

Abstract
Renal fibrosis is a common pathological feature of chronic kidney disease (CKD) with various etiologies, which seriously affects the structure and function of the kidney. Pregnane X receptor (PXR) is a member of the nuclear receptor superfamily and plays a critical role in regulating the genes related to xenobiotic and endobiotic metabolism in mammals. Previous studies show that PXR is expressed in the kidney and has protective effect against acute kidney injury (AKI). In this study, we investigated the role of PXR in CKD. Adenine diet-induced CKD (AD) model was established in wild-type and PXR humanized (hPXR) mice, respectively, which were treated with pregnenolone-16α-carbonitrile (PCN, 50 mg/kg, twice a week for 4 weeks) or rifampicin (RIF, 10 mg·kg−1·d−1, for 4 weeks). We showed that both PCN and RIF, which activated mouse and human PXR, respectively, improved renal function and attenuated renal fibrosis in the two types of AD mice. In addition, PCN treatment also alleviated renal fibrosis in unilateral ureter obstruction (UUO) mice. On the contrary, PXR gene deficiency exacerbated renal dysfunction and fibrosis in both adenine- and UUO-induced CKD mice. We found that PCN treatment suppressed the expression of the profibrotic Wnt7a and β-catenin in AD mice and in cultured mouse renal tubular epithelial cells treated with TGFβ1 in vitro. We demonstrated that PXR was colocalized and interacted with p53 in the nuclei of tubular epithelial cells. Overexpression of p53 increased the expression of Wnt7a, β-catenin and its downstream gene fibronectin. We further revealed that p53 bound to the promoter of Wnt7a gene to increase its transcription and β-catenin activation, leading to increased expression of the downstream profibrotic genes, which was inhibited by PXR. Taken together, PXR activation alleviates renal fibrosis in mice via interacting with p53 and inhibiting the Wnt7a/β-catenin signaling pathway.
Keywords: chronic kidney disease, renal fibrosis, PXR, Wnt7a, β-catenin, p53
Introduction
Chronic kidney disease (CKD) is a clinical syndrome secondary to renal functional or concomitant structural damages. The main causes of CKD include diabetes mellitus, hypertension, glomerulonephritis, pyelonephritis, autoimmune diseases, polycystic kidney disease, Alport syndrome, congenital malformations, long-term acute kidney disease, and chronic use of anti-inflammatory drugs [1]. CKD afflicts ~10%–13% population worldwide [2]. Most patients with CKD are asymptomatic in the early stage of the disease, but as the disease progresses, a considerable proportion of patients will eventually develop end-stage renal disease (ESRD), a disease requiring lifelong dialysis or kidney transplant. Numerous epidemiological studies have shown that the prevalence of ESRD is constantly increasing globally, especially in the developed countries [3–5]. The high cost of treatment and increasing prevalence have made CKD a major internationally public health problem, imposing a huge socioeconomic burden on affected individuals, families, and societies.
Renal fibrosis, a common pathological feature and final manifestation of CKD with various etiologies, is characterized by inflammatory cell infiltration, innate cell mesenchymal transformation, increased extracellular matrix protein production and decreased matrix degradation [6]. It is well known that several pathways are closely related to the occurrence and development of renal fibrosis including the transforming growth factor (TGF)-β/Smad, Notch, Hedgehog and yes-associated protein 1 (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) signaling pathways [7–9]. Notably, recent studies have revealed that Wingless/Int (Wnt) signaling pathway, which was first reported for its important role in embryonic development, plays a critical role in the progression of renal fibrosis [10]. In adult kidneys, this signaling pathway is usually silenced, but is reactivated when the kidney is damaged. Numerous studies have shown that persistent abnormal activation of the Wnt/β-catenin signaling pathway can accelerate the progression of renal fibrosis, while inhibition of this pathway can effectively alleviate renal fibrosis via decreasing expressions of fibronectin, fibroblast-specific protein 1, Snail1, matrix metalloproteinase7 (MMP-7), plasminogen activator inhibitor-1 (PAI-1) [11–13]. These findings demonstrate that targeting the Wnt/β-catenin signaling pathway may represent an attractive strategy to treat renal fibrosis [7, 14, 15]. However, there is hitherto no effective and safe treatment for renal fibrosis and CKDs in clinical practice.
As a member of the nuclear receptor superfamily, pregnane X receptor (PXR, Nr1i2) is a ligand-activated transcription factor which plays an important role in metabolizing endogenous substances and exogenous drugs [16]. After activated by a ligand, PXR relocalizes into the nucleus where it forms a heterodimer with the retinoid X receptor (RXR), recruiting multiple co-activators to increase the transcription of its downstream genes encoding the type I and type II detoxification enzymes. Unlike most of other nuclear receptors, PXR exhibits species-specific responses to ligand activation. For example, rifampicin does not significantly activate mouse PXR, but is a very potent activator of human and rabbit PXR, while pregnenolone-16a-carbonitrile (PCN) only weakly activates human PXR but is a very efficacious activator of mouse and rat PXR. In addition to regulating the metabolism of endogenous and exogenous substances, increasing evidence has demonstrated that PXR has anti-fibrotic effect in the liver. Pregnenolone-16α-carbonitrile (PCN), a PXR agonist, was reported to significantly inhibit the differentiation of hepatic stellate cells and liver fibrosis induced by carbon tetrachloride in rats and mice, but the anti-fibrosis effect of PCN was eliminated after PXR knockout [17, 18]. In addition, treatment of human stellate cells for 16 h by the PXR agonist rifampicin decreased the expressions of fibrosis-related genes including TGFβ1, α smooth muscle actin (αSMA) and Wnt signaling-associated genes (catenin and desmocollin) [19]. In the kidney, previous studies have demonstrated that PXR has a protective effect in acute kidney injury (AKI) [20, 21]. However, the role of PXR in CKD and renal fibrosis remains unclear. Therefore, the aim of the present study was to explore the role and mechanism of PXR in CKD.
Materials and methods
Animal study
All animal experiments in the current study were approved by the Ethical Committee of Dalian Medical University and conformed to international guidelines for animal usage in research. Wild-type (WT) C57BL/6J mice (male, 8 weeks old) were obtained from Beijing HFK Bioscience Co., Ltd (Beijing, China). PXR knockout (PXR−/−) mice on C57BL/6J background and humanized PXR (hPXR) mice on 129/SvJ backgound were generated by Shanghai Model Organisms Center, Inc (Shanghai, China) and were routinely bred at the Advanced Institute for Medical Sciences, Dalian Medical University (Dalian, China). All mice were housed in a temperature of 22 ± 1 °C with a 12 h light-dark cycle (8:00–20:00) and 65% ±5% humidity and fed an ad libitum diet.
Establishment of mouse renal fibrosis models
To establish an adenine diet-induced CKD model, mice were fed with 0.2% adenine diet for 28 days, while the control mice were fed with normal diet. To generate an unilateral ureteral obstruction (UUO) model, the mice were anesthetized with sodium pentobarbital (40 mg/kg, Sigma-Aldrich, USA) intraperitoneally and placed on an electronic heating blanket to maintain body temperature at 36.5–37 °C during surgery. The left ureters of the mice were ligated with 4-0 silk thread for 8 days. The sham operation group of mice was used as the control.
Chemicals and reagents
Pregnenolone-16α-carbonitrile (PCN, CAS: C3884) and rifampin (RIF, B2021) were purchased from APExBIO (Houston, TX, USA). Adenine was purchased from Sigma-Aldrich (St. Louis, MO, USA). Recombinant human TGFβ1 was purchased from Pepro Tech (P01137, NJ, USA). The 0.2% adenine diet is produced by Medicience Biopharmaceutical Co., Ltd (Yangzhou, China). TRIzol was purchased from Invitrogen (Carlsbad, CA, USA). The bicinchoninic acid (BCA) protein assay kit was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Transient transfection reagent (Lipofectamine 3000) was purchased from Invitrogen (Carlsbad, CA, USA). Dual-Luciferase® Reporter Assay System 10-Pack was purchased from Promoga (E1910, WI, USA). DAPI (c0065) and normal goat serum (SL038) were purchased from Beijing Solarbio Science and Technology Co., Ltd (Beijing, China). The antibodies include those against PXR (ab192579, Abcam, Cambridge, UK; orb385455, Biorbyt, Wuhan, China; 67912-1-lg, Proteintech, Wuhan, China), p53 (#2524, CST, Danvers, MA, USA; 10442-1-AP, Proteintech, Wuhan, China), β-actin (#66009, Proteintech, Wuhan, China), β-catenin (610154, BD Transduction Laboratories, NJ, USA), Non-phospho (Active) β-Catenin (Ser45) (#19807, CST, Danvers, MA, USA), Wnt7a (sc-365665, Santa Cruz, Dallas, TX, USA), fibronectin (15613-1-AP, Proteintech, Wuhan, China), collagen I (sc-293182, Santa Cruz, Dallas, USA), αSMA (SAB4200689, Sigma-Aldrich, St. Louis, MO, USA), TGFβ1 (ab215715, abcam, Cambridge, UK), Samd3 (#9523, CST, Danvers, MA, USA), p-Smad3 (#9520, CST, Danvers, MA, USA), Histon-H3 (D2B12, 96C10, CST, Danvers, MA, USA) and IgG (#2729, CST, Danvers, MA, USA; sc-2025, Santa Cruz, Dallas, TX, USA). All secondary antibodies were from ABclonal Technology Co., Ltd (Wuhan, China). Alexa Fluor-488 or Alexa Fluor-594 conjugated secondary antibodies were from Life Technologies (Grand Island, NY, USA).
Measurements of BUN, sCr and albuminuria
The mice with adenine diet-induced CKD were euthanized after 28 days, while the UUO model mice were euthanized on the eighth postoperative day. Kidney tissues, blood samples and urine samples were collected for further analysis. Blood urea nitrogen (BUN) and serum creatinine (sCr) levels were determined using commercially available kits (C011-2-1, C013-2) obtained from Nanjing Jiancheng Bioengineering Research Institute (Nanjing, China). Urinary microalbumin levels were examined by an enzyme-linked immunosorbent assay (ELISA) kit (D721120, Sangon Biotech Co., Ltd, Shanghai, China).
Cell culture and treatment
Mouse tubular epithelial cell line (MTEC, purchased from ScienCell, San Diego, CA, USA) was cultured with DME/F-12 medium (HyClone) containing 10% fetal bovine serum (FBS). Human embryonic kidney cell line 293T (HEK293T, Fuheng Cell Center, Shanghai, China) was cultured with DMEM basic medium containing 10% FBS. Cells were placed in a 37 °C, 5% CO2 incubator. To test whether PXR inhibits the effect of TGFβ1, MTEC cells were treated with PCN or infected with an adenovirus expressing a full-length mouse PXR (HanBio, Shanghai, China) or human p53 expression vector (a gift from Prof. Nan-hong Tang at Fujian Medical University, Fuzhou, China).
Real-time PCR
Total RNA was extracted from the kidneys and cultured MTEC cells. Quantitative real-time PCR was performed with a routine procedure using specific primers described in Supplementary Table S1. β-Actin was used as an internal control.
Western blot analysis and immunoprecipitation
Western blotting assay was performed as described previously [22, 23]. An amount of 40 µg protein samples was separated by SDS-PAGE and transferred to PVDF membranes (Millipore, MA, USA). The membrane was blocked with 5% nonfat milk for 1 h at room temperature and then incubated with primary antibodies overnight at 4 °C. Membranes were washed 3 times for 15 min in Tris-buffered saline 5% Tween 20 (TBST) solution and incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. After being washed three times for 30 min, the membrane was transferred to ECL reagent and images were collected with a Tanon-5200 (Tanon, Shanghai, China).
Renal pathological assessments
The kidney tissues were formalin-fixed and paraffin-embedded. The tissue sections were stained with hematoxylin and eosin (H&E) or Masson trichrome according to the manufacturer’s protocol. Ten fields under 200× microscopes were randomly selected in the cortical area for each kidney section. Histopathologic scoring was performed in a blinded fashion in ten consecutive fields per section from five different mice in each group. The tubulointerstitial (TI) damage score is based on the percentage of total cortical area affected by tubular dilation, tubular atrophy, interstitial fibrosis, and interstitial inflammation. The semiquantitative scoring of the lesions was carried out, and the scoring results were as follows: 0, no change; 1, lesions involved 0–25% area; 2, lesions involved 25%–50% area; 3, lesions involved 50%–75% area; 4, lesions involved 75%–100% area; 5, lesions involved the entire area [24]. The percentage of interstitial fibrotic area to the selected field was analyzed with ImageJ software, and an average percentage of kidney fibrotic area for each section was calculated. All slides were investigated and reviewed blindly by two pathologists.
Immunostaining
Immunohistochemical staining was performed on 5 μm tissue sections. 3% H2O2 was used to remove endogenous peroxidase activity. 5% BSA was used to block the non-specific binding site. Diaminobenzidine (DAB) was used to display peroxidase and hematoxylin was used to label the nucleus. For immunofluorescence assay, staining was performed on kidney sections and cultured MTEC. The non-specific binding sites were blocked by 5% BSA containing 10% goat serum.
Immunoprecipitation assay (IP)
The IP experiments were performed according to the instructions of the Thermo Fisher Scientific Chromatin Immunoprecipitation Kit (88805, Thermo Fisher Scientific, Waltham, MA, USA). Briefly, Pierce Protein A/G magnetic beads were washed twice with 1× modified cross-linking buffer, and then incubated with 5 μg of antibody (specific antibody against p53 or corresponding non-specific IgG antibody) at 4 °C for 4 h. After incubation, the beads were washed 3 times with 1× modified cross-linking buffer and cross-linked with antibody using DSS for 30 min. The beads were washed 3 times with the elution buffer, followed by 2 washes with the lysis/wash buffer and then stored at 4 °C. HEK293T cells (1 × 107) were co-transfected with a p53 expression vector with or without a PXR expression vector, and then treated with 40 μM PCN for 12 h. The cells were lysed with 800 μL of ice-cold lysis/wash buffer on ice for 20 min and then centrifuged at 15,000 r/min for 15 min at 4 °C. Protein concentration was determined with a BCA kit. Protein lysate (3 μg) in a final volume of 500 μL was incubated with pre-crosslinked magnetic beads overnight at 4 °C, and the remainder was used as input for immunoblot analysis. After washing twice with the lysis/wash buffer and once with ultrapure water, the antigen was eluted with the elution buffer for immunoblotting assay.
Identification of putative transcription factor binding sites
A computer-based search for potential transcription factor binding motifs was carried out on the TRANSFAC 8.3 professional database by the of TFSEARCH (https://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) [25, 26]. The sequences of classical p53 response elements were obtained from the JSAPAR database.
Chromatin immunoprecipitation assay (ChIP)
According to the manufacturer’s protocol, ChIP assay was performed with the ChIP-IT EXPress Enzymatic Kit (53009, Active Motif, Shanghai, China). Specific binding to target DNA fragment was analyzed by PCR. The sequences of the primers used are shown in Supplementary Table S1. PCR products were electrophoresed on 2% agarose gels and stained with Gold View II Nuclear Staining Dyes (G8142, Solarbio, Beijing, China).
Construction of a mouse Wnt7a gene promoter-driven luciferase reporter
The mouse genomic DNA extracted by Genomic DNA Purification Kit (Promega, Madison, WI, USA) was used as a template for polymerase chain reaction amplification (PCR). The PCR reaction was carried out with a pair of specific primers carrying two restriction enzyme sequences of Kpn I and Mlu I. The PCR product of a Wnt7a gene promoter fragment (−454 bp ~+53 bp) was validated and inserted into the pGL3.0-Basic vector with T4 ligase (Trans Gen Biotech, Beijing, China) to obtain the Wnt7a gene promoter-driven luciferase reporter. The primers used for PCR amplification are shown in Supplementary Table S1 and the construct was confirmed by DNA sequencing.
Luciferase activity assay
The HEK293T cells were seeded on 96-well culture plates to reach 70%–80% confluency in DMEM basic medium containing 10% FBS. After replacing with a serum-free medium, the cells were co-transfected with the indicated Wnt7a gene promoter-driven luciferase reporter vector and a pRL-TK vector (Promega) encoding Renilla luciferase, with or without a mPXR expression vector in the presence or absence of a pcDNA3.0-p53 expression vector (a generous gift of Prof. Nan-hong Tang, Research Center for Molecular Medicine, Fujian Medical University, Fuzhou, China) using Lipofectamine 3000 (L3000015, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instruction. After transfection for 7 h, the medium was changed to 5% FBS and the cells were treated with PCN (40 μM) or an equivalent volume of DMSO for 24 h. The luciferase assay was performed using the dual luciferase assay system kit according to the manufacturer’s protocol (E1910, Promoga, WI, USA).
Statistical analysis
All statistical analyses were performed using Student’s t test or one-way ANOVA with the GraphPad Prism 8.0 (GraphPad Software Inc., La Jolla, CA, USA). Results were presented as means ± standard deviation error (SEM). Results were considered significant at two tailed P < 0.05.
Results
PXR activation by PCN alleviates mouse renal damage induced by adenine
In the present study, we used the adenine-induced CKD mouse model which can mimic most of renal structural and functional changes in human CKD and require no surgery or genetic manipulation [27, 28]. To explore whether PXR plays a protective role in CKD, we constructed the adenine-induced CKD model in C57BL/6J mice and treated CKD mice with or without PCN, a specific exogenous agonist of mouse PXR, for 4 weeks [29]. The mice were divided into 4 groups: (i) Ctrl (mice were fed with normal diet), (ii) PCN (mice were fed with normal diet and treated with PCN at 50 mg/kg, twice a week), (iii) AD (mice were fed with 0.2% adenine diet), and (iv) AD + PCN (mice were fed with 0.2% adenine diet and treated with PCN at 50 mg/kg, twice a week) (Fig. 1a) [30]. Compared with the Ctrl mice, adenine treatment resulted in a significant reduce in body size, granular changes on the surface of the kidneys, and a marked increase in the levels of serum creatinine (sCr), blood urea nitrogen (BUN) and 24-h urinary albumin excretion, all of which was improved by PCN treatment (Fig. 1b–e). In addition, consistent with previous studies, the adenine diet led to a sharp decrease in body weight, kidney weight and urine osmolality (Supplementary Fig. S1a, b and f), a significant increase in relative kidney weight (the ratio of kidney weight to body weight), 24-h water intake and urine output in mice (Supplementary Fig. S1c–e) [24, 31]. To assess the effect of PCN on renal structural damage induced by adenine diet, we visualized paraffin sections with H&E staining and scored the renal tubulointerstitial (TI) damage (Fig. 1f, g). Mice in the Ctrl and PCN groups showed normal renal morphology, while mice in the AD group exhibited various renal structural damages, including renal tubular atrophy, tubule dilatation with lumenal necrotic material, tubular epithelium thinning, interstitial inflammatory cell infiltration. Consistently, PCN treatment significantly improved those renal tissue morphological changes induced by adenine (Fig. 1f, g). These findings demonstrate that the PXR activator PCN can exert renoprotective effect on adenine-induced renal functional and structural damages.
Fig. 1. PXR activation by PCN alleviates mouse renal damage induced by adenine.

a Schematic diagram of adenine-induced CKD with PCN treatment. b The gross view of the mice and the macroscopic appearance of the kidneys were photographed at the end of 4-week treatment. PCN treatment significantly improved adenine-induced body size reduction and granular appearance of the kidneys. c–e PCN treatment significantly attenuated adenine-induced increase in serum creatinine and BUN levels and urinary albumin excretion (n = 5–9). f Representative images of H&E staining. PCN significantly alleviated adenine-induced renal tubular atrophy, dilatation with crystal deposition and cellular casts, and tubulointerstitial fibrosis. Scale bar: 100 μm. g Tubulointerstitial (TI) damage score of histopathologic features in H&E staining (n = 5–6). Results are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
PXR activation by PCN attenuates mouse renal fibrosis induced by adenine
Fibrosis is the final manifestation of CKD [6]. Masson trichrome staining and semiquantitative measurement results showed that PCN reduced adenine-induced deposition of extracellular matrix proteins (Fig. 2a, b). Similarly, the immunostaining analysis demonstrated that PCN treatment markedly improved adenine-induced renal fibrosis as reflected by reduced expression of fibronectin, αSMA and collagen I (Fig. 2a, c–e). Transforming growth factor β1 (TGFβ1), as a key factor in the progression of renal fibrosis, induces the epithelial to mesenchymal transformation (EMT), fibroblast to myofibroblast transdifferentiation and extracellular matrix production by activating the downstream Smad proteins, and ultimately promotes renal fibrosis by increasing expression of its target genes including fibronectin and αSMA. Western blot analysis showed that PCN treatment significantly reduced adenine-induced protein expression of p-Smad3, fibronectin and αSMA (Fig. 2f, g). Consistently, immunofluorescence staining showed that PCN markedly inhibited nuclear translocation of p-Smad3 induced by adenine (Fig. 2h, i). As expected, PCN treatment also markedly decreased mRNA expression of TGFβ1 and fibronectin as assessed by real-time PCR (Supplementary Fig. S2a, b). These results suggest that PXR activation can alleviate renal fibrosis by inhibiting the activation of Smad3 and extracellular matrix protein production.
Fig. 2. PXR activation by PCN attenuates renal fibrosis induced by adenine.

a Masson trichrome staining and immunostaining showing that PCN treatment significantly attenuated renal extracellular matrix protein deposition and the protein expression of fibronectin, αSMA and collagen I. Scale bar: 100 μm. b–e Semiquantitative analysis of a (n = 4–6). f Western blot analysis demonstrating that PCN treatment markedly reduced adenine-induced expression of fibrosis-related proteins including p-Smad3, fibronectin and αSMA (n = 3). g Semiquantitative measurement for the protein levels in f. h Immunofluorescence assay showing that PCN treatment significantly inhibited adenine-induced activation of p-Smad3. Scale bar: 50 μm. i Semiquantitative measurement for the protein levels in h (n = 3). Data represents the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
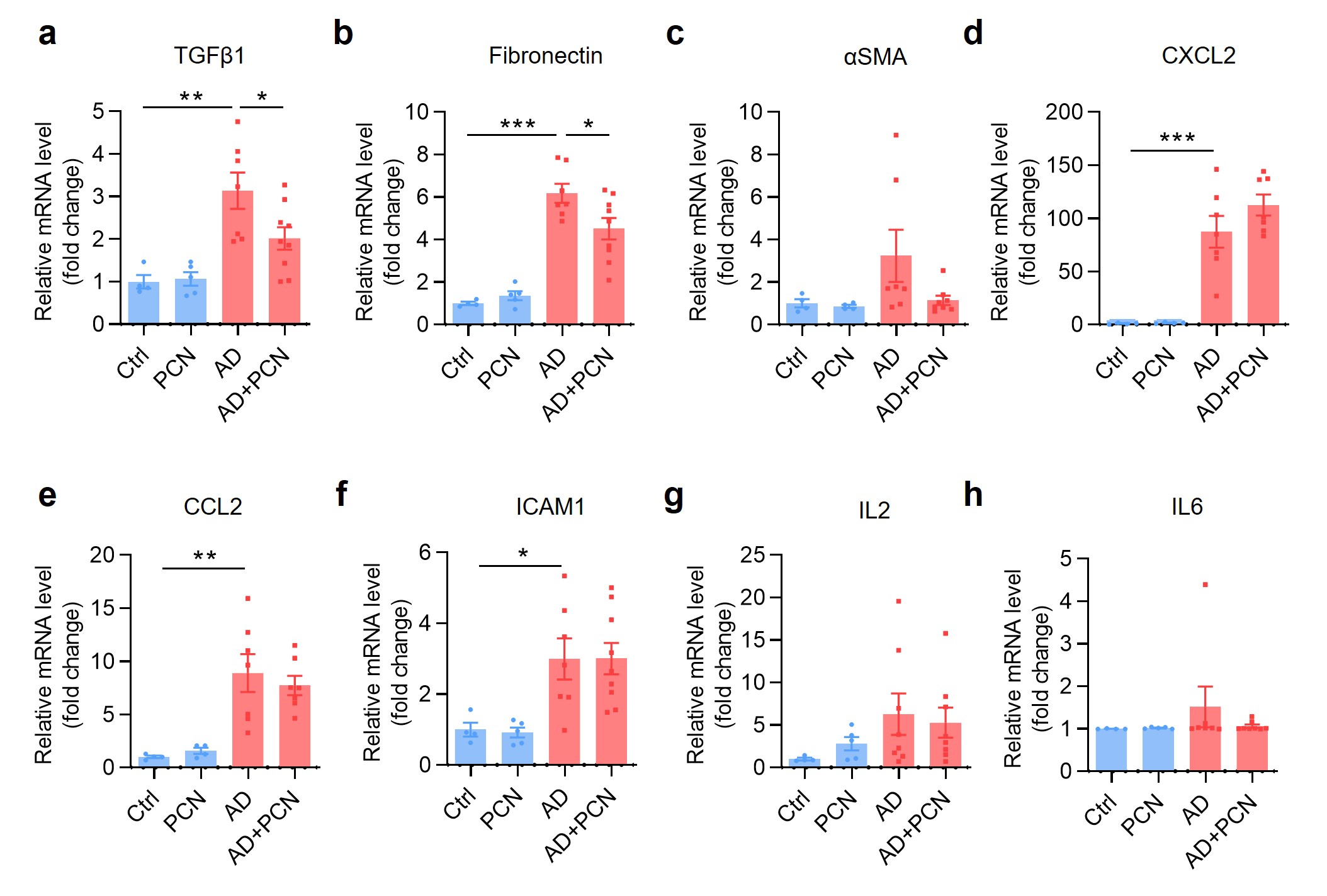
It is well documented that inflammation plays an important role in the progression and prognosis of CKD [32, 33]. We utilized real-time PCR assay to determine the effect of PCN on the expression of adenine-induced profibrotic and proinflammatory cytokines. The results showed that the mRNA levels of TGFβ1, fibronectin, CXCL2, CCL2 and ICAM1 in the AD group were significantly upregulated compared with the Ctrl group, with insignificant changes in gene expressions of αSMA, IL-2 and IL-6. However, PCN treatment did not affect the expression of adenine-induced inflammatory gene expression (Supplementary Fig. S2d–h). Together, these findings demonstrate that treatment of the PXR activator PCN can downregulate profibrotic gene expression with little effect on adenine-induced inflammation.
PXR activation by PCN ameliorates renal fibrosis in a mouse UUO model
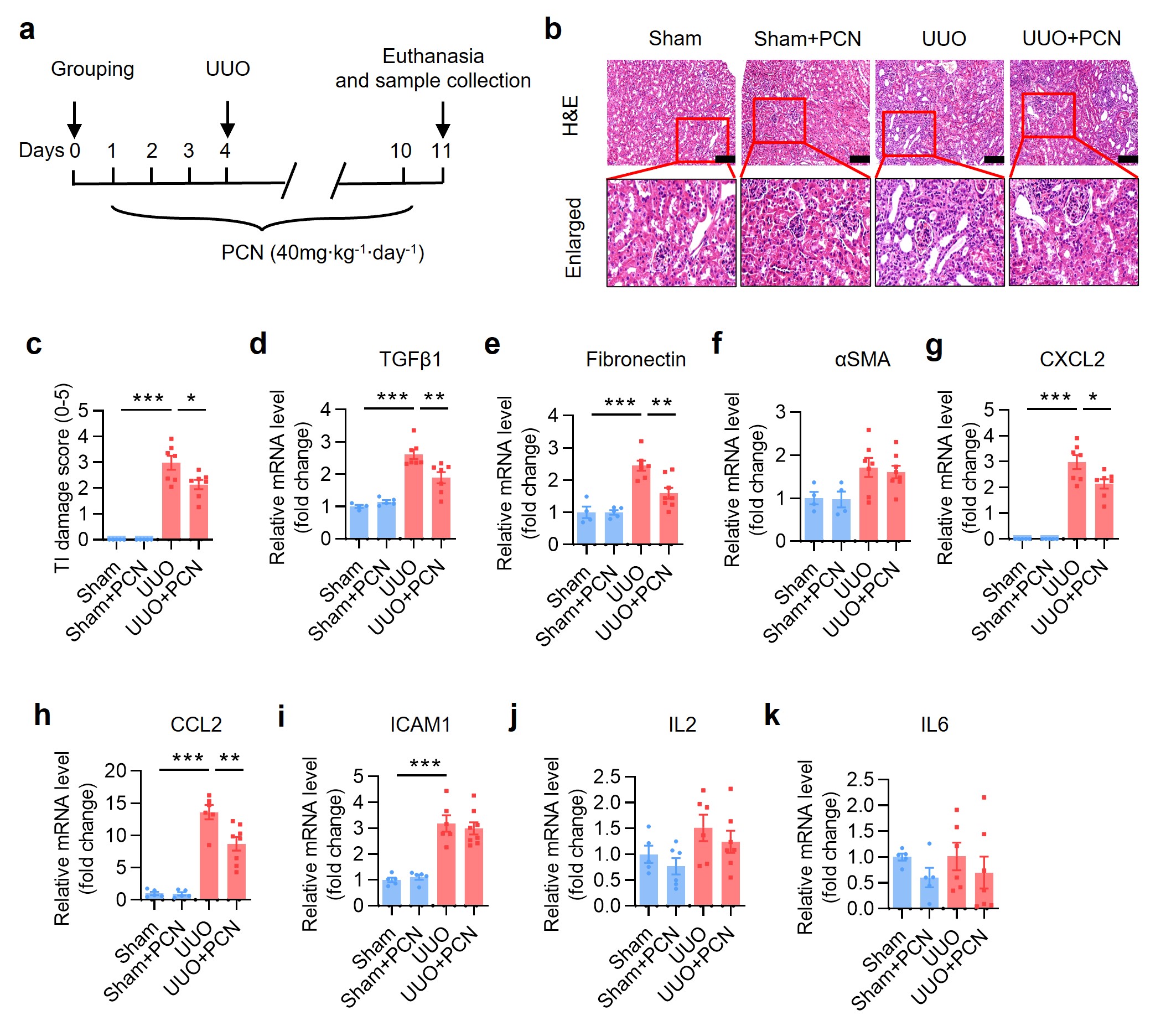
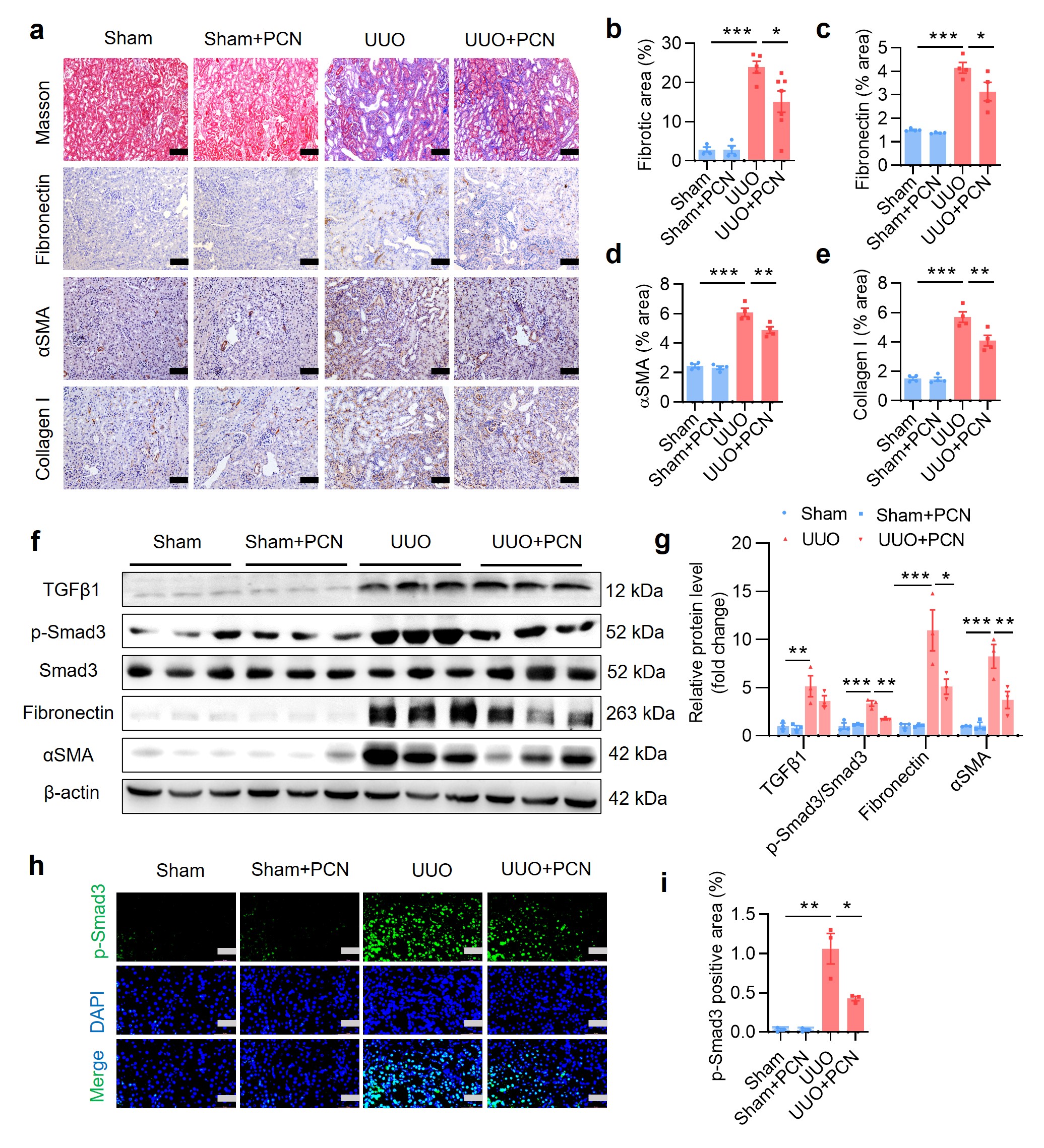
To further determine the protective effect of PXR on renal fibrosis, we also generated a mouse model of unilateral ureteral obstruction (UUO), a commonly used animal model to study renal fibrosis [34]. The experiment was composed of following 4 groups: (i) Sham, (ii) Sham+PCN, (iii) UUO, and (iv) UUO+PCN (Supplementary Fig. S3a). In the PCN-treated groups, mice received daily intraperitoneal injection of PCN at 40 mg/kg 3 days before and 6 days after the establishment of UUO. The PCN concentration and treatment times were selected according to the literature and our previous experience [20, 35]. Results of the H&E staining and tubulointerstitial (TI) damage score analysis demonstrated that the kidneys in the Sham group and Sham+PCN group showed normal tissue morphology. However, the obstructed kidneys in the UUO group exhibited many pathological changes such as lumen enlargement, epithelial thinning and tubular atrophy, all of which was significantly attenuated by the PCN treatment (Supplementary Fig. S3b, c). Semi-quantitative measurement of Masson trichrome staining and immunostaining (fibronectin, αSMA and collagen I) indicated that PCN treatment markedly alleviated renal fibrosis caused by UUO (Supplementary Fig. S4a–e). Western blot analysis further showed that PCN treatment significantly inhibited the protein expression levels of fibrosis-related genes including TGFβ1, p-Smad3, fibronectin and αSMA in the obstructed kidneys (Supplementary Fig. S4f, g). Immunofluorescence assay further indicated that renal p-Smad3 expression in the nuclei was significantly increased in the UUO group, which was markedly attenuated after PCN treatment (Supplementary Fig. S4h, i). In addition, PCN treatment also significantly reduced the mRNA expression of TGFβ1 and fibronectin (Supplementary Fig. S3d, e). In addition, PCN treatment significantly reduced UUO-induced CXCL2 and CCL2 expression (Supplementary Fig. S3g, h), with little effect on other genes (Supplementary Fig. S3f, i–k). Collectively, these findings demonstrate that treatment with the PXR activator PCN can ameliorate UUO-associated renal fibrosis in mice.
PXR activation by rifampicin alleviates adenine-induced renal damage in PXR-humanized mice
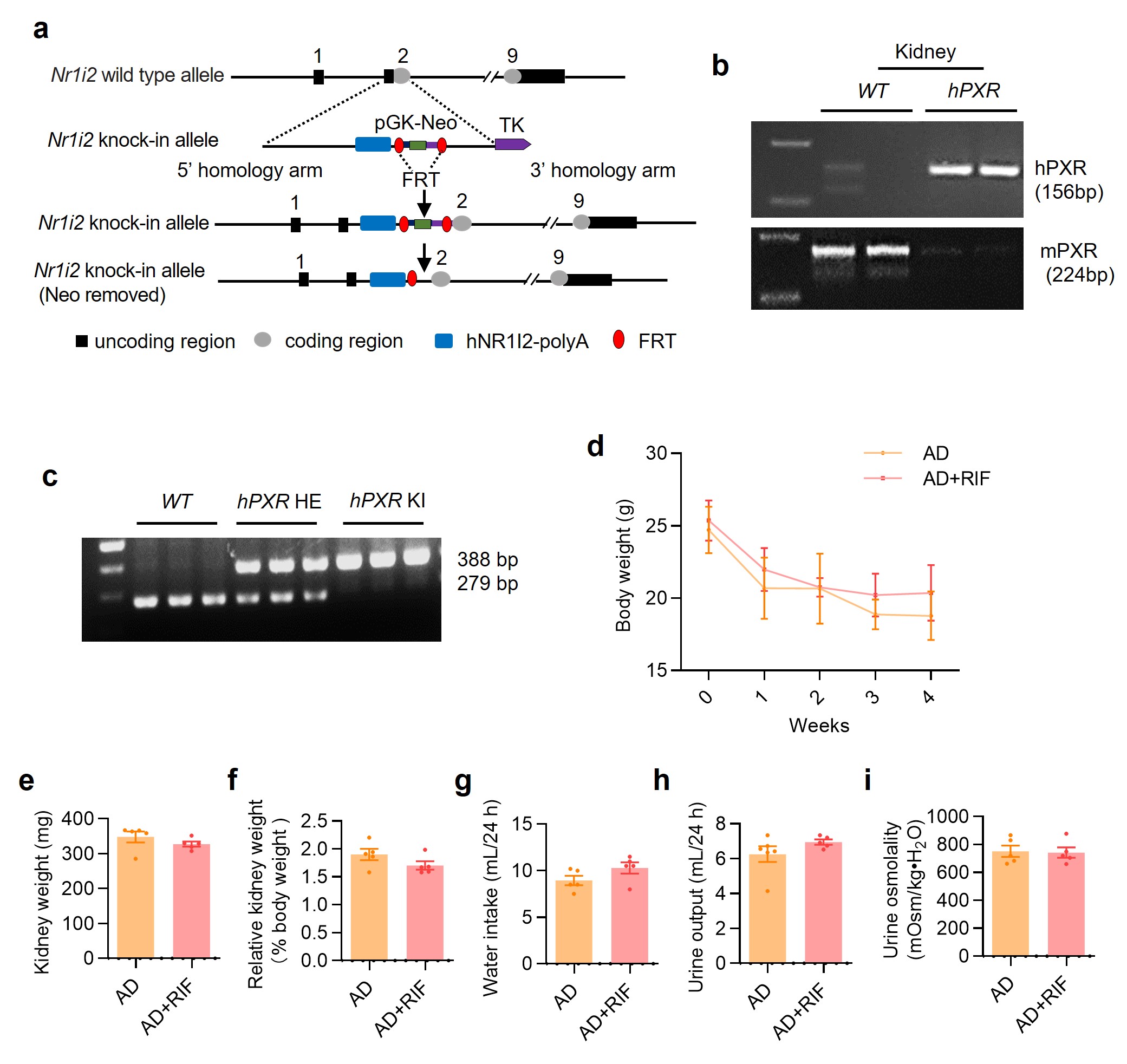
The DNA binding domain of mouse and human PXR shares 96% amino acid identity, but the ligand-binding domains are only 76% identical [36]. Therefore, mouse exogenous agonist PCN is unable to activate human PXR. To test whether human PXR has similar renal protective effect, we constructed a PXR-humanized mouse and genotyped by PCR (Supplementary Fig. S5a–c). Eight-week-old male PXR-humanized (hPXR) mice were divided into two groups: (i) AD (mice were fed with 0.2% adenine diet for 4 weeks), and (ii) AD + RIF (mice were fed with 0.2% adenine diet and treated with 10 mg·kg−1·d−1 rifampicin for 4 weeks) (Fig. 3a). Renal gross images showed that RIF treatment alleviated adenine-induced the granular changes on the surface of the kidneys (Fig. 3b). Serum biochemistry analysis demonstrated that RIF treatment decreased sCr and BUN levels compared to the vehicle controls (Fig. 3c, d). RIF also significantly reduced 24-h urinary albumin excretion in hPXR mice (Fig. 3e). H&E staining further showed that RIF markedly attenuated adenine-induced renal histological damages (Fig. 3f, g). However, RIF did not affect body weight, kidney weight, relative kidney weight, water intake, urine output and urine osmolality in adenine-treated hPXR mice (Supplementary Fig. S5d–i). These results indicate that activation of PXR can exert renoprotective effect on adenine-induced renal functional and histological changes not only in wild-type but also in PXR-humanized mice.
Fig. 3. PXR activation by rifampicin alleviates adenine-induced renal damage in PXR-humanized mice.

a Schematic diagram of adenine-induced CKD with or without rifampicin (RIF) treatment. Adenine diet-fed PXR-humanized (hPXR) mice were treated with or without daily intragastric administration of RIF (10 mg/kg) for 28 days. b The gross view of the mice and the macroscopic appearance of the kidneys were photographed on day 29. RIF treatment reduced adenine-induced renal atrophy. RIF treatment significantly reduced the levels of sCr (c), BUN (d) and urinary albumin excretion (e) (n = 5). f H&E staining showed that RIF treatment mitigated renal tubular atrophy, dilatation with crystal deposition and cellular casts, and tubulointerstitial fibrosis. Scale bar: 100 μm. g The TI damage score of histopathologic features in f (n = 5). Results are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
PXR activation by rifampicin attenuates adenine-induced renal fibrosis in PXR-humanized mice
Masson staining was used to detect extracellular matrix (ECM) protein accumulation in the kidneys of mice and immunostaining was used to assess renal expression levels of fibrosis markers. The results showed that RIF treatment significantly abolished ECM deposition and accumulation of fibronectin, αSMA and collagen I in renal interstitium compared with the AD group (Fig. 4a–e). Similarly, immunoblot assay found that RIF treatment significantly inhibited adenine-induced protein expression of multiple fibrosis-related genes including p-Smad3, fibronectin and αSMA (Fig. 4f, g). In addition, RIF markedly attenuated adenine-induced p-Smad3 protein nuclear translocation in the kidney (Fig. 4h, i) and mRNA expression of TGFβ1 (Supplementary Fig. S6a), with little effect on the mRNA levels of other profibrotic and proinflammatory factors (Supplementary Fig. S6b–h). These findings suggest that PXR activation likely also attenuates renal fibrosis in humans.
Fig. 4. PXR activation by rifampicin alleviates renal fibrosis induced by adenine in PXR-humanized mice.

a Masson trichrome staining and immunostaining showing that rifampicin (RIF) treatment significantly alleviated renal extracellular matrix protein deposition and reduced the protein expression of fibronectin, αSMA and collagen I. Scale bar: 100 μm. Semiquantitative analysis of fibrotic area (b), fibronectin (c), αSMA (d) and collagen I (e) positive staining area (n = 4–5). f Western blot assay demonstrating that RIF treatment inhibited protein expression of p-Smad3, fibronectin and αSMA. g Semiquantitative measurement for the protein levels in f (n = 4). h Immunofluorescence analysis showed that RIF treatment markedly reduced p-Smad3 expression. Scale bar: 50 μm. i Semiquantitative measurement of p-Smad3 levels in h (n = 3). Data represents the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
PXR gene ablation exacerbates mouse renal damages caused by adenine
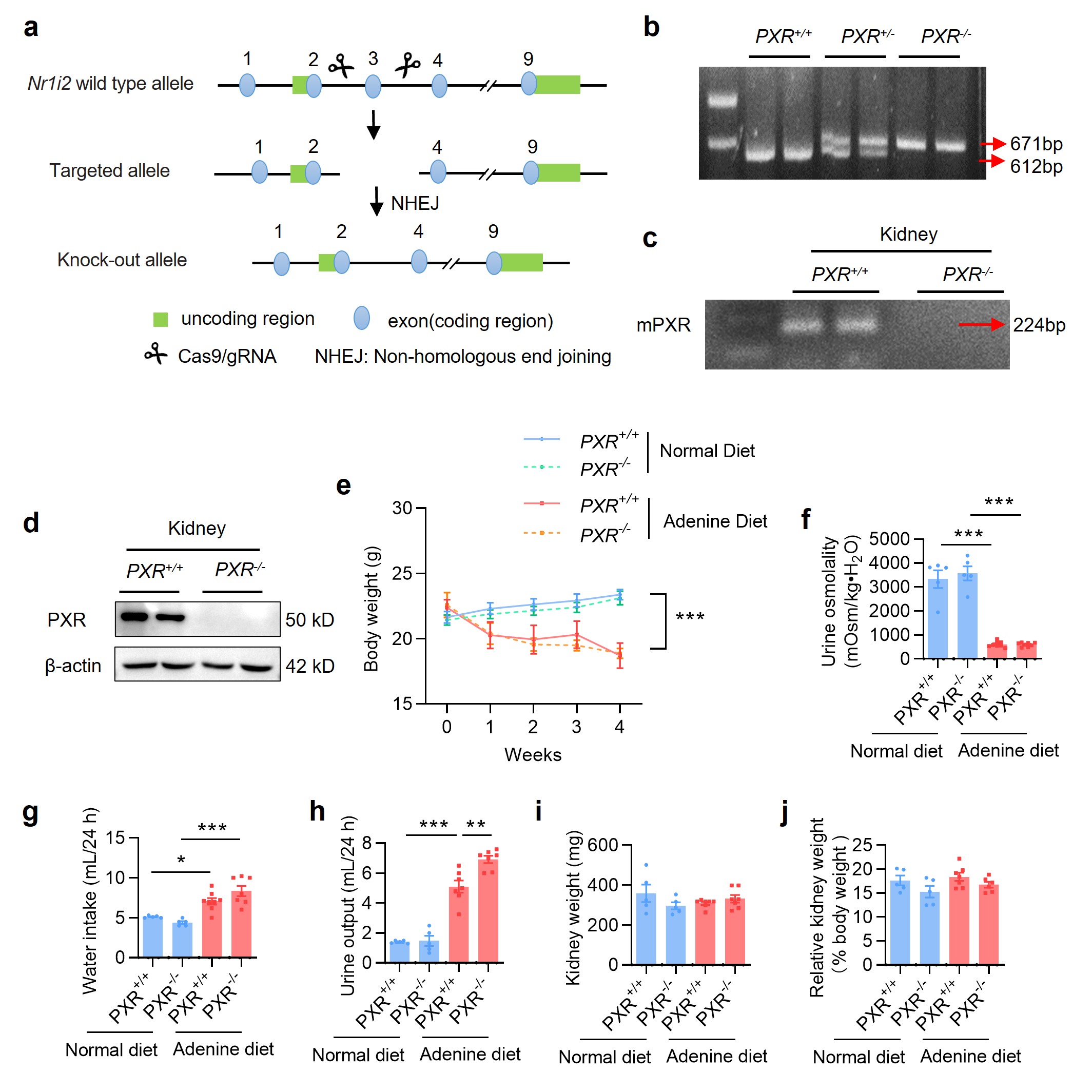
To further ascertain the protective role of PXR in renal fibrosis, we generated a PXR knockout mouse induced by a frame-shift mutation (Supplementary Fig. S7a). We verified the successful construction of knockout mice by genotyping mouse tail DNA sequence and showing the absence of PXR mRNA and protein expression (Supplementary Fig. S7b–d). Eight-week-old male wild-type (PXR+/+) and PXR knockout (PXR−/−) mice were fed with either adenine diet or normal diet for 4 weeks. PXR gene deficiency markedly aggravated the granular changes on the surface of the kidneys of mice fed with adenine (Fig. 5a). Furthermore, PXR gene ablation significantly increased the levels of sCr, BUN and urinary albumin excretion induced by adenine diet (Fig. 5b–d). Furthermore, the TI damage score and H&E staining showed that PXR gene knockout aggravated adenine-induced tubular epithelial thinning, lumen enlargement and tubulointerstitial sclerosis (Fig. 5e, f). Consistent with previous results, the adenine diet led to a sharp decrease in body weight and urinary osmolality, an increase in water intake and urine output in both genotypes (Supplementary Fig. S7e–h). However, PXR gene knockout only increased urine output in adenine diet groups (Supplementary Fig. S7h). In addition, there were no changes in absolute and relative weights of kidneys among four groups (Supplementary Fig. S7i, j). Together, these findings demonstrate that PXR gene deficiency exacerbates adenine-induced functional and structural damages in mouse kidneys.
Fig. 5. PXR gene ablation exacerbates adenine-induced renal damages.

a Gross view of the mice and the macroscopic appearance of the kidneys of mice treated with adenine or vehicle for 4 weeks. PXR gene knockout exacerbated adenine-induced body size reduction and granular appearance of the kidneys. PXR gene deficiency aggravated adenine-induced increase in sCr (b), BUN (c) and urinary albumin excretion (d) (n = 5–6). e The TI damage score of histopathologic features (n = 4–6). f H&E staining showing that PXR gene knockout aggravated renal tubular atrophy, dilatation with crystal deposition and cellular casts, and tubulointerstitial fibrosis. Scale bar: 100 μm. Results are expressed as mean ± SEM. **P < 0.01, ***P < 0.001.
PXR gene ablation aggravates adenine-induced renal fibrosis in mice
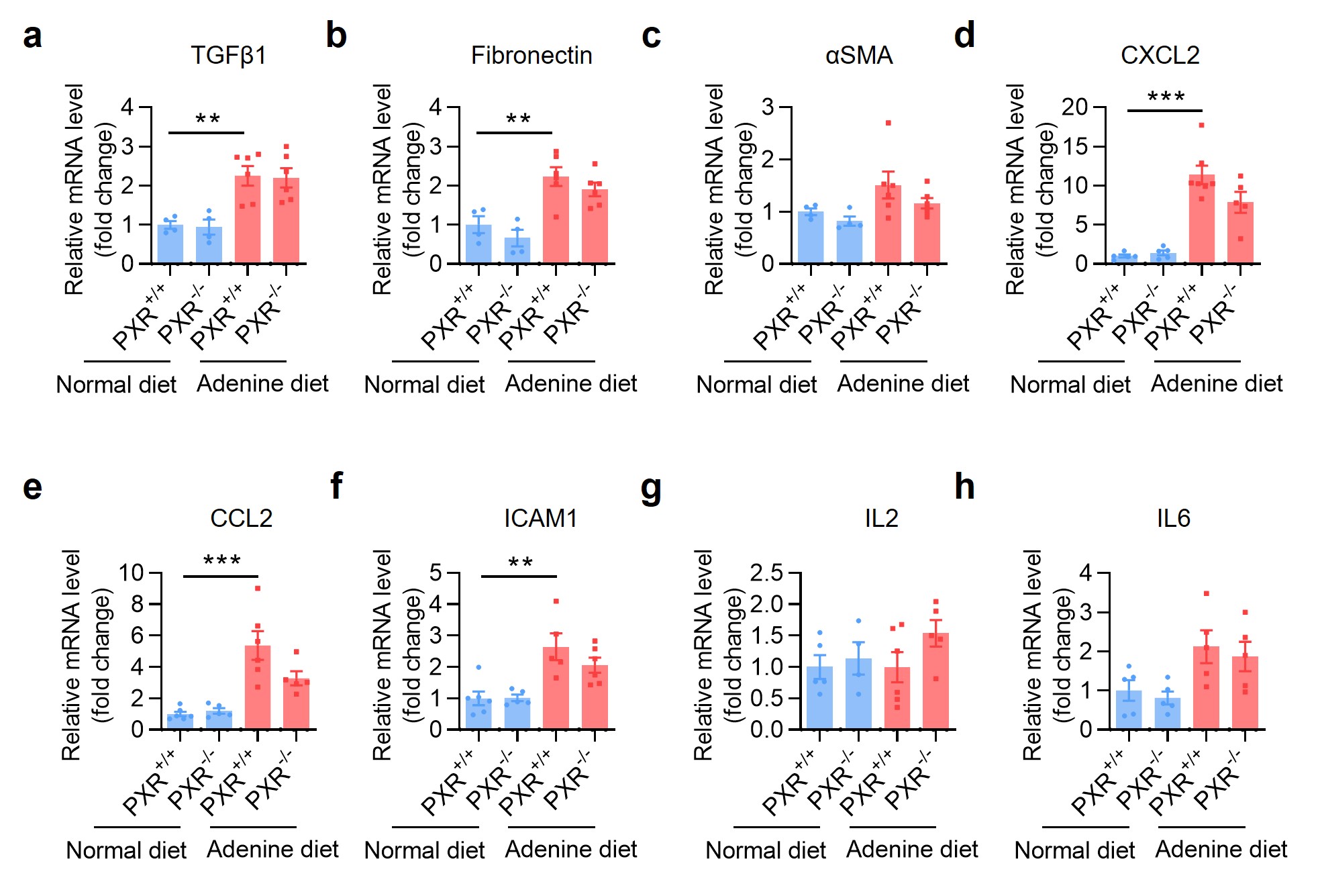
Masson trichrome staining showed that PXR gene knockout increased adenine-induced ECM deposition (Fig. 6a). Similarly, immunostaining assays demonstrated that PXR gene deficiency aggravated adenine-induced the accumulation of fibronectin, αSMA and collagen I proteins (Fig. 6a–e). Although PXR gene deficiency had little effect on mRNA expression of profibrotic and proinflammatory genes in mouse kidney (Supplementary Fig. S8a–h), adenine-induced protein expression of p-Smad3, fibronectin and αSMA was further increased in mice deficient for PXR (Fig. 6f, g). Consistently, by using immunofluorescence assay we found that adenine-elicited activation of p-Smad3 was significantly enhanced in mice with PXR gene deficiency (Fig. 6h, i). Collectively, these findings demonstrate that PXR gene deficiency aggravates adenine-induced renal fibrosis in mice, suggesting an antifibrotic action of PXR agonist.
Fig. 6. PXR gene ablation exacerbates adenine-induced renal fibrosis.

a Masson trichrome staining and immunostaining demonstrating that PXR gene knockout increased adenine-induced extracellular matrix protein deposition and renal protein expression of fibronectin, αSMA and collagen I. Scale bar: 100 μm. b–e Semiquantitative analysis of fibrotic area and protein expression levels in a (n = 4). f Western blot analysis demonstrating that PXR gene knockout aggravated adenine-induced p-Smad3, fibronectin and αSMA expression. g Semiquantitative measurement for the protein levels in f (n = 3). h Immunofluorescence analysis showing that PXR gene knockout increased adenine-induced p-Smad3 activation. Scale bar: 50 μm. i Semiquantitative measurement of p-Smad3 in h (n = 3). Data represents the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
PXR gene ablation exacerbates UUO-related renal fibrosis in mice
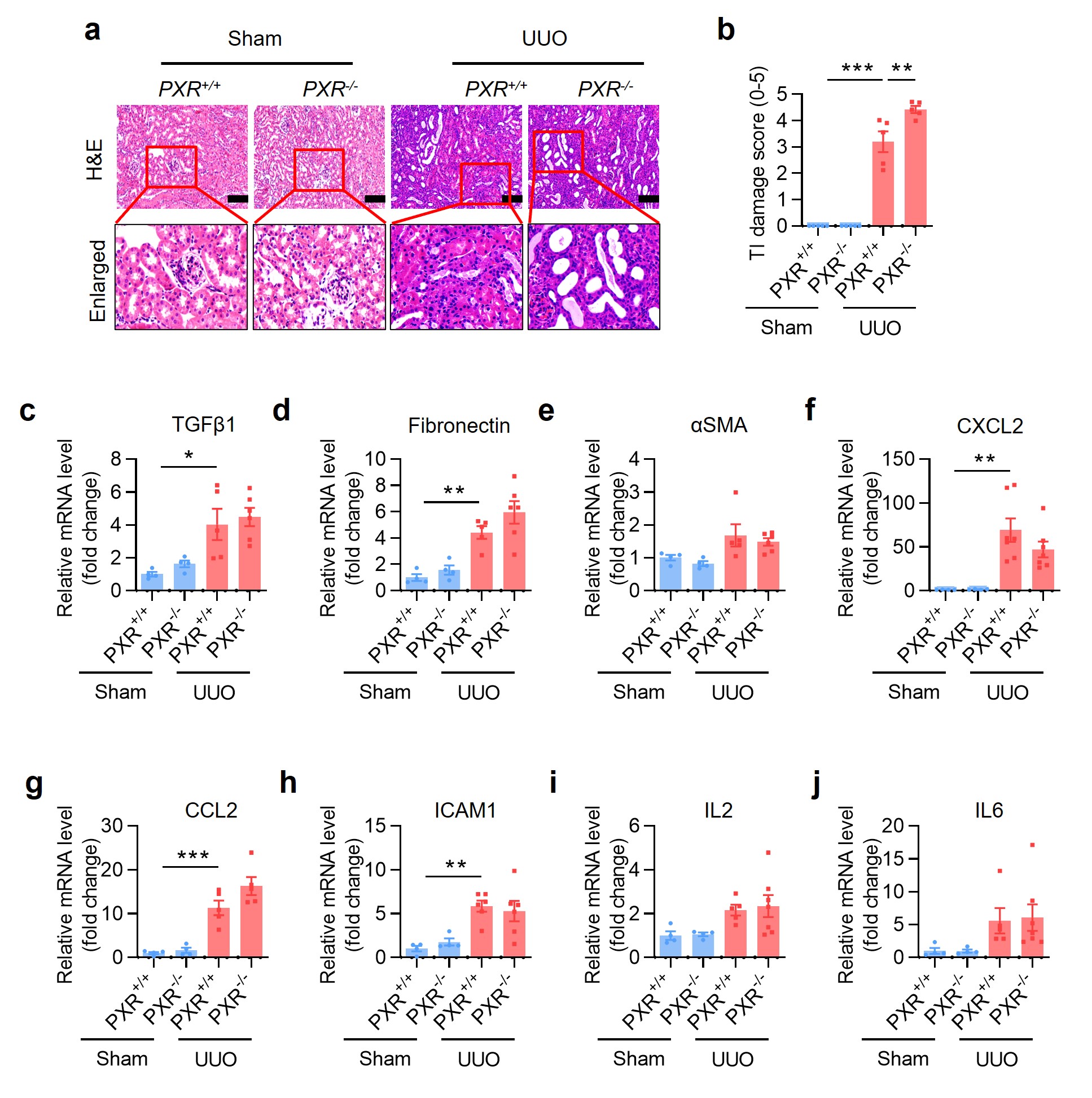
To confirm the renoprotective role of PXR in renal fibrosis, 8-week-old male PXR+/+ and PXR−/− mice were divided into the Sham and UUO group, respectively. H&E staining and the TI damage score showed that PXR gene deficiency markedly increased the number of dilated renal tubules and the sclerotic area of the tubulointerstitium caused by UUO (Supplementary Fig. S9a, b). Masson trichrome staining and immunohistochemical analysis revealed that PXR gene deficiency aggravated UUO-induced renal ECM deposition and protein accumulation of fibronectin, αSMA and collagen I (Supplementary Fig. S10a–e). Although no significant effect on renal mRNA expression of fibrotic (Supplementary Fig. S9c–e) and inflammatory factors (Supplementary Fig. S9f–j), PXR gene deficiency increased UUO-induced protein expression of p-Smad3, fibronectin and αSMA (Supplementary Fig. S10f, g). Consistently, PXR gene deficiency further increased UUO-induced nuclear expression of p-Smad3 (Supplementary Fig. S10h, i). Together, these results indicate that PXR dysfunction accelerates the progression of renal fibrosis.
PXR activation inhibits the Wnt7a/β-catenin signaling in vivo
To elucidate the underlying mechanism by which PXR attenuates renal fibrosis, we focused on the Wnt/β-catenin signaling pathway given its important role in the pathogenesis of fibrosis [37–39]. We measured the mRNA levels of all 19 Wnt molecules in the kidneys of mice receiving adenine diet with or without PCN treatment for 4 weeks (Fig. 1a). The results revealed that adenine treatment increased the mRNA expression of Wnt7a about ~13-fold in the kidney, where adenine-induced Wnt7a expression can be significantly inhibited by PCN (Fig. 7a). Consistently, adenine markedly increased protein expression of Wnt7a and its downstream effector β-catenin, both of which were markedly suppressed by PCN treatment (Fig. 7b, c), suggesting that PXR might exert its anti-fibrotic effect by inhibiting the Wnt7a/β-catenin pathway. In support, immunofluorescence study showed that PCN treatment markedly inhibited adenine-induced expression and nuclear translocation of active β-catenin protein (Fig. 7d). In line with these findings, in another renal fibrosis model, PCN treatment also markedly suppressed UUO-induced activation of the Wnt7a/β-catenin pathway (Supplementary Fig. S11a–d). These results demonstrate that the protective role of PXR in renal fibrosis is likely mediated by inhibiting the Wnt7a/β-catenin pathway.
Fig. 7. PXR activation inhibits the Wnt7a/β-catenin signaling in the kidneys of adenine-treated mice.

a Real-time PCR analysis demonstrating the effect of PCN treatment on the expression of all 19 Wnt members in mice treated with adenine for 4 weeks. Adenine induced ~13-fold increase in Wnt7a expression, which was significantly attenuated by PCN treatment (n = 4–8). b, c Western blot assay and semiquantitative analysis indicate that PCN treatment significantly decreased adenine-induced protein levels of Wnt7a and its downstream effector β-catenin (n = 3). d Immunofluorescence assay showed that PCN treatment inhibited adenine-induced activation of β-catenin. Scale bar: 50 μm. Results are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
PXR activation and overexpression inhibit TGFβ1-induced fibrotic gene expression in cultured renal tubular cells
To further confirm the protective effect of PXR on renal fibrosis, mouse renal tubular epithelial cell lines (MTEC) were cultured and treated with TGFβ1 to induce fibrotic gene expression in vitro. PCN treatment blocked TGFβ1-induced fibronectin, αSMA and collagen I mRNA expression (Supplementary Fig. S12a–c) and reversed TGFβ1-elicted activation of p-Smad3 and protein expression of fibronectin and αSMA protein (Fig. 8a–c). Similarly, PXR overexpression by infecting the cells with an adenovirus carrying a full-length mouse PXR cDNA (Ad-PXR) (Supplementary Fig. S12d, e) also markedly inhibited the activation of the TGFβ1/Smad3 signaling pathway and the expression of its downstream fibrotic genes (Fig. 8d–f, Supplementary Fig. S12f–h). Collectively, PXR activation and overexpression inhibit TGFβ1-induced expression of fibrosis-related genes in cultured renal tubular cells.
Fig. 8. PXR activation and overexpression inhibit TGFβ1-induced fibrotic gene expression in cultured MTEC.

a Western blot analysis demonstrating that PXR activation decreased TGFβ1-induced upregulation of p-Smad3, fibronectin and αSMA protein expression in MTEC. b Semiquantitative analysis of the protein levels in a (n = 4). c Immunofluorescence assay showed that PXR activation attenuated TGFβ1-induced activation of p-Smad3. Scale bar: 25 μm. d Western blot analysis demonstrated that adenovirus-mediated PXR overexpression decreased TGFβ1-induced p-Smad3, fibronectin and αSMA expression in MTEC. e Semiquantitative analysis of the protein levels in d (n = 4). f Immunofluorescence assay showed that PXR overexpression abolished the activation of p-Smad3 induced by TGFβ1. Scale bar: 25 μm. Results are expressed as mean ± SEM. **P < 0.01, ***P < 0.001.
PXR activation and overexpression inhibit the Wnt7a/β-catenin signaling in cultured renal tubular cells
To characterize the underlying mechanism by which PXR suppresses renal fibrosis, we determined the effect of PXR on the expression of the Wnt7a/β-catenin signaling in mouse renal tubular epithelial cell. As expected, Wnt7a mRNA (Supplementary Fig. S13a, b) and protein expression (Fig. 9a, b) was induced by TGFβ1, which was accompanied by upregulation and activation of its downstream target β-catenin (Fig. 9a–c). PXR activation by PCN (Fig. 9a–c) or PXR overexpression (Fig. 9d–f) significantly abolished TGFβ1-induced upregulation and activation of the Wnt7a/β-catenin pathway. Given the critical role of the Wnt/β-catenin pathway in organ fibrosis, these findings demonstrate that PXR attenuates renal fibrosis by suppressing the activation of this pathway.
Fig. 9. PXR activation and overexpression inhibit the Wnt7a/β-catenin signaling in cultured MTEC.

a Western blot analysis demonstrating that PXR activation inhibited Wnt7a and β-catenin expression after TGFβ1 treatment in MTEC. b Semiquantitative analysis of the protein levels in a (n = 4). c Immunofluorescence assay showed that PXR activation inhibited the activation of β-catenin induced by TGFβ1 (5 ng/mL) for 24 h. Scale bar: 25 μm. d Western blot analysis demonstrated that PXR overexpression inhibited Wnt7a and β-catenin expression after TGFβ1 treatment in MTEC. e Semiquantitative analysis of the protein levels in d (n = 4). f Immunofluorescence assay showed that PXR activation inhibited the activation of β-catenin induced by TGFβ1 (5 ng/mL) for 24 h. Scale bar: 25 μm. Results are expressed as mean ± SEM. **P < 0.01, ***P < 0.001.
PXR inhibits transcription of Wnt7a by interacting with p53
Previous studies have reported that p53 promotes the progression of renal fibrosis [40]. The Wnt/β-catenin signaling pathway is the main target of p53 in regulating anti-differentiation of embryonic stem cells and induction of Wnt signaling by p53 is critical for driving mesendodermal differentiation of pluripotent cells [41, 42]. In addition, PXR protein can physically interact with p53 protein [43]. Based on these findings, we speculated that PXR may inhibit the Wnt7a transcription through interacting with p53 protein. In line with previous report, co-immunoprecipitation (Co-IP) assay confirmed the physical interaction between PXR and p53 in HEK293T cells (Fig. 10a). Furthermore, immunofluorescence staining revealed that the PCN treatment increased the nuclear translocation of PXR and co-localization with p53 protein in the nuclei of MTEC (Fig. 10b). To test whether p53 directly regulates the transcription of Wnt7a gene, we searched the JASPAR website and the PROMO database and found a classic binding element of p53 between −257 bp and −243 bp in the promoter region of mouse Wnt7a (Fig. 10c). By using the ChIP assay, we found p53 can directly bind to the sequence between −504 bp and −227 bp within the Wnt7a promoter, which was significantly inhibited by overexpression of a PXR expression vector (Fig. 10d, e), suggesting PXR can block p53-induced Wnt7a gene transcription. Subsequently, we constructed a luciferase reporter containing a 507 bp DNA fragment spanning the region of −454 bp and +53 bp and co-transfected with the p53 expression vector in renal 293T cells. The results showed that p53 can significantly enhance the promoter activity of the Wnt7a gene, which was markedly attenuated by PXR (Fig. 10f). Consistently, an adenovirus-mediated p53 overexpression (Supplementary Fig. S14a, b) induced Wnt7a and β-catenin and their downstream fibronectin expression (Fig. 10g–i). Furthermore, p53-induced activation of the Wnt7a/β-catenin pathway was significantly inhibited by siRNA-mediated Wnt7a gene knockdown (Fig. 10g–i, Supplementary Fig. S14c, d). Together, these findings demonstrate that PXR physically interacts with p53 to inhibit Wnt7a expression, thereby attenuating fibrosis via blocking the Wnt7a/β-catenin signaling pathway.
Fig. 10. PXR physically interacts with p53 protein and inhibits p53-induced Wnt7a gene transcription and β-catenin activation.

a Co-IP assay showing protein-protein interaction between PXR and p53 in HEK293T cells co-transfected with both mouse PXR and human p53 expression vector. b Immunofluorescence assay demonstrating that PCN treatment increased nuclear co-localization of p53 and PXR protein. Scale bar: 5 μm. c Identification of a putative p53 binding element using the JASPAR website. The predicted p53 binding site in mouse Wnt7a promoter is located between −257 bp and −243 bp. ChIP assay (d) and quantitative analysis (e) revealed that PXR markedly blocked the binding of p53 to mouse Wnt7a gene promoter. Mouse IgG served as a negative control (n = 3). f PXR overexpression significantly inhibited p53-induced Wnt7a promoter-driven luciferase reporter activity in HEK293T cells (n = 4–7). g Western blot analysis demonstrated that adenovirus-mediated p53 overexpression increased expression of Wnt7a, β-catenin and fibronectin. Wnt7a siRNA treatment eliminated p53-induced β-catenin activation and fibronectin expression in MTEC. h Quantitative analysis of the protein levels in g (n = 4). i Immunofluorescence assay showed that p53 overexpression increased the expression and activation of β-catenin, which was abolished by Wnt7a siRNA. Scale bar: 25 μm Results are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Given the high morbidity and mortality of CKD, identification and validation of novel therapeutic targets are critical in the development of effective drugs for the treatment of CKD. Renal fibrosis is the ultimate manifestation of CKD with various etiologies. Therefore, alleviating renal fibrosis is an attractive strategy for the treatment of progressive renal disease [44]. In the present study, we demonstrate a potential therapeutic effect of PXR on renal fibrosis in two murine models of CKD. We found that the mouse PXR activator PCN significantly improved renal function and histological damages in both adenine diet-induced and unilateral ureter obstruction (UUO)-associated renal fibrosis models. The antifibrotic effect of the human PXR activator rifampicin was further validated in a PXR-humanized mouse line. In contrast, PXR gene deficiency markedly accelerated renal fibrosis in both CKD models. Mechanistically, PXR physically interacts with p53 to suppress p53-mediated gene transcription of the profibrotic Wnt7a, thereby inhibiting the Wnt7a/β-catenin pathway (Fig. 11). Collectively, our findings demonstrate that PXR can alleviate renal fibrosis by inhibiting the Wnt7a/β-catenin signaling via interacting with p53.
Fig. 11. Proposed mechanisms by which PXR alleviates kidney fibrosis through interacting with p53.
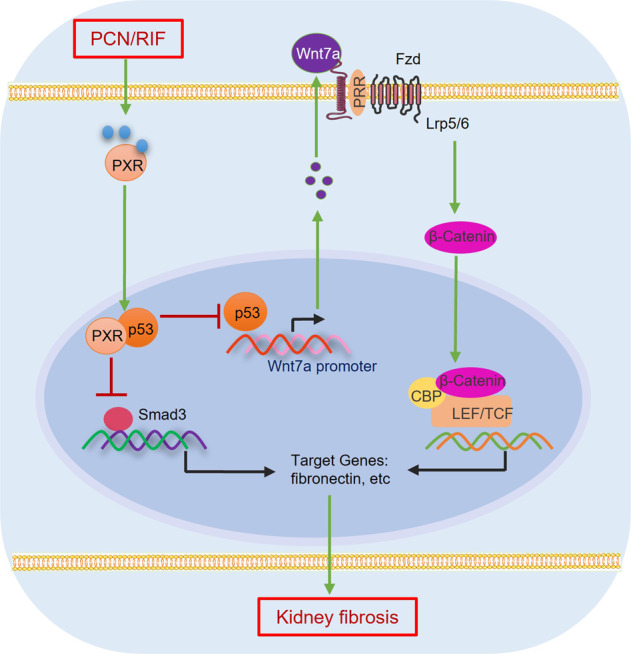
Fibrotic kidney exhibits increased expression of p53, which promotes the assembly of the p53/p-Smad3 complex to induce profibrotic gene expression. In addition, p53 can bind to the promoter region of Wnt7a to increase Wnt7a transcription and β-catenin activation, leading to increased expression of the downstream profibrotic genes. By physically interacting with p53, PXR reduces the binding of p53 to p-Smad3 protein and Wnt7a gene promoter to suppress both the p-Smad3 and β-catenin signaling, thereby attenuating renal fibrosis. Fzd Frizzled, LRP5/6 low density lipoprotein receptor related protein-5 or −6, PRR prorenin receptor, CBP cAMP response element binding protein (CREB)-binding protein, TCF T-cell factor, LEF lymphoid enhancer-binding factor.
PXR is a member of the ligand-activated nuclear receptor transcription factor superfamily, with high expression in the liver, small intestine and kidney [29]. In 2018, it was reported that the expression of PXR was significantly increased in the kidneys of CKD patients and mice with diabetic nephropathy [45]. Similarly, PXR expression level was reported to be inversely associated with the severity of liver fibrosis in patients with chronic HCV infection and its activation exerts an anti-fibrosis effect in a model of CCl4-induced liver injury [46–48]. These findings suggest a possible association between PXR and the pathogenesis of CKD. By using wild-type and PXR humanized or gene deficient mice, the present study provides direct evidence that PXR can alleviate renal function decline, improve structural damages and suppress the activity of the TGFβ1/Smad3 signaling and gene expression of profibrotic genes in two well-documented mouse CKD models, suggesting PXR may represent an attractive novel therapeutic target for the treatment of renal fibrosis in mice and humans. Whether the human PXR activator especially rifampicin is effective in treating CKD patients warrants further investigation.
The Wnt/β-catenin signaling pathway plays a crucial role in driving kidney fibrosis after injury. It is silenced in normal adult kidneys and reactivated in various forms of CKD [49, 50]. A recent study has shown that severe ischemia-reperfusion injury (IRI) resulted in sustained and exaggerated Wnt/β-catenin activation in the kidney, which was accompanied by development of renal fibrotic lesions characterized by interstitial myofibroblast activation and excessive extracellular matrix deposition [51]. By screening all 19 members of the Wnt family, we identified Wnt7a as a dominant isoform induced in the adenine diet-induced renal fibrosis model. We further found that the expression of Wnt7a and activity of its downstream signaling molecule β-catenin were significantly upregulated in both adenine diet-induced and UUO-associated fibrotic kidney disease models, which was markedly suppressed by the treatment with PXR activator PCN. These findings suggest that PXR can suppress the Wnt7a/β-catenin pathway to attenuate renal fibrosis.
Although Wnt protein is the main activator of the β-catenin signaling pathway, β-catenin can also be activated by other factors, such as transforming growth factor β1 (TGFβ1), a central profibrotic cytokine which binds and phosphorylates LRP6 to induce Wnt7a expression and activate classic Wnt signaling [52, 53]. In the present study, we found that PXR activation by PCN or overexpression via an adenovirus-mediated approach inhibits TGFβ1-induced activation of the Wnt/β-catenin signal pathway accompanied by a marked reduction in Smad3 phosphorylation and fibrotic gene expression in cultured renal tubular epithelial cells. Consistent with the in vitro results, PXR activation also significantly ameliorated renal fibrosis and reduced the expression and activity of the Wnt7a/β-catenin pathway, as well as phosphorylated Smad3 and the production of extracellular matrix proteins. Together, these findings demonstrate that PXR activation-mediated attenuation of renal fibrosis is associated with a marked inhibition of the TGFβ-Smad and Wnt/β-catenin signaling pathways.
TGFβ1 is the principal driver of tissue scarring mainly through the canonical TGFβ1/Smad3 pathway leading to renal interstitial fibrosis and eventual renal failure [54]. Increasing evidence suggests that TGFβ1 also promotes renal fibrosis via the non-canonical signaling involving p53 and Wnt/β-catenin networks [40]. p53 is a prominent tumor suppressor and plays a central role in DNA damage repair, growth arrest, senescence, and apoptosis. However, in recent years, p53 has been found to co-operate with other signaling pathways to promote renal fibrosis [55–57]. Its expression and activity are significantly upregulated in many renal diseases and contribute to the pathogenesis of acute renal injury and CKD [54, 58]. It has been previously reported that TGFβ1 stimulates the assembly of a p53-Smad3 protein complex, which is required for transcription of the renal fibrotic genes [40, 59]. In addition, the Wnt/β-catenin signaling pathway is the main target of p53 and molecular interaction and reciprocal transactivation between the p53 and Wnt pathway have been revealed [41, 60–62]. These findings raise a possibility that p53 may promote the development of renal fibrosis by activating the TGFβ-Smad3 and Wnt/β-catenin signaling pathways. In the present study, we found that PXR is colocalized with p53 in the nucleus where it physically interacts with p53 proteins. The molecular interaction between PXR and p53 reduces the binding of p53 to the promoter region of the Wnt7a gene, thereby suppressing p53-induced Wnt7a expression and the activity of the downstream effector β-catenin. In addition, the crosstalk between PXR and p53 also diminishes the assembly of p53-Smad3 complex, leading to the inhibition of Smad3-induced profibrotic gene transcription. Collectively, these results demonstrate that PXR alleviates renal fibrosis through suppressing both TGFβ/Smad3 and Wnt7a/β-catenin pathways via physically interacting with p53 in the kidney (Fig. 11).
In summary, the present study reports evidence that PXR activation improves kidney function and attenuates renal fibrosis in two murine fibrotic kidney models generated in wild-type, PXR gene knockout and PXR-humanized mice. Global deletion of PXR markedly worsens renal fibrosis following adenine diet feeding or ureter obstruction. The renoprotective effect of PXR is associated with a marked reduction of Smad3 and β-catenin expression and activity. The underlying mechanism appears to be related to the protein interaction between PXR and p53, which suppresses p53-induced Wnt7a gene transcription and Smad3-elicited profibrotic gene expression. Therefore, PXR plays a critical role in the pathogenesis of renal fibrosis and pharmacological targeting the PXR-p53 interaction represents an attractive therapeutic strategy for the treatment of CKD.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Prof. Nan-hong Tang for providing pcDNA3.0-p53 expression vector. This work was supported by the National Key R&D Program of China (2020YFC2005000 to XYZ and YFG); the National Natural Science Foundation of China (82270703 to XYZ, 81970606 to XYZ, and 81970595 to YFG); and the East China Normal University Medicine and Health Joint Fund (2022JKXYD03001 to XYZ).
Author contributions
WHM, YFG and XYZ conceived the experiments. WHM, YY, HCL, SYH, CZ, YHZ, YZH, XWS and RFQ performed the experiments and acquired the data. ZLL, HX and CXD analyzed the data. WHM drafted the original manuscript. YFG and XYZ contributed to the conception of the study and revised the manuscript. All authors approved the final manuscript and agreed to be accountable for all aspects of the work.
Competing interests
The authors declare no competing interests.
Ethics approval
The use of animals and the study protocols were reviewed and approved by the Animal Care and Use Review Committee of Dalian Medical University (AEE22128).
Contributor Information
You-fei Guan, Email: guanyf@dmu.edu.cn.
Xiao-yan Zhang, Email: xyzhang@hsc.ecnu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-023-01113-7.
References
- 1.Stenvinkel P, Chertow GM, Devarajan P, Levin A, Andreoli SP, Bangalore S, et al. Chronic inflammation in chronic kidney disease progression: role of Nrf2. Kidney Int Rep. 2021;6:1775–87. doi: 10.1016/j.ekir.2021.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neovius M, Jacobson SH, Eriksson JK, Elinder CG, Hylander B. Mortality in chronic kidney disease and renal replacement therapy: a population-based cohort study. BMJ Open. 2014;4:e004251. doi: 10.1136/bmjopen-2013-004251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al. Chronic kidney disease: global dimension and perspectives. Lancet. 2013;382:260–72. doi: 10.1016/s0140-6736(13)60687-x. [DOI] [PubMed] [Google Scholar]
- 4.Tonelli M, Wiebe N, Culleton B, House A, Rabbat C, Fok M, et al. Chronic kidney disease and mortality risk: a systematic review. J Am Soc Nephrol. 2006;17:2034–47. doi: 10.1681/asn.2005101085. [DOI] [PubMed] [Google Scholar]
- 5.Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124:2333–40. doi: 10.1172/jci72271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miguel V, Tituana J, Herrero JI, Herrero L, Serra D, Cuevas P, et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J Clin Invest. 2021;131. 10.1172/jci140695. [DOI] [PMC free article] [PubMed]
- 7.Edeling M, Ragi G, Huang S, Pavenstadt H, Susztak K. Developmental signalling pathways in renal fibrosis: the roles of Notch, Wnt and Hedgehog. Nat Rev Nephrol. 2016;12:426–39. doi: 10.1038/nrneph.2016.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piersma B, Bank RA, Boersema M. Signaling in fibrosis: TGF-beta, WNT, and YAP/TAZ converge. Front Med. 2015;2:59. doi: 10.3389/fmed.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urban ML, Manenti L, Vaglio A. Fibrosis–a common pathway to organ injury and failure. N Engl J Med. 2015;373:95–6. doi: 10.1056/NEJMc1504848. [DOI] [PubMed] [Google Scholar]
- 10.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 11.Zuo Y, Liu Y. New insights into the role and mechanism of Wnt/beta-catenin signalling in kidney fibrosis. Nephrology. 2018;23:38–43. doi: 10.1111/nep.13472. [DOI] [PubMed] [Google Scholar]
- 12.Feng Y, Ren J, Gui Y, Wei W, Shu B, Lu Q, et al. Wnt/beta-catenin-promoted macrophage alternative activation contributes to kidney fibrosis. J Am Soc Nephrol. 2018;29:182–93. doi: 10.1681/asn.2017040391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Surendran K, Schiavi S, Hruska KA. Wnt-dependent beta-catenin signaling is activated after unilateral ureteral obstruction, and recombinant secreted frizzled-related protein 4 alters the progression of renal fibrosis. J Am Soc Nephrol. 2005;16:2373–84. doi: 10.1681/asn.2004110949. [DOI] [PubMed] [Google Scholar]
- 14.Breyer MD, Susztak K. The next generation of therapeutics for chronic kidney disease. Nat Rev Drug Discov. 2016;15:568–88. doi: 10.1038/nrd.2016.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dolman ME, Harmsen S, Storm G, Hennink WE, Kok RJ. Drug targeting to the kidney: advances in the active targeting of therapeutics to proximal tubular cells. Adv Drug Deliv Rev. 2010;62:1344–57. doi: 10.1016/j.addr.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 16.Luan ZL, Zhang C, Ming WH, Huang YZ, Guan YF, Zhang XY. Nuclear receptors in renal health and disease. EBioMedicine. 2022;76:103855. doi: 10.1016/j.ebiom.2022.103855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marek CJ, Tucker SJ, Konstantinou DK, Elrick LJ, Haefner D, Sigalas C, et al. Pregnenolone-16alpha-carbonitrile inhibits rodent liver fibrogenesis via PXR (pregnane X receptor)-dependent and PXR-independent mechanisms. Biochem J. 2005;387:601–8. doi: 10.1042/bj20041598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright MC. The impact of pregnane X receptor activation on liver fibrosis. Biochem Soc Trans. 2006;34:1119–23. doi: 10.1042/bst0341119. [DOI] [PubMed] [Google Scholar]
- 19.Haughton EL, Tucker SJ, Marek CJ, Durward E, Leel V, Bascal Z, et al. Pregnane X receptor activators inhibit human hepatic stellate cell transdifferentiation in vitro. Gastroenterology. 2006;131:194–209. doi: 10.1053/j.gastro.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 20.Luan Z, Wei Y, Huo X, Sun X, Zhang C, Ming W, et al. Pregnane X receptor (PXR) protects against cisplatin-induced acute kidney injury in mice. Biochim Biophys Acta Mol Basis Dis. 2021;1867:165996. doi: 10.1016/j.bbadis.2020.165996. [DOI] [PubMed] [Google Scholar]
- 21.Yu X, Xu M, Meng X, Li S, Liu Q, Bai M, et al. Nuclear receptor PXR targets AKR1B7 to protect mitochondrial metabolism and renal function in AKI. Sci Transl Med. 2020;12. 10.1126/scitranslmed.aay7591. [DOI] [PubMed]
- 22.Luan ZL, Ming WH, Sun XW, Zhang C, Zhou Y, Zheng F, et al. A naturally occurring FXR agonist, alisol B 23-acetate, protects against renal ischemia-reperfusion injury. Am J Physiol Ren Physiol. 2021;321:F617–F28. doi: 10.1152/ajprenal.00193.2021. [DOI] [PubMed] [Google Scholar]
- 23.Tie L, Xiao H, Wu DL, Yang Y, Wang P. A brief guide to good practices in pharmacological experiments: Western blotting. Acta Pharmacol Sin. 2021;42:1015–7. doi: 10.1038/s41401-020-00539-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schley G, Klanke B, Kalucka J, Schatz V, Daniel C, Mayer M, et al. Mononuclear phagocytes orchestrate prolyl hydroxylase inhibition-mediated renoprotection in chronic tubulointerstitial nephritis. Kidney Int. 2019;96:378–96. doi: 10.1016/j.kint.2019.02.016. [DOI] [PubMed] [Google Scholar]
- 25.Messeguer X, Escudero R, Farre D, Nunez O, Martinez J, Alba MM. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics. 2002;18:333–4. doi: 10.1093/bioinformatics/18.2.333. [DOI] [PubMed] [Google Scholar]
- 26.Farre D, Roset R, Huerta M, Adsuara JE, Rosello L, Alba MM, et al. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003;31:3651–3. doi: 10.1093/nar/gkg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ali BH, Al-Salam S, Al Za’abi M, Waly MI, Ramkumar A, Beegam S, et al. New model for adenine-induced chronic renal failure in mice, and the effect of gum acacia treatment thereon: comparison with rats. J Pharmacol Toxicol Methods. 2013;68:384–93. doi: 10.1016/j.vascn.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Q, Liu L, Lin W, Yin S, Duan A, Liu Z, et al. Rhein reverses Klotho repression via promoter demethylation and protects against kidney and bone injuries in mice with chronic kidney disease. Kidney Int. 2017;91:144–56. doi: 10.1016/j.kint.2016.07.040. [DOI] [PubMed] [Google Scholar]
- 29.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 30.Ma Y, Liu D. Activation of pregnane X receptor by pregnenolone 16 alpha-carbonitrile prevents high-fat diet-induced obesity in AKR/J mice. PLoS One. 2012;7:e38734. doi: 10.1371/journal.pone.0038734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ali BH, Al Salam S, Al Suleimani Y, Al Za’abi M, Abdelrahman AM, Ashique M, et al. Effects of the SGLT-2 inhibitor canagliflozin on adenine-induced chronic kidney disease in rats. Cell Physiol Biochem. 2019;52:27–39. doi: 10.33594/000000003. [DOI] [PubMed] [Google Scholar]
- 32.Akchurin OM, Kaskel F. Update on inflammation in chronic kidney disease. Blood Purif. 2015;39:84–92. doi: 10.1159/000368940. [DOI] [PubMed] [Google Scholar]
- 33.Mihai S, Codrici E, Popescu ID, Enciu AM, Albulescu L, Necula LG, et al. Inflammation-related mechanisms in chronic kidney disease prediction, progression, and outcome. J Immunol Res. 2018;2018:2180373. doi: 10.1155/2018/2180373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez-Klimova E, Aparicio-Trejo OE, Tapia E, Pedraza-Chaverri J. Unilateral ureteral obstruction as a model to investigate fibrosis-attenuating treatments. Biomolecules. 2019;9. 10.3390/biom9040141. [DOI] [PMC free article] [PubMed]
- 35.Hassani-Nezhad-Gashti F, Rysa J, Kummu O, Napankangas J, Buler M, Karpale M, et al. Activation of nuclear receptor PXR impairs glucose tolerance and dysregulates GLUT2 expression and subcellular localization in liver. Biochem Pharmacol. 2018;148:253–64. doi: 10.1016/j.bcp.2018.01.001. [DOI] [PubMed] [Google Scholar]
- 36.Kliewer SA. Nuclear receptor PXR: discovery of a pharmaceutical anti-target. J Clin Invest. 2015;125:1388–9. doi: 10.1172/jci81244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miao J, Liu J, Niu J, Zhang Y, Shen W, Luo C, et al. Wnt/beta-catenin/RAS signaling mediates age-related renal fibrosis and is associated with mitochondrial dysfunction. Aging Cell. 2019;18:e13004. doi: 10.1111/acel.13004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo C, Zhou S, Zhou Z, Liu Y, Yang L, Liu J, et al. Wnt9a promotes renal fibrosis by accelerating cellular senescence in tubular epithelial cells. J Am Soc Nephrol. 2018;29:1238–56. doi: 10.1681/asn.2017050574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen X, Tan H, Xu J, Tian Y, Yuan Q, Zuo Y, et al. Klotho-derived peptide 6 ameliorates diabetic kidney disease by targeting Wnt/beta-catenin signaling. Kidney Int. 2022;102:506–20. doi: 10.1016/j.kint.2022.04.028. [DOI] [PubMed] [Google Scholar]
- 40.Higgins SP, Tang Y, Higgins CE, Mian B, Zhang W, Czekay RP, et al. TGF-beta1/p53 signaling in renal fibrogenesis. Cell Signal. 2018;43:1–10. doi: 10.1016/j.cellsig.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee KH, Li M, Michalowski AM, Zhang X, Liao H, Chen L, et al. A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc Natl Acad Sci USA. 2010;107:69–74. doi: 10.1073/pnas.0909734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Zou Y, Nowotschin S, Kim SY, Li QV, Soh CL, et al. The p53 family coordinates Wnt and nodal inputs in mesendodermal differentiation of embryonic stem cells. Cell Stem Cell. 2017;20:70–86. doi: 10.1016/j.stem.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan L, Chen Z, Wu L, Su Y, Wang X, Tang N. Inhibitory effect of PXR on ammonia-induced hepatocyte autophagy via P53. Toxicol Lett. 2018;295:153–61. doi: 10.1016/j.toxlet.2018.06.1066. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7:684–96. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watanabe A, Marumo T, Kawarazaki W, Nishimoto M, Ayuzawa N, Ueda K, et al. Aberrant DNA methylation of pregnane X receptor underlies metabolic gene alterations in the diabetic kidney. Am J Physiol Ren Physiol. 2018;314:F551–F60. doi: 10.1152/ajprenal.00390.2017. [DOI] [PubMed] [Google Scholar]
- 46.Congiu M, Mashford ML, Slavin JL, Desmond PV. Coordinate regulation of metabolic enzymes and transporters by nuclear transcription factors in human liver disease. J Gastroenterol Hepatol. 2009;24:1038–44. doi: 10.1111/j.1440-1746.2009.05800.x. [DOI] [PubMed] [Google Scholar]
- 47.Hanada K, Nakai K, Tanaka H, Suzuki F, Kumada H, Ohno Y, et al. Effect of nuclear receptor downregulation on hepatic expression of cytochrome P450 and transporters in chronic hepatitis C in association with fibrosis development. Drug Metab Pharmacokinet. 2012;27:301–6. doi: 10.2133/dmpk.dmpk-11-rg-077. [DOI] [PubMed] [Google Scholar]
- 48.Wallace K, Cowie DE, Konstantinou DK, Hill SJ, Tjelle TE, Axon A, et al. The PXR is a drug target for chronic inflammatory liver disease. J Steroid Biochem Mol Biol. 2010;120:137–48. doi: 10.1016/j.jsbmb.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol. 2009;20:765–76. doi: 10.1681/asn.2008060566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou L, Li Y, Zhou D, Tan RJ, Liu Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/beta-catenin signaling. J Am Soc Nephrol. 2013;24:771–85. doi: 10.1681/asn.2012080865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao L, Zhou D, Tan RJ, Fu H, Zhou L, Hou FF, et al. Sustained activation of Wnt/beta-catenin signaling drives AKI to CKD progression. J Am Soc Nephrol. 2016;27:1727–40. doi: 10.1681/asn.2015040449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rooney B, O’Donovan H, Gaffney A, Browne M, Faherty N, Curran SP, et al. CTGF/CCN2 activates canonical Wnt signalling in mesangial cells through LRP6: implications for the pathogenesis of diabetic nephropathy. FEBS Lett. 2011;585:531–8. doi: 10.1016/j.febslet.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 53.Ren S, Johnson BG, Kida Y, Ip C, Davidson KC, Lin SL, et al. LRP-6 is a coreceptor for multiple fibrogenic signaling pathways in pericytes and myofibroblasts that are inhibited by DKK-1. Proc Natl Acad Sci USA. 2013;110:1440–5. doi: 10.1073/pnas.1211179110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Overstreet JM, Gifford CC, Tang J, Higgins PJ, Samarakoon R. Emerging role of tumor suppressor p53 in acute and chronic kidney diseases. Cell Mol Life Sci. 2022;79:474. doi: 10.1007/s00018-022-04505-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma Z, Li L, Livingston MJ, Zhang D, Mi Q, Zhang M, et al. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J Clin Invest. 2020;130:5011–26. doi: 10.1172/jci135536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patel S, Tang J, Overstreet JM, Anorga S, Lian F, Arnouk A, et al. Rac-GTPase promotes fibrotic TGF-beta1 signaling and chronic kidney disease via EGFR, p53, and Hippo/YAP/TAZ pathways. FASEB J. 2019;33:9797–810. doi: 10.1096/fj.201802489RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li C, Xie N, Li Y, Liu C, Hou FF, Wang J. N-acetylcysteine ameliorates cisplatin-induced renal senescence and renal interstitial fibrosis through sirtuin1 activation and p53 deacetylation. Free Radic Biol Med. 2019;130:512–27. doi: 10.1016/j.freeradbiomed.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 58.Ying Y, Kim J, Westphal SN, Long KE, Padanilam BJ. Targeted deletion of p53 in the proximal tubule prevents ischemic renal injury. J Am Soc Nephrol. 2014;25:2707–16. doi: 10.1681/asn.2013121270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Overstreet JM, Samarakoon R, Meldrum KK, Higgins PJ. Redox control of p53 in the transcriptional regulation of TGF-beta1 target genes through SMAD cooperativity. Cell Signal. 2014;26:1427–36. doi: 10.1016/j.cellsig.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xiao Q, Werner J, Venkatachalam N, Boonekamp KE, Ebert MP, Zhan T. Cross-talk between p53 and Wnt signaling in cancer. Biomolecules. 2022;12. 10.3390/biom12030453. [DOI] [PMC free article] [PubMed]
- 61.Han D, Xu Y, Peng WP, Feng F, Wang Z, Gu C, et al. Citrus alkaline extracts inhibit senescence of A549 cells to alleviate pulmonary fibrosis via the beta-catenin/P53 pathway. Med Sci Monit. 2021;27:e928547. doi: 10.12659/msm.928547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gu Z, Tan W, Feng G, Meng Y, Shen B, Liu H, et al. Wnt/beta-catenin signaling mediates the senescence of bone marrow-mesenchymal stem cells from systemic lupus erythematosus patients through the p53/p21 pathway. Mol Cell Biochem. 2014;387:27–37. doi: 10.1007/s11010-013-1866-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.