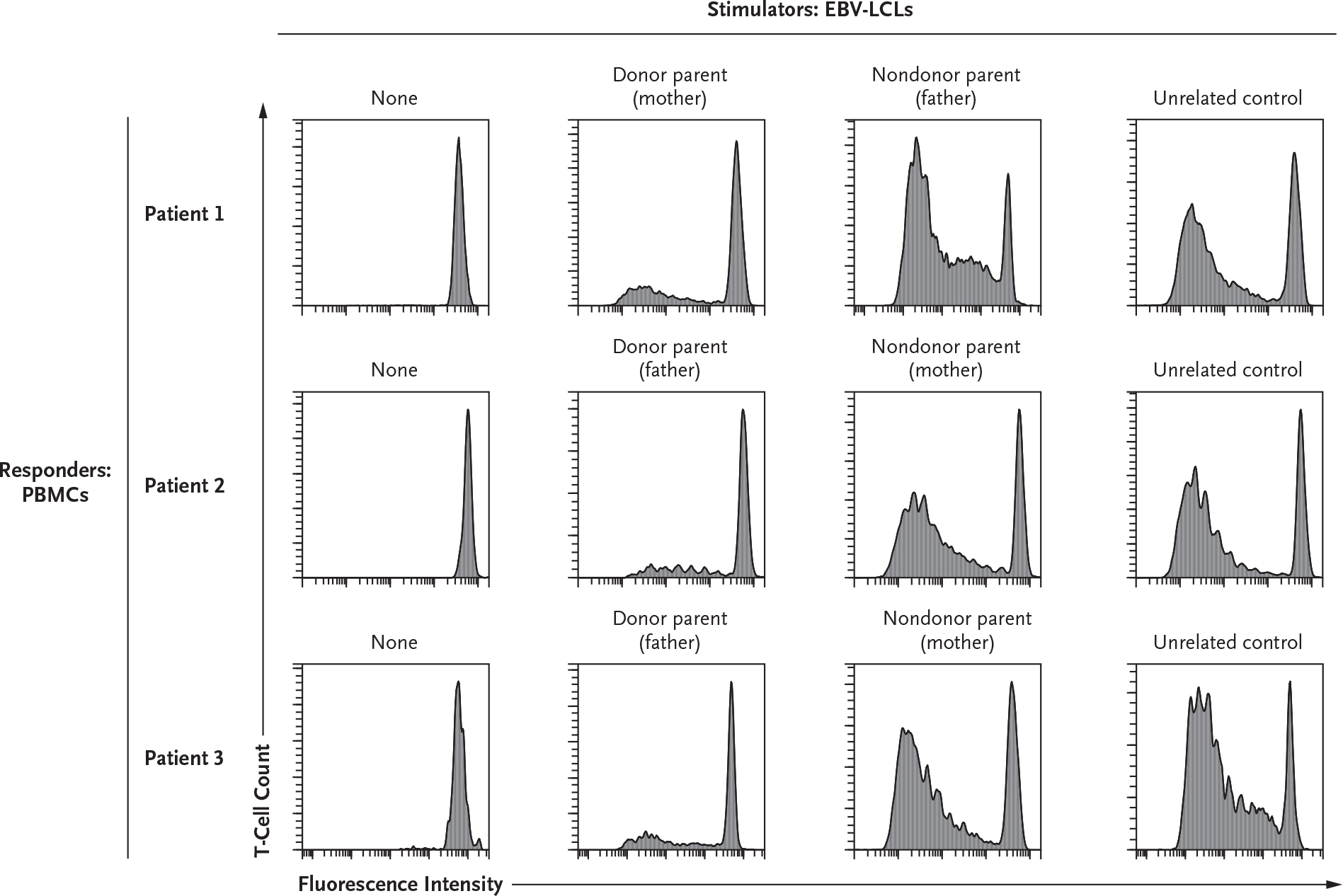
Peripheral blood mononuclear cells (PBMCs), which were used as responder cells, were isolated from the three patients at least 1 year after their kidney transplantation (KT). The PBMCs were stained with CellTrace Violet (Invitrogen) before coculture with stimulator Epstein–Barr virus (EBV)–transduced B lymphoblastoid cell lines (EBV-LCLs) previously generated from the parents of the patients or from an unrelated healthy third-party control. CellTrace Violet fluorescence intensity (on a logarithmic scale) for CD3+CD19− cells (x axis) is shown plotted against T-cell count (y axis); a loss of fluorescence indicates cell proliferation. T-cell proliferation was measured after 6 days of culture and obtained at a responder-to-stimulator ratio of 2:1. T cells of both of the two siblings (Patients 1 and 2) and of Patient 3 showed functional tolerance to EBV-LCLs derived from their respective donors, whereas they were immune competent and proliferated in the presence of EBV-LCLs derived from nondonor parents or a healthy, unrelated control. The plots in the first column show the absence of T-cell proliferation when PBMCs were cultured in medium alone. The results shown are representative of three replicates for each combination of stimulator and responder cells.
CASE REPORTS
Patient 1 received a maternal αβ–haploidentical HSCT (8/10 HLA-matched) after pharmacokinetically guided pre-HSCT conditioning. (Details of the pharmacokinetic modeling for all three patients are provided in Fig. S2.) Dialysis was continued during HSCT to control fluid and electrolyte balance. Engraftment occurred on day 12 after HSCT, when the absolute neutrophil count was greater than 500 per microliter and showed full donor chimerism of both CD15+ myeloid and CD3+ T cells (Table 1). On day 27, the patient received 1×106 donor-derived, iC9 genetically modified T cells per kilogram.22 His post-HSCT course was complicated by asymptomatic reactivation of human herpesvirus 6 (HHV-6), which resolved with 2 weeks of ganciclovir therapy, and by grade II cutaneous acute GVHD (onset on day 52 after HSCT), which was treated with prednisone (1 mg per kilogram per day for 1 week followed by a rapid taper). CD4+ and CD8+ T cells, including their naive subsets, progressively increased after HSCT, with levels of 800 per microliter and 400 per microliter, respectively, present at 1 year after HSCT. Five months after HSCT, he received a kidney transplant from his mother followed by immunosuppression with prednisone and tacrolimus for 30 days. At the time of this report 34 months later, the patient had full donor hematopoietic chimerism, was free of clinical rejection without any immunosuppressive drugs, and had a serum creatinine level of 0.25 mg per deciliter (22 μmol per liter).
Patient 2, the sibling of Patient 1, received paternal αβ–haploidentical HSCT (5/10 HLA-matched) after pharmacokinetically guided pre-HSCT conditioning. Because of increasing creatinine levels and fluid overload, she began to undergo hemodialysis on day 21 after HSCT. Donor-derived iC9 genetically modified T cells (1×106 per kilogram) were given on day 24. The patient did not have any viral reactivation or signs of acute or chronic GVHD. Chimerism studies performed at the time of hematopoietic engraftment on day 14 showed mixed chimerism (71% donor cells in whole blood) that became fully donor by day 150. Because of the coronavirus disease 2019 (Covid-19) pandemic, paternal kidney transplantation was postponed until 10 months after HSCT, by which time the patient had normal T-cell function (Table 1). At the time of this report, 23 months after kidney transplantation, the patient had full hematopoietic donor chimerism, was free of rejection without any immunosuppressive drugs, and had a serum creatinine level of 0.29 mg per deciliter (26 μmol per liter).
Patient 3 received αβ–haploidentical HSCT (7/10 HLA-matched) from her father after having received pharmacokinetically adjusted pre-HSCT conditioning while undergoing peritoneal dialysis. Her post-HSCT course was uneventful, and she reached full donor chimerism with hematopoietic engraftment at day 15. Five and a half months after HSCT, she received a kidney transplant from her father. As a result of hyperglycemia, her post–kidney transplantation treatment with tacrolimus and methylprednisolone was stopped on days 5 and 12, respectively. At the time of this report, 22 months after kidney transplantation, she had full donor hematopoietic chimerism, was free of rejection without any immunosuppressive drugs, and had a serum creatinine level of 0.32 mg per deciliter (28 μmol per liter).
DISCUSSION
To achieve full donor chimerism while avoiding the risk of severe GVHD and to fully reconstitute the adaptive immune systems of three pediatric patients with SIOD, we successfully transplanted HLA-haploidentical hematopoietic stem cells depleted of αβ T cells and CD19 B cells after reduced-intensity conditioning. Each patient then received a kidney transplant from the same parent who served as the donor for haploidentical HSCT, and all immune suppression was withdrawn by 30 days after kidney transplantation. The αβ–haploidentical HSCT resulted in full donor hematopoietic engraftment, lymphocyte recovery, and immune reconstitution without any post-HSCT immunosuppression. One patient had transient grade II cutaneous acute GVHD, which responded to a short course of glucocorticoids. Only one patient had HHV-6 viremia. Cytomegalovirus reactivation after either HSCT or the kidney transplantation was not detected in any of the patients.
Previous attempts to induce tolerance by establishing transient mixed chimerism required revisions of the conditioning regimen to overcome engraftment syndrome and improve the consistency of the results.2,3 Persistent mixed chimerism in the context of an HLA mismatch has allowed the reduction of immunosuppression after kidney transplantation to a single agent but has not allowed complete discontinuation of immunosuppression in those cases.4 The establishment of 100% donor chimerism has been associated with HSCT-related toxic effects.5,24 In contrast, our strategy resulted in rapid and persistent full donor lymphoid and myeloid engraftment and complete discontinuation of all immunosuppression by day 30 after kidney transplantation, without severe GVHD. SIOD includes both impaired T-cell immunity and impaired hematopoiesis, which might have favored the rapid appearance of functional tolerance to the kidney allograft. These results are also consistent with previous preclinical studies in mice showing functional tolerance to skin allografts after haploidentical HSCT25 and the previously unplanned clinical use of kidney transplantation after HSCT.18–20 Further studies will be needed to determine whether these outcomes can be achieved in allograft recipients with intact pretransplantation T-cell immunity and hematopoiesis.
We successfully transplanted kidneys into three patients with SIOD, a syndrome characterized by variable T-cell immunodeficiency; each patient underwent αβ T-cell–depleted and CD19 B-cell–depleted haploidentical HSCT followed by kidney transplantation from the same donor and continued to have normal renal function without immunosuppression at 22 to 34 months after kidney transplantation. This regimen has resulted in the correction of their primary immune deficiency.
Supplementary Material
Acknowledgments
Supported by the Kruzn for a Kure Foundation (to Drs. Bertaina and Lewis).
We thank Dr. Cornelius Boerkoel III (Sanford USD Medical Center and Hospital, Sioux Falls, SD) for useful discussions.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
Contributor Information
Alice Bertaina, Division of Hematology, Oncology, Stem Cell Transplantation, and Regenerative Medicine, Center for Definitive and Curative Medicine, Stanford Children’s Health, Department of Pediatrics, Stanford University School of Medicine, Stanford, CA
Paul C. Grimm, Division of Nephrology, Department of Pediatrics, Stanford University School of Medicine, Stanford, CA
Kenneth Weinberg, Division of Hematology, Oncology, Stem Cell Transplantation, and Regenerative Medicine, Center for Definitive and Curative Medicine, Stanford Children’s Health, Department of Pediatrics, Stanford University School of Medicine, Stanford, CA
Robertson Parkman, Division of Hematology, Oncology, Stem Cell Transplantation, and Regenerative Medicine, Center for Definitive and Curative Medicine, Stanford Children’s Health, Department of Pediatrics, Stanford University School of Medicine, Stanford, CA
Karen M. Kristovich, Division of Hematology, Oncology, Stem Cell Transplantation, and Regenerative Medicine, Center for Definitive and Curative Medicine, Stanford Children’s Health, Department of Pediatrics, Stanford University School of Medicine, Stanford, CA
Giulia Barbarito, Division of Hematology, Oncology, Stem Cell Transplantation, and Regenerative Medicine, Center for Definitive and Curative Medicine, Stanford Children’s Health, Department of Pediatrics, Stanford University School of Medicine, Stanford, CA
Elizabeth Lippner, Division of Allergy, Immunology, and Rheumatology, Departments of Surgery, Stanford University School of Medicine, Stanford, CA
Girija Dhamdhere, Division of Allergy, Immunology, and Rheumatology, Departments of Surgery, Stanford University School of Medicine, Stanford, CA
Vasavi Ramachandran, Division of Allergy, Immunology, and Rheumatology, Departments of Surgery, Stanford University School of Medicine, Stanford, CA
Jordan M. Spatz, Division of Allergy, Immunology, and Rheumatology, Departments of Surgery, Stanford University School of Medicine, Stanford, CA
Sahar Fathallah-Shaykh, Division of Pediatric Nephrology, Department of Pediatrics, University of Alabama, Birmingham
T. Prescott Atkinson, Division of Pediatric, Department of Pediatrics Allergy and Immunology, University of Alabama, Birmingham
Amira Al-Uzri, Department of Pediatrics, University of Alabama, Birmingham; the Division of Nephrology, Department of Pediatrics, Oregon Health Sciences University, Portland
Geraldine Aubert, Terry Fox Laboratory, BC Cancer Agency, Vancouver, Canada
Kim van der Elst, Department of Clinical Pharmacy, University Medical Center Utrecht, Utrecht University, Utrecht, the Netherlands
Sean G. Green, Department of Pharmacy, Stanford Children’s Health, Stanford, CA
Rajni Agarwal, Division of Hematology, Oncology, Stem Cell Transplantation, and Regenerative Medicine, Center for Definitive and Curative Medicine, Stanford Children’s Health, Department of Pediatrics, Stanford University School of Medicine, Stanford, CA
Priscila F. Slepicka, Division of Hematology, Oncology, Stem Cell Transplantation, and Regenerative Medicine, Center for Definitive and Curative Medicine, Stanford Children’s Health, Department of Pediatrics, Stanford University School of Medicine, Stanford, CA
Ami J. Shah, Division of Hematology, Oncology, Stem Cell Transplantation, and Regenerative Medicine, Center for Definitive and Curative Medicine, Stanford Children’s Health, Department of Pediatrics, Stanford University School of Medicine, Stanford, CA
Maria G. Roncarolo, Division of Hematology, Oncology, Stem Cell Transplantation, and Regenerative Medicine, Center for Definitive and Curative Medicine, Stanford Children’s Health, Department of Pediatrics, Stanford University School of Medicine, Stanford, CA
Amy Gallo, Department of Surgery, Pediatrics, Stanford University School of Medicine, Stanford, CA
Waldo Concepcion, Department of Surgery, Pediatrics, Stanford University School of Medicine, Stanford, CA Department of Pediatrics, Stanford University School of Medicine, Stanford, CA.
David B. Lewis, Division of Allergy, Immunology, and Rheumatology, Department of Surgery, Stanford University School of Medicine, Stanford, CA
REFERENCES
- 1.Poggio ED, Augustine JJ, Arrigain S, Brennan DC, Schold JD. Long-term kidney transplant graft survival — making progress when most needed. Am J Transplant 2021;21:2824–32. [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Cosimi AB, Spitzer TR, et al. HLA-mismatched renal transplantation without maintenance immunosuppression. N Engl J Med 2008;358:353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawai T, Sachs DH, Sprangers B, et al. Long-term results in recipients of combined HLA-mismatched kidney and bone marrow transplantation without maintenance immunosuppression. Am J Transplant 2014;14:1599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busque S, Scandling JD, Lowsky R, et al. Mixed chimerism and acceptance of kidney transplants after immunosuppressive drug withdrawal. Sci Transl Med 2020;12(528):e aax8863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leventhal JR, Ildstad ST. Tolerance induction in HLA disparate living donor kidney transplantation by facilitating cell-enriched donor stem cell Infusion: the importance of durable chimerism. Hum Immunol 2018;79:272–6. [DOI] [PubMed] [Google Scholar]
- 6.Lippner E, Lücke T, Salgado C, Boerkoel C, Lewis DB. Schimke immunoosseous dysplasia. Seattle: GeneReviews, April 14, 2022. (https://www.ncbi.nlm.nih.gov/books/NBK1376/). [Google Scholar]
- 7.Boerkoel CF, Takashima H, John J, et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immunoosseous dysplasia. Nat Genet 2002;30:215–20. [DOI] [PubMed] [Google Scholar]
- 8.Poole LA, Cortez D. Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Crit Rev Biochem Mol Biol 2017;52:696–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarin S, Javidan A, Boivin F, et al. Insights into the renal pathogenesis in Schimke immuno-osseous dysplasia: a renal histological characterization and expression analysis. J Histochem Cytochem 2015;63:3 2–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lücke T, Kanzelmeyer N, Baradaran-Heravi A, et al. Improved outcome with immunosuppressive monotherapy after renal transplantation in Schimke-immuno-osseous dysplasia. Pediatr Transplant 2009;13:482–9. [DOI] [PubMed] [Google Scholar]
- 11.Baradaran-Heravi A, Lange J, Asakura Y, Cochat P, Massella L, Boerkoel CF. Bone marrow transplantation in Schimke immuno-osseous dysplasia. Am J Med Genet A 2013;161A:2 609–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petty EM, Yanik GA, Hutchinson RJ, et al. Successful bone marrow transplantation in a patient with Schimke immunoosseous dysplasia. J Pediatr 2000;137:882–6. [DOI] [PubMed] [Google Scholar]
- 13.Baradaran-Heravi A, Raams A, Lubieniecka J, et al. SMARCAL1 deficiency predisposes to non-Hodgkin lymphoma and hypersensitivity to genotoxic agents in vivo. Am J Med Genet A 2012;158A:2204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertaina A, Merli P, Rutella S, et al. HLA-haploidentical stem cell transplantation after removal of αβ+ T and B cells in children with nonmalignant disorders. Blood 2014;124:8 22–6. [DOI] [PubMed] [Google Scholar]
- 15.Merli P, Pagliara D, Galaverna F, et al. TCRαβ/CD19 depleted HSCT from an HLA-haploidentical relative to treat children with different nonmalignant disorders. Blood Adv 2022;6:281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strocchio L, Pagliara D, Algeri M, et al. HLA-haploidentical TCRαβ+/CD19+-depleted stem cell transplantation in children and young adults with Fanconi anemia. Blood Adv 2021;5 :1333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhattacharyya R, Tan AM, Chan MY, Jamuar SS, Foo R, Iyer P. TCR αβ and CD19-depleted haploidentical stem cell transplant with reduced intensity conditioning for Hoyeraal-Hreidarsson syndrome with RTEL1 mutation. Bone Marrow Transplant 2016;51:753–4. [DOI] [PubMed] [Google Scholar]
- 18.Faraci M, Bertaina A, Dalissier A, et al. Solid organ transplantation after hematopoietic stem cell transplantation in childhood: a multicentric retrospective survey. Am J Transplant 2019;1 9:1798–805. [DOI] [PubMed] [Google Scholar]
- 19.Miano M, Ginevri F, Nocera A, et al. Successful double bone marrow and renal transplantation in a patient with Fanconi anemia. Blood 2002;99:3 482–3. [DOI] [PubMed] [Google Scholar]
- 20.Eder M, Schwarz C, Kammer M, et al. Allograft and patient survival after sequential HSCT and kidney transplantation from the same donor — a multicenter analysis. Am J Transplant 2019;19:475–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abatay-Sel F, Savran-Oguz F, Kalayoglu-Besisik S, et al. Short tandem repeat-polymerase chain reaction (STR-PCR) with quantitative real time-polymerase chain reaction (qRT-PCR) method using for chimerism analysis. Clin Lab 2019; 65. [DOI] [PubMed] [Google Scholar]
- 22.Zhou X, Naik S, Dakhova O, Dotti G, Heslop HE, Brenner MK. Serial activation of the inducible caspase 9 safety switch after human stem cell transplantation. Mol Ther 2016;24:8 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kharfan-Dabaja MA, Kumar A, Ayala E, et al. Standardizing definitions of hematopoietic recovery, graft rejection, graft failure, poor graft function, and donor chimerism in allogeneic hematopoietic cell transplantation: a report on behalf of the American Society for Transplantation and Cellular Therapy. Transplant Cell Ther 2021;27:642–9. [DOI] [PubMed] [Google Scholar]
- 24.Leventhal JR, Mathew JM. Outstanding questions in transplantation: tolerance. Am J Transplant 2020;2 0: 348–54. [DOI] [PubMed] [Google Scholar]
- 25.Aversa F, Bachar-Lustig E, Or-Geva N, et al. Immune tolerance induction by nonmyeloablative haploidentical HSCT combining T-cell depletion and posttransplant cyclophosphamide. Blood Adv 2017;1:2166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.