

Abstract
A large number of proteins are capable of inserting themselves into lipids, and interacting with membranes, such as transmembrane proteins and apolipoproteins. Protein–lipid interactions have been identified as one of the keys in understanding biological processes, while the structure of proteins at the lipid-binding stage can provide evidence to help identify their roles and critical functions. However, structure determination of proteins at the lipid-binding stage is rather difficult, because conformational and compositional heterogeneities of the protein–lipid complexes are major barriers to unravel their structures using traditional methods, such as X-ray crystallography. Electron microscopy (EM) is an alternative approach to determine protein structure and has demonstrated a capability in visualizing lipid–protein interactions directly. Among various EM techniques, negative-staining (NS) is an easy, rapid, qualitative approach that is a well-established technique, frequently used in research laboratories. Conventional NS protocols, unfortunately, often generate artifacts with lipid-related proteins, such as the rouleau formation of lipoproteins. To overcome this artifact formation, Ren and his colleagues recently developed an optimized NS protocol that was validated by comparing images of lipoproteins from cryo-electron microscopy (cryo-EM). The optimized NS protocol could produce “near native-state” particle images and high contrast images of the protein in its lipid-binding state that is favorable for three-dimensional (3D) reconstruction by single-particle analysis and individual-particle electron tomography (IPET), suggesting this optimized protocol can be used widely to examine the structure of proteins at lipid-binding stage.
Keywords: Lipoprotein structure, Lipoprotein morphology, Electron microscopy, Optimized negative-staining protocol, Negative-staining electron microscopy
1. Introduction
Protein–lipid interactions can be found in membrane proteins and apolipoproteins with lipids, which can function as pumps, transporters, cell-to-cell communication messengers (1), or lipid transfer vehicles for lipid metabolism (2). To understand the mechanisms of these biological complexes, studying the structure of a protein at the lipid-bound stage is crucial. To determine their structures, however, difficulties arise due to their dynamic heterogeneity in size, shape, and component. For example, the dynamic nature of lipoproteins plays a vital, functional role for cholesteryl transport in cardiovascular disease (CVD).
Structure determination of proteins at the lipid-binding stage, however, is rather difficult by X-ray crystallography because of conformational and compositional heterogeneity. Electron microscopy (EM), as an alternative approach, has been used more frequently to determine protein structure, and among various EM techniques, cryo-crystallography has been successful in determining a “native-state” structure of proteins in the lipid-bound form under frozen hydrated cryogenic conditions (3, 4). For example, Walz et al. used cryo-crystallography uncovered details of the interaction between a lipid and a membrane protein, aquaporin (AQP0), at a resolution of 1.9 Å (3). This technique is quite difficult, requires the most advanced level of equipment and considerable amount of expertise, and, most importantly, requires two-dimensional crystallization, which is still considered an art.
Single-particle cryo-EM is another alternative approach that has become a most popular method to study protein structure. This approach, used as the only technology capable of directly visualizing proteins at the native state, involves imaging hundreds to thousands of particles embedded in vitreous ice. By this method, the images of the particles, which have the same space orientation, are grouped and averaged to reduce the noise level and increase image contrast prior to reconstruction into a 3D density map (4, 5). Usually, such averaging and classification can improve contrast (6, 7); however, it has limited accuracy with heterogeneous particle populations (8), such as high-density lipoprotein (HDL). Another restriction of the cryo-EM approach is radiation damage of the sample. Hence, imaging is performed under a low-illumination dose and under low-temperature conditions. As a result, cryo-EM images contain a low signal-to-noise ratio with high amounts of noise (9).
Negative-staining (NS) is an easy, rapid, qualitative approach and a well-established technique, frequently used in many research laboratories. NS-EM is an approach that directly visualizes individual particles, such as proteins, viruses, and even very thin cell slice specimens. It also permits the study of morphology and structure of these particles, along with lipid-bound forms of apolipoprotein (8, 10); hence, NS-EM has been frequently used (11–13). NS has improved resistance to radiation when compared to cryo-EM (10), and due to the protein being fixed and coated with a layer of heavy metal ions in an emulsion of high ionic strength, it provides exceedingly high contrast (11, 12). However, in many experiments artifacts from NS have been noticed (14–17). For example, a rouleau forms in lipid-bound forms of apoE4 with the conventional NS protocol, and it is believed to be an artifact, because rouleaux are not observed in serum or solutions of phosphate-buffered or Tris-buffered saline (18–20). Phosphotungstic acid (PTA) is used with conventional NS at high salt concentrations in buffer, but NS experiments with the apoE4•palmitoyl-oleoylphosphatidylcholine (POPC) phospholipid particle and liposome vesicles showed that the particles were stacked to each other by PTA, connected by lipid surfaces of neighboring particles (8). As a result, Ren and his colleagues developed an optimized NS protocol that minimizes rouleau formation usually seen in conventional NS-EM studies, and this method was utilized to report structure and morphology of apoE4•POPC reconstituted HDL (rHDL) (8); apoA-I 7.8, 8.4, and 9.6 nm discoidal rHDL (10); 9.3 nm spherical rHDL (10); human plasma HDL (10); low-density lipoprotein (LDL) (10); intermediate-density lipoprotein (IDL) (10); very-low-density lipoprotein (VLDL) (10); a hydrophobic glycoprotein such as cholesteryl ester transfer protein (CETP) (21, 22); and the antibody IgG (23–25) (Fig. 1). Benefits of the optimized NS protocol allow for the study of morphology and structure of individual particles due to minimization of rouleaux, and most of all images from EM with optimized NS provide increased structural resolution that is highly cooperative for 3D reconstruction models.
Fig. 1.
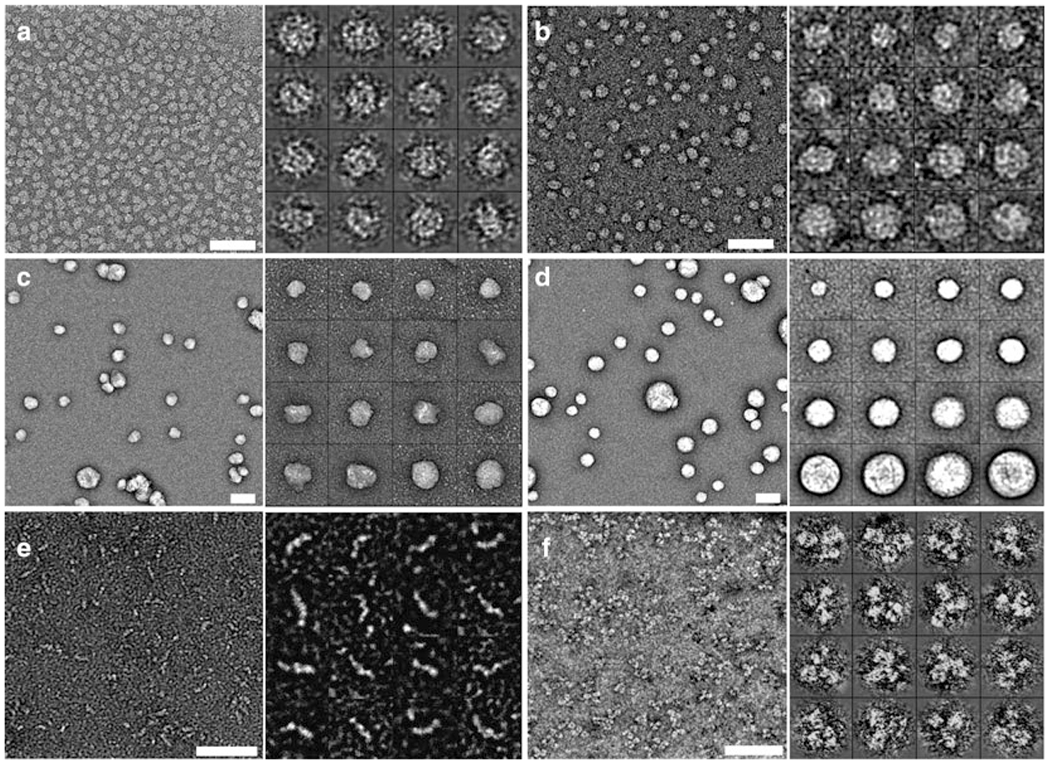
Morphologies of proteins by optimized negative-staining EM. Micrographs of apoE4-POPC (a) (8), α-HDL from plasma (b) (10), IDL (c) (10), VLDL (d) (10), cholesteryl ester transfer protein (CETP, 53 kDa) (e) (22), and the IgG antibody (f) (25), obtained by EM using the optimized negative-staining protocol. Micrographs by EM for all specimens (a-f) mentioned above are shown individually (left), and 16 selected individual particles are shown from a larger EM micrograph for all specimens (a-f) mentioned above (right). Bar = 50 nm.
The optimized NS protocol involves placing a drop of the lipoprotein solution on a glow-discharged carbon-coated copper grid and then removing excess solution by blotting with filter paper (Fig. 2). Immediately after, three washes with deionized water on the EM carbon-coated grid are performed (Fig. 2), and then it is stained with uranyl formate (UF) (Fig. 2). Following staining, the grid is blotted again with filter paper and then dried in air. Finally, it is stored at room temperature before being used for EM (8, 10) (Fig. 2).
Fig. 2.
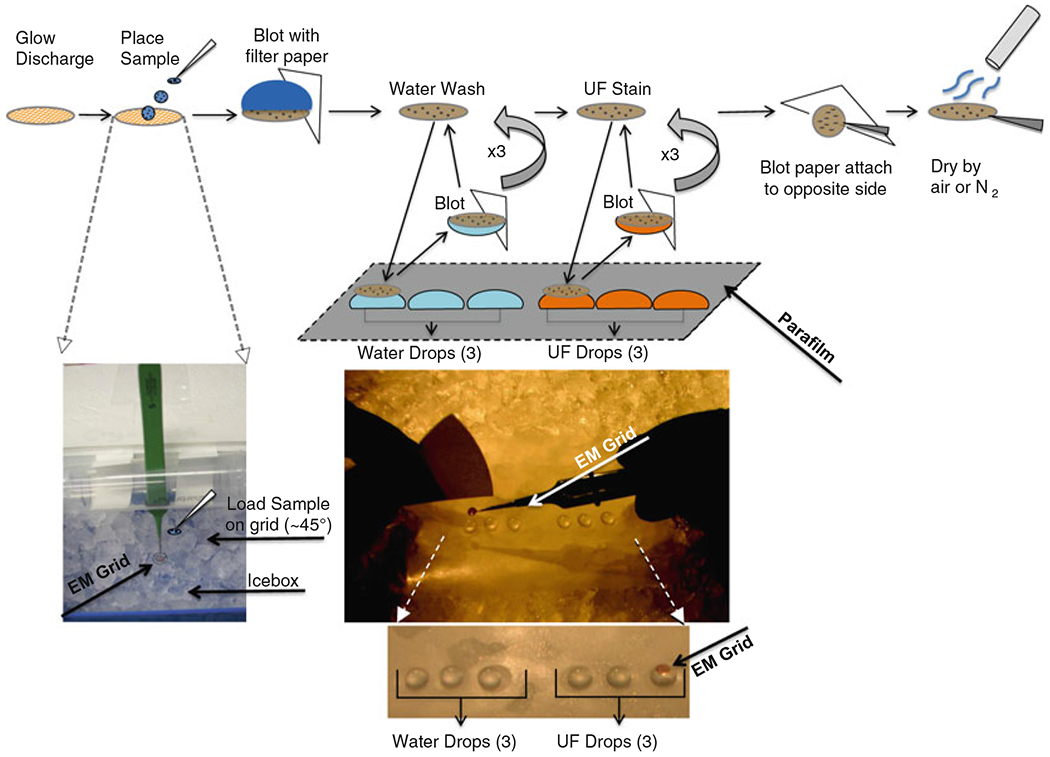
Diagram of the optimized negative-staining protocol procedure. EM grid manipulations with stain, filter paper, and water contacts in chronological order (top cartoon), grid in icebox incubation at 4°C (left bottom), rapid contact with stain or water with filter paper from Parafilm in flat icebox chamber (middle top), 3 drops of water followed by 3 drops of stain on Parafilm in flat icebox chamber with EM grid lying on top of last UF stain drop (middle bottom).
2. Materials
Uranyl formate: UO2(CHO2)2•H2O.
1 NORM-JECT 1 ml tuberculin syringe, Luer.
Sterile syringe filter: pore size of 0.02 μm (Anotop 10).
Protein sample, 2.5 μl (~0.005 mg/ml, protein).
Dulbecco’s Phosphate-Buffered Saline.
Parafilm: 4 × 4″.
Ice.
Cu-300 CN: Thin carbon-coated 300 mesh copper grids (Pacific Grid-Tech, San Francisco, CA).
Dumont style#5 medical tweezers with clamping ring.
Ultrapure water: Obtained from Millipore Synthesis unit.
Filter papers: Qualitative circles, 90 mm (Whatman).
Aluminum foil.
Petri dishes.
EMS 100: Glow discharge unit.
Icebox: 4″ × 5″, with lid, insulated.
Flat ice chamber: leveled, insulated, uniformly flat, large enough to hold the 4″ × 4″ Parafilm, and contains a lid.
3. Methods
Prepare 100 ml of a 1% (w/v) solution of UF powder in deionized water and stir it overnight in a dark room at room temperature. Cover the bottle used to prepare the solution with aluminum foil. Filter 5 ml of the 1% solution with the NORM-JECT syringe and the Anotop filter of 0.02 μm, and aliquot it into 2 ml vials, wrapped in aluminum foil to keep the solution in the dark. Immediately after aliquoting the 1% UF solution, place the vials into liquid nitrogen by using a long-handled forceps (see Notes 1, 2).
Store the 2 ml vials of the 1% solution in an −80°C freezer until use.
Before use, thaw a vial in a 4°C water bath, and make sure it remains wrapped (cover it) in aluminum foil to keep the vial in the dark.
Once the UF is thawed and in liquid form, filter the UF again, using a 1 ml NORM-JECT syringe and the Anotop filter of 0.02 μm pore size, cover it with aluminum foil, and store it on ice or at 4°C (see Note 2).
Place ice in a uniformly leveled manner into the flat ice chamber, and cover it.
Designate 3 rows of 6 small circular regions in Parafilm. Place the Parafilm in the flat ice chamber and then place ~35 μl drops of deionized water in the first three circle regions. Subsequently place ~35 μl drops of the filtered UF in the next three small circle regions in each row (see Note 3).
Fill the icebox with ice, cover it, and let it stand for ~10 min.
Obtain carbon-coated grids with Dumont #5 medical tweezers with clamping ring, perform glow discharge with an EMS 100, and place the grids on a clean filter paper in a petri dish and cover it (see Note 4).
Open the icebox and hold the grid with tweezers at a 45° angle; place ~2.5 μl of the lipoprotein sample on the grid and incubate for 1 min (see Note 5).
After ~1 min, remove excess solution by gently touching the edge of the grid with filter paper. Wash the grid by briefly placing the surface of the grid with a drop (~35 μl) of deionized water on Parafilm and then blot with filter paper to remove the excess solution. The touching and blotting steps are to be performed three times, each with a clean drop of deionized water. Perform the same touching and blotting steps with three successive drops (~35 μl) of 1% UF solution applied on Parafilm, and remove the excess solution by blotting similarly with water. Contact the grid with the last UF drop with the sample side down for 1–3 min in the dark (close the lid of the flat ice chamber) before removing excess stain by blotting again with the entire backside parallel to the grid (non-carbon side) with filter paper. Subsequently, air-dry the sample by a low flow of nitrogen gas at room temperature (see Fig. 2, Note 6).
Store the grid on filter paper in a petri dish, and partially cover it for ~30 min.
Send the grid to EM or store it in a grid storage box (see Note 7).
Fig. 3.

The best EM imaging area. The best imaging area of the protein was generally from the area that contained the thicker stain. Micrographs showing cloudy areas to locate and obtain images by EM for lipoproteins. Cloud (highlighted by box) of lipoprotein to designate lipoprotein location at 80× magnification (left), same designated area of cloud (highlighted by box) of lipoprotein further magnified at 4Kx (middle), and same designated cloud area further magnified at 80Kx (right).
4. Notes
UF is sensitive to low light. Hence, this procedure should be performed in the dark. UF is also radioactive and should be handled accordingly. The waste of the UF should be placed in an appropriate waste container in compliance with the appropriate waste management system guidelines of the lab.
During filtration, filter very slowly, and ensure the UF is covered by aluminum foil. Be careful to use the correct side of the filter when attaching it to the syringe. Discard the filter and the syringe as radioactive waste components.
Ensure that the Parafilm is leveled when placing it into the flat ice chamber. Use paperweights as necessary to hold the Parafilm down onto the ice. Make circles/wells with care and do not place them too close to each other. Rows of circles should be made according to the number of samples, but to have a few extra is always best practice in case of errors. Divide circles into two sections of 3, one for water and the other for UF. Again, UF is light sensitive, so close the lid whenever possible.
Before handling carbon-coated grids, clean the tweezers by lightly wiping them on unused filter paper. Handle grids with tweezers so that the tweezers grip the metal edge of the grid, do not bend grids, and ensure carbon-coated grids are always faced up. Refer to your instrument manual to perform the glow discharge appropriately with the EMS 100. Alternate glow dischargers or plasma cleaners maybe used, but ensure that glow discharge occurs by validating your process. Be careful not to clamp down too hard on grids, as they bend easily.
Ensure the sample (protein portion) concentration is ~0.01-0.005 mg/ml, and use Dulbecco’s Phosphate-Buffered Saline to dilute if necessary. When placing a sample on the grid, be careful to approach the grid at an angle and close lid when possible in an icebox to slow down any reactivity of the grid with air (hence the low-temperature placement of the grid at ~4°C or less). Multiple grids can be held at a time in an icebox if desired, but contact with ice must be avoided at all times.
Prepare filter papers beforehand, cut them into small sections to avoid confusion, and designate one paper for water and one for UF. Properly discard UF-contaminated paper according to lab guidelines. Be extremely careful to blot by touching the side of grid with filter paper, and ensure the carbon side of the grid is contacting deionized water and UF stain. It is important to note that the thickness of the stain of the carbon-coated grid is not even. In some areas the stain is thicker than in other areas. However, the best images of proteins have generally been obtained from areas of thicker staining (Fig. 3). An area of thicker stain looks like a “cloud” on the grid when using low magnification (<400×).
For storage of the grids, excess heat, dryness, and low humidity conditions must be avoided.
References
- 1.Reichow SL, Gonen T (2009) Lipid–protein interactions probed by electron crystallography. Curr Opin Struct Biol 19:560–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gennis RB, Jonas A (1977) Protein-lipid interactions. Annu Rev Biophys Bioeng 6:195–238 [DOI] [PubMed] [Google Scholar]
- 3.Gonen T, Cheng YF, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T (2005) Lipid–protein interactions in double-layered two-dimensional AQPO crystals. Nature 438:633–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ren G, Rudenko G, Ludtke SJ, Deisenhofer J, Chiu W, Pownall HJ (2010) Model of human low-density lipoprotein and bound receptor based on CryoEM. Proc Natl Acad Sci U S A 107:1059–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar V, Butcher SJ, Oorni K, Engelhardt P, Heikkonen J, Kaski K, Ala-Korpela M, Kovanen PT (2011) Three-dimensional cryoEM Reconstruction of native LDL particles to 16 Å resolution at physiological body temperature. PloS One 6:e18841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ludtke SJ, Baldwin PR, Chiu W (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128:82–97 [DOI] [PubMed] [Google Scholar]
- 7.Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A (1996) SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol 116:190–199 [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Song J, Newhouse Y, Zhang S, Weisgraber KH, Ren G (2010) An optimized negative-staining protocol of electron microscopy for apoE4•POPC lipoprotein. J Lipid Res 51:1228–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orlova EV, Sherman MB, Chiu W, Mowri H, Smith LC, Gotto AM (1999) Three-dimensional structure of low density lipoproteins by electron cryomicroscopy. Proc Natl Acad Sci U S A 96:8420–8425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang L, Song J, Cavigiolio G, Ishida BY, Zhang S, Kane JP, Weisgraber KH, Oda MN, Rye K-A, Pownall HJ, Ren G (2011) Morphology and structure of lipoproteins revealed by an optimized negative-staining protocol of electron microscopy. J Lipid Res 52:175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohi M, Li Y, Cheng Y, Walz T (2004) Negative-staining and image classification—powerful tools in modern electron microscopy. Biol Proced Online 6:23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliver RM (1973) Negative stain electron microscopy of protein macromolecules. Methods Enzymol 27:616–672 [DOI] [PubMed] [Google Scholar]
- 13.Woeste S, Demchick P (1991) New version of the negative stain. Appl Environ Microbiol 57:1858–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bradley DE (1962) A study of the negative-staining process. J Gen Microbiol 29:503–516 [DOI] [PubMed] [Google Scholar]
- 15.Cunningham WP, Staehelin LA, Rubin RW, Wilkins R, Bonneville M (1974) Effects of phosphotungstate negative-staining on the morphology of the isolated Golgi apparatus. J Cell Biol 62:491–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egelman EH, Amos LA (2009) Electron microscopy of helical filaments: rediscovering buried treasures in negative stain. Bioessays 31:909–911 [DOI] [PubMed] [Google Scholar]
- 17.Melchior V, Hollingshead CJ, Cahoon ME (1980) Stacking in lipid vesicle-tubulin mixtures is an artifact of negative-staining. J Cell Biol 86:881–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Innerarity TL, Pitas RE, Mahley RW (1979) Binding of arginine-rich (E) apoprotein after recombination with phospholipid vesicles to the low density lipoprotein receptors of fibroblasts. J Biol Chem 254:4186–4190 [PubMed] [Google Scholar]
- 19.Peng D, Song C, Reardon CA, Liao S, Getz GS (2003) Lipoproteins produced by ApoE−/− astrocytes infected with adenovirus expressing human ApoE. J Neurochem 86:1391–1402 [DOI] [PubMed] [Google Scholar]
- 20.Schneeweis LA, Koppaka V, Lund-Katz S, Phillips MC, Axelsen PH (2005) Structural analysis of lipoprotein E particles. Biochemistry 44:12525–12534 [DOI] [PubMed] [Google Scholar]
- 21.Ren G, Zhang S, Cavigiolio G, Lei D, Krauss RM, Oda M, Weisgraber KH, Rye KA, Pownall HJ, Qiu X (2010) Cholesteryl ester transfer protein penetrates lipoproteins for cholesteryl ester transfer. Biophys J 98:36a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Yan F, Zhang S, Lei D, Charles MA, Cavigiolio G, Oda M, Krauss RM, Weisgraber KH, Rye K-A, Pownall HJ, Qiu X, Ren G (2012) Structural basis of transfer between lipoproteins by cholesteryl ester transfer protein. Nat Chem Biol 8:342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Ren G (2010) Determining the dynamic protein structure by individual-particle electron tomography: an individual antibody structure at a nanometer resolution. Biophys J 98:441a–441a [Google Scholar]
- 24.Zhang L, Kaspar A, Woodnutt G, Ren G (2010) Monitoring the structural changes of conjugated antibodies by high-resolution electron microscopy and individual-particle electron tomography. Biophys J 98:440a–441a [Google Scholar]
- 25.Zhang L, Ren G (2012) IPET and FETR: Experimental Approach for Studying Molecular Structure Dynamics by Cryo-Electron Tomography of a Single-Molecule Structure. PLoS One 7:e30249. [DOI] [PMC free article] [PubMed] [Google Scholar]