

Abstract
Background
Ulcerative colitis (UC) and Crohn’s disease are 2 types of inflammatory bowel disease (IBD), a group of chronic digestive disorders caused by aberrant immune responses to intestinal microbes. Although changes in the composition of immune cell subsets in the context of IBD have been previously described, the interactions and communication among cells are less well understood. Moreover, the precise mechanisms of action underlying many biologic therapies, including the anti-α4β7 integrin antagonist vedolizumab, remain incompletely understood. Our study aimed to explore possible additional mechanisms through which vedolizumab acts.
Methods
We performed cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) on peripheral blood and colon immune cells derived from patients with ulcerative colitis treated with the anti-α4β7 integrin antagonist vedolizumab. We applied a previously published computational approach, NicheNet, to predict immune cell-cell interactions, revealing putative ligand-receptor pairs and key transcriptional changes downstream of these cell-cell communications (CCC).
Results
We observed decreased proportions of T helper 17 (TH17) cells in UC patients who responded to vedolizumab and therefore focused the study on identifying cell-cell communications and signals of TH17 cells with other immune cells. For example, we observed that colon TH17 cells from vedolizumab nonresponders were predicted to have a greater degree of interactions with classical monocytes compared with responders, whereas colon TH17 cells from vedolizumab responders exhibited more interactions with myeloid dendritic cells compared with nonresponders.
Conclusions
Overall, our results indicate that efforts to elucidate cell-cell communications among immune and nonimmune cell types may increase the mechanistic understanding of current and investigational therapies for IBD.
Keywords: single-cell RNA-sequencing, cell-cell communications, T cells, ulcerative colitis, vedolizumab
Key Messages.
What is already known?
Vedolizumab, a monoclonal antibody that binds to ⍺4β7 integrin, is thought to work primarily by blocking leukocyte trafficking into the gut; however, changes in innate immunity are consistently reported following treatment.
What is new here?
Inferred cell-cell communications show that signals from innate immune cells that promote TH17 differentiation are decreased in vedolizumab responders.
How can this study help patient care?
This study provides evidence of a possible mechanism of action for vedolizumab that may decrease innate cell signaling promoting TH17 cell differentiation, which may contribute to its clinical efficacy.
Introduction
Ulcerative colitis (UC) is a type of inflammatory bowel disease (IBD) characterized by chronic intestinal inflammation that is limited to the mucosal layer of the colon. By contrast, Crohn’s disease, another subtype of IBD, is characterized by transmural inflammation that can affect any part of the alimentary tract. Inflammatory bowel disease is thought to arise from a dysregulation of innate and adaptive immune responses to gut microbiota, but the pathophysiology is still not completely understood.1
Many types of immune and nonimmune cells have been implicated in the development of IBD, including CD4+ and CD8+ T lymphocytes,2-4 stromal cells,5 macrophages,6 innate lymphoid cells,7 and epithelial cells.8 Many groups have sought to better characterize changes in the number, composition, and phenotype of immune cell changes within the peripheral blood and intestine in IBD patients.2-5,8,9 However, the cell-cell interactions and signaling events that drive observed alterations of specific cell types are less well understood. Moreover, the precise mechanism of action of effective therapies for IBD, including the α4β7 integrin antagonist vedolizumab, remains poorly understood.
Vedolizumab is a humanized monoclonal antibody targeting the ⍺4β7 integrin heterodimer expressed on the surface of lymphocytes that mediates migration into intestinal Peyer’s patches and lamina propria via interaction with the mucosal vascular adhesin MAdCAM-1.10 In vitro studies of vedolizumab show that the humanized antibody results in internalization of ⍺4β7 integrin in lymphocytes without subsequent lymphocyte activation and does not activate antibody-dependent cellular cytotoxicity or complement-dependent cytotoxicity.11 Vedolizumab’s clinical potency has been presumed to be due to exclusion of gut-homing ⍺4β7 integrin-expressing lymphocytes from intestinal tissue; its effect on innate immune cells is becoming increasingly appreciated. Recent studies reveal additional functions for ⍺4β7 integrin signaling, including promoting T-cell proliferation and upregulating T helper 17 (TH17) cell cytokine production and gene expression.12 Single-cell analyses of gut tissues from IBD patients receiving vedolizumab have suggested significant interactions between TH17 cells and inflammatory monocytes/macrophages.4,13,14 These findings, combined with the observation that the innate immune response is also affected by ⍺4β7 blockade, reinforce the notion that vedolizumab may exert actions beyond simply inhibiting lymphocyte trafficking.
Here, we used cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq),15 which integrates transcriptional and proteomic profiling in single-cells, to analyze peripheral blood and colon immune cells from UC patients following treatment with vedolizumab, some of whom responded to therapy and others who did not. We annotated the cells using a published RNA-seq reference data set16 and applied Differential NicheNet, an algorithm that infers ligand-receptor relationships from differential gene expression,17 in order to identify putative cell-cell communications (CCCs) and predict secondary downstream transcriptional changes. We identified decreased proportions of colonic TH17 cells in UC patients who responded to vedolizumab compared with those who failed to respond. We therefore focused the study on characterizing cell-cell communications of TH17 cells with other immune cells. For example, we observed that colon TH17 cells from vedolizumab nonresponders were predicted to have a greater degree of interactions with classical monocytes compared with responders, whereas colon TH17 cells from vedolizumab responders exhibited more interactions with myeloid dendritic cells. Overall, our results indicate that efforts to elucidate cell-cell communications among immune and nonimmune cell types may advance the mechanistic understanding of current and investigational therapies for IBD.
Materials and Methods
Patient Criteria and Ethics
This study was reviewed and approved by the Human Research Protections Program at the University of California, San Diego and the San Diego VA Healthcare. Patients with a diagnosis of ulcerative colitis and active endoscopic disease on minimal or no medical therapy were consented into the study; patient demographics and individual disease characteristics and prior treatment regimens are provided in Supplementary Tables 1 and 2. Intestinal biopsies and peripheral blood were obtained from patients undergoing routine colonoscopy for clinical management of their disease.
Human Peripheral Blood Mononuclear and Intestinal Cell Isolation
Blood was collected directly into BD Vacutainer CPT cell preparation tubes (BD Biosciences) for isolation of CD45+ immune cells, as previously described.18 Prior to single-cell sequencing, frozen cells were thawed, washed, and passed through 70-µm filters prior to antibody labeling and fluorescence-activated cell sorting (FACS) described below. Intestinal biopsies were obtained with endoscopic biopsy forceps from the rectum and collected in conical tubes with Hank’s buffered saline solution (HBSS, Corning); they were then transferred into freezing buffer (10% [v/v] dimethyl sulfoxide [DMSO], 40% [v/v] complete Roswell Park Memorial Institute media [RPMI], 50% [v/v] fetal bovine serum [FBS]) and stored at −80°C, as previously described.18
Purification of CD45+ Immune Cells and 10x Genomics Library Preparation and Sequencing
Single-cell suspensions of peripheral blood mononuclear cell (PBMC) and intestinal cell preparations were incubated with a Fixable Viability Dye eFluor 780 (eBioscience, Invitrogen) on ice for 10 minutes and then incubated with BioLegend Human TruStain FcX Block for 10 minutes to block nonspecific binding of the Fc receptor. An antibody mixture containing antihuman CD45 (2D1; BioLegend) and a 58-plex TotalSeq antibody cocktail generated using 1 ug of each TotalSeq antibody was prepared in Cell Staining Buffer (BioLegend). Samples were incubated for 60 minutes on ice, then washed, and resuspended in complete RPMI. The CD45+ immune cells were sorted on a FACSAria2 (BD Biosciences) and collected in fresh complete RPMI. The FACS-purified CD45+ immune cells were then washed and resuspended in PBS + 0.04% (w/v) bovine serum albumin (UltraPure BSA, Thermo Fisher Scientific). Single-cell libraries were prepared according to the protocol for 10x Genomics for Single Cell 3’ Gene Expression v3.1, as previously described.18 Single-cell RNA and protein libraries were pooled at equimolar amounts for a final 10 nM concentration and sequenced on a NovaSeq 6000 S4 Flow Cell (Illumina), as previously described.18
Computational Analyses
Single-cell analyses and computational analyses using Seurat and NicheNet were performed as previously published.18 Raw sequencing data were fed through CellRanger pipeline (10x Genomics), and downstream processing was done using Seurat v4.1.1.19 The data were filtered to remove low quality cells and doublets with the following parameters: <200 features, >10% mitochondrial features, and >2500 features. Protein and RNA data sets were normalized, and then immune cell types were assigned labels using SingleR v.1.4.120 and manually verified with canonical protein markers according to Supplementary Table 3. NicheNet, a cell-cell communications workflow, compares differentially expressed (DE) genes in designated receiver cells to the DE genes of possible sender cells to infer key ligand-receptor pairs between the senders and receivers. We used Differential Nichenet, the most updated version of Nichenet, to rank upstream ligands and receptors in senders and receivers that had at least 25 identified cells within their respective tissue and expressed by at least 10% of the cell population, as previously described.18
Pathway Enrichment Analyses
The top 50 prioritized ligand-receptor pairs were inputs for ligand-receptor pathway analysis. For ligand-target pathway analysis, the ligand-targets were required to share the same highly ranked ligand-receptors upstream of targets. The top 2 ligand-sender pairs with the highest ligand target weights served as pathway analysis inputs.
Statistical Analysis
GraphPad Prism 9 (GraphPad Software) was used for statistical analysis of cell type proportions and for subsequent graphical representations of data. Descriptive statistics are presented as mean ± standard deviation. Continuous variables were analyzed using Mann-Whitney U tests. A 2-tailed P < .05 was considered statistically significant.
Results
Study Population
A cohort of 13 patients with ulcerative colitis treated with the anti-α4β7 integrin antibody vedolizumab and evaluated by colonoscopy 5 to 9 months following initiation of drug therapy (Figure 1A, Supplementary Table 1, Supplementary Table 2) was recruited for the study. Drug response was determined by subsequent endoscopic Mayo score following vedolizumab: 6 patients were categorized as nonresponders with moderate or severe disease, and 7 patients were categorized as responders with no to mild disease. There were 7 females and 6 males, with an approximately equal sex distribution in the nonresponders and nonresponders (57% vs 50%). The mean age was 41.4 years for all patients, 41.5 years for nonresponders, and 41.3 for responders. The mean age at diagnosis was 33.8 years for all patients, 34.5 years for nonresponders, and 33.3 years for responders. The average body mass index was 28.15 for all patients, 25.75 for nonresponders, and 30.21 for responders. With regards to disease extent, 8 of 13 patients (62%) had pancolitis seen on prior endoscopy, 3 of 13 (23%) had left-sided colitis, and 2 of 13 (15%) had proctitis. One of 6 nonresponders was taking concomitant systemic corticosteroids; another nonresponder was taking concomitant methotrexate; and a third nonresponder was taking concomitant azathioprine (Supplementary Table 2). Two of 7 responders in this cohort were biologic-naïve, but all 6 nonresponders had prior anti-tumor necrosis factor (TNF) therapy with infliximab or adalimumab. The average duration of vedolizumab exposure before endoscopic evaluation for nonresponders was 228.5 days compared with 153.4 days for responders.
Figure 1.
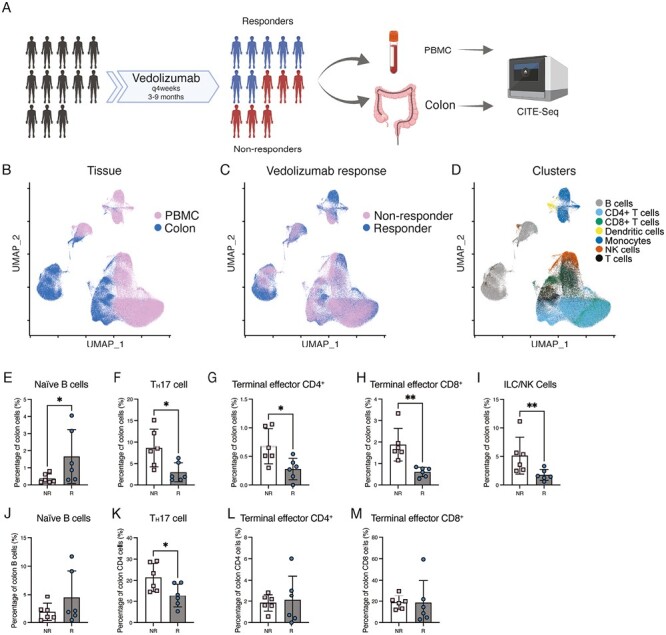
CITE-seq analyses of immune cells of vedolizumab nonresponders and responders. A, Experimental design. Immune cells from blood and colon derived from subjects with ulcerative colitis (UC) treated with vedolizumab (VDZ) for 5 to 9 months were stained with the TotalSeq antibody cocktail and fluorescently labeled antibodies, isolated by FACS, and prepared for CITE-seq using the 10x Genomics Chromium platform. Image created with BioRender.com. B, UMAP plots colored by tissue compartment identity, (C) by nonresponder (NR) vs responder (R), and (D) major immune cell type. E-I, Proportions of naïve B cells, TH17 cells, terminal effector CD4+ T cells, terminal effector CD8+ T cells, and innate lymphoid cell/natural killer cell (ILC/NK) in colon tissue. J, Naïve B cells as a proportion of total colon B cells. K, TH17 cells as a proportion of total colon CD4+ T cells. L, Terminal effector CD4+ T cells as a proportion of total colon CD4+ T cells. M, Terminal effector CD8+ T cells as a proportion of total colon CD8+ T cells.
CITE-seq Analysis of Immune Cells from Vedolizumab Responders and Nonresponders
Cells from mucosal biopsies and peripheral blood were processed into single-cell suspensions, FACS-purified on the basis of CD45, a pan-immune cell marker, and processed for CITE-seq using the 10x Genomics Chromium platform (Figure 1A). Antibodies targeting 58 proteins were selected for inclusion in the antibody panel (Supplementary Table 3). Uniform manifold approximation and projection (UMAP) analysis showed that cells from peripheral blood and colon clustered separately (Figure 1B), whereas cells from nonresponders and responders generally clustered together (Figure 1C). We broadly annotated the immune cells into 7 major immune cell types and observed that B cells and myeloid cells clustered distinctly from the other immune cell types, including CD4+ T cells, CD8+ T cells, unconventional T cells, and natural killer (NK) cells (Figure 1D). To further refine our annotations, we used a previously published set of peripheral blood immune cell transcriptional signatures derived from bulk RNA-seq of FACS-purified cells.16 Because these immune cell subsets were derived from the peripheral blood rather than tissue, we revised the annotations of these subsets to account for cell populations found in the tissues exhibiting transcriptional signatures similar to those of cells circulating in the peripheral blood (Supplementary Table 4). For example, the original annotation “NK cells” was revised to “ILC/NK cells,” as NK cells are considered a subset of the innate lymphoid cell (ILC) family and exhibit similar transcriptional signatures.21
Immune profiling demonstrated changes in cell abundances in colon tissue but not in the peripheral blood. We observed that T cells were the most abundant immune cell type in the peripheral blood, though there were no differences in the proportions of T cells between responders and nonresponders (Supplementary Figure 1A). Within colon tissue, responders exhibited an increase in the proportions of cells annotated as naïve B cells compared with that in nonresponders (Figure 1E), whereas there were no significant changes observed in the proportions of plasmablasts (Supplementary Figure 1B). Moreover, vedolizumab responders exhibited decreased proportions in TH17, terminal effector CD4+, terminal effector CD8+, and innate lymphoid cells/natural killer cells (ILC/NK cells; Figures 1F–I), all of which have been previously implicated in the pathogenesis of UC.22-24 The TH1 and TH1/17 subsets were not significantly different between responders and nonresponders (Supplemental Figures 2A and B). There were no significant differences in the proportions of innate immune cells such as myeloid dendritic cells or monocyte subsets within the peripheral blood or colon tissue (Supplemental Figures 2C-F). However, there was a trend towards increased nonclassical monocytes in responders, corresponding to a decrease in the proportion of classical monocytes as a subset of all monocytes within peripheral blood (Supplemental Figure 2G, H). Vedolizumab has been previously reported to alter lymphocyte populations within the lamina propria, though no significant alterations in lymphocyte populations in peripheral blood were observed in the current study.14 The absence of significant alterations to most lymphocyte populations suggested that an alternative mechanism, such as differentiation or cell signaling, may play a role in clinical responders.
We confirmed the accuracy of the modified annotations using the protein and gene expression data (Figure 2A and B). For example, the “tissue-resident memory T cells/MAIT (TRM/MAIT)” cluster expressed high levels of CD69 and CD103, which are markers associated with tissue-resident memory T (TRM) cells. Cells expressing CD3 were annotated as CD4+ and CD8+ T cells and included TRM cells (Figure 2C). The 8 clusters annotated as CD4+ T cells generally had higher expression of CD4 protein (Figure 2D), whereas the 4 clusters annotated as CD8+ T cells had higher expression of CD8⍺ protein (Figure 2E). The CD19 protein was most highly expressed in cells annotated as B cells, confirming the accuracy of the annotations (Figure 2F).
Figure 2.
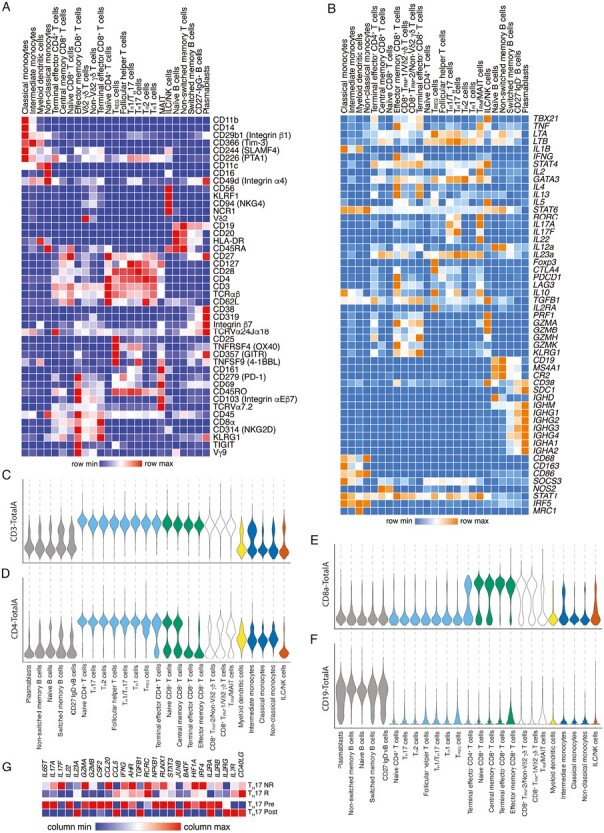
Expression of canonical genes and proteins in annotated immune cell subsets. A, Relative expression of selected proteins included in the CITE-seq antibody panel, represented as a hierarchically clustered summary heatmap; columns represent each of the 25 annotated immune cell subsets, and rows represent individual proteins. B, Relative expression of selected canonical genes, represented as a hierarchically clustered summary heatmap; columns represent each of the 25 annotated immune cell subsets and rows represent selected genes. C-F, Expression of selected proteins in each of the 25 annotated immune cell subsets, represented as violin plots, ordered, and colored by major immune cell type (gray, B cells; light blue, CD4+ T cells; green, CD8+ T cells; black outline, tissue-resident/unconventional T cells; yellow, myeloid cells; blue, monocytes; brown, ILC/NK cells. G, Relative expression of selected genes related to TH17 function and regulation comparing colon TH17 from vedolizumab nonresponders, vedolizumab responders, single subject pretreatment, and single subject post-treatment, represented as a summary heat map; columns represent selected genes and rows represent the samples.
Vedolizumab Responders Exhibit a Decrease in the Inflammatory TH17 Cell Effector Program
Interleukin (IL)-17 signaling and T helper 17 (TH17) cells have been associated with the pathogenesis and disease activity of UC.3,22 As mentioned above, vedolizumab responders exhibited a decrease in the proportions of TH17 cells compared with nonresponders (Figure 1F). When we compared selected genes expressed by colon TH17 cells in the responder, nonresponder, pre-treatment, and post-treatment samples (Figure 2G), we observed decreased expression of inflammatory cytokines and granzymes such as IL6ST, IL17A, IL17F, GZMA, GZMB, CCL20, and IFNG, consistent with quiescent disease, along with increased expression of IL23A, CCL5, and TGFB1. Similarly, comparison of colon TH17 cells from the pre- vs post-treatment conditions revealed increased expression of IL6ST, IL17A, IL22, GZMA, GZMB, CCL20, and IFNG, along with decreased IL23A, CCL5, and TGFB1 expression in pretreatment colon TH17 cells. With respect to transcription factors involved in TH17 effector function in nonresponders vs responders, we observed decreased expression of AHR, NFKB1, RUNX1, STAT3, BATF, HIF1A, and IRF4. Comparison of cytokine receptor expression revealed increased expression of IL2RB (CD122), which controls responsiveness to IL-15, in colon TH17 cells from vedolizumab responders and the pretreatment sample. Conversely, increased expression of IL7R (CD127), which controls responsiveness to IL-7, was observed in colon TH17 cells from vedolizumab responders and the pretreatment sample. These findings suggest that the clinical response to vedolizumab may be associated with changes in the gene expression and cellular interactions of TH17 cells.
TH17 From Vedolizumab Nonresponders Have Increased Signaling From Pro-inflammatory Innate Immune Cells
To explore CCCs that might regulate changes in TH17 cells in vedolizumab nonresponders vs responders, we applied NicheNet,17 a previously published computational algorithm that infers ligand-receptor and ligand-target relationships from single-cell gene expression data. The approach involves the following steps: (1) defining the “sender” cell types, “receiver” cell types, and gene set that will guide the selection of prioritized ligands; (2) defining a set of potential ligands expressed by the defined sender cell types known to bind potential receptors expressed by defined receiver cell types; (3) ranking ligands based on transcriptional signatures downstream of the putative ligand; and (4) inferring target genes in receiver cells based on specific ligand-receptor pairs. Thus, putative ligand-receptor pairs can be identified between sender-receiver cell types, and gene expression changes downstream of engagement of ligand-receptor interactions can be assessed.
We designated TH17 cells as receivers and compared ligand-receptor and ligand-target interactions inferred in vedolizumab responders vs nonresponders (Figure 3). We observed that colon TH17 cells from vedolizumab nonresponders had a greater degree of interactions with classical monocytes compared with responders (Figures 3A, B). Conversely, colon TH17 cells from vedolizumab responders exhibited more interactions with myeloid dendritic cells compared to nonresponders. Moreover, colon TH17 cells from vedolizumab nonresponders exhibited interactions with TH1/TH17 cells and ILC/NK cells that were strikingly absent in responders, whereas colon TH17 cells from vedolizumab responders exhibited a number of interactions that were absent in nonresponders, including interactions with follicular T helper cells, naïve B cells, naïve CD8+ T cells, and central memory CD8+ T cells (Figures 3A, B).
Figure 3.
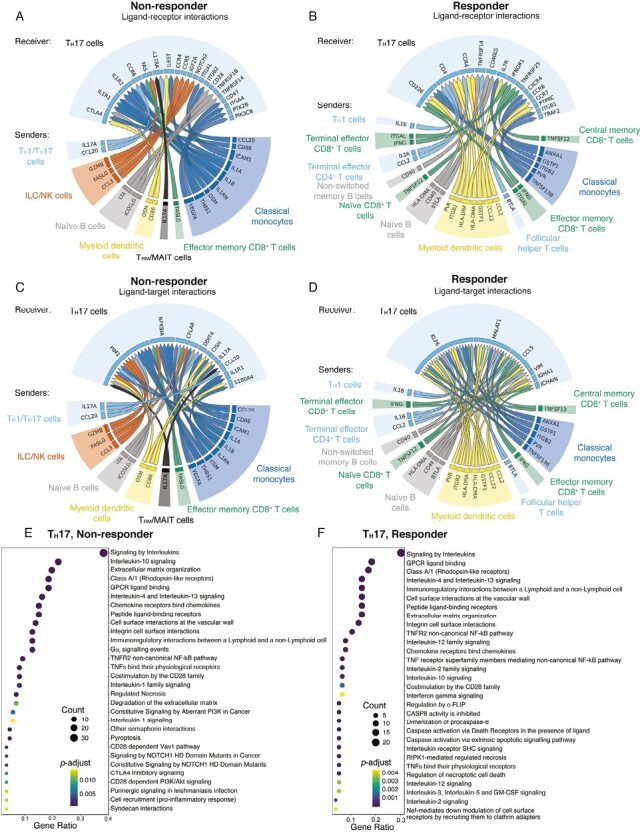
NicheNet analyses of colon TH17 cell interactions with other immune cells in vedolizumab nonresponders vs responders. A-B, Putative ligand-receptor pairs on “sender” immune cells and “receiver” cell (TH17), represented as Circos plots, in nonresponders and responders. Colors indicate major immune cell type. Specific immune cell subsets are labeled around the Circos plot. C-D, Genes predicted to be induced in TH17 cells by predicted ligand-receptor interactions, represented as Circos plots, in nonresponders and responders. E-F, Pathway analyses of enriched genes downstream of predicted ligand-receptor pairs represented as reactome plots in nonresponders and responders.
Focusing next on specific ligand-receptor interactions in vedolizumab nonresponders, we observed that IL1A and IL1B produced by classical monocytes (Supplemental Figure 3A) were predicted to bind to IL1RA and IL1RB on colonic TH17 cells (Figure 3A, Supplemental Figure 3A), suggesting a possible increase in inflammatory interleukin (IL)-1 signaling, which has been associated with active inflammation and TH17 cell differentiation in the colons of UC patients.25,26 Expression of oncostatin M (OSM), an IL-6 family cytokine associated with treatment responsiveness, was elevated in both classical monocytes and myeloid dendritic cells.27 Moreover, IL17A was predicted to be produced by TH1/TH17 and TRM/MAIT cells and received by TH17 cells in nonresponders (Figure 3A, Supplemental Figure 3B). This observation may represent a compensatory response to limit inflammation by TH17 cells since IL-17A signaling in TH17 cells has been reported to function in a cell-intrinsic negative feedback loop that limits production of other pathogenic TH17-lineage cytokines28 like IL-17F, IL-22, and GM-CSF (Figure 2G). Lastly, FASLG expressed by ILC/NK and effector memory CD8+ T cells was predicted to bind to FAS on TH17 cells, whereas GZMB produced by ILC/NK cells was predicted to bind to its receptor IGF2R on TH17 cells (Figure 3A). These observations raised the possibility of killing of TH17 cells by ILC/NK cells or effector memory CD8+ T cells as a compensatory mechanism to dampen inflammation.
We next examined inferred ligand-receptor interactions with colon TH17 cells in vedolizumab responders. The pleiotropic cytokine interleukin 16, encoded by IL16, which has been shown to act as an inhibitor of antigen-specific CD4+ T cell activation,29 was expressed by TH1 and terminal effector CD4+ T cells and predicted to act on TH17 cells (Figure 3B, Supplemental Figure 3B), which may result in a dampening of intestinal inflammation. Glutathione S-transferase P, encoded by GSTP1, which plays an important role in detoxification by catalyzing the conjugation of many hydrophobic and electrophilic compounds with reduced glutathione, was predicted to be expressed by myeloid dendritic cells; notably, GSTP1 polymorphisms have been associated with an increased risk of ulcerative colitis.30 Naïve B cells and TFH cells exhibited increased expression of B and T lymphocyte attenuator, encoded by BTLA, which was predicted to bind to TNFRSF14, also known as the herpesvirus entry mediator (HVEM) on TH17 cells. The complex costimulatory receptor HVEM transmits an activating signal when bound to its ligand LIGHT but an inhibitory signal when bound by BTLA; intriguingly, TNFRSF14 has been identified as an IBD risk gene.31 Classical monocytes and myeloid dendritic cells produced ANXA1 (Supplemental Figure 3A), a potent anti-inflammatory protein that has been shown to reduce TH17 cytokine production32 and was predicted to bind to CXCR4 and CCR6 on TH17 cells in vedolizumab responders. Taken together, these findings suggest specific CCC in vedolizumab responders that play a role in mediating its effects.
Analyses of predicted gene expression downstream of inferred ligand-receptor interactions in vedolizumab nonresponders revealed induction of a number of genes in colon TH17 cells. Induction of IL17A may reflect uncontrolled inflammation in vedolizumab nonresponders, whereas production of CCL20 may recruit additional TH17 cells into the tissue (Figures 3C and 2G).33 In addition, induction of IL1R1, S100A4, DDIT4, and PIM1, factors that promote TH17 differentiation, may contribute to increased proportions of colonic Th17 cells in vedolizumab nonresponders. Increased interleukin-1 signaling through IL1R1 may favor the differentiation of TH17 over TREG cells by inhibiting TGFβ-induced FOXP3 expression.26 The calcium binding protein encoded by S100A4 has been shown to regulate STAT3 activity and expression of the TH17 lineage-defining factor RORɣt,30 DNA-damage-inducible transcript 4, encoded by DDIT4, has been shown to promote human TH17 cell differentiation in autoimmune disease.34Induction of PIM1, which encodes for a serine/threonine kinase, has been shown to promote TH17 cell differentiation and pathogenicity.35 In vedolizumab responders, analyses of predicted gene expression downstream of inferred ligand-receptor interactions revealed induction of a distinct set of genes (Figure 3D). For example, MALAT1 encodes for a long noncoding RNA that has been shown to repress the TH17 cell effector program and has been associated with reduced disease severity in murine experimental autoimmune encephalomyelitis.36 Pathway analyses revealed enrichment of pathways involved in IL-1 signaling and pro-inflammatory cell recruitment responses in vedolizumab nonresponders (Figure 3E), whereas enrichment of IL-3 signaling, which has been shown to inhibit TH17 differentiation and promote TREG cell differentiation,37 was enriched in vedolizumab responders (Figure 3F).
Increased Pro-inflammatory Chemokines and Cytokines Pre- vs Post-therapy
We next performed analyses of a responder patient, corresponding to patient 1 in Supplemental Table 2, for whom we had collected peripheral blood and colon samples prior to and following vedolizumab therapy. We observed decreased proportions of classical and intermediate monocytes along with increased proportions of naïve T cells in the blood following therapy (Supplementary Figure 1C). In the colon, we observed decreased proportions of plasmablasts and increased proportions of TREG cells (Supplementary Figure 1D). We designated TH17 cells as receivers and compared ligand-receptor and ligand-target interactions inferred in pre- vs post-therapy samples (Figure 4). Compared with cells from the post-therapy sample, we observed that colon TH17 cells from the pre-therapy sample exhibited a greater degree of interactions with ILC/NK cells (Figure 4A). By contrast, colon TH17 cells from the post-therapy sample exhibited more interactions with terminal effector CD8+ T cells, which was strikingly absent in cells derived from the pre-therapy sample.
Figure 4.
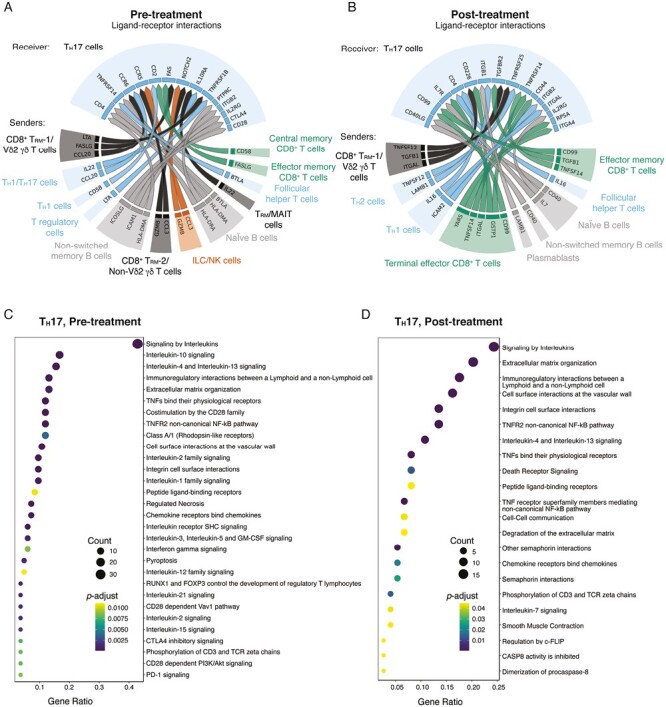
NicheNet analyses of colon TH17 cell interactions with other immune cells in a single subject pre- and post-vedolizumab therapy. A-B, Putative ligand-receptor pairs on “sender” immune cells and “receiver” cells (TH17), represented as Circos plots, in pre-treatment and post-treatment samples. Colors indicate major immune cell type. Specific immune cell subsets are labeled around the Circos plot. C-D, Pathway analyses of enriched genes downstream of predicted ligand-receptor pairs, represented as reactome plots, in pre-treatment and post-treatment samples.
Focusing on specific ligand-receptor interactions in the pre-therapy sample, we observed that the chemokines CCL3 and CCL20 were predicted to bind to colon TH17 cells (Figure 4A, Supplemental Figure 3C); these chemokines were notably absent in the post-therapy sample (Figure 4B). In the pre-therapy sample, the pro-inflammatory FASLG expressed by CD8+ TRM-1/Vδ2 γδ T cells and effector memory CD8+ T cells was predicted to bind to FAS on TH17 cells, whereas GZMB produced by ILC/NK cells was predicted to bind to its receptor IGF2R on TH17 cells (Figure 4A). Moreover, the TNF superfamily ligand LTA produced by CD8+ TRM-1/Vδ2 γδ T cells and TREG cells was predicted to bind to its receptor TNFRSF1B (TNFR2) in colon TH17 cells in the pre-therapy sample (Supplemental Figure 3C); signaling through the inflammatory cytokine LTA can induce apoptosis and inflammation, much like TNF.38 Lastly, IL22 was predicted to be produced by TH1/TH17 cells and TRM/MAIT cells (Figure 4A, Supplemental Figure 3D). Interleukin-22 signaling, which was elevated in inflamed mucosa, has been shown to stimulate epithelial tissue repair,39 and its induction in the pre-therapy colon TH17 cells may represent a compensatory effect to dampen inflammation.
In post-treatment colon samples, LAMB1, which encodes for the extracellular matrix protein laminin B1, was expressed by TH2 cells and predicted to bind to colon TH17 cells (Figure 4B). Polymorphisms in LAMB1 have been previously linked to UC risk.40 The immunoregulatory cytokine TGFB1 was produced by CD8+ TRM-1/Vδ2 γδ T cells and effector memory CD8+ T cells (Supplemental Figure 3C). The homeostatic cytokine IL7, which promotes the survival of T cells, was also produced by cells annotated as naïve B cells and predicted to bind to its corresponding receptor complex components IL7R and IL2RG produced by TH17 cells. Additional ligand-receptor interactions identified in post-treatment colon TH17 cells, such as GSTP1-IL7R and IL16-CD4, were also observed in vedolizumab responders, as described above.
Analyses of pathway analyses of gene expression downstream of inferred ligand-receptor interactions in pretreatment TH17 cells revealed an enrichment of pathways involved in IL-1 signaling and pro-inflammatory cell recruitment responses, along with signaling of several other cytokines, including IL-12, IL-2, and IL-15 (Figure 4C). Analyses of pathway analyses of gene expression in post-treatment TH17 cells revealed an enrichment of pathways associated with inhibition of CASP8 activity along with IL-7 signaling (Figure 4D). Taken together, these findings suggest that the clinical response to vedolizumab may be associated with changes in the gene expression and cellular interactions of TH17 cells and provide a resource data set for the field to investigate other immune cells that may influence the mechanism of action of vedolizumab.
Discussion
Targeted therapy against ⍺4β7 integrins has been beneficial for IBD patients with a favorable risk-efficacy profile and is mechanistically distinct from TNF and JAK inhibitors and IL-12/23 blockade.41 The main mechanism of action of vedolizumab is thought to involve blocking ⍺4β7-MAdCAM-1 binding, which limits lymphocyte migration into the gut. However, prior studies have suggested that vedolizumab may also alter gene expression of TH17 cells and that active UC is associated with increased production of TH17 cell-associated cytokines.4,13,14 We hypothesized that TH17 cells from vedolizumab nonresponders and responders might receive distinct cell signals that could contribute to these previously reported changes. In this study, we bioinformatically inferred CCC using single-cell RNA-sequencing data to identify interactions and signals between TH17 and other immune cells.
Consistent with previously reported findings, we observed alterations in immune cell populations within colonic tissue in vedolizumab nonresponders vs responders. In particular, we found increased relative proportions of TH17 cells, terminal effector CD4 and CD8 cells, and ILC/NK cells in nonresponders, along with decreased relative proportions of naïve B cells. This increase in the TH17 cell compartment in active UC mucosa has been previously reported, though it has not been a consistently identified feature.3,4,14 We did not observe differences in peripheral immune cell populations between nonresponders and responders.
We found that TH17 cells in vedolizumab nonresponders exhibited increased expression of TH17 cell cytokines IL17A, IL17F, and IFNG. This upregulation of an inflammatory IL-17 response has been reported in active UC tissue compared with noninflamed and healthy tissue3 and is likely a reflection of decreased disease activity.
Although we did not observe differences in the proportions of innate immune cell subsets in gut tissues of vedolizumab nonresponders and responders, we found differences in TH17 cell-associated signaling pathways. In vedolizumab nonresponders, we identified inflammatory monocytes/macrophages expressing IL1A, IL1B, IL1RN, OSM, and CCL20; OSM-expressing myeloid dendritic cells; and ILC/NK cells expressing GZMB, FASLG, and CCL3. Reduction of pretreatment expression levels of OSM following TNF inhibition or vedolizumab has been recognized as a marker of treatment responsiveness27,42; the elevated OSM expression in the nonresponders observed in the current study is likely a reflection of persistent disease activity and a failure to adequately suppress OSM. Ligand-target analysis identified PIM1 and IL1R1, factors that promote TH17 cell differentiation, as well as CCL20, a chemokine that recruits additional TH17 cells. Expression of PIM1 has been reported in pro-inflammatory TH17 cells and is necessary for TH17 cell differentiation in experimental models of uveitis.35 Additionally, IL-1 signaling has been shown to promote TH17 cell differentiation and expansion,26 and increased IL-1 signaling from pro-inflammatory macrophages has also been demonstrated in other studies.3,13 The signaling environment for vedolizumab responders was notably less inflammatory and reflected genes and pathways upregulated during homeostasis. For example, ANXA1, which is associated with resolution of inflammation and decreased lymphocyte migration,32 and MALAT1, which has been shown to inhibit inflammatory macrophage polarization and TH17 cell differentiation,36 were expressed in more highly expressed in vedolizumab responders. Thus, innate immune cells in vedolizumab nonresponders appear to express molecules that promote TH17 cell differentiation, expansion, and retention that may contribute to the observed increased TH17 cell proportions and activity. Taken together, the data raise the possibility that vedolizumab may decrease inflammatory TH17 cell activity indirectly through effects on innate immune cells.
All of the nonresponders had prior TNF therapy, but only 2 of the responders were biologic-experienced (Supplemental Table 2). Although not statistically significant, this trend is consistent with the clinical observation that previous exposure to biologics is associated with poorer response to subsequent types of therapy and raises the possibility that the observations reported here may have been influenced by prior immunosuppressive therapy. Other groups have observed that responders to anti-TNF therapy exhibited decreased monocyte numbers and activation compared with nonresponders,43,44 though the long-term durability of changes following discontinuation of anti-TNF therapy on immune profiles has not been established. In our cohort, we observed that within the peripheral monocyte compartment of vedolizumab nonresponders, there was a greater proportion of classical monocytes even after previously having received anti-TNF therapy (Supplemental Figure 2G). This supports the notion that a differential monocyte response is a marker of drug responsiveness for both anti-TNF therapy and vedolizumab.45 While the conventional synthetic disease modifying anti-rheumatic drugs methotrexate and azathioprine also have broad immunosuppressive effects on monocyte maturation and function, they appeared to be ineffective in the nonresponders who still exhibited evidence of inflammatory macrophage and dendritic cell gene expression profiles (Supplemental Figure 3A). Our study was not designed to detect differences in absolute cell number or other drug-specific effects.
There were several limitations of the study. Our study cohort was small and had limited representation from diverse ethnic backgrounds; as such, we were unable to draw any significant conclusions regarding drug response and immune profile based on ethnic backgrounds. Furthermore, our cohort lacked non-vedolizumab-treated patients in remission, limiting our ability to distinguish whether the changes we observed were a direct consequence of vedolizumab therapy or simply due to reduced UC disease activity. There was only 1 patient with paired pre- and post-treatment samples, precluding us from inferring cause and effect of vedolizumab therapy. Indirect comparison of our findings with a previously published set of differentially expressed genes in colonic tissue prior to vedolizumab treatment42 showed minimal overlap, with the exception of increased CD40L expression in responder TH17 cells (Figure 2G); as noted previously, nonresponders in the current study had increased expression of OSM in contrast to the decreased OSM expression seen in vedolizumab nonresponders in a previously published data set.42 As our study also did not analyze nonimmune cells, we were unable to infer contributions from nonimmune cells in the colonic tissue, thereby narrowing the scope of our investigation of CCC, and we were unable to make direct comparisons to prior studies using bulk tissue. Future investigations with mechanistic studies will be needed to further elucidate the interactions between TH17 and innate immune cells in UC and whether vedolizumab acts through mechanisms that are independent of lymphocyte trafficking.
Supplementary Material
Contributor Information
Paul Hsu, Department of Medicine, University of California San Diego, La Jolla, California, USA.
Eunice J Choi,, Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, California, USA.
Shefali A Patel,, Department of Medicine, University of California San Diego, La Jolla, California, USA.
William H Wong,, Department of Medicine, University of California San Diego, La Jolla, California, USA.
Jocelyn G Olvera,, Department of Medicine, University of California San Diego, La Jolla, California, USA.
Priscilla Yao,, Department of Medicine, University of California San Diego, La Jolla, California, USA.
Yi Chia Liu, Department of Medicine, University of California San Diego, La Jolla, California, USA.
Matthew S Tsai, Department of Medicine, University of California San Diego, La Jolla, California, USA.
Wei Wang, Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, California, USA; Department of Cellular and Molecular Medicine, University of California San Diego, La Jolla, California, USA.
Brigid S Boland, Department of Medicine, University of California San Diego, La Jolla, California, USA.
John T Chang, Department of Medicine, University of California San Diego, La Jolla, California, USA; Department of Medicine, Jennifer Moreno Department of Veteran Affairs Medical Center, San Diego, CA, 92161, USA.
Author Contributions
Conceptualization: B.S.B., J.T.C; investigation and analysis: P.H., E.Y.C., C.S.I., P.Y., Y.L., S.A.V., W.H.W., J.G.O., B.S.B., M.S.T; supervision and funding acquisition: W.W., J.T.C.
Funding
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases-funded San Diego Digestive Diseases Research Center (P30DK120515) and funded by grants from Takeda (IISR-2016-101852 to J.T.C.) and the National Institutes of Health: (DK123406 to B.S.B.; AI129973, AI123202, BX005106, and CX002396 to J.T.C.; AI150282 to W.W.; and AI132122 to W.W. and J.T.C.). P.H. was supported by AR064194; M.S.T. was supported by DK007202. CITE-seq using the 10x Genomics platform were performed at the UCSD IGM Genomics Center and supported by National Institute of Health (P30KC063491, P30CA023100, and S10OD026929).
Conflicts of Interest
J.T.C. received research grant funding from Takeda to support, in part, research presented in the current manuscript. B.S.B. has received institutional research grant from Prometheus Biosciences and Gilead and has received institutional consulting fees from Bristol Myers Squibb, Takeda, and Pfizer. P.H., E.J.C., S.A.P., W.H.W., J.G.O., P.Y., Y.C.L., M.S.T., and W.W. have no conflicts of interest to report.
Data Availability
The data sets generated for this study are available at Gene Expression Omnibus under accession number GSE228165.
References
- 1. Graham DB, Xavier RJ.. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature. 2020;578(7796):527-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boland BS, He Z, Tsai MS, et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci Immunol. 2020;5(50):eabb4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smillie CS, Biton M, Ordovas-Montanes J, et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell. 2019;178(3):714-730.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mitsialis V, Wall S, Liu P, et al. Boston Children’s Hospital Inflammatory Bowel Disease Center . Single-cell analyses of colon and blood reveal distinct immune cell signatures of ulcerative Colitis and Crohn’s disease. Gastroenterology. 2020;159(2):591-608.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kinchen J, Chen HH, Parikh K, et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell. 2018;175(2):372-386.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Na YR, Stakenborg M, Seok SH, Matteoli G.. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol. 2019;16(9):531-543. [DOI] [PubMed] [Google Scholar]
- 7. Zhou L, Chu C, Teng F, et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature. 2019;568(7752):405-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parikh K, Antanaviciute A, Fawkner-Corbett D, et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature. 2019;567(7746):49-55. [DOI] [PubMed] [Google Scholar]
- 9. Mayer AT, Holman DR, Sood A, et al. A tissue atlas of ulcerative colitis revealing evidence of sex-dependent differences in disease-driving inflammatory cell types and resistance to TNF inhibitor therapy. Sci Adv. 2023;9(3):eadd1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berlin C, Berg EL, Briskin MJ, et al. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74(1):185-195. [DOI] [PubMed] [Google Scholar]
- 11. Lichnog C, Klabunde S, Becker E, et al. Cellular mechanisms of etrolizumab treatment in inflammatory bowel disease. Front Pharmacol. 2019;10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DeBerg HA, Konecny AJ, Shows DM, Lord JD.. MAdCAM-1 costimulates T cells through integrin alpha(4)beta(7) to cause gene expression events resembling costimulation through CD28. Immunohorizons. 2022;6(3):211-223. [DOI] [PubMed] [Google Scholar]
- 13. Devlin JC, Axelrad J, Hine AM, et al. Single-cell transcriptional survey of ileal-anal pouch immune cells from ulcerative colitis patients. Gastroenterology. 2021;160(5):1679-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zeissig S, Rosati E, Dowds CM, et al. Vedolizumab is associated with changes in innate rather than adaptive immunity in patients with inflammatory bowel disease. Gut. 2019;68(1):25-39. [DOI] [PubMed] [Google Scholar]
- 15. Stoeckius M, Hafemeister C, Stephenson W, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Monaco G, Lee B, Xu W, et al. RNA-Seq Signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 2019;26(6):1627-1640.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Browaeys R, Saelens W, Saeys Y.. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods. 2020;17(2):159-162. [DOI] [PubMed] [Google Scholar]
- 18. Duong HG, Choi EJ, Hsu P, et al. Identification of CD8+ T cell - immune cell communications in ileal Crohn’s disease [published online ahead of print March 1, 2023]. Clin Transl Gastroenterol. doi:10.14309/ctg.0000000000000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888-1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aran D, Looney AP, Liu L, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vivier E, Artis D, Colonna M, et al. Innate lymphoid cells: 10 years on. Cell. 2018;174(5):1054-1066. [DOI] [PubMed] [Google Scholar]
- 22. Kobayashi T, Okamoto S, Hisamatsu T, et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut. 2008;57(12):1682-1689. [DOI] [PubMed] [Google Scholar]
- 23. Muller S, Lory J, Corazza N, et al. Activated CD4+ and CD8+ cytotoxic cells are present in increased numbers in the intestinal mucosa from patients with active inflammatory bowel disease. Am J Pathol. 1998;152(1):261-268. [PMC free article] [PubMed] [Google Scholar]
- 24. Ng SC, Plamondon S, Al-Hassi HO, et al. A novel population of human CD56+ human leucocyte antigen D-related (HLA-DR+) colonic lamina propria cells is associated with inflammation in ulcerative colitis. Clin Exp Immunol. 2009;158(2):205-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones GR, Bain CC, Fenton TM, et al. Dynamics of colon monocyte and macrophage activation during colitis. Front Immunol. 2018;9:2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ikeda S, Saijo S, Murayama MA, Shimizu K, Akitsu A, Iwakura Y.. Excess IL-1 signaling enhances the development of Th17 cells by downregulating TGF-beta-induced Foxp3 expression. J Immunol. 2014;192(4):1449-1458. [DOI] [PubMed] [Google Scholar]
- 27. Toedter G, Li K, Marano C, et al. Gene expression profiling and response signatures associated with differential responses to infliximab treatment in ulcerative colitis. Am J Gastroenterol. 2011;106(7):1272-1280. [DOI] [PubMed] [Google Scholar]
- 28. Chong WP, Mattapallil MJ, Raychaudhuri K, et al. The cytokine IL-17A limits Th17 pathogenicity via a negative feedback loop driven by autocrine induction of IL-24. Immunity. 2020;53(2):384-397.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cruikshank W, Little F.. lnterleukin-16: the ins and outs of regulating T-cell activation. Crit Rev Immunol. 2008;28(6):467-483. [DOI] [PubMed] [Google Scholar]
- 30. Ananthakrishnan AN, Nguyen DD, Sauk J, Yajnik V, Xavier RJ.. Genetic polymorphisms in metabolizing enzymes modifying the association between smoking and inflammatory bowel diseases. Inflamm Bowel Dis. 2014;20(5):783-789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shui JW, Kronenberg M.. HVEM is a TNF receptor with multiple regulatory roles in the mucosal immune system. Immune Netw. 2014;14(2):67-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yazid S, Gardner PJ, Carvalho L, et al. Annexin-A1 restricts Th17 cells and attenuates the severity of autoimmune disease. J Autoimmun. 2015;58:1-11. [DOI] [PubMed] [Google Scholar]
- 33. Wang C, Kang SG, Lee J, Sun Z, Kim CH.. The roles of CCR6 in migration of Th17 cells and regulation of effector T-cell balance in the gut. Mucosal Immunol. 2009;2(2):173-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang F, Liu G, Li D, Wei C, Hao J.. DDIT4 and associated lncDDIT4 modulate Th17 differentiation through the DDIT4/TSC/mTOR pathway. J Immunol. 2018;200(5):1618-1626. [DOI] [PubMed] [Google Scholar]
- 35. Li H, Xie L, Zhu L, et al. Multicellular immune dynamics implicate PIM1 as a potential therapeutic target for uveitis. Nat Commun. 2022;13(1):5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Masoumi F, Ghorbani S, Talebi F, et al. Malat1 long noncoding RNA regulates inflammation and leukocyte differentiation in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2019;328:50-59. [DOI] [PubMed] [Google Scholar]
- 37. Rani L, Kumar A, Karhade J, et al. IL-3 regulates the differentiation of pathogenic Th17 cells. Eur J Immunol. 2022;52(11):1842-1858. [DOI] [PubMed] [Google Scholar]
- 38. Etemadi N, Holien JK, Chau D, et al. Lymphotoxin alpha induces apoptosis, necroptosis and inflammatory signals with the same potency as tumour necrosis factor. FEBS J. 2013;280(21):5283-5297. [DOI] [PubMed] [Google Scholar]
- 39. Patnaude L, Mayo M, Mario R, et al. Mechanisms and regulation of IL-22-mediated intestinal epithelial homeostasis and repair. Life Sci. 2021;271:119195. [DOI] [PubMed] [Google Scholar]
- 40. Silverberg MS, Cho JH, Rioux JD, et al. Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat Genet. 2009;41(2):216-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Innocenti T, Roselli J, Lynch EN, et al. Infectious risk of vedolizumab compared with other biological agents in the treatment of inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2021;33(1S):e574-e579. [DOI] [PubMed] [Google Scholar]
- 42. Gazouli M, Dovrolis N, Bourdakou MM, et al. Response to anti-alpha4beta7 blockade in patients with ulcerative colitis is associated with distinct mucosal gene expression profiles at baseline. Inflamm Bowel Dis. 2022;28(1):87-95. [DOI] [PubMed] [Google Scholar]
- 43. Magnusson MK, Strid H, Isaksson S, et al. Response to infliximab therapy in ulcerative colitis is associated with decreased monocyte activation, reduced CCL2 expression and downregulation of Tenascin C. J Crohns Colitis. 2015;9(1):56-65. [DOI] [PubMed] [Google Scholar]
- 44. Chara L, Sanchez-Atrio A, Perez A, et al. Monocyte populations as markers of response to adalimumab plus MTX in rheumatoid arthritis. Arthritis Res Ther. 2012;14(4):R175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu H, Dasgupta S, Fu Y, et al. Subsets of mononuclear phagocytes are enriched in the inflamed colons of patients with IBD. BMC Immunol. 2019;20(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated for this study are available at Gene Expression Omnibus under accession number GSE228165.