

Summary
The rapid accumulation of single-cell RNA-seq data has provided rich resources to characterize various human cell populations. However, achieving accurate cell-type annotation using public references presents challenges due to inconsistent annotations, batch effects, and rare cell types. Here, we introduce SELINA (single-cell identity navigator), an integrative and automatic cell-type annotation framework based on a pre-curated reference atlas spanning various tissues. SELINA employs a multiple-adversarial domain adaptation network to remove batch effects within the reference dataset. Additionally, it enhances the annotation of less frequent cell types by synthetic minority oversampling and fits query data with the reference data using an autoencoder. SELINA culminates in the creation of a comprehensive and uniform reference atlas, encompassing 1.7 million cells covering 230 distinct human cell types. We substantiate its robustness and superiority across a multitude of human tissues. Notably, SELINA could accurately annotate cells within diverse disease contexts. SELINA provides a complete solution for human single-cell RNA-seq data annotation with both python and R packages.
Keywords: single-cell RNA sequencing, cell-type prediction, deep learning, domain adaptation, reference atlas, pretrained model
Graphical abstract
Highlights
-
•
SELINA combines SMOTE, MADA, and an autoencoder to improve annotation accuracy
-
•
SELINA pre-builds a reference atlas with 1.7 million cells covering 230 human cell types
-
•
SELINA annotates cell types with high accuracy in various disease scenarios
Motivation
Cell-type annotation is a crucial step for interpreting cell-type functions in scRNA-seq data processing. There are two main methods for cell-type annotation, marker based and reference based. Reference-based methods transfer cell-type labels from reference datasets to query datasets using machine learning techniques, resulting in improved accuracy and broader applications. However, challenges remain, including difficulty in leveraging large-scale public data, cell number imbalances, batch effects, and reliance on reference data quality. Addressing these challenges is essential to improve the accuracy of cell-type annotation and enable the full potential of scRNA-seq data.
Ren et al. develop SELINA (single-cell identity navigator), an integrative and automatic cell-type annotation framework based on a multiple-adversarial domain adaptation network and a pre-curated reference atlas of various tissues. SELENA enables accurate and robust cell-type annotation.
Introduction
Single-cell RNA sequencing (scRNA-seq) can profile thousands of cells to reveal heterogeneity within complex tissues. The key step in scRNA-seq data processing is cell-type annotation, which is vital for interpreting function features for certain cell types and is required for many downstream analyses, including trajectory analysis or cell-cell interactions. Cell-type annotation methods can be roughly divided into two categories. Marker-based methods such as Garnett1 and SCINA2 rely on clustering performance and the quality of cell-type-specific marker genes. In contrast, reference-based methods, such as scmap,3 scPred,4 SingleR,5 CHETAH,6 SingleCellNet,7 ACTINN,8 mtSC,9 Cell BLAST,10 CellO,11 scCATCH,12 scMatch13 scDeepSort,14 and CellTypist15 transfer cell-type labels from reference datasets to query datasets using various machine learning techniques. As the reference-based methods do not require prior knowledge,16 they show improved accuracy and broader applications compared to marker-based methods with the continuous accumulation and increasing throughput of scRNA-seq datasets.
Although reference-based methods have the above advantages in cell-type annotation, several challenges remain to be resolved. First, current tools are often designed for transferring cell-type assignments between single reference data and single query data; hence, they cannot leverage the wealthy information hidden in the enormous public data. Second, the cell numbers of different cell types are often imbalanced; therefore, the minority cell types are always ignored in the modeling process. Third, the underlying batch effects between reference data and query data are often overlooked, which may hinder accurate label transfer. Last, all these methods heavily rely on the quality and quantity of reference datasets. ScCATCH was pre-trained on cell-type-specific markers. SingleR, CellO, and scMatch provided references from bulk RNA-seq samples. ScDeepSort and CellTypist only provide scRNA-seq references with limited lineages such as immune cells or embryonic cells. Even though great efforts in systematically collecting and curating public datasets have been made to build scRNA-seq data portals that involve millions of cells, which have spawned Human Cell Atlas17 (HCA), Animal Cell Atlas18 (ACA), Single Cell Portal from the Broad Institute,19 Human Cell Landscape20 (HCL), and Single Cell Expression Atlas from European Bioinformatics Institute21 (EMBL-EBL), a uniform and comprehensive reference atlas is still lacking due to the inconsistent annotation and large batch effects between datasets.
To address these challenges, we built a comprehensive single-cell transcriptomics data atlas consisting of 35 human tissues across 7 different sequencing platforms from 136 datasets. The datasets have been curated and made accessible through the HUSCH22 website. Based on 1,706,710 uniformly processed cells from 230 cell types, we proposed an algorithm that can effectively utilize multiple datasets for single-cell assignment. It applies the synthetic minority oversampling technique23 (SMOTE) to boost the number of rare cell types and employs multi-adversarial domain adaptation24 (MADA) to update the parameters of the supervised deep learning framework in the pre-training stage. Furthermore, it utilizes an autoencoder to adaptively adjust the pre-trained parameters based on the distribution of query data. We demonstrated the power of SELINA (single-cell identity navigator) in batch removal and systematically evaluated the performance of SELINA with existing tools on 95 datasets from 17 tissues. In addition, we proved that SELINA could take other unified databases as references by testing on datasets from the Allen Institute.25,26,27 Finally, the benchmark in multiple disease scenarios demonstrated that SELINA could annotate cell types with a higher accuracy than the mainstream methods. The comprehensive cell-type references of SELINA and its superior ability in transferring annotations pave the way for users to accurately annotate single cells.
Results
Overview of SELINA
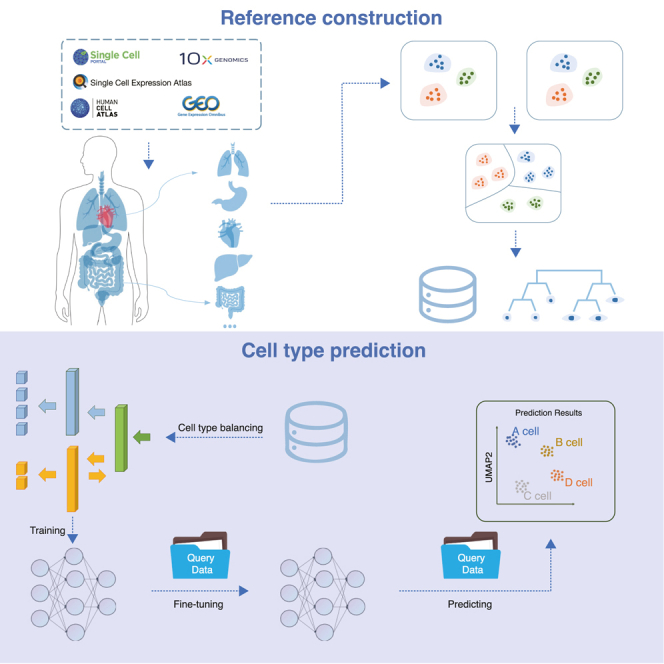
The SELINA workflow is mainly composed of two steps: reference construction and cell-type prediction. For reference construction, public scRNA-seq datasets were collected from multiple databases and processed with a standardized pipeline from MAESTRO28 including quality control, principal component analysis (PCA), batch removal within each dataset, unsupervised clustering, and annotation based on the original labels or cell-type-specific gene markers from the original study (Figure S1A). Next, the inconsistent annotations across datasets were manually unified and assigned to the major lineage and minor lineage based on the Cell Ontology29 and literature (Figure S1B). After annotation unification, all datasets within a tissue were merged, and the batch effects across datasets were removed using harmony.30 For each lineage, the outlier cells were removed to correct the potential misannotations. Finally, the curated cell types were organized into a cell-type ontology tree.
Based on the uniformed reference, we developed a cell-type prediction algorithm consisting of three steps: cell-type balancing, pre-training, and fine-tuning. Usually, the classifier will achieve a higher training accuracy at the cost of the minority cell types being misclassified. Therefore, to increase the sensitivity of the classifier to the minority cell types, SELINA utilizes SMOTE23 to generate synthetic samples to increase the weights of the minority cell types. Then, SELINA takes datasets from one tissue as input and employs a MADA-based network24 to obtain a pre-trained model. By training the supervised deep learning framework in an adversarial way, the underlying common information of the same cells from different sequencing platforms is uncovered. To further remove the batch noise between the reference and query data, an autoencoder is used to adaptively fine-tune the pre-trained parameters according to the distribution of query data. Finally, the labels from reference datasets are transferred to the query data based on the fully trained model (Figure 1).
Figure 1.

Overview of SELINA
SELINA consists of two sections: reference construction and cell-type prediction. Public datasets were collected from various databases and uniformly processed based on a two-step approach containing within datasets processing and across datasets unification. For each tissue, a well-organized cell-type classification tree was built, and the expression profiles were merged as the annotation algorithm input. The algorithm consists of three steps: cell type balancing, training, and fine-tuning. First, the rare cell types from the merged training data are oversampled. Second, the training data is pre-trained with a supervised deep learning framework containing a gradient reversal layer. Third, the parameters of the pre-trained model are adjusted according to the distribution of query data. Finally, the adjusted model takes query data as input and assigns the cells with cell types from reference data.
SELINA provides a large-scale and well-annotated human single-cell expression reference
A total of 1,706,710 cells from 136 datasets were collected to build the single-cell transcriptomic data portal, which covered 230 human cell types and 7 different sequencing platforms (Table S1). The reference atlas was expanded based on HCL20 to include more datasets from other sequencing platforms. All the datasets were categorized into 35 major tissues according to the definition in HCL. We first summarized the features of the datasets in the reference. The blood (n = 14), intestine (n = 14), and bone marrow (n = 13) tissues have the largest dataset numbers (Figure 2A). Blood and bone marrow tissues also have the largest number of cells (Figure S2B), indicating a better characterization of immune cells for our reference. The kidney has the most abundant cell types (Figure S2A). Importantly, 27 out of the 35 tissues have two or more datasets in the reference, suggesting good coverage and depth of our reference. The majority of the data was generated using 10x genomics and Microwell-seq, which included 60 and 58 datasets, respectively. Data from Smart-seq, InDrop, Drop-seq, and snDrop-seq only account for a small proportion of all the datasets (Figure 2A).
Figure 2.

Reference data and annotation algorithm of SELINA
(A) Dataset number of different tissues collected in SELINA reference. Different colors represent different sequencing platforms.
(B) Cell-type classification tree from the liver with two levels of annotations.
(C) Annotation algorithm of SELINA. First, the rare cell types are oversampled with SMOTE. The green dots are cells from the original minority cell types, and the orange dots are the synthetic cells. The balanced data are trained with a MADA-based pre-training framework. Each bar represents one layer of the model. Then the pre-trained model is fine-tuned using an autoencoder. The feature extractor and cell-type discriminator are extracted to construct the encoder, and the decoder is randomly initialized with a structure symmetrical to that of the encoder. The decoder is trained with the encoder fixed; subsequently, the encoder is trained with the decoder fixed. Finally, the decoder is removed, and the encoder is used to classify query cells.
For each tissue, the inconsistent cell-type names between different datasets were unified and subsequently divided into the major level and minor level based on the literature and the Cell Ontology.29 Taking the cell-type names from the liver as an example, the major lineage has 14 different cell-type categories, and only dendritic cells (DCs), endothelial cells, and epithelial cells have sublineages (Figure 2B). Aggregating all tissues together, we constructed a comprehensive human cell-type ontology tree to describe the parent-child relationships of cells within scRNA-seq data. By incorporating and curating cell types from published studies, SELINA affords users a more unified standard that dictates cell-type landscapes in the scRNA-seq data and organizes this landscape for annotating input scRNA-seq data.
SELINA combines SMOTE, MADA, and an autoencoder to improve the annotation accuracy
The annotation algorithm in SELINA comprises three steps, including cell-type balancing, pre-training, and fine-tuning (Figure 2C). In most classification algorithms, the classifier will misclassify the minority samples to achieve a higher training accuracy with a lower learning cost. Especially, when the minority samples exist both in the training and testing data, it is hard for the classifier to correctly assign the minority samples in testing data as the training samples only afford limited information. Data augmentation techniques can resolve the problem of data imbalance, of which SMOTE23 is a classical algorithm. The scRNA-seq data exhibited a strong category imbalance. In our reference, the minority cell types only contain dozens of cells, while the majority cell types can be characterized by tens of thousands of cells. Therefore, SELINA first adopts SMOTE to oversample the minority cell types. For each cell in a pair of randomly selected cells from the rare cell types, SELINA multiplies its gene expression vector by a random weight and then sums the pair of weighted vectors to obtain a synthetic cell. Colloquially, the generated cell is the linear combination of the original cells and locates randomly on the line connecting the pair of cells. This process will proceed until the cell numbers of rare cell types reach the same magnitude as the majority of cell types or are not less than 1,000.
In the pre-training phase, SELINA applies MADA24 to remove the batch noise induced by different sequencing platforms. The architecture of the pre-training framework consists of three components: a feature extractor, a cell-type discriminator, and a sequencing platform discriminator. The feature vector generated by the feature extractor will flow into the cell-type discriminator and platform discriminator simultaneously. Unlike a conventional adversarial neural network,31 the platform discriminator in our pre-training framework contains multiple classifiers of which the number is equal to the cell types. For a certain platform classifier, the input feature vector will be multiplied by the probability of the input cell being assigned as the cell type paired with this platform classifier, and the probability is calculated by the cell-type discriminator. During the backward propagation of platform predicting errors, the gradient of the feature vector will be reversed so that the feature extractor is trained to maximize the loss of the platform discriminator, while the platform discriminator is trained to minimize the loss. Thus, as the training proceeds, features generated by the feature extractor become worse for the platform discriminator to classify; however, even though the difference between the input features from different platforms is slight, the platform discriminator can always manage to distinguish the platform sources until the difference is nearly eliminated. This strategy can enable fine-grained alignment of expression distributions from different sequencing platforms by capturing the batch information of each cell type separately and training the feature extractor with the platform discriminator in an adversarial way.
In the original application scenario of MADA, the domain discriminator is used to help with extracting the common features between reference data and query data. However, in SELINA, MADA is used to uncover the common information between different platforms within the reference data. Therefore, the difference between reference data and query data still exists. To reduce such differences, an autoencoder is employed in the fine-tuning step. The encoder, combining the feature extractor and cell-type discriminator of the pre-trained model, has learned the transformation from a large amount of reference data. By contrast, the decoder still needs to be adjusted due to the random initialization of the parameters. SELINA first freezes the encoder and updates the parameters of the decoder. Freezing the encoder can prevent the parameters of the encoder from changing considerably so that the learned transformation in the pre-trained model can be well preserved. Once the total loss decreases to convergence, SELINA will freeze the decoder and then update the parameters of the encoder. The reconstruction loss will be further decreased after encoder training so that the encoder is shifted based on the distribution of query data, which can reduce the batch noise between reference data and query data.
Data augmentation for rare cell types and batch removal in both data integration and querying processes improve annotation accuracy
To validate the improvement of SMOTE in rare cell-type annotation, we selected 4 datasets from the liver as reference data, including 15,859 cells,20,32 and 1 dataset as query data.33 Seven cell types with limited cell numbers in the training data were defined as rare cell types, including CD4T (n = 98), plasma (n = 462), mast (n = 12), pDC (n = 32), cholangiocyte (n = 391), PL (periportal LSEC, n = 358), and PE (portal endothelial cell, n = 212), and these cell types were also presented in the query data. The implementation of SMOTE increased the number of rare cell types that were correctly assigned. Before SMOTE, almost all CD4T cells were misannotated as CD8T cells, and no mast cells were correctly annotated (Figure 3A). Nearly 1/3 of CD4 T cells and half of the mast cells were correctly annotated after implementing SMOTE (Figure 3A). The distribution of synthetic cells may still be slightly different from the query cells due to the limited number of original cells and the randomness of the synthesis process. Therefore, the enhancement contributed by SMOTE might be limited compared to collecting more data. We calculated the F1 of these cell types and found a large improvement except for the PL, of which some were probably misannotated in the reference (Figure 3B). To test the effectiveness of oversampling, we benchmarked SELINA with and without SMOTE in three tissues: intestine, stomach and liver. For each tissue we selected the dataset with the most abundant cell types as the test data, and all the remaining datasets within the tissue were used as reference data. We then performed downsampling on each cell type in the reference data with a gradient proportion (10%, 20%, 30%, 40%, and 50%). SMOTE was utilized to oversample each downsampled dataset, and we recorded the benchmark results after the oversampling. Comparing the results with and without oversampling, we found that most cell types achieved more accurate predictions after oversampling (Figures S2D–S2F). In conclusion, SMOTE can remedy the undesirable training results and overfitting of the classifier caused by the limited number of training cells.
Figure 3.

Evaluation of computational techniques used in SELINA
(A) The prediction results of rare cell types with SMOTE (SMOTE) and without SMOTE (Base) synthesizing new cells in reference data. The heights of the bars and linkage lines represent the cell number. The bars on the left are the original labels provided by the corresponding paper, and the bars on the right represent the prediction results of SELINA. PL, periportal liver sinusoidal endothelial cell; PE, portal endothelial; CVL, central venous liver sinusoidal endothelial cell.
(B) Bar plot for the F1 scores of rare cell types with SMOTE and without SMOTE (Base) oversampling training data before pre-training.
(C) Boxplot for the LISI scores of cells that are specific to one sequencing platform (top) and common cells between different platforms (bottom). Original embedding represents dimension reduction results from PCA. Feature extractor represents the results from the output layer of the feature extractor. Cell-type classifier shows the results from the first hidden layer of the cell-type classifier. Each dot corresponds to the LISI score of one cell. Cell coordinates were calculated by UMAP. The sequencing platform was taken as the batch information when calculating the LISI scores. Center lines indicate the median value, and lower and upper hinges represent the 25th and 75th percentiles, respectively. p values were determined by pairwise Wilcoxon rank-sum test, one-sided, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(D–F) 2D-UMAP representation of liver data based on dimension reduction results from PCA, cell-type classifier, and PCA followed by harmony. Graphs on the top and bottom show the sequencing platform labels and cell-type labels for each cell respectively.
(G) Boxplots for LISI scores of reference and query cells in the liver. Graph on the top shows the LISI scores of the common cell types between reference and query data. The graph on the bottom shows the LISI scores of the cell types that are unique to reference or query data. Cell coordinates were calculated by UMAP based on the output from the first hidden layer of the pre-trained model and tuned model, respectively. The cell source (reference/query) was taken as batch information when calculating the LISI scores. Each dot represents the LISI score of one cell. p values were determined by pairwise Wilcoxon rank-sum test, one-sided, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(H) Mean accuracy and MacroF1 (3 repeats) of different strategies. Base represents the situation in which the data were not oversampled, and the pre-trained model was used without fine-tuning. +SMOTE represents the results when SMOTE was additionally implemented to oversample rare cell types. +AE shows the results when SMOTE and autoencoder were both implemented. Each dot represents the testing result of one dataset (n = 17). p values were determined by pairwise Wilcoxon rank-sum test, one-sided, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To further confirm that SELINA can eliminate batch effects existing in reference data, we applied uniform manifold approximation and projection34 (UMAP) to display the features in the output layer of the feature extractor and the first hidden layer of the cell-type discriminator. We also used local inverse Simpson’s index (LISI)30 to quantitively assess the batch effects (see STAR Methods). A higher LISI score means cells are well mixed across different platforms. Ideally, after batch removal for sequencing platforms, the same cell types sequenced by different platforms will be clustered closer, and the cell types that are unique to specific platforms will be further separated. Thus, we calculated the LISI scores for platform-common cells and platform-specific cells separately. A total of five liver datasets20,32,33 were merged and used in the following batch removal evaluation. Both the feature extractor transformation and the cell-type classifier transformation increased the LISI scores of platform-common cells and decreased the LISI scores of platform-specific cells (Figure 3C). Cells of the same cell type from different platforms were clustered closer compared to the original embedding, suggesting that with the cell-type-specific platform discriminators, the feature extractor can uncover the underlying common features for each cell type. Our pre-training framework significantly removed the batch effects and also showed better separation in similar cell types like CD4+ T cells, CD8+ T cells, and NK cells compared to the conventional batch correction tool Harmony30 (Figures 3D–3F and S3A).
We next tested whether fine-tuning using an autoencoder could remove the batch effects between the reference data and query data using the liver data. We calculated the LISI scores of cell types possessed by both the reference and query data (QR-common) and cell types that are unique to reference or query data (QR-specific) separately. The increased LISI scores showed that common cell types from reference data and query data mixed better, and the decreased LISI scores showed that QR-specific cell types clustered more independently after fine-tuning (Figures 3G, S3B, and S3C). Similar results were also observed on two query datasets from the lung and intestine, indicating the necessity of the fine-tuning step (Figure S3D).
Finally, we investigated whether the performance improvement of oversampling and fine-tuning is robust and tested it on three different tissues, including the bladder, brain, and liver. Each time, we selected one dataset as query data, and the remaining datasets were merged as reference data. Accuracy and MacroF1 are gradually improved with the implementation of SMOTE and autoencoder (Figure 3H). We took the test in the liver as an example to show how the metrics of each cell type changed with the gradual implementation of SMOTE and autoencoder, and we found a higher improvement for the rare cell types (Figure S3E). Taken together, these results demonstrate that the techniques implemented in SELINA can help to utilize multiple reference datasets to annotate unlabeled datasets, and the improvement is consistent and robust across different tissues.
SELINA outperforms other existing tools in the comprehensive performance evaluation
We systematically compared the performance of SELINA with existing annotation tools and traditional machine learning methods including support vector machine (SVM), random forest (RF), and k-nearest neighbor (kNN) using data from a single sequencing platform and multiple sequencing platforms respectively. The single-platform evaluation was performed on 9 tissues containing multiple datasets from the same sequencing platform, including 479,740 cells. The multi-platform evaluation was carried out on 14 tissues containing datasets from multiple sequencing platforms, covering 658,270 cells (Table S2). For each tissue, one dataset was picked out as query data, while the others were merged as the reference. The training and testing process was repeated until each dataset was tested. The average accuracy and MacroF1 of all datasets in one tissue represent the performance of one certain method. SELINA achieves the best accuracy and MacroF1 in the evaluation on both the single-platform and multi-platform data. For single-platform tests (Figure 4A), SELINA is the top method with an average accuracy of 61.51%, followed by SELINA-Base (60.57%), ACTINN8 (59.35%), SingleCellNet7 (58.34%), mtSC9 (58.01%), scibet35 (57.97%), CellTypist (57.28%), SVM (57.24%), Seurat (56.24%), SingleR5 (55.45%), RF (55.10%), CelliD (48.87%), scmap3 (47.43%), kNN (46.00%), and Cell BLAST10 (42.51%), and SELINA also performs best with the highest MacroF1 (0.5126), followed by SELINA-Base (0.4857), mtSC (0.4808), scibet (0.4808), ACTINN (0.4739), Seurat (0.4694), SVM (0.4633), SingleCellNet (0.4574), CellTypist (0.4510), CelliD (0.4409), SingleR (0.4406), RF (0.3937), scmap (0.3492), Cell BLAST (0.3363), and kNN (0.3356).
Figure 4.

Performance evaluation of SELINA and existing annotation tools
(A and B) The prediction accuracy and MacroF1 using datasets from the same sequencing platform (A) and multiple sequencing platforms (B) as reference. The number in each cell represents the average performance of all tested datasets within one tissue. The top row shows the average performance of all tissues for each method. The number of datasets included in each tissue is listed behind the tissue names.
(C) Mean training time (3 repeats) for increasing training cell numbers.
(D) Mean testing time (3 repeats) for increasing testing cell numbers.
In terms of the multi-platform evaluation, SELINA ranks first with a highest average accuracy (64.42%), followed by SELINA-Base (62.28%), ACTINN (61.31%), SingleCellNet (61.08%), CellTypist (60.99%), mtSC (59.76%), SVM (58.76%), scibet (58.46%), RF (56.20%), Seurat (55.71%), singleR (53.97%), CelliD36 (52.41%), scmap (52.15%), kNN (48.67%), and Cell BLAST (42.99%). Besides, SELINA is the top-ranked method with an average MacroF1 of 0.5082, followed by mtSC (0.4811), SELINA-Base (0.4785), ACTINN (0.4678), scibet (0.4528), CellTypist (0.4487), SingleCellNet (0.4476), SVM (0.4365), CelliD (0.4238), Seurat (0.4105), SingleR (0.4051), scmap (0.3750) RF (0.3710), kNN (0.3169), and Cell BLAST (0.3118). Specifically, for the multi-platform evaluation, SELINA ranks first in 9 tissues and second in 1 tissue, and in terms of MacroF1, SELINA ranks first in 11 tissues and second in 2 tissues (Figure 4B). Both the base version and the full version of SELINA showed improved performance over existing methods, with the full version incorporating the cell balancing and query fine-tuning modules ranking the best, indicating the importance of adding these features. The performance varies between tissues, which is probably caused by the different overlapping ratios of cell types in different tissues (Figure S4A, see STAR Methods). The detailed performance comparisons for 5 representative tissues are shown in Figures S4B–S4F. All these results suggest the robustness and superiority of SELINA in annotating unlabeled datasets.
The 5-fold cross-validation was employed in the single-sample evaluation. Since all individual parts used in one test were from the same sample and had minimal batch effects, which limited the potential for deep learning methods to demonstrate their advantages on batch removal, many non-deep learning methods have shown significant improvements in performance (Figures S4G and S4H). SELINA performs well in the single-sample testing with an average accuracy of 94.03%.
Moreover, we compared the training time and querying time of SELINA with the other tools. As SELINA, ACTINN, and mtSC are deep learning-based methods, they were trained with a GPU and tested using a CPU, and the rest of the methods were trained and tested using the same CPU as the deep learning model tests used. We set various reference cell numbers and query cell numbers to investigate the dynamic change in time consumption and found it was positively correlated with the cell number in all methods. Compared to other methods, SELINA exhibits moderate computational efficiency. It takes SELINA approximately 3 min to train a model on a dataset with 25,000 cells and less than 1 min to fine-tune the parameters on a dataset with 10,000 cells (Figures 4C and 4D). Additionally, the fine-tuning step can be largely accelerated by the GPU, with a more obvious effect as the cell number increases (Figure S4I). In conclusion, SELINA can realize more accurate annotation than published tools with an acceptable runtime.
Expandable references of SELINA
The traditional strategy took a single annotated dataset as reference data. With the explosion of data volume, tools utilizing integrated information from multiple datasets have now become mainstream. Since the cell types and cell numbers within a single dataset may be limited, the single reference cannot afford sufficient information for a model to learn. To ensure that the effective integration of multiple references can improve the annotation result, we evaluated the performance of SELINA with an increasing number of reference datasets on the pancreas, liver, and lung tissues. (Figures 5A, S5A, and S5B). The performance improves stably as the training data continues to increase.
Figure 5.

Expandable references of SELINA
(A) Performance evaluation of SELINA with an increasing number of datasets being used as a reference in the lung.
(B) Performance evaluation on datasets from the Allen Institute. Each dot represents the testing result of one dataset (n = 6).
(C and D) 2D-UMAP representation of two examples from the Allen Institute. Graphs on the left and right show the original annotation and prediction results, respectively.
To further prove that SELINA supports the use of other harmonized scRNA-seq databases or user custom references, we tested SELINA on 2 datasets from the Allen Brain Atlas, of which one includes single-nucleus RNA-seq (snRNA-seq) data from 76,533 total nuclei derived from 2 postmortem human brain specimens using the 10x platform, and the other includes snRNA-seq data from 49,495 nuclei across multiple human cortical areas profiled with Smart-seq. We selected cell types with cell numbers larger than 1,000, which are 13 subsets of excitatory neurons, 5 subsets of inhibitory neurons, and 1 subset of oligodendrocyte. After benchmarking multiple tools using these data, SELINA ranks second out of the 9 tested tools with an average accuracy of 93.32% and an average MacroF1 of 0.9336. ACTINN, Scibet, SingleCellNet, and SingleR also achieve a higher accuracy and MacroF1 compared to scmap, mtSC, CelliD, and Cell BLAST (Figure 5B). We present two examples to show the annotation results of SELINA and compare them with the original annotation. The high prediction accuracy indicates the enormous capacity of SELINA in annotating scRNA-seq datasets with more fine-grained cell types (Figures 5C, 5D, S5C, and S5D). In summary, increasing the number of reference datasets can provide more comprehensive cell types and expression information, which can be utilized by SELINA to annotate datasets more accurately. Furthermore, SELINA can also employ datasets from users or consortiums with expert knowledge37,38,39,40 as references.
Application of SELINA in annotating disease datasets
The scRNA-seq technique is widely used in characterizing mechanisms in the development of various diseases. Therefore, we next investigate whether SELINA could be used to annotate the cells in different disease scenarios. We constructed a reference using normal immune and tissue-specific cells and compared the performance of SELINA with other annotation tools in disease scenarios on type 2 diabetes (T2D),35 non-small-cell lung carcinoma (NSCLC),36 and Alzheimer’s disease (AD)37 datasets (Table S4). Cells in the disease datasets might be annotated as unknown as they may have altered expression levels compared to the normal status. A higher filtering threshold will result in a higher percentage of cells being predicted as unknown. For the T2D dataset, almost all cells were correctly predicted (Figure 6A). SELINA ranked first out of five tools with accuracy near 1 compared to ACTINN, mtSC, scmap, and SingleR (Figure 6B). For the NSCLC dataset, although a tiny portion of immune cells was predicted incorrectly, the majority of other cells had accurate predictions (Figures 6C and 6D). SELINA also showed remarkably improved performance compared to other tools under different thresholds in the AD dataset (Figures 6E and 6F). These results collectively suggest that SELINA could accurately annotate the cells in the disease scenario.
Figure 6.

Application of SELINA in annotating disease datasets
(A) The SELINA prediction results of T2D. The heights of the bars and linkage lines represent the cell number. The bars on the left are the original labels provided by the corresponding paper, and the bars on the right represent the prediction results of SELINA.
(B) Performance of SELINA and other tools on T2D data with an increasing percentage of cells to be predicted as unknown.
(C) The SELINA prediction results of NSCLC.
(D) Performance of SELINA and other tools in NSCLC data with an increasing percentage of cells to be predicted as unknown.
(E) The SELINA prediction results of AD.
(F) Performance of SELINA and other tools in AD data with an increasing percentage of cells to be predicted as unknown.
(G–I) The percentage of correctly predicted cells in patients diagnosed with different disease stages. For tests on T2D (G) and NSCLC (H), each dot represents one patient. All stages are aligned in ascending order of disease stages.
The alteration of gene expression in disease-associated cells might be correlated with their disease stages. To figure out whether SELINA can track the expression difference in diseased-associated cells, we chose the beta cells from T2D data, malignant cells from NSCLC, and neuron cells from AD to evaluate as they are the major abnormal cells in the corresponding disease. We first tested on one T2D dataset with 5 patients. The HbA1c value is a measurement of the risk of developing type 2 diabetes. The proportion of accurately predicted beta cells decreases further when the HbA1c value rises (Figure 6G). Then we tested SELINA’s performance on one NSCLC dataset with 3 patients, which covers the tumor region and tumor-adjacent region. A similar trend is observed, as the percentage of accurately predicted cells dropped during the progression of the disease (Figure 6H). Finally, in the AD data, the percentage of accurately predicted neurons also decreased along with the Braak stage (Figure 6I). The above analyses suggest that SELINA is sensitive to the disease stages and can accurately tackle the difference between normal and diseased cells.
Additionally, we explore the possibility of using SELINA to annotate the cells in different disease scenarios with disease data as a reference. To distinguish between normal and diseased cells, we added a cell source classifier and a few platform classifiers on the basis of the original pre-training framework (Figure S6A). In addition, we adjusted the loss function so that the parameter update will be first dominated by platform batch within cell sources (normal/abnormal) and then by the batch in cell types. We merged the datasets from T2D,35,38,39,40 AD,37,41 and NSCLC,42 respectively (Table S4), and benchmarked using a 5-fold cross-validation strategy. Compared to the training with only cell-type information, the added cell source classifier improves prediction accuracy and MacroF1 for cell-type annotation (Figure S6B). The improvement indicates that the cell source information can help SELINA to characterize cells from different sources separately. Thus, the classifier can match the reference and query cells of the same source more accurately. Next, we compared SELINA with other annotation tools. The tools are ranked by the average performance on three diseases, and SELINA is the most accurate predictor for both cell sources and cell types (Figures S6C–S6F).
Taken together, all the findings demonstrate that SELINA can be effectively used to annotate datasets with diseases.
Discussion
Substantial amounts of well-labeled human scRNA-seq data have been generated in the public domain. Previous reference data-based studies utilized various strategies to achieve automatic annotation based on annotated scRNA-seq data. However, existing algorithms do not solve the problems of imbalanced cell types and batch effects between reference and query datasets. In addition, due to the huge amount of public scRNA-seq data, a comprehensive reference atlas with uniformed cell types is still not available. In this study, we developed an accurate deep learning-based framework, SELINA, for single-cell assignment along with a large-scale reference data portal covering 1,706,710 cells and 35 tissues. SELINA can handle the imbalance of cell types existing in reference data using SMOTE. In addition, SELINA can remove not only the batch effects across reference datasets but also the batch noise between the query dataset and the reference dataset. We systematically benchmarked the performance of SELINA on 17 different human tissues and demonstrated its superiority for accurate cell-type annotation compared to existing tools. In our evaluation, the deep learning-based methods, such as SELINA, ACTINN, and mtSC, generally outperformed traditional machine learning-based methods, such as SVM and SingleCellNet, contradicting previous studies16,41 when the benchmark included more datasets and cell types, and it is reasonable since deep learning models usually have stronger nonlinear modeling ability, which can better mimic the underlying function between the input and output when the training data are more complex. We also confirmed that SELINA can learn to classify cells from other unified databases, which was validated using data from the Allen Brain Atlas. Furthermore, we demonstrated that SELINA could be used to annotate the cells in various disease scenarios. In conclusion, our method, combined with the curated reference, provides a one-stop solution for human scRNA-seq data annotation.
For reference-based approaches, the performance of data annotation depends heavily on the quality of the reference. Therefore, we have invested huge efforts in building a comprehensive and high-quality reference. To accelerate the data collection, we built a semi-automatic processing pipeline, including data crawling, standardized processing, and harmonization. We have put great efforts into systematized operations such as format unification of raw data, cell-type harmonization, lineage division, and misannotation detection and exclusion. The quality of reference is extremely vital for automatic annotation. A reference with abundant cell types and enough training samples for each cell type can enable huge improvements in annotation accuracy, which can be confirmed by the results of our study. We observed that insufficient training cells and a limited number of datasets and cell types will lead to low annotation accuracy. It should be mentioned that the reference-based approaches can only predict the cell type presented in the reference. Cells that do not exist in the training set will be predicted as unknown. Despite our reference containing relatively large-scale datasets, the number of datasets in different tissues and the cell-type abundance in each tissue is still imbalanced. This issue will be solved as the amount of data continues to grow or data-generation algorithms42,43 develop. In the future, SELINA will be updated to include more experimentally validated cell types. Apart from incomplete reference, different cell types with similar transcriptomic profiles are often misannotated in the reference data, e.g., CD4+ T cells and CD8+ T cells. Recently, CITE-seq44 and REAP-seq45 capturing surface proteins could better distinguish cells with similar transcriptomic profiles. By integrating the data from CITE-seq and REAP-seq, we can obtain more accurately annotated cell-type references. Additionally, although SELINA achieved a better performance in disease data annotation using cross-validation, it is difficult to distinguish normal cells from abnormal cells for one unlabeled disease dataset with other independent datasets as references due to the diversity of the expression profiles for abnormal cells (Figures S6G–S6I).46 For example, pancreatic β cells can behave very differently in different datasets due to different levels of cellular stress. Therefore, it is difficult to build a comprehensive database for disease scRNA-seq data.47
Finally, the deep learning model we used only excels at clustering and classification tasks; it still lacks biological insights, such as the ability to identify key factors for one cell type. In the future, we will use graph-based algorithms48 to extract the structural information so that the association between genes and cell types can be preserved to present more explainable knowledge and be used for novel cell-type identifications and even automatic annotation correction. With the presence of the above features in the SELINA algorithm and continuous expansion of the SELINA reference, we anticipate SELINA to accurately characterize all human cell types with the potential to transfer to other species.
Limitations of the study
In our study, we observed that insufficient training cells and the limited number of datasets and cell types will lead to low annotation accuracy. In addition, although reference-based methods can predict cell types present in the reference, our relatively large-scale reference still suffers from imbalances in tissue-specific datasets and cell-type abundance. Finally, while our deep learning model performed well in clustering and classification, it lacks biological insights such as identifying key factors for a cell type.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Benchmark datasets | This paper | Table S1 |
| Software and algorithms | ||
| SELINA | This paper |
https://zenodo.org/badge/latestdoi/671793671 https://github.com/wanglabtongji/SELINA.py |
| SELINA-reference | This paper | https://github.com/wanglabtongji/SELINA-reference |
| SMOTE | Chawla, N. V. et al.23 | https://arxiv.org/pdf/1106.1813.pdf |
| MAESTRO | Wang, C et al.28 | https://github.com/liulab-dfci/MAESTRO |
| conventional correlation analysis | Stuart, T et al.49 | https://satijalab.org/seurat/ |
| multi-adversarial domain adaptation | Pei et al.24 | https://arxiv.org/pdf/1809.02176.pdf |
| LISI | Korsunsky, I. et al.30 | https://github.com/immunogenomics/harmony |
Resource availability
Lead contact
Resource-related questions may be directed to the lead contact at 08chenfeiwang@tongji.edu.cn.
Materials availability
This study did not generate new unique reagents.
Method details
Reference construction
We have built a semi-automatic data collection and processing workflow based on MAESTRO, which could automatically download and process the scRNA-seq data from public repositories such as GEO, EMBL-EBI, GSA, and HCA. We have applied the workflow to collect 136 normal human scRNA-seq data from 35 tissues, the details for the workflow are as follows.
The semi-automatic workflow has two major components, data processing and unification (Figure S1A). For the data processing components, dataset records in public databases were first automatically crawled based on keywords such as scRNA-seq, single-cell RNA-seq. Only the records that include scRNA-seq data were kept. While different studies provided datasets with various formats, we first unified them into data formats that can be automatically processed using the workflow. Then the workflow performed quality control, normalization, dimension reduction, batch effect removal within each dataset, and clustering analysis. To systematically measure the batch effects, each dataset was quantified with a metric based on information entropy and the Euclidean distance between cell coordinates in the UMAP graph. The information entropy can reflect the complexity of a system, and a higher entropy value means that different batches of cells are mixed more evenly. The entropy was calculated by
where represents the number of batches and represents the proportion of cells belonging to a particular batch among the 30 cells in the neighborhood. The threshold was set based on the ratio of the theoretically maximum entropy in a set of data to the median of all entropy values. If this ratio is larger than 4, it proves that most of the entropy values are distributed on the smaller side, which means that for most of the cells, the surrounding cells are not evenly mixed among different batches. For data with entropy levels below the threshold, the batch effects were removed using the conventional correlation analysis49 (CCA). Specifically, in the annotation step, the cells were either named directly from the original studies to assure authenticity or annotated using the marker genes from the original studies (Table S3). The average logFC of each cell type marker gene in each cell cluster was calculated as the cell type score, and the cell type with the highest score was assigned to that cell cluster. The annotation results were manually validated based on the expression distribution of the marker genes after the automatic annotation.
The second unification component aimed to remove low-quality cells and inconsistent annotations. The labels for the same cell type were first unified using Cell Ontology. For example, pancreatic polypeptide cells and gamma cells were used in different studies and referred to as the same cell type, we unified all of them to pancreatic polypeptide cells according to the literature. Next, the cell types within each tissue were divided into major lineage (for example, DC cell and epithelial cell) and minor lineage (for example, cDC1 cell, cDC2 cell, ciliated cell, and club cell). We take liver endothelial cells as an example, as Figure S1B left panel shows, there are four types of annotations for endothelial cells from the liver, of which three types belong to the minor level and one type belongs to the major level. Data with unclear labels may lead to a lower training accuracy of models, therefore we reassigned sub-lineage annotations to the endothelial cells based on the marker genes that the papers provided. Cells without expression of any known cell type’s marker genes were assigned with major lineage labels attached with the suffix Subset. For each tissue type, the workflow merged all the datasets and performed batch removal with harmony and re-clustering within each tissue. Mislabeled cells, for example, the tissue-specific cells mixed with immune cells, were simply removed from the reference. We deposited the workflow at https://github.com/SELINA-team/SELINA-reference_construction. We intend to maintain our SELINA reference in the long run. Besides, to promote the usage of SELINA, we have also provided an online annotation function for users to annotate their scRNA-seq dataset (http://selina.compbio.cn/#/Annotation).
Data augmentation of rare cell types
SMOTE was applied in the high dimensional space formed by all the genes within the reference data. The minority cell types were defined based on the ratio of cell numbers of them to that of the cell type with the maximum cell number.
The criteria for selecting minority cell types are listed as follows. Let be the cell number of minority cell types, and let be the maximum cell number.
If is within the interval (100,500), the cell types (n < 100) are defined as rare cell types and will be oversampled to 100 cells. If is in the interval (500,1000), then cells types (n < 500) are defined as rare cell types and will be oversampled to 500 cells. If is greater than 1000, then cell types (n < 1000) are defined as rare cell types and will be oversampled to 1000 cells. 1000 is the limit for oversampling cells as excessively oversampling may reduce the quality.
During the synthesis, a cell and its k nearest neighbors from one rare cell type are randomly chosen to generate synthetic cells at random points on the line connecting the anchor cell and the neighbor cells. We denote the gene expression profiles of the anchor cell and one certain neighbor cell as and , respectively. The new cell can be calculated using the following formula:
where is a random number between 0 and 1.
SELINA training with normal data as reference
The pre-training framework is composed of a feature extractor, a cell type discriminator and a platform discriminator (Figure 2C). We denote the feature extractor as and the cell type discriminator as . Suppose we have cell types, as the platform discriminator has an equal number of classifiers to cell types, these classifiers are denoted as , each one is responsible for matching cells from different platforms associated with one cell type. Suppose we have cells, for each cell we have the gene expression vector , we denote the probability of one cell being assigned to each of the K cell types as , which is a vector calculated by
The attention of one certain platform discriminator to one cell is calculated as the corresponding prediction probability , which is used to weight the extracted features of that cell. The cost function of the platform discriminator is calculated as the average loss of all classifiers within it, and the formula is shown as follows:
where is the -th platform discriminator with as its cross-entropy loss and is the platform label. The objective function is the sum of balanced loss from the cell type discriminator and platform discriminator, which is calculated using the formula listed below:
is the cross-entropy loss of the cell type discriminator, is the cell type label, and is a hyperparameter that balances the two objectives in the optimization problem, the formula is as follows:
where represents the index of one specific batch being trained in the current epoch, is the total number of training iterations on the entire sample, is the index of current iteration on the entire sample, is the number of batches in the training set. First, is calculated as a value representing the progress of the training process, ranging from 0 to 1. Then, is calculated using a sigmoid function with a scaling factor of 10. The optimization problem is to find the parameters that simultaneously satisfy
The feature extractor and cell type discriminator of the pre-trained model are connected as the encoder of the autoencoder. The structure of the decoder is symmetrical to that of encoder. We denote the encoder as and the decoder as We first freeze the encoder and train the decoder and then fix the decoder and train the encoder. The objective function is
where is the mean squared error (MSE) loss of the autoencoder. The optimization problem is to find the parameters satisfying Once the fine-tuning step is finished, the decoder is removed, and the encoder is used to predict for unlabeled datasets.
SELINA training with disease data as reference
The disease datasets were collected from published papers (Table S4), annotated with original cell type labels, merged and trained using the adjusted framework described in the following section.
The pre-training framework is expanded to leverage the cell source information. A cell source classifier and a few platform classifiers of which the number is equal to cell sources are added to the pre-training framework (Figure S6A). The platform classifiers now can be categorized into two types, one pairs with cell source classifier and the other pairs with cell type classifier. Besides, the cost function was adjusted as:
where , , represent numbers of cell sources, cell types and cells in training data respectively, , , and represent loss of cell source discriminator (), cell type discriminator (), the -th platform classifier () responsible for -th cell source and the -th platform classifier () responsible for -th cell type respectively. , , , , and represent prediction results and true label for cell source, cell type, and platform label for the -th cell respectively. Besides, the fine-tuning step was removed when SELINA taking the disease data as reference.
Model parameters
The neural network was implemented with PyTorch (Figure S2C). The feature extractor contains three layers: one input layer with the same number of nodes as the gene number and one output layer with 100 nodes followed by a dropout layer. The cell type discriminator has four layers: a 100-node input layer, a 50-node hidden layer followed by a dropout layer, and an output layer with a number of nodes equal to cell types. Each platform discriminator unit contains three layers: a 100-node input layer, a 25-node hidden layer, and an output layer with a number of nodes equal to sequencing platforms. The output layer of feature extractor and input layer of cell type discriminator, cell source discriminator and platform classifiers were expanded to 500 nodes when training with disease data.
The Rectified Linear Units (ReLU) was used as activation function. The Adam optimizer was used as the optimizer with default settings. The learning rates of pre-training and encoder training were set to 0.0001. For decoder training, the learning rate was set to 0.0005. The epoch numbers of pre-training and decoder training were set to 50, whereas the epoch number of encoder training was set to 20. The dropout layers' parameters were set to default.
Calculation of LISI score
LISI was proposed to quantitively assess the mixing degree of samples from different batches.30 LISI first selects an anchor cell and assigns each cell in the neighborhood a weight based on the Euclidean distance to the anchor cell. The weight is calculated using Gaussian kernel-based distribution with a fixed perplexity (30), the formula is as follows:
is the weight of the n-th neighbor cell, and are the coordinates of the anchor cell and the n-th neighbor cell, is the parameter of the Gaussian kernel-based distribution which can be inferred using the perplexity, and represents the total number of selected neighbor cells. The weights are used to calculate Inverse Simpson Index that represents the expected number of cells need to be sampled if cells from one batch are observed twice. The Inverse Simpson Index is calculated as:
represents the number of neighbor cells belonging to one specific batch and represents the total number of batches.
Benchmark of SELINA and existing tools
For all datasets in each tissue, we iteratively took one dataset as query data and merged the remaining datasets as a reference dataset. The extremely large datasets that consume vast amounts of memory were downsampled with all cell types intact. The expression profiles of the reference data and query data were scaled to 10000 and log-transformed. The querying process used common genes between query and reference data. The annotations of each tested dataset were from the minor lineage. Different methods have different ways of identifying unknown cell types and different thresholds for the unknown cell types, and some of them even do not have the settings to identify unknown cell types. For a fair comparison, we set a threshold of 0 for all the methods, which means that all cells will be forced to be given an annotation label, and there is no unknown type of cells for all the methods. For all benchmarks, scmap, singleR, SingleCellNet, scibet and CelliD were trained and tested with CPU AMD EPYC 7552 2.2 GHz. SELINA, ACTINN, mtSC and Cell BLAST, which are deep learning-based frameworks, were trained with GPU GTX960 and tested with AMD EPYC 7552 2.2 GHz. All the parameters were the defaults or set as recommended in the corresponding documentations.
The accuracy was defined as the ratio of corrected assigned cells over all cells. The MacroF1 score was calculated as listed below:
K represents the number of cell types in the query dataset. and are the precision and recall of the k-th cell type.
Calculation of overlapping score
The overlapping score represents the degree of overlap of cell types between different datasets. For each tissue, we selected one dataset as the query dataset and found the common cell types between the query dataset and all the other datasets. The corresponding overlapping ratio for this dataset was calculated as the proportion of cells from the shared cell types. This process was repeated until all the datasets had been taken as query dataset, and the overlapping ratio of this tissue was the averaged ratio of all datasets.
Quantification and statistical analysis
Count data normalization
In reference construction, feature counts for each cell are divided by the total counts for that cell firstly. After scaling, the values are natural-log transformed using log1p to stabilize the data and reduce the impact of outliers. The count data normalization was implemented using the NormalizeData function in the R package MAESTRO.
Pairwise wilcoxon rank-sum test
In Figures 3C, 3G and 3H, we compared p values using the one-sided pairwise Wilcoxon rank-sum test. Significance levels were indicated by asterisks, where ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. The significance calculation was implemented using the geom_signif function in the R package ggsignif.50
Acknowledgments
This work was supported by the National Key R&D Program of China (2022YFA1106000), the National Natural Science Foundation of China (32222026, 32170660, 92168205, 81872290, 81972551, 81991502), Shanghai Rising Star Program (21QA1408200), Natural Science Foundation of Shanghai (21ZR1467600), Natural Science Foundation of Sichuan Province (2022NSFSC0054), and the Young Elite Scientist Sponsorship Program by CAST (2021QNRC001).
Author contributions
C.W., T.L., and J.Z. conceived the project. P.R. designed the SELINA algorithm and evaluated the performance with X.S. X.S., X.D., Z.Y., X.X.D., J.W., Y.Y., and J.H. collected the data. X.S. processed the data with help from P.R., Z.Y., and X.X.D., and X.S. cleaned and built the SELINA reference. P.R., X.S., J.Z., T.L., and C.W. wrote the manuscript with help from the other authors. C.W. supervised the whole project.
Declaration of interests
The authors declare no competing interests.
Published: August 31, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2023.100577.
Contributor Information
Jing Zhang, Email: zhangjing@tongji.edu.cn.
Taiwen Li, Email: litaiwen@scu.edu.cn.
Chenfei Wang, Email: 08chenfeiwang@tongji.edu.cn.
Supplemental information
Data and code availability
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table. The pre-trained models covering 136 human datasets are available from https://github.com/wanglabtongji/SELINA-reference. R version of SELINA and the automatic data collection workflow, as well as the benchmark code used in the analyses, can be found at https://github.com/SELINA-team/.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Pliner H.A., Shendure J., Trapnell C. Supervised classification enables rapid annotation of cell atlases. Nat. Methods. 2019;16:983–986. doi: 10.1038/s41592-019-0535-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Z., Luo D., Zhong X., Choi J.H., Ma Y., Wang S., Mahrt E., Guo W., Stawiski E.W., Modrusan Z., et al. SCINA: A Semi-Supervised Subtyping Algorithm of Single Cells and Bulk Samples. Genes. 2019;10 doi: 10.3390/genes10070531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiselev V.Y., Yiu A., Hemberg M. scmap: projection of single-cell RNA-seq data across data sets. Nat. Methods. 2018;15:359–362. doi: 10.1038/nmeth.4644. [DOI] [PubMed] [Google Scholar]
- 4.Alquicira-Hernandez J., Sathe A., Ji H.P., Nguyen Q., Powell J.E. scPred: accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol. 2019;20:264. doi: 10.1186/s13059-019-1862-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R., et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Kanter J.K., Lijnzaad P., Candelli T., Margaritis T., Holstege F.C.P. CHETAH: a selective, hierarchical cell type identification method for single-cell RNA sequencing. Nucleic Acids Res. 2019;47:e95. doi: 10.1093/nar/gkz543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan Y., Cahan P. SingleCellNet: A Computational Tool to Classify Single Cell RNA-Seq Data Across Platforms and Across Species. Cell Syst. 2019;9:207–213.e2. doi: 10.1016/j.cels.2019.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma F., Pellegrini M. ACTINN: automated identification of cell types in single cell RNA sequencing. Bioinformatics. 2020;36:533–538. doi: 10.1093/bioinformatics/btz592. [DOI] [PubMed] [Google Scholar]
- 9.Duan B., Chen S., Chen X., Zhu C., Tang C., Wang S., Gao Y., Fu S., Liu Q. Integrating multiple references for single-cell assignment. Nucleic Acids Res. 2021;49:e80. doi: 10.1093/nar/gkab380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao Z.-J., Wei L., Lu S., Yang D.-C., Gao G. Searching large-scale scRNA-seq databases via unbiased cell embedding with Cell BLAST. Nat. Commun. 2020;11:3458–3513. doi: 10.1038/s41467-020-17281-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernstein M.N., Ma Z., Gleicher M., Dewey C.N. CellO: Comprehensive and hierarchical cell type classification of human cells with the Cell Ontology. iScience. 2021;24 doi: 10.1016/j.isci.2020.101913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shao X., Liao J., Lu X., Xue R., Ai N., Fan X. scCATCH: automatic annotation on cell types of clusters from single-cell RNA sequencing data. iScience. 2020;23 doi: 10.1016/j.isci.2020.100882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hou R., Denisenko E., Forrest A.R.R. scMatch: a single-cell gene expression profile annotation tool using reference datasets. Bioinformatics. 2019;35:4688–4695. doi: 10.1093/bioinformatics/btz292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shao X., Yang H., Zhuang X., Liao J., Yang P., Cheng J., Lu X., Chen H., Fan X. scDeepSort: a pre-trained cell-type annotation method for single-cell transcriptomics using deep learning with a weighted graph neural network. Nucleic Acids Res. 2021;49:e122. doi: 10.1093/nar/gkab775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu C., Prete M., Webb S., Jardine L., Stewart B., Hoo R., He P., Teichmann S.A. Automatic cell type harmonization and integration across Human Cell Atlas datasets. bioRxiv. 2023 doi: 10.1101/2023.05.01.538994. Preprint at. [DOI] [PubMed] [Google Scholar]
- 16.Abdelaal T., Michielsen L., Cats D., Hoogduin D., Mei H., Reinders M.J.T., Mahfouz A. A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol. 2019;20:194. doi: 10.1186/s13059-019-1795-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regev A., Teichmann S.A., Lander E.S., Amit I., Benoist C., Birney E., Bodenmiller B., Campbell P., Carninci P., Clatworthy M., et al. The Human Cell Atlas. Elife. 2017;6 doi: 10.7554/eLife.27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao Z.J., Wei L., Lu S., Yang D.C., Gao G. Searching large-scale scRNA-seq databases via unbiased cell embedding with Cell BLAST. Nat. Commun. 2020;11:3458. doi: 10.1038/s41467-020-17281-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding J., Adiconis X., Simmons S.K., Kowalczyk M.S., Hession C.C., Marjanovic N.D., Hughes T.K., Wadsworth M.H., Burks T., Nguyen L.T., et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020;38:737–746. doi: 10.1038/s41587-020-0465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han X., Zhou Z., Fei L., Sun H., Wang R., Chen Y., Chen H., Wang J., Tang H., Ge W., et al. Construction of a human cell landscape at single-cell level. Nature. 2020;581:303–309. doi: 10.1038/s41586-020-2157-4. [DOI] [PubMed] [Google Scholar]
- 21.Papatheodorou I., Moreno P., Manning J., Fuentes A.M.P., George N., Fexova S., Fonseca N.A., Füllgrabe A., Green M., Huang N., et al. Expression Atlas update: from tissues to single cells. Nucleic Acids Res. 2020;48:D77–D83. doi: 10.1093/nar/gkz947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi X., Yu Z., Ren P., Dong X., Ding X., Song J., Zhang J., Li T., Wang C. HUSCH: an integrated single-cell transcriptome atlas for human tissue gene expression visualization and analyses. Nucleic Acids Res. 2023;51:D1029–D1037. doi: 10.1093/nar/gkac1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chawla N.V., Bowyer K.W., Hall L.O., Kegelmeyer W.P. SMOTE: Synthetic Minority Over-sampling Technique. J. Artif. Intell. Res. 2002;16:321–357. doi: 10.1613/jair.953. [DOI] [Google Scholar]
- 24.Pei Z.Y., Cao Z.J., Long M.S., Wang J.M. 2018. Multi-Adversarial Domain Adaptation. Thirty-Second AAAI Conference on Artificial Intelligence. [Google Scholar]
- 25.Tasic B., Yao Z., Graybuck L.T., Smith K.A., Nguyen T.N., Bertagnolli D., Goldy J., Garren E., Economo M.N., Viswanathan S., et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature. 2018;563:72–78. doi: 10.1038/s41586-018-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hodge R.D., Bakken T.E., Miller J.A., Smith K.A., Barkan E.R., Graybuck L.T., Close J.L., Long B., Johansen N., Penn O., et al. Conserved cell types with divergent features in human versus mouse cortex. Nature. 2019;573:61–68. doi: 10.1038/s41586-019-1506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bakken T.E., Jorstad N.L., Hu Q., Lake B.B., Tian W., Kalmbach B.E., Crow M., Hodge R.D., Krienen F.M., Sorensen S.A., et al. Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature. 2021;598:111–119. doi: 10.1038/s41586-021-03465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang C., Sun D., Huang X., Wan C., Li Z., Han Y., Qin Q., Fan J., Qiu X., Xie Y., et al. Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol. 2020;21:198. doi: 10.1186/s13059-020-02116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jupp S., Burdett T., Leroy C., Parkinson H.E. 2015. A New Ontology Lookup Service at EMBL-EBI. [Google Scholar]
- 30.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ganin Y., Ustinova E., Ajakan H., Germain P., Larochelle H., Laviolette F., Marchand M., Lempitsky V.S. 2016. Domain-Adversarial Training of Neural Networks. [Google Scholar]
- 32.MacParland S.A., Liu J.C., Ma X.Z., Innes B.T., Bartczak A.M., Gage B.K., Manuel J., Khuu N., Echeverri J., Linares I., et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018;9:4383. doi: 10.1038/s41467-018-06318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramachandran P., Dobie R., Wilson-Kanamori J.R., Dora E.F., Henderson B.E.P., Luu N.T., Portman J.R., Matchett K.P., Brice M., Marwick J.A., et al. Resolving the fibrotic niche of human liver cirrhosis at single cell level. Nature. 2019;575:512–518. doi: 10.1038/s41586-019-1631-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McInnes L., Healy J., Saul N., Großberger L., Flberger L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018;3:861. [Google Scholar]
- 35.Li C., Liu B., Kang B., Liu Z., Liu Y., Chen C., Ren X., Zhang Z. SciBet as a portable and fast single cell type identifier. Nat. Commun. 2020;11:1818. doi: 10.1038/s41467-020-15523-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cortal A., Martignetti L., Six E., Rausell A. Gene signature extraction and cell identity recognition at the single-cell level with Cell-ID. Nat. Biotechnol. 2021;39:1095–1102. doi: 10.1038/s41587-021-00896-6. [DOI] [PubMed] [Google Scholar]
- 37.Tabula Sapiens Consortium∗. Jones R.C., Karkanias J., Krasnow M.A., Pisco A.O., Quake S.R., Salzman J., Yosef N., Bulthaup B., Brown P., et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022;376 doi: 10.1126/science.abl4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Domínguez Conde C., Xu C., Jarvis L.B., Rainbow D.B., Wells S.B., Gomes T., Howlett S.K., Suchanek O., Polanski K., King H.W., et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science. 2022;376 doi: 10.1126/science.abl5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eraslan G., Drokhlyansky E., Anand S., Fiskin E., Subramanian A., Slyper M., Wang J., Van Wittenberghe N., Rouhana J.M., Waldman J., et al. Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science. 2022;376 doi: 10.1126/science.abl4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suo C., Dann E., Goh I., Jardine L., Kleshchevnikov V., Park J.-E., Botting R.A., Stephenson E., Engelbert J., Tuong Z.K., et al. Mapping the developing human immune system across organs. Science. 2022;376:eabo0510. doi: 10.1126/science.abo0510. eabo0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Köhler N.D., Büttner M., Andriamanga N., Theis F.J. Deep learning does not outperform classical machine learning for cell-type annotation. bioRxiv. 2021 doi: 10.1101/653907. Preprint at. [DOI] [Google Scholar]
- 42.Goodfellow I.J., Pouget-Abadie J., Mirza M., Xu B., Warde-Farley D., Ozair S., Courville A.C., Bengio Y. 2014. Generative Adversarial Nets. [Google Scholar]
- 43.Kingma D.P., Welling M. Vol. 1312. 2014. Auto-Encoding Variational Bayes. CoRR abs/ p. 6114. [Google Scholar]
- 44.Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H., Satija R., Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peterson V.M., Zhang K.X., Kumar N., Wong J., Li L., Wilson D.C., Moore R., McClanahan T.K., Sadekova S., Klappenbach J.A. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 2017;35:936–939. doi: 10.1038/nbt.3973. [DOI] [PubMed] [Google Scholar]
- 46.Bardou P., Mariette J., Escudié F., Djemiel C., Klopp C. jvenn: an interactive Venn diagram viewer. BMC Bioinf. 2014;15:1–7. doi: 10.1186/1471-2105-15-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma L., Zheng J. Single-cell gene expression analysis reveals β-cell dysfunction and deficit mechanisms in type 2 diabetes. BMC Bioinf. 2018;19:515–548. doi: 10.1186/s12859-018-2519-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao Z.-J., Gao G. Multi-omics single-cell data integration and regulatory inference with graph-linked embedding. Nat. Biotechnol. 2022;40:1458–1466. doi: 10.1038/s41587-022-01284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahlmann-Eltze C., Patil I. PsyArxiv; 2021. Ggsignif: R Package for Displaying Significance Brackets for 'ggplot2. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table. The pre-trained models covering 136 human datasets are available from https://github.com/wanglabtongji/SELINA-reference. R version of SELINA and the automatic data collection workflow, as well as the benchmark code used in the analyses, can be found at https://github.com/SELINA-team/.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.