

Abstract
Activation of NLR family pyrin domain-containing 3 (NLRP3) inflammasome plays important role in defending against infections, but its aberrant activation is causally linked to many inflammatory diseases, thus being a therapeutic target for these diseases. Theaflavin, one major ingredient of black tea, exhibits potent anti-inflammatory and anti-oxidative activities. In this study, we investigated the therapeutic effects of theaflavin against NLRP3 inflammasome activation in macrophages in vitro and in animal models of related diseases. We showed that theaflavin (50, 100, 200 μM) dose-dependently inhibited NLRP3 inflammasome activation in LPS-primed macrophages stimulated with ATP, nigericin or monosodium urate crystals (MSU), evidenced by reduced release of caspase-1p10 and mature interleukin-1β (IL-1β). Theaflavin treatment also inhibited pyroptosis as shown by decreased generation of N-terminal fragment of gasdermin D (GSDMD-NT) and propidium iodide incorporation. Consistent with these, theaflavin treatment suppressed ASC speck formation and oligomerization in macrophages stimulated with ATP or nigericin, suggesting reduced inflammasome assembly. We revealed that theaflavin-induced inhibition on NLRP3 inflammasome assembly and pyroptosis resulted from ameliorated mitochondrial dysfunction and reduced mitochondrial ROS production, thereby suppressing interaction between NLRP3 and NEK7 downstream of ROS. Moreover, we showed that oral administration of theaflavin significantly attenuated MSU-induced mouse peritonitis and improved the survival of mice with bacterial sepsis. Consistently, theaflavin administration significantly reduced serum levels of inflammatory cytokines including IL-1β and attenuated liver inflammation and renal injury of mice with sepsis, concomitant with reduced generation of caspase-1p10 and GSDMD-NT in the liver and kidney. Together, we demonstrate that theaflavin suppresses NLRP3 inflammasome activation and pyroptosis by protecting mitochondrial function, thus mitigating acute gouty peritonitis and bacterial sepsis in mice, highlighting a potential application in treating NLRP3 inflammasome-related diseases.
Keywords: NLRP3 inflammasome, theaflavin, mitochondrial ROS, pyroptosis, sepsis, organ injury
Introduction
Inflammasomes are multiprotein signaling platforms that are formed in response to environmental stresses or pathogenic infections [1]. One of the most extensively studied inflammasomes is NLR family pyrin domain-containing 3 (NLRP3) inflammasome. NLRP3 is a cytosolic sensor that is activated by stresses or “danger signals”, such as nigericin, ATP, and monosodium urate crystals (MSU), released from pathogens or injured tissues. All these signals trigger the assembly of NLRP3 inflammasome that acts as a platform for the activation of pro-caspase-1, the latter of which in turn leads to the maturation of pro-inflammatory cytokines including interleukin (IL)-1β [2]. In addition to mediating IL-1β maturation, active caspase-1 also cleaves the pore-forming protein gasdermin D (GSDMD) to generate its N-terminal fragment (GSDMD-NT) [3, 4]. The latter binds distinct phospholipids, such as phosphatidylinositol phosphates and phosphatidylserine, in the inner leaflet of the plasma membrane to form pores [5–7]. Such membranous pores not only mediate the release of cellular contents including mature IL-1β but also induce a lytic form of cell death known as pyroptosis [8, 9], which is an inflammatory cell death that can trigger robust inflammation [10, 11]. Apart from binding to the plasma membrane, GSDMD-NT also forms pores in the mitochondrial membrane thereby leading to the release mitochondrial reactive oxygen species (ROS) and mitochondrial DNA, which in turn augment inflammasome activation [12, 13].
Normally, NLRP3 inflammasome activation plays critical roles in maintaining tissue homeostasis by eliminating stressed cells and in defensing against infections by triggering potent inflammatory responses [14]. Pyroptosis following the inflammasome activation leads to the release of intracellular pathogens, which can then be eliminated by other phagocytes such as recruited neutrophils [15]. However, excessive activation of NLRP3 inflammasome also contributes to the pathogenesis and progression of various inflammatory diseases, such as acute gouty arthritis [16], inflammatory bowel disease [17], atherosclerosis [18], type 2 diabetes [19], Alzheimer’s disease [20], and cancers [21]. In addition, excessive inflammasome activation and pyroptosis are associated with multiple organ dysfunction in septic patients [22–24], which is currently only to be treated with antibiotics and supportive care [25, 26]. On the other hand, pharmacologic inhibition of NLRP3 inflammasome activation and pyroptotic cell death dampens the severity and the progression of various inflammatory diseases [27–30], suggesting that agents targeting NLRP3 inflammasome assembly are promising candidates for the treatment of inflammasome-related diseases [31].
Aside from chemically synthesized agents such as MCC950 that target NLRP3 [22, 32], several natural products from medicinal herbs have been shown to modulate NLRP3 inflammasome activation [33–38], For example, parthenolide, a sesquiterpene lactone isolated from feverfew (Tanacetum parthenium), can directly target the ATPase activity of NLRP3 protein to exert anti-inflammatory properties [38]. Oridonin, an active component of the medicinal herb Rabdosia rubescens, has been demonstrated to covalently interact with the cysteine residue 279 of NLRP3, thereby interrupting NLRP3-NEK7 interaction and consequently inhibiting the activation of NLRP3 inflammasome [36]. Our previous study also showed that scutellarin, a flavonoid purified from Erigeron breviscapus, inhibited NLRP3 inflammasome activation and pyroptosis in macrophages through augmenting PKA signaling [34]. These studies suggest that phytochemicals from edible and herbal plants are a preferable source for identifying therapeutic agents, considering that a small clinical study of the well-known MCC950 has been discontinued due to its liver toxicity [39, 40].
Black tea is widely consumed because of its health benefits, of which theaflavin is a major ingredient [41]. Theaflavin has been shown to possess various pharmacological activities such as anti-inflammatory, anticancer, antibacterial, antioxidative, and hypolipidemic activities [42–47]. For its anti-inflammatory activity, theaflavin has been shown to significantly reduce the mRNA levels of the lipopolysaccharide (LPS)-induced inflammatory cytokines, such as IL-6, monocyte chemoattractant protein-1, and intercellular adhesion molecule-1 in bone marrow-derived macrophages by blocking NF-κB signaling [48]. Theaflavin can also inhibit cerebral hemorrhage-induced inflammatory responses and brain injury by suppressing NF-κB and mitogen-activated protein kinase pathways in vivo [44]. Despite these studies, it is not known whether theaflavin has any influence on NLRP3 inflammasome activation and pyroptosis. Given the beneficial effects and low toxicity of dietary theaflavins [49], it is of interest to explore theaflavin’s effects on the activation of NLRP3 inflammasomes and in animal models of NLRP3-related diseases.
In this study, we demonstrated that theaflavin inhibited NLRP3 inflammasome activation in macrophages upon ATP, nigericin, or MSU treatment. Theaflavin decreased mitochondrial damage and mitochondrial ROS production, thereby reducing NLRP3 inflammasome assembly and pyroptosis. Moreover, theaflavin was able to mitigate MSU-induced peritonitis. And it also ameliorated septic organ injury and prolonged the survival of mice with bacterial sepsis, which was associated with decreased inflammasome activation in vivo. Our data reveal a potential application of theaflavin as an agent to treat inflammatory diseases associated with aberrant NLRP3 activation.
Materials and methods
Reagents and antibodies
Theaflavin (B20140) was bought from Yuan-Ye Bio-Technology (Shanghai, China), dissolved in dimethyl sulfoxide (DMSO) at 100 mM and stored at −80 °C. Monosodium urate crystal (MSU) (tlrl-msu), nigericin (tlrl-nig), Pam3CysSerLys4 (Pam3CSK4, tlrl-pms), flagellin (tlrl-pafla), and poly(dA:dT) (tlrl-patn) were obtained from InvivoGen (San Diego, CA, USA). FuGENE HD transfection reagent (E2311) was obtained from Promega (Madison, WI, USA). Dulbecco’s modified Eagle’s medium (DMEM) with high glucose (11965092), fetal bovine serum (FBS) (10091148), penicillin-streptomycin (15140122), Opti-MEM (11058021), Lipofectamine 2000 (11668-030), the antibody against β-actin (PA5-85291), and MitoSOX Red (M36008) were obtained from Thermo Fisher (Carlsbad, CA, USA). DMSO (D8418), propidium iodide (PI) (P4170), Hoechst 33342 (B2261), disuccinimidyl suberate (D8418), LPS (Escherichia coli O111:B4) (L4391), Tween-80 (P8074), Tween-20 (P1379), adenosine triphosphate (ATP) (A6419), and DL-dithiothreitol (DTT) (D0632) were bought from Sigma-Aldrich (St. Louis, MO, USA). MCC950 (S8930) was purchased from Selleck Chemical (Houston, TX, USA). Phorbol 12-myristate 13-acetate (PMA) (S1819), cell lysis buffer (P0013), and phenylmethanesulfonyl fluoride (PMSF) (ST505) were obtained from Beyotime (Shanghai, China). Specific antibodies against IL-1β (12242), ASC (67824), Alexa Flour 488-conjugated anti-ASC (17507), horse-radish peroxidase (HRP)-linked goat anti-rabbit IgG (7074) were purchased from Cell Signaling Technology (Danvers, MA, USA). The anti-NLRP3 antibody (AG-20B-0014) was purchased from AdipogenAG (Liestal, Switzerland). The antibodies against pro-Caspase-1 + p10 + p12 (ab179515), GSDMD (ab209845), and NEK7 (ab133514) were purchased from Abcam (Cambridge, UK). CF568-conjugated goat-anti-rabbit IgG (H + L), highly cross-adsorbed (20103) was obtained from Biotium (Hayward, CA, USA). Anti-mouse CD11b FITC (101205) and anti-mouse Ly-6G PE (127607) were obtained from BioLegend (San Diego, CA, USA).
Animals
Female C57BL/6J mice (6–8 weeks of age) were bought from the Laboratory Animal Center of Southern Medical University (Guangzhou, China). C57BL/6J Nlrp3+/− mice were generated by CRISPR/Cas9 system and obtained from GemPharmatech Co. (Nanjing, China) and the homozygote genotype was identified as previously described [50]. All the mice were acclimatized for 1 week before experiments. The animal study was reviewed and approved by the Committee on the Ethics of Animal Experiments at Jinan University. Animal experiments were conducted in accordance with the guidelines of the Jinan University Laboratory Animal Center.
Cell culture
Mouse macrophage cell line J774A.1 was obtained from the Kunming Cell Bank of Type Culture Collection, Chinese Academy of Sciences (Kunming, China). The cells were maintained in complete DMEM medium (containing 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin) and cultured at 37 °C in a humidified incubator with 5% CO2. The cells were sub-cultured every 2–3 days by using a cell scraper (541070, Greiner, Frickenhausen, Germany) to split cells. Before experiments, cells were cultured in complete DMEM medium overnight in 24-well plates at 9 × 104 cells/well (0.5 mL) or in 6-well plates at 3.5 × 105 cells/well (1.7 mL). Human THP-1 cells (kindly provided by Dr. Yao Wang of Sun Yat-sen University, Guangzhou, China) were cultured in RPMI-1640 supplemented with 10% FBS and 50 µM β-mercaptoethanol. THP-1 cells were differentiated into macrophages by adding 500 nM of PMA for 16 h.
Bone marrow-derived macrophages differentiation
Bone marrow-derived macrophages (BMDMs) were differentiated as described previously [51]. Briefly, C57BL/6J mice were sacrificed by cervical dislocation and sterilized with 75% ethanol. Bone marrow cells from hind femora and tibias of mice were cultured in BM-Mac medium (80% DMEM supplemented with 10% FBS and 1% penicillin–streptomycin plus 20% M-CSF-contained medium from L929 cells) at 37 °C in a humidified incubator of 5% CO2. Bone marrow cells were differentiated into BMDMs after 6 days. BMDMs were then collected by using a cell-scraper and incubated in six-well plates at 1.6 × 106 cells/well (1.7 mL) or 24-well plates at 2.5 × 105 cells/well (0.5 mL) with complete DMEM medium. The cells were ready for experiments after overnight incubation.
Cell death assay
Cell death was measured by PI incorporation as previously described [50, 52]. Briefly, cells were incubated for 4 h with LPS (500 ng/mL) and then were pre-treated with or without indicated concentrations of theaflavin for 1 h followed by stimulation with ATP, nigericin, or MSU for indicated time periods. After indicated treatment, PI (2 µg/mL) and Hoechst 33342 (5 µg/mL) solutions were added into medium to stain the dying cells and the nuclei, respectively. To trigger the activation of NLR family CARD domain-containing protein 4 (NLRC4) and absent in melanoma 2 (AIM2) inflammasome, BMDMs were primed with 1 μg/mL Pam3CSK4 in Opti-MEM for 4 h. Subsequently, the cells were treated with various concentrations of theaflavin for 1 h and then transfected with 0.5 μg/mL flagellin plus 0.25% (v/v) FuGENE HD for 16 h, or 2 μg/mL poly(dA:dT) plus 0.1% (v/v) Lipofectamine 2000 for 16 h, respectively [50]. The stained cells were observed by live imaging using Zeiss Axio Observer D1 microscope equipped with a Zeiss LD Plan-Neofluar 20×/0.4 Korr M27 objective lens (Carl Zeiss MicroImaging GmbH, Göttingen, Germany). Fluorescence images were captured with a Zeiss Axiocam MR R3-cooled CCD camera controlled with ZEN software (Carl Zeiss).
Precipitation of soluble proteins
Soluble proteins in culture supernatants were precipitated with 7.2% trichloroacetic acid plus 0.15% sodium deoxycholate as previously reported [50]. The precipitated pellets were then washed thrice with cold acetone, and re-dissolved in 2× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer and subjected to Western blot analysis of mature IL-1β and caspase-1p10.
Western blot analysis
Western blotting was performed as previously described [51]. Briefly, after indicated treatments, total proteins were separated by SDS-PAGE and electro-transferred to PVDF membranes (03010040001; Roche Diagnostics GmbH, Mannheim, Germany). The membrane was then blocked and incubated with a specific primary antibody at 4 °C overnight, followed by incubation with appropriate HRP-conjugated secondary antibody. Target bands were revealed with an enhanced chemiluminescence kit (BeyoECL Plus; P0018S, Beyotime) and captured on X-ray films. Blots were recorded by FluorChem 8000 imaging system (AlphaInnotech, San Leandro, CA, USA) and analyzed with the AlphaEaseFC 4.0 software (AlphaInnotech).
Co-immunoprecipitation
The interaction between NEK7 and NLRP3 was analyzed by using a co-immunoprecipitation (Co-IP) assay. Briefly, BMDMs were seeded in a 6-well plate overnight and treated routinely to trigger NLRP3 inflammasome activation. After removing culture medium, the cells were lysed with freshly prepared IP lysis/wash buffer containing PMSF (P0013, Beyotime) in an ice bath for 5 min. Then, the cell lysates were scraped, transferred to cold tubes and sheared 10 times through a 21-gauge needle. After being centrifuged at 13,000 r/min (4 °C) for 10 min, the supernatants were transferred to ice-cold tubes. The protein concentration was measured with a Pierce BCA protein assay kit (#23227, Thermo Scientific), and equal amounts of lysates (150 µg) were subjected to Co-IP with a Pierce classic IP kit (#26146, Thermo Fisher) according to the manufacturer’s protocol. In brief, after being precleared with control agarose resin, the lysates were incubated with mouse anti-NLRP3 antibodies or isotype control IgG (0.75 µg antibody/100 µg lysates) at 4 °C overnight. The protein-antibody complexes were captured with Protein A/G agarose resin and eluted with 2× reducing sample buffer containing DTT in boiling water for 10 min. The abundance of NLRP3 and NEK7 in the elution was analyzed by Western blotting.
Immunofluorescence microscopy
Immunofluorescence analysis was performed as previously described [50]. In brief, BMDMs were seeded in 24-well plates overnight at 37 °C. After indicated treatments, cells were fixed with 4% paraformaldehyde for 15 min and permeabilized with 2 mL of cold methanol at −20 °C for 10 min. After permeabilization, the cells were blocked by blocking buffer, followed by incubation with anti-ASC antibodies overnight and staining with CF568-conjugated goat-anti-rabbit IgG for 1 h. Finally, the nuclei were stained with 5 µg/mL Hoechst 33342 solution for 10 min. The images were acquired through a Zeiss Axio Observer D1 microscope with a Zeiss LD Plan-Neofluar 40×/0.6 Korr M27 objective (Carl Zeiss) with Zeiss Axiocam MR R3 cooled CCD camera controlled with the ZEN software (Carl Zeiss).
ASC oligomer cross-linking
The cross-linking of ASC oligomers was carried out as previously described [35]. Briefly, BMDMs were seeded in six-well plates overnight at 37 °C in an incubator with 5% CO2. After indicated treatments, cells were lysed with cold PBS containing 0.5% Triton X-100, and the cell lysates were centrifuged at 6000 × g for 15 min at 4 °C. The pellets were washed twice with PBS and then resuspended in 200 µL of PBS and cross-linked with disuccinimidyl suberate (2 mM) for 30 min at room temperature. The crosslinked pellets were centrifuged at 6000 × g for 15 min at 4 °C and redissolved in 20 µL of 2× SDS-PAGE sample loading buffer. Samples were boiled for 5 min and subjected to Western blot analysis.
Detection of cellular reactive oxygen species
Reactive oxygen species (ROS) assay kit (S0033, Beyotime) was used to detect total intracellular ROS generated in live cells according to the instructions of the supplier, and the fluorescence of DCFH-DA within the cells was observed and images were captured with an Axio Observer D1 microscope (Carl Zeiss). Mean fluorescence intensity was quantified with the ZEN software (Carl Zeiss).
Mitochondrial ROS and mitochondrial membrane potential
For detection of mitochondrial ROS, after indicated treatments, MitoSOX red (3 μM) was used to detect the mitochondrial superoxide levels (indicative of mitochondrial ROS) in live cells according to the instructions of the supplier, and the fluorescence of MitoSOX within the cells were observed and captured with an Axio Observer D1 microscope (Carl Zeiss). Mitochondrial membrane potential was detected with TMRE assay kit (C2001, Beyotime) or JC-1 assay kit (C2006, Beyotime) according to the instructions of the manufacturer. Mean fluorescence intensity was quantified with the ZEN software (Carl Zeiss).
Mitochondrial morphology
To detect the morphology of mitochondria after indicated treatments, BMDMs were stained with Mito-Tracker Green (C1048, Beyotime) at a final concentration of 200 nM and Hoechst 33342 (10 µg/mL) for 30 min and images were captured with an Axio Observer D1 microscope (Carl Zeiss).
Mitochondrial protein release assay
Separation of the cytosol and mitochondrial fractions was performed according to the instructions of the cell mitochondria isolation kit (C3601, Beyotime). Immunoblotting was used to detect the levels of cytochrome c in the cytosol and mitochondria.
Mitochondrial DNA release assay
Measurement of mtDNA in the cytosol of BMDMs was performed according to the protocol described previously [53–55]. Briefly, 1.6 × 106 cells in 2 mL of complete medium were seeded in a six-well plate. After indicated treatments, 1% NP-40 (100 µL) was added to each well. After 15 min of lysis on ice, the lysates were spined at 13,000 r/min (16,000 × g) for 15 min at 4 °C to pellet the insoluble fraction. The supernatant (the cytosolic fraction) was transferred to a new tube and the pellet was discarded. DNA was isolated from the cytosolic fraction using a Universal Genomic DNA Purification Mini Spin kit (D0063, Beyotime). Quantitative PCR (qPCR) was employed to measure mtDNA using TB Green Premix Ex Taq (Tli RNaseH Plus) (RR420A, Takara, Dalian, China) on a CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). mtDNA was quantified by qPCR using primers specific for cytochrome c oxidase 1 (Cox-1), the mitochondrial D-loop region, or a specific region of mtDNA that is not inserted into nuclear DNA (non-NUMT). Nuclear DNA encoding 18S rDNA, Tert, or B2m were used for normalization [53–55]. Primer sequences are listed in Table 1.
Table 1.
Sequences of primers for quantitative PCR
| Gene name | Primer sequence |
|---|---|
| D-loop | Forward: 5′-AATCTACCATCCTCCGTGAAACC-3′ |
| Reverse: 5′-TCAGTTTAGCTACCCCCAAGTTTAA-3′ | |
| Tert | Forward: 5′-CTAGCTCATGTGTCAAGACCCTCTT-3′ |
| Reverse: 5′-GCCAGCACGTTTCTCTCGTT-3′ | |
| Cox I | Forward: 5′- GCCCCAGATATAGCATTCCC -3′ |
| Reverse: 5′- GTTCATCCTGTTCCTGCTCC -3′ | |
| 18s rDNA | Forward: 5′- TAGAGGGACAAGTGGCGTTC -3′ |
| Reverse: 5′- GTTCATCCTGTTCCTGCTCC -3′ | |
| non-NUMT | Forward: 5′- CTAGAAACCCCGAAACCAAA -3′ |
| Reverse: 5′- CCAGCTATCACCAAGCTCGT -3′ | |
| B2m | Forward: 5′- ATGGGAAGCCGAACATACTG -3′ |
| Reverse: 5′- CAGTCTCAGTGGGGGTGAAT -3′ |
MSU-induced peritonitis
A mouse model of MSU-induced peritonitis was performed essentially as described previously [29, 30, 35]. Briefly, eight-week-old mice were administered intragastrically (i.g.) with theaflavin (250 mg/kg body weight, which was referred to a previous publication [56] and our preliminary experiments, or vehicle (2% Tween-80 in PBS) once a day for three consecutive days prior to intraperitoneal injection with 0.2 mL of sterile PBS alone or with 1 mg MSU (in 0.2 mL sterile PBS). One hour later, mice were treated with vehicle or theaflavin (i.g.) once again. After four hours, mice were sacrificed and peritoneal lavage was collected with 5 mL of cold PBS. The peritoneal lavage fluid was harvested and centrifuged at 300 × g for 10 min at 4 °C to remove cells, and the supernatant was collected for the measurement of cytokines. IL-1β and IL-6 in the peritoneal lavage fluids were determined by using cytometric bead array (CBA) mouse IL-1β FlexSet (#558266) and mouse inflammation kit (#552364) (BD Biosciences, San Jose, CA, USA), respectively, according to the manufacturer’s instructions. Neutrophils in the peritoneal exudate were stained with anti-mouse CD11b FITC and anti-mouse Ly-6G PE and analyzed by flow cytometry.
Bacterial infection
A murine model of bacterial sepsis was established according to the protocol described previously [57, 58]. Briefly, fifty C57BL/6 J mice were acclimated for 1 week, randomly divided into five groups (10 mice/group), and intragastrically administered with a theaflavin solution (250 mg/kg or 500 mg/kg body weight) or vehicle (2% Tween-80 in PBS) for three consecutive days, which was referred to previous studies [56, 59, 60] and our preliminary experiments. Three hours after the gavage on the third day, viable E. coli (DH5α) cells (2.0 × 109 colony-forming units (CFU)/mouse, in 0.5 mL of PBS), were injected into the peritoneal cavity of each mouse. One hour after bacterial infection, mice were intragastrically administered once again with theaflavin solution or vehicle, respectively. Mouse survival was monitored every 6 h for 5 consecutive days.
In a separate experiment, mice (5 mice/group) were treated similarly as above, except that they were injected (i.p.) with viable E. coli (1.0 × 109 CFU/mouse). The mice were sacrificed 8 h after bacterial infection and their sera were collected for measurement of serum IL-1β and other inflammatory cytokines by CBA. Serum blood urea nitrogen (BUN) and creatinine were also analyzed by spectrophotometry using respective assay kits (E2020, and E2038, Applygen Technologies, Beijing, China) according to the manufacturer’s instructions. The liver and intestine were isolated and fixed in 4% neutral formaldehyde or extracted with 2× SDS-PAGE loading buffer for Western blot analysis, respectively. Paraffin sections of the tissues were stained with hematoxylin and eosin (H&E). Images were captured under the Zeiss Axio Observer D1 microscope (ZEISS).
Statistical analysis
Each experiment was performed three times independently. Data were expressed as mean ± standard deviation (SD) and analyzed for statistical significance using GraphPad Prism 6.0 (GraphPad Software Inc, San Diego, CA, USA). One-way analysis of variance (ANOVA) followed by Bonferroni post hoc test and unpaired Student’s t test was used to analyze the statistical significance among multiple groups and between two groups, respectively. Significance was defined as follows: *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Theaflavin inhibits ATP- or nigericin-induced NLRP3 inflammasome activation in macrophages
Although theaflavin (Fig. 1a) has been reported to exhibit anti-inflammatory activities [44, 47, 48], whether it has any effects on the activation of NLRP3 inflammasome is not known. To address this issue, we pretreated LPS-primed mouse macrophage cell line J774A.1 cells and primary BMDMs with or without theaflavin and then stimulated them with ATP or nigericin as a secondary signal to activate NLRP3 inflammasome. Western blot analysis was used to analyze protein levels in culture supernatants and cell lysates. LPS priming induced the expression of NLRP3 and pro-IL-1β proteins in J774A.1 cells and BMDMs (Fig. 1b-d), whereas pro-caspase-1 and ASC were constitutively expressed in these cells irrespective of LPS priming, which is in line with published studies [28]. Upon ATP or nigericin stimulation, both caspase-1p10 and mature IL-1β (hallmarks of inflammasome activation) were detected in the culture supernatants (Fig. 1b–d), which were fully inhibited by the NLRP3-specific inhibitor MCC950 [22], thus verifying the activation of the NLRP3 inflammasome. Notably, theaflavin treatment strikingly inhibited the release of caspase-1p10 and mature IL-1β into the culture supernatants of either J774A.1 cells stimulated with ATP (Fig. 1b) or BMDMs stimulated with ATP or nigericin (Fig. 1c, d). Furthermore, we detected the effects of theaflavin on inflammasome activation in Nlrp3-/- BMDMs treated with ATP or nigericin. Western blotting showed that ATP or nigericin alone or in combination with theaflavin did not induce caspase-1p10 generation (Supplementary Fig. S1a), indicating that there was no inflammasome activation. Besides, theaflavin had minimal effects on the generation of caspase-1p10 during NLRC4 (stimulated by flagellin) and AIM2 [by poly(dA:dT)] inflammasome activation (Supplementary Fig. S2a). Together, these data demonstrate that theaflavin preferably inhibits NLRP3 inflammasome activation in macrophages.
Fig. 1. Theaflavin inhibits ATP- or nigericin-induced NLRP3 inflammasome activation in macrophages.

J774A.1 cells or bone-marrow-derived macrophages (BMDMs) were first primed with LPS (500 ng/mL) for 4 h and then pretreated with indicated doses of theaflavin for 1 h, followed by incubation with ATP (4 mM) for 1 h (J774A.1) (b), ATP (4 mM) for 30 min (BMDMs) (c) or nigericin (5 µM) for 1 h (BMDMs) (d) in the absence of LPS. a The chemical structure of theaflavin. Western blot analysis of caspase-1 and IL-1β in the culture supernatants and indicated proteins in cell lysates from J774A.1 cells treated with ATP (b) or BMDMs treated with ATP (c) or nigericin (d). β-Actin was used as a loading control for cell lysates. TF theaflavin, MCC MCC950, CASP1 caspase-1.
Theaflavin suppresses ASC speck formation in macrophages stimulated with ATP or nigericin
To further explore the effects of theaflavin on NLRP3 inflammasome, we next examined the influence of theaflavin on ASC speck formation upon ATP or nigericin stimulation, which is a hallmark of NLRP3 inflammasome activation [22, 61]. Western blot analysis showed that ASC levels were largely unaffected in macrophages by either ATP or nigericin stimulation in the absence or presence of theaflavin treatment (Fig. 1b–d). Immunofluorescence microscopy revealed that, while ASC distributed diffusely in untreated cells, ATP or nigericin treatment induced ASC aggregates that indicate ASC speck formation; as compared to ATP or nigericin alone, pretreatment of 50, 100, and 200 μM of theaflavin dramatically decreased the formation of ASC aggregates (Fig. 2a–d), indicating that theaflavin dose-dependently inhibits ATP- or nigericin-induced ASC speck assembly. Apart from ASC speck formation, NLRP3 inflammasome assembly can be revealed by ASC oligomerization after disuccinimidyl suberate cross-linking [22, 62]. Western blotting showed that oligomerized ASC was observed in cells stimulated with ATP or nigericin and that theaflavin dose-dependently inhibited the formation of oligomerized ASC (Fig. 2e, f), consistent with the inhibition of ASC speck assembly (Fig. 2a–d) and NLRP3 activation (Fig. 1b–d). These results indicate that theaflavin disrupts NLRP3 inflammasome assembly by blocking the formation of ASC specks, suggesting its action on ASC speck assembly or upstream steps of ASC speck formation.
Fig. 2. Theaflavin blocks ASC speck formation and oligomerization upon NLRP3 activation.

BMDMs were primed with LPS (0.5 µg/mL) for 4 h and then pretreated with indicated doses of theaflavin for 1 h, followed by stimulation with ATP (4 mM) for 30 min (a, c, e) or nigericin (5 μM) for 1 h in the absence of LPS (b, d, f). Immunofluorescence microscopy analysis of ASC distribution (red) in BMDMs treated with ATP (a) or nigericin (b). Nuclei (blue) were revealed by Hoechst 33342 staining. A set of representative images are shown with ASC specks being indicated by yellow arrows. Scale bars, 20 μm. Percentages of cells with ASC specks were quantified in BMDMs treated with ATP (c) or nigericin (d). Western blot analysis of ASC oligomers in Triton X-100 insoluble, disuccinimidyl suberate-cross-linked pellets from BMDMs stimulated with ATP (e) or nigericin (f). Data are shown as mean ± SD (n = 5). ***P < 0.001; ns not significant, TF theaflavin.
Theaflavin suppresses pyroptosis by inhibiting GSDMD cleavage
Following NLRP3 inflammasome activation, GSDMD is cleaved by active caspase-1 to liberate its N-terminal fragment, GSDMD-NT, which then oligomerizes and forms pores in the plasma membrane to execute pyroptosis [4, 6, 63]. We thus examined the effect of theaflavin on such lytic cell death by using propidium iodide (PI) incorporation. Fluorescence microscopy showed that theaflavin pretreatment dose-dependently decreased PI incorporation into the cells treated with ATP or nigericin, indicating that ATP or nigericin-induced lytic cell death was inhibited by theaflavin (Fig. 3a, b, d, e, g, h). As expected, Western blot analysis showed that GSDMD was cleaved to generate GSDMD-NT in cells treated with ATP or nigericin, which was blocked by MCC950, thus confirming that this process was NLRP3-dependent. Consistent with PI staining results, theaflavin significantly reduced the GSDMD-NT generation in ATP- or nigericin-treated cells (Fig. 3c, f, i). In contrast, ATP or nigericin alone or in combination with theaflavin did not induce GSDMD-NT generation and PI incorporation in Nlrp3−/− BMDMs (Supplementary Fig. S1a, b), indicating that there was no pyroptosis. Besides, theaflavin had barely affected flagellin- and poly(dA:dT)-induced generation of GSDMD-NT and PI incorporation (Supplementary Fig. S2a, b), suggesting that it did not inhibit NLRC4- and AIM2-mediated pyroptosis. Together with the aforementioned data, these results indicate that theaflavin inhibits GSDMD-NT generation by suppressing NLRP3-mediated caspase-1 activation, thereby preventing macrophages from pyroptosis.
Fig. 3. Theaflavin suppresses pyroptosis by inhibiting GSDMD-NT generation.

Cells were first primed with LPS (500 ng/mL) for 4 h and then pretreated with indicated doses of theaflavin for 1 h, followed by incubation with ATP (4 mM) for 1 h (J774A.1), ATP (4 mM) for 30 min (BMDMs) or nigericin (5 µM) for 1 h in the absence of LPS. a, d, g After theaflavin and ATP or nigericin treatment, cells were stained with 2 μg/mL propidium iodide (PI) (red; staining dying cells) and 5 μg/mL Hoechst 33342 (blue; staining all nuclei) for 10 min, and then observed by fluorescence microscopy. Bright-field images are also shown in merged ones. Scale bars, 40 μm. b, e, h PI-positive cells in 5 randomly chosen fields were quantified. The percentage of lytic cell death is defined as the ratio of PI-positive cells relative to all (revealed by Hoechst 33342). Western blot analysis was used to determine the expression levels of GSDMD-NT in J774A.1 (c) or BMDM (f, i) cell lysates. Data are shown as mean ± SD (n = 5). *P < 0.05; ***P < 0.001; GSDMD full-length GSDMD, GSDMD-NT N-terminal fragment of GSDMD, NI nigericin, TF theaflavin, MCC MCC950.
Inhibition of NLRP3 assembly by theaflavin is associated with amelioration of mitochondrial damage and mitochondrial ROS production
We next sought to explore how theaflavin inhibited NLRP3 inflammasome activation and pyroptosis. Previous studies have shown that mitochondrial dysfunction and the production of mitochondrial ROS are important upstream events for triggering NLRP3 activation [55, 64, 65]. Theaflavin has been shown to display potent antioxidant activities [66–68]. Based on these previous studies, we speculated that theaflavin might have affected the function of mitochondria during NLRP3 inflammasome activation. To this end, we detected mitochondrial membrane potential (MMP) using the potentiometric dye JC-1. JC-1 forms aggregates (emitting red fluorescence) in cells containing mitochondria with normal MMP, but becomes monomers (emitting green fluorescence) in those cells containing damaged mitochondria with decreased MMP. Fluorescence microscopy showed that ATP or nigericin decreased the percentages of cells with JC-1 aggregates (red fluorescence) but an increase of cells with JC-1 monomer (green fluorescence), while theaflavin pretreatment prevented these changes (Fig. 4a–d), indicating that theaflavin could reverse the ATP- or nigericin-induced reduction of MMP. We further stained the cells with another dye TMRE that can stain mitochondria with normal MMP (emitting red fluorescence) but not with decreased MMP. Fluorescence microscopy showed that theaflavin significantly reversed the reduction of TMRE fluorescence in cells treated with ATP or nigericin, indicative of protective effects of theaflavin on MMP (Supplementary Fig. S3a-d). These results suggest that theaflavin protects mitochondria in macrophages from ATP- or nigericin-induced damage.
Fig. 4. Theaflavin alleviates mitochondrial damage induced by ATP or nigericin.

BMDMs were first primed with LPS (500 ng/mL) for 4 h, and then pretreated with indicated doses of theaflavin for 1 h, followed by incubation with ATP (4 mM) for 30 min or nigericin (5 μM) for 1 h. JC-1 was used to assay the mitochondrial membrane potential in the cells treated with ATP (a, b) or nigericin (c, d). One set of representative fluorescence microscopy images show JC-1 aggregates (red) and monomers (blue) (a, c). Scale bars, 20 μm. Quantitative analyses of JC-1 aggregates-containing cells treated with ATP (b) or nigericin (d), respectively. e Fluorescence microscopy was used to reveal mitochondria in BMDMs stained with Mito-Tracker Green (staining mitochondria; green) and Hoechst 33342 (staining nuclei; blue). Scale bars, 2 µm. f Cytosolic release of mitochondrial DNA (mtDNA) was determined by qPCR with primers specific for mtDNA (cytochrome c oxidase 1, Cox-1) and nucleic DNA (nDNA) (18 s rDNA) in LPS-primed BMDMs treated ATP or nigericin. Western blotting of cytochrome c in mitochondria and cytosol enriched fractions from BMDMs treated with ATP (g) or nigericin (h). α-Tubulin and Tom20 were used as a loading control for cytosolic and mitochondrial fractions, respectively. Data are shown as mean ± SD (n = 5). ***P < 0.001; NI, nigericin; Cyto c, cytochrome c; TF, theaflavin.
We next verified the protective effect of theaflavin on ATP- or nigericin-induced mitochondrial damage by observing the mitochondrial morphology. Normal functional mitochondria appear as long tubular network, whereas damaged mitochondria are fragmented [69]. Mitochondrial morphology was observed by using Mito-Tracker green (staining mitochondria independent of MMP) in this study. Fluorescence microscopy showed that the mitochondria in LPS- or theaflavin-treated cells displayed filamentous morphology. Upon ATP or nigericin treatment, the mitochondria became fragmented. However, theaflavin pretreatment effectively inhibited ATP- or nigericin-induced mitochondrial fragmentation (Fig. 4e). Furthermore, qPCR analysis showed that ATP or nigericin treatment increased the levels of mtDNA in the cytosol, whereas theaflavin pretreatment significantly decreased the levels of mtDNA in the cytosol of BMDMs treated with ATP or nigericin (Fig. 4h, Supplementary Fig. S3e, f), indicating that theaflavin reduced the ATP- or nigericin-induced mtDNA release. In addition, we also detected cytosolic cytochrome c, which is sequestered in mitochondrial cristae in normal cells but is released into the cytosol when the mitochondria were damaged [70–72]. Western blotting showed that ATP or nigericin treatment increased the levels of cytochrome c in the cytosol, whereas theaflavin pretreatment decreased the levels of cytochrome c in the cytosol (Fig. 4e, f). Together, these results indicate that theaflavin can prevent mitochondrial dysfunction induced by NLRP3 inflammasome activators, suggesting that the inhibitory effects of theaflavin on NLRP3 inflammasome activation and pyroptosis are associated with the protection of mitochondrial function.
Considering the critical role of mitochondrial ROS in NLRP3 inflammasome activation [73] and GSDMD-NT oligomerization to form pores [74], we examined whether theaflavin inhibited ROS production upon ATP or nigericin stimulation. Intracellular ROS were examined by DCFH-DA staining. Fluorescence microscopy showed that ATP or nigericin treatment induced a marked increase in the DCFH green fluorescence indicating increased levels of total intracellular ROS, while theaflavin significantly decreased DCFH green fluorescence indicative of reduced levels of ROS in ATP- or nigericin-treated cells (Fig. 5a–d). In addition, we used MitoSOX staining to detect the levels of mitochondrial superoxide which is reflective of mitochondrial ROS production. Fluorescence microscopy showed that ATP or nigericin treatment increased MitoSOX red fluorescence whereas theaflavin pretreatment markedly decreased the red fluorescence, indicating that the mitochondrial ROS levels were increased upon ATP or nigericin treatment while theaflavin markedly reduced the mitochondrial ROS levels within the cells (Fig. 5e–h). These results indicate that theaflavin can effectively reduce ROS production from mitochondria of macrophages during NLRP3 inflammasome activation.
Fig. 5. Theaflavin inhibits ROS production induced by ATP or nigericin.

BMDMs were first primed with LPS (500 ng/mL) for 4 h, and then pretreated with indicated doses of theaflavin for 1 h, followed by incubation with ATP (4 mM) for 30 min or nigericin (5 μM) for 1 h. Reactive oxygen species (ROS) in cells were detected by using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) and observed by fluorescence microscopy. (b) and (d) show quantitative analyses of (a) and (c), respectively. Mitochondrial superoxide was assayed by staining with MitoSOX and observed by fluorescence microscopy. (g) and (h) are quantitative analyses of (e) and (f), respectively. Data are shown as mean ± SD (n = 5). Scale bars, 20 μm. *P < 0.05; ***P < 0.001; NI nigericin, TF theaflavin.
As NLRP3 inflammasome assembly requires the binding of NEK7 to its leucine-rich repeat domain downstream of induction of mtROS [65, 75] while theaflavin suppressed ROS production as mentioned above, we next examined whether theaflavin could interfere with the interaction between NEK7 and NLRP3. Co-IP was performed to precipitate NLRP3, and NEK7 in the precipitate was detected by Western blotting. When LPS plus ATP or nigericin but no theaflavin was added, there was a band at 32 kDa as revealed by an anti-NEK7 antibody; with the addition of 200 μM of theaflavin, the level of NEK7 band was markedly decreased (Fig. 6a, b), indicating that the interaction between NLRP3 and NEK7 was inhibited by theaflavin.
Fig. 6. Theaflavin inhibits NEK7-NLRP3 interaction during ATP- or nigericin-induced NLRP3 inflammasome activation.
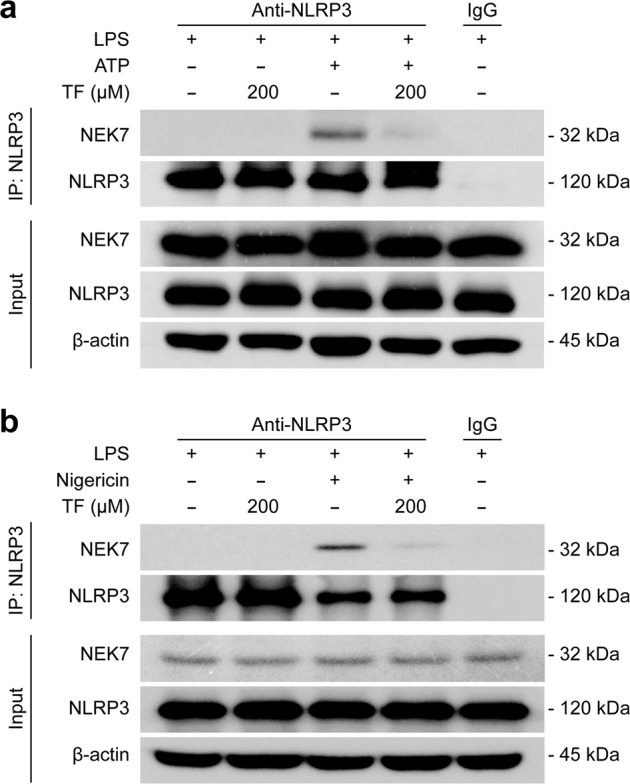
BMDMs were first primed with LPS (500 ng/mL) for 4 h, and then pretreated with indicated doses of theaflavin for 1 h, followed by incubation with ATP (4 mM) for 30 min (a) or nigericin (5 μM) for 1 h (b). A Co-IP assay was performed to assess the interaction between NEK7 and NLRP3 after the indicated treatments. Indicated proteins were revealed by Western blotting. TF theaflavin.
Taken together, these results indicate that theaflavin prevents mitochondrial dysfunction, and inhibits the mitochondrial ROS production and the interaction between NLRP3 and NEK7, thus inhibiting the NLRP3 inflammasome assembly.
MSU-induced NLRP3 inflammasome assembly in macrophages is blocked by theaflavin treatment
The deposition of monosodium urate crystals (MSU) in joints and periarticular tissues has long been recognized as the cause of acute gouty arthritis [76, 77], and it has been shown that MSU is a potent inducer of NLRP3 inflammasome [78]. We thus next explored whether theaflavin could affect MSU-induced NLRP3 inflammasome activation. Western blotting showed that MSU significantly increased the levels of caspase-1p10 (10 kDa) and mature IL-1β (17 kDa) in the culture supernatants of BMDMs, whereas theaflavin was able to inhibit the release of these inflammasome activation markers into the culture supernatants (Fig. 7a), indicating that NLRP3 inflammasome activation was suppressed by theaflavin. Immunofluorescence microscopy showed that the MSU-induced formation of ASC specks was dose-dependently inhibited by theaflavin (Fig. 7c, d). Western blot analysis of cross-linked samples showed that theaflavin also inhibited the formation of ASC oligomers induced by MSU (Fig. 7b), indicating suppression of ASC recruitment and assembly. Besides, Co-IP results showed that theaflavin also blocked the interaction between NLRP3 and NEK7 in BMDMs treated with MSU (Supplementary Fig. S4). Together, these data indicate that theaflavin inhibits MSU-induced activation of NLRP3 inflammasome.
Fig. 7. Theaflavin inhibits NLRP3 inflammasome assembly and necrosis in macrophages treated with MSU.

BMDMs were first primed with LPS (500 ng/mL) for 4 h, and then pretreated with indicated doses of theaflavin for 1 h, followed by incubation with MSU (200 μg/mL) for 6 h in the presence or absence of theaflavin. a The expression levels of indicated proteins in cell lysates and culture supernatants were analyzed by Western blotting. β-Actin was adopted as a loading control for cell lysates. b Western blotting analysis of ASC oligomers in Triton X-100 insoluble pellets cross-linked with disuccinimidyl suberate. c Immunofluorescence microscopy analysis of ASC distribution (red) in BMDMs treated with MSU. Nuclei (blue) were revealed by Hoechst 33342 staining. A set of representative images are shown with ASC specks being indicated by yellow arrows. Scale bars, 20 μm. d Percentages of cells with ASC specks were quantified in BMDMs treated with MSU. e Representative images showing PI (red, staining dying cells) combined with Hoechst 33342 (blue, staining all nuclei) fluorescence. Scale bars, 40 μm. f Quantification of PI-positive cells in 5 randomly chosen fields and ratios of PI-positive over all cells (revealed by Hoechst 33342) are defined as the percentages of lytic cell death. Data are shown as mean ± SD (n = 5). **P < 0.01; ***P < 0.001; MSU monosodium urate crystals, TF theaflavin.
We also tested the effect of theaflavin on MSU-induced lytic cell death, which has been revealed in our previous study to be a complex form of necrosis that is not solely reliant on NLRP3 inflammasome-mediated pyroptosis [35]. PI uptake assay revealed that theaflavin dose-dependently suppressed MSU-induced necrosis (Fig. 7e, f). Together, these results indicate that theaflavin not only inhibits NLRP3 inflammasome activation but also suppresses necrosis in macrophages treated with MSU.
Theaflavin attenuates MSU-induced inflammation in a mouse model of acute gouty peritonitis
As theaflavin inhibits gout-associated MSU-induced NLRP3 inflammasome activation and necrosis, we next examined whether it could inhibit MSU-induced inflammatory responses in vivo. Intraperitoneal injection of MSU is a commonly-used mouse model of acute gouty arthritis and can cause NLRP3-dependent inflammation characterized by IL-1β production and massive neutrophil influx into the peritoneal cavity [29, 30, 78]. Mice were orally administered with theaflavin or vehicle once a day for three consecutive days prior to intraperitoneal injection of MSU. The results showed that MSU administration for 6 h significantly increased the peritoneal levels of IL-1β and IL-6 (as measured by CBA assay) and the frequency of CD11b+Ly6G+ neutrophils (as analyzed by flow cytometry) in the peritoneal cavity; theaflavin pretreatment significantly reduced the amounts of peritoneal IL-1β and IL-6 and diminished the frequency of CD11b+Ly6G+ neutrophils compared to the vehicle control (Fig. 8a–e). These data indicate that theaflavin attenuates MSU-induced NLRP3 inflammasome activation and mitigates MSU-induced acute gouty peritonitis in vivo.
Fig. 8. MSU-induced peritonitis is alleviated by theaflavin treatment.

C57BL/6 J mice were administered intragastrically (i.g.) with theaflavin (250 mg/kg body weight) or vehicle (2% Tween 80 in PBS) once a day for three consecutive days before intraperitoneal injection with sterile PBS (0.2 mL) or with 1 mg MSU (in 0.2 mL sterile PBS). One hour later, mice were administered with vehicle or theaflavin (i.g.) once again. Neutrophil and soluble cytokine levels in the peritoneal lavage fluids were quantified 6 h after MSU administration (n = 6 mice per group). a Representative flow cytometric dot-plots of peritoneal exudate cells. b, c Quantification of neutrophils in the peritoneal lavage fluids. d, e IL-1β and IL-6 levels in peritoneal lavage fluid supernatants were measured by cytometric bead array (CBA) assay. Data are shown as mean ± SD (n = 6). ***P < 0.001; TF theaflavin.
Theaflavin alleviates sepsis and septic shock organ injuries in mice
Apart from its involvement in MSU-induced peritonitis, the NLRP3 inflammasome has also been associated with many other inflammatory diseases including sepsis [3, 14, 79–81]. Considering that theaflavin exhibits inhibitory effects on NLRP3 inflammasome activation and pyroptosis in vitro, we explored whether theaflavin could mitigate sepsis in a mouse model of bacterial sepsis. Mice were administered with theaflavin or vehicle by gavage for three consecutive days. Viable E. coli cells (2.0×109 CFU/mouse) were injected into the peritoneal cavity of mice to induce sepsis. The results showed that mice treated with low-dose (250 mg/kg) and high-dose (500 mg/kg) theaflavin protected 20% and 40% of bacterial-infected mice from septic death during the period of observation (120 h), respectively, whereas all infected mice treated with vehicle died within 30 h (Fig. 9a). The protective effect of theaflavin on the septic death of mice is of significance.
Fig. 9. Theaflavin alleviates sepsis in mice.

a C57BL/6 J mice were administered intragastrically (i.g.) with theaflavin (250 or 500 mg/kg body weight) or vehicle (2% Tween 80 in PBS) once a day for three consecutive days prior to intraperitoneal (i.p.) bacterial infection with viable Escherichia coli (2.0 × 109 CFU/mouse). Mice were administered (i.g.) with theaflavin or vehicle once again at 1 h after the bacterial injection. Mice survival was monitored every 6 h for 5 consecutive days. Kaplan–Meier survival curves were used to analyze the data (10 mice per group). The significance was evaluated by the log-rank (Mantel-Cox) test. b–d Mice were treated as in (a) except that mice were injected (i.p.) with viable E. coli (1.0 × 109 CFU/mouse). The serum levels of IL-1β (b), IL-6 (c), and TNF-α (d) at 8 h post bacterial infection were measured by CBA assay (4 or 5 mice per group). Data are shown as mean ± SD (n = 5). ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; TF theaflavin.
During bacterial sepsis, inflammasome activation and subsequent pyroptosis not only trigger the release of multiple inflammatory cytokines but also cause organ injury [82, 83]. To test the effect of theaflavin, we injected a sublethal dose (1 × 109 CFU/mouse) of viable E. coli into the peritoneal cavity of mice with or without theaflavin treatment in a separate experiment. Eight hours after infection, serum samples, and liver and kidney tissues were collected. The results showed that bacterial infection induced a marked increase in the serum levels of IL-1β, IL-6, and TNF-α, whereas theaflavin treatment at the tested doses significantly reduced the serum levels of these cytokines with the low-dose having induced a trend decrease of IL-1β and IL-6 levels (Fig. 9b–d).
In addition to assaying inflammatory cytokines in serum, we also examined inflammatory cell infiltration in the liver. Histochemical analysis showed that bacterial infection caused marked infiltration of inflammatory immune cells in the liver, which was attenuated by theaflavin administration (Fig. 10a). As kidney is one common organ undergoing injury during sepsis [84], we also explored the effect of theaflavin on the kidney injury during bacterial sepsis. Histopathological analysis revealed that bacterial infection caused glomerulus vacuolar degeneration and even shedding of renal tubular epithelial cells in the kidney, while theaflavin administration ameliorated the damage (Fig. 10b). Consistent with this, bacterial infection increased the serum levels of BUN and creatinine, whereas theaflavin treatment decreased the levels of these renal function markers (Fig. 10c, d), indicating amelioration of bacterial-induced acute kidney injury. Lastly, we examined NLRP3 inflammasome activation markers in both the liver and kidney. Western blotting showed that NLRP3 expression was increased in the liver (Fig. 10e) and kidney (Fig. 10f) of bacterial-infected mice but theaflavin only minimally affected its expression. Neither theaflavin treatment nor bacterial infection affected ASC expression. However, the cleavage of pro-caspase-1 (producing a p10 fragment) and GSDMD (generating a GSDMD-NT fragment), two hallmarks for inflammasome activation, were increased in the bacterial-infected group but were significantly reduced by theaflavin treatment (Fig. 10e, f). Together, these data suggest that theaflavin attenuates bacterial-induced sepsis and organ injury by reducing NLRP3 inflammasome activation and pyroptosis in vivo.
Fig. 10. Theaflavin protects against septic shock organ injury in mice.

C57BL/6J mice were administered intragastrically (i.g.) with theaflavin (250 or 500 mg/kg body weight) or vehicle (2% Tween 80 in PBS) once a day for three consecutive days prior to intraperitoneal (i.p.) bacterial infection with viable E. coli (1.0 × 109 CFU/mouse). Mice were administered (i.g.) with theaflavin or vehicle once again at 1 h after the bacterial injection. Mice were sacrificed 8 h after bacterial infection. Hematoxylin and eosin staining of the liver (a) and kidney (b) tissue sections of mice infected with live E. coli for 8 h. Scale bars, 100 μm (scale bars in enlarged insets, 20 μm). Kidney injury was also measured by serum blood urea nitrogen (BUN) (c) and creatinine (d) levels in different mice groups (4 or 5 mice per group). Western blotting of NLRP3, ASC, caspase-1 (CASP1), and GSDMD in the liver (e) or kidney lysates (f) from 3 separate mice of each group with E. coli treatment. β-Actin was used as a loading control for tissue lysates. Data are shown as mean ± SD (n = 5). ns, not significant; *P < 0.05; ***P < 0.001; TF theaflavin.
Discussion
Theaflavin is a major component presented in black tea, formed by oxidation of catechins in the leaves of Camellia sinensis during fermentation [43]. Previous studies have reported that theaflavin has anti-inflammatory activities in different experimental settings [44, 47]. In this study, we showed that theaflavin was able to inhibit NLRP3 inflammasome activation and pyroptosis in vitro and in vivo, which was associated with reduced mitochondrial dysfunction and ROS generation. Theaflavin had minimal effects on NLRC4 and AIM2 activation. Thus, our finding adds a new layer of action mechanism for theaflavin in displaying its anti-inflammatory activities by preferably regulating NLRP3 inflammasome activation and pyroptotic cell death in macrophages.
NLRP3 is unique among the NLR family of sensors as it is the only known member to be activated by numerous pathogenic and sterile inflammatory signals [85]. The activators that can be sensed by NLRP3 include pore-forming bacterial toxins like nigericin and gramicidin, extracellular ATP, various metabolic crystals (such as MSU and hemozoin), environmental nanoparticles (such as silica dioxide particles), among others [86]. It is therefore likely that NLRP3 senses these activators indirectly through some common signaling pathways they triggered. It has been proposed that NLRP3 can be activated by several signals including potassium ion (K+) efflux [87], cathepsin released from disrupted lysosomes [88], and ROS released from damaged mitochondria [89–91], etc. The critical role of mitochondria in triggering NLRP3 activation has been further strengthened by a study on mtDNA released from damaged mitochondria [55]. Notably, one recent study reveals that Ragulator-RAG-regulated mitochondrial ROS production is required for GSDMD-NT oligomerization to promote pore formation after GSDMD cleavage, thereby mediating pyroptosis [74]. Consistent with those studies, we found in this study that both NLRP3 inflammasome assembly and pyroptosis were robustly inhibited by theaflavin, which was associated with the amelioration of mitochondrial damage and a decrease in mitochondrial ROS production. Moreover, theaflavin-mediated suppression of pyroptosis was associated with reduced generation of GSDMD-NT resulting from decreased activation of caspase-1. Due to the critical role of GSDMD-NT in mediating pyroptosis by other inflammasomes [92–94], our data suggest that theaflavin may also inhibit pyroptotic cell death during non-canonical inflammasome activation aside from canonical NLRP3 inflammasome assembly.
The role of ROS in promoting NLRP3 activation is well known, but how ROS affects NLRP3 inflammasome assembly is largely unclear until recently. Recent studies showed that NEK7 is critical for the activation of NLRP3 inflammasome and that NEK7 directly interacts with the leucine-rich repeat domain of NLRP3 to promote inflammasome assembly [65, 95, 96]. Although it is known that deubiquitination of NLRP3 by BRCC3 regulates inflammasome activity [97], a recent study revealed further mechanism underlying this process by showing that NLRP3-NEK7 interaction is also essential for NLRP3 deubiquitination by BRCC3 and subsequently inflammasome assembly [98]. Of note, ROS scavenger N-acetyl-L-cysteine diminishes both the phosphorylation of NEK7 and the interaction between NEK7 and NLRP3, indicating that ROS play an important role in the binding of NEK7 to NLRP3 [65]. Consistent with these findings, our data showed that theaflavin inhibited the interaction between NEK7 and NLRP3 likely by preventing mitochondrial ROS production, thereby leading to the blockade of NLRP3 inflammasome assembly.
It is yet unknown how theaflavin prevents mitochondrial dysfunction and decreases ROS production. As the main source of cellular ROS is mitochondria, one possible explanation is that theaflavin acts as an antioxidative agent to protect mitochondria from damage and to remove ROS generated from mitochondria, considering that it is a polyphenolic compound [46, 99]. Another possibility is that theaflavin may indirectly regulate the expression of major components of cellular antioxidative system. Nuclear factor erythroid 2-related factor 2 (Nrf2), which regulates the production of antioxidant proteins and peptides, including glutathione, has been reported to regulate ROS production by mitochondria and NADPH oxidase [100]. Recent studies have shown that theaflavin ameliorates murine osteoarthritis, renal ischemia/reperfusion injury, atherosclerosis progress, and hematopoietic stem cell damage via regulating Nrf2 [66, 67, 101, 102]. Although this possibility appears low due to the short period of incubation times in our in vitro experimental setting, we cannot exclude the possible role of the Nrf2 pathway in mediating the effect of theaflavin on regulating inflammasome assembly in vivo.
The link between lytic cell death including pyroptosis and inflammation has long been recognized. NLRP3 inflammasome overactivation, accompanied by pyroptotic cell death, results in excessive release of a variety of proinflammatory cytokines and intracellular inflammatory components, which is one important mechanism underlying the pathogenesis of multiple inflammatory disorders [31]. Acute gouty arthritis is one such disorder that is caused by MSU deposition in joints and periarticular tissues [76], and MSU has been causally linked to gouty arthritis by aberrantly activating NLRP3 inflammasome [78]. Thus, blocking NLRP3 activation by colchicine has been used for the treatment of acute gouty arthritis, yet colchicine is toxic and can cause severe side effects such as diarrhea [103]. Notably, one previous study revealed that MSU-induced necrosis cannot be blocked by genetic deletion of GSDMD [104]. Consistent with this, our previous study showed that neither NLRP3-specific inhibitor MCC950 nor GSDMD inhibitor disulfiram could significantly inhibit MSU-induced necrosis in macrophages even though MCC950 could effectively block NLRP3 inflammasome assembly [35], suggesting that the necrosis is a complex form of lytic cell death that may have involved multiple necrotic signaling pathways. In the present study, we found that theaflavin not only inhibited MSU-triggered NLRP3 inflammasome assembly but also markedly suppressed MSU-induced necrosis, suggesting that it may be a potent agent to prevent MSU-induced inflammation in vivo. Indeed, we found that theaflavin markedly attenuated MSU-induced peritonitis, indicating its potential application in treating acute gouty arthritis.
Sepsis is characterized by multiple organ dysfunction resulting from the host’s injurious response to infection. Systemic inflammatory cytokines, particularly IL-1β, IL-6, and TNF-α, are significantly increased during sepsis in both patients and animal models [105, 106]. Although it was believed that the inflammatory cytokines, including IL-1β, play a major role in septic shock and septic death [105, 106], recent studies revealed that caspase-11 (in mice) or caspase-4/-5 (in human) may be critical factors in bacterial sepsis, as loss of caspase-11 but not caspase-1 protected mice against LPS-induced cell death [93, 107, 108]. It is known that caspase-11/-4/-5 can be activated by cytosolic LPS to cleave GSDMD to generate GSDMD-NT, the latter of which executes pyroptosis [3, 4, 109]. Notably, this non-canonical pyroptosis induced by caspase-11/-4/-5 can further cause non-canonical NLRP3 inflammasome activation through GSDMD-NT-dependent K+ efflux, thereby leading to caspase-1 activation and the conversion of pro-IL-1β to mature IL-1β [3, 109, 110]. Consistent with these findings, in the current study, we showed that theaflavin significantly prolonged the survival of mice with bacterial sepsis. Importantly, theaflavin administration markedly mitigated liver inflammation and acute kidney injury accompanied by decreased caspase-1 activation and GSDMD-NT generation in both organs, concomitant with reduced serum levels of inflammatory cytokines including IL-1β, suggesting that theaflavin-mediated inhibition of NLRP3 inflammasome and pyroptotic cell death confers protection against septic organ injury.
Tea pigments including theaflavin are relatively low toxic polyphenolic compounds. The doses of theaflavin used in this study were based on our preliminary experiments and previous studies showing that 250 mg/kg body weight of theaflavin for 2 weeks [56] or 500 mg/kg body weight of tea pigments orally administered did not have overt toxicity [59, 60]. Specifically, oral theaflavin gavage of 250 mg ·kg−1 ·d−1 for 2 weeks did not significantly affect the body weight or tissue weight of liver, heart, kidney, adrenal gland or spleen but instead inhibited the progress of disuse muscle atrophy in the experimental mice [56]. Consistent with these reports, in our study, orally administration of 500 mg ·kg−1 ·d−1 of theaflavin alone for 3 consecutive days did not cause overt changes in the liver and kidney of mice, confirming low toxicity of theaflavin. Yet future studies are needed to evaluate the toxicity of theaflavin and its interaction with other drugs in animals and humans.
The question is whether the doses used in mice could be achieved in humans. According to the guidelines for dose conversion from animals to humans [111], conversion of the effective dose of theaflavin (250 mg/kg) in acute gouty peritonitis into human equivalent dose would be ~20 mg/kg body weight. This means that for a person with the average human body weight (60 kg), the dose should be ~1200 mg/day. As the total theaflavins in black tea is around 2–20 g/kg [49], it appears that such doses of theaflavin are reasonably unachievable through daily black tea consumption, but this may be achieved by supplementation of purified theaflavin or tea pigments. However, drinking black tea may still be beneficial to human beings if the inflammatory stimulation is relatively weak (but still harmful for humans) or for chronic inflammatory diseases. Supporting this, a randomized pilot clinical study showed that oral administration of theaflavin at 100 mg/day for ten weeks had a beneficial effect on body fat and muscle in healthy individuals [112]. Yet further investigations are needed for translating animal studies of theaflavin into clinical applications.
One limitation of this study is a lack of positive drugs in the animal experiments. Previous identified NLRP3-specific inhibitors, such as MCC950 [22] and dapansutrile (OLT1177) [113], have been investigated in clinical trials. Whereas a clinical study of MCC950 had been discontinued due to its liver toxicity [40], OLT1177 has been proved to be safe at 1000 mg daily doses for 8 days in humans [113] and is currently undergoing a phase 2 clinical trial for acute gout flare [114]. Thus, OLT1177 may be preferably used as a positive control in comparison with theaflavin in vivo, which awaits further research.
In summary, our study demonstrates that theaflavin is an inhibitor of NLRP3 inflammasome activation triggered by multiple stimuli, including ATP, nigericin, or MSU crystals. We also provide evidence showing that inhibition of NLRP3 inflammasome assembly by theaflavin is associated with its effects on protecting mitochondrial function thereby reducing the interaction between NEK7 and NLRP3. Interestingly, theaflavin inhibited both MSU-induced peritonitis and bacterial sepsis, highlighting its potential application in the treatment of acute gouty arthritis and septic shock, which warrants future investigations.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82274167, 81873064, and 81773965) and by Funding of Science and Technology Projects in Guangzhou (202201020083). We also thank Prof. Yong-tang Zheng (Kunming Institute of Zoology, Chinese Academy of Sciences) for his kind help in this study.
Author contributions
XHH, DYOY, and QBZ conceived and supervised the research; SYC, YPL, YPY, and HRZ performed in vitro experiments; ZJS, QQL, TY, RX, and LHX performed animal experiments; SYC and XHH analyzed the data; XHH, DYOY, and SYC wrote the paper.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Si-yuan Chen, Ya-ping Li, Yi-ping You
Contributor Information
Qing-bing Zha, Email: zhaqingbb@sina.com.
Dong-yun Ou-Yang, Email: dongyun1967@aliyun.com.
Xian-hui He, Email: thexh@jnu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-023-01105-7.
References
- 1.Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. 2016;165:792–800. doi: 10.1016/j.cell.2016.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wen H, Miao EA, Ting JP. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity. 2013;39:432–41. doi: 10.1016/j.immuni.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 4.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 5.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–6. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 6.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766–78. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P. The gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol. 2018;48:584–92. doi: 10.1002/eji.201747404. [DOI] [PubMed] [Google Scholar]
- 9.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25:1285–98. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42:245–54. doi: 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27:673–84. doi: 10.1016/j.tcb.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity. 2020;52:475–86. doi: 10.1016/j.immuni.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weindel CG, Martinez EL, Zhao X, Mabry CJ, Bell SL, Vail KJ, et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell. 2022;185:3214–31. doi: 10.1016/j.cell.2022.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–61. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- 15.Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin D in pyroptosis. Acta Pharm Sin B. 2021;11:2768–82.. doi: 10.1016/j.apsb.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisenstein A, Hilliard BK, Pope SD, Zhang C, Taskar P, Waizman DA, et al. Activation of the transcription factor NRF2 mediates the anti-inflammatory properties of a subset of over-the-counter and prescription NSAIDs. Immunity. 2022;55:1082–95. doi: 10.1016/j.immuni.2022.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen X, Liu G, Yuan Y, Wu G, Wang S, Yuan L. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-kappaB signaling. Cell Death Dis. 2019;10:906. doi: 10.1038/s41419-019-2157-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orecchioni M, Kobiyama K, Winkels H, Ghosheh Y, McArdle S, Mikulski Z, et al. Olfactory receptor 2 in vascular macrophages drives atherosclerosis by NLRP3-dependent IL-1 production. Science. 2022;375:214–21. doi: 10.1126/science.abg3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim SR, Lee SG, Kim SH, Kim JH, Choi E, Cho W, et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun. 2020;11:2127. doi: 10.1038/s41467-020-15983-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–8. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ju M, Bi J, Wei Q, Jiang L, Guan Q, Zhang M, et al. Pan-cancer analysis of NLRP3 inflammasome with potential implications in prognosis and immunotherapy in human cancer. Brief Bioinform. 2020;22:bbaa345. doi: 10.1093/bib/bbaa345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21:736–45.. doi: 10.1038/s41590-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang P, Liu Y, Hu L, Huang K, Hong M, Wang Y, et al. NLRC4 inflammasome dependent cell death occurs by a complementary series of three death pathways and determines lethality in mice. Sci Adv. 2021;7:eabi9471. doi: 10.1126/sciadv.abi9471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steinhagen F, Schmidt SV, Schewe JC, Peukert K, Klinman DM, Bode C. Immunotherapy in sepsis—brake or accelerate? Pharmacol Ther. 2020;208:107476. doi: 10.1016/j.pharmthera.2020.107476. [DOI] [PubMed] [Google Scholar]
- 26.Liu VX, Fielding-Singh V, Greene JD, Baker JM, Iwashyna TJ, Bhattacharya J, et al. The timing of early antibiotics and hospital mortality in sepsis. Am J Respir Crit Care Med. 2017;196:856–63. doi: 10.1164/rccm.201609-1848OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263–9. doi: 10.1038/nm.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang F, Ye XJ, Chen MY, Li HC, Wang YF, Zhong MY, et al. Inhibition of NLRP3 inflammasome activation and pyroptosis in macrophages by taraxasterol is associated with its regulation on mTOR signaling. Front Immunol. 2021;12:632606. doi: 10.3389/fimmu.2021.632606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. 2015;160:62–73. doi: 10.1016/j.cell.2014.11.047. [DOI] [PubMed] [Google Scholar]
- 30.Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science. 2020;369:eaas8995. doi: 10.1126/science.aas8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coll RC, Schroder K, Pelegrin P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci. 2022;43:653–68. doi: 10.1016/j.tips.2022.04.003. [DOI] [PubMed] [Google Scholar]
- 32.Corcoran SE, Halai R, Cooper MA. Pharmacological inhibition of the nod-like receptor family pyrin domain containing 3 inflammasome with MCC950. Pharmacol Rev. 2021;73:968–1000. doi: 10.1124/pharmrev.120.000171. [DOI] [PubMed] [Google Scholar]
- 33.Li CG, Yan L, Mai FY, Shi ZJ, Xu LH, Jing YY, et al. Baicalin inhibits nod-like receptor family, pyrin containing domain 3 inflammasome activation in murine macrophages by augmenting protein kinase A signaling. Front Immunol. 2017;8:1409. doi: 10.3389/fimmu.2017.01409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Jing YY, Zeng CY, Li CG, Xu LH, Yan L, et al. Scutellarin suppresses NLRP3 inflammasome activation in macrophages and protects mice against bacterial sepsis. Front Pharmacol. 2017;8:975. doi: 10.3389/fphar.2017.00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhong CS, Zeng B, Qiu JH, Xu LH, Zhong MY, Huang YT, et al. Gout-associated monosodium urate crystal-induced necrosis is independent of NLRP3 activity but can be suppressed by combined inhibitors for multiple signaling pathways. Acta Pharmacol Sin. 2022;43:1324–36. doi: 10.1038/s41401-021-00749-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun. 2018;9:2550. doi: 10.1038/s41467-018-04947-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang YD, Bai WJ, Li CG, Xu LH, Wei HX, Pan H, et al. Piperine suppresses pyroptosis and interleukin-1beta release upon ATP triggering and bacterial infection. Front Pharmacol. 2016;7:390. doi: 10.3389/fphar.2016.00390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW, et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem. 2010;285:9792–802. doi: 10.1074/jbc.M109.082305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El-Sharkawy LY, Brough D, Freeman S. Inhibiting the NLRP3 inflammasome. Molecules. 2020;25:5533. [DOI] [PMC free article] [PubMed]
- 40.Coss R. Could an NLRP3 inhibitor be the one drug to conquer common diseases. C EN Glob Enterp. 2020;98:26–31. [Google Scholar]
- 41.Shan Z, Nisar MF, Li M, Zhang C, Wan CC. Theaflavin chemistry and its health benefits. Oxid Med Cell Longev. 2021;2021:6256618. doi: 10.1155/2021/6256618. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Tong T, Liu YJ, Kang J, Zhang CM, Kang SG. Antioxidant activity and main chemical components of a novel fermented tea. Molecules. 2019;24:2917. [DOI] [PMC free article] [PubMed]
- 43.Chakrabarty S, Nag D, Ganguli A, Das A, Ghosh Dastidar D, Chakrabarti G. Theaflavin and epigallocatechin-3-gallate synergistically induce apoptosis through inhibition of PI3K/Akt signaling upon depolymerizing microtubules in HeLa cells. J Cell Biochem. 2019;120:5987–6003. doi: 10.1002/jcb.27886. [DOI] [PubMed] [Google Scholar]
- 44.Fu G, Wang H, Cai Y, Zhao H, Fu W. Theaflavin alleviates inflammatory response and brain injury induced by cerebral hemorrhage via inhibiting the nuclear transcription factor kappa beta-related pathway in rats. Drug Des Dev Ther. 2018;12:1609–19. doi: 10.2147/DDDT.S164324. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Betts JW, Wareham DW, Haswell SJ, Kelly SM. Antifungal synergy of theaflavin and epicatechin combinations against candida albicans. J Microbiol Biotechnol. 2013;23:1322–6. doi: 10.4014/jmb.1303.03010. [DOI] [PubMed] [Google Scholar]
- 46.Peluso I, Serafini M. Antioxidants from black and green tea: from dietary modulation of oxidative stress to pharmacological mechanisms. Br J Pharmacol. 2017;174:1195–208. doi: 10.1111/bph.13649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu Y, Jin F, Wang Y, Li F, Wang L, Wang Q, et al. In vitro and in vivo anti-inflammatory effects of theaflavin-3,3’-digallate on lipopolysaccharide-induced inflammation. Eur J Pharmacol. 2017;794:52–60. doi: 10.1016/j.ejphar.2016.11.027. [DOI] [PubMed] [Google Scholar]
- 48.Kim S, Joo YE. Theaflavin inhibits LPS-induced IL-6, MCP-1, and ICAM-1 expression in bone marrow-derived macrophages through the blockade of NF-kappaB and MAPK signaling pathways. Chonnam Med J. 2011;47:104–10. doi: 10.4068/cmj.2011.47.2.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi M, Lu Y, Wu J, Zheng Z, Lv C, Ye J, et al. Beneficial effects of theaflavins on metabolic syndrome: from molecular evidence to gut microbiome. Int J Mol Sci. 2022;23:7595. [DOI] [PMC free article] [PubMed]
- 50.Zeng B, Huang YT, Chen SY, Xu R, Xu LH, Qiu JH, et al. Dextran sodium sulfate potentiates NLRP3 inflammasome activation by modulating the KCa3.1 potassium channel in a mouse model of colitis. Cell Mol Immunol. 2022;19:925–43. doi: 10.1038/s41423-022-00891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang YT, Liang QQ, Zhang HR, Chen SY, Xu LH, Zeng B, et al. Baicalin inhibits necroptosis by decreasing oligomerization of phosphorylated MLKL and mitigates caerulein-induced acute pancreatitis in mice. Int Immunopharmacol. 2022;108:108885. doi: 10.1016/j.intimp.2022.108885. [DOI] [PubMed] [Google Scholar]
- 52.Zhong MY, Huang YT, Zeng B, Xu LH, Zhong CS, Qiu JH, et al. Induction of multiple subroutines of regulated necrosis in murine macrophages by natural BH3-mimetic gossypol. Acta Biochim Biophys Sin. 2022;54:64–76. doi: 10.3724/abbs.2021004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bronner DN, O’Riordan MX. Measurement of mitochondrial DNA release in response to ER stress. Bio Protoc. 2016;6:e1839. [DOI] [PMC free article] [PubMed]
- 54.Bronner DN, Abuaita BH, Chen X, Fitzgerald KA, Nunez G, He Y, et al. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity. 2015;43:451–62. doi: 10.1016/j.immuni.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560:198–203. doi: 10.1038/s41586-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki K, Hirashima N, Fujii Y, Fushimi T, Yamamoto A, Ueno T, et al. Theaflavins decrease skeletal muscle wasting in disuse atrophy induced by hindlimb suspension in mice. J Clin Biochem Nutr. 2021;68:228–34. doi: 10.3164/jcbn.20-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wegiel B, Larsen R, Gallo D, Chin BY, Harris C, Mannam P, et al. Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation. J Clin Invest. 2014;124:4926–40. doi: 10.1172/JCI72853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.He XH, Ouyang DY, Xu LH. Injection of escherichia coli to induce sepsis. Methods Mol Biol. 2021;2321:43–51. doi: 10.1007/978-1-0716-1488-4_5. [DOI] [PubMed] [Google Scholar]
- 59.Xie G, He RR, Feng X, Yan T, Lan F, Wu MZ, et al. The hypoglycemic effects of camellia assamica var. kucha extract. Biosci Biotechnol Biochem. 2010;74:405–7. doi: 10.1271/bbb.90618. [DOI] [PubMed] [Google Scholar]
- 60.Igarashi M, Satoh T, Yamashita H, Watanabe K. Black tea inhibits small intestinal α-glucosidase activity in db/db mouse. Jpn J Complem Altern Med. 2014;11:25–33. [Google Scholar]
- 61.Stutz A, Horvath GL, Monks BG, Latz E. ASC speck formation as a readout for inflammasome activation. Methods Mol Biol. 2013;1040:91–101. doi: 10.1007/978-1-62703-523-1_8. [DOI] [PubMed] [Google Scholar]
- 62.He Y, Varadarajan S, Muñoz-Planillo R, Burberry A, Nakamura Y, Núñez G. 3,4-methylenedioxy-beta-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J Biol Chem. 2014;289:1142–50. doi: 10.1074/jbc.M113.515080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Erratum: Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;540:150. doi: 10.1038/nature20106. [DOI] [PubMed] [Google Scholar]
- 64.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–40. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 65.Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. 2016;17:250–8. doi: 10.1038/ni.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zeng J, Deng Z, Zou Y, Liu C, Fu H, Gu Y, et al. Theaflavin alleviates oxidative injury and atherosclerosis progress via activating microRNA-24-mediated Nrf2/HO-1 signal. Phytother Res. 2021;35:3418–27. doi: 10.1002/ptr.7064. [DOI] [PubMed] [Google Scholar]
- 67.Han X, Zhang J, Xue X, Zhao Y, Lu L, Cui M, et al. Theaflavin ameliorates ionizing radiation-induced hematopoietic injury via the NRF2 pathway. Free Radic Biol Med. 2017;113:59–70. doi: 10.1016/j.freeradbiomed.2017.09.014. [DOI] [PubMed] [Google Scholar]
- 68.Adhikary A, Mohanty S, Lahiry L, Hossain DM, Chakraborty S, Das T. Theaflavins retard human breast cancer cell migration by inhibiting NF-kappaB via p53-ROS cross-talk. FEBS Lett. 2010;584:7–14. doi: 10.1016/j.febslet.2009.10.081. [DOI] [PubMed] [Google Scholar]
- 69.Connolly NMC, Theurey P, Adam-Vizi V, Bazan NG, Bernardi P, Bolanos JP, et al. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ. 2018;25:542–72. doi: 10.1038/s41418-017-0020-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, et al. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/S1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 71.Gottlieb E, Armour SM, Harris MH, Thompson CB. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003;10:709–17. doi: 10.1038/sj.cdd.4401231. [DOI] [PubMed] [Google Scholar]
- 72.Frey TG, Mannella CA. The internal structure of mitochondria. Trends Biochem Sci. 2000;25:319–24. doi: 10.1016/S0968-0004(00)01609-1. [DOI] [PubMed] [Google Scholar]
- 73.Poudel B, Gurung P. An update on cell intrinsic negative regulators of the NLRP3 inflammasome. J Leukoc Biol. 2018;103:1165–77. [DOI] [PMC free article] [PubMed]
- 74.Evavold CL, Hafner-Bratkovic I, Devant P, D’Andrea JM, Ngwa EM, Borsic E, et al. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell. 2021;184:4495–511.e19. doi: 10.1016/j.cell.2021.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Groß CJ, Mishra R, Schneider KS, Médard G, Wettmarshausen J, Dittlein D, et al. K+ efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity. 2016;45:761–73. doi: 10.1016/j.immuni.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 76.Desai J, Steiger S, Anders HJ. Molecular pathophysiology of gout. Trends Mol Med. 2017;23:756–68. doi: 10.1016/j.molmed.2017.06.005. [DOI] [PubMed] [Google Scholar]
- 77.Mulay SR, Anders H-J. Crystallopathies. N Engl J Med. 2016;374:2465–76. doi: 10.1056/NEJMra1601611. [DOI] [PubMed] [Google Scholar]
- 78.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 79.Moon JS, Lee S, Park MA, Siempos II, Haslip M, Lee PJ, et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest. 2015;125:665–80. doi: 10.1172/JCI78253. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 80.Wang D, Zhang Y, Xu X, Wu J, Peng Y, Li J, et al. YAP promotes the activation of NLRP3 inflammasome via blocking K27-linked polyubiquitination of NLRP3. Nat Commun. 2021;12:2674. doi: 10.1038/s41467-021-22987-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li T, Sun H, Li Y, Su L, Jiang J, Liu Y, et al. Downregulation of macrophage migration inhibitory factor attenuates NLRP3 inflammasome-mediated pyroptosis in sepsis-induced AKI. Cell Death Discov. 2022;8:61. doi: 10.1038/s41420-022-00859-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hao H, Cao L, Jiang C, Che Y, Zhang S, Takahashi S, et al. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsis. Cell Metab. 2017;25:856–67. doi: 10.1016/j.cmet.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17:588–606. doi: 10.1038/nrd.2018.97. [DOI] [PubMed] [Google Scholar]
- 84.Poston JT, Koyner JL. Sepsis associated acute kidney injury. BMJ. 2019;364:k4891. doi: 10.1136/bmj.k4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang L, Hauenstein AV. The NLRP3 inflammasome: Mechanism of action, role in disease and therapies. Mol Asp Med. 2020;76:100889. doi: 10.1016/j.mam.2020.100889. [DOI] [PubMed] [Google Scholar]
- 86.He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41:1012–21. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–53. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–56. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–5. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 90.Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–9. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–3. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–9. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 94.Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8 dependent cleavage of gasdermin D and cell death. Science. 2018;362:1064–9. doi: 10.1126/science.aau2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354–7. doi: 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature. 2019;570:338–43. doi: 10.1038/s41586-019-1295-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. 2013;49:331–8. doi: 10.1016/j.molcel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 98.Niu T, De Rosny C, Chautard S, Rey A, Patoli D, Groslambert M, et al. NLRP3 phosphorylation in its LRR domain critically regulates inflammasome assembly. Nat Commun. 2021;12:5862. doi: 10.1038/s41467-021-26142-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ye T, Yang X, Liu H, Lv P, Lu H, Jiang K, et al. Theaflavin protects against oxalate calcium-induced kidney oxidative stress injury via upregulation of SIRT1. Int J Biol Sci. 2021;17:1050–60. doi: 10.7150/ijbs.57160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kovac S, Angelova PR, Holmström KM, Zhang Y, Dinkova-Kostova AT, Abramov AY. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta. 2015;1850:794–801. doi: 10.1016/j.bbagen.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu XX, Zheng G, Tang SK, Liu HX, Hu YZ, Shang P. Theaflavin protects chondrocytes against apoptosis and senescence via regulating Nrf2 and ameliorates murine osteoarthritis. Food Funct. 2021;12:1590–602. doi: 10.1039/D0FO02038A. [DOI] [PubMed] [Google Scholar]
- 102.Li Z, Zhu J, Wan Z, Li G, Chen L, Guo Y. Theaflavin ameliorates renal ischemia/reperfusion injury by activating the Nrf2 signalling pathway in vivo and in vitro. Biomed Pharmacother. 2021;134:111097. doi: 10.1016/j.biopha.2020.111097. [DOI] [PubMed] [Google Scholar]
- 103.McKenzie BJ, Wechalekar MD, Johnston RV, Schlesinger N, Buchbinder R. Colchicine for acute gout. Cochrane Database Syst Rev. 2021;8:CD006190. doi: 10.1002/14651858.CD006190.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rashidi M, Simpson DS, Hempel A, Frank D, Petrie E, Vince A, et al. The pyroptotic cell death effector gasdermin D is activated by gout-associated uric acid crystals but is dispensable for cell death and IL-1beta release. J Immunol. 2019;203:736–48. doi: 10.4049/jimmunol.1900228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. 2017;17:407–20. doi: 10.1038/nri.2017.36. [DOI] [PubMed] [Google Scholar]
- 106.Deutschman CS, Tracey KJ. Sepsis: current dogma and new perspectives. Immunity. 2014;40:463–75. doi: 10.1016/j.immuni.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 107.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 108.Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase-11, an ICE-interacting protease, is essential for the a ctivation of ICE. Cell. 1998;92:501–9. doi: 10.1016/S0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 109.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–92. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 110.Rühl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K+ efflux. Eur J Immunol. 2015;45:2927–36. doi: 10.1002/eji.201545772. [DOI] [PubMed] [Google Scholar]
- 111.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–61. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 112.Aizawa T, Yamamoto A, Ueno T. Effect of oral theaflavin administration on body weight, fat, and muscle in healthy subjects: a randomized pilot study. Biosci Biotechnol Biochem. 2017;81:311–5. doi: 10.1080/09168451.2016.1246170. [DOI] [PubMed] [Google Scholar]
- 113.Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci USA. 2018;115:E1530–e9. doi: 10.1073/pnas.1716095115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Clinicaltrials.gov identifier: NCT05658575. https://clinicaltrials.gov/ct2/show/NCT05658575.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.