

Abstract
The search for selective opioid ligands with desired pharmacological potency and improved safety profile has always been an area of interest. Our previous effort yielded a potent opioid modulator, NAN, a 6α-N-7′-indolyl-substituted naltrexamine derivative, which exhibited promising pharmacological activities both in vitro and in vivo. However, significant human ether-a-go-go-related gene (hERG) liability limited its further development. Therefore, a systematic structural modification on NAN was conducted in order to alleviate hERG toxicity while preserving pharmacological properties, which led to the discovery of 2′-methylindolyl derivative compound 21. Compared to NAN, compound 21 manifested overall improved pharmacological profiles. Follow-up hERG channel inhibition evaluation revealed a seven-fold decreased potency of compound 21 compared to NAN. Furthermore, several fundamental drug-like property evaluations suggested a reasonable ADME profile of 21. Collectively, compound 21 appeared to be a promising opioid modulator for further development as a novel therapeutic agent toward opioid use disorder treatments.
Graphical Abstract

INTRODUCTION
Currently, the United States is in the midst of an opioid crisis because of illicitly non-medical use of opioids. The latest statistics show that the number of drug overdose deaths was four times higher in 2018 than in 1999.1 Meanwhile, in 2018, the United States alone reported 128 deaths per day due to opioid overdose.2 It is also important to note that approximately 2.1 million Americans live with opioid use disorder (OUD) nowadays,3 which not only negatively affects public health but also poses tremendous welfare burden on our society and economy. In an attempt to address OUD, a considerable number of opioid ligands with antagonistic properties have been developed over past decades. Among them, the mu opioid receptor (MOR) ligands possessing high binding selectivity and low receptor efficacy have shown prominence because of their pivotal roles in treating OUD, for example, buprenorphine. Despite this encouraging progress, some of them still suffer unwanted side effects and potential toxicity such as sedation, vomiting, nausea, and respiratory depression, which have in a large part resulted in low patient compliance and in turn preventing proper prescribing.4–6 Therefore, novel MOR ligands with high binding affinity and low efficacy as well as improved drug-like properties are still in high demand.
Through years of continuous efforts to develop selective opioid receptor ligands, we have identified several lead compounds with highly promising potency and selectivity by incorporating various heteroaromatic ring systems onto the C6-position of the epoxymorphinan skeleton, largely based on the “message-address” concept.7–14 Most recently, we described the discovery of a potent MOR modulator, 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(indole-7-carboxamido)morphinan (NAN), via employing an indole moiety as an address portion (Figure 1).13,15 Further computational chemistry and modeling studies suggested that NAN may act as a bitopic MOR ligand, that is, its “message” portion or the epoxymorphinan moiety may interact with the MOR orthosteric site, while the indole side chain, referred to as the “address” portion, may bind to the allosteric binding site of the MOR. Following in vivo assays revealed that NAN did not induce significant withdrawal signs in morphine-pelleted mice and neither did elicit fentanyl-like discriminative stimulus effects in rats.13,16 We reasoned that these favorable pharmacological profiles of NAN could be due to the combination of the advantages of orthosteric and allosteric ligands as proposed previously.17–21 In all, these results demonstrated the potential of NAN as a candidate to develop novel therapeutic agents in the treatment of OUD.
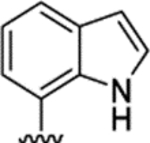
Figure 1.

Chemical structure of MOR-targeted bitopic ligand NAN.
With these promising results in hand, we thus evaluated the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of NAN in an effort to assess its potential for further development. These follow-up studies revealed that NAN possessed several favorable profiles (data not shown). However, it was also observed that NAN manifested high inhibition of human ether-a-go-go related gene (hERG) channel with an IC50 value of 0.021 μM. This may potentially lead to drug-induced long QT syndrome (LQTS) with the increased risks of cardiac arrest,22–24 making it a less than ideal candidate for further development. In terms of this, we decided to conduct further optimization in order to overcome the hERG liability of NAN.
The blockade of the hERG channel has emerged as a significant hurdle in early drug discovery campaigns. Over years, enormous efforts have been prompted in attenuating hERG affinity of drug candidates.25,26 Of these proposed strategies and tactics, control of cLogP is regarded to be particularly important to avoid hERG activity, through which a considerable number of successful optimizations have been previously reported.25,27–29 Some empirical guidelines are also proposed for the reduction of unwanted hERG activity. For example, if clogP < 3.0, subtle structural modifications may offer the highest probability of success; if clogP ≥ 3.0, reduction of cLogP by incorporation of heteroatoms or polar groups or removal of lipophilic functions could be a preferable approach.26,30 Nevertheless, the distinctive nature of the hERG channel renders the rational circumvention of liability while simultaneously optimizing compounds for desirable pharmacological efficacy and safety profile, a challenging undertaking.
Meanwhile, designing therapeutic agents that possess a potential central nervous system (CNS) acting profile is one of the most challenging endeavors in drug discovery.31–33 Several physicochemical properties have long been proven to be critical to achieve optimal CNS exposure, mainly including lipophilicity (cLogP), ionization state (pKa), molecular weight (MW), topological polar surface area (tPSA), hydrogen bond donor count (HBD), and hydrogen bond acceptor count (HBA).31 Based on several retrospective analyses of required physicochemical properties for optimal brain exposure in marked CNS drugs and clinical candidates, a guideline has been proposed as a useful aid for designing molecules with desirable blood–brain barrier (BBB) permeability (e.g., cLogP 2–5; cLogD 2–5; pKa 7.5–10.5).30,33,34 Of note, while the BBB holds highly restrictive nature via varying passive and active mechanisms to support CNS homeostasis, multiple components in the interactions between a drug and BBB have positively or negatively contributed to its permeability.32 As a consequence, even subtle structural changes of a drug can cause substantial permeability changes, and in some cases, drugs with similar functions were not equipotent to penetrate the BBB.35,36
Herein, we report our most recent work on the systematic structural modification of NAN focusing on the 2′ and 3′ positions of its indole side chain. A total of 34 new ligands were synthesized, structurally characterized, and biologically evaluated, and one novel compound was identified with overall improved drug-like profiles compared to NAN warranting further characterization and optimization.
RESULTS AND DISCUSSION
Molecular Design.
To guide the structural optimization of NAN, we first calculated its physicochemical parameters by using ACD/Percepta (v2020.2.0) software,37 which were found to be well within the aforementioned range for reasonable BBB permeability (cLogP = 2.62, cLogD = 1.58, and pKa = 7.96). Such a predicted centrally acting profile was also corroborated by our previously reported in vivo study results.13,15 Considering the fact that NAN possessed a cLogP value less than 3, we hypothesized subtle structural modifications, particularly for its address portion that confers receptor selectivity and functionality, may possibly fine-tune its physicochemical property to reduce the hERG liability without compromising its pharmacological efficacy.
Based on the previous molecular modeling studies of NAN at the MOR, the indole ring of NAN, namely the address portion, may form hydrophobic interactions with the allosteric binding site of the MOR, formed by residues L219ECL2, F221ECL2, E2295.35, L2325.38, and K2335.39.13,15 The structural analysis of the binding modes indicated that this binding site appeared to be able to accommodate a relatively larger address portion than that of NAN. Meanwhile, it was also observed that the C2 and C3 of the indole ring pointed straightly into the binding pocket, making them the preferable sites for structural modifications. Based on these observations, we thus decided to initiate our exploration by modifying the 2′ and 3′ positions of the indole side chain.
To cover the structural variability of the substituents at these two positions as much as possible, different functional groups with a broad range of the partition constant π, the Hammett substituent constant σ, and the resonance constant ER values from all the quadrants of the Craig plots were chosen as the substituents.38 Moreover, steric properties of substituents were also considered in our molecular design seeking to investigate the contributions of substituent size increment to receptor selectivity profiles of the ligands. Furthermore, in light of the fact that the stereochemistry at the C6-position of the epoxymorphinan skeleton appeared to bring significant influence on the pharmacological profiles of opioid ligands,39 analogues with either an α or a β amide linkage were synthesized and biologically evaluated for comparative purposes (Figure 2).
Figure 2.

Schematic representative of NAN derivatives (compounds 1–34) explored in this study.
Additionally, the physicochemical parameters of newly designed compounds were predicted by ACD/Percepta (v2020.2.0) (Table S1). It was found that all relevant parameters of the newly designed ligands were in line with previously proposed values for desirable BBB permeability.33,34 (cLogP = 2.21–4.44, cLogD = 1.18–3.82, and pKa = 7.5–8.0).
Chemical Synthesis.
As discussed in the previous section, the proposed structural modification of NAN mainly focused on the indole moiety while keeping the epoxymorphinan skeleton intact. Therefore, we commenced our study with the synthesis of 2′ or 3′-substituted indole-7-carboxylic acids. Although the synthesis of indole and its derivatives have been extensively investigated in the past decades, there are very limited reports on the synthesis of substituted indole-7-carboxylic acids according to our literature search, particularly for the analogues bearing substituents on the 2′ or 3′ positions. Hence, considering previously described procedures and other reported similar functional group transformations as well as synthetic challenges, 16 2′ or 3′-substituted indole-7-carboxylic acids, including 12 new compounds, were in-house synthesized and structurally characterized (Schemes 1 and 2, see details in Supporting Information). Briefly, commercially available methyl oxindole-7-carboxylate was employed as the start material to prepare the 2′-substituted carboxylic acids, while the 3′-substituted carboxylic acids were furnished by using methyl indole-7-carboxylate as start material followed by electrophilic substitution reactions and other functional transformations. Additionally, 6α- and 6β-naltrexamine were obtained via a modified stereoselective method.40 Subsequently, all prepared indole-7-carboxylic acids were coupled with 6α- or 6β-naltrexamine. After stirring for 2 days, all solvent was removed under vacuum, and the resulted crude products were then treated with K2CO3 in methanol (Scheme 3). After purification by flash column chromatography (FCC), the title free bases were obtained in reasonable yields, which were finally converted to their hydrochloride salts before being submitted to further biological evaluation.
Scheme 1.

Synthesis of 2′-Substituted Indole-7-Carboxylic Acids for the Newly Designed NAN Derivatives: (a) POBr3, Imidazole, DCE, 80%; (b) LiOH, THF/H2O, 34–99%; (c) POCl3, Diethylaniline, Toluene, 90%; (d) Pd(t-Bu3P)2, Zn(CN)2, Zn(TFA)2, K3PO4, Toluene/DMA, 61%; and (e) Phenylboronic Acid, Pd(PPh3)4, Na2CO3, Toluene/EtOH/H2O, 67%
Scheme 2.

Synthesis of 3′-Substituted Indole-7-Carboxylic Acids for the Newly Designed NAN Derivatives: (a) NBS, DCE, −20 °C, 72%; (b) LiOH, THF/H2O, 68–98%; (c) NCS, MeOH, 73%; (d) AgNO3, BzCl, ACN, 33%; (e) POCl3, DMF, 70%; (f) Hydroxylamine Hydrochloride, DMF, 110 °C, 45%; (g) Pd(OH)2/C, H2, THF/MeOH, 70%; (h) Acetone/Cyclopentanone/Cyclohexanone, Et3SiH, Cl3CCOOH, Toluene, 41–75%; (i) (Boc)2O, DMAP, TEA, DCM, 80%; (j) Phenylboronic Acid, Pd(dppf)Cl2, Na2CO3, THF/H2O, 82%; (k) TFA, DCM, 99%; (l) POCl3, DMA, 20%; (m) Hydroxylamine Hydrochloride, NaOAc, EtOH/H2O, 93%; and (n) CH3COOH, Reflux, 76%
Scheme 3.

Synthetic Routes of NAN Derivatives 1–34
In Vitro Pharmacological Studies.
We first subjected all synthesized NAN derivatives to radioligand competition binding assay to evaluate their binding affinity and selectivity on the three opioid receptors as reported previously,14 in which monoclonal mouse opioid receptor-expressed Chinese hamster ovary (CHO) cell lines were adopted. Afterward, [35S]-GTPγS assay was conducted to assess whether every target compound acted as an agonist, partial agonist, or antagonist at the MOR by measuring its efficacy relative to a full agonist DAMGO. The binding affinity, selectivity, potency, and efficacy data of 6β-configuration NAN derivatives are summarized in Table 1, and the results of 6α-configuration NAN derivatives are summarized in Table 2.
Table 1.
Binding Affinity, Selectivity, and MOR [35S]-GTPγS Functional Assay Results for NAN Derivatives with 6β-Configuration

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compds | -R | Ki (nM) | Selectivity | MOR [35S]-GTPγS binding | ||||
| MOR | KOR | DOR | κ/μ | δ/μ | EC50 (nM) | % Emax of DAMGO | ||
| NLX a | - | 0.79 ± 0.02 | 1.1 ± 0.03 | 76 ± 2 | 1.0 | 69 | ND | 13 ± 1 |
| NTX b | - | 0.26 ± 0.02 | 117.1 ± 8.9 | 5.15 ± 0.26 | 20 | 450 | ND | 7.75 ± 0.20 |
| 1 |

|
0.35 ± 0.01 | 0.46 ± 0.04 | 3.04 ± 0.27 | 1.3 | 8.8 | 0.88 ± 0.05 | 36.19 ± 2.11 |
| 2 |

|
0.63 ± 0.06 | 1.28 ± 0.22 | 2.42 ± 0.44 | 2.0 | 3.8 | 1.40 ± 0.15 | 38.52 ± 2.02 |
| 3 |

|
0.73 ± 0.09 | 1.66 ± 0.19 | 12.05 ± 0.62 | 2.3 | 16.5 | 3.78 ± 0.16 | 21.53 ± 1.09 |
| 4 |

|
0.31 ± 0.07 | 0.80 ± 0.15 | 15.89 ± 0.38 | 2.6 | 51.1 | 1.51 ± 0.22 | 21.79 ± 1.44 |
| 5 |

|
3.26 ± 0.17 | 1.14 ± 0.03 | 23.85 ± 3.96 | 0.4 | 7.3 | 5.44 ± 1.98 | 10.19 ± 1.01 |
| 6 |

|
0.70 ± 0.05 | 1.08 ± 0.14 | 11.61 ± 2.68 | 1.6 | 16.7 | 1.05 ± 0.08 | 70.40 ± 1.26 |
| 7 |

|
0.62 ± 0.08 | 0.33 ± 0.05 | 4.43 ± 1.06 | 0.5 | 7.2 | 1.05 ± 0.23 | 59.51 ± 5.14 |
| 8 |

|
0.38 ± 0.04 | 1.00 ± 0.33 | 25.40 ± 3.39 | 2.7 | 67.4 | 3.07 ± 0.87 | 38.84 ± 0.36 |
| 9 |

|
0.54 ± 0.07 | 0.80 ± 0.13 | 25.62 ± 8.06 | 1.5 | 47.5 | 1.13 ± 0.27 | 60.87 ± 1.94 |
| 10 |

|
0.69 ± 0.04 | 4.03 ± 0.85 | 24. 28 ± 4.07 | 5.8 | 35.1 | 1.19 ± 0.49 | 21.33 ± 1.85 |
| 11 c |

|
>50 | 15.57 ± 1.68 | 81.13 ± 9.98 | <0.3 | < 1.6 | 203.99 ± 77.95 | 10.88 ± 2.39 |
| 12 |

|
0.27 ± 0.04 | 6.52 ± 0.73 | 31.04 ± 4.96 | 24.4 | 116.3 | 0.88 ± 0.09 | 8.84 ± 0.79 |
| 13 |

|
0.46 ± 0.04 | 1.01 ± 0.25 | 27.92 ± 5.93 | 2.2 | 61.3 | 1.22 ± 0.18 | 43.95 ± 1.48 |
| 14 |

|
0.89 ± 0.09 | 0.98 ± 0.19 | 19.75 ± 2.72 | 1.1 | 22.2 | 1.73 ± 0.40 | 36.23 ± 2.83 |
| 15 |

|
1.45 ± 0.11 | 10.84 ± 3.42 | 9.88 ± 1.13 | 7.5 | 6.8 | 2.28 ± 0.59 | 21.32 ± 1.71 |
| 16 |

|
5.57 ± 0.64 | 41.74 ± 7.38 | 63.99 ± 1.99 | 7.5 | 11.5 | 20.84 ± 4.47 | 6.23 ± 0.54 |
| 17 |

|
1.46 ± 0.16 | 26.06 ± 2.39 | 49.33 ± 10.14 | 17.9 | 33.8 | 1.78 ± 0.79 | 8.47 ± 0.32 |
Table 2.
Binding Affinity, Selectivity, and MOR [35S]-GTPγS Functional Assay Results for NAN Derivatives with 6α-Configuration

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compds | -R | Ki (nM) | Selectivity | MOR [35S]-GTPγS binding | ||||
| MOR | KOR | DOR | κ/μ | δ/μ | EC50 (nM) | % Emax of DAMGO | ||
| NLX a | - | 0.79 ± 0.02 | 1.1 ± 0.03 | 76 ± 2 | 1.0 | 69 | ND | 13 ± 1 |
| NTX b | - | 0.26 ± 0.02 | 117.1 ± 8.9 | 5.15 ± 0.26 | 20 | 450 | ND | 7.75 ± 0.20 |
| NAN c |
|
0.23 ± 0.02 | 1.69 ± 0.35 | 13.69 ± 1.39 | 7.3 | 59.5 | 3.85 ± 2.32 | 19.11 ± 3.31 |
| 18 |

|
0.48 ± 0.04 | 1.50 ± 0.12 | 6.73 ± 1.00 | 3.1 | 14.0 | 2.36 ± 0.70 | 13.35 ± 1.89 |
| 19 |

|
0.65 ± 0.05 | 2.73 ± 0.45 | 1.82 ± 0.06 | 4.2 | 2.8 | 1.82 ± 0.26 | 14.80 ± 0.40 |
| 20 |

|
0.87 ± 0.13 | 7.10 ± 1.65 | 10.65 ± 0.93 | 8.2 | 12.2 | 3.58 ± 0.38 | 14.38 ± 1.41 |
| 21 |

|
0.59 ± 0.03 | 2.10 ± 0.27 | 16.01 ± 1.09 | 3.6 | 27.3 | 3.86 ± 1.25 | 12.04 ± 1.87 |
| 22 |

|
2.45 ± 0.11 | 6.42 ± 1.57 | 5.36 ± 1.00 | 2.6 | 2.2 | 2.07 ± 0.13 | 7.44 ± 1.11 |
| 23 |

|
0.93 ± 0.13 | 5.28 ± 0.36 | 19.19 ± 3.99 | 5.7 | 20.6 | 2.46 ± 0.59 | 14.93 ± 0.55 |
| 24 |

|
0.98 ± 0.20 | 5.96 ± 0.48 | 37.13 ± 9.46 | 6.1 | 37.9 | 3.67 ± 0.68 | 13.22 ± 0.87 |
| 25 |

|
0.87 ± 0.08 | 5.46 ± 0.92 | 12.18 ± 2.2 | 6.3 | 14.0 | 2.54 ± 0.50 | 11.45 ± 0.61 |
| 26 |
|
1.07 ± 0.20 | 9.84 ± 0.74 | 21.60 ± 2.57 | 9.2 | 20.3 | 2.32 ± 0.23 | 11.27 ± 0.25 |
| 27 |

|
0.25 ± 0.04 | 2.82 ± 0.5 | 11.90 ± 1.91 | 11.3 | 47.5 | 0.89 ± 0.17 | 8.24 ± 0.58 |
| 28 |

|
0.43 ± 0.03 | 6.39 ± 1.07 | 10.88 ± 1.58 | 14.9 | 25.4 | 5.21 ± 0.48 | 9.81 ± 0.89 |
| 29 |

|
0.23 ± 0.02 | 2.10 ± 0.38 | 5.19 ± 0.35 | 9.4 | 23.0 | 1.48 ± 0.16 | 9.97 ± 0.24 |
| 30 |

|
0.69 ± 0.09 | 4.20 ± 0.90 | 13.81 ± 4.02 | 6.1 | 20.1 | 2.68 ± 0.89 | 15.65 ± 1.76 |
| 31 |

|
2.78 ± 0.17 | 11.02 ± 1.66 | 25.87 ± 5.69 | 4.0 | 9.3 | 2.48 ± 0.73 | 8.37 ± 1.66 |
| 32 |

|
2.66 ± 0.49 | 21.11 ± 2.89 | 28.23 ± 5.40 | 7.9 | 10.6 | 2.05 ± 0.68 | 12.35 ± 1.06 |
| 33 |

|
2.11 ± 0.17 | 9.65 ± 1.82 | 18.63 ± 7.24 | 4.6 | 8.9 | 2.62 ± 0.14 | 20.53 ± 0.13 |
| 34 |

|
2.37 ± 0.23 | 13.39 ± 1.28 | 15.02 ± 2.71 | 5.7 | 6.3 | 1.85 ± 0.05 | 5.57 ± 0.09 |
As seen in Table 1, compounds 1–5 with substituents at 2′-position of the indole ring showed subnanomolar affinity for the MOR and kappa opioid receptor (KOR) while having a lower affinity at the delta opioid receptor (DOR) except for compound 5 which bound to the MOR with nanomolar affinity. The noticeably decreased binding affinity at the MOR as seen for compound 5 could be due to an impaired aromatic stacking interaction between ligand and MOR allosteric binding site caused by the steric hindrance of the phenyl group, as such an interaction was critical to maintain high MOR binding affinity.13,15 Introduction of substituents with different electronic and steric characteristics at 3′-position on the indole ring basically resulted in the similar binding and selectivity profiles as observed in 2′-substituted derivatives. In details, except for 11, compounds 6–14 retained subnanomolar-to-nanomolar affinity for the MOR and the KOR, indicating that the substituents with different electronic characteristics at 3′-position on the indole ring only had a marginal effect on the binding affinity to these two receptors. It is interesting that compound 11 carrying an acetyl group at the 3′-position exhibited drastically decreased binding affinity at all three receptors, which could be ascribed to its poor solubility as we observed during the testing. On the other hand, increasing the size of substituents at 3′-position of the indole ring in compounds 13–17 potentially hampered the interactions between these ligands and all three receptors. The considerably decreased binding affinities as seen in compound 15–17 suggested that the address portions in all three receptors were not tolerant to bulky functional groups. This observation could serve as a rationale to omit oversized functional groups in the address portion in future molecular design.
Regarding the 6α-configuration analogues in Table 2, it seemed that the introduction of different substituents at 2′-position on the indole ring resulted in no significant change in the binding affinity on the MOR and KOR while slightly lowered selectivity to the MOR over the KOR and DOR (except for compound 22). Interestingly, compound 22, bearing a phenyl group at 2′-position, showed considerably decreased binding affinity at both the MOR and KOR while partially improved binding affinity at the DOR. For 3′-substituted compounds 23–30 containing functional groups with varying electronic characteristics, it appeared that the structural modifications retained subnanomolar affinity on the MOR and moderate affinity on the KOR and slightly decreased selectivity to the MOR over the DOR in comparison to NAN. Again, the increment in size of the substituents at the 3′-position, as seen in compounds 31–34, significantly diminished their binding affinity at all three receptors, which corroborated the finding that the address portions in opioid receptors were seemingly intolerant of bulky address portions.
Collectively, it was found that most of NAN derivatives possessed similar MOR selectivity profiles to those of NAN. This indicated that the binding of these ligands to the MOR was not noticeably influenced by the introduction of different substituents on the indole ring. More specifically, it has been previously proposed that the possible additional hydrogen bounding interactions between the 1′-position of indole ring of NAN and a hydrogen bond acceptor in the binding pocket could play a critical role in conferring greater binding affinity at the MOR and KOR over the DOR.13,15 Thus, we speculated that such hydrogen bonding interactions could still be intact in the ligand–receptor recognition process in the context of proposed structural modifications, which may provide a possible explanation of why most of NAN derivatives primarily behaved in a similar way to NAN in radioligand binding assays.
Afterward the [35S]-GTPγS functional assay was performed, as described previously.7 Based on the data shown in Tables 1 and 2, it appeared that all NAN derivatives primarily acted as partial agonists with low-to-moderate efficacy and one-digit nanomolar potency except compounds 11 and 16. While the substitution position and the electronic characteristic seemed to have variable impact on their potency and efficacy at the MOR, the introduction of bulky substituents appeared to favor MOR antagonism (e.g., compounds 5, 16, 17, 31, and 34).
Bitopic ligands are compounds designed by tethering two distinct pharmacophores through an optimal linker, which can simultaneously bind to the orthosteric binding site and allosteric binding site to achieve high affinity and selectivity. Considering the pivotal role of the linker in modulating allosteric profile and enhancing functional selectivity17,41 in the sense of the influence of configurational arrangement of the amide bond linker at the C6-position of the epoxymorphinan skeleton on binding affinity and efficacy profiles of these ligands, comparisons between one target ligand and its corresponding C-6 epimer were carried out accordingly. It was observed that switching the linkage stereochemistry from the β-configuration to α-configuration basically maintained their subnanomolar-to-nanomolar binding affinity for the MOR while producing slight reduction in the binding affinity for the KOR. With regard to the potency and efficacy, such structural alterations did not pose dramatic effect on their MOR potency but seemed to primarily result in a decrease in efficacy on the MOR (Tables 1 and 2).
In the [35S]-GTPγS binding assay, we have routinely measured MOR levels represented by the value of Bmax, and they range from ~2.5 to 4 pmol/mg, which are relatively higher than those in the rodent CNS.42 Previously, to further characterize the competitive antagonist property of our compounds of interest, additional functional assays had been conducted on low MOR-expressing CHO cells with Bmax values of 0.2–0.4 pmol/mg, which were similar to previously reported MOR Bmax values from multiple regions of mouse brain, as well as in mouse thalamus. These results indicated that all of our lead compounds of interest, for example, NAN, NAP, and NAQ, could produce a parallel rightward shift in the concentration effect curve of MOR full agonist DAMGO, suggesting that they basically behaved as high affinity MOR competitive antagonists in MOR-CHO cells and mouse thalamus.15,43 Since compound 21 carrying very subtle structural change retained the overall pharmacological properties similar to those of NAN, we thus anticipated that it may also act as a high affinity MOR antagonist in multiple testing models with varying MOR expression levels while further verifications are warranted.
In Vivo Warm-Water Tail Immersion Assay.
All compounds were then tested by warm-water tail immersion assays to evaluate their capacity to produce antinociception or block the antinociception elicited by morphine in mice following previously described protocol.14 In this assay, a single dose (10 mg/kg) was applied because it has been proven to be a practical and efficient screening approach in our laboratories to sort out the ligands with agonism or antagonism in vivo.7,11,14,45 The outcomes were presented as the percentage of maximum possible effect (% MPE). As seen in Figure 3A,B, compounds 7, 9, and 13 (10 mg/kg) showed noticeable antinociception with relatively high % MPE values, which appeared to correspond to their moderate efficacy observed in the [35S]-GTPγS functional assay. The compounds that did not manifest significant antinociception were subsequently tested as antagonists at a single dose of 10 mg/kg. As depicted in Figure 3C, three compounds (18, 21, and 30) exhibited different degrees of inhibition of morphine’s antinociceptive effect while the remainder of tested compounds only had marginal-to-moderate influence on the antinociceptive effect induced by morphine at 10 mg/kg. Notably, compound 21, which showed a high potency and low efficacy in the [35S]-GTPγS functional assay, stood out as the most potent member in this series with excellent antagonism. Its AD50 value was determined as 10.56 mg/kg from the following dose–response study. Although it was relatively less potent in comparison to the original hit NAN (2.07 mg/kg),15 these in vivo results did suggest that compound 21 potentially had the ability to cross the BBB and block the antinociceptive effect of morphine in CNS. Collectively, the in vitro efficacies of target ligands basically matched with the warm-water immersion assay results except for compound 19, suggesting that the agonism and antagonism efficacy observed in tail-immersion study could be largely stemmed from the MOR pharmacology. As for compound 19, since it showed no significantly improved binding selectivity among all three receptors, we decided not to pursue it any further.
Figure 3.

Warm-water tail immersion assays results of target compounds in mice (n = 5) at 56 ± 0.1 °C. (A) Antinociception effects of compounds 1–17 (10 mg/kg). (B) Antinociception effects of compounds 18–34 (10 mg/kg). (C) Blockage of the morphine-mediated antinociception of selected compounds (10 mg/kg) in the presence of morphine (10 mg/kg). The tested compound was injected subcutaneously at time 0. Morphine was administered after 5 min, and 20 min after the morphine injection, tail flick was conducted using warm water. Saline and morphine were the negative and positive controls. Data are presented as mean values ± SD. *P < 0.05 compared to vehicle; **P < 0.01 compared to vehicle.
As discussed above, multiple factors have been proposed to contribute either beneficial or deleterious effects on the CNS permeability of administrated drugs, and even minor structural changes could lead to significant permeability alterations. More specifically, it is well recognized that physicochemical properties of a compound have a pivotal influence on its brain exposure.46 In our case, while preparing the stock solution of tested compounds in the in vivo assay, we observed that some target compounds, particularly for the ones with bulky substituents, showed surprisingly poor solubilities in water and testing buffers, which could largely compromise their CNS acting activities. This observation further highlighted the necessity of evading oversized functional groups in the address portion in forthcoming opioid ligand design campaign. On the other hand, it is well documented that recognition by P-glycoprotein (P-gp) significantly limits the passive diffusion of compounds into the CNS.47 Several physicochemical parameters, including tPSA, HBD, and HBA, have been suggested to pose additive effect to reduce passive permeability while simultaneously facilitating interactions with P-gp transporter.48 According to the analysis of the physicochemical properties of compound data base from Amgen, tPSA and HBD appeared to play dominant roles in modulating the P-gp efflux ratio, that is, compounds with tPSA > 90 Å2 and >2 HBDs were more likely to act as P-gp efflux substrates.34 A revisit of the calculated physicochemical parameters of our synthesized compounds showed that they had tPSA > 97 Å2 and ≥4 HBDs (Table S1), indicating that they may possess P-gp liability when applied in in vivo studies. This could be another possible explanation why most of target compounds did not show potential CNS activity, though such a posit needs to be validated through further experimental assessment.
To further evaluate the overall CNS property of these compounds, we also utilized the CNS multiparameter optimization (CNS MPO) desirability tool,49 which consisted of six parameters including cLogP, cLogD, MW, tPSA, HBDs, and pKa, to generate the collective score ranged from 0 to 6. A higher CNS MPO score is considered to be more desirable, and it is generally accepted that a CNS-acting compound has MPO score greater than 4. For the purpose of comparison, the CNS MPO score of the original hit NAN was also calculated accordingly. As depicted in Table S1, the MPO score of NAN was 3.8, which was very close to the cutoff value of 4. Since the centrally acting profile of NAN has been validated by our prior in vivo study, we reasoned that the CNS MPO desirability tool would be applicable in understanding the CNS profiles of our newly synthetized compounds. As expected, most NAN derivatives had MPO scores lower than 3, suggesting that they may not be CNS-penetrant compounds. This trend was basically consistent with their lack of high potency in the in vivo assay. It is worthwhile noting that compound 21 was one of these tested compounds that had a highest MPO score, with a value of 3.8. This observation was in line with our conclusion that compound 21 acted as a CNS-acting agent as it had potent capability to antagonize the antinociception induced by morphine.
Through our in vivo assay, compound 21 bearing a methyl group at the 2′ position of the indole ring was found to be the most potent member that antagonizes the antinociception elicited by morphine. It is generally accepted that insertion of a methyl group would make a molecule more lipophilic and theoretically less water-soluble. However, several studies also revealed that installation of a methyl group may also lead to increased solubility through varying mechanisms, including additional hydrophobic interactions, alterations in the ionization state of functional groups, and lower energy of the crystalline network.50,51 Meanwhile, the slightly increased lipophilicity stemming from the addition of the methyl group to a compound is likely to increase its solubility in the biomembrane, which could in turn facilitate the transport through the bloodstream to target tissues.52,53 In this context, we reasoned that incorporation of a methyl group may fine-tune the physicochemical property of compound 21 to the point where it could pass through the BBB efficiently to block morphine-induced analgesia. Actually, such a “Magic Methyl” group has been widely applied at a strategic site in a lead compound due to the potential for profound changes to physicochemical and pharmacological properties.51,54,55
KOR and DOR [35S]GTPγS Functional Assays.
In order to further understand the underlying mechanism of the observed effect of compound 21 in the warm-water tail immersion assay, the potency and efficacy of compound 21 at the KOR and DOR were evaluated by employing the KOR and DOR [35S]GTPγS functional assays. To induce a maximum stimulation at the KOR and DOR, the KOR full agonist U50,488H and the DOR full agonist SNC80 were adopted as the reference compounds, respectively. As outlined in Table 3, compound 21 exhibited enhanced potency at the KOR as compared with NAN with an EC50 value of ~7 nM. It was also observed that compound 21 acted as a low efficacy partial agonist (Emax ~ 48%) at the KOR. On the other hand, compound 21 behaved as a moderate efficacy DOR agonist (Emax ~ 77%) with high potency (EC50 ~ 5 nM). Collectively, the functional assay results demonstrated that compound 21 is a low efficacy KOR agonist, an intermediate efficacy DOR agonist besides a MOR low efficacy agonist.
Table 3.
KOR and DOR [35S]GTPγS Functional Assays Results for Compound 21a
| KOR | DOR | |||
|---|---|---|---|---|
| compds | EC50 (nM) | Emax(% max of U50,488H) | EC50 (nM) | Emax(% max of SNC80) |
| NANb | 17.79 ± 3.24 | 73.90 ± 3.69 | 22.81 ± 3.95 | 26.67 ± 1.17 |
| 21 | 6.65 ± 0.78 | 48.28 ± 1.45 | 4.99 ± 0.21 | 76.82 ± 4.82 |
The values are the mean ± SEM of at least three independent experiments.
Data were taken from ref 15.
In the past few years, mounting evidence have highlighted the prominence of developing multifunctional opioid receptor ligands as novel medications.56,57 It is well-documented that the severe adverse effects of opioid agonists could primarily ascribe to the modulation of the MOR.58,59 While it is generally accepted that KOR agonists might cause unwanted effects such as diuresis and dysphoria,60,61 the stimulation of DOR appears to elicit fewer side effects.62–64 Therefore, the fact that compound 21 manifested a relatively higher efficacy at the DOR but a lower one at the KOR comparing to those of NAN indicated that it may hold promise of being a candidate with less undesirable side effects toward OUD treatments.
Calcium Flux Assay.
It has been documented that the cytosolic calcium concentration can be rapidly enhanced upon activation of the Gq/calcium pathway in CHO cells.65 The measurement of calcium flux has become a widely accepted method to assess GPCR functions. To characterize the influence of compound 21 in this G protein-mediated signaling pathway, a calcium flux assay was carried out, in which chimeric G proteins (Gαqi5) were used to facilitate the measurement.14 As shown in Figure S1a, compound 21 showed no obvious agonist activity to increase the intracellular calcium concentration. After that, compound 21 was tested to concentration-dependently antagonize DAMGO-induced calcium flux with an IC50 value of 87.53 ± 32.77 nM, which was equipotent with NAN (IC50 = 50.29 ± 1.62 nM) (Figure S1b).15 Considering that compound 21 had very low efficacy in GTPγS functional assay (Emax = 12.04 ± 1.87%), it is postulated that it may function as a neutral MOR antagonist in this pathway, resulting in the lack of agonism to enhance the intracellular calcium concentration.
hERG Inhibitory Activity Evaluation.
As mentioned above, the hERG-related cardiotoxicity has been regarded as a necessary safety evaluation index in early drug discovery campaigns. Drugs that have hERG liability are likely to blockade the inward rectifying voltage-gated K+ channel (IKr) in the heart, resulting in QT interval prolongation with potential risk of fatal arrhythmias.25 Our parent hit NAN was shown to manifest significant inhibition against the hERG channel, which limited its further development. Therefore, the hERG inhibitory activity of compound 21 was further assessed to see whether the potential risk for cardiotoxicity can be alleviated. E-4031, an antiarrhythmic drug that selectively blocks hERG potassium channel, was adopted as the positive control in the automated patch-clamp assay.66 It turned out that compound 21 exhibited a moderate hERG inhibition with an IC50 value of 0.14 μM and possessed a seven-fold improved hERG profile in comparison to NAN (IC50 = 0.021 μM), suggesting the less likelihood of cardiotoxicity (Table 4). It was intriguing how structurally similar MOR ligands, NAN and compound 21, with comparable cLogP values result in considerably different hERG inhibitory activity (Table 4). Previous mutagenesis data and homology modeling of hERG potassium channel suggested that the π–π stacking interactions between key residues Phe656 and Tyr652 situating in the large channel cavity and the aromatic moiety in a drug molecule are the primary determining factors of hERG–drug interaction.67,68 We thus postulated that the indole ring of NAN, a moiety with aromatic nature, may possibly form putative interactions with the aromatic residue-rich hERG channel cavity to exert significant inhibitory activity. However, the subtle structural alteration by installing a methyl group at the 2′ position of the indole ring (e.g., compound 21) may potentially disrupt these interactions and consequently impair the affinity for the hERG channel. Despite such an improvement, it is generally accepted that the favorable lead compounds are in the submicromolar-to-micromolar range of potency at inhibiting the hERG channel.69 Therefore, next round of structural optimization on compound 21, for example, introduction of heteroatoms in the indole moiety to further mitigate hERG toxicity, is still necessary to further improve its safety profiles.
Table 4.
hERG Toxicity Assay Results for Compound 21 and NANa
| compds | hERG IC50 (μM) | cLogPb |
|---|---|---|
| NAN | 0.021 | 2.62 |
| 21 | 0.14 | 2.77 |
| E-4031 | 0.029 | 2.12 |
This assay was performed on the hERG-expressing CHO-K1 cell line.
The given values were predicted by ACD/Percepta (v2020.2.0) software.
In Vitro Metabolic Stability Profiles and Other Drug-like Properties.
To further profile compound 21, several foundational ADMET assays were conducted accordingly. Its plasma protein binding was moderately high across species (99% for human, 96% for rat). Caco-2 permeability assay suggested that compound 21 manifested an acceptable efflux ratio of 2.2. In addition, the metabolic stability profiles in human and rat liver microsomes were also evaluated, indicating that it has decent metabolic stability in both species (t1/2 > 60 min, CLint(liver) < 38.5 mL/min/kg for human; t1/2 > 60 min, CLint(liver) < 38.5 mL/min/kg for rat). In light of these results, we reasoned that compound 21 holds promise of being a long-acting agent, though such a posit needs to be validated through further experimental assessment. Additionally, considering that the 3-hydroxy group of the epoxymorphinan core structure has been shown to play a key role in the metabolic process for most opioid compounds,70 we are working on preparing compound 21-based prodrug by esterification at the 3-hydroxy group in order to increase the bioavailability as well as to prolong the half-life. These study results will be reported in due course. To summarize, these reasonable metrics, as observed above, warrant further structural optimization campaign and PK profile characterization for compound 21.
In Vivo Opioid Withdrawal Studies.
Studies have revealed that naltrexone and naloxone may induce serious opioid withdrawal signs when administrated to opioid-dependent patients.71–73 Given that compound 21 antagonized morphine-mediated antinociception in vivo assay and possessed reasonable drug-like properties, we thus assessed whether it would produce opioid withdrawal symptoms in chronic morphine-pelleted mice. In this study, three somatic signs of opioid withdrawal in mice, including wet-dog shakes, paw tremors, and jumps, were recorded over a 20 min time period after every injection of compound 21. As shown in Figure 4, naloxone (1 mg/kg) induced significant withdrawal symptoms after injection in morphine-pelleted mice (Figure 4, the first columns), which were similar to our previous report.14 In contrast, compound 21 did not precipitate more wet-dog shakes in morphine-pelleted mice in all the doses tested (Figure 4A). Moreover, a 40 mg/kg dose of compound 21 produced jumps that were not significantly different from those at 1 mg/kg naloxone (Figure 4B). Given that NAN did not induce significant wet-dog shakes and jumps at 50 mg/kg,15 it seemed that the profiles of compound 21 in these two observations were similar to those of NAN. More interestingly, compared to naloxone (1 mg/kg), 21 produced significantly fewer paw tremors for all the doses tested up to 80 mg/kg (Figure 4C). NAN, on the other hand, precipitated seemingly more paw tremors at a lower dose of 50 mg/kg.15 Based on these observations, it appeared that compound 21 possessed comparable or overall improved profiles in withdrawal studies in comparison with the parent hit NAN. Since the KOR and DOR [35S]GTPγS functional assays indicated that compound 21 may act as a multifunctional opioid receptor ligand with high efficacy at the DOR but moderate efficacy at the KOR, we thus speculated that this could be the primary reason that compound 21 precipitated less withdrawal signs than NAN and naloxone. It is worth noting that although short-acting agent naloxone was utilized as a standard compound in the opioid withdrawal studies, we have not validated the particular therapeutic application for compound 21 as structural optimization and pharmacological assessment are still being performed aiming to further fine-tune its physicochemical properties and drug-like properties. In summary, our results suggested that compound 21 would produce less withdrawal signs than naloxone, which may deem as an advantage in the treatment of OUD over naloxone.
Figure 4.

In vivo withdrawal assays of compound 21 in chronic morphine-pelleted mice (n = 5). (A) Wet-dog shakes, (B) jumps, and (C) paw tremors. All columns in figures represent morphine-pelleted mice. * indicates P < 0.05, ** indicates P < 0.005, and **** indicates P < 0.0001, compared to 1 mg/kg naloxone (NLX, s.c.).
CONCLUSIONS
Amidst the nation-wide opioid crisis, developing selective MOR modulators with favorable pharmacological efficacy and improved safety profile is still highly desirable. Our previous efforts have demonstrated NAN as a potent MOR modulator with promising potency in the treatment of OUD. However, subsequent ADMET property evaluation revealed its significant hERG liability that may pose a vast challenge for its further development. Therefore, NAN-based structural modification was conducted with an aim to address this issue. 34 opioid ligands bearing various substituted indole rings were prepared and biologically evaluated. Among them, 2′-methylindolyl NAN derivative compound 21, which showed comparable antagonism against DAMGO-induced intracellular calcium flux in comparison with NAN, also possessed potency in blocking the antinociceptive effect of morphine at a somehow lower potency. Nevertheless, its pharmacological profile seemed improved, as shown by fewer withdrawal symptoms at similar doses compared to those of NAN. Furthermore, hERG inhibition assessment indicated that compound 21 possessed a largely improved hERG profile with about seven-fold improvement compared to NAN. Taken together, our current study seems a successful paradigm to illustrate how to reduce hERG liability while maintaining the overall pharmacological activity in the early stage drug discovery campaign. The systematic structural modification on parent hit NAN enabled the identification of compound 21 exhibiting improved overall pharmacological and safety profiles, which appeared to serve as a more promising opioid receptor modulator for the development of new therapeutic agents toward OUD.
EXPERIMENTAL SECTION
Chemistry.
All solvents and reagents were purchased from either Sigma-Aldrich or Alfa Aesar and were used as received without further purification. Melting points were measured on a MPA100 OptiMelt automated melting point apparatus without correction. IR spectra were recorded on a Thermo Scientific Nicolet iS10 FT-IR spectrometer. Analytical thin-layer chromatography (TLC) analyses were carried out on Analtech Uniplate F254 plates, and FCC was performed over silica gel (230–400 mesh, Merck). 1H (400 MHz) and 13C (100 MHz) nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Ultrashield 400 Plus spectrometer, and chemical shifts were expressed in ppm. Mass spectra were obtained on an Applied BioSystems 3200 Q trap with a turbo V source for TurbolonSpray. Analytical reversed-phase high-performance liquid chromatography (HPLC) was performed on a Varian ProStar 210 system using the Agilent Microsorb-MV 100–5 C18 column (250 × 4.6 mm). All analyses were conducted at ambient temperature with a flow rate of 0.8 mL/min. Mobile phase is acetonitrile/water (90/10) with 0.1% trifluoroacetic acid (TFA). The UV detector was set up at 210 nm. Compound purities were calculated as the percentage peak area of the analyzed compound, and retention times (Rt) were presented in minutes. The purity of all newly synthesized compounds was identified as ≥95%.
General Procedure for the Amide Formation Reaction.
A carboxylic acid (2.5 equiv) dissolved in dry DMF (1.5 mL) was added with 1-hydroxybenzotriazole (HOBt, 3 equiv), N-(3-dimethylamino-propyl)-N′-ethylcarbodiimide hydrochloride (EDCI, 3 equiv), 4 Å molecular sieves, and triethylamine (5 equiv) at 0 °C degree. After 1 h, a solution of 6α-naltrexamine or 6β-naltrexamine (1 equiv) in predried DMF (1.5 mL) was added dropwise. The reaction mixture was then stirred at room temperature. Once TLC showed a complete consumption of naltrexamine (typically in 2 days), the reaction was diluted by methanol and filtered through celite. The filtrate was then concentrated to dryness and dissolved in anhydrous methanol (5 mL), then K2CO3 (2.5 equiv) was added. The reaction mixture was stirred overnight at room temperature and filtered again over celite. After concentration, the residue was purified by FCC with CH2Cl2/MeOH/1% NH3·H2O as the eluent to give the free base. After structural confirmation by 1H NMR, the corresponding free base was then converted into a hydrochloride salt, which was fully characterized by 1H NMR, 13C NMR, IR, MS, and HPLC.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(2-bromo-indole-7-carboxamido)morphinan (1).
The title compound was prepared following the general procedure as a white solid in 43% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 11.35 (s, 1H, exchangeable), 9.34 (s, 1H, exchangeable), 8.86 (s, 1H, exchangeable), 8.78 (d, J = 8.0 Hz, 1H, exchangeable), 7.73 (d, J = 7.1 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.13 (t, J = 7.7 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.67 (d, J = 8.1 Hz, 1H), 6.63 (d, J = 2.2 Hz, 1H), 6.18 (s, 1H, exchangeable), 4.90 (d, J = 7.8 Hz, 1H), 3.88 (d, J = 5.0 Hz, 1H), 3.78 (m, 1H), 3.36 (m, 1H), 3.28 (m, 1H), 3.12 (m, 1H), 3.06 (m, 1H), 2.86 (m, 1H), 2.48–2.43 (m, 2H), 1.95 (m, 1H), 1.80 (m, 1H), 1.66 (m, 1H), 1.49 (m, 1H), 1.42 (m, 1H), 1.08 (m, 1H), 0.69 (m, 1H), 0.61 (m, 1H), 0.52 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.2, 142.1, 141.3, 134.9, 129.7, 129.6, 122.8, 120.6, 120.2, 119.3, 119.1, 117.9, 116.6, 110.4, 103.9, 89.8, 69.8, 61.8, 56.7, 51.0, 46.5, 45.7, 29.5, 27.4, 23.8, 23.0, 5.7, 5.1, 2.6. HRMS calcd for C29H31N3O4Br [M + H]+, 564.1492; found, 564.1503. mp 235 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(2-chloro-indole-7-carboxamido)morphinan (2).
A white solid was obtained in 48% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 11.45 (s, 1H, exchangeable), 9.34 (s, 1H, exchangeable), 8.87 (s, 1H, exchangeable), 8.77 (d, J = 8.0 Hz, 1H, exchangeable), 7.74 (d, J = 7.7 Hz, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.14 (t, J = 7.7 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 8.2 Hz, 1H), 6.54 (d, J = 2.2 Hz, 1H), 6.20 (s, 1H, exchangeable), 4.90 (d, J = 7.8 Hz, 1H), 3.88 (d, J = 4.2 Hz, 1H), 3.78 (m, 1H), 3.38 (m, 1H), 3.35 (m, 1H), 3.10 (m, 1H), 3.05 (m, 1H), 2.87 (m, 1H), 2.47–2.42 (m, 2H), 1.96 (m, 1H), 1.80 (m, 1H), 1.66 (m, 1H), 1.49 (m, 1H), 1.42 (m, 1H), 1.08 (m, 1H), 0.68 (m, 1H), 0.61 (m, 1H), 0.52 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.1, 142.1, 141.3, 133.5, 129.7, 128.8, 124.3, 122.9, 120.6, 120.2, 119.3, 119.1, 117.8, 116.7, 99.7, 89.8, 69.8, 61.7, 56.7, 51.0, 46.5, 45.6, 29.5, 27.4, 23.8, 23.0, 5.7, 5.1, 2.6. HRMS calcd for C29H31N3O4Cl [M + H]+, 520.1998; found, 520.2008. mp 270 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(2-cyano-indole-7-carboxamido)morphinan (3).
A white solid was obtained in 75% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 12.22 (s, 1H, exchangeable), 9.34 (s, 1H, exchangeable), 8.87 (s, 1H, exchangeable), 8.88 (d, J = 8.0 Hz, 1H, exchangeable), 7.97 (d, J = 7.0 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.43 (d, J = 2.1 Hz, 1H), 7.27 (t, J = 7.7 Hz, 1H), 6.74 (d, J = 8.1 Hz, 1H), 6.67 (d, J = 8.1 Hz, 1H), 6.21 (s, 1H, exchangeable), 4.90 (d, J = 7.8 Hz, 1H), 3.89 (d, J = 5.3 Hz, 1H), 3.78 (m, 1H), 3.37 (m, 1H), 3.28 (m, 1H), 3.12 (m, 1H), 3.06 (m, 1H), 2.87 (m, 1H), 2.54 (m, 1H), 2.45 (m, 1H), 1.98 (m, 1H), 1.81 (m, 1H), 1.66 (m, 1H), 1.49 (m, 1H), 1.42 (m, 1H), 1.09 (m, 1H), 0.69 (m, 1H), 0.60 (m, 1H), 0.52 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 165.6, 142.1, 141.3, 135.0, 129.7, 127.4, 125.5, 124.0, 120.5, 120.2, 119.3, 118.2, 117.8, 114.1, 113.5, 107.6, 89.8, 69.7, 61.7, 56.7, 51.1, 45.6, 29.5, 28.6, 27.4, 23.8, 23.0, 5.7, 5.1, 2.6. HRMS calcd for C30H31N4O4 [M + H]+, 511.2340; found, 511.2348. mp 251 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(2-methyl-indole-7-carboxamido)morphinan (4).
A white solid was obtained in 72% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.80 (s, 1H, exchangeable), 9.35 (s, 1H, exchangeable), 8.87 (s, 1H, exchangeable), 8.68 (d, J = 8.0 Hz, 1H, exchangeable), 7.63 (d, J = 7.4 Hz, 1H), 7.58 (d, J = 7.7 Hz, 1H), 7.02 (t, J = 7.6 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 8.1 Hz, 1H), 6.20 (s, 1H, exchangeable), 6.17 (dd, J = 1.9, 0.9 Hz, 1H), 4.92 (d, J = 7.8 Hz, 1H), 3.88 (d, J = 4.6 Hz, 1H), 3.78 (m, 1H), 3.39 (m, 1H), 3.28 (m, 1H), 3.11 (m, 1H), 3.05 (m, 1H), 2.86 (m, 1H), 2.48–2.46 (m, 2H), 2.40 (s, 3H), 1.96 (m, 1H), 1.80 (m, 1H), 1.64 (m, 1H), 1.49 (m, 1H), 1.42 (m, 1H), 1.08 (m, 1H), 0.69 (m, 1H), 0.61 (m, 1H), 0.52 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.8, 142.2, 141.3, 137.1, 134.7, 130.0, 129.7, 122.5, 120.5, 119.3, 118.6, 117.9, 117.8, 115.9, 99.1, 89.9, 69.8, 61.8, 56.7, 50.9, 46.5, 45.7, 29.5, 27.4, 23.9, 23.0, 13.4, 5.7, 5.1, 2.6. HRMS calcd for C30H34N3O4 [M + H]+, 500.2544; found, 500.2570. mp 280 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(2-phenyl-indole-7-carboxamido)morphinan (5).
A white solid was obtained in 72% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.93 (d, J = 1.9 Hz, 1H, exchangeable), 9.34 (s, 1H, exchangeable), 8.88 (s, 1H, exchangeable), 8.85 (d, J = 8.1 Hz, 1H, exchangeable), 7.87–7.83 (m, 2H), 7.81–7.76 (m, 2H), 7.46 (t, J = 7.7 Hz, 2H), 7.35 (t, J = 7.4 Hz, 1H), 7.15 (t, J = 7.7 Hz, 1H), 7.00 (d, J = 2.4 Hz, 1H), 6.74 (d, J = 8.1 Hz, 1H), 6.68 (d, J = 8.1 Hz, 1H), 6.19 (s, 1H, exchangeable), 4.93 (d, J = 7.9 Hz, 1H), 3.88 (d, J = 4.7 Hz, 1H), 3.82 (m, 1H), 3.37 (m, 1H), 3.27 (m, 1H), 3.12 (m, 1H), 3.06 (m, 1H), 2.87 (m, 1H), 2.47–2.44 (m, 2H), 1.95 (m, 1H), 1.81 (m, 1H), 1.70 (m, 1H), 1.51 (m, 1H), 1.45 (m, 1H), 1.08 (m, 1H), 0.69 (m, 1H), 0.61 (m, 1H), 0.52 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.8, 142.2, 141.3, 138.3, 135.9, 131.4, 130.0, 129.7, 129.0 (×2), 128.0, 125.2 (×2), 124.0, 120.6, 120.3, 119.0, 117.9, 116.2, 99.1, 89.8, 69.8, 61.8, 59.8, 56.7, 51.0, 46.5, 45.7, 29.5, 27.3, 23.8, 23.0, 5.7, 5.1, 2.6. HRMS calcd for C35H36N3O4 [M + H]+, 562.2700; found, 562.2717. mp 244 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3-bromo-indole-7-carboxamido)morphinan (6).
A white solid was obtained in 78% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 11.38 (d, J = 2.2 Hz, 1H, exchangeable), 9.36 (s, 1H, exchangeable), 8.89 (s, 1H, exchangeable), 8.84 (d, J = 8.1 Hz, 1H, exchangeable), 7.86 (d, J = 7.3 Hz, 1H), 7.62 (d, J = 7.9 Hz, 1H), 7.47 (d, J = 2.2 Hz, 1H), 7.23 (t, J = 7.7 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 8.1 Hz, 1H), 6.22 (s, 1H, exchangeable), 4.90 (d, J = 7.8 Hz, 1H), 3.89 (d, J = 5.0 Hz, 1H), 3.78 (m, 1H), 3.39–3.35 (m, 2H), 3.11 (m, 1H), 3.06 (m, 1H), 2.87 (m, 1H), 2.47–2.41 (m, 2H), 1.96 (m, 1H), 1.81 (m, 1H), 1.64 (m, 1H), 1.50–1.46 (m, 2H), 1.08 (m, 1H), 0.68 (m, 1H), 0.60 (m, 1H), 0.51 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.0, 142.1, 141.4, 133.7, 129.7, 127.6, 126.2, 121.8, 121.1, 120.6, 119.4, 119.2, 117.9, 117.3, 89.8, 89.0, 69.8, 61.7, 56.7, 50.92, 46.5, 45.7, 29.5, 27.4, 23.9, 23.1, 5.8, 5.2, 2.6. HRMS calcd for C29H31N3O4Br [M + H]+, 564.1492; found, 564.1522. mp 280 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3-chloro-indole-7-carboxamido)morphinan (7).
A white solid was obtained in 82% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 11.28 (d, J = 2.3 Hz, 1H, exchangeable), 9.36 (s, 1H, exchangeable), 8.90 (s, 1H, exchangeable), 8.85 (d, J = 8.1 Hz, 1H, exchangeable), 7.86 (d, J = 7.2 Hz, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.44 (d, J = 2.3 Hz, 1H), 7.23 (t, J = 7.7 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 8.1 Hz, 1H), 6.25 (s, 1H, exchangeable), 4.91 (d, J = 7.8 Hz, 1H), 3.90 (d, J = 5.1 Hz, 1H), 3.78 (m, 1H), 3.39–3.35 (m, 2H), 3.11 (m, 1H), 3.06 (m, 1H), 2.87 (m, 1H), 2.49–2.42 (m, 2H), 1.97 (m, 1H), 1.81 (m, 1H), 1.63 (m, 1H), 1.48 (m, 1H), 1.42 (m, 1H), 1.09 (m, 1H), 0.68 (m, 1H), 0.58 (m, 1H), 0.52 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.0, 142.1, 141.2, 133.2, 129.7, 126.0, 123.7, 121.1, 120.9, 120.6, 119.4, 119.1, 117.9, 117.4, 103.4, 89.9, 69.8, 61.7, 56.7, 50.9, 46.5, 45.7, 29.5, 27.4, 23.9, 23.1, 5.8, 5.2, 2.6. HRMS calcd for C29H31N3O4Cl [M + H]+, 520.1998; found, 520.2015. mp 281 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3-cyano-indole-7-carboxamido)morphinan (8).
A white solid was obtained in 71% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 12.08 (d, J = 2.8 Hz, 1H, exchangeable), 9.37 (s, 1H, exchangeable), 8.96 (d, J = 8.1 Hz, 1H, exchangeable), 8.90 (s, 1H, exchangeable), 8.17 (d, J = 2.8 Hz, 1H), 7.95 (d, J = 7.1 Hz, 1H), 7.85 (d, J = 7.9 Hz, 1H), 7.36 (t, J = 7.7 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 8.1 Hz, 1H), 6.25 (s, 1H, exchangeable), 4.90 (d, J = 7.8 Hz, 1H), 3.89 (d, J = 5.2 Hz, 1H), 3.79 (m, 1H), 3.37 (m, 1H), 3.31 (m, 1H), 3.11 (m, 1H), 3.04 (m, 1H), 2.86 (m, 1H), 2.48–2.42 (m, 2H), 1.97 (m, 1H), 1.82 (m, 1H), 1.64 (m, 1H), 1.49 (m, 1H), 1.42 (m, 1H), 1.08 (m, 1H), 0.68 (m, 1H), 0.60 (m, 1H), 0.52 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 165.5, 142.1, 141.4, 135.7, 133.4, 129.7, 128.2, 122.2, 122.0, 121.2, 121.0, 119.4, 118.5, 117.9, 116.0, 89.8, 84.5, 69.8, 61.7, 56.6, 51.0, 46.5, 45.7, 29.5, 27.4, 23.9, 23.0, 5.7, 5.2, 2.6. HRMS calcd for C30H31N4O4 [M + H]+, 511.2340; found, 511.2350. mp 273 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3-nitro-indole-7-carboxamido)morphinan (9).
A white solid was obtained in 60% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 12.38 (d, J = 3.3 Hz, 1H, exchangeable), 9.36 (s, 1H, exchangeable), 9.03 (d, J = 8.1 Hz, 1H, exchangeable), 8.89 (s, 1H, exchangeable), 8.39 (d, J = 3.3 Hz, 1H), 8.30 (d, J = 7.9 Hz, 1H), 8.00 (d, J = 7.0 Hz, 1H), 7.50 (t, J = 7.8 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.67 (d, J = 8.1 Hz, 1H), 6.21 (s, 1H, exchangeable), 4.89 (d, J = 7.8 Hz, 1H), 3.88 (d, J = 5.0 Hz, 1H), 3.80 (m, 1H), 3.40–3.35 (m, 2H), 3.12 (m, 1H), 3.04 (m, 1H), 2.86 (m, 1H), 2.48–2.41 (m, 2H), 1.97 (m, 1H), 1.81 (m, 1H), 1.64 (m, 1H), 1.49 (m, 1H), 1.43 (m, 1H), 1.06 (m, 1H), 0.68 (m, 1H), 0.60 (m, 1H), 0.51 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 165.3, 142.1, 141.4, 133.1, 131.0, 129.6, 128.4, 123.3, 123.1, 122.7, 121.1, 120.6, 119.4, 118.6, 117.9, 89.8, 69.7, 61.7, 56.7, 51.1, 46.5, 45.7, 29.5, 27.4, 23.8, 23.0, 5.7, 5.2, 2.6. HRMS calcd for C29H31N4O6 [M + H]+, 531.2238; found, 531.2253. mp 262 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3-formyl-indole-7-carboxamido)morphinan (10).
A white solid was obtained in 30% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 12.00 (s, 1H, exchangeable), 9.98 (s, 1H, exchangeable), 9.34 (s, 1H, exchangeable), 8.91 (d, J = 8.0 Hz, 1H, exchangeable), 8.87 (s, 1H, exchangeable), 8.29 (d, J = 7.8 Hz, 1H), 8.22 (d, J = 3.2 Hz, 1H), 7.91 (d, J = 7.5 Hz, 1H), 7.34 (t, J = 7.7 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.67 (d, J = 8.1 Hz, 1H), 6.19 (s, 1H, exchangeable), 4.90 (d, J = 7.8 Hz, 1H), 3.88 (d, J = 5.0 Hz, 1H), 3.81 (m, 1H), 3.41–3.36 (m, 2H), 3.14–3.04 (m, 2H), 2.86 (m, 1H), 2.48–2.45 (m, 2H), 1.97 (m, 1H), 1.79 (m, 1H), 1.66 (m, 1H), 1.51–1.41 (m, 2H), 1.07 (m, 1H), 0.69 (m, 1H), 0.59 (m, 1H), 0.51 (m, 1H), 0.43 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 185.5, 165.8, 142.1, 141.3, 139.5, 135.3, 129.6, 125.5, 124.5, 121.9, 121.6, 120.6, 119.4, 117.9, 117.8, 117.6, 89.8, 69.7, 61.7, 56.7, 51.0, 46.5, 45.6, 29.5, 27.4, 23.0, 22.9, 5.7, 5.1, 2.6. HRMS calcd for C30H32N3O5 [M + H]+, 514.2336; found, 514.2339. mp 280 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3-acetyl-indole-7-carboxamido)morphinan (11).
A white solid was obtained in 53% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 11.82 (s, 1H, exchangeable), 9.35 (s, 1H, exchangeable), 8.89 (s, 1H, exchangeable), 8.87 (d, J = 7.9 Hz, 1H, exchangeable), 8.38 (d, J = 7.8 Hz, 1H), 8.18 (d, J = 3.0 Hz, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.29 (t, J = 7.7 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.67 (d, J = 8.1 Hz, 1H), 6.19 (s, 1H, exchangeable), 4.90 (d, J = 7.8 Hz, 1H), 3.87 (d, J = 5.2 Hz, 1H), 3.79 (m, 1H), 3.37 (m, 1H), 3.13 (m, 1H), 3.08–3.04 (m, 3H), 2.86 (m, 1H), 2.52 (m, 1H), 2.47 (s, 3H), 1.95 (m, 1H), 1.80 (m, 1H), 1.66 (m, 1H), 1.52–1.39 (m, 2H), 1.06 (m, 1H), 0.68 (m, 1H), 0.60 (m, 1H), 0.53 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 192.9, 166.0, 142.1, 141.3, 135.2, 134.9, 129.7, 126.7, 125.0, 121.3, 121.1, 120.6, 119.4, 117.9, 117.5, 116.3, 89.8, 69.7, 61.2, 58.6, 50.9, 46.5, 45.5, 30.1, 29.5, 27.4, 23.0, 19.0, 5.7, 5.1, 2.6. HRMS calcd for C31H34N3O5 [M + H]+, 528.2493; found, 528.2472. mp 255 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3-acet-amido-indole-7-carboxamido)morphinan (12).
A white solid was obtained in 74% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.74 (s, 1H, exchangeable), 9.90 (s, 1H, exchangeable), 9.33 (s, 1H, exchangeable), 8.86 (s, 1H, exchangeable), 8.72 (d, J = 8.1 Hz, 1H, exchangeable), 8.01 (d, J = 7.9 Hz, 1H), 7.76 (d, J = 7.5 Hz, 1H), 7.74 (d, J = 2.5 Hz, 1H), 7.10 (t, J = 7.7 Hz, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 8.1 Hz, 1H), 6.15 (s, 1H, exchangeable), 4.90 (d, J = 7.8 Hz, 1H), 3.86 (d, J = 4.8 Hz, 1H), 3.78 (m, 1H), 3.38–3.35 (m, 2H), 3.13–3.04 (m, 3H), 2.85 (m, 1H), 2.46 (m, 1H), 2.10 (s, 3H), 1.96 (m, 1H), 1.79 (m, 1H), 1.63 (m, 1H), 1.50–1.40 (m, 2H), 1.06 (m, 1H), 0.69 (m, 1H), 0.60 (m, 1H), 0.51 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.9, 166.5, 142.2, 141.3, 131.7, 129.7, 121.9, 121.8, 120.5, 120.2, 119.3, 117.9, 117.2, 116.3, 116.2, 115.1, 89.9, 69.8, 61.8, 54.9, 50.8, 45.6, 45.5, 30.5, 29.5, 26.5, 23.9, 23.0, 5.7, 5.1, 2.6. HRMS calcd for C31H35N4O5 [M + H]+, 543.2602; found, 543.2584. mp 245 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3-methyl-indole-7-carboxamido)morphinan (13).
A white solid was obtained in 91% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.73 (d, J = 0.8 Hz, 1H, exchangeable), 9.36 (s, 1H, exchangeable), 8.89 (s, 1H, exchangeable), 8.71 (d, J = 8.1 Hz, 1H, exchangeable), 7.75 (d, J = 7.4 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.11 (d, J = 0.8 Hz, 1H), 7.08 (t, J = 7.7 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 8.1 Hz, 1H), 6.22 (s, 1H, exchangeable), 4.91 (d, J = 7.8 Hz, 1H), 3.82 (d, J = 5.1 Hz, 1H), 3.78 (m, 1H), 3.32–3.29 (m, 2H), 3.15–3.02 (m, 2H), 2.87 (m, 1H), 2.49–2.39 (m, 2H), 2.27 (d, J = 0.8 Hz, 3H), 1.95 (m, 1H), 1.80 (m, 1H), 1.63 (m, 1H), 1.48 (m, 1H), 1.41 (m, 1H), 1.08 (m, 1H), 0.68 (m, 1H), 0.60 (m, 1H), 0.51 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.7, 142.2, 141.3, 134.7, 129.7, 129.5, 124.1, 122.0, 120.6, 119.7, 119.3, 117.9, 117.4, 116.3, 109.1, 89.9, 69.8, 61.7, 56.7, 50.8, 46.5, 45.6, 29.5, 27.4, 23.9, 23.0, 9.4, 5.7, 5.1, 2.6. HRMS calcd for C30H34N3O4 [M + H]+, 500.2544; found, 500.2545. mp 281 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3-iso-propyl-indole-7-carboxamido)morphinan (14).
A white solid was obtained in 80% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.75 (d, J = 1.8 Hz, 1H, exchangeable), 9.36 (s, 1H, exchangeable), 8.88 (s, 1H, exchangeable), 8.70 (d, J = 8.1 Hz, 1H, exchangeable), 7.76 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 7.6 Hz, 1H), 7.07 (t, J = 7.7 Hz, 1H), 7.06 (d, J = 1.8 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 8.1 Hz, 1H), 6.20 (s, 1H, exchangeable), 4.91 (d, J = 7.8 Hz, 1H), 3.82 (d, J = 5.1 Hz, 1H), 3.78 (m, 1H), 3.33–3.29 (m, 2H), 3.16 (m, 1H), 3.09 (m, 1H), 3.06 (m, 1H), 2.86 (m, 1H), 2.48–2.43 (m, 2H), 1.95 (m, 1H), 1.80 (m, 1H), 1.64 (m, 1H), 1.49 (m, 1H), 1.42 (m, 1H), 1.30 (d, J = 2.2 Hz, 3H), 1.29 (d, J = 2.2 Hz, 3H), 1.06 (m, 1H), 0.70 (m, 1H), 0.60 (m, 1H), 0.51 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.7, 142.2, 141.3, 134.9, 129.7, 127.8, 122.4, 121.6, 121.5, 120.5, 119.6, 119.3, 117.8, 117.3, 116.4, 89.9, 69.8, 61.7, 56.7, 50.8, 46.5, 45.6, 29.5, 27.4, 24.7, 23.9, 23.5, 23.4, 23.0, 5.7, 5.1, 2.6. HRMS calcd for C32H38N3O4 [M + H]+, 528.2857; found, 528.2877. mp 233 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3-cyclo-pentyl-indole-7-carboxamido)morphinan (15).
A white solid was obtained in 84% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.72 (s, 1H, exchangeable), 9.35 (s, 1H, exchangeable), 8.89 (s, 1H, exchangeable), 8.69 (d, J = 8.0 Hz, 1H, exchangeable), 7.74 (t, J = 7.1 Hz, 2H), 7.09 (d, J = 2.3 Hz, 1H), 7.06 (d, J = 7.7 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 8.1 Hz, 1H), 6.21 (s, 1H, exchangeable), 4.92 (d, J = 7.8 Hz, 1H), 3.89 (d, J = 5.2 Hz, 1H), 3.78 (m, 1H), 3.32–3.27 (m, 2H), 3.22 (m, 1H), 3.14–3.03 (m, 2H), 2.87 (m, 1H), 2.48–2.42 (m, 2H), 2.12–2.05 (m, 2H), 1.95 (m, 1H), 1.82–1.75 (m, 2H), 1.72–1.56 (m, 6H), 1.50–1.40 (m, 2H), 1.07 (m, 1H), 0.68 (m, 1H), 0.60 (m, 1H), 0.52 (m, 1H), 0.43 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.7, 142.2, 141.3, 135.0, 129.7, 128.4, 122.6, 121.9, 120.5, 119.7, 119.3, 118.9, 117.9, 117.3, 116.4, 89.9, 69.8, 61.7, 56.7, 50.8, 46.5, 45.6, 36.2, 33.0, 32.9, 29.5, 27.4, 24.7 (×2), 23.7, 23.0, 5.7, 5.1, 2.6. HRMS calcd for C34H40N3O4 [M + H]+, 554.3013; found, 554.3011. mp 235 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3-cyclo-hexyl-indole-7-carboxamido)morphinan (16).
A white solid was obtained in 85% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.73 (s, 1H, exchangeable), 9.35 (s, 1H, exchangeable), 8.89 (s, 1H, exchangeable), 8.69 (d, J = 8.0 Hz, 1H, exchangeable), 7.75 (d, J = 7.8 Hz, 1H), 7.72 (d, J = 7.5 Hz, 1H), 7.06 (t, J = 7.7 Hz, 1H), 7.05 (d, J = 1.7 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 8.1 Hz, 1H), 6.22 (s, 1H, exchangeable), 4.91 (d, J = 7.8 Hz, 1H), 3.89 (d, J = 4.9 Hz, 1H), 3.77 (m, 1H), 3.30 (m, 1H), 3.11 (m, 1H), 3.06–3.00 (m, 2H), 2.87 (m, 1H), 2.79 (m, 1H), 2.49–2.41 (m, 2H), 1.99–1.90 (m, 3H), 1.83–1.76 (m, 3H), 1.72 (m, 1H), 1.64 (m, 1H), 1.50–1.37 (m, 6H), 1.27 (m, 1H), 1.07 (m, 1H), 0.68 (m, 1H), 0.60 (m, 1H), 0.52 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.7, 142.2, 141.3, 134.7, 129.7, 127.9, 122.4, 121.7, 120.8, 120.6, 119.6, 119.3, 117.8, 117.3, 116.5, 89.9, 69.8, 61.7, 56.7, 50.8, 46.5, 45.6, 34.6, 33.8, 33.7, 29.4, 27.45, 26.4 (×2), 26.0, 23.9, 23.0, 5.7, 5.1, 2.6. HRMS calcd for C35H42N3O4 [M + H]+, 568.3170; found, 568.3157. mp 236 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3-phenyl-indole-7-carboxamido)morphinan (17).
A white solid was obtained in 85% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 11.32 (d, J = 2.1 Hz, 1H, exchangeable), 9.37 (s, 1H, exchangeable), 8.89 (s, 1H, exchangeable), 8.81 (d, J = 8.1 Hz, 1H, exchangeable), 8.06 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 7.5 Hz, 1H), 7.70–7.66 (m, 2H), 7.63 (d, J = 2.1 Hz, 1H), 7.44 (t, J = 7.8 Hz, 2H), 7.26 (t, J = 7.4 Hz, 1H), 7.21 (t, J = 7.7 Hz, 1H), 6.74 (d, J = 8.1 Hz, 1H), 6.67 (d, J = 8.1 Hz, 1H), 6.22 (s, 1H, exchangeable), 4.93 (d, J = 7.8 Hz, 1H), 3.85 (d, J = 5.2 Hz, 1H), 3.81 (m, 1H), 3.38 (m, 1H), 3.29 (m, 1H), 3.11 (m, 1H), 3.05 (m, 1H), 2.87 (m, 1H), 2.47–2.43 (m, 2H), 1.97 (m, 1H), 1.81 (m, 1H), 1.67 (m, 1H), 1.50 (m, 1H), 1.44 (m, 1H), 1.07 (m, 1H), 0.68 (m, 1H), 0.60 (m, 1H), 0.52 (m, 1H), 0.43 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.5, 142.2, 141.3, 135.2, 135.2, 129.7, 128.8 (×2), 126.8 (×2), 126.5, 125.6, 124.6, 122.8, 120.6, 120.3, 119.3, 118.9, 117.9, 117.0, 115.6, 89.9, 69.8, 61.8, 56.7, 50.9, 46.5, 45.6, 29.5, 27.4, 23.9, 23.0, 5.7, 5.1, 2.6. HRMS calcd for C35H36N3O4 [M + H]+, 562.2700; found, 562.2684. mp 240 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(2-bromo-indole-7-carboxamido)morphinan (18).
A white solid was obtained in 53% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 11.48 (s, 1H, exchangeable), 9.18 (s, 1H, exchangeable), 8.88 (s, 1H, exchangeable), 8.18 (d, J = 7.6 Hz, 1H, exchangeable), 7.67 (d, J = 7.7 Hz, 2H), 7.12 (t, J = 7.7 Hz, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.64 (s, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.32 (s, 1H, exchangeable), 4.87 (d, J = 3.6 Hz, 1H), 4.69 (m, 1H), 3.93 (d, J = 6.9 Hz, 1H), 3.38 (m, 1H), 3.11 (m, 1H), 3.06 (m, 1H), 2.96 (m, 1H), 2.74 (m, 1H), 2.54 (m, 1H), 2.46 (m, 1H), 1.93 (m, 1H), 1.68 (m, 1H), 1.57 (m, 1H), 1.48 (m, 1H), 1.21 (m, 1H), 1.08 (m, 1H), 0.70 (m, 1H), 0.62 (m, 1H), 0.50 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.2, 146.1, 138.8, 134.6, 129.4, 128.7, 122.6, 122.1, 120.7, 119.2, 119.0, 118.2, 117.2, 110.4, 103.8, 87.2, 79.2, 69.4, 61.0, 57.0, 45.8, 45.2, 30.2, 29.2, 23.4, 19.3, 5.7, 5.1, 2.5. HRMS calcd for C29H31N3O4Br [M + H]+, 564.1492; found, 564.1491. mp 233 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(2-chloro-indole-7-carboxamido)morphinan (19).
A white solid was obtained in 40% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 11.55 (s, 1H, exchangeable), 9.16 (s, 1H, exchangeable), 8.86 (s, 1H, exchangeable), 8.17 (d, J = 7.6 Hz, 1H, exchangeable), 7.68 (t, J = 7.0 Hz, 2H), 7.13 (t, J = 7.7 Hz, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.55 (d, J = 2.1 Hz, 1H), 6.28 (s, 1H, exchangeable), 4.87 (d, J = 3.7 Hz, 1H), 4.68 (m, 1H), 3.92 (d, J = 5.0 Hz, 1H), 3.40 (m, 1H), 3.11 (m, 1H), 3.05 (m, 1H), 2.95 (m, 1H), 2.75 (m, 1H), 2.45–2.43 (m, 2H), 1.92 (m, 1H), 1.68 (m, 1H), 1.57 (m, 1H), 1.48 (m, 1H), 1.20 (m, 1H), 1.08 (m, 1H), 0.70 (m, 1H), 0.62 (m, 1H), 0.49 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.2, 146.1, 138.8, 133.3, 128.7, 128.7, 124.3, 122.8, 122.1, 120.8, 119.2, 119.1, 118.3, 117.2, 99.8, 87.3, 69.5, 61.1, 57.1, 54.9, 45.8, 45.2, 30.3, 29.3, 23.5, 19.3, 5.7, 5.2, 2.6. HRMS calcd for C29H31N3O4Cl [M + H]+, 520.1998; found, 520.2012. mp 254 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(2-cyano-indole-7-carboxamido)morphinan (20).
A white solid was obtained in 36% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 12.29 (s, 1H, exchangeable), 9.18 (s, 1H, exchangeable), 8.89 (s, 1H, exchangeable), 8.32 (d, J = 7.5 Hz, 1H, exchangeable), 7.94 (d, J = 6.8 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.45 (d, J = 2.1 Hz, 1H), 7.26 (t, J = 7.7 Hz, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.36 (s, 1H, exchangeable), 4.87 (d, J = 3.7 Hz, 1H), 4.70 (m, 1H), 3.94 (d, J = 6.7 Hz, 1H), 3.27 (m, 1H), 3.11 (m, 1H), 3.05 (m, 1H), 2.95 (m, 1H), 2.75 (m, 1H), 2.54 (m, 1H), 2.47 (m, 1H), 1.96 (m, 1H), 1.68 (m, 1H), 1.57 (m, 1H), 1.49 (m, 1H), 1.24 (m, 1H), 1.09 (m, 1H), 0.70 (m, 1H), 0.63 (m, 1H), 0.50 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 165.7, 146.1, 138.8, 134.9, 128.7, 127.2, 125.3, 124.6, 122.1, 120.2, 119.1, 118.5, 118.2, 114.2, 113.5, 107.5, 87.1, 69.4, 61.2, 57.0, 45.9, 45.2, 30.3, 29.2, 23.5, 19.2, 5.7, 5.2, 2.6. HRMS calcd for C30H31N4O4 [M + H]+, 511.2340; found, 511.2360. mp 235 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(2-methyl-indole-7-carboxamido)morphinan (21).
A white solid was obtained in 88% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.86 (s, 1H, exchangeable), 9.21 (s, 1H, exchangeable), 8.88 (s, 1H, exchangeable), 8.06 (d, J = 7.6 Hz, 1H, exchangeable), 7.59 (dd, J = 7.6, 3.4 Hz, 2H), 7.02 (t, J = 7.6 Hz, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.34 (s, 1H, exchangeable), 6.19 (m, 1H), 4.88 (d, J = 3.7 Hz, 1H), 4.67 (m, 1H), 3.94 (d, J = 6.9 Hz, 1H), 3.38 (m, 1H), 3.11 (m, 1H), 3.05 (m, 1H), 2.96 (m, 1H), 2.74 (m, 1H), 2.54 (m, 1H), 2.46 (m, 1H), 2.43 (s, 3H), 1.95 (m, 1H), 1.68 (m, 1H), 1.56 (m, 1H), 1.48 (m, 1H), 1.21 (m, 1H), 1.08 (m, 1H), 0.70 (m, 1H), 0.63 (m, 1H), 0.50 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.8, 146.1, 138.8, 137.1, 134.5, 129.9, 128.7, 123.3, 122.5, 122.7, 119.2, 118.2, 117.8, 116.2, 99.2, 87.3, 69.4, 61.1, 57.0, 45.8, 45.2, 30.3, 29.3, 23.5, 19.3, 13.4, 5.7, 5.2, 2.6. HRMS calcd for C30H34N3O4 [M + H]+, 500.2544; found, 500.2540. mp 290 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(2-phenyl-indole-7-carboxamido)morphinan (22).
A white solid was obtained in 82% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.94 (s, 1H, exchangeable), 9.20 (s, 1H, exchangeable), 8.90 (s, 1H, exchangeable), 8.21 (d, J = 7.5 Hz, 1H, exchangeable), 7.90 (d, J = 7.4 Hz, 2H), 7.77 (d, J = 7.7 Hz, 2H), 7.48 (t, J = 7.7 Hz, 2H), 7.36 (t, J = 7.4 Hz, 1H), 7.14 (t, J = 7.6 Hz, 1H), 7.01 (d, J = 2.3 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 6.36 (s, 1H, exchangeable), 4.89 (d, J = 3.7 Hz, 1H), 4.74 (m, 1H), 3.96 (d, J = 6.7 Hz, 1H), 3.39 (m, 1H), 3.12 (m, 1H), 3.06 (m, 1H), 2.99 (m, 1H), 2.74 (m, 1H), 2.58–2.51 (m, 2H), 1.97 (m, 1H), 1.68 (m, 1H), 1.59 (m, 1H), 1.50 (m, 1H), 1.24 (m, 1H), 1.09 (m, 1H), 0.71 (m, 1H), 0.63 (m, 1H), 0.51 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.6, 146.0, 138.8, 138.4, 135.6, 131.4, 129.9, 128.8 (×2), 128.6, 127.8, 125.3 (×2), 123.7, 122.0, 120.8, 119.1, 118.9, 118.3, 116.7, 99.1, 87.2, 69.4, 61.1, 57.0, 45.7, 45.2, 45.2, 30.2, 29.3, 23.5, 19.4, 5.6, 5.1, 2.5. HRMS calcd for C35H36N3O4 [M + H]+, 562.2700; found, 562.2711. mp 233 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(3-bromo-indole-7-carboxamido)morphinan (23).
A white solid was obtained in 84% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 11.45 (d, J = 2.0 Hz, 1H, exchangeable), 9.24 (s, 1H, exchangeable), 8.90 (s, 1H, exchangeable), 8.28 (d, J = 7.5 Hz, 1H, exchangeable), 7.83 (d, J = 7.0 Hz, 1H), 7.62 (d, J = 7.9 Hz, 1H), 7.50 (d, J = 2.0 Hz, 1H), 7.23 (t, J = 7.7 Hz, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.37 (s, 1H, exchangeable), 4.87 (d, J = 3.8 Hz, 1H), 4.68 (m, 1H), 3.94 (d, J = 6.9 Hz, 1H), 3.31–3.26 (m, 2H), 3.14–3.03 (m, 2H), 2.96 (m, 1H), 2.73 (m, 1H), 2.47 (m, 1H), 1.95 (m, 1H), 1.68 (m, 1H), 1.55 (m, 1H), 1.48 (m, 1H), 1.22 (m, 1H), 1.07 (m, 1H), 0.70 (m, 1H), 0.62 (m, 1H), 0.49 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.1, 146.1, 138.8, 133.6, 128.7, 127.4, 126.1, 122.1, 121.7, 121.7, 119.2, 119.2, 118.2, 117.7, 99.5, 89.0, 87.2, 69.4, 61.1, 57.1, 45.9, 45.2, 30.3, 29.2, 23.5, 19.2, 5.6, 5.2, 2.6. HRMS calcd for C29H31N3O4Br [M + H]+, 564.1492; found, 564.1492. mp 234 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(3-chloro-indole-7-carboxamido)morphinan (24).
A white solid was obtained in 95% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 11.35 (d, J = 2.1 Hz, 1H, exchangeable), 9.24 (s, 1H, exchangeable), 8.90 (s, 1H, exchangeable), 8.28 (d, J = 7.5 Hz, 1H, exchangeable), 7.83 (d, J = 7.1 Hz, 1H), 7.70 (d, J = 7.9 Hz, 1H), 7.47 (d, J = 2.1 Hz, 1H), 7.22 (t, J = 7.7 Hz, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.38 (s, 1H, exchangeable), 4.87 (d, J = 3.8 Hz, 1H), 4.68 (m, 1H), 3.94 (d, J = 6.8 Hz, 1H), 3.38 (m, 1H), 3.30 (m, 1H), 3.13–3.02 (m, 2H), 2.96 (m, 1H), 2.74 (m, 1H), 2.46 (m, 1H), 1.95 (m, 1H), 1.67 (m, 1H), 1.56 (m, 1H), 1.48 (m, 1H), 1.22 (m, 1H), 1.07 (m, 1H), 0.69 (m, 1H), 0.62 (m, 1H), 0.49 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.1, 146.1, 138.8, 133.1, 128.7, 125.8, 123.6, 122.1, 121.7, 120.8, 119.2, 119.0, 118.2, 117.7, 103.5, 87.2, 69.4, 61.1, 57.0, 45.9, 45.2, 30.2, 29.2, 23.5, 19.2, 5.7, 5.2, 2.6. HRMS calcd for C29H31N3O4Cl [M + H]+, 520.1998; found, 520.1996. mp 236 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(3-cyano-indole-7-carboxamido)morphinan (25).
A white solid was obtained in 72% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 12.14 (d, J = 2.4 Hz, 1H, exchangeable), 9.22 (s, 1H, exchangeable), 8.89 (s, 1H, exchangeable), 8.40 (d, J = 7.5 Hz, 1H, exchangeable), 8.20 (d, J = 2.4 Hz, 1H), 7.92 (d, J = 7.0 Hz, 1H), 7.85 (d, J = 7.9 Hz, 1H), 7.35 (t, J = 7.7 Hz, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.34 (s, 1H, exchangeable), 4.87 (d, J = 3.8 Hz, 1H), 4.69 (m, 1H), 3.93 (d, J = 6.8 Hz, 1H), 3.28 (m, 1H), 3.10 (m, 1H), 3.04 (m, 1H), 2.95 (m, 1H), 2.73 (m, 1H), 2.48–2.43 (m, 2H), 1.93 (m, 1H), 1.68 (m, 1H), 1.56 (m, 1H), 1.49 (m, 1H), 1.24 (m, 1H), 1.08 (m, 1H), 0.70 (m, 1H), 0.62 (m, 1H), 0.49 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 165.6, 146.1, 138.8, 135.7, 133.3, 128.7, 128.1, 122.7, 122.1, 122.0, 121.1, 119.2, 118.8, 118.3, 116.0, 87.1, 84.6, 69.4, 61.1, 57.0, 52.8, 46.0, 45.2, 30.4, 29.2, 23.5, 19.1, 5.7, 5.2, 2.6. HRMS calcd for C30H31N4O4 [M + H]+, 511.2340; found, 511.2325. mp 263 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(3-nitro-indole-7-carboxamido)morphinan (26).
A white solid was obtained in 58% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 12.46 (d, J = 2.9 Hz, 1H, exchangeable), 9.24 (s, 1H, exchangeable), 8.90 (s, 1H, exchangeable), 8.51 (d, J = 7.5 Hz, 1H, exchangeable), 8.44 (d, J = 2.9 Hz, 1H), 8.29 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 6.9 Hz, 1H), 7.48 (t, J = 7.8 Hz, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 6.37 (s, 1H, exchangeable), 4.88 (d, J = 3.8 Hz, 1H), 4.70 (m, 1H), 3.94 (d, J = 6.7 Hz, 1H), 3.28 (m, 1H), 3.14–3.03 (m, 2H), 2.96 (m, 1H), 2.74 (m, 1H), 2.53 (m, 1H), 2.45 (m, 1H), 1.95 (m, 1H), 1.68 (m, 1H), 1.57 (m, 1H), 1.49 (m, 1H), 1.25 (m, 1H), 1.08 (m, 1H), 0.69 (m, 1H), 0.62 (m, 1H), 0.49 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 165.5, 146.0, 138.8, 133.0, 130.9, 128.7, 128.4, 123.4, 123.1, 122.9, 122.1, 120.9, 119.2, 119.0, 118.2, 87.0, 69.4, 61.0, 57.0, 51.4, 46.1, 45.2, 30.3, 29.2, 23.5, 19.1, 5.7, 5.2, 2.6. HRMS calcd for C29H31N4O6 [M + H]+, 531.2238; found, 531.2248. mp 260 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(3-formyl-indole-7-carboxamido)morphinan (27).
A white solid was obtained in 59% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 12.05 (s, 1H, exchangeable), 9.99 (s, 1H, exchangeable), 9.20 (s, 1H, exchangeable), 8.87 (s, 1H, exchangeable), 8.34 (d, J = 7.5 Hz, 1H, exchangeable), 8.29 (d, J = 7.8 Hz, 1H), 8.25 (d, J = 3.2 Hz, 1H), 7.89 (d, J = 7.1 Hz, 1H), 7.33 (t, J = 7.8 Hz, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 6.30 (s, 1H, exchangeable), 4.88 (d, J = 3.8 Hz, 1H), 4.70 (m, 1H), 3.92 (d, J = 6.6 Hz, 1H), 3.37–3.34 (m, 2H), 3.12–3.04 (m, 2H), 2.94 (m, 1H), 2.74 (m, 1H), 2.53 (m, 1H), 1.93 (m, 1H), 1.69 (m, 1H), 1.58–1.47 (m, 2H), 1.24 (m, 1H), 1.05 (m, 1H), 0.70 (m, 1H), 0.62 (m, 1H), 0.49 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 185.4, 165.9, 146.0, 139.3, 138.8, 135.2, 128.6, 125.4, 124.3, 122.5, 122.0, 121.5, 119.2, 118.2, 118.1, 117.7, 87.0, 69.4, 61.1, 57.0, 50.5, 47.1, 45.2, 30.3, 29.2, 22.1, 19.2, 5.6, 5.1, 2.5. HRMS calcd for C30H32N3O5 [M + H]+, 514.2336; found, 514.2314. mp 260 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(3-acetyl-indole-7-carboxamido)morphinan (28).
A white solid was obtained in 70% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 11.88 (s, 1H, exchangeable), 9.21 (s, 1H), 8.88 (s, 1H, exchangeable), 8.38 (d, J = 7.9 Hz, 1H), 8.31 (d, J = 7.4 Hz, 1H, exchangeable), 8.21 (d, J = 3.0 Hz, 1H), 7.84 (d, J = 7.5 Hz, 1H), 7.28 (t, J = 7.7 Hz, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 6.34 (s, 1H, exchangeable), 4.88 (d, J = 3.7 Hz, 1H), 4.69 (m, 1H), 3.93 (d, J = 6.5 Hz, 1H), 3.29 (m, 1H), 3.15–3.00 (m, 3H), 2.95 (m, 1H), 2.74 (m, 1H), 2.52 (m, 1H), 2.46 (s, 3H), 1.94 (m, 1H), 1.67 (m, 1H), 1.59–1.46 (m, 2H), 1.24 (m, 1H), 1.07 (m, 1H), 0.69 (m, 1H), 0.62 (m, 1H), 0.49 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 192.9, 166.1, 146.0, 138.8, 135.2, 134.8, 128.7, 126.6, 124.9, 122.1, 122.0, 121.0, 119.2, 118.2, 117.8, 116.4, 87.1, 69.4, 61.1, 57.0, 50.6, 46.0, 45.2, 30.3, 29.2, 27.4, 23.5, 19.2, 5.7, 5.2, 2.6. HRMS calcd for C31H34N3O5 [M + H]+, 528.2493; found, 528.2477. mp 251 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(3-acet-amido-indole-7-carboxamido)morphinan (29).
A white solid was obtained in 80% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.77 (s, 1H, exchangeable), 9.92 (s, 1H, exchangeable), 9.22 (s, 1H, exchangeable), 8.87 (s, 1H, exchangeable), 8.13 (d, J = 7.5 Hz, 1H, exchangeable), 8.02 (d, J = 7.9 Hz, 1H), 7.77 (d, J = 2.5 Hz, 1H), 7.73 (d, J = 7.3 Hz, 1H), 7.10 (t, J = 7.7 Hz, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.31 (s, 1H, exchangeable), 4.88 (d, J = 3.8 Hz, 1H), 4.67 (m, 1H), 3.92 (d, J = 6.8 Hz, 1H), 3.28–3.24 (m, 2H), 3.12–3.03 (m, 2H), 2.93 (m, 1H), 2.75 (m, 1H), 2.54 (m, 1H), 2.10 (s, 3H), 1.95 (m, 1H), 1.66 (m, 1H), 1.58–1.46 (m, 2H), 1.22 (m, 1H), 1.06 (m, 1H), 0.69 (m, 1H), 0.62 (m, 1H), 0.49 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 167.0, 166.6, 146.0, 138.8, 131.6, 128.7, 122.1, 121.7, 121.7, 120.8, 119.2, 118.2, 117.2, 116.6, 116.3, 115.2, 87.2, 69.4, 64.9, 61.1, 57.0, 45.8, 45.2, 30.3, 29.2, 25.6, 23.5, 23.0, 5.7, 5.2, 2.6. HRMS calcd for C31H35N4O5 [M + H]+, 543.2602; found, 543.2577. mp 259 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(3-methyl-indole-7-carboxamido)morphinan (30).
A white solid was obtained in 91% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.77 (d, J = 0.9 Hz, 1H, exchangeable), 9.23 (s, 1H, exchangeable), 8.88 (s, 1H, exchangeable), 8.09 (d, J = 7.5 Hz, 1H, exchangeable), 7.70 (t, J = 7.5 Hz, 2H), 7.14 (d, J = 0.9 Hz, 1H), 7.08 (t, J = 7.6 Hz, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.32 (s, 1H, exchangeable), 4.87 (d, J = 3.8 Hz, 1H), 4.67 (m, 1H), 3.92 (d, J = 6.8 Hz, 1H), 3.28 (m, 1H), 3.10 (m, 1H), 3.04 (m, 1H), 2.95 (m, 1H), 2.75 (m, 1H), 2.47–2.43 (m, 2H), 2.28 (d, J = 0.9 Hz, 3H), 1.93 (m, 1H), 1.67 (m, 1H), 1.56 (m, 1H), 1.48 (m, 1H), 1.22 (m, 1H), 1.08 (m, 1H), 0.70 (m, 1H), 0.62 (m, 1H), 0.49 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.7, 146.1, 138.8, 134.5, 129.4, 128.7, 124.1, 122.1, 122.0, 120.3, 119.2, 118.2, 117.3, 116.6, 109.3, 87.3, 69.4, 61.1, 57.0, 53.2, 45.8, 45.2, 30.3, 29.3, 23.5, 19.3, 9.4, 5.7, 5.2, 2.6. HRMS calcd for C30H34N3O4 [M + H]+, 500.2544; found, 500.2544. mp 242 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(3-iso-propyl-indole-7-carboxamido)morphinan (31).
A white solid was obtained in 94% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.80 (s, 1H, exchangeable), 9.24 (s, 1H, exchangeable), 8.90 (s, 1H, exchangeable), 8.10 (d, J = 7.6 Hz, 1H, exchangeable), 7.76 (d, J = 7.8 Hz, 1H), 7.70 (d, J = 7.2 Hz, 1H), 7.10 (d, J = 2.0 Hz, 1H), 7.07 (t, J = 7.7 Hz, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.36 (s, 1H, exchangeable), 4.87 (d, J = 3.8 Hz, 1H), 4.67 (m, 1H), 3.94 (d, J = 6.8 Hz, 1H), 3.29 (m, 1H), 3.17 (m, 1H), 3.10 (m, 1H), 3.04 (m, 1H), 2.96 (m, 1H), 2.74 (m, 1H), 2.53 (m, 1H), 2.46 (m, 1H), 1.96 (m, 1H), 1.67 (m, 1H), 1.55 (m, 1H), 1.47 (m, 1H), 1.31 (d, J = 2.2 Hz, 3H), 1.30 (d, J = 2.2 Hz, 3H), 1.21 (m, 1H), 1.08 (m, 1H), 0.70 (m, 1H), 0.62 (m, 1H), 0.49 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.8, 146.1, 138.8, 134.8, 128.7, 127.7, 122.4, 122.1, 121.7, 121.5, 120.2, 119.1, 118.2, 117.3, 116.8, 87.2, 69.4, 61.1, 57.0, 45.8, 45.2, 30.3, 29.3, 27.4, 24.7, 23.5, 23.4, 19.3, 5.7, 5.2, 2.6. HRMS calcd for C32H38N3O4 [M + H]+, 528.2857; found, 528.2867. mp 220 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(3-cyclo-pentyl-indole-7-carboxamido)morphinan (32).
A white solid was obtained in 81% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.77 (s, 1H, exchangeable), 9.22 (s, 1H, exchangeable), 8.89 (s, 1H, exchangeable), 8.08 (d, J = 7.5 Hz, 1H, exchangeable), 7.75 (d, J = 7.8 Hz, 1H), 7.70 (d, J = 7.4 Hz, 1H), 7.11 (d, J = 1.9 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.35 (s, 1H, exchangeable), 4.87 (d, J = 3.8 Hz, 1H), 4.67 (m, 1H), 3.94 (d, J = 6.6 Hz, 1H), 3.28 (m, 1H), 3.24 (m, 1H), 3.10 (m, 1H), 3.04 (m, 1H), 2.95 (m, 1H), 2.73 (m, 1H), 2.57–2.51 (m, 2H), 2.14–2.05 (m, 2H), 1.95 (m, 1H), 1.81–1.72 (m, 2H), 1.72–1.58 (m, 6H), 1.48 (m, 1H), 1.22 (m, 1H), 1.09 (m, 1H), 0.70 (m, 1H), 0.62 (m, 1H), 0.49 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.7, 146.0, 138.8, 134.8, 128.7, 128.3, 122.6, 122.1, 121.9, 120.3, 119.1, 119.0, 118.2, 117.3, 116.8, 87.2, 69.4, 61.1, 57.5, 57.0, 45.8, 45.2, 36.2, 33.0, 32.9, 30.3, 29.2, 24.8 (×2), 23.5, 19.3, 5.7, 5.2, 2.6. HRMS calcd for C34H40N3O4 [M + H]+, 554.3013; found, 554.3006. mp 239 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(3-cyclo-hexyl-indole-7-carboxamido)morphinan (33).
A white solid was obtained in 91% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 10.79 (d, J = 1.7 Hz, 1H, exchangeable), 9.24 (s, 1H, exchangeable), 8.89 (s, 1H, exchangeable), 8.08 (d, J = 7.6 Hz, 1H, exchangeable), 7.76 (d, J = 7.8 Hz, 1H), 7.69 (d, J = 7.1 Hz, 1H), 7.08 (d, J = 1.7 Hz, 1H), 7.05 (d, J = 7.7 Hz, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.35 (s, 1H, exchangeable), 4.87 (d, J = 3.8 Hz, 1H), 4.67 (m, 1H), 3.94 (d, J = 6.7 Hz, 1H), 3.27 (m, 1H), 3.10 (m, 1H), 3.04 (m, 1H), 2.96 (m, 1H), 2.83–2.70 (m, 2H), 2.53 (m, 1H), 2.47 (m, 1H), 2.01–1.97 (m, 2H), 1.92 (m, 1H), 1.83–1.77 (m, 2H), 1.75–1.66 (m, 2H), 1.57–1.48 (m, 2H), 1.47–1.38 (m, 4H), 1.31–1.17 (m, 2H), 1.07 (m, 1H), 0.69 (m, 1H), 0.61 (m, 1H), 0.49 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.8, 146.0, 138.8, 134.6, 128.7, 127.7, 122.3, 122.1, 121.6, 121.0, 120.2, 119.1, 118.2, 117.3, 116.8, 87.2, 69.4, 61.1, 57.0, 50.7, 45.8, 45.2, 34.6, 33.8 (×2), 30.3, 29.3, 26.4 (×2), 26.0, 23.5, 19.3, 5.7, 5.2, 2.6. HRMS calcd for C35H42N3O4 [M + H]+, 568.3170; found, 568.3186. mp 251 °C, dec.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(3-phenyl-indole-7-carboxamido)morphinan (34).
A white solid was obtained in 84% yield. Hydrochloride salt: 1H NMR (400 MHz, DMSO-d6): δ 11.37 (d, J = 1.9 Hz, 1H, exchangeable), 9.24 (s, 1H, exchangeable), 8.90 (s, 1H, exchangeable), 8.22 (d, J = 7.5 Hz, 1H, exchangeable), 8.06 (d, J = 8.0 Hz, 1H), 7.80 (d, J = 7.2 Hz, 1H), 7.69 (dd, J = 8.0, 1.0 Hz, 2H), 7.66 (d, J = 1.9 Hz, 1H), 7.45 (t, J = 7.8 Hz, 2H), 7.26 (t, J = 7.4 Hz, 1H), 7.20 (t, J = 7.7 Hz, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 6.37 (s, 1H, exchangeable), 4.90 (d, J = 3.8 Hz, 1H), 4.71 (m, 1H), 3.95 (d, J = 6.8 Hz, 1H), 3.28 (m, 1H), 3.11 (m, 1H), 3.05 (m, 1H), 2.97 (m, 1H), 2.75 (m, 1H), 2.55–2.53 (m, 2H), 1.96 (m, 1H), 1.69 (m, 1H), 1.58 (m, 1H), 1.49 (m, 1H), 1.24 (m, 1H), 1.09 (m, 1H), 0.70 (m, 1H), 0.62 (m, 1H), 0.50 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.6, 146.1, 138.8, 135.3, 135.1, 128.9 (×2), 128.7, 126.8 (×2), 126.4, 125.6, 124.6, 122.7, 122.1, 120.9, 119.2, 118.9, 118.3, 117.4, 115.8, 87.2, 69.4, 61.1, 57.0, 50.6, 45.9, 45.2, 30.3, 29.3, 23.5, 19.3, 5.7, 5.2, 2.6. HRMS calcd for C35H36N3O4 [M + H]+, 562.2700; found, 562.2705. mp 278 °C, dec.
Biological Evaluation.
Drugs.
Morphine (morphine sulfate pentahydrate salt) was purchased from Mallinckrodt (St. Louis, MO) or provided by the National Institute of Drug Abuse (NIDA). Naltrexone was purchased from Sigma-Aldrich (St. Louis, MO). All other reagents and radioligands were purchased from either Sigma-Aldrich or Thermo Fisher.
Animals.
Male Swiss-Webster mice (23–35 g, 7–8 weeks, Harlan Laboratories, Indianapolis, IN) were housed five to a cage in animal care quarters maintained at 22 °C on a 12 h light/dark cycle with food and water available ad libitum. Protocols and procedures were approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University Medical Center and complied with the recommendations of the International Association for the Study of Pain.
In Vitro Competitive Radioligand Binding Assay and Functional Assay.
Competition binding was performed using the monoclonal opioid receptor expressed in CHO, as previously described.14
Calcium Flux Assay.
The ligands were first tested with various concentrations (0.3 nM to 3 μM) for possible agonist activity in MOR-CHO cells. MOR-CHO cells were transfected with Gqi5 pcDNA1 using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s recommended procedure. Cells were incubated for 4 h at 37 °C and 5% CO2 and then trypsinized and transferred to a clear bottom, black 96-well plate (Greiner Bio-one) at 3 × 106 cells per well in their respective growth media and incubated until confluent. 48 h after transfection, the growth media was decanted, and cells were then incubated with 50 μL of fluo-4 AM loading buffer [24 μL 2 mM fluo-4 AM solution (Invitrogen), 12 μL 250 mM probenecid, in 6 mL assay buffer (HBSS–HEPES–Ca–Mg-probenecid)] for 45 min. Loading buffer was then decanted, and cells were incubated for an additional 15 min in 20 μL of each compound in varying concentrations and 60 μL assay buffer. Ca2+ concentrations were monitored by RFU for 90 s right after addition of 20 μL of agonist (DAMGO) to each well in the microplate reader (FlexStation3, Molecular Devices). Peak values were obtained using SoftMaxPro software (Molecular Devices), and non-linear regression curves were generated using GraphPad Prism to calculate IC50 values. All doses were tested with triplicates. All experiments were repeated at least four times to obtain standard error values.
Warm-Water Immersion Assay.
Antinociception studies for all synthesized derivatives were conducted using male Swiss-Webster mice, as previously reported.14
Desirability Functions and MPO Score Calculation.
The calculated physicochemical properties employed herein were obtained from ACD/Percepta (v2020.2.0) software. The CNS MPO scores were calculated using the previously published method.49
Foundational ADMET Assessment.
The ADMET assessments including the hERG inhibitory activity, metabolic stability profiles, and other drug-like properties were conducted by Eurofins Panlabs, Inc. following previously reported protocols.66,74–76
Assay Procedures for hERG Activity.66
Inhibition of the hERG potassium channel was examined in CHO-K1 (Chinese Hamster Ovary) cells stably transfected with hERG cDNA. CHO-K1 cells expressing hERG were plated in 35 mm dishes at least 24 h prior to the experiment and maintained at 37 °C under 5% CO2. A micropipette was drawn out from borosilicate glass to give a tip resistance between 3 and 5 MΩ. For each experiment, a single dish of cells was removed from the incubator, washed twice with extracellular solution at room temperature before being applied to the automated patch-clamp sites.
After whole cell configuration is achieved, the cell is held at −80 mV. A 50 ms pulse to −40 mV is delivered to measure the leaking current, which is subtracted from the tail current online. Then, the cell is depolarized to +20 mV for 2 s, followed by a 1 s pulse to −40 mV to reveal the hERG tail current. This paradigm is delivered once every 5 s to monitor the current amplitude. The assay is conducted at room temperature. The extracellular solution is applied first, and the cell is stabilized in the solution for 5 min. Then, the tested compound is applied from low to high concentration sequentially on the same cell. Peak tail amplitudes were then plotted as a function of the sweep number. Five peak tail current measurements at the steady state before the test compound application were averaged and used as the control current amplitude. Four or five peak tail current measurements at the steady state after the test compound application were averaged and used as the remaining current amplitude after inhibition by the test compound. Percent inhibition was calculated according to the following equation
Opioid-Withdrawal Studies.
This experiment was conducted, as previously reported.14 The data were expressed as mean ± SEM.
Statistical Analysis.
One-way ANOVA followed by the posthoc Dunnett test were performed to assess significance using Prism 6.0 software (GraphPad Software, San Diego, CA).
Supplementary Material
Funding
This work was partially supported by NIH DA024022 and DA050311 (Y.Z.). The authors are grateful to NIDA Drug Supply Program for providing the free base of naltrexone. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Drug Abuse or the National Institutes of Health.
ABBREVIATIONS
- ADMET
absorption, distribution, metabolism, excretion, and toxicity
- CHO
Chinese hamster ovary
- CL
confidence level
- CNS
central nervous system
- DAMGO
[D-Ala2-MePhe4-Gly(ol)5]enkephalin
- DOR
delta opioid receptor
- GPCRs
G protein-coupled receptors
- HBA
hydrogen bond acceptor count
- HBD
hydrogen bond donor count
- hERG
human ether-a-go-go-related gene
- KOR
kappa opioid receptor
- LQTS
long QT syndrome
- MOR
mu opioid receptor
- % MPE
percentage maximum possible effect
- NAN
17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(indole-7-carboxamido)morphinan
- NIDA
National Institute of Drug Abuse
- OUD
opioid use disorder
- tPSA
topological polar surface are
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01499?goto=supporting-info.
Spectra data for newly reported compounds (1H NMR, 13C NMR, and HPLC graphs); synthetic procedure for 2′ and 3′-substituted indole-7-carboxylic acids; and in silico physicochemical property prediction of target compounds (PDF)
Molecular formula strings and some data (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.2c01499
The authors declare no competing financial interest.
Contributor Information
Hongguang Ma, Department of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University, Richmond, Virginia 23298, United States.
Piyusha P. Pagare, Department of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University, Richmond, Virginia 23298, United States
Mengchu Li, Department of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University, Richmond, Virginia 23298, United States.
Logan T. Neel, Department of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University, Richmond, Virginia 23298, United States
Rolando E. Mendez, Department of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, Virginia 23298, United States
James C. Gillespie, Department of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, Virginia 23298, United States
David L. Stevens, Department of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, Virginia 23298, United States
William L. Dewey, Department of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, Virginia 23298, United States
Dana E. Selley, Department of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, Virginia 23298, United States
Yan Zhang, Department of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University, Richmond, Virginia 23298, United States; Institute for Drug and Alcohol Studies, Richmond, Virginia 23298-0059, United States.
REFERENCES
- (1).Wilson N; Kariisa M; Seth P; Smith H; Davis NL Drug and Opioid-Involved Overdose Deaths — United States, 2017–2018. Morb. Mortal. Wkly. Rep 2020, 69, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).CDC/NCHS. National Vital Statistics System, Mortality. CDC Wonder 2018; US Department of Health and Human Services: Atlanta, GA, 2018. [Google Scholar]
- (3).Ahmad FB; Rossen LM; Sutton P Provisional Drug Overdose Death Counts 2020; National Center for Health Statistics: Atlanta, 2020. [Google Scholar]
- (4).Isbell H; Vogel VH The addiction liability of methadon (amidone, dolophine, 10820) and its use in the treatment of the morphine abstinence syndrome. Am. J. Psychiatry 1949, 105, 909–914. [DOI] [PubMed] [Google Scholar]
- (5).Walsh SL; Preston KL; Stitzer ML; Cone EJ; Bigelow GE Clinical pharmacology of buprenorphine: ceiling effects at high doses. Clin. Pharmacol. Ther 1994, 55, 569–580. [DOI] [PubMed] [Google Scholar]
- (6).O’brien CP; Greenstein RA; Mintz J; Woody GE Clinical experience with naltrexone. Am. J. Drug Alcohol Abuse 1975, 2, 365–377. [DOI] [PubMed] [Google Scholar]
- (7).Li G; Aschenbach LC; Chen J; Cassidy MP; Stevens DL; Gabra BH; Selley DE; Dewey WL; Westkaemper RB; Zhang Y Design, Synthesis, and Biological Evaluation of 6α- and 6β-N-Heterocyclic Substituted Naltrexamine Derivatives as μ Opioid Receptor Selective Antagonists. J. Med. Chem 2009, 52, 1416–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yuan Y; Elbegdorj O; Chen J; Akubathini SK; Beletskaya IO; Selley DE; Zhang Y Structure selectivity relationship studies of 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4′-pyridyl)-carboxamido]morphinan derivatives toward the development of the mu opioid receptor antagonists. Bioorg. Med. Chem. Lett 2011, 21, 5625–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yuan Y; Elbegdorj O; Chen J; Akubathini SK; Zhang F; Stevens DL; Beletskaya IO; Scoggins KL; Zhang Z; Gerk PM; Selley DE; Akbarali HI; Dewey WL; Zhang Y Design, Synthesis, and Biological Evaluation of 17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4′-pyridyl)carboxamido]morphinan Derivatives as Peripheral Selective μ Opioid Receptor Agents. J. Med. Chem 2012, 55, 10118–10129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yuan Y; Zaidi SA; Stevens DL; Scoggins KL; Mosier PD; Kellogg GE; Dewey WL; Selley DE; Zhang Y Design, syntheses, and pharmacological characterization of 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(isoquinoline-3′-carboxamido)morphinan analogues as opioid receptor ligands. Bioorg. Med. Chem 2015, 23, 1701–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yuan Y; Zaidi SA; Elbegdorj O; Aschenbach LC; Li G; Stevens DL; Scoggins KL; Dewey WL; Selley DE; Zhang Y Design, synthesis, and biological evaluation of 14-heteroaromatic-substituted naltrexone derivatives: pharmacological profile switch from mu opioid receptor selectivity to mu/kappa opioid receptor dual selectivity. J. Med. Chem 2013, 56, 9156–9169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zheng Y; Obeng S; Wang H; Jali AM; Peddibhotla B; Williams DA; Zou C; Stevens DL; Dewey WL; Akbarali HI; Selley DE; Zhang Y Design, Synthesis, and Biological Evaluation of the Third Generation 17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4′-pyridyl)carboxamido]morphinan (NAP) Derivatives as μ/κ Opioid Receptor Dual Selective Ligands. J. Med. Chem 2019, 62, 561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Obeng S; Wang H; Jali A; Stevens DL; Akbarali HI; Dewey WL; Selley DE; Zhang Y Structure-Activity Relationship Studies of 6α- and 6β-Indolylacetamidonaltrexamine Derivatives as Bitopic Mu Opioid Receptor Modulators and Elaboration of the “Message-Address Concept” To Comprehend Their Functional Conversion. ACS Chem. Neurosci 2018, 10, 1075–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ma H; Obeng S; Wang H; Zheng Y; Li M; Jali AM; Stevens DL; Dewey WL; Selley DE; Zhang Y Application of Bivalent Bioisostere Concept on Design and Discovery of Potent Opioid Receptor Modulators. J. Med. Chem 2019, 62, 11399–11415. [DOI] [PubMed] [Google Scholar]
- (15).Obeng S; Jali A; Zheng Y; Wang H; Schwienteck KL; Chen C; Stevens DL; Akbarali HI; Dewey WL; Banks ML; Liu-Chen LY; Selley DE; Zhang Y Characterization of 17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(indole-7-carboxamido)morphinan (NAN) as a Novel Opioid Receptor Modulator for Opioid Use Disorder Treatment. ACS Chem. Neurosci 2019, 10, 2518–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Obeng S; Yuan Y; Jali A; Selley DE; Zhang Y In vitro and in vivo functional profile characterization of 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(isoquinoline-3-carboxamido)-morphinan (NAQ) as a low efficacy mu opioid receptor modulator. Eur. J. Pharmacol 2018, 827, 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Fronik P; Gaiser BI; Sejer Pedersen D Bitopic Ligands and Metastable Binding Sites: Opportunities for G Protein-Coupled Receptor (GPCR) Medicinal Chemistry. J. Med. Chem 2017, 60, 4126–4134. [DOI] [PubMed] [Google Scholar]
- (18).Gentry PR; Sexton PM; Christopoulos A Novel Allosteric Modulators of G Protein-coupled Receptors. J. Biol. Chem 2015, 290, 19478–19488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Feng Z; Hu G; Ma S; Xie XQ Computational Advances for the Development of Allosteric Modulators and Bitopic Ligands in G Protein-Coupled Receptors. AAPS J. 2015, 17, 1080–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Christopoulos A Advances in G protein-coupled receptor allostery: from function to structure. Mol. Pharmacol 2014, 86, 463–478. [DOI] [PubMed] [Google Scholar]
- (21).Burford NT; Watson J; Bertekap R; Alt A Strategies for the identification of allosteric modulators of G-protein-coupled receptors. Biochem. Pharmacol 2011, 81, 691–702. [DOI] [PubMed] [Google Scholar]
- (22).Redfern WS; Carlsson L; Davis AS; Lynch WG; Mackenzie I; Palethorpe S; Siegl PKS; Strang I; Sullivan AT; Wallis R; Camm AJ; Hammond TG Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res 2003, 58, 32–45. [DOI] [PubMed] [Google Scholar]
- (23).Chaudhary AMD; Khan MF; Dhillon SS; Naveed S A Review of Samidorphan: A Novel Opioid Antagonist. Cureus 2019, 11, No. e5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mahida S; Hogarth AJ; Cowan C; Tayebjee MH; Graham LN; Pepper CB Genetics of congenital and drug-induced long QT syndromes: current evidence and future research perspectives. J. Intervent. Card Electrophysiol 2013, 37, 9–19. [DOI] [PubMed] [Google Scholar]
- (25).Garrido A; Lepailleur A; Mignani SM; Dallemagne P; Rochais C hERG toxicity assessment: Useful guidelines for drug design. Eur. J. Med. Chem 2020, 195, 112290. [DOI] [PubMed] [Google Scholar]
- (26).Jamieson C; Moir EM; Rankovic Z; Wishart G Medicinal chemistry of hERG optimizations: Highlights and hang-ups. J. Med. Chem 2006, 49, 5029–5046. [DOI] [PubMed] [Google Scholar]
- (27).Vaz RJ; Gao Z; Pribish J; Chen X; Levell J; Davis L; Albert E; Brollo M; Ugolini A; Cramer DM; Cairns J; Sides K; Liu F; Kwong J; Kang J; Rebello S; Elliot M; Lim H; Chellaraj V; Singleton RW; Li Y Design of bivalent ligands using hydrogen bond linkers: synthesis and evaluation of inhibitors for human β-tryptase. Bioorg. Med. Chem. Lett 2004, 14, 6053–6056. [DOI] [PubMed] [Google Scholar]
- (28).Fraley ME; Arrington KL; Buser CA; Ciecko PA; Coll KE; Fernandes C; Hartman GD; Hoffman WF; Lynch JJ; McFall RC; Rickert K; Singh R; Smith S; Thomas KA; Wong BK Optimization of the indolyl quinolinone class of KDR (VEGFR-2) kinase inhibitors. Bioorg. Med. Chem. Lett 2004, 14, 351–355. [DOI] [PubMed] [Google Scholar]
- (29).Fletcher SR; Burkamp F; Blurton P; Cheng SK; Clarkson R; O’Connor D; Spinks D; Tudge M; van Niel MB; Patel S; Chapman K; Marwood R; Shepheard S; Bentley G; Cook GP; Bristow LJ; Castro JL; Hutson PH; MacLeod AM 4-(Phenylsulfonyl)piperidines: Novel, Selective, and Bioavailable 5-HT2A Receptor Antagonists. J. Med. Chem 2002, 45, 492–503. [DOI] [PubMed] [Google Scholar]
- (30).Wager TT; Chandrasekaran RY; Hou X; Troutman MD; Verhoest PR; Villalobos A; Will Y Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem. Neurosci 2010, 1, 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Rankovic Z CNS drug design: balancing physicochemical properties for optimal brain exposure. J. Med. Chem 2015, 58, 2584–2608. [DOI] [PubMed] [Google Scholar]
- (32).Viscusi ER; Viscusi AR Blood-brain barrier: mechanisms governing permeability and interaction with peripherally acting μ-opioid receptor antagonists. Reg. Anesth. Pain Med 2020, 45, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Pajouhesh H; Lenz GR Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hitchcock SA; Pennington LD Structure–Brain Exposure Relationships. J. Med. Chem 2006, 49, 7559–7583. [DOI] [PubMed] [Google Scholar]
- (35).Oldendorf WH; Hyman S; Braun L; Oldendorf SZ Blood-brain barrier: penetration of morphine, codeine, heroin, and methadone after carotid injection. Science 1972, 178, 984–986. [DOI] [PubMed] [Google Scholar]
- (36).Bouw MR; Gårdmark M; Hammarlund-Udenaes M Pharmacokinetic-pharmacodynamic modelling of morphine transport across the blood-brain barrier as a cause of the antinociceptive effect delay in rats–a microdialysis study. Pharm. Res 2000, 17, 1220–1227. [DOI] [PubMed] [Google Scholar]
- (37).ACD Percepta; Advanced Chemistry Development, Inc., Toronto, ON, Canada, 2021. www.acdlabs.com. [Google Scholar]
- (38).Craig PN Interdependence between physical parameters and selection of substituent groups for correlation studies. J. Med. Chem 1971, 14, 680–684. [DOI] [PubMed] [Google Scholar]
- (39).Griffin JF; Larson DL; Portoghese PS Crystal structures of .alpha.- and .beta.-funaltrexamine: conformational requirement of the fumaramate moiety in the irreversible blockage of .mu. opioid receptors. J. Med. Chem 1986, 29, 778–783. [DOI] [PubMed] [Google Scholar]
- (40).Sayre LM; Portoghese PS Stereospecific synthesis of the 6.alpha.- and 6.beta.-amino derivatives of naltrexone and oxy-morphone. J. Org. Chem 1980, 45, 3366–3368. [Google Scholar]
- (41).Newman AH; Battiti FO; Bonifazi A 2016 Philip S. Portoghese Medicinal Chemistry Lectureship: Designing Bivalent or Bitopic Molecules for G-Protein Coupled Receptors. The Whole Is Greater Than the Sum of Its Parts. J. Med. Chem 2020, 63, 1779–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Sim-Selley LJ; Scoggins KL; Cassidy MP; Smith LA; Dewey WL; Smith FL; Selley DE Region-dependent attenuation of μ opioid receptor-mediated G-protein activation in mouse CNS as a function of morphine tolerance. Br. J. Pharmacol 2007, 151, 1324–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Yuan Y; Li G; He H; Stevens DL; Kozak P; Scoggins KL; Mitra P; Gerk PM; Selley DE; Dewey WL; Zhang Y Characterization of 6α- and 6β-N-Heterocyclic Substituted Naltrexamine Derivatives as Novel Leads to Development of Mu Opioid Receptor Selective Antagonists. ACS Chem. Neurosci 2011, 2, 346–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Peng X; Knapp BI; Bidlack JM; Neumeyer JL Pharmacological Properties of Bivalent Ligands Containing Butor-phan Linked to Nalbuphine, Naltrexone, and Naloxone at μ, δ, and κ Opioid Receptors. J. Med. Chem 2007, 50, 2254–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Pagare PP; Li M; Zheng Y; Kulkarni AS; Obeng S; Huang B; Ruiz C; Gillespie JC; Mendez RE; Stevens DL; Poklis JL; Halquist MS; Dewey WL; Selley DE; Zhang Y Design, Synthesis, and Biological Evaluation of NAP Isosteres: A Switch from Peripheral to Central Nervous System Acting Mu-Opioid Receptor Antagonists. J. Med. Chem 2022, 65, 5095–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gleeson MP Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem 2008, 51, 817–834. [DOI] [PubMed] [Google Scholar]
- (47).Hitchcock SA Structural modifications that alter the P-glycoprotein efflux properties of compounds. J. Med. Chem 2012, 55, 4877–4895. [DOI] [PubMed] [Google Scholar]
- (48).Fridén M; Winiwarter S; Jerndal G; Bengtsson O; Wan H; Bredberg U; Hammarlund-Udenaes M; Antonsson M Structure–Brain Exposure Relationships in Rat and Human Using a Novel Data Set of Unbound Drug Concentrations in Brain Interstitial and Cerebrospinal Fluids. J. Med. Chem 2009, 52, 6233–6243. [DOI] [PubMed] [Google Scholar]
- (49).Wager TT; Hou X; Verhoest PR; Villalobos A Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci 2016, 7, 767–775. [DOI] [PubMed] [Google Scholar]
- (50).Bazzini P; Wermuth CG The Practice of Medicinal Chemistry; Academic Press: San Diego, 2008; pp 431–463. [Google Scholar]
- (51).Barreiro EJ; mmerle AE; Fraga CA The methylation effect in medicinal chemistry. Chem. Rev 2011, 111, 5215–5246. [DOI] [PubMed] [Google Scholar]
- (52).Mälkiä A; Murtomäki L; Urtti A; Kontturi K Drug permeation in biomembranes. Eur. J. Pharm. Sci 2004, 23, 13–47. [DOI] [PubMed] [Google Scholar]
- (53).Fagerholm U The Role of Permeability in Drug ADME/PK, Interactions and Toxicity-Presentation of a Permeability-Based Classification System (PCS) for Prediction of ADME/PK in Humans. Pharm. Res 2008, 25, 625–638. [DOI] [PubMed] [Google Scholar]
- (54).Aynetdinova D; Callens MC; Hicks HB; Poh CYX; Shennan BDA; Boyd AM; Lim ZH; Leitch JA; Dixon DJ Installing the “magic methyl” - C-H methylation in synthesis. Chem. Soc. Rev 2021, 50, 5517–5563. [DOI] [PubMed] [Google Scholar]
- (55).Schönherr H; Cernak T Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions. Angew. Chem., Int. Ed. Engl 2013, 52, 12256–12267. [DOI] [PubMed] [Google Scholar]
- (56).Olson KM; Lei W; Keresztes A; LaVigne J; Streicher JM Novel Molecular Strategies and Targets for Opioid Drug Discovery for the Treatment of Chronic Pain. Yale J. Biol. Med 2017, 90, 97–110. [PMC free article] [PubMed] [Google Scholar]
- (57).nther T; Dasgupta P; Mann A; Miess E; Kliewer A; Fritzwanker S; Steinborn R; Schulz S Targeting multiple opioid receptors - improved analgesics with reduced side effects? Br. J. Pharmacol 2018, 175, 2857–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Zimmerman DM; Leander JD Selective opioid receptor agonists and antagonists: research tools and potential therapeutic agents. J. Med. Chem 1990, 33, 895–902. [DOI] [PubMed] [Google Scholar]
- (59).Schmidhammer H Opioid receptor antagonists. Prog. Med. Chem 1998, 35, 83–132. [PubMed] [Google Scholar]
- (60).White KL; Robinson JE; Zhu H; DiBerto JF; Polepally PR; Zjawiony JK; Nichols DE; Malanga CJ; Roth BL The G Protein-Biased κ-Opioid Receptor Agonist RB-64 Is Analgesic with a Unique Spectrum of Activities In Vivo. J. Pharmacol. Exp. Ther 2015, 352, 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Land BB; Bruchas MR; Lemos JC; Xu M; Melief EJ; Chavkin C The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J. Neurosci 2008, 28, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Rapaka RS; Porreca F Development of delta opioid peptides as nonaddicting analgesics. Pharm. Res 1991, 8, 1–8. [DOI] [PubMed] [Google Scholar]
- (63).Dondio G; Ronzoni S; Farina C; Graziani D; Parini C; Petrillo P; Giardina GA Selective δ opioid receptor agonists for inflammatory and neuropathic pain. Farmaco 2001, 56, 117–119. [DOI] [PubMed] [Google Scholar]
- (64).Gallantine EL; Meert TF A comparison of the antinociceptive and adverse effects of the mu-opioid agonist morphine and the delta-opioid agonist SNC80. Basic Clin. Pharmacol. Toxicol 2005, 97, 39–51. [DOI] [PubMed] [Google Scholar]
- (65).Coward P; Chan SD; Wada HG; Humphries GM; Conklin BR Chimeric G proteins allow a high-throughput signaling assay of Gi-coupled receptors. Anal. Biochem 1999, 270, 242–248. [DOI] [PubMed] [Google Scholar]
- (66).Mathes C QPatch: the past, present and future of automated patch clamp. Expert Opin. Ther. Targets 2006, 10, 319–327. [DOI] [PubMed] [Google Scholar]
- (67).Mitcheson JS; Chen J; Lin M; Culberson C; Sanguinetti MC A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. U.S.A 2000, 97, 12329–12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Pearlstein RA; Vaz RJ; Kang J; Chen XL; Preobrazhenskaya M; Shchekotikhin AE; Korolev AM; Lysenkova LN; Miroshnikova OV; Hendrix J; Rampe D Characterization of HERG potassium channel inhibition using CoMSiA 3D QSAR and homology modeling approaches. Bioorg. Med. Chem. Lett 2003, 13, 1829–1835. [DOI] [PubMed] [Google Scholar]
- (69).Yu HB; Zou BY; Wang XL; Li M Investigation of miscellaneous hERG inhibition in large diverse compound collection using automated patch-clamp assay. Acta Pharmacol. Sin 2016, 37, 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Lötsch J Opioid metabolites. J. Pain Symptom Manag 2005, 29, S10–S24. [DOI] [PubMed] [Google Scholar]
- (71).Resnick RB; Kestenbaum RS; Washton A; Poole D Naloxone-precipitated withdrawal: a method for rapid induction onto naltrexone. Clin. Pharmacol. Ther 1977, 21, 409–413. [DOI] [PubMed] [Google Scholar]
- (72).Charney DS; Eugene Redmond DE Jr.; Galloway MP; Kleber HD; Heninger GR; Murberg M; Roth RH Naltrexone precipitated opiate withdrawal in methadone addicted human subjects: evidence for noradrenergic hyperactivity. Life Sci. 1984, 35, 1263–1272. [DOI] [PubMed] [Google Scholar]
- (73).Wightman RS; Nelson LS; Lee JD; Fox LM; Smith SW Severe opioid withdrawal precipitated by Vivitrol(R). Am. J. Emerg. Med 2018, 36, 1128.e1–1128.e2. [DOI] [PubMed] [Google Scholar]
- (74).Banker MJ; Clark TH; Williams JA Development and validation of a 96-well equilibrium dialysis apparatus for measuring plasma protein binding. J. Pharm. Sci 2003, 92, 967–974. [DOI] [PubMed] [Google Scholar]
- (75).Hidalgo IJ; Raub TJ; Borchardt RT Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterol 1989, 96, 736–749. [PubMed] [Google Scholar]
- (76).Obach RS; Baxter JG; Liston TE; Silber BM; Jones BC; MacIntyre F; Rance DJ; Wastall P The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther 1997, 283, 46–58. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.