

Abstract
Nei-like glycosylase 1 (NEIL1) is a DNA repair enzyme that initiates the base excision repair (BER) pathway to cleanse the human genome of damage. The substrate specificity of NEIL1 includes several common base modifications formed under oxidative stress conditions, as well as the imidazole ring open adducts that are induced by alkylating agents following initial modification at N7 guanine. An example of the latter is the persistent and mutagenic 8,9-dihydro-8-(2,6-diamino-4-oxo-3,4-dihydropyrimid-5-yl-formamido)-9-hydroxyaflatoxin (AFB1-FapyGua) adduct, resulting from the alkylating agent aflatoxin () exo-8-9-epoxide. Naturally occurring single nucleotide polymorphic (SNP) variants of NEIL1 are hypothesized to be associated with an increased risk for development of early-onset hepatocellular carcinoma (HCC), especially in environments with high exposures to aflatoxins and chronic inflammation from viral infections and alcohol consumption. Given that exposures and hepatitis B viral (HBV) infections represent a major problem in the developing countries of sub-Saharan Africa, it is pertinent to study SNP NEIL1 variants that are present in this geographic region. In this investigation, we characterized the three most common NEIL1 variants found in this region: P321A, R323G, and I182M. Biochemical analyses were conducted to determine the proficiencies of these variants in initiating the repair of DNA lesions. Our data show that damage recognition and excision activities of P321A and R323G were near that of wild-type (WT) NEIL1 for both thymine glycol (ThyGly) and AFB1-FapyGua. The substrate specificities of these variants with respect to various oxidatively-induced base lesions were also similar to that of WT. In contrast, the I182M variant was unstable, such that it precipitated under a variety of conditions and underwent rapid inactivation at a biologically relevant temperature, with partial stabilization being observed in the presence of undamaged DNA. This study provides insight regarding the potential increased risk for early-onset HCC in human populations carrying the NEIL1 I182M variant.
Keywords: Aflatoxin, Chronic inflammation, Hepatocellular carcinoma, DNA damage, Base excision repair
1. Introduction
The combination of environmental exposures, chronic inflammation, and genetic variations are known to modulate susceptibility to multiple human diseases, including cancer, neurodegeneration, and metabolic syndrome. There has been great interest in gaining in-depth understandings of these factors, including the identification of the principal drivers of disease, with core purposes being to adopt preventative measures and mitigate disease initiation and progression. Implementation of such a comprehensive strategy has been attempted in the case of hepatocellular carcinoma (HCC), which is responsible for over 850,000 annual deaths worldwide [1,2], with over 80% of new cases in East Asia and sub-Saharan Africa [3,4]. The primary focus of these efforts has been directed toward the best-established drivers for HCC: hepatitis B viral (HBV) infection and ingestion of foods contaminated with aflatoxin-producing molds [5–7].
Aflatoxin () is a mycotoxin produced by the mold species Aspergillus flavus and Aspergillus parasiticus that grow on staple foods such as maize, nuts, rice, and wheat [8]. Aflatoxins are estimated to affect 5 billion people worldwide, with the distribution clustering primarily in East Asia, South Asia, and sub-Saharan Africa [5,7,9,10]. The combination of tropical/sub-tropical climates, poor food storage practices, and poverty in these geographical regions exacerbate the risk for aflatoxin exposure [8,10]. In sub-Saharan Africa, besides the carcinogenic effects from chronic exposure, there have been multiple accounts of acute aflatoxicosis outbreaks that resulted in an unprecedented number of casualties (Kenya 2004: [11]; Tanzania 2016: [12]).
When ingested, the aflatoxins are absorbed within the gastrointestinal tract and transported to the liver, where they undergo metabolic activation to the exo-8,9-epoxides by members of the cytochrome P450 family, including CYP1A2 and CYP3A4 [13,14]. can also be activated in human lung by CYP2A13 [15,16]. The exo-8,9-epoxide primarily reacts with guanines in DNA, initially forming the cationic 8, 9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin (AFB1-N7-Gua) [17]. This species either undergoes spontaneous depurination resulting in an apurinic (AP) site or converts into the imidazole ring-opened 8,9-dihydro-8-(2,6-diamino-4-oxo-3,4-dihydropyrimid-5-yl-formamido)-9-hydroxyaflatoxin (AFB1-FapyGua) adduct [17]. In contrast to the cationic AFB1-N7-Gua adduct, the AFB1-FapyGua adduct is chemically stable and persistent in cellular DNA [18–20]. Solution structures of duplex DNA containing AFB1-FapyGua reveal intercalation of the moiety within the base stack, which results in increased stability of the modified DNA to thermal denaturation [21,22]. AFB1-FapyGua is a strong block to replication, and both biochemical and cell biology data suggest that this lesion is bypassed in a highly error-prone manner by DNA polymerase [23,24]. Replication of DNAs containing AFB1-FapyGua was shown to induce high-frequency base substitutions, predominantly transversions, with lower levels of transitions and transversions [23,25]. Consistent with the key role of this adduct in AFB1-induced mutagenesis and carcinogenesis, transversions are frequently found in the tumor-suppressing TP53 gene in HCCs associated with exposure [26–28] and are most common in the mutational signature of (COSMIC signature SBS24) [29–33].
Nei-like glycosylase 1 (NEIL1) is a DNA repair enzyme that initiates the base excision repair (BER) pathway. This bifunctional glycosylase/lyase excises the damaged base through hydrolysis of the N-glycosidic bond and then cleaves the phosphodiester bond in sequential,-elimination reactions, which following removal of the 3′ phosphate [34], creates a substrate appropriate for gap-filling DNA synthesis. NEIL1 is able to recognize a broad range of hydroxyl and other radical-induced lesions including the pyrimidine-derived thymine glycol (ThyGly), 5-hydroxy-5-methylhydantoin (5-OH-5-MeHyd), and 5-hydroxycytosine (5-OH-Cyt) and purine-derived 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua) and 4,6-diamino-5-formamidopyrimidine (FapyAde) (Fig. 1) [35–43]. The substrate specificity of NEIL1 also extends to alkylated ring-fragmented purine lesions, including methyl- and nitrogen mustard-induced FapyGua [37,44], as well as alkylated pyrimidines such as psoralen-induced interstrand cross-links [45].
Fig. 1.
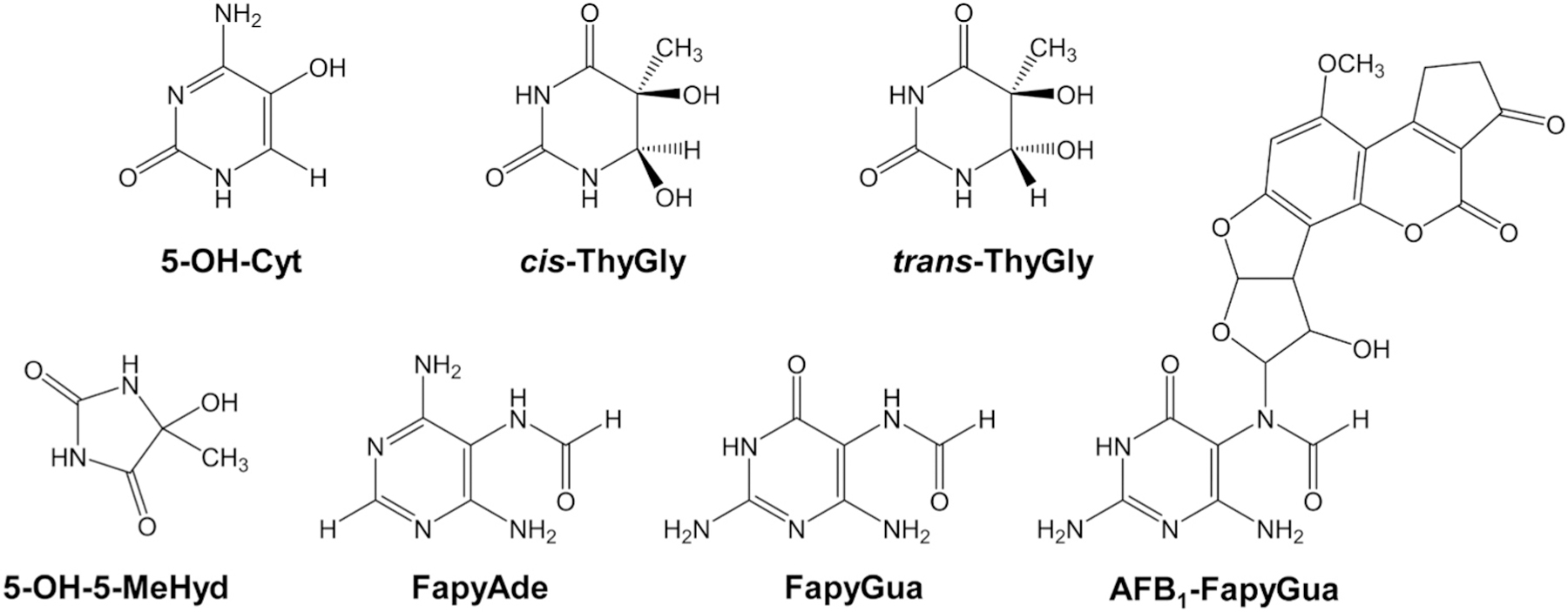
Substrates for NEIL1 used in this study. Pyrimidine-derived lesions: 5-OH-Cyt, cis-ThyGly, trans-ThyGly, 5-OH-5-MeHyd; Purine-derived lesions: FapyAde, FapyGua; AFB1-FapyGua.
Removal of AFB1-FapyGua was initially attributed exclusively to nucleotide excision repair (NER) [46,47]. However, our studies have demonstrated that NEIL1 can efficiently excise this adduct from synthetic oligodeoxynucleotides in vitro as well as from genomes of mouse liver [19]. The biological significance of this activity was evidenced by Neil1-deficient mice having a 3.2-fold increased risk of developing at least one tumor relative to wild-type (WT) mice following a single aflatoxin dose [19]. The effect of NER deficiency was significantly less, with the risk estimated for XPA mice being only 1.2-fold elevated relative to WT [19,46]. Thus, NEIL1 is anticipated to play a critical role in limiting HCC induction by cleansing the genome of lesions that are induced by both aflatoxin exposures and chronic inflammation associated with HBV infection.
Given the role of NEIL1 in the repair of DNA damage caused by both and inflammation, it is hypothesized that naturally occurring, pathogenic SNP variants of NEIL1 could increase human susceptibility to the development of early-onset HCC, especially in regions of the world with high aflatoxin exposure and HBV infection. Since these represent a major problem in sub-Saharan Africa [5,7,9,10], it is important to study the most prevalent SNP NEIL1 variants in this region: I182M, P321A, and R323G. Implications of this research may provide proactive strategies for individuals and populations that are discovered to be at higher genetic risk for HCC.
2. Materials and methods
2.1. Expression constructs for creation of NEIL1 variants
The starting plasmid for creation of constructs to express I182M, P321A, and R323G NEIL1 variants was the pET22b(+) vector, encoding for the full-length edited version of NEIL1 (K242R) [39]. Single nucleotide mutations were introduced using the Q5 Site-Directed Mutagenesis kit (New England BioLabs), using primers designed according to the kit’s guidelines. Following PCR in the presence of the template vector and primers, the products were introduced into DH5-α Escherichia coli (New England BioLabs), and colonies were selected at 37 °C on Luria-Bertani (LB) (1% tryptone, 0.5% yeast extract, 1% NaCl (w/v), pH 7.0) agar plates containing 50 μg/mL ampicillin. Individual clones were grown overnight in LB media at 37 °C and plasmids were isolated with QIAprep Spin Miniprep kit (Qiagen). The open reading frame sequences with a C-terminal 6-histidine tag were analyzed and mutations were confirmed using the Sanger Sequencing method (DNA Sequencing Core, Vollum Institute, Oregon Health & Science University).
2.2. Expression and purification of human NEIL1
The vectors described above were introduced into BL21(DE3) E. coli (New England Biolabs), and individual clones were obtained on LB-agar plates containing 50 μg/mL ampicillin. Colonies were inoculated into 20 mL LB media containing 50 μg/mL ampicillin and grown overnight at 37 °C. The overnight culture was used to inoculate 2 L of LB media and the culture was incubated at 37 °C until the reached 0.8. Expression of NEIL1 was induced by addition of isopropyl 1-thio-β-D-galactopyranoside to a final concentration of 0.5 mmol/L and incubation continued for 4 h. The temperature of the induction/expression step was optimized for each variant, with the WT NEIL1 and P321A grown at 30 °C, I182M at 20 °C, and R323G at 25 °C. Cells were pelleted by centrifugation at x g for 10 min and stored at −80 °C. The frozen pellet was resuspended on ice in a binding buffer for Ni2+ chromatography (50 mmol/L sodium phosphate (pH 8), 300 mmol/L sodium chloride, 50 mmol/L imidazole, 5 mmol/L β-mercaptoethanol), followed by lysis using a French pressure cell at 96.5 MPa (14,000 psi), and brief sonication (3 times 10 s each). Cellular debris was removed by centrifugation at x g for 20 min, and the supernatant was loaded onto a 10 mL Qiagen Ni-NTA agarose column that was pre-equilibrated with binding buffer. The column was washed with column volumes using binding buffer until the optical density at 280 nm was < 0.05. WT and variant NEIL1 enzymes were eluted with 150 mL binding buffer containing a gradient of 50–500 mmol/L imidazole. Fractions (3 mL) were collected, and the protein concentration of each fraction was measured with the Bradford reagent (Bio-Rad) using an Infinite M200 Tecan system. The purity of NEIL1 in each fraction was evaluated by gel electrophoresis with a NuPAGE 4–12% Bis-Tris gel under denaturing conditions. The gel was stained with Bio-Safe Coomassie Blue (Bio-Rad). WT NEIL1 went through an additional purification step via SP Sepharose Fast Flow column chromatography (Sigma-Aldrich). All fractions comprising visibly pure protein were pooled for long-term storage. The storage buffers were optimized for each protein as follows. The WT NEIL1 and the P321A variant were dialyzed against 10 mmol/L histidine, 10 mmol/L reduced glutathione, 150 mmol/L sodium chloride, 0.1 mmol/L ethylenediamine-tetraacetic acid (EDTA), and 10% (v/v) glycerol. The R323G variant was dialyzed against 20 mmol/L Tris-HCl (pH 7.4), 100 mmol/L KCl, 1 mmol/L β-mercaptoethanol, 0.1 mmol/L EDTA, and 15% (v/v) glycerol. The I182M variant rapidly precipitated in these buffers and was soluble only at concentrations < 2 μmol/L. Empirically, it was found that I182M could remain soluble in 50 mmol/L sodium phosphate (pH 8.0), 300 mmol/L sodium chloride, 150 mmol/L imidazole, 5 mmol/L β-mercaptoethanol, and 10% (v/v) glycerol. The aliquots of NEIL1 proteins were flash-frozen and stored at −80 °C.
2.3. Glycosylase activity assays using site-specifically modified DNA substrates
The activities of NEIL1 variants were assayed using site-specifically modified DNA substrates by either plate reader- or gel-based methods. The ThyGly-containing, uracil-containing, and undamaged oligodeoxynucleotides were obtained from Integrative DNA Technologies. The AFB1-FapyGua-containing oligodeoxynucleotide was prepared as previously described [22]. For the plate reader-based method [48], ThyGly- and AP-containing substrates were created using 5′ TAMRA-labeled 17-mer oligodeoxynucleotides (5′-TAMRA-TCACCT(ThyGly) CGTACGACTC-3′ and 5′-TAMRA-TCACC(U)TCGTACGACTC-3′). These were annealed to a complementary strand containing a Black Hole Quencher (BHQ2) on the 3′ end (5′-GAGTCGTACGAAGGTGA-BHQ2-3′). To create the AP site, the double-stranded, uracil-containing DNA (100 nmol/L) was incubated with uracil DNA glycosylase (UDG) (New England BioLabs) to release the base. The reactions were carried out in 100 μL of NEIL1 reaction buffer (20 mmol/L Tris-HCl (pH 7.4), 100 mmol/L KCl, 100 μg/mL BSA, and 0.01% (v/v) Tween 20) using 10 units of UDG. To test for the glycosylase or AP lyase activities, DNA substrate (50 nmol/L) was combined with NEIL1 enzymes (50 nmol/L or 100 nmol/L, as shown in the corresponding figure legends) in a 20 μL reaction volume in a black 384-well plate (Corning). Fluorescence signal was monitored with the Infinite M200 Tecan plate reader with parameters set at 37 °C with a 525/9 nm excitation filter and 598/20 nm emission filter. The relative fluorescence values were recorded every 2 min. The rates of the linear phase of reactions were obtained by fitting the data to a linear equation using GraphPad Prism.
The gel-based assays were performed using a ThyGly-containing substrate (analogous to that described above, but without the BHQ2 moiety) or a 3′ TAMRA-labeled 20-mer oligodeoxynucleotide containing an AFB1-FapyGua lesion (5′-CCATA(AFB1-FapyGua)CTAC-CATCGCTGGA-TAMRA-3′), annealed to a complementary strand (5′-TCCAGCGATGGTAGCTATGG-3′). For single-turnover experiments, DNA and NEIL1 were pre-incubated separately at 37 °C for 3 min in NEIL1 reaction buffer and combined to initiate the reaction. The final concentration of substrate was 50 nmol/L; enzyme concentrations were as indicated in the corresponding figure legends. For each time point, aliquots of the master reaction mixture were quenched with 80% (v/v) formamide and 16 mmol/L EDTA, and heated to 95 °C for 2 min. The DNA fragments were separated on a 15% polyacrylamide gel containing 8 mol/L urea (DNA denaturing conditions). The substrate and product bands were visualized and analyzed on the FluorChem M system (Protein Simple) with a 534 nm LED light source and a 607/36 nm emission filter. The percentage of product values (P) were plotted as a function of time (t) and fitted to a one-phase exponential equation using the GraphPad Prism software to calculate the rate constant (), non-specific product (), and the maximal substrate used (S). Stimulation of NEIL1 activity by PCNA was tested using a TAMRA-labeled 51-mer oligodeoxynucleotide containing a ThyGly (5′-TAMRA-GCTTAGCTTGGAATCGTATCATGTA(ThyGly)ACTCGTGTGCCGTGTAGACCGTGCC-3′) that was annealed with complementary strands that produced either a duplex (5′-GGCACGGTCTACACGGCACACGAGTGTACATGATACGATTCCAAGCTAAGC-3′) or forked (5′-GGCACGGTCTACACGGCACACGAGTCATGTACTTAG-3′) substrate. The sequences of these substrates were as used in a prior investigation [49]. The substrate was pre-incubated for 3 min with PCNA or BSA (control) at 37 °C. To initiate the reactions, 20 μL of enzyme was mixed with 20 μL substrate. The final reaction concentrations were 250 nmol/L PCNA or BSA, 25 nmol/L substrate, and 25 nmol/L or 50 nmol/L enzyme. For each time point, aliquots of the master reaction mixture were quenched with 80% (v/v) formamide and 16 mmol/L EDTA, and heated to 95 °C for 2 min. The products were loaded and run on a 15% polyacrylamide gel containing 8 mol/L urea. The substrate and product bands were visualized and analyzed on the FluorChem M system (Protein Simple) with a 534 nm LED light source and a 607/36 nm emission filter.
2.4. NEIL1-catalyzed reactions using high-molecular weight DNA substrate and detection of excised DNA base lesions by gas chromatography-tandem mass spectrometry (GC-MS/MS)
Calf thymus DNA in a N2O-saturated buffered aqueous solution was γ-irradiated in a source at a dose of 5 Gy and then dialyzed as described previously [50]. Unirradiated control samples were also dialyzed. Aliquots of 50 μg DNA were dried in a SpeedVac. For each data point, a triplicate of 50 μg DNA were supplemented with aliquots of ThyGly-2H4, 5-OH-Cyt-13C,15N2, 5-OH-5-MeHyd-13C,15N2, FapyAde-13C,15N2, 8-OH-Ade-13C,15N2, FapyGua-13C,15N2 and 8-OH-Gua-15N5 as internal standards. The samples were dissolved in 50 μL of an incubation buffer (50 mmol/L phosphate buffer (pH 7.4), 100 mmol/L KCl, 1 mmol/L EDTA, and 0.1 mmol/L dithiothreitol). The samples were incubated with 2 μg of NEIL1 WT, P321A, R323G or I182M at 37 °C for 60 min. Control samples without enzymes were also incubated for 60 min. After incubations, an aliquot of 125 μL ethanol was added to the samples to precipitate DNA and stop the reaction. The samples were kept at −25 °C for 60 min. After centrifugation, the supernatant fractions were separated, lyophilized, and following derivatization by trimethylsilylation, analyzed by GC-MS/MS using multiple reaction monitoring as described previously [51].
3. Results
3.1. NEIL1 variants in sub-Saharan Africa
The goal of this study was to assess the abilities of the most common NEIL1 variants that are present in populations in sub-Saharan Africa to initiate repair of oxidatively-induced base lesions and AFB1-FapyGua adducts. According to the Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org), variants with the highest allelic frequencies in sub-Saharan Africa are I182M (1.24%, variant ID: 15-75644563), P321A (0.26%, variant ID: 15-75647018), and R323G (0.55%, variant ID: 15-75647024). Two of these variants, P321A and R323G, lie in the C-terminal domain of NEIL1 (Fig. 2), which was shown to be critical for protein stability and interactions with partners, including PCNA, FEN-1, and RFA [49,52–56]. I182M is in the DNA binding domain, H2TH [57] (Fig. 2).
Fig. 2.

Crystal structure (5ITT) of human NEIL1 bound to duplex DNA containing THF (https://www.rcsb.org/structure/5ITT, released 7/6/2016, accessed 9/16/2022). X-ray diffraction was used to obtain a crystallized structure of human NEIL1 (green) bound to DNA (pink) [57]. Residue I182M lies in an alpha helical structure within the DNA binding domain, H2TH. Beyond residue K291 to the C-terminus, the structure remains unresolved, and this portion of the sequence is generally not conserved between organisms [49,55]. Residues P321A and R323G lie within this unstructured area, indicated with the blue dashed line.
3.2. Expression and purification of NEIL1 variants
The variant enzymes, I182M, P321A, and R323G, were the edited form of NEIL1 (K242R). This form results from the pre-mRNA AAA codon conversion of the central adenosine to inosine via adenosine deaminase, ADAR1 [58]. All NEIL1 gene variants were sequence confirmed prior to expression. The recombinant, 6-histidine-tagged proteins were overexpressed in E. coli, purified using affinity Ni2+ chromatography, and the level of purity for each fraction was visualized by an SDS protein gel (Fig. S1). These analyses showed prominent bands at a molecular mass of kDa, consistent with the calculated molecular mass of full-length NEIL1 containing the tag and linker (45.3 kDa for WT NEIL1). The highly purified fractions were pooled and used for kinetic studies. Due to challenges associated with differential solubility and stability of the variants, storage buffers were optimized for each variant as described in the Material and Methods section. The effect of different storage conditions on the NEIL1 activity was investigated on a synthetic oligodeoxynucleotide containing a site-specific ThyGly. These reactions demonstrated no measurable buffer-dependent differences (data not shown). To ensure that the measured activities were due to NEIL1, a separate purification was carried out from E. coli cells that contained the empty expression vector. The procedures of cell growth, breakage, and purification were identical to that for NEIL1 WT. Comparable fractions were found to possess no activity on either a ThyGly- or AP-containing oligodeoxynucleotides, thus ruling out trace contaminants from the E. coli host contributing to measurable activities (data not shown).
3.3. Survey of NEIL1 variant activities
Following purification of each NEIL1 variant, activities were measured using site-specifically modified double-stranded oligodeoxynucleotides containing either a ThyGly or an AP site (Fig. S2A). The damage-containing strand had a 5′-TAMRA-label, and the complementary strand had a 3′ Black Hole Quencher 2 (BHQ2). Using the ThyGly substrate, the combined activity of the glycosylase and AP lyase resulted in incision of the modified strand and thermally-induced melting of the TAMRA-labeled DNA fragment into bulk solution. With TAMRA no longer in the vicinity of the BHQ2, this resulted in an increase in fluorescence proportional to the amount of product formed (Fig. S2B). The same detection principle was used for the AP-containing substrate (generated by excision of uracil by UDG immediately preceding the NEIL1 nicking reaction), except this experimental design modification only measured the lyase activity (Fig. S2C). Fig. S2B shows the ability of each variant to cleave the ThyGly-containing DNA, with steeper slopes indicating higher quantities of substrate to product conversion per min. While the apparent rate at which product was generated differs between variants, all displayed the ability to recognize the lesions and complete the chemical steps necessary to release the fluorescently-labeled DNA fragment. All variants reached a maximum level of product formation within 30 min Fig. S2C presents the cleavage data using the AP-containing DNA. The rate of product formation on this substrate was considerably faster relative to ThyGly-containing DNA. This is consistent with previous investigations using the edited form of NEIL1 on the ThyGly substrate, which demonstrated that the glycosylase reaction is the rate-limiting step [39,59]. Compared to WT and the other variants, the I182M variant showed incomplete conversion of substrate to product both on ThyGly- and AP-containing oligodeoxynucleotides.
3.4. Rates of excision of AFB1-FapyGua and ThyGly by NEIL1 variants
To characterize the catalytic efficiencies of each of the NEIL1 variants, gel-based assays were carried out using excess NEIL1 on DNA containing site-specific ThyGly or AFB1-FapyGua. For WT, P321A and R323G, reactions rates were comparable in the presence of 10-, 15-, or 20-fold molar excesses of enzyme over DNA, demonstrating that conditions of single turnover were satisfied. As discussed later, the I182M variant was the exception in this regard. As shown in Fig. 3A, the values for incision of the AFB1-FapyGua adduct by the P321A and R323G NEIL1 variants were 0.34 0.05 min−1 and 0.25 0.01 min−1, respectively. These values were comparable to the rate of AFB1-FapyGua excision by WT NEIL1, which was 0.36 0.03 min−1. Similar analyses using the ThyGly-containing substrate (Fig. 3B) revealed values of 1.5 0.1 min−1 and 1.5 0.1 min−1 for the P321A and R323G NEIL1 variants, respectively. WT NEIL1 activity on ThyGly-containing DNA yielded a value of 1.3 min−1, which is consistent with previous investigations having established rates for WT at 1.2 min−1 to 1.4 min−1 [19,40,44].
Fig. 3.

Incision of site-specifically modified oligodeoxynucleotides by NEIL1 variants. Reaction rates were measured under single turnover conditions using 50 nmol/L double-stranded TAMRA-labeled substrates, with 500 nmol/L NEIL1 on AFB1-FapyGua (A) and 750 nmol/L NEIL1 on ThyGly (B). The reactions were incubated at 37 °C in a master solution and each time point was collected by quenching with 80% formamide, 16 mmol/L EDTA and heating at 95 °C for 2 min. The samples were run on a 15% polyacrylamide gel under DNA denaturing conditions. The product percentages for three individual repeats were plotted and fitted to a one phase exponential equation using GraphPad Prism. The uncertainties are standard deviations.
3.5. Substrate specificity of NEIL1 variants
The kinetic analyses using synthetic oligodeoxynucleotides were informative for comparison of NEIL1 variants with respect to their efficiencies of excising ThyGly and AFB1-FapyGua lesions. To examine each variant’s substrate specificity, genomic calf-thymus DNA was exposed to 5 Gy of γ-irradiation, creating a range of hydroxyl radical-induced DNA damage. The purified NEIL1 variants were incubated with γ-irradiated DNA for 1 h, and the released damaged bases were identified and quantified using GC-MS/MS with isotope-dilution. Fig. 4 displays the levels of each DNA damage type that were released following incubation of DNA with NEIL1 enzymes. Fig. 4A presents the base-release data by variant. NEIL1 WT (Fig. 4Aa), P321A (Fig. 4Ab) and R323G (Fig. 4Ac) showed similar specificities, with the exception that P321A demonstrated a slightly reduced release of FapyAde relative to other lesions. In contrast, the I182M (Fig. 4Ad) variant exhibited a very different substrate specificity, with strongest preference for 5-OH-5-MeHyd and substantially decreased activity on FapyGua and FapyAde lesions. Fig. 4B presents the data grouped by damage type. The differences between the non-specific base hydrolysis and base release in the presence of these enzymes were statistically significant in most cases (). However, the I182M variant showed no statistical difference from the no enzyme control on FapyGua and 5-OH-Cyt substrates (Table S1). All NEIL1 variants showed reduced amounts of bases released as compared to WT, with the exception of trans-ThyGly (Fig. 4Be); only the I182M variant showed a significant reduction of incision of trans-ThyGly. With all four enzymes tested, the reactions were not limited by the concentrations of substrates available. As shown in Fig. 4B, the levels of damaged bases released by either Nth or Fpg glycosylases significantly exceeded that released by NEIL1 enzymes.
Fig. 4.

Release of oxidatively-induced DNA base lesions from high-molecular weight DNA by WT and variants of NEIL1. Aliquots (50 μg) of γ-irradiated calf thymus DNA were incubated for 1 h at 37 °C in the presence of NEIL1 enzymes (2 μg) or reaction buffer. Panel A shows the amounts of released DNA base lesions by NEIL1 WT (a), P321A (b), R323G (c), and I182M (d) were plotted after subtraction of background. Panel B shows the amounts of FapyGua (a), FapyAde (b), 5-OH-5-MeHyd (c), cis-ThyGly (d), trans-ThyGly (e), 5-OH-Cyt (f) released for WT and each NEIL1 variant relative to no enzyme controls. For reference, the amounts of DNA base lesions released by E. coli Fpg and Nth (2 μg each) are shown [51]. Statistically significant differences are displayed for NEIL1 variant versus WT (* = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001). All uncertainties are standard deviations.
3.6. PCNA and FEN1 did not affect rates of ThyGly excision by NEIL1
The high-resolution X-ray crystal structures of NEIL1 do not include the C-terminal portion of the enzyme [57,60–62]. It has been previously shown that this unstructured domain is important for interactions with a variety of replication-associated proteins, including PCNA, and that these interactions stimulate NEIL1 activity [49,52–54,56]. Thus, experiments were designed to test if mutations located in this portion of NEIL1 (P321A and R323G) affect catalytic efficiencies in the presence of these interacting proteins. In vitro reactions with WT NEIL1 were performed on a ThyGly substrate in the presence or absence of PCNA. Incision analyses using a 10:1 molar ratio of PCNA:DNA revealed no stimulation of NEIL1 (Fig. S4). Two different preparations of PCNA and varying concentrations of NEIL1 yielded comparable data, and increasing the PCNA:DNA molar ratio showed activity inhibition. Similar data were also obtained using FEN-1, another replication-associated factor that has been shown to modestly stimulate NEIL1 activity [52]. We speculate that the difference between the results of our study and previous studies [49,52], i.e., lack of NEIL1 stimulation by PCNA and FEN-1 versus stimulation, can be explained by differences in either protein sources or substrates used (ThyGly versus 5-OH-Ura). Since no stimulation of WT NEIL1 activity by either PCNA or FEN-1 was observed, it was not possible to investigate the effect of the P321A and R323G substitutions on interactions with these proteins.
3.7. Temperature sensitivity of the NEIL1 I182M variant
The expression of the I182M variant was successful and the enzyme remained soluble and catalytically stable in the elution buffer associated with the Ni2+ chromatography purification. However, we found that all attempts to change buffer, improve protein purity, or increase its concentration resulted in protein aggregation and total loss of activity. Evidence of instability was reinforced during gel-based activity assays on site-specifically modified oligodeoxynucleotides. Specifically, while the kinetic analyses of the WT NEIL1 and the two other variants revealed reproducible data under single turnover conditions (Fig. 3), analyses using the I182M variant showed that even in vast excess of input enzyme (up to 20-fold over substrate), the initial rates of the reaction positively correlated with enzyme concentrations, indicating that these conditions were not truly single turnover (Fig. S3). In addition, the premature leveling of product formation by this variant were inconsistent and uncorrelated with enzyme concentration.
Based on the above observations, we hypothesized that the I182M variant may have folding instability. This hypothesis guided the design of investigations to test temperature sensitivity of this variant. Separately stored aliquots of the I182M variant were pre-incubated at 37 °C for increasing amounts of time and then added to the TAMRA/BHQ2 ThyGly substrate, also pre-incubated at 37 °C (Fig. 5A). Kinetics of the reactions were measured using a plate-reader as previously described. The slopes from the initial linear phase of the reactions were obtained, and the relative activities for each pre-incubation time point were calculated against the initial slope of a reaction using the enzyme without pre-incubation. While the WT NEIL1 retained the majority of its original activity throughout a 60 min pre-incubation time course, the I182M variant exhibited negligible activity, even after a 5 min pre-incubation. Interestingly, when the I182M variant was added directly to substrate, significant activity was observed 5 min into the reaction (Fig. S2). Thus, we hypothesized that the I182M variant may retain structural integrity in the presence of duplex DNA. Experiments were designed to measure the activity of this variant following pre-incubation of enzyme with undamaged duplex DNA, which was identical to ThyGly-containing DNA but had no modified base or fluorescent labels. A single aliquot of the I182M variant was diluted to 1 μmol/L with reaction buffer containing various amounts of undamaged DNA or no DNA to obtain final DNA:NEIL1 molar ratios of 0:1, 1:1, 2.5:1, and 10:1. These were subject to 15 min pre-incubation at 37 °C, diluted to 100 nmol/L enzyme concentration, and added to the ThyGly-containing substrate. The kinetics of the reactions were measured, and the relative activities calculated as described above. Data shown in Fig. 5B demonstrates that following pre-incubation, there was a positive correlation between concentration of undamaged DNA and enzyme activity. While there was extreme reduction of ThyGly cleavage when I182M was pre-incubated in the absence of DNA, almost 40% of its activity was retained following pre-incubation with 500 nmol/L undamaged DNA.
Fig. 5.

Assessment of NEIL1 I182M temperature sensitivity. WT NEIL1 and the I182M variant were pre-incubated at 37 °C for 0–60 min and the activities were tested (A). I182M was pre-incubated at 37 °C for 0 min or 15 min with varying molar ratios of undamaged DNA (0x, 1x, 2.5x, 10x). The final concentrations of undamaged DNA are shown on the x-axis (B). The reactions were conducted using 50 nmol/L enzymes and 50 nmol/L duplex TAMRA/BHQ2-conjugated ThyGly substrate. The increase in fluorescence signal was monitored in the Tecan M200 plate reader and the rates were calculated from initial linear phase of the reactions. The residual activities were calculated against the enzyme activity at 0 min pre-incubation. Three independent experiments were performed, and the average relative activities were plotted. Uncertainties are standard deviations.
4. Discussion
Given the scope and magnitude of human suffering associated with the induction of HCC globally, the adoption of multi-pronged approaches to limit its incidence is well justified. These strategies have their foundations rooted in understanding the fundamental drivers that contribute to increasing the risk of developing this cancer. Since it is well established that the major risks for HCC induction are HBV infection and consumption of foods contaminated with aflatoxin-producing molds [5–7], the combination of vaccination against HBV and limiting aflatoxin ingestion are central in efforts to mitigate tumor induction. Further, risk analyses have demonstrated that there was a strong correlation between cancer rates and the source of household water: up to a 12-fold increase in liver cancer rates was associated with exposure to microcystins, the blue-green algae-derived hepatoxic tumor promoters [63]. Proactive public health projects have made considerable progress toward improving the quality of household waters, with an anticipated positive effect on reducing overall HCC incidence.
However, given that aflatoxin exposures are not completely controllable, experimental strategies have been implemented that limit the overall bioavailability of activated aflatoxin. Toward this end, randomized clinical interventions have been designed and conducted to enhance detoxication of aflatoxin using oltipraz, sulforaphane-containing broccoli-spout tea, and green-tea polyphenols [64–66]. Analyses of these data revealed that in addition to minimizing exposure, dietary intake of compounds that reduce the potential for DNA adduct formation may significantly contribute to reductions in overall HCC rates. It is anticipated that additional factors, such as SNP variants in genes that regulate the bioactivation/detoxication of aflatoxin and variable expression of these genes, may also affect susceptibility to HCC.
Other potential sources of increased carcinogenesis risk are SNP variants in DNA repair genes that could diminish repair of both inflammation- and AFB1-induced DNA damages, especially when these variants are present in human populations that reside in geographical regions with high levels of HBV infection and aflatoxin exposure. Although it has been long-established that the NER pathway is a significant contributor in repairing AFB1-induced DNA adducts [46,47], to the best of our knowledge, no studies have been conducted to investigate risk associated with polymorphic variants in the numerous genes in this pathway. However, more recently, studies have shown that in murine models, the BER pathway is crucial in maintaining genomic stability and protecting against exposures to mutagenetic and carcinogenic compounds [67]. This includes our findings that Neil1 deficiency results in increased risk for AFB1-induced HCC [19]. Not only is NEIL1 important for genomic stability, there is also evidence suggesting its role in maintaining healthy metabolic function. Studies from this lab have discovered that Neil1 knockout mice develop metabolic syndrome, the symptoms of which include severe obesity, dyslipidemia, fatty liver disease, and hyperinsulinemia [68].
Since NEIL1 is crucial for initiating the removal of DNA lesions caused by and chronic inflammation associated with HBV infection, it is of interest to determine how naturally occurring single amino acid changes modulate biochemical and cellular functions. In this regard, SNP variants of NEIL1, P68H (variant ID: 15-75641449), G245R (variant ID: 15-75646094), and A51V (variant ID: 15-75641398), that are present in populations of East Asia who experience both of the above exposures have been functionally characterized on genomic DNA containing a spectrum of oxidatively-induced base damage and site-specifically modified oligodeoxynucleotides [42]. These variants showed a range of activities, from being catalytically comparable to WT NEIL1 (G245R) or modestly affected (P68H) to severely compromised (A51V). A51V was discovered to be temperature sensitive, such that its half-life at 37 °C was min [42]. To determine whether there is a correlation between the incidence of HCCs and the presence of this or other potentially deleterious variants, the NEIL1 gene was sequenced in HCC patients from Qidong County in China, a region with documented exposure [31,42]. All variant alleles A51V, P68H, and G245R were found in the 49 sequenced samples. Given that these variants are present in East Asia at much lower frequencies of, 1.4%, and 0.65%, respectively, this study suggests that there may be an enrichment of functionally limited NEIL1 variants in humans with HCC. Although the P68H and G245R variants were shown in vitro to have near-normal activities, their presence in this small cohort of HCC patients may indicate that these mutations are not neutral under conditions of lifetime exposures.
Other NEIL1 variants that have been biochemically characterized include G83D (variant ID: 15-75641494) and C136R (variant ID: 15-75641652), which were both reported to be glycosylase inactive [39, 41]. Although the C136R variant is very rare (frequency in African/African American populations), the G83D variant is significantly more common, with the highest presence found in Ashkenazi Jewish populations). Biochemical analyses of G83D suggest that this amino acid change does not lead to protein degradation or interfere with damage recognition, but prevents initiation of glycosylase reaction [39,41]. In cells, expression of this variant leads to replication fork stress, chromosomal aberrations, and cellular transformation. Thus, G83D may exert a dominant negative phenotype by being able to bind but not excise the damage [69]. This hypothesis is currently being tested in an animal model.
Through this study of examining activities of SNP NEIL1 variants present in sub-Saharan Africa, it was found that variants P321A and R323G possessed WT-like activity. These enzymes were able to efficiently recognize and cleave a variety of oxidatively-induced DNA lesions and the AFB1-FapyGua adduct. While P321A and R323G variants were similar to WT with regard to their stabilities, efficiencies of damage excision, and substrate specificities, the I182M variant exhibited extreme instability at 37 °C in the absence of DNA, with partial stabilization being observed in the presence of DNA. Biological significance of this amino acid substitution is currently being addressed through the creation of cells and mice that either exclusively express the I182M variant or are heterozygous. These experimental models correspond to recorded instances in humans carrying one or both alleles of I182M. In addition to observing cellular responses to aflatoxins and oxidizing agents, in vivo investigations of this variant’s stability will be explored. If these analyses demonstrate evidence for an increased mutagenic and carcinogenic risk, it may suggest that individuals carrying this SNP variant could be more susceptible for early-onset HCC associated with oxidatively-induced DNA damage and exposure.
4.1. Disclaimer
Certain equipment, instruments, software, or materials are identified in this paper in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement of any product or service by NIST, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose.
Supplementary Material
Acknowledgements
The authors wish to thank Dr. Andrew H. Kellum, Jr for synthesis and purification of the aflatoxin B1-adducted oligodeoxynucleotides and Dr. Dmitri Rozanov for preparation of the NEIL1 P321A plasmid. The authors also thank Dr. Michael O’Donnell, Rockefeller University and Dr. Muralidhar L. Hegde, The University of Texas Medical Branch for supplying purified partner proteins of NEIL1. This work was supported by National Institutes of Health (NIH) grants R01 CA-55678, R01 ES-029357, and P01 CA-160032 (M.P.S., A.K.M., and R.S.L.). The Vanderbilt-Ingram Cancer Center is funded by NIH grant P30 CA-068485. R.S.L. and AKM acknowledge support from the National Institute of Environmental Health Sciences (R01 ES-031086) and from the Oregon Institute of Occupational Health Sciences at Oregon Health & Science University via funds from the Division of Consumer and Business Services of the State of Oregon (ORS 656.630). Funding for open access charge: NIH.
Footnotes
CRediT authorship contribution statement
JTZ, IGM, MPS, MD, AKM, and RSL conceived and designed the study and wrote and revised the manuscript. JTZ, IGM, MK, and PJ generated the experimental data. All authors have read and approved the final manuscript.
Declaration of Competing Interest
All authors have no conflicts of interest to declare.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.dnarep.2023.103544.
Data Availability
Data will be made available on request.
References
- [1].Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F, Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012, Int J. Cancer 136 (2015) E359–E386. [DOI] [PubMed] [Google Scholar]
- [2].Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J. Clin 68 (2018) 394–424. [DOI] [PubMed] [Google Scholar]
- [3].El-Serag HB, Rudolph KL, Hepatocellular carcinoma: epidemiology and molecular carcinogenesis, Gastroenterology 132 (2007) 2557–2576. [DOI] [PubMed] [Google Scholar]
- [4].Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR, A global view of hepatocellular carcinoma: trends, risk, prevention and management, Nat. Rev. Gastroenterol. Hepatol 16 (2019) 589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Liu Y, Chang CC, Marsh GM, Wu F, Population attributable risk of aflatoxin-related liver cancer: systematic review and meta-analysis, Eur. J. Cancer 48 (2012) 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].McGlynn KA, Petrick JL, El-Serag HB, Epidemiology of hepatocellular carcinoma, Hepatology 73 (Suppl 1) (2021) 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Amponsah-Dacosta E, Hepatitis B virus infection and hepatocellular carcinoma in sub-Saharan Africa: Implications for elimination of viral hepatitis by 2030? World J. Gastroenterol 27 (2021) 6025–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cotty PJ, Jaime-Garcia R, Influences of climate on aflatoxin producing fungi and aflatoxin contamination, Int J. Food Microbiol 119 (2007) 109–115. [DOI] [PubMed] [Google Scholar]
- [9].Wu F, Mitchell NJ, Male D, Kensler TW, Reduced foodborne toxin exposure is a benefit of improving dietary diversity, Toxicol. Sci 141 (2014) 329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Benkerroum N, Aflatoxins: producing-molds, structure, health issues and incidence in Southeast Asian and Sub-Saharan African countries, Int J. Environ. Res Public Health 17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Azziz-Baumgartner E, Lindblade K, Gieseker K, Rogers HS, Kieszak S, Njapau H, Schleicher R, McCoy LF, Misore A, DeCock K, Rubin C, Slutsker L, Aflatoxin G, Investigative, case-control study of an acute aflatoxicosis outbreak, Kenya, 2004, Environ. Health Perspect 113 (2005) 1779–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kamala A, Shirima C, Jani B, Bakari M, Sillo H, Rusibamayila N, Saeger SD, Kimanya M, Gong YY, Simba A, t.i. team, Outbreak of an acute aflatoxicosis in Tanzania during 2016, World Mycotoxin J 11 (2018) 311–320. [Google Scholar]
- [13].Ueng YF, Shimada T, Yamazaki H, Guengerich FP, Oxidation of aflatoxin B1 by bacterial recombinant human cytochrome P450 enzymes, Chem. Res Toxicol 8 (1995) 218–225. [DOI] [PubMed] [Google Scholar]
- [14].Gallagher EP, Kunze KL, Stapleton PL, Eaton DL, The kinetics of aflatoxin B1 oxidation by human cDNA-expressed and human liver microsomal cytochromes P450 1A2 and 3A4, Toxicol. Appl. Pharm 141 (1996) 595–606. [DOI] [PubMed] [Google Scholar]
- [15].He XY, Tang L, Wang SL, Cai QS, Wang JS, Hong JY, Efficient activation of aflatoxin B1 by cytochrome P450 2A13, an enzyme predominantly expressed in human respiratory tract, Int J. Cancer 118 (2006) 2665–2671. [DOI] [PubMed] [Google Scholar]
- [16].Zhang Z, Yang X, Wang Y, Wang X, Lu H, Zhang X, Xiao X, Li S, Wang X, Wang SL, Cytochrome P450 2A13 is an efficient enzyme in metabolic activation of aflatoxin G1 in human bronchial epithelial cells, Arch. Toxicol 87 (2013) 1697–1707. [DOI] [PubMed] [Google Scholar]
- [17].Croy RG, Essigmann JM, Reinhold VN, Wogan GN, Identification of the principal aflatoxin B1-DNA adduct formed in vivo in rat liver, Proc. Natl. Acad. Sci. USA 75 (1978) 1745–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Croy RG, Wogan GN, Temporal patterns of covalent DNA adducts in rat liver after single and multiple doses of aflatoxin B1, Cancer Res 41 (1981) 197–203. [PubMed] [Google Scholar]
- [19].Vartanian V, Minko IG, Chawanthayatham S, Egner PA, Lin YC, Earley LF, Makar R, Eng JR, Camp MT, Li L, Stone MP, Lasarev MR, Groopman JD, Croy RG, Essigmann JM, McCullough AK, Lloyd RS, NEIL1 protects against aflatoxin-induced hepatocellular carcinoma in mice, Proc. Natl. Acad. Sci. USA 114 (2017) 4207–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Coskun E, Jaruga P, Vartanian V, Erdem O, Egner PA, Groopman JD, Lloyd RS, Dizdaroglu M, Aflatoxin-guanine DNA adducts and oxidatively induced DNA damage in aflatoxin-treated mice in vivo as measured by liquid chromatography-tandem mass spectrometry with isotope dilution, Chem. Res Toxicol 32 (2019) 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mao H, Deng Z, Wang F, Harris TM, Stone MP, An intercalated and thermally stable FAPY adduct of aflatoxin B1 in a DNA duplex: structural refinement from 1H NMR, Biochemistry 37 (1998) 4374–4387. [DOI] [PubMed] [Google Scholar]
- [22].Tomar R, Minko IG, Kellum AH Jr., Voehler MW, Stone MP, McCullough AK, Lloyd RS, DNA sequence modulates the efficiency of NEIL1-catalyzed excision of the aflatoxin B1-induced formamidopyrimidine guanine adduct, Chem. Res Toxicol 34 (2021) 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lin YC, Li L, Makarova AV, Burgers PM, Stone MP, Lloyd RS, Molecular basis of aflatoxin-induced mutagenesis-role of the aflatoxin B1-formamidopyrimidine adduct, Carcinogenesis 35 (2014) 1461–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lin YC, Owen N, Minko IG, Lange SS, Tomida J, Li L, Stone MP, Wood RD, McCullough AK, Lloyd RS, DNA polymerase zeta limits chromosomal damage and promotes cell survival following aflatoxin exposure, Proc. Natl. Acad. Sci. USA 113 (2016) 13774–13779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Minko IG, Kellum AH Jr., Stone MP, Lloyd RS, The aflatoxin B1-induced imidazole ring-opened guanine adduct: High mutagenic potential that is minimally affected by sequence context, Environ. Mol. Mutagen (2023). [DOI] [PMC free article] [PubMed]
- [26].Bressac B, Kew M, Wands J, Ozturk M, Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa, Nature 350 (1991) 429–431. [DOI] [PubMed] [Google Scholar]
- [27].Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC, Mutational hotspot in the p53 gene in human hepatocellular carcinomas, Nature 350 (1991) 427–428. [DOI] [PubMed] [Google Scholar]
- [28].Smela ME, Hamm ML, Henderson PT, Harris CM, Harris TM, Essigmann JM, The aflatoxin B1 formamidopyrimidine adduct plays a major role in causing the types of mutations observed in human hepatocellular carcinoma, Proc. Natl. Acad. Sci. USA 99 (2002) 6655–6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chawanthayatham S, Valentine CC 3rd, Fedeles BI, Fox EJ, Loeb LA, Levine SS, Slocum SL, Wogan GN, Croy RG, Essigmann JM, Mutational spectra of aflatoxin B1 in vivo establish biomarkers of exposure for human hepatocellular carcinoma, Proc. Natl. Acad. Sci. USA 114 (2017) E3101–E3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Huang MN, Yu W, Teoh WW, Ardin M, Jusakul A, Ng AWT, Boot A, Abedi-Ardekani B, Villar S, Myint SS, Othman R, Poon SL, Heguy A, Olivier M, Hollstein M, Tan P, Teh BT, Sabapathy K, Zavadil J, Rozen SG, Genome-scale mutational signatures of aflatoxin in cells, mice, and human tumors, Genome Res 27 (2017) 1475–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang W, He H, Zang M, Wu Q, Zhao H, Lu LL, Ma P, Zheng H, Wang N, Zhang Y, He S, Chen X, Wu Z, Wang X, Cai J, Liu Z, Sun Z, Zeng YX, Qu C, Jiao Y, Genetic features of aflatoxin-associated hepatocellular carcinoma, Gastroenterology 153 (2017) 249–262, e242. [DOI] [PubMed] [Google Scholar]
- [32].Fedeles BI, Essigmann JM, Impact of DNA lesion repair, replication and formation on the mutational spectra of environmental carcinogens: Aflatoxin B1 as a case study, DNA Repair (Amst. ) 71 (2018) 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, Islam SMA, Lopez-Bigas N, Klimczak LJ, McPherson JR, Morganella S, Sabarinathan R, Wheeler DA, Mustonen V, Group PMSW, Getz G, Rozen SG, Stratton MR, Consortium P, The repertoire of mutational signatures in human cancer, Nature 578 (2020) 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK, AP endonuclease-independent DNA base excision repair in human cells, Mol. Cell 15 (2004) 209–220. [DOI] [PubMed] [Google Scholar]
- [35].Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S, Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA, Proc. Natl. Acad. Sci. USA 99 (2002) 3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bandaru V, Sunkara S, Wallace SS, Bond JP, A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII, DNA Repair (Amst. ) 1 (2002) 517–529. [DOI] [PubMed] [Google Scholar]
- [37].Rosenquist TA, Zaika E, Fernandes AS, Zharkov DO, Miller H, Grollman AP, The novel DNA glycosylase, NEIL1, protects mammalian cells from radiation-mediated cell death, DNA Repair (Amst. ) 2 (2003) 581–591. [DOI] [PubMed] [Google Scholar]
- [38].Jaruga P, Birincioglu M, Rosenquist TA, Dizdaroglu M, Mouse NEIL1 protein is specific for excision of 2,6-diamino-4-hydroxy-5-formamidopyrimidine and 4,6-diamino-5-formamidopyrimidine from oxidatively damaged DNA, Biochemistry 43 (2004) 15909–15914. [DOI] [PubMed] [Google Scholar]
- [39].Roy LM, Jaruga P, Wood TG, McCullough AK, Dizdaroglu M, Lloyd RS, Human polymorphic variants of the NEIL1 DNA glycosylase, J. Biol. Chem 282 (2007) 15790–15798. [DOI] [PubMed] [Google Scholar]
- [40].Krishnamurthy N, Zhao X, Burrows CJ, David SS, Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1, Biochemistry 47 (2008) 7137–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dizdaroglu M, Coskun E, Jaruga P, Repair of oxidatively induced DNA damage by DNA glycosylases: mechanisms of action, substrate specificities and excision kinetics, Mutat. Res Rev. Mutat. Res 771 (2017) 99–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Minko IG, Vartanian VL, Tozaki NN, Linde OK, Jaruga P, Coskun SH, Coskun E, Qu C, He H, Xu C, Chen T, Song Q, Jiao Y, Stone MP, Egli M, Dizdaroglu M, McCullough AK, Lloyd RS, Characterization of rare NEIL1 variants found in East Asian populations, DNA Repair 79 (2019) 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Minko IG, Vartanian VL, Tozaki NN, Coskun E, Coskun SH, Jaruga P, Yeo J, David SS, Stone MP, Egli M, Dizdaroglu M, McCullough AK, Lloyd RS, Recognition of DNA adducts by edited and unedited forms of DNA glycosylase NEIL1, DNA Repair (Amst. ) 85 (2020), 102741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Minko IG, Christov PP, Li L, Stone MP, McCullough AK, Lloyd RS, Processing of N5-substituted formamidopyrimidine DNA adducts by DNA glycosylases NEIL1 and NEIL3, DNA Repair (Amst. ) 73 (2019) 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Couve-Privat S, Mace G, Rosselli F, Saparbaev MK, Psoralen-induced DNA adducts are substrates for the base excision repair pathway in human cells, Nucleic Acids Res 35 (2007) 5672–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Takahashi Y, Nakatsuru Y, Zhang S, Shimizu Y, Kume H, Tanaka K, Ide F, Ishikawa T, Enhanced spontaneous and aflatoxin-induced liver tumorigenesis in xeroderma pigmentosum group A gene-deficient mice, Carcinogenesis 23 (2002) 627–633. [DOI] [PubMed] [Google Scholar]
- [47].Alekseyev YO, Hamm ML, Essigmann JM, Aflatoxin B1 formamidopyrimidine adducts are preferentially repaired by the nucleotide excision repair pathway in vivo, Carcinogenesis 25 (2004) 1045–1051. [DOI] [PubMed] [Google Scholar]
- [48].Jacobs AC, Calkins MJ, Jadhav A, Dorjsuren D, Maloney D, Simeonov A, Jaruga P, Dizdaroglu M, McCullough AK, Lloyd RS, Inhibition of DNA glycosylases via small molecule purine analogs, PLoS One 8 (2013), e81667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dou H, Theriot CA, Das A, Hegde ML, Matsumoto Y, Boldogh I, Hazra TK, Bhakat KK, Mitra S, Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes, J. Biol. Chem 283 (2008) 3130–3140. [DOI] [PubMed] [Google Scholar]
- [50].Reddy PT, Jaruga P, Nelson BC, Lowenthal M, Dizdaroglu M, Stable isotope-labeling of DNA repair proteins, and their purification and characterization, Protein Expr. Purif 78 (2011) 94–101. [DOI] [PubMed] [Google Scholar]
- [51].Kant M, Quintana V, Coskun E, Jaruga P, Lloyd RS, Sweasy JB, Dizdaroglu M, Polymorphic variant Asp239Tyr of human DNA glycosylase NTHL1 is inactive for removal of a variety of oxidatively-induced DNA base lesions from genomic DNA, DNA Repair (Amst. ) 117 (2022), 103372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hegde ML, Theriot CA, Das A, Hegde PM, Guo Z, Gary RK, Hazra TK, Shen B, Mitra S, Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1, J. Biol. Chem 283 (2008) 27028–27037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Theriot CA, Hegde ML, Hazra TK, Mitra S, RPA physically interacts with the human DNA glycosylase NEIL1 to regulate excision of oxidative DNA base damage in primer-template structures, DNA Repair (Amst. ) 9 (2010) 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hegde ML, Banerjee S, Hegde PM, Bellot LJ, Hazra TK, Boldogh I, Mitra S, Enhancement of NEIL1 protein-initiated oxidized DNA base excision repair by heterogeneous nuclear ribonucleoprotein U (hnRNP-U) via direct interaction, J. Biol. Chem 287 (2012) 34202–34211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hegde PM, Dutta A, Sengupta S, Mitra J, Adhikari S, Tomkinson AE, Li GM, Boldogh I, Hazra TK, Mitra S, Hegde ML, The C-terminal domain (CTD) of human DNA glycosylase NEIL1 is required for forming BERosome repair complex with DNA replication proteins at the replicating genome: dominant negative function of the CTD, J. Biol. Chem 290 (2015) 20919–20933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sharma N, Chakravarthy S, Longley MJ, Copeland WC, Prakash A, The C-terminal tail of the NEIL1 DNA glycosylase interacts with the human mitochondrial single-stranded DNA binding protein, DNA Repair (Amst. ) 65 (2018) 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Doublie S, Bandaru V, Bond JP, Wallace SS, The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity, Proc. Natl. Acad. Sci. USA 101 (2004) 10284–10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yeo J, Goodman RA, Schirle NT, David SS, Beal PA, RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1, Proc. Natl. Acad. Sci. USA 107 (2010) 20715–20719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Prakash A, Carroll BL, Sweasy JB, Wallace SS, Doublie S, Genome and cancer single nucleotide polymorphisms of the human NEIL1 DNA glycosylase: activity, structure, and the effect of editing, DNA Repair (Amst. ) 14 (2014) 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhu C, Lu L, Zhang J, Yue Z, Song J, Zong S, Liu M, Stovicek O, Gao YQ, Yi C, Tautomerization-dependent recognition and excision of oxidation damage in base-excision DNA repair, Proc. Natl. Acad. Sci. USA 113 (2016) 7792–7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Liu M, Zhang J, Zhu C, Zhang X, Xiao W, Yan Y, Liu L, Zeng H, Gao YQ, Yi C, DNA repair glycosylase hNEIL1 triages damaged bases via competing interaction modes, Nat. Commun 12 (2021) 4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tomar R, Minko IG, Sharma P, Kellum AH, Lei L, Harp JM, Iverson TM, Lloyd RS, Egli M, Stone MP, Base excision repair of the N-(2-deoxy-d-erythro-pentofuranosyl)-urea lesion by the hNEIL1 glycosylase, Nucleic Acids Res 51 (2023) 3754–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chen J, Zhu J, Wang G, Groopman JD, Kensler TW, Qidong: a crucible for studies on liver cancer etiology and prevention, Cancer Biol. Med 16 (2019) 24–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wang JS, Shen X, He X, Zhu YR, Zhang BC, Wang JB, Qian GS, Kuang SY, Zarba A, Egner PA, Jacobson LP, Munoz A, Helzlsouer KJ, Groopman JD, Kensler TW, Protective alterations in phase 1 and 2 metabolism of aflatoxin B1 by oltipraz in residents of Qidong, People’s Republic of China, J. Natl. Cancer Inst 91 (1999) 347–354. [DOI] [PubMed] [Google Scholar]
- [65].Kensler TW, Chen JG, Egner PA, Fahey JW, Jacobson LP, Stephenson KK, Ye L, Coady JL, Wang JB, Wu Y, Sun Y, Zhang QN, Zhang BC, Zhu YR, Qian GS, Carmella SG, Hecht SS, Benning L, Gange SJ, Groopman JD, Talalay P, Effects of glucosinolate-rich broccoli sprouts on urinary levels of aflatoxin-DNA adducts and phenanthrene tetraols in a randomized clinical trial in He Zuo township, Qidong, People’s Republic of China, Cancer Epidemiol. Biomark. Prev 14 (2005) 2605–2613. [DOI] [PubMed] [Google Scholar]
- [66].Tang L, Tang M, Xu L, Luo H, Huang T, Yu J, Zhang L, Gao W, Cox SB, Wang JS, Modulation of aflatoxin biomarkers in human blood and urine by green tea polyphenols intervention, Carcinogenesis 29 (2008) 411–417. [DOI] [PubMed] [Google Scholar]
- [67].Zhao S, Tadesse S, Kidane D, Significance of base excision repair to human health, Int Rev. Cell Mol. Biol 364 (2021) 163–193. [DOI] [PubMed] [Google Scholar]
- [68].Vartanian V, Lowell B, Minko IG, Wood TG, Ceci JD, George S, Ballinger SW, Corless CL, McCullough AK, Lloyd RS, The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase, Proc. Natl. Acad. Sci. USA 103 (2006) 1864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Galick HA, Marsden CG, Kathe S, Dragon JA, Volk L, Nemec AA, Wallace SS, Prakash A, Doublie S, Sweasy JB, The NEIL1 G83D germline DNA glycosylase variant induces genomic instability and cellular transformation, Oncotarget 8 (2017) 85883–85895. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.