

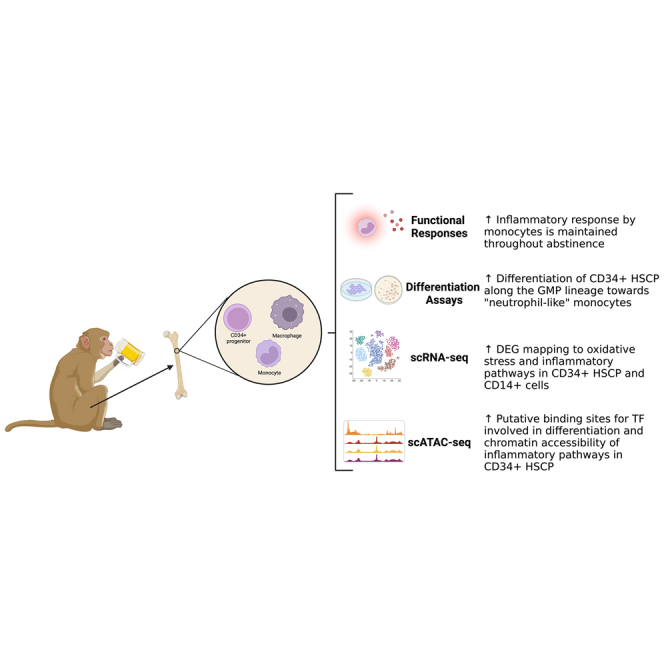
Summary
Chronic heavy alcohol drinking (CHD) rewires monocytes and macrophages toward heightened inflammatory states with compromised antimicrobial defenses that persist after 1-month abstinence. To determine whether these changes are mediated through alterations in the bone marrow niche, we profiled monocytes and hematopoietic stem cell progenitors (HSCPs) from CHD rhesus macaques using a combination of functional assays and single cell genomics. CHD resulted in transcriptional profiles consistent with increased activation and inflammation within bone marrow resident monocytes and macrophages. Furthermore, CHD resulted in transcriptional signatures associated with increased oxidative and cellular stress in HSCP. Differentiation of HSCP in vitro revealed skewing toward monocytes expressing “neutrophil-like” markers with greater inflammatory responses to bacterial agonists. Further analyses of HSCPs showed broad epigenetic changes that were in line with exacerbated inflammatory responses within monocytes and their progenitors. In summary, CHD alters HSCPs in the bone marrow leading to the production of monocytes poised to generate dysregulated hyper-inflammatory responses.
Keywords: alcohol, monocytes, progenitors, monopoiesis, myelopoiesis, non-human primates, bone marrow
Graphical abstract
Highlights
-
•
Chronic drinking increases inflammatory responses that persist after abstinence
-
•
Bone marrow progenitors, monocytes, and macrophages are altered by chronic drinking
-
•
Chronic drinking skews monopoiesis to produce neutrophil-like inflammatory monocytes
In this study, Lewis and colleagues leveraged a rhesus macaque model of voluntary ethanol self-administration to investigate the effects of chronic heavy drinking (CHD) on monopoiesis. Data presented herein show that CHD is associated with functional, transcriptional, and epigenetic changes in progenitor cells in the bone marrow resulting in the generation of monocytes poised toward generating heightened inflammatory responses.
Introduction
Alcohol drinking is widespread with >2 billion current drinkers worldwide (WHO, 2018). Alcohol and its metabolites induce organ damage and increase the incidence of cardiovascular disease (Mukamal and Rimm, 2001; O'Keefe et al., 2014), cancer (Rumgay et al., 2021), liver cirrhosis (Bruha et al., 2009), and sepsis (O'Brien et al., 2011). Moreover, heavy alcohol drinking increases susceptibility to bacterial and viral infections (Baum et al., 2010; Bhattacharya and Shuhart, 2003; Saitz et al., 1997). Increased vulnerability to infections is hypothesized to be mediated by functional, transcriptomic, and epigenomic changes in monocytes and tissue-resident macrophages leading to increased inflammation but compromised antimicrobial responses (Lewis et al., 2021; Sureshchandra et al., 2016, 2019a; Szabo and Saha, 2015). Whether these changes are limited to the periphery or can be traced to progenitor cells has yet to be determined.
Monocytes arise from hematopoietic stem cell progenitors (HSCPs) in the bone marrow through progressively restricted lineage committed progenitors (Kawamura and Ohteki, 2018; Wolf et al., 2019). Two pathways of monocyte production are described in mice starting from common myeloid progenitors (CMP) and proceeding through granulocyte-monocyte progenitors (GMP) to monocyte progenitors (MP) or monocyte-DC progenitors (MDP) to common monocyte progenitors (cMoP) (Wolf et al., 2019; Yanez et al., 2017). Myelopoiesis in humans is not as well characterized, but CMP, GMP, MDP, and cMoP populations have been identified (Kawamura and Ohteki, 2018; Lee et al., 2015). Mature monocytes can be classified into classical, intermediate, and non-classical subsets, which can be found in the bone marrow and in circulation (Teh et al., 2019). Infection, inflammation, or other stress factors can alter monocyte production and induce “emergency monopoiesis” from the bone marrow (Baldridge et al., 2011; Takizawa et al., 2012; Wolf et al., 2019).
Chronic heavy drinking (CHD) is known to affect hematopoiesis. Specifically, lymphopenia, anemia, and thrombocytopenia are observed in patients with alcohol use disorder (Ballard, 1997; Latvala et al., 2004; Liu, 1980; Panasiuk and Kemona, 2001; Shi et al., 2019; Smith et al., 2015). Studies in rodent models of alcohol exposure show impaired hematopoietic precursor cell activation as well as perturbation of granulocyte precursor responses and differentiation resulting in reduced bacterial clearance (Raasch et al., 2010; Shi et al., 2017). Studies in non-human primates (NHPs) have reported increased numbers of mature macrophages and osteoclasts in the bone marrow with concurrent alcohol use and simian immune deficiency virus infection (Siggins et al., 2014). Another study using an NHP model of chronic voluntary drinking followed by several bouts of abstinence and ethanol re-exposure showed impaired mitochondrial function and ability to form progenitor colonies from bone marrow HSCPs (Varlamov et al., 2020). While these studies have identified a significant impact of alcohol on the functions of bone marrow progenitor cells, none of them examined monopoiesis or connected alterations in HSCPs to dysregulated inflammatory responses in peripheral myeloid cells.
To address this gap in knowledge, we leverage an NHP model of voluntary ethanol consumption to assess the impact of CHD on monocytes and their progenitors in the bone marrow compartment. We collected bone marrow cells from the femurs of male and female animals engaged in CHD (average daily consumption of >3 g ethanol per kilogram body weight) for 12 months. We performed phenotypic, functional, and single cell transcriptomic/epigenomic assays on both monocytes/macrophages and HSCPs within the bone marrow. We report that CHD is associated with disruption in HSCPs, altered monopoiesis, and the production of monocytes poised toward hyper-inflammatory responses.
Results
CHD-induced heightened inflammatory state of circulating monocytes persists after 1-month abstinence
We have previously shown that CHD in NHPs leads to increased tumor necrosis factor (TNF)-⍺ production in response to lipopolysaccharide (LPS) stimulation by circulating monocytes and tissue-resident macrophages that is mediated by epigenetic and transcriptional changes (Lewis et al., 2021; Sureshchandra et al., 2019b). As monocytes circulate for only 5–7 days, we determined if circulating monocytes would revert to a control state after a 1-month abstinence period (Teh et al., 2019). We collected peripheral blood mononuclear cells (PBMCs) from male NHPs after 12 months of CHD and following a 1-month abstinence period (n = 4) and control animals (n = 3) (Allen et al., 2018) (Figures 1A and 1B). The frequency of TNF-⍺/interleukin (IL)-6-secreting monocytes in response to bacterial Toll-like receptor (TLR) ligands remained increased following 1-month abstinence indicating long-lasting rewiring of the circulating monocytes with CHD (Figure 1C).
Figure 1.

Shifts in the single cell transcriptional profiles of bone marrow resident CD14+ cells with CHD
(A) Experimental design for study generated on Biorender.com.
(B) Timeline for blood collected after 1 month of abstinence (top) and bone marrow collected after 12 months of CHD (bottom).
(C) Bar plots showing the percent of TNF-α/IL-6+ monocytes measured using intracellular cytokine staining after 16 h of stimulation with bacterial TLR ligands performed on total PBMCs after 12 months of chronic drinking and 1-month abstinence for CHD animals and time-matched housing controls. Frequency of responding cells was corrected for the unstimulated condition. Error bars indicate SEM. ∗∗∗p < 0.001.
(D–M) scRNA-seq of 26,270 CD14+ bone marrow resident cells obtained from n = 3 female controls pooled (due to low cell numbers), three female CHDs pooled, four male controls, four male CHDs.
(D) UMAP clustering of cells.
(E) Violin plot showing expression of marker genes.
(F) Heatmap of average expression for highly expressed marker genes for classical monocyte clusters.
(G) Bar plots showing percentage of total cells contributing to each cluster for controls and CHDs. Error bars indicate SEM. ∗p < 0.05.
(H) Dot plot showing expression of CXCR4 across each cluster. Size of the dot represents percent of cells expressing the gene and the color is the average expression value.
(I) Dot plot showing expression of up- and downregulated DEGs with CHD common to all monocyte subsets. Size of the dot represents percent of cells expressing the gene and the color is the average expression value.
(J) Violin plots representing module scores for indicated pathways in total monocytes. Statistical significance was tested using the Mann-Whitney test.
(K) Dot plot showing gene expression of CCR2 across each monocyte subset split by CHD and control. Size of the dot represents percent of cells expressing the gene and the color is the average expression value.
(L) Bar plot of CCR2 surface protein expression MFI on total monocytes in bone marrow. Error bars indicate SEM.
(M) Bar plot representing −log10(q value) enrichment scores for genes upregulated in classical monocyte clusters with CHD.
(N) Bar plot of CD80 MFI on classical monocytes. Error bars indicate SEM.
(O) Percentage of IL-6+ cells within each monocyte and macrophage population in bone marrow following stimulation with a bacterial TLR cocktail corrected for the unstimulated condition. Error bars indicate SEM. Statistical significance was tested by t test with Welch’s correction where ∗p < 0.05. See also Figures S1 and S2.
scRNA-seq of bone marrow monocytes reveals increased oxidative stress and inflammatory transcriptional signatures with CHD
The persistence of an inflammatory phenotype after 1-month abstinence suggests that CHD may impact the progenitors in the bone marrow compartment. To test this hypothesis, we first assessed the abundance of classical (CD14+CD16−), intermediate (CD14+CD16+), and non-classical monocyte (CD14−CD16+) and macrophage (CD169+) populations by flow cytometry and found no changes after 12 months of CHD (Figures 1A, 1B, S1A, and S1B). To determine if CHD induced transcriptional changes, we performed small cell RNA sequencing (scRNA-seq) on CD14+ bone marrow cells after 12 months of CHD (Figure S1C). After integrating all datasets, 11 clusters were identified by the expression of canonical markers: eight classical monocyte clusters (C1-8; CD14, LYZ), one intermediate monocyte cluster (Int.; MAMU-DRA, S100A10), one non-classical monocyte cluster (N.C.; FCGR3), and one macrophage cluster (Mac.; FABP4, SIGLEC1) (Figures 1D, 1E, and S1D). Coinciding with flow cytometry data, no changes in the relative abundance of classical, intermediate, and non-classical subsets were identified with CHD (Figure S1D); however, the resolution of scRNA-seq data allowed us to identify eight classical monocyte states defined by unique expression profiles of alarmins (S100), chemokines, and interferon responsive genes (Figure 1F and Table S1). CHD was associated with a significant increase in classical cluster C8 (Figure 1G), which was defined by high expression of CREBRF, HERPUD1, FOSB, and CXCR4, the receptor for SCF-1 shown to be expressed on pro-monocytes (Chong et al., 2016) (Figure 1H).
We next identified differentially expressed genes (DEGs) within the monocyte clusters. We noted decreased expression of HSPA8, CCL4L1, EGR1, MAMU-DQA1, and SELENOP and increased expression of IL1B, S100A9, THBS1, HIF1A, and IFI27 across classical, intermediate, and non-classical clusters with CHD (Figure 1I). This was accompanied by increased module scores associated with oxidative stress and chronic inflammation in all CHD monocyte clusters (Figure 1J). Interestingly, both RNA and protein expression of CCR2, associated with monocyte egress from bone marrow (Tsou et al., 2007), was increased on classical monocytes after CHD, indicating potential dysregulation of monocyte export (Figures 1K and 1L). DEGs within classical clusters include upregulation of genes mapping to chemotaxis (CSF3R, VEGFA), response to wounding (CD44, FN1), and chronic inflammatory response (S100A8, S100A9) with CHD (Figures 1M and S1E). Functionally, expression of the co-stimulatory molecule CD80 was increased on classical monocytes suggesting higher activation with CHD (Figure 1N). Moreover, a greater percentage of classical monocytes from CHD animals produced IL-6, albeit a small percentage of the cells (Figure 1O).
In the non-classical subset, DEGs important for defense response (MRC1, STAT1) and stem cell differentiation (MEF2C, CITED2) were reduced, while genes involved in wound healing (SERPINA1, THBS1) and migration (MIF, FN1) increased with CHD (Figure S1F). These data suggest CHD alters the transcriptome of bone marrow-resident monocytes, with the potential to impact monocyte export and activation.
CHD alters phenotype and transcriptome of bone marrow resident macrophages
Bone marrow macrophages have critical functions in homeostasis of the bone marrow niche (Mitroulis et al., 2020; Winkler et al., 2010). While the frequency of this cluster did not differ between control and CHD animals (Figures 1G and S1B), considerable transcriptional changes were noted with CHD. DEGs downregulated with CHD mapped to GO terms associated with cellular responses to stimuli (CCL8, CCL13, CCL24, CCL4L1) and developmental processes (DDX5, EGR1, FOS, BTG2) (Figures S2A and S2B). Conversely, DEGs upregulated with CHD mapped to mitochondrial respiration (MT-ATP8, MT-CO2, MT-ND2, NDUFA1) and leukocyte migration (IFI27, TREM1, VEGFA, VCAN) (Figures S2A and S2C). Coinciding with these findings, the number of macrophages and the expression intensity of CD86 decreased with CHD (Figure S2D). These data suggest resident macrophage dysfunction as a potential mechanism of monocyte inflammatory profiles in the bone marrow.
CHD skews monocyte differentiation from CD34+ progenitors
As monocytes are constantly produced by and stored in the bone marrow, we investigated the impact of CHD on CD34+ progenitor cell differentiation into monocytes. We used a colony-forming unit (CFU) assay to assess the functional ability of CD34+ cells from CHD bone marrow to produce myeloid and erythroid colonies. No differences were noted in the total number of colonies after 7 or 10 days of culture (Figure S3A), but progenitors skewed toward granulocyte/monocyte-containing colonies (CFU-GM and CFU-GEMM) and away from erythroid only colonies (CFU-E and BFU-E) in CHD cultures (Figures 2A–2C). This suggests CHD alters the differentiation trajectory of CD34+ progenitors toward granulocyte/monocyte lineage.
Figure 2.

CHD alters CD34+ progenitor cell differentiation to monocytes
(A) Stacked bar plot showing percentages of indicated colonies.
(B and C) Bar plots showing the percentage of granulocyte/monocyte (B) and erythroid (C) colonies from total colonies across control and CHD groups. Error bars indicate SEM.
(D) Experimental design created on Biorender.com. Sorted CD34+ cells from control and CHD macaque bone marrow were cultured in monocyte differentiation media supplement for 7 days.
(E) Example gating strategy showing CD34+ versus CD14+ cells.
(F) Bar plots showing quantification of the culture output by flow cytometry. Error bars indicate SEM.
(G–K) scRNA-seq of 12,781 cells from pooled controls (n = 2 male + 2 female) and pooled CHD (n = 2 male + 2 female).
(G) UMAP with overlapped Slingshot pseudotime trajectory lines.
(H) Dot plot showing expression of marker genes. Size of the dot represents percent of cells expressing the gene and the color represents an average expression value.
(I) Log expression of AZU1, MPO, MAMU-DRA, and CD74 plotted for each cell across the indicated scaled Slingshot pseudotime trajectory (trendline shown).
(J) Bar plots showing representative percentages of each cluster across control and CHD groups.
(K) Cell density plots for MDP (left) and GMP (right) lineage determined by Slingshot. Statistical significance was tested by t test with Welch’s correction where ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001. See also Figure S3.
Additionally, we sorted and cultured CD34+ cells for 7 days in media with a monocyte skewing supplement (Figure 2D). Interestingly, cultures from CHD animals showed altered capacity to differentiate into CD14+CD34− cells compared with control animals, with a higher percentage of cells retaining CD34 expression (Figures 2E and 2F). Monocyte maturation markers CD115 and CD11C increased similarly in the cultured CD14+ populations regardless of CHD, indicating differentiation is occurring (Figure S3B).
To further assess differences in monocyte differentiation, we performed scRNA-seq on the differentiated cells (Figure 2D and Table S1). Using highly expressed gene markers, we identified progenitor cells (CD34, STMN1, CD38), GMP-derived monocytes (S100A8/9), monocyte-dendritic cell (MDP)-derived monocytes (IRF8, CX3CR1), monocyte-derived macrophages (SIGLEC1, S100A11), and two clusters of megakaryocyte/erythroid progenitors (PLEK, HBA, GPX4) (Figures 2G and 2H and Table S1). Pseudotime analysis identified a GMP lineage defined by increasing expression of AZU1 and MPO, and an MDP lineage defined by increasing expression of MAMU-DRA and CD74 (Figures 2G and 2I). While CD34+ cells from controls differentiated primarily along the MDP lineage into dendritic cell (DC) monocytes, CD34+ cells from the CHD group differentiated along the GMP lineage toward “neutrophil-like” monocytes (Figures 2J and 2K) (Yanez et al., 2017). Given these observations, we investigated whether the “neutrophil-like” phenotype was evident in mature CD14+ monocytes. Module scores of genes associated with neutrophil differentiation and function as well as expression of GMP lineage markers S100A8/A9 were increased in the monocytes from the CHD group, while that of MDP lineage markers CD74 and MAMU-DRA were decreased (Figures 3A and 3B).
Figure 3.

CHD-mediated alteration in differentiation results in pro-inflammatory monocytes
(A) Violin plots showing module scores of genes associated with neutrophil differentiation and function across monocyte subsets from Figure 1.
(B) Violin plots depicting expression of S100A8, S100A9, CD74, and MAMU-DRA across monocyte subsets from Figure 1.
(C) Bar plots showing the log10(fold change +1) concentration of each of the indicated analytes measured by Luminex after 6-h stimulation with a bacterial TLR cocktail. Error bars indicate SEM. Statistical significance was tested by t test with Welch’s correction where ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001.
These observations suggest that CHD induces CD34+ progenitor cells differentiation into monocytes poised toward inflammatory responses and away from antigen-presentation functions. Furthermore, monocyte-differentiated CD34+ cells stimulated with a bacterial TLR ligand cocktail had increased production of TNF-α, IL-6, IL1b, G-CSF, CCL3 (MIP-1a), and IL-2 in CHD cultures (Figure 3C). This indicates that CHD alters the differentiation potential of CD34+ progenitors and skews monocyte differentiation toward the inflammatory GMP-derived lineage.
scRNA-seq of CD34+ progenitors reveals reduced proliferation but increased oxidative stress and inflammatory pathways in myeloid progenitors with CHD
To uncover molecular underpinnings of the CD34+ cells dysregulation, we assessed the abundance of progenitor populations by flow cytometry (Figure S3C). Frequencies of HSCP and CMP were modestly increased with CHD, while those of multipotent progenitors were significantly increased (Figure S3D). To assess the transcriptional impact of CHD on these progenitors, we profiled CD34+ bone marrow cells by scRNA-seq (Table S1). UMAP clustering revealed 17 unique clusters distinguished by highly expressed gene markers (Figures S4A–S4C and Table S1). Identification of CMP, GMP, and MDP subsets was further confirmed using module scoring based on gene lists from the Human Cell Atlas bone marrow single cell dataset (Hay et al., 2018) (Figure S4D).
We then focused on the myeloid subsets to interrogate the effects of CHD on this lineage. Contaminating mature bone marrow monocytes were removed from the myeloid clusters using the CD14+ scRNA-seq data described in Figure 2 (Figure S5A). The myeloid progenitor CD34+ cells were then re-clustered and annotated via expression of HSC and myeloid lineage marker genes (Figures 4A and 4B). These included populations from less mature HSCs (HOPX, CD164) to more mature pro-neutrophils (ELANE, MPO, AZU1), monocyte progenitors (LYZ, S100A8/9), and pro-DCs (FCER1A, IRF8) (Figures 4A and 4B). CHD was associated with a significantly reduced proliferating (MKI67) MP/GMP cluster (Figure 4C). Slingshot trajectory analysis identified three major paths (Figure 4A) for pro-monocyte, pro-DC, and pro-neutrophil lineages that were comparable between CHD and controls (Figures 3D, 3E, S5B, and S5C).
Figure 4.

CHD CD34+ bone marrow myeloid progenitor single cell transcriptional profiles
scRNA-seq of 16,610 cells from three female and four male controls, and three female and four male CHD animals.
(A) UMAP with Slingshot trajectory lines.
(B) Stacked violin plot of marker gene expression.
(C) Cluster percentages between CHD and control groups. Compared using two-way ANOVA with correction for multiple comparisons. Error bars indicate SEM.
(D) Heatmap of genes that dictate the monocyte lineage trajectory.
(E) Cell density plot for control and CHD groups across the monocyte trajectory lineage.
(F and H) Functional enrichment terms for DEGs upregulated with CHD from less (F) and more (H) mature clusters split by CHD and control groups.
(G and I) Heatmap of averaged gene expression for upregulated DEGs from less (G) and more (I) mature clusters split by CHD and control groups.
(J) Violin plots for indicated module scores across groups. Statistical analysis was performed by the Mann-Whitney test. Unless indicated, statistical significance was tested by t test with Welch’s correction. ∗p < 0.05, ∗∗∗∗p < 0.0001. See also Figures S4 and S5.
The cells along the monocyte differentiation pseudotime were grouped into less mature (HSC 1, HSC 2, CMP/GMP, GMP) and more mature (GMP/MP 1, GMP/MP 2, MP/GMP [Ki67], MP, MP [Ki67]) monocyte progenitor cells (Figure 4D). Upregulated DEGs within the less mature progenitor cells were involved in “oxidative phosphorylation” and “leukocyte aggregation” processes after CHD (Figures 4F and 4G), but downregulated DEGs were involved in “cellular oxidant detoxification” and “regulation of chemokine production” processes (Figures S5D and S5E). Similarly, expression of genes enriching to “oxidative phosphorylation” and “regulation of cell activation” processes was increased in the more mature progenitors with CHD (Figures 4H and 4I) while genes enriching to “cytoplasmic translation” and “peptide biosynthetic process” were downregulated (Figures S5F and S5G). Finally, module scores of oxidative stress and chronic inflammation were significantly upregulated across all CD34+ cells with CHD (Figure 4J). These data suggest that CHD upregulates the transcriptional signatures of oxidative stress and inflammation/activation in the CD34+ progenitor cells and throughout the monocyte differentiation lineage.
scATAC-seq of CD34+ and CD14+ cells reveal broad changes in the epigenome of MPs
To determine if epigenetic mechanisms were driving transcriptional changes, we sorted CD14+ and CD34+ cells from the bone marrow and performed single cell ATAC sequencing (Table S3). Initial UMAP clustering revealed 13 clusters, defined by gene scores of canonical lineage markers (Figures S6A–6C and Table S3). We then extracted HSC, CMP-GMP, and CD14+ subsets based on expression of established markers (HPOX8, IRF78, ELANE, MPO, LYZ, MAMU-DRA, CD14) and re-clustered them (Figure 5A and Table S3). This analysis revealed six clusters: HSC (HOPX), GMP 1 and 2 (ELANE), MP/GMP (LYZ), cMoP (LYZ), and CD14+ (Figures 5A and 5B). Cluster annotation was confirmed by transcription factor (TF) motif deviation scores, showing a greater abundance of motif binding sites for FOSL2, CEBPA, and JUNB in more mature progenitors, while HSCs contained more HOXB8 motif binding sites (Figure S6D).
Figure 5.

CD14+ and CD34+ bone marrow cell scATAC-seq
scATAC-seq of 32,200 cells from n = 3 male controls and n = 2 female and 2 male CHD animals.
(A) UMAP with pseudotime lineage trajectories.
(B) Feature plots of marker genes from the gene score matrix.
(C) Integrative analysis of TF gene scores and motif accessibility across the monocyte lineage pseudotime. TFs unique to the indicated group are bolded and colored (green for controls and blue for CHD).
(D) A plot of TF motif accessibility along pseudotime for TF unique to CHD.
(E–H) Functional enrichment from EnrichR (E and G) and Metascape (F and H) databases for differentially accessible regions (DARs) more open in the indicated group for HSC (E and F) and CD14+ (G and H) clusters. Color indicates the −log10(P) value and length indicates the odds ratio from EnrichR (E and G) or the number of genes mapping to the term from Metascape (F and H).
(I) Functional enrichment determined by GO Biological Process within GREAT database for differentially accessible regions more open control (left) and CHD (right) groups for the pro-monocyte cluster. Color indicates the −log10(P) value and length indicates the log2(Fold Enrichment). See also Figure S6.
As the frequency of cell subsets was not changed with CHD (Figure S6E), we performed trajectory analysis using Slingshot to uncover potential differences in monocyte differentiation between groups (Figures 5A and 5C). Motif accessibility for key TFs including REL, BACH1, PPARD, SPI1, HIVEP1, MAGF, ZIC4, and KLF1, increased with CHD, indicative of changes in monocyte differentiation (Figures 5C and 5D). Next, we compared differentially accessible regions (DARs) within each cluster. We observed decreased accessibility in promoter, 5′ UTR, downstream, and intronic regions in HSC and CD14+ clusters with CHD (Figure S6F). Functional enrichment of promoters, 5′ UTR, and downstream regions that were more open in HSC of control animals (closed in CHD group) harbored genes important for cytokine production, differentiation and NF-κB signaling, while those more open with CHD played a role in defense response, Th1, and type 1 interferon (IFN) signaling, indicating increased inflammation with CHD (Figures 5E and 5F).
Functional enrichment of these regions in CD14+ cells from controls mapped to terms such as differentiation, MAPK activity, and IL-12 production, while those open after CHD mapped to apoptotic processes, protein localization, and IL-6 signaling (Figures 5G and 5H). GREAT analysis of DARs in distal intergenic regions open in CD14+ cells from controls enriched to cell-cell adhesion, “negative regulation of MAPK cascade,” and IFN-γ signaling, while those more open in CHD mapped to “negative regulation of catabolic process” (Figure 5I). These observations indicate that CHD impacts monocytes and their progenitors in the bone marrow on an epigenetic level, altering the differentiation trajectory and leading to the functional dysregulation observed in the periphery.
Discussion
CHD can disrupt antimicrobial defenses and exacerbate inflammatory responses of monocytes and macrophages (Lewis et al., 2021; Sureshchandra et al., 2019a, 2019b; Szabo and Saha, 2015). Using an NHP model of voluntary ethanol consumption, we have shown increased relative abundance of blood monocytes and splenic macrophages with CHD (Lewis et al., 2021; Sureshchandra et al., 2019b). Moreover, we and others have noted CHD increased inflammatory responses following stimulation with LPS (Lewis et al., 2021; Sureshchandra et al., 2019a, 2019b; Szabo and Saha, 2015). Here, we show that increased inflammatory responses to TLR stimulation persist following a 1-month abstinence period, suggesting that CHD could dysregulate monocyte production/maturation in the bone marrow. Other studies have suggested ethanol and its metabolites modulate monocyte function via epigenetic modification and oxidative stress of both mature cells and HSCPs, but exact mechanisms remain poorly defined (Lewis et al., 2021; Sureshchandra et al., 2019b; Varlamov et al., 2020). In this study, we assessed the phenotypic, functional, transcriptomic, and epigenomic profiles of bone marrow resident monocytes/macrophages and CD34+ HSC collected from NHP after 12 months of CHD.
While we noted no differences in relative abundance of mature monocytes in the bone marrow with CHD, scRNA-seq showed increased expression of inflammatory genes across all bone marrow resident CD14+ cell subsets. Interestingly, the frequency of CXCR4hi monocytes, transitional pre-monocytes that help maintain the mature monocyte pool in the bone marrow (Chong et al., 2016), increased with CHD. Expression of CCR2, which plays a critical role in monocyte egress from the bone marrow (Tsou et al., 2007), also increased on bone marrow resident monocytes with CHD. Collectively, these observations suggest CHD-induced disruption of bone marrow monocyte trafficking between the bone marrow and blood.
We also report skewing of CD34+ HSCs toward the GMP (“neutrophil-like” monocyte) lineage and away from MDP (DC-monocyte) lineage that was accompanied by an increase in inflammatory mediator production. We previously reported increases in “neutrophil-like” markers (S100A8, S100A9) with CHD in peripheral monocytes (Lewis et al., 2021), suggesting long-lasting reprogramming of monocytes with CHD. These data suggest CHD-induced shifts in myelopoiesis and monopoiesis from CD34+ progenitors, giving rise to monocytes poised toward a heightened inflammatory state. Indeed, bone marrow resident classical monocytes from CHD animals produced higher levels of IL-6 than those found in controls. While the frequency of IL-6-producing cells was rather low, classical monocytes are the predominant cluster and therefore the overall increase in production of IL-6 could be substantial. We also observed decreased expression of genes important for antigen processing and presentation, which is in line with reduced antimicrobial function noted in vivo with CHD (Barr et al., 2016; Lewis et al., 2021).
CHD metabolism produces acetaldehyde and increases NADH levels, promoting reactive oxygen species production (Di Rocco et al., 2019). These products induce DNA damage and oxidative stress on cells, directly affecting cellular function (Di Rocco et al., 2019; Garaycoechea et al., 2012). Indeed, differential expression analysis from scRNA-seq data revealed increases in transcriptional signatures of oxidative stress and chronic inflammation in bone marrow resident monocytes/macrophages and HSCPs. Additionally, expression of mitochondrial genes increased in MPs, suggesting CHD-induced shifts in the cellular metabolism of monocytes/macrophages and HSCPs. These observations are in line with an earlier study that reported altered mitochondrial function in NHP bone marrow cells after 12 months of chronic drinking followed by another 12 months of repeated abstinence/re-exposure periods (Varlamov et al., 2020) and a recent study reporting increased expression of mitochondrial genes in alveolar macrophages with CHD (Lewis et al., 2022). These data suggest that the hyper-inflammatory profiles of circulating monocytes with CHD could be attributed to CHD-induced oxidative stress and mitochondrial function on CD34+ progenitors in the bone marrow.
Previous work showed considerable epigenetic changes in peripheral monocytes and tissue-resident macrophages with CHD (Lewis et al., 2021, 2022; Sureshchandra et al., 2019b). In this study, we identified modest epigenetic changes with CHD in bone marrow resident cells. The larger epigenetic changes detected in the periphery may be driven by the larger concentration of ethanol and its metabolites in circulation. We did observe altered DAR in cis-regulatory regions and an increase in terms associated with inflammation in HSCPs and bone marrow resident CD14+ cells. Most notably, accessibility of regions overlapping genes that regulate or respond to IL-6 increased.
This study suggests that CHD alters monopoiesis from the bone marrow compartment in NHPs. CD14+ cells that reside in the bone marrow compartment have inflammatory-skewed transcriptional profiles, suggesting CHD remodeling of this tissue compartment. Mechanistically, CHD disrupted the differentiation of CD34+ cells into monocytes leading to the in vitro production of hyper-inflammatory monocytes. These disruptions are potentially mediated by compromised niche-maintaining macrophages as indicated by decreased expression of chemokines and heightened signatures of oxidative phosphorylation.
Some of the limitations of this work include small sample sizes and lack of analyses using sex as a biological variable due to the low number of cells available from female drinking macaques and complex design of the voluntary NHP drinking model. The low cell availability limited the number of functional assays performed as well. Future research efforts will focus on determining a timeline for alcohol-induced bone marrow remodeling, how alcohol alters monocyte release from the bone marrow compartment, and the impact of abstinence and relapse on these findings.
Experimental procedures
Resource availability
Corresponding author
Additional information and requests for resources should be directed to Dr. Ilhem Messaoudi (ilhem.messaoudi@uky.edu).
Materials availability
This study did not generate new unique reagents.
Animal studies and sample collection
This study was approved by the Oregon National Primate Research Center (ONPRC) Institutional Animal Care and Use Committee. ONPRC is an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-approved institute.
These studies used samples obtained from an NHP model of ethanol self-administration as described in Baker et al. (2014), Grant et al. (2008), and Jimenez et al. (2015).
Blood (Cohort 14) and bone marrow (Cohorts 6a and 7a) samples were acquired through the Monkey Alcohol Tissue Research Resource (www.matrr.com) after 12 months of CHD (Cohorts 6a and 7a) and 1-month abstinence (Cohort 14) (Table 1).
Table 1.
Summary of samples used in this study
| MATRR ID | Sex | Drinking status | Mean daily ethanol intake (g/kg/d) ± SD | Blood ethanol content (Avg mg%) |
|---|---|---|---|---|
| 10068 | F | Control | 0 | 0 |
| 10071 | F | Control | 0 | 0 |
| 10076 | F | Control | 0 | 0 |
| 10186 | F | Control | 0 | 0 |
| 10187 | F | Control | 0 | 0 |
| 10093 | M | Control | 0 | 0 |
| 10094 | M | Control | 0 | 0 |
| 10095 | M | Control | 0 | 0 |
| 10096 | M | Control | 0 | 0 |
| 10245 | M | Control | 0 | 0 |
| 10250 | M | Control | 0 | 0 |
| 10241 | M | Control | 0 | 0 |
| 10080 | F | EtOH | 3.3 ± 1.0 | 41 |
| 10070 | F | EtOH | 3.9 ± 1.0 | 46 |
| 10081 | F | EtOH | 4.0 ± 1.1 | 57 |
| 10079 | F | EtOH | 4.0 ± 1.3 | 65 |
| 10078 | F | EtOH | 5.0 ± 1.4 | 81 |
| 10069 | F | EtOH | 5.2 ± 1.2 | 103 |
| 10088 | M | EtOH | 2.9 ± 1.3 | 74 |
| 10091 | M | EtOH | 3.2 ± 0.9 | 79 |
| 10097 | M | EtOH | 3.0 ± 0.6 | 79 |
| 10098 | M | EtOH | 3.3 ± 1.0 | 98 |
| 10252 | M | EtOH | 4.17 ± 0.9 | 91 |
| 10242 | M | EtOH | 3.2 ± 0.7 | 113 |
| 10244 | M | EtOH | 2.8 ± 0.5 | 97 |
| 10243 | M | EtOH | 2.2 ± 0.5 | 30 |
Mean daily ethanol (EtOH) intake reflects the average dose consumed during the period of 12-month self-administration period. Blood for ethanol concentration was taken at 7 h into the 22-h daily session every 5–7 days throughout the 12 consecutive months of alcohol access and analyzed by gas chromatography.
Phenotype
A total of 0.5 to 1 × 106 PBMCs were stained, acquired with an Attune NxT Flow Cytometer (ThermoFisher Scientific, Waltham, MA), and analyzed using FlowJo software (Ashland, OR). Percentage of live cells or median fluorescence intensities (MFI) were assessed for each marker. Staining panels and progenitor population definitions are in the supplemental methods.
PBMC and bone marrow stimulation assays
A total of 1 × 106 PBMCs were cultured in RPMI supplemented with 10% fetal bovine serum (FBS) with or without a bacterial agonist cocktail and Brefeldin A for 16 h in 96-well tissue culture plates at 37 °C in a 5% CO2 environment. They were next stained, fixed, and permeabilized using Fixation buffer (BioLegend) and incubated overnight with intracellular antibodies IL-6 (Invitrogen, MQ2-13A5, FITC) and TNF-ɑ (BD Biosciences, MAb11, APC).
Monocyte differentiation assay
A total of 1 × 103 sorted CD34+ cells were plated in a 96-well plate in 100 μL StemSpan SFEM Media supplemented with StemSpan Myeloid Expansion Supplement II containing TPO, SCF, Flt3, GM-CSF, and M-CSF and incubated at 37 °C in a 5% CO2 environment. On day 4, 100 μL of the same media was added. On day 7, some of the cells were stained with antibodies for phenotype determination. Some of the cells were used for scRNA-seq analysis and the remaining cells were incubated with or without a bacterial agonist cocktail for 6 h. Supernatants were collected and stored short-term at −80°C.
Luminex assay
Immune mediators in the supernatants cell stimulations were measured using a Multiplex Luminex assay panel (Millipore, Burlington, MA). Differences in induction of proteins post stimulation were calculated using log10 (pg/mL fold change +1). Undiluted samples were run in duplicate on the Magpix Instrument (Luminex, Austin, TX). Data were fit using a 5P-logistic regression on xPONENT software (version 7.0c).
CFU assay
MethoCult CFU assay was performed with MethoCult H4435-enriched medium (STEMCELL Technologies, Vancouver, Canada) following manufacturer’s protocol. Briefly, CD34+ bone marrow cells were sorted using fluorescence-activated cell sorting (FACS) and resuspended in Iscove’s modified Dulbecco’s medium +2% FBS to achieve a final plating concentration of 1,000 cells/culture dish. Cells were mixed with 3 mL MethoCult and plated into 35-mm culture dishes in triplicate then incubated at 37°C in a 5% CO2 environment. Total colonies were counted on days 7 and 10, and colony identification was performed on day 10.
Single cell RNA library preparation, sequencing, and analysis
Specific parameters for each scRNA-seq library can be found in Table S1. Cells were thawed, surface stained, and sorted using a BD FACSAria Fusion before being loaded into the 10X Chromium Controller. Library preparations were performed for samples according to manufacturer protocol (10X v3.1 dual index chemistry) and sequenced on a NovaSeq S4 (Illumina).
Sequencing reads were aligned to the Mmul_8.0.1 reference genome using cellranger v6.0.1 (Zheng et al., 2017) using the count function for single sample libraries and the multi function for multiplexed samples. Quality control steps were performed prior to downstream analysis with Seurat (Butler et al., 2018). Data normalization was performed using SCTransform (Hafemeister and Satija, 2019). Sample integration was performed using the SelectIntegrationFeatures (using 3000 features), PrepSCTIntegration, FindIntegrationAnchors, and IntegrateData functions. Contaminating clusters with B or T cell gene expression were removed. Clusters were characterized using the FindAllMarkers function and all marker gene lists are available in Table S1. Figures were generated using Seurat, ggplot2, and pheatmap and each dot corresponds to an individual animal.
Differential expression analysis was performed using MAST with significant genes defined as Log10(Fold change) ≥ 0.25; false discovery rate (FDR) ≤ 0.05.
Pseudotime trajectory were reconstructed using Slingshot (Street et al., 2018) with the UMAP dimensional reduction from Seurat as input and the progenitor/HSC population selected as the start. Temporally expressed genes were identified by ranking all genes by their variance across pseudotime and then further fit using the generalized additive model with pseudotime as an independent variable.
Gene lists from the Kyoto Encyclopedia of Genes and Genomes pathway dataset and the Human Cell Atlas bone marrow single cell analysis (Hay et al., 2018) were used with the AddModuleScore function. A list of all module scores can be found in Table S2. Values for module scores were tested for significance in Prism 7 and functional enrichment was done using Metascape (Zhou et al., 2019). All plots were generated using ggplot2 and Seurat and each dot corresponds to an individual animal.
Single cell ATAC-seq library preparation, sequencing, and analysis
Bone marrow cells were stained for CD34, CD14, and Sytox Green, and sorted on a BD FACSAria Fusion before being lysed. Nuclei were isolated and counted, and transposition reaction performed according to the manufacturer’s instructions (10X Genomics). A total of 3,000–20,000 nuclei were loaded into the 10X Chromium Controller. Library preparation (10X v2 chemistry) was performed according to the manufacturer’s protocol and sequenced on a NovaSeq S4 (Illumina) to a depth of >25,000 paired reads/cell.
Sequencing reads were pre-processed using the cellranger-atac pipeline (v2.1.0) (10X Genomics) and aligned to the Mmul_8.0.1 reference genome. The ArchR package (Granja et al., 2021) (v1.0.1) was used for downstream analysis. Arrow files were created and low-quality cells were filtered out (<3,162 fragments, <8 transcriptional start site (TSS) enrichment, doublets calculated by addDoubletScores). An ArchR project was created by combining all Arrow files. Iterative latent semantic indexing was performed as the first dimensional reduction followed by the addHarmony function to correct batch effects. UMAP was used for the final dimensional reduction and clusters were added using the addClusters function. Marker features were identified using the getMarkerFeatures function. Myeloid cell lineage clusters were defined by accessible marker genes, extracted, and the above dimensional reduction and clustering steps were performed again. Pseudo-bulk replicates were created for peak calling using MACS2. Per-cell deviations across motif annotations were computed using the addDeviationsMatrix function. The getMarkerFeatures function was used to determine DAR between CHD and control.
Trajectory analysis was performed using the addTrajectory function in ArchR. To identify positive TF regulators, we performed an integrative analysis of gene scores and motif accessibility across the identified psuedotime trajectory.
Genomic annotation of open chromatin regions related to the promoter, 5′ UTR, downstream, and distal intergenic regions in DAR analysis was assigned using ChIPSeeker. Promoters were defined as −1,000 base pairs (bp) to +100 bp around the TSS. Genes with no annotations were excluded from downstream analyses. Functional enrichment analysis was performed using Metascape and Enrichr. The distal intergenic regions of the macaque were converted to the human genome (hg38) coordinates using the UCSC liftOver tool due to the lack of available macaque annotation databases (Prescott et al., 2015). The functions of cis-regulatory regions were predicted using GREAT.
Statistical analysis
All statistical analyses were conducted in Prism 7. Datasets were first tested for normality (Gaussian distribution) and outliers (ROUT method). Two group comparisons were carried out using an unpaired t test with Welch’s correction (parametric) or Mann-Whitney (non-parametric) test. Differences among three groups were tested using one-way ANOVA (α = 0.05) followed by Holm Sidak’s multiple comparisons tests. Error bars for all graphs are defined as ± standard error of the mean (SEM) and each dot corresponds to an individual animal. FDR p values <0.05 were considered statistically significant. Values between 0.05 and 0.1 are reported as trending patterns.
Acknowledgments
We thank the members of the Grant laboratory for expert animal care and sample procurement; Dr. Jennifer Atwood for assistance with sorting in the flow cytometry core at the Institute for Immunology, UCI; Dr. Melanie Oakes from the UCI Genomics and High-Throughput Facility for assistance with sequencing; and Dr. Helen Goodridge for assistance with CFU assay protocols and analysis. This study was supported by NIH 1R01AA028735-01 (I.M.), 5U01AA013510-20 (K.A.G.), 2R24AA019431-11 (K.A.G.), and P-51OD011092 (ONPRC core grant support). S.A.L. was supported by NIH 1F31A028704-01. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author contributions
S.A.L., K.A.G., and I.M. conceived and designed the experiments. S.A.L., B.M.D., and M.B.B. performed the experiments. S.A.L., B.M.D., M.B.B., and Q.Q. analyzed the data. S.A.L. and I.M. wrote the paper. All authors have read and approved the final draft of the manuscript.
Conflict of interests
The authors declare no competing interests.
Published: August 31, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2023.08.001.
Supplemental information
Data and code availability
The datasets supporting the conclusions of this article are available on NCBI’s Sequence Read Archive (SRA: PRJNA947630).
References
- Allen D.C., Gonzales S.W., Grant K.A. Effect of repeated abstinence on chronic ethanol self-administration in the rhesus monkey. Psychopharmacology (Berl) 2018;235:109–120. doi: 10.1007/s00213-017-4748-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker E.J., Farro J., Gonzales S., Helms C., Grant K.A. Chronic alcohol self-administration in monkeys shows long-term quantity/frequency categorical stability. Alcohol Clin. Exp. Res. 2014;38:2835–2843. doi: 10.1111/acer.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge M.T., King K.Y., Goodell M.A. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 2011;32:57–65. doi: 10.1016/j.it.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard H.S. The hematological complications of alcoholism. Alcohol Health Res. World. 1997;21:42–52. [PMC free article] [PubMed] [Google Scholar]
- Barr T., Helms C., Grant K., Messaoudi I. Opposing effects of alcohol on the immune system. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2016;65:242–251. doi: 10.1016/j.pnpbp.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum M.K., Rafie C., Lai S., Sales S., Page J.B., Campa A. Alcohol use accelerates HIV disease progression. AIDS Res. Hum. Retroviruses. 2010;26:511–518. doi: 10.1089/aid.2009.0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya R., Shuhart M.C. Hepatitis C and alcohol: interactions, outcomes, and implications. J. Clin. Gastroenterol. 2003;36:242–252. doi: 10.1097/00004836-200303000-00012. [DOI] [PubMed] [Google Scholar]
- Bruha R., Dvorak K., Dousa M., Petrtyl J., Svestka T. Alcoholic liver disease. Prague Med. Rep. 2009;110:181–190. [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S.Z., Evrard M., Devi S., Chen J., Lim J.Y., See P., Zhang Y., Adrover J.M., Lee B., Tan L., et al. CXCR4 identifies transitional bone marrow premonocytes that replenish the mature monocyte pool for peripheral responses. J. Exp. Med. 2016;213:2293–2314. doi: 10.1084/jem.20160800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rocco G., Baldari S., Pani G., Toietta G. Stem cells under the influence of alcohol: effects of ethanol consumption on stem/progenitor cells. Cell. Mol. Life Sci. 2019;76:231–244. doi: 10.1007/s00018-018-2931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaycoechea J.I., Crossan G.P., Langevin F., Daly M., Arends M.J., Patel K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489:571–575. doi: 10.1038/nature11368. [DOI] [PubMed] [Google Scholar]
- Granja J.M., Corces M.R., Pierce S.E., Bagdatli S.T., Choudhry H., Chang H.Y., Greenleaf W.J. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021;53:403–411. doi: 10.1038/s41588-021-00790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant K.A., Leng X., Green H.L., Szeliga K.T., Rogers L.S.M., Gonzales S.W. Drinking typography established by scheduled induction predicts chronic heavy drinking in a monkey model of ethanol self-administration. Alcohol Clin. Exp. Res. 2008;32:1824–1838. doi: 10.1111/j.1530-0277.2008.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay S.B., Ferchen K., Chetal K., Grimes H.L., Salomonis N. The Human Cell Atlas bone marrow single-cell interactive web portal. Exp. Hematol. 2018;68:51–61. doi: 10.1016/j.exphem.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez V.A., Helms C.M., Cornea A., Meshul C.K., Grant K.A. An ultrastructural analysis of the effects of ethanol self-administration on the hypothalamic paraventricular nucleus in rhesus macaques. Front. Cell. Neurosci. 2015;9:260. doi: 10.3389/fncel.2015.00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura S., Ohteki T. Monopoiesis in humans and mice. Int. Immunol. 2018;30:503–509. doi: 10.1093/intimm/dxy063. [DOI] [PubMed] [Google Scholar]
- Latvala J., Parkkila S., Niemelä O. Excess alcohol consumption is common in patients with cytopenia: studies in blood and bone marrow cells. Alcohol Clin. Exp. Res. 2004;28:619–624. doi: 10.1097/01.alc.0000122766.54544.3b. [DOI] [PubMed] [Google Scholar]
- Lee J., Breton G., Oliveira T.Y.K., Zhou Y.J., Aljoufi A., Puhr S., Cameron M.J., Sékaly R.P., Nussenzweig M.C., Liu K. Restricted dendritic cell and monocyte progenitors in human cord blood and bone marrow. J. Exp. Med. 2015;212:385–399. doi: 10.1084/jem.20141442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis S.A., Doratt B.M., Sureshchandra S., Jankeel A., Newman N., Shen W., Grant K.A., Messaoudi I. Ethanol consumption induces non-specific inflammation and functional defects in alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 2022;67:112–124. doi: 10.1165/rcmb.2021-0346OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis S.A., Sureshchandra S., Doratt B., Jimenez V.A., Stull C., Grant K.A., Messaoudi I. Transcriptional, epigenetic, and functional reprogramming of monocytes from non-human primates following chronic alcohol drinking. Front. Immunol. 2021;12:724015. doi: 10.3389/fimmu.2021.724015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.K. Effects of alcohol on granulocytes and lymphocytes. Semin. Hematol. 1980;17:130–136. [PubMed] [Google Scholar]
- Mitroulis I., Kalafati L., Bornhäuser M., Hajishengallis G., Chavakis T. Regulation of the bone marrow niche by inflammation. Front. Immunol. 2020;11:1540. doi: 10.3389/fimmu.2020.01540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamal K.J., Rimm E.B. Alcohol's effects on the risk for coronary heart disease. Alcohol Res. Health. 2001;25:255–261. [PMC free article] [PubMed] [Google Scholar]
- O'Brien J.M., Jr., Lu B., Ali N.A., Levine D.A., Aberegg S.K., Lemeshow S. Insurance type and sepsis-associated hospitalizations and sepsis-associated mortality among US adults: a retrospective cohort study. Crit. Care. 2011;15:R130. doi: 10.1186/cc10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe J.H., Bhatti S.K., Bajwa A., DiNicolantonio J.J., Lavie C.J. Alcohol and cardiovascular health: the dose makes the poison.or the remedy. Mayo Clin. Proc. 2014;89:382–393. doi: 10.1016/j.mayocp.2013.11.005. [DOI] [PubMed] [Google Scholar]
- Panasiuk A., Kemona A. Bone marrow failure and hematological abnormalities in alcoholic liver cirrhosis. Rocz. Akad. Med. Bialymst. 2001;46:100–105. [PubMed] [Google Scholar]
- Prescott S.L., Srinivasan R., Marchetto M.C., Grishina I., Narvaiza I., Selleri L., Gage F.H., Swigut T., Wysocka J. Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell. 2015;163:68–83. doi: 10.1016/j.cell.2015.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raasch C.E., Zhang P., Siggins R.W., 2nd, LaMotte L.R., Nelson S., Bagby G.J. Acute alcohol intoxication impairs the hematopoietic precursor cell response to pneumococcal pneumonia. Alcohol Clin. Exp. Res. 2010;34:2035–2043. doi: 10.1111/j.1530-0277.2010.01291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumgay H., Murphy N., Ferrari P., Soerjomataram I. Alcohol and cancer: epidemiology and biological mechanisms. Nutrients. 2021;13:3173. doi: 10.3390/nu13093173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitz R., Ghali W.A., Moskowitz M.A. The impact of alcohol-related diagnoses on pneumonia outcomes. Arch. Intern. Med. 1997;157:1446–1452. [PubMed] [Google Scholar]
- Shi X., DeLucia A.L., Bao J., Zhang P. Alcohol abuse and disorder of granulopoiesis. Pharmacol. Ther. 2019;198:206–219. doi: 10.1016/j.pharmthera.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X., Lin Y.P., Gao B., Zhang P. Impairment of hematopoietic precursor cell activation during the granulopoietic response to bacteremia in mice with chronic-plus-binge alcohol administration. Infect. Immun. 2017;85:e00369-17. doi: 10.1128/IAI.00369-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggins R.W., Molina P., Zhang P., Bagby G.J., Nelson S., Dufour J., LeCapitaine N.J., Walsh C., Welsh D.A. Dysregulation of myelopoiesis by chronic alcohol administration during early SIV infection of rhesus macaques. Alcohol Clin. Exp. Res. 2014;38:1993–2000. doi: 10.1111/acer.12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C., Gasparetto M., Jordan C., Pollyea D.A., Vasiliou V. The effects of alcohol and aldehyde dehydrogenases on disorders of hematopoiesis. Adv. Exp. Med. Biol. 2015;815:349–359. doi: 10.1007/978-3-319-09614-8_20. [DOI] [PubMed] [Google Scholar]
- Street K., Risso D., Fletcher R.B., Das D., Ngai J., Yosef N., Purdom E., Dudoit S. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018;19:477. doi: 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureshchandra S., Rais M., Stull C., Grant K., Messaoudi I. Transcriptome profiling reveals disruption of innate immunity in chronic heavy ethanol consuming female rhesus macaques. PLoS One. 2016;11:e0159295. doi: 10.1371/journal.pone.0159295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureshchandra S., Raus A., Jankeel A., Ligh B.J.K., Walter N.A.R., Newman N., Grant K.A., Messaoudi I. Dose-dependent effects of chronic alcohol drinking on peripheral immune responses. Sci. Rep. 2019;9:7847. doi: 10.1038/s41598-019-44302-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureshchandra S., Stull C., Ligh B.J.K., Nguyen S.B., Grant K.A., Messaoudi I. Chronic heavy drinking drives distinct transcriptional and epigenetic changes in splenic macrophages. EBioMedicine. 2019;43:594–606. doi: 10.1016/j.ebiom.2019.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G., Saha B. Alcohol's effect on host defense. Alcohol Res. 2015;37:159–170. [PMC free article] [PubMed] [Google Scholar]
- Takizawa H., Boettcher S., Manz M.G. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood. 2012;119:2991–3002. doi: 10.1182/blood-2011-12-380113. [DOI] [PubMed] [Google Scholar]
- Teh Y.C., Ding J.L., Ng L.G., Chong S.Z. Capturing the fantastic voyage of monocytes through time and space. Front. Immunol. 2019;10:834. doi: 10.3389/fimmu.2019.00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou C.L., Peters W., Si Y., Slaymaker S., Aslanian A.M., Weisberg S.P., Mack M., Charo I.F. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varlamov O., Bucher M., Myatt L., Newman N., Grant K.A. Daily ethanol drinking followed by an abstinence period impairs bone marrow niche and mitochondrial function of hematopoietic stem/progenitor cells in rhesus macaques. Alcohol Clin. Exp. Res. 2020;44:1088–1098. doi: 10.1111/acer.14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . World Health Organization; 2018. Global Status Report on Alcohol and Health 2018.https://apps.who.int/iris/handle/10665/274603 [Google Scholar]
- Winkler I.G., Sims N.A., Pettit A.R., Barbier V., Nowlan B., Helwani F., Poulton I.J., van Rooijen N., Alexander K.A., Raggatt L.J., Lévesque J.P. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010;116:4815–4828. doi: 10.1182/blood-2009-11-253534. [DOI] [PubMed] [Google Scholar]
- Wolf A.A., Yáñez A., Barman P.K., Goodridge H.S. The ontogeny of monocyte subsets. Front. Immunol. 2019;10:1642. doi: 10.3389/fimmu.2019.01642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez A., Coetzee S.G., Olsson A., Muench D.E., Berman B.P., Hazelett D.J., Salomonis N., Grimes H.L., Goodridge H.S. Granulocyte-monocyte progenitors and monocyte-dendritic cell progenitors independently produce functionally distinct monocytes. Immunity. 2017;47:890–902.e94. doi: 10.1016/j.immuni.2017.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the conclusions of this article are available on NCBI’s Sequence Read Archive (SRA: PRJNA947630).