

Abstract
Copy number variants (CNVs), duplications and deletions of genomic sequences, contribute to evolutionary adaptation but can also confer deleterious effects and cause disease. Whereas the effects of amplifying individual genes or whole chromosomes (i.e., aneuploidy) have been studied extensively, much less is known about the genetic and functional effects of CNVs of differing sizes and structures. Here, we investigated Saccharomyces cerevisiae (yeast) strains that acquired adaptive CNVs of variable structures and copy numbers following experimental evolution in glutamine-limited chemostats. Although beneficial in the selective environment, CNVs result in decreased fitness compared with the euploid ancestor in rich media. We used transposon mutagenesis to investigate mutational tolerance and genome-wide genetic interactions in CNV strains. We find that CNVs increase mutational target size, confer increased mutational tolerance in amplified essential genes, and result in novel genetic interactions with unlinked genes. We validated a novel genetic interaction between different CNVs and BMH1 that was common to multiple strains. We also analyzed global gene expression and found that transcriptional dosage compensation does not affect most genes amplified by CNVs, although gene-specific transcriptional dosage compensation does occur for ∼12% of amplified genes. Furthermore, we find that CNV strains do not show previously described transcriptional signatures of aneuploidy. Our study reveals the extent to which local and global mutational tolerance is modified by CNVs with implications for genome evolution and CNV-associated diseases, such as cancer.
Evolution occurs through changes to an organism's genome and selection on the functional effects of those changes. Genomes can evolve in many ways, including through single-nucleotide changes, structural rearrangements, and the deletion or duplication of segments of DNA. Amplification of segments of DNA sequence, a type of copy number variation (CNV), is an important source of rapid adaptive evolution. In the short term, gene amplification can result in increased gene expression, which provides a selective advantage facilitating adaptation (Kondrashov 2012; Myhre et al. 2013). In the long term, amplification of genes may relax selective constraints, allowing accumulation of mutations in the additional gene copies and gene evolution through subfunctionalization or neofunctionalization (Ohno 1970; Innan and Kondrashov 2010; Freeling et al. 2015). Rapid adaptation through gene amplification is prevalent throughout the tree of life and has been shown to mediate adaptation to a variety of selective pressures from nutrient limitation to antibiotics in both natural and experimental populations of microbes (Gresham et al. 2008; Nair et al. 2008; Selmecki et al. 2009; Paulander et al. 2010; Pränting and Andersson 2011; Hong and Gresham 2014; Dhami et al. 2016; Lauer and Gresham 2019; Todd and Selmecki 2020). Gene amplification is also common in cancers and can promote tumorigenesis (Ben-David and Amon 2020). For example, oncogene amplification confers enhanced proliferation properties to cells, driving their aberrant growth. Thus, understanding the evolutionary, genetic, and functional consequences of CNVs is of central importance to our understanding of genome evolution and disease.
CNVs can range from small (50-bp) amplifications and deletions of a region within a chromosome to the gain or loss of whole chromosomes, known as aneuploidy. Historically, aneuploidy has been considered as distinct from CNVs as the scale and underlying mechanisms that generate them are very different from CNVs (Compton 2011). However, for our purpose, we define CNVs as any variation that alters existing DNA copy number. Previous studies have investigated the effect of amplifying individual genes primarily using plasmid libraries with native or inducible promoters (Moriya 2015). These studies have found that in commonly used laboratory strains ∼10%–20% of genes are deleterious when overexpressed, whereas 0%–5% are beneficial (Sopko et al. 2006; Douglas et al. 2012; Arita et al. 2021; Ascencio et al. 2021). However, these effects are dependent on genetic background as a recent study found significant variation in which genes are deleterious when overexpressed in 15 genetically diverse yeast lineages (Robinson et al. 2021). Fitness effects of gene amplification tend to be dependent on both the particular gene amplified and the environmental context, although most amplified genes have neutral effects regardless of environment (Payen et al. 2016; Ascencio et al. 2021). To date, there is conflicting evidence for the various models of CNV-associated fitness costs, including those based on global differences in gene expression (Rice and McLysaght 2017; Birchler and Veitia 2022), with some studies finding the amplification of dosage-sensitive genes to be the main driver (Makanae et al. 2013; Robinson et al. 2021), whereas other studies do not (Sopko et al. 2006; Arita et al. 2021; Ascencio et al. 2021).
Aneuploidy is frequent (∼20%) in strains of yeast isolated from diverse ecologies (Hose et al. 2015; Zhu et al. 2016; Peter et al. 2018; Scopel et al. 2021), and these aneuploids grow similarly to their euploid counterparts in most conditions (Hose et al. 2015; Gasch et al. 2016). Aneuploids also frequently arise in evolution experiments and are often associated with increased fitness (Gresham et al. 2008; Rancati et al. 2008; Yona et al. 2012; Hong and Gresham 2014; Sunshine et al. 2015; Lauer et al. 2018). However, adaptive aneuploids can show antagonistic pleiotropy such that they are deleterious in other environments (Sunshine et al. 2015; Linder et al. 2017). Seminal studies of one laboratory strain, W303, found that aneuploids grow more slowly than euploids, regardless of karyotype (Torres et al. 2007; Sheltzer et al. 2012; Beach et al. 2017); show a transcriptional signature characteristic of the yeast environmental stress response (ESR) (Torres et al. 2007; Sheltzer et al. 2012); and result in a variety of cellular stresses, including proteotoxic, metabolic, and mitotic stress (Zhu et al. 2018). These effects may also be background dependent as one study mapped differences in aneuploidy tolerance between W303 and wild yeast strains to a single gene, SSD1, which has a truncating mutation in W303 (Hose et al. 2020). SSD1 is an RNA-binding translational regulator, whose targets include mitochondrial transcripts. Loss of SSD1 function results in defects in mitochondrial function and proteostasis that enhances sensitivity to aneuploidy (Hose et al. 2020). In addition to observing different fitness effects, studies of aneuploids in different genetic backgrounds have found differing results in transcriptomic dosage compensation, ESR, and proteotoxic stress (Torres et al. 2007; Pavelka et al. 2010; Dephoure et al. 2014; Hose et al. 2015; Gasch et al. 2016; Zhu et al. 2018; Larrimore et al. 2020; Muenzner et al. 2022).
Whereas numerous studies have investigated the effects of single-gene amplifications and whole-chromosome aneuploidy, little is known about the effects of CNVs that vary in size, structure, and copy number. One survey sought to study the fitness effects of a diverse set of synthetic amplicons extending from the telomere and ranging in size from 0.4–1000 kb in diploid yeast cells (Sunshine et al. 2015). Through comparison to single-gene amplifications (Payen et al. 2016), it was found that the distribution of fitness effects for telomeric amplicons was broader than that of single-gene amplifications. Notably, of the telomere-amplified regions that affected fitness, 94% had condition-dependent effects. Research using mutation accumulation lines, wherein selection against deleterious mutations is relaxed by extreme bottlenecking (Katju and Bergthorsson 2019), has generated numerous estimates of CNV formation rates (Lynch et al. 2008; Liu and Zhang 2019), but efforts at understanding the effect CNVs have on gene expression in these backgrounds is ongoing (Hine et al. 2018; Konrad et al. 2018; Liu and Zhang 2019). Finally, it remains unknown whether there are common fitness effects, genetic interactions, or transcriptomic states associated with nonengineered, or adaptive, CNVs.
In this study, we investigated seven yeast strains containing diverse CNV structures. The strains all contain amplification of the GAP1 locus and additional proximate sequence and were previously isolated from evolution experiments in glutamine-limited chemostats (Lauer et al. 2018). Unlike engineered or mutation accumulation strains, these evolved strains are part of the adaptive process of evolutionary changes that includes CNVs as well as single-nucleotide variants (SNVs). The CNVs in this study are extremely young and lack the signatures of paralog divergence that typify most gene amplifications (Schrider and Hahn 2010; Sanchez et al. 2017), such as genomic reconfiguration and divergent mutations between copies. We used transposon mutagenesis to investigate alterations in mutational tolerance within GAP1 CNV strains. Using RNA-seq, we investigated the altered transcriptome of GAP1 CNV strains. To better understand fitness differences between the GAP1 CNV strains, we evaluated the potential role of several fitness cost models that are often used to explain growth deficits in aneuploids. Finally, we tested for common patterns of gene expression shared between the GAP1 CNV strains and other gene expression signatures previously observed in aneuploid strains.
Results
GAP1 CNVs confer variable fitness effects
Previously, we performed experimental evolution using budding yeast cells in glutamine-limited chemostats for approximately 270 generations (Lauer et al. 2018). The yeast strain (a haploid derivative of S288c) used to initiate the evolution experiments contained a fluorescent reporter gene adjacent to the general amino acid permease gene, GAP1, on Chr XI, which functions as a GAP1 CNV reporter. The proximity of the fluorescent gene (∼1 kilobase from the 5′ end of GAP1) ensures that it is typically coamplified with GAP1 enabling efficient detection and isolation of CNV-containing strains. Using the CNV reporter dynamics and simulation-based inference, we have determined that GAP1 CNVs form at a high rate and confer large fitness increases in glutamine-limited environments (Avecilla et al. 2022). The structures of some GAP1 CNVs are frequently complex and can only be imperfectly estimated using short-read sequencing and pulse-field electrophoresis (Lauer et al. 2018). Therefore, we resolved the CNV structures of seven strains using long-read sequencing (Spealman et al. 2022), enabling accurate determination of gene copy number (Supplemental Table S1) as well as SNVs (Supplemental Tables S2, S3). The GAP1 copy numbers range from two copies (Aneu) to three (Trip1, Trip2, ComTrip, Sup) and four (ComQuad, ComSup) copies (Fig. 1A; Supplemental Fig. S1A–I); the number of amplified genes ranges from 20 to 315; and the total amount of amplified DNA ranges from about 79,000 to about 667,000 additional nucleotides (Supplemental Table S4), including changes in the copy number of the rDNA locus (Supplemental Table S5). The GAP1 CNVs have a variety of structures, including an aneuploid (Aneu), inverse triplications characteristic of origin-dependent inverted repeat amplification (ODIRA; Trip1, Trip2), a supernumerary chromosome (Sup), and more complex structures probably resulting from multiple mutational events (ComTrip, ComQuad, ComSup). The diversity in GAP1 CNVs is also reflected in which genes are amplified, with only 17 common genes amplified in all evolved strains (Supplemental Fig. S2; Supplemental Table S6).
Figure 1.

Strains with GAP1 CNVs differ in structure and fitness. (A) We previously evolved a euploid Saccharomyces cerevisiae strain in glutamine-limited chemostats and isolated seven strains that have CNVs on Chr XI that include GAP1. The structure of each GAP1 CNV was resolved using long-read sequencing and is summarized here. The amplified region is shown as a colored block with arrows. Arrows pointing right represent copies that maintain their original orientation, whereas arrows pointing left represent copies that are inverted. The number of genes amplified and the number of additional base pairs (bp) are annotated. (B) The relative fitness (compared with the ancestral strain) of strains containing GAP1 CNVs was determined by pairwise competition experiments in glutamine-limited chemostats. Error bars are 95% confidence intervals for the slope of the linear regression used to compute fitness (see Methods). (C) Average and standard deviation (error bars) growth rate relative to the ancestral, euploid strain in YPGal batch culture. Horizontal black lines in B and C denote the ancestral euploid fitness. Note that panels B and C do not show the same type of measurement.
All CNV strains have fitness greater than or equal to the ancestral euploid strain in the glutamine-limited environment in which they evolved (Fig. 1B). However, we find that the majority of CNV strains grow slower than the euploid strain in a rich-media nonselective environment: yeast-peptone-galactose (YPGal) batch culture (Fig. 1C). The fitness benefit in the environment in which they evolved and the fitness deficit in the alternative environment differ between strains. Fitness benefits and costs do not correlate with the number of additional bases or the number of open reading frames in the CNV region (Supplemental Fig. S3).
Common features of CNVs and CNV-amplified genes
Genes contained within CNVs show no significant enrichment in Gene Ontology terms (Benjamini–Hochberg [BH] adj.P ≤ 0.1). The breakpoints that define the CNV boundaries are unique to each strain (Supplemental Fig. S1) and occur in both coding (10 of 18) and noncoding (eight of 10) regions. An analysis of noncoding RNA (ncRNA) in GAP1 CNVs found no significant enrichment.
We performed transcriptome analysis of all seven CNV strains and the euploid ancestor in YPGal batch culture (Methods). A core set of 17 genes (here referred to as the CNV core set) is present within each GAP1 CNV (Supplemental Fig. S2). Although these genes show higher expression than the euploid, their expression varies greatly between strains (Supplemental Fig. S4). The CNV core set is not significantly enriched for known gene functions (BH adj.P ≤ 0.1). KAE1 is the only essential gene within the CNV core set. There are five genes of unknown function (GMH1, YKR041W, UIP5, YKR045C, FMP46) and one transcription factor, DAL80, a negative regulator of GAP1. Notably, although GAP1 is amplified in every CNV, it is lowly expressed in rich-media conditions, having less than the median abundance of expression in every background. The observed growth defects (Fig. 1C) are unlikely to be the product of the CNV core set shared by all CNV strains as the slowest growing strains in YPGal, Aneu, Trip1, and ComQuad only have the CNV core set in common.
One potential consequence of CNV amplification that could have a large-scale effect on gene expression and fitness would be the amplification of transcription factor genes. In addition to DAL80 in the CNV core set, PUT3 (ComSup) and BAS1 (Sup, ComSup) are amplified in a subset of strains, and IXR1, RGT1, MSN4, HAP4, ABF1, and ASH1 are duplicated in the aneuploid strain. An analysis of the transcript abundance of these genes (Supplemental Table S7) suggests that CNV amplification is only weakly associated with changes in transcript abundance, with many having significant differences in transcript abundance, relative to the ancestor, even when unamplified (Methods). In terms of downstream effects, an analysis of the targets of these transcription factors finds only one case (IXR1 in Aneu) in which an increase in transcription factor gene copy number corresponds with a significant increase in expression of a target gene (Fisher's exact test [FET], 1.67-fold increase, P = 2 × 10−4). This suggests that compensatory changes in the gene regulatory network may act to mitigate changes in transcription factor gene copy number, either by reducing the transcription of the transcription factor gene itself or by lowering the activity of the transcription factor protein.
SNVs are rare, strain specific, and associated with adaptation to glutamine-limited environments
In addition to GAP1 CNVs, each strain contains a small number (median, three) of unique SNVs compared with the ancestor (Supplemental Table S1). These variants have the potential to epistatically interact with each other and the CNVs. After filtering for SNVs with a low likelihood of having a strong fitness effect, including those in intergenic regions, those transposon associated, or those causing low impact missense mutations (Methods), five SNVs remained across all strains that have a high likelihood of having a strong fitness effect (Supplemental Table S3). Each SNV is found in a distinct gene (SSO1, POM152, SSK2, DAL5, PBS2) and only in one strain each (Aneu, Trip1, ComTrip, Trip2, Sup, respectively). However, they potentially contribute to increased fitness in glutamine-limited environments as they are associated with nitrogen catabolite repression (NCR), mitophagy, the electron transport chain, and retrograde (RTG) signaling (Giannattasio et al. 2005; Jazwinski and Kriete 2012).
Two strains contain smaller CNVs at the GLC7 locus
Two strains, ComSup and Sup, also contain independent amplifications of the same locus on Chromosome V (Supplemental Table S6; Supplemental Fig. S1F,I). This amplification is composed of GLC7, YER134C, GDI1, YER137C, and the transposon genes YER137C-A and YER138C. These genes are not significantly enriched in any functions. Unlike GAP1 CNVs, the GLC7-YER137C CNV is significantly enriched in tRNAs as it contains extra copies of tRNA-His, tRNA-Ile, tRNA-Lys, and tRNA-Val (Supplemental Table S7).
Transposon mutagenesis defines mutational tolerance in CNV strains
We sought to investigate the genetic impact of CNVs using high-throughput genetics. Previous studies using transposon mutagenesis in bacteria and yeast have shown that transposon insertion density reflects tolerance to mutation and is an efficient means of identifying genomic regions essential for cell survival in a specific environment or genetic background (Guo et al. 2013; Michel et al. 2017; Segal et al. 2018; Grech et al. 2019; Gale et al. 2020; Levitan et al. 2020). We generated Hermes insertion libraries in each CNV strain and in two independent replicates of the ancestral euploid strain using modifications of published methods (Fig. 2A; Gangadharan et al. 2010; Caudal et al. 2022). Briefly, Hermes transposition was induced in YPGal media using batch cultures undergoing serial transfer, and transposition events were selected using an antibiotic marker. Insertion sites were identified by targeted PCR, followed by library preparation and deep sequencing (Methods). Unique insertion sites and the number of reads per insertion site were identified using a custom bioinformatic pipeline (see Methods) (Supplemental Table S8). As our sequencing and analysis pipeline cannot differentiate between recurrent insertion events and PCR duplicates, we quantified the number of unique insertion sites per gene, unless otherwise noted (Supplemental Tables S9, S10). The nine libraries showed variation in the number of unique insertion sites, which scaled with the total number of reads sequenced (Supplemental Fig. S5; Supplemental Tables S11, S12). To normalize for differences in sequencing depth, we determined the number of insertions per million reads (Methods). The frequency of normalized insertions only weakly correlated with CDS length (Adj.R-squared = 0.198, P < 0.01, Supplemental Fig. S6). With a lower boundary of CDS length sensitivity empirically estimated between 78 and 87 nucleotides (nt) long (the smallest CDS identified in the majority of samples and the smallest CDS identified in all samples, respectively). To estimate the false-negative rate of zero insertions, we binned each CDS by 100 nt and derived an empirical FDR less than 0.05 (Methods) (Supplemental Fig. S7).
Figure 2.
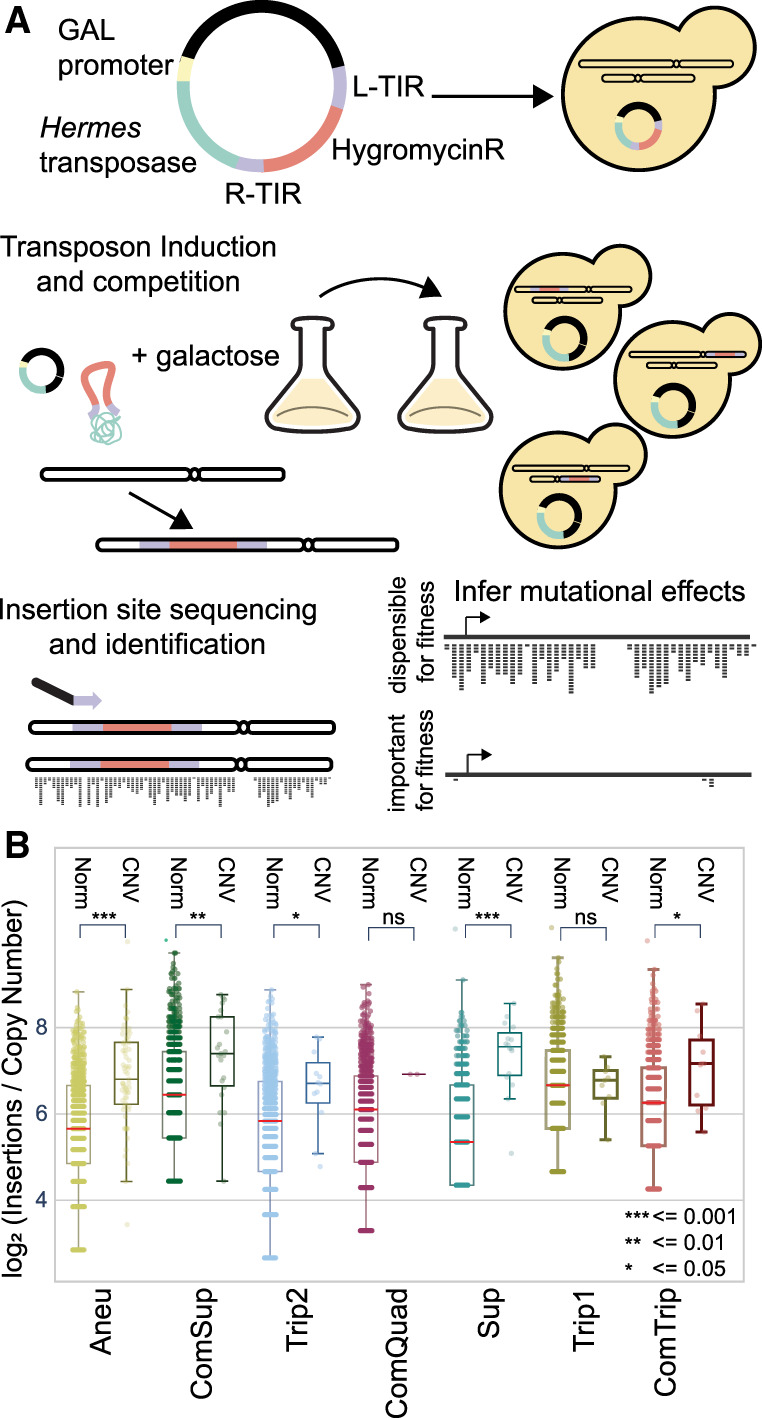
Profiling mutation tolerance in CNV strains using insertional mutagenesis. (A) Plasmids containing the Hermes transposase regulated by the GALS promoter (a truncated GAL1 promoter) and a hygromycin-resistance gene flanked by the Hermes terminal inverted repeats (TIRs) were transformed into each yeast strain. Upon addition of galactose, the transposase is expressed, and the hygromycin-resistance gene flanked by the TIRs is excised from the plasmid and inserted in the yeast genome. DNA is extracted, digested with restriction enzymes, and circularized. Insertion sites are identified by inverse PCR and amplicon sequencing. Mutational tolerance for a gene is inferred using the number of unique insertion sites over the protein-coding region. (B) Unique insertion sites per gene copy number (CN) for essential genes. Genes are defined as either CNV associated (CNV) or not (Norm). A Mann–Whitney U statistic was used to compare distributions between the CNV and Norm essential genes, P-values are indicated by the following: (ns) P > 0.05, (*) P ≤ 0.05, (**) P ≤ 0.01, (***) P ≤ 0.001.
As our transposition protocol entails serial propagation with periodic bottlenecking, the mutation frequency per gene reflects the tolerance to mutation in the rich-media condition (YPGal). We compared our transposon insertion data to a list of essential genes generated in YPD (Supplemental Fig. S8; Winzeler et al. 1999) and to relative fitness measurements of genes grown on YPGal (Supplemental Fig. S9; Costanzo et al. 2021) that were defined using complete open reading frame deletions. In all CNV and euploid strains, essential genes have fewer insertions than nonessential genes (Mann–Whitney U [MWU], P ≤ 0.0001). GAL genes in the CNV strains also had no significant change in insertion frequency relative to the euploid. These results confirm that transposon insertion density is a reliable predictor of sequence tolerance to disruptive mutation in CNV strains.
Gene amplification increases mutational target size
We investigated how gene amplification affects insertion density by considering only coding sequences within the CNV regions of each strain. We find that in six of seven CNV strains, amplified genes have a higher insertion frequency than in the euploid (paired t-test, P < 0.0001), consistent with increased target size resulting in increased mutation frequency. The single exception, ComQuad, is likely owing to insufficient statistical power, because it has the fewest amplified genes. The genes amplified in the GLC7-YER137C CNV also have higher insertions relative to the euploid ancestors, although not statistically significant (DESeq2, adj.P > 0.05).
We find that, for unamplified genes, essential genes (Winzeler et al. 1999) have significantly fewer insertions than nonessential genes (Welch's t-test, P < 0.0001) (Supplemental Fig. S10) and a notable 3′ insertion bias (Supplemental Fig. S9). After normalizing for copy number, we find that CNV-amplified essential genes have significantly higher (MWU, P ≤ 0.05) insertion frequencies than essential genes in unamplified regions for five of seven CNV strains (Fig. 2B), consistent with relaxed selection upon increases in copy number. The two exceptions, ComQuad and Trip1, have the fewest CNV-amplified essential genes (two and nine) and thus are likely underpowered for observing this effect. Mutation frequencies in essential genes within the CNV are also increased in these strains (Fig. 2B).
To study the impact of CNVs on mutational tolerance across the entire genome, we compared genome-wide mutation frequencies in the CNV strains with the euploid ancestor. The number of insertions in a gene in the euploid strains is positively correlated with the number of insertions in a CNV strain (Supplemental Fig. S11). We find a clear relationship between increased copy number and increased insertion frequency (Supplemental Fig. S12). The strength of this relationship, assessed using the slope of the regression line, is less than that expected on the basis of copy number for all strains. This is likely owing to the saturation of unique insertion calls, as the difference in the slope of the regression increases with increasing copy number (Supplemental Fig. S13).
We sought to identify genes that show evidence of dosage sensitivity by identifying genes with insertion frequencies higher than the predicted value. We found several candidates (genes with standardized residuals > 2) in CNV-associated genes (FET, P ≤ 0.05). For example, UTH1 is amplified in all CNV strains and has significantly higher copy number normalized insertions in each strain (Supplemental Tables S13, S14), suggesting that amplification of UTH1 is deleterious. This is consistent with UTH1 having been shown to lead to cell death when overexpressed in the W303 genetic background (Camougrand et al. 2003). Notably, UTH1 is a mitochondrial protein involved in regulating both mitochondrial biogenesis and degradation (Camougrand et al. 2004) and is known to be regulated by SSD1, whose loss of function is associated with fitness defects in aneuploid strains of W303 (Hose et al. 2020), suggesting UTH1 may function as part of the SSD1 regulatory response. Indeed, we see reduced insertion frequencies in SSD1 in all CNV-containing backgrounds and significant reductions in Aneu, Trip1, Trip2, and Sup (Adj.R-squared, standardized residual < −2) (Supplemental Table S13). Two other genes, VPS1 and ECM9, also show this same pattern of significantly higher insertion frequencies, suggesting they are also dosage-sensitive genes. Only one non-CNV gene has a similar profile: PDR5, which is significantly enriched in four of the seven strains. Conversely, significantly reduced insertional frequency may reflect an advantage owing to increased copy number. We find this to be rare, however, with only YKR005C having significantly lower mutation frequency in three of four CNV strains in which it is amplified (Supplemental Table S13). An analysis for genes whose expressions significantly covaried with amplified genes found no significant evidence beyond that expected at random (Supplemental Table S15).
We also observed that 18 genes have significantly higher rates of insertion in the euploid ancestor than the CNV strains, suggesting that they show lower mutational tolerance in the CNV strains (Supplemental Table S13). These include both GPB1 and GPB2, which are multistep regulators involved in the cAMP-PKA signaling and RAS signaling pathways, and the set of 18 genes is enriched for functions in GTPase activity (GO:0007264), negative regulation of RAS (GO:0046580), and cAMP (GO:2000480; GSEA, adj.P ≤ 0.05). This suggests that the RAS/PKA pathway may be a critical pathway in the adaptation to glutamine-limited media or in the adaptation to some CNVs.
GAP1 CNVs result in common and strain-specific genetic interactions
To establish a genome-wide view of differences in mutational tolerance between CNV strains and the euploid strain, we first identified 327 genes that have no insertions in either replicate of the euploid strain. Of these, 136 (42%) have previously been annotated as essential or as having low fitness in galactose (Supplemental Tables S16, S17). We define these 327 genes as “euploid intolerant.” Genes that have no insertion events may be owing to intolerance of mutation or owing to chance (i.e., a false negative). To account for this, we empirically calculated a false-negative rate of less than 0.05 (Methods).
Many euploid-intolerant genes had insertions in one or more of the CNV strains, and seven euploid-intolerant genes had insertions in all CNV strains (Fig. 3A). These seven genes have previously been annotated as essential or as having low fitness in galactose. Although four of these genes were amplified in one or more CNV strains, none are amplified in all CNV strains (Fig. 3A), suggesting that increased mutational tolerance in the CNV strains is not simply attributable to increased target size. Furthermore, it is unlikely that they arise because of a common set of genes amplified in all strains. An illustrative example of this is BMH1, which has significantly higher insertion frequencies in the Aneu, Trip1, and ComQuad strains. These strains share 17 amplified core genes between them (Supplemental Fig. S2), but these 17 genes are also amplified in every CNV strain. As BMH1 only displays increased mutational tolerance in three strains, it is unlikely that the core set of 17 genes in the CNVs underlies this effect.
Figure 3.

CNV strains have common and allele-specific genetic interactions. (A) Seven genes have no insertions in either replicate of the euploid strain, whereas insertions are identified in these genes in all CNV strains. The gray line represents the minimum insertion count per gene to be considered significant. The numbers above each bar of each strain indicate the copy number of the gene if it is contained within a CNV. If no number is given, it is a single-copy gene. (B) Shared enriched GO terms identified using gene set enrichment analysis (GSEA) of log2 fold-changes obtained from differential analysis comparing each CNV insertion profile to the euploid insertion profiles (Q-value, circle size). For all enriched gene sets, see Supplemental Figure S14. Positive enrichment scores (red) indicate functions that have increased insertion frequencies in the CNV strain, whereas negative enrichment scores (blue) indicate the inverse. ComSup had no significant enrichment of any gene sets. (C) Significant genes (P.adjust < 0.05) from differential analysis comparing each CNV insertion profile to the euploid insertion profiles. Positive values (red) indicate an increase in frequency in the CNV strain; negative values (blue) indicate the inverse. If a gene is amplified, the copy number is annotated. (D) Mean and standard deviation (error bars) of growth rate relative to the ancestral, euploid strain in YPGal batch culture. P-values from two-sample t-test are indicated by the following: (ns) not significant, (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001. (E) The mean strength and standard deviation (error bars) of the genetic interaction (ε) were determined based on the deviation of expected fitness based on a multiplicative fitness model (Methods) for GAP1 CNV and BMH1 double mutants, calculated from growth rates shown in D.
To assess genetic interactions between the CNVs and the rest of the genome, we determined differential insertion frequency using the number of unique insertion sites per gene between each CNV strain and the euploid replicates (Supplemental Table S18). To assess the global trend, we performed gene set enrichment analysis (GSEA) using the ranked list of fold-change in number of insertions (Fig. 3B; Supplemental Table S19). We found Aneu, Trip1, and ComQuad have increased mutational tolerance in genes annotated with terms related to mitochondrial translation, RNA metabolism, and gene expression, whereas ComTrip had decreased mutational tolerance in mitochondrial translation and gene expression. Genes with functions in amide metabolism also had increased mutational tolerance in Aneu, Trip1, Trip2, and ComQuad, whereas mutational tolerance in genes involved in amide metabolism was decreased in ComTrip. Aneu, Trip1, and ComQuad shared only one gene set, organelle localization, with decreased tolerance. Indeed, for the decreased gene sets Aneu had more in common with Trip2 and Sup. We also observed that some strains had gene sets enriched for terms that were not enriched in other strains (Supplemental Fig. S14). For example, the supernumerary chromosome containing strain (Sup) shows a wide range of gene sets specific to only that strain.
We identified individual genes with differences in insertional tolerance in CNV strains compared with the ancestral euploid strain (Fig. 3C; Supplemental Table S18). Most genes that are significant in one strain tend to have similar trends in other CNV strains, with few exceptions (Supplemental Fig. S15A). The Aneu, ComQuad, and Trip1 strains all have significantly more insertions than the euploid ancestor in BMH1, which is involved in many processes, including regulation of mitochondrial-nuclear signaling (Liu et al. 2003), carbon metabolism (Dombek et al. 2004), and transcription and chromatin organization (Kumar 2017; Jain et al. 2021). Notably, we found these strains also showed greatly reduced growth when plated on glycerol media, suggesting impaired mitochondrial function or dysregulation in retrograde signaling (Roca-Portoles and Tait 2021).
Unlinked genes that are not associated with CNVs and show differences in insertion tolerance may reflect novel genetic interactions. To test this, we generated complete deletions of the coding sequence of BMH1 in all strains except ComSup, for which we could not obtain a transformant, and measured growth rates of the single and double mutants in YPGal (Fig. 3D). We find that deletion of BMH1 does not significantly reduce growth rate in Aneu, ComQuad, and Trip1, whereas it results in reduced growth rate in all other strains. We calculated the strength of the genetic interaction with BMH1 (Mani et al. 2008) and confirmed the existence of a novel positive genetic interaction with BMH1 and the GAP1 CNV for these three strains (Fig. 3E; Supplemental Fig. S15B).
Gene amplification results in increased mRNA expression
Transcriptional dosage compensation, in which the amplification of a gene is buffered by changes in the regulation of gene expression such that the transcription abundance is unchanged, is observed in some studies of aneuploids (Kojima and Cimini 2019; Birchler and Veitia 2022) but has not been broadly studied for other CNVs. To evaluate the role of gene expression in fitness and mutational tolerance, we performed RNA-seq on each strain and the euploid ancestor in YPGal (Supplemental Tables S20–S22). First, we investigated genes encoded on Chr XI for evidence of dosage compensation (Supplemental Table S23). We found that in each CNV strain, GAP1 CNV-amplified genes have significantly higher mRNA expression than in the euploid ancestor (MWU, P < 0.001) (Fig. 4A; Supplemental Table S24), and expression in amplified genes is highly correlated with euploid expression (Supplemental Fig. S16), with many amplified genes being significantly higher than their euploid counterparts (Supplemental Table S23). After correcting for copy number (Supplemental Table S25), we find the mRNA expression is, broadly, in agreement with the abundances observed in the ancestor (Fig. 4B; Supplemental Tables S26, S27), suggesting that there is not widespread dosage compensation acting on CNVs.
Figure 4.

Amplified genes result in increased mRNA expression. (A) Replicate averaged log2 fold-change of all genes on Chr XI in each CNV strain compared with the euploid ancestor. Numbers indicate the copy number of genes within the CNV. (B) Log2 of the fold-change in mRNA expression in the CNV compared with the ancestral euploid strain normalized by gene copy number for all genes amplified in each CNV strain.
mRNA expression is not correlated with transposon insertion frequency
Several models of fitness cost involving gene copy number and gene expression have been proposed (Makanae et al. 2013; Rice and McLysaght 2017). The dosage burden model considers the baseline expression of genes as optimal, with deviations from this expression being detrimental to fitness (Dekel and Alon 2005; Wagner 2005). Under this model, the greater the difference in gene expression, the greater the fitness cost (Wagner 2007; Makanae et al. 2013; Bonney et al. 2015). To test whether dosage burden explains insertional frequency, we compared the log2 fold-change of transposon insertion frequency with the log2 fold-change for mRNA expression for each CNV strain compared with the euploid strain. If the CNV cost is related to dosage burden, then we would expect that the fold-change in transposon insertions would positively correlate with the fold-change in mRNA expression. However, we do not observe a significant correlation between mRNA abundance and transposon insertion in any of the strains (Supplemental Fig. S17). This suggests that adverse effects of CNVs do not stem from dosage burden or a cost of gene expression.
Conversely, the dosage balance model proposes that fitness costs stem from dosage-sensitive genes, wherein the CNV results in a stoichiometric imbalance between genes that encode components of macromolecular complexes (Wagner 2005; Veitia et al. 2008). Although more than 100 genes have been previously identified as dosage sensitive in Saccharomyces cerevisiae (Makanae et al. 2013), only two of them are amplified in our strains (SPC42 and TPK3) and only in the aneuploid strain, making their utility limited. More broadly, essential genes are often central components in protein interaction complexes (Jeong et al. 2001; Ning et al. 2010; Ascencio et al. 2021). However, as previously described, CNV-amplified essential genes already show significantly higher numbers of inserts relative to the single-copy euploid ancestor (Fig. 2B), making it difficult to separate the effect of relaxed purifying selection from negative selection.
Evidence of gene-specific dosage compensation
Although increases in copy number are generally proportional to increases in mRNA expression, we find evidence of gene-specific dosage compensation. By comparing the observed mRNA abundance against what would be expected given their amplified copy number and expression in the ancestor (Supplemental Table S25), we find 22 genes with significantly lower mRNA expression (DESeq2, adj.P-value < 0.05), suggesting dosage compensation acts to reduce the expression of these amplified genes (Supplemental Table S26). However, down-regulation is prevalent on numerous unamplified genes as well (Supplemental Table S27).
To account for changes in expression separate from amplification, we compared changes in expression of genes that were amplified in only some strains with the expression of those genes in strains in which the gene was not amplified within the CNV, using MWU (Methods) (Supplemental Fig. S18). We identified 39 out of 313 genes for which the CNV-associated gene had significantly lower expression than expected when it occurred within a CNV compared with when it was not in a CNV (Supplemental Table S28). This set of genes is also potentially subject to dosage compensation. Taken together, our results are consistent with a minority of genes (∼12%) being subject to gene-specific, strain-specific dosage compensation effects.
CNV strains do not show previously described transcriptional signatures of aneuploidy
Although previous studies have reported evidence of different stress responses induced by aneuploidy, it is not known if similar stress response pathways are induced by CNVs. To evaluate this, we compared the expression of our strains with previously published data. One previous study of a laboratory strain of yeast (W303) identified a transcriptomic signature of aneuploidy that is independent of which chromosome is duplicated (Torres et al. 2007; Terhorst et al. 2020) and comprises 868 genes characteristic of the yeast ESR (Gasch et al. 2000). The expression of genes in the ESR are correlated with growth rate (Brauer et al. 2008), and several studies have shown that strains with higher degrees of aneuploidy (i.e., more additional base pairs) show lower growth rates and stronger ESR expression (Torres et al. 2007; Terhorst et al. 2020).
We compared the expression profiles of CNV strains with the expression response to aneuploidy (Torres et al. 2007) in both batch and chemostat conditions. To compare the results of Torres et al. (2007) to our own, we subset the ESR genes from both our data sets (798 of 868, for which we had complete data). We calculated the log2 fold-change in mRNA for each of our evolved strains relative to the euploid ancestor and the mean log2 fold-change in mRNA for each aneuploid strain from Torres et al. (2007) relative to their euploid strain. Using these log2 ratios for each gene, we then calculate Pearson coefficient between our strains and that of Torres et al. (2007). We find a negative correlation between the ESR genes compared with the batch grown culture (Fig. 5A; Supplemental Fig. S19) and positive correlation with ESR expression of aneuploids in chemostats (Fig. 5B; Supplemental Fig. S20). This suggests that ESR gene expression in CNVs strains is similar to that of aneuploids when growth rate is controlled using chemostats.
Figure 5.

Global gene expression signatures in CNV strains in relation to aneuploidy signatures. Previous research has identified ESR-like signatures of gene expression using W303 aneuploid strains grown in batch (A) (Torres et al. 2007) and chemostat (B) (Torres et al. 2007) conditions. To evaluate how similar the CNV responses are to aneuploid responses, we calculated the mean log2 fold-change for all evolved strains relative to their ancestors. We used only the subset of ESR genes and calculated Pearson's correlation between the mean aneuploid expression and each of our CNV strains (vertical axis). Because ESR has previously been shown to correlate with growth rate (Torres et al. 2007), we then ordered these correlation values by the relative YPGal growth rates for each CNV strain (horizontal). Correlation with the CAGE signature (Tsai et al. 2019) identified in BY1747 aneuploid strains grown in rich media (C). Heatmaps for each data set show the log2 fold-change in expression for 436 genes (rows) that were significantly differentially expressed in at least one CNV strain in this study (D) and the expression of those genes of Torres et al. (2007) aneuploids in batch (E), Torres et al. (2007) aneuploids in chemostat (F), and Tsai et al. (2019) (G).
Recently, a common aneuploidy gene expression (CAGE) signature was described that has similarities to the transcriptional response to hypo-osmotic shock (Tsai et al. 2019). We find that expression of CAGE genes are moderately positively correlated between Tsai et al. (2019) and CNV strains (Fig. 5C; Supplemental Fig. S21), with the exception of ComTrip. Notably, we find a positive correlation between CAGE gene expression in Tsai et al. (2019) and in the growth rate controlled aneuploid strains in Torres et al. (2007) (Supplemental Fig. S22).
Genome-wide gene expression effects of GAP1 CNVs
We identified 436 genes, 341 of which are not located on Chr XI, that had significantly altered expression in one or more CNV strains compared with the euploid strain (log2 fold-change > 1.5, BH adjusted P < 0.05) (Fig. 5D; Supplemental Table S29). Of these, 73 are ESR genes, and 13 are in the CAGE signature. The 436 genes fall into two major clusters: Genes that have decreased expression are involved in cellular respiration, nucleoside biosynthetic processes, and small-molecule metabolism, and genes that have increased expression are involved in transposition, nucleic acid metabolic processes, and siderophore transport (hypergeometric test P < 0.0001) (Supplemental Table S30). Similarly, wild yeast strains that are aneuploidy tolerant show down-regulation of mitochondrial ribosomal proteins and genes involved in respiration, and up-regulation of oxidoreductases (Hose et al. 2015). The clusters and enrichment patterns remain even when excluding genes on Chr XI (Supplemental Table S31). Additionally, we see similar functional enrichment for gene expression differences of individual strains (Supplemental Table S26).
There are nine genes that have significantly different expression than the euploid in all strains and are not on Chr XI (Supplemental Fig. S23). Three genes have increased expression: Two are retrotransposons, and the third, RGI2, is involved in energy metabolism under respiratory conditions (Domitrovic et al. 2010). Repressed genes include the paralogs MRH1 and YRO2, both of which localize to the mitochondria (Reinders et al. 2006, 2007); OPT2, an oligopeptide transporter (Wiles et al. 2006); YGP1, a cell wall–related secretory glycoprotein (Destruelle et al. 1994); and two proteins of unknown function, PNS1 and RTC3.
We compared significantly differentially expressed genes in one or more CNV strains to the data generated from aneuploid strains (Fig. 5E; Torres et al. 2007; Tsai et al. 2019). As with comparison to the ESR and CAGE signatures, we see that CNV strains are more similar to the aneuploids grown in chemostats (Fig. 5F; Torres et al. 2007) and the S288c aneuploids (Fig. 5G; Tsai et al. 2019) than the aneuploids growing in batch culture. Hose et al. (2020) compared gene expression in aneuploid wild yeast strains that are tolerant of CNV to their euploid counterparts and to aneuploid wild yeast with SSD1 deleted. Although SSD1 may be an important gene in the modulation of aneuploidy, it has no significant association to the CNV signature described in our data (Supplemental Fig. S24).
Low fitness is associated with mitochondrial dysfunction
Several lines of evidence suggested that the slower relative growth rates of strains Aneu, Trip1, and ComQuad (Fig. 3D) may be the product of mitochondrial dysfunction. Aneu, Trip1, and ComQuad strains have shared expression signatures (Fig. 3B) linked to mitochondrial function and translation activity (da Cunha et al. 2015) (Supplemental Fig. S25). The slow growing Aneu, Trip1, and ComQuad strains also have increased tolerance to BMH1 insertions (Fig. 3A) and a positive genetic interaction with deletion of BMH1 (Fig. 3E). BMH1 is a 14-3-3 protein involved in post-transcriptional gene regulation of numerous pathways, including Ras/MAPK and rapamycin-sensitive signaling, and is also a negative regulator of retrograde signaling (da Cunha et al. 2015) along with many other functions. One possible explanation of these signatures is that the mitochondria are dysfunctional, and therefore, retrograde signaling is constitutively activated, allowing a tolerance of mutations in BMH1.
Because yeast simultaneously ferment and respire galactose (Fendt and Sauer 2010), we tested impairment of mitochondrial function by evaluating growth in the presence of carbonyl-cyanide 3-chlorophenylhydrazone (CCCP), a mitochondrial uncoupling agent. We found that treating with CCCP greatly reduced growth (>75% reduced) in Trip1 and ComQuad (Supplemental Fig. S26) but not in Aneu. This result is suggestive of a defect in mitochondrial function in at least two of the CNV strains.
Discussion
In this study, we sought to understand the effect of adaptive CNVs on fitness, genetic interactions, and gene expression. We used yeast strains with different CNVs at the GAP1 locus that arose through the adaptation to glutamine-limited growth conditions in chemostats over hundreds of generations (Lauer et al. 2018). Previous studies have suggested that epistasis between CNVs and other mutations is an important contributor to evolutionary dynamics (Lauer et al. 2018; Pavani et al. 2021), but few studies have systematically investigated genetic interactions with CNVs (Dodgson et al. 2016).
Transposon mutagenesis is a powerful tool to investigate genetic interactions genome-wide in strains with large and complex mutations. Unlike synthetic genetic array (SGA) analysis, which is commonly used to investigate genetic interactions, transposon mutagenesis does not require mating the query strain to the deletion collection. Transposon mutagenesis therefore avoids some of the issues that are encountered using SGA: inaccuracies in the deletion collection (Ben-Shitrit et al. 2012; Giaever and Nislow 2014), secondary mutations including aneuploidy (Hughes et al. 2000), and the impact on gene expression of neighboring genes owing to the deletion (Baryshnikova and Andrews 2012; Ben-Shitrit et al. 2012). Despite these benefits, transposon mutagenesis has some shortcomings, including differing insertion efficiency between genetic backgrounds (Caudal et al. 2022). However, our study used CNV strains in a single genetic background and therefore was not impacted by this issue.
Applying genome-wide insertional mutagenesis in seven different CNV strains allowed us to draw three general conclusions regarding the genetic consequences of CNVs. First, increases in gene copy number result in increased insertional frequencies. This observation is consistent with CNVs increasing the mutational target size. Second, essential genes that are intolerant of mutation in the haploid ancestral state become tolerant of mutations through the acquisition of additional gene copies. This is consistent with gene amplification relaxing selective constraints on gene evolution. Third, CNVs can result in increased mutational tolerance in unlinked genes located on other chromosomes. Thus, our study shows that the genetic consequences of a CNV are widespread throughout the genome impacting both the genes that lie within the CNV as well as genes that have no apparent physical relationship with the CNV.
We observed different levels of relative fitness and growth rate between CNV strains when grown in different conditions. We do not find that these fitness effects correlate significantly with the number of amplified nucleotides or with the number of genes in the CNV. Other models of fitness cost involving gene copy number have been proposed, namely the “dosage burden” and “dosage balance” models. The dosage burden model proposes that the baseline expression of a gene is optimal, with deviations from this expression being detrimental to fitness (Dekel and Alon 2005; Wagner 2005; Makanae et al. 2013; Bonney et al. 2015). This model predicts that insertional frequency should positively correlate with mRNA expression. However, this was not observed in our study (Supplemental Fig. S17). Conversely, the dosage balance model posits that stoichiometric imbalance of genes underlies fitness costs, as the imbalance of proteins involved in macromolecular complexes is detrimental to fitness (Wagner 2005; Veitia et al. 2008). The dosage balance model would predict that amplified essential genes, which are enriched for protein–protein interaction hubs (Ning et al. 2010), would have higher numbers of insertions than expected given the background frequency. Although we do see a significant increase relative to the single-copy strain, it is not in excess of what is expected given the background frequency. Taken together, these findings suggest that the observed differences in fitness and growth rate observed in this study are not primarily driven by the mechanisms proposed in these two models.
Dosage compensation is one method of mitigating the gene expression costs of increased gene copy number (Veitia et al. 2008), although its role in aneuploid yeast strains is unresolved (Springer et al. 2010; Hose et al. 2015; Gasch et al. 2016; Torres et al. 2016; Kojima and Cimini 2019). We find that gene amplification broadly results in increased mRNA expression. However, for a small number of CNV-amplified genes (12%, 39 out of 313 possible), we do find evidence of gene-specific dosage compensation. Although this is less than some previous reports (57%–61% [Table 1 of Hose et al. 2015, Class 2a and Class 3a]), it is consistent with others (13% [Gasch et al. 2016] and 14% [Springer et al. 2010]), although methodological differences make direct comparisons difficult. Our finding suggests that gene-specific dosage compensation may play a role in mitigating CNV gene expression costs.
Consistent with a recent study of aneuploids, we do not find activation of the ESR (Larrimore et al. 2020) as described previously for aneuploid W303 strains grown in batch culture (Torres et al. 2007). However, we did observe weak correlation with ESR genes from aneuploid W303 strains grown in chemostat and with the CAGE response (Tsai et al. 2019). We found the correlation with ESR genes varied in relation to the growth rates of our strains, suggesting that the activation of the ESR pathway may be primarily growth rate dependent and not driven by the GAP1 CNVs. We found that GAP1 CNV strains showed a unique shared gene expression signature comprising increased expression of genes involved in transposition, nucleic acid metabolic processes, and siderophore transport, and decreased expression of those involved in cellular respiration, nucleoside biosynthetic processes, and small-molecule metabolism, although the extent to which the expression differed from the euploid varied between CNV strains.
Additional lines of evidence suggest that some of the CNV strains have altered mitochondrial activity or function, with two strains showing sensitivity to the mitochondrial uncoupling agent CCCP, increased enrichment in mutational tolerance in mitochondria-associated GO terms, and altered patterns of interaction with BMH1. This may explain some of the overlap in expression observed with the hypo-osmotic stress-response-like CAGE response (Tsai et al. 2019), as HOG1 pathway is also involved in mitochondrial regulation via the mitophagy pathway. The relationship between increased gene copy number and mitochondrial functions warrants further investigation.
Although we do find commonalities between strains in terms of changes in mutational tolerance, CNV-associated gene expression, and global patterns of gene expression, we also find numerous instances of strain-specific differences. One limitation of this work is that all CNV-containing strains used in this study share amplifications of the GAP1 locus gained through adaptation to glutamine-limited conditions. Further studies of CNVs of various structures from different regions of the genome would be of interest to determine the generalizability of our findings.
Methods
Yeast strains
The euploid ancestral GAP1 CNV reporter and the evolved GAP1 CNV strains were previously described and characterized by Lauer et al. (2018). The CNV strains are clonal isolates that evolved for 150 or 250 generations in glutamine-limited chemostats (Lauer et al. 2018).
Each strain was transformed with the Hermes-containing plasmid, pSG36_HygMX, using the EZ-Yeast transformation kit (MP Biomedicals 2100200). Transformants were recovered on YPG agar + 200 µg/mL hygromycin B. A single transformant colony was picked to perform each transposon mutagenesis experiment. Separate transformation and colony selection were performed for each replicate.
To generate BMH1 mutants, we transformed frozen competent yeast cells for each strain (Gietz and Schiestl 2007) with an mCherry gene under control of the constitutively expressed ACT1 promoter (ACT1pr::mCherry::ADH1term) and marked by the HphMX hygromycin B–resistance cassette (TEFpr::HygR::TEFterm). The plasmid DGP363, containing this construct, was used as template for PCR using primers containing the same BMH1-specific targeting homology, and transformation resulted in a complete deletion of the BMH1 open reading frame. Transformants were recovered on YPD agar + 400 µg/mL G418 + 200 µg/mL hygromycin B, and BMH1 deletion positive transformants were confirmed using BMH1 specific primers and a HygR primer. We verified mCitrine and mCherry fluorescence using a Cytek Aurora flow cytometer.
Evaluation of SNVs identified in CNV strains.
Strains were sequenced and SNVs identified as described by Lauer et al. (2018). We evaluated the potential impact of each SNV (Supplemental Table S2) using Ensembl's VEP (McLaren et al. 2016) and then classified SNVs into low probability severity and high probability severity groups (Supplemental Table S3).
Competitive fitness in chemostats
Fitness measurements of strains in chemostats were originally performed by Lauer et al. (2018). Strains were cocultured in glutamine-limited chemostats with FY4, a nonfluorescent reference strain, from which all the strains derived. The relative abundance of each strain was measured using flow cytometry analysis of samples collected every two to three generations for approximately 15 generations. Estimated relative fitness was calculated using a linear regression of the natural log of the ratio of the two genotypes against time. Ninety-five-percent confidence intervals were determined for the estimated fitness using the standard error of the regression and the appropriate t-statistic, depending on the degrees of freedom.
Growth analysis in batch culture
To evaluate growth rates under the same conditions as those used for transposon mutagenesis, we performed growth rate analysis in YPGal batch cultures. For each experiment, we inoculated three colonies per strain into 3–5 mL YPGal and grew them overnight at 30°C. We then back diluted 5 µL of culture into 195 µL fresh YPGal. We collected OD600 data over ∼48 h using a Tecan Spark and growthcurver used to fit the data to a logistic equation.
Transposon mutagenesis
All incubation steps were conducted at 30°C with agitation for 24 h. A single transformant for each strain was used to inoculate 30 mL of YPD containing 200 µg/mL hygromycin B and incubated until OD5 was reached. To induce transposition, the culture was then diluted to an OD of 0.05 in YPGal with 200 µg/mL hygromycin B to a final volume of 50 mL and incubated. The culture was then diluted to OD of 0.05 in 50 mL YPGal + 200 µg/mL hygromycin B and incubated; this was repeated three more times for a total of four serial transfers. The culture was then pelleted by centrifugation, resuspended to an OD of 0.5 in 50 mL YPD without hygromycin B, and incubated for 24 h. This was performed twice to promote plasmid loss. The cultures were then diluted to an OD of 0.5 in 100 mL YPD with 200 µg/mL hygromycin B and incubated for 24 h to select cells with the transposon inserted in the genome. The final culture was pelleted by centrifugation, and the cells were frozen at −20°C for storage until DNA extraction was performed (see Supplemental Methods).
Insertion site sequencing
DNA was extracted from cell pellets using the MasterPure yeast DNA purification kit (Lucigen MPY80200), incubated with zymolyase at 37°C to enhance cell lysis, and then precipitated using a glycogen/sodium acetate/ethanol DNA precipitation (Green and Sambrook 2016). For each sample, 2 μg of DNA was digested using DpnII (NEB R0543L), and 2 μg of DNA was digested with NlaIII (NEB R0125L). The reactions were heat inactivated, circularized by ligation using T4 Ligase (Thermo Fisher Scientific EL0011), and then precipitated. Inverse PCR was performed using Hermes_F and Hermes_R primers (Supplemental Table S32) and DreamTaq (Thermo Fisher Scientific EP0701). The PCR products were confirmed on 2% agarose gels, and the concentration was quantified using Qubit dsDNA BR assay kit.
Library preparation and sequencing were performed using two different library preparation and sequencing methods (Beijing Genomics Institute [BGI] and New York City [NYC]). For samples from 1728, 1736, and 1740, 35 PCR reactions using primers (Supplemental Table S32) were performed as described above, and the PCR products were pooled and cleaned by precipitation. For each sample, at least 6 µg at a minimum concentration of 30 ng/µL was sent to BGI for library preparation and sequenced using a paired-end (2 × 100) protocol on an Illumina HiSeq 4000 or DNBSEQ platform. For all other samples, four PCR reactions were performed as described above, and the PCR products were pooled by sample and cleaned by precipitation. Five nanograms of each PCR product pool was used as input into a modified Nextera XT library preparation (for more details, see Supplemental Methods). The fragment size of each library was measured with an Agilent TapeStation 2200, and qPCR was performed to determine the library concentration. The libraries were pooled at equimolar concentrations and sequenced using a single-end (1 × 150) protocol on an Illumina NextSeq 500 at CGSB in NYC. Libraries were prepared once and sequenced twice for increased coverage.
Transposon insertion sequencing site identification and annotation
We used cutadapt v1.16 (Martin 2011) to trim the Hermes TIR sequence on the 5′ end. Trimmed reads with fewer than 20 bases, non-TIR containing reads, plasmid sequences, and Nextera transposase sequences were discarded. Reads were aligned to a modified reference genome (Supplemental File S1) using BWA-MEM v.0.7.15 (Li and Durbin 2010). BAMs were generated using SAMtools v1.9 (Li et al. 2009). Samples prepared by both BGI and NYC methods had high Pearson's correlation (0.85–0.94) for unique insertions identified per gene (Supplemental Table S8) and were combined for downstream analysis. Insertion positions' coordinates were calculated using the first aligned base that was read. Positions were annotated using BEDTools v2.26.0 (Quinlan and Hall 2010) and a modified reference GFF (Supplemental File S1). All downstream analyses used unique insertion positions and did not take into account the number of reads per unique insertion position. The libraries have between 85,327 and 329,624 unique insertion sites identified, and each site is supported by an average of 18.6 sequencing reads (Supplemental Table S4). We normalized for sequencing depth by calculating insertions per million—number of unique insertion sites per feature/(total unique insertion sites/1,000,000) (Levitan et al. 2020)—and required a minimum of 50 insertions per million per feature for all comparisons. Importantly, the Hermes transposon method has a preference for nucleosome-free regions, which tend to be right before and right after genes. However, as we consider only coding regions and do not perform comparisons between genes within the genome, nucleosome occupancy bias is unlikely to impact our analysis.
Differential analysis
To determine differential expression (RNA-seq) or insertion abundance (Tn) of genes, we used DESeq2 (version 1.30.1) (Love et al. 2014) with default parameters: Wald with LFC shrinkage, BH P-value correction ≤ 0.05.
GSEA for transposon insertions
We used clusterProfiler version 3.18.1 (Yu et al. 2012) to perform GSEA (Korotkevich et al. 2016) using the ranked log2 fold-change in insertions generated by DESeq2. GO terms with Q-values ≤ 0.05 were combined by semantic similarity using Revigo (Supplemental Table S19; Yu et al. 2010).
Genetic interaction analysis
To calculate genetic interactions based on growth rates, we first calculate the relative fitness of each single mutant by
where m is the intrinsic growth rate of the strain (parameter r from logistic equation used to fit growth curves). We calculated the expected fitness of the CNV mutant using either the additive model:
or a multiplicative model:
and we calculated the strength of the genetic interaction as the deviation from this expectation: (Mani et al. 2008).
Error bars were calculated using the propagation of the standard deviation of each measured population.
RNA sequencing
Overnight cultures were grown from three replicate colonies per strain in 5 mL YPGal, pelleted, resuspended in 5 mL fresh YPGal, incubated for 3 h, and then harvested by vacuum filtration and rapidly frozen using liquid nitrogen. RNA was extracted using hot acid phenol/chloroform and Phase Lock Gels (Neymotin et al. 2014). Samples were enriched for polyadenylated RNA using the Lexogen Poly(A) RNA selection kit V1.5 (157.96), and stranded RNA-seq libraries were prepared using the Lexogen CORALL Total RNA-seq library prep kit (095.96) according to the manufacturer's protocol. The libraries were pooled at equimolar concentrations and sequenced using a paired-end (2 × 150) protocol on an Illumina NextSeq 500. The resulting FASTQs were trimmed, aligned, and UMI deduplicated, and coverage per feature was calculated using an in-house pipeline. Correlation between replicates was high, with the exception of one replicate of ComQuad, which was excluded from further analysis (Supplemental Table S8).
Comparison of gene transcript abundances to previous aneuploid stress response studies
To compare the gene expression results to Torres et al. (2007), we subset the ESR genes from both our data sets (798 of 868, for which we had complete data), calculated the log2 fold-change in mRNA for each of our evolved strains relative to our euploid ancestor, and calculated the mean log2 fold-change in mRNA for each aneuploid strain from Torres et al. (2007) relative to their euploid strain. Using these log2 ratios, we calculated Pearson correlation coefficient between our strains and Torres et al. (2007). A similar approach was performed for Tsai et al. (2019) for the 222 CAGE genes.
Gene copy number determination and transcript abundance copy number correction
The determination of copy number for each gene in each strain (Supplemental Table S6) was performed using the reconstructed CNV topologies (Spealman et al. 2022, 2023); ODIRA CNVs were resolved as described previously (Spealman et al. 2020). Gene copy numbers were then used to estimate expected mRNA abundance assuming no dosage compensation, such that the expected abundance of a CNV gene would be equal to the euploid expression multiplied by the copy number of the gene in the CNV strain (Supplemental Table S25). The difference between the observed and expected expression was evaluated using DESeq2 (Supplemental Table S26).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA910831. All computational analyses are publicly available as Supplemental Code (Supplemental File S2) and on GitHub (https://github.com/pspealman/CNV_essentiality).
Supplementary Material
Acknowledgments
We thank members of the Gresham, Schacherer, and Vogel laboratories. This work was made possible by grants from the National Institutes of Health (National Institute of General Medical Sciences) to P.S. (F32-GM131573) and D.G. (R01-GM134066, R01-GM107466) and from the National Science Foundation to G.A. (DGE1342536) and D.G. (MCB1818234). J.S. is supported by the European Research Council (ERC) Consolidator grant (772505). J.S. is a fellow of the University of Strasbourg Institute for Advanced Study (USIAS) and a member of the Institut Universitaire de France.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277625.122.
Freely available online through the Genome Research Open Access option.
Competing interest statement
The authors declare no competing interests.
References
- Arita Y, Kim G, Li Z, Friesen H, Turco G, Wang RY, Climie D, Usaj M, Hotz M, Stoops EH, et al. 2021. A genome-scale yeast library with inducible expression of individual genes. Mol Syst Biol 17: e10207. 10.15252/msb.202110207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascencio D, Diss G, Gagnon-Arsenault I, Dubé AK, DeLuna A, Landry CR. 2021. Expression attenuation as a mechanism of robustness against gene duplication. Proc Natl Acad Sci 118: e2014345118. 10.1073/pnas.2014345118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avecilla G, Chuong JN, Li F, Sherlock G, Gresham D, Ram Y. 2022. Neural networks enable efficient and accurate simulation-based inference of evolutionary parameters from adaptation dynamics. PLoS Biol 20: e3001633. 10.1371/journal.pbio.3001633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baryshnikova A, Andrews B. 2012. Neighboring-gene effect: a genetic uncertainty principle. Nat Methods 9: 341– 343. 10.1038/nmeth.1936 [DOI] [PubMed] [Google Scholar]
- Beach RR, Ricci-Tam C, Brennan CM, Moomau CA, Hsu P-H, Hua B, Silberman RE, Springer M, Amon A. 2017. Aneuploidy causes non-genetic individuality. Cell 169: 229–242.e21. 10.1016/j.cell.2017.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David U, Amon A. 2020. Context is everything: aneuploidy in cancer. Nat Rev Genet 21: 44–62. 10.1038/s41576-019-0171-x [DOI] [PubMed] [Google Scholar]
- Ben-Shitrit T, Yosef N, Shemesh K, Sharan R, Ruppin E, Kupiec M. 2012. Systematic identification of gene annotation errors in the widely used yeast mutation collections. Nat Methods 9: 373–378. 10.1038/nmeth.1890 [DOI] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA. 2022. One hundred years of gene balance: how stoichiometric issues affect gene expression, genome evolution, and quantitative traits. Cytogenet Genome Res 161: 529–550. 10.1159/000519592 [DOI] [PubMed] [Google Scholar]
- Bonney ME, Moriya H, Amon A. 2015. Aneuploid proliferation defects in yeast are not driven by copy number changes of a few dosage-sensitive genes. Genes Dev 29: 898–903. 10.1101/gad.261743.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, Botstein D. 2008. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol Biol Cell 19: 352–367. 10.1091/mbc.e07-08-0779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camougrand N, Grelaud-Coq A, Marza E, Priault M, Bessoule J-J, Manon S. 2003. The product of the UTH1 gene, required for Bax-induced cell death in yeast, is involved in the response to rapamycin. Mol Microbiol 47: 495–506. 10.1046/j.1365-2958.2003.03311.x [DOI] [PubMed] [Google Scholar]
- Camougrand N, Kiššová I, Velours G, Manon S. 2004. Uth1p: a yeast mitochondrial protein at the crossroads of stress, degradation and cell death. FEMS Yeast Res 5: 133–140. 10.1016/j.femsyr.2004.05.001 [DOI] [PubMed] [Google Scholar]
- Caudal E, Friedrich A, Jallet A, Garin M, Hou J, Schacherer J. 2022. Loss-of-function mutation survey revealed that genes with background-dependent fitness are rare and functionally related in yeast. Proc Natl Acad Sci 119: e2204206119. 10.1073/pnas.2204206119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton DA. 2011. Mechanisms of aneuploidy. Curr Opin Cell Biol 23: 109–113. 10.1016/j.ceb.2010.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Hou J, Messier V, Nelson J, Rahman M, VanderSluis B, Wang W, Pons C, Ross C, Ušaj M, et al. 2021. Environmental robustness of the global yeast genetic interaction network. Science 372: eabf8424. 10.1126/science.abf8424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cunha FM, Torelli NQ, Kowaltowski AJ. 2015. Mitochondrial retrograde signaling: triggers, pathways, and outcomes. Oxid Med Cell Longev 2015: 482582. 10.1155/2015/482582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekel E, Alon U. 2005. Optimality and evolutionary tuning of the expression level of a protein. Nature 436: 588–592. 10.1038/nature03842 [DOI] [PubMed] [Google Scholar]
- Dephoure N, Hwang S, O'Sullivan C, Dodgson SE, Gygi SP, Amon A, Torres EM. 2014. Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. eLife 3: e03023. 10.7554/eLife.03023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destruelle M, Holzer H, Klionsky DJ. 1994. Identification and characterization of a novel yeast gene: the YGP1 gene product is a highly glycosylated secreted protein that is synthesized in response to nutrient limitation. Mol Cell Biol 14: 2740–2754. 10.1128/mcb.14.4.2740-2754.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhami MK, Hartwig T, Fukami T. 2016. Genetic basis of priority effects: insights from nectar yeast. Proc Biol Sci 283: 20161455. 10.1098/rspb.2016.1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodgson SE, Kim S, Costanzo M, Baryshnikova A, Morse DL, Kaiser CA, Boone C, Amon A. 2016. Chromosome-specific and global effects of aneuploidy in Saccharomyces cerevisiae. Genetics 202: 1395–1409. 10.1534/genetics.115.185660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombek KM, Kacherovsky N, Young ET. 2004. The Reg1-interacting proteins, Bmh1, Bmh2, Ssb1, and Ssb2, have roles in maintaining glucose repression in Saccharomyces cerevisiae. J Biol Chem 279: 39165–39174. 10.1074/jbc.m400433200 [DOI] [PubMed] [Google Scholar]
- Domitrovic T, Kozlov G, Freire JCG, Masuda CA, da Silva Almeida M, Montero-Lomeli M, Atella GC, Matta-Camacho E, Gehring K, Kurtenbach E. 2010. Structural and functional study of Yer067w: a new protein involved in yeast metabolism control and drug resistance. PLoS One 5: e11163. 10.1371/journal.pone.0011163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas AC, Smith AM, Sharifpoor S, Yan Z, Durbic T, Heisler LE, Lee AY, Ryan O, Göttert H, Surendra A, et al. 2012. Functional analysis with a barcoder yeast gene overexpression system. G3 (Bethesda) 2: 1279–1289. 10.1534/g3.112.003400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt S-M, Sauer U. 2010. Transcriptional regulation of respiration in yeast metabolizing differently repressive carbon substrates. BMC Syst Biol 4: 12. 10.1186/1752-0509-4-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M, Scanlon MJ, Fowler JE. 2015. Fractionation and subfunctionalization following genome duplications: mechanisms that drive gene content and their consequences. Curr Opin Genet Dev 35: 110–118. 10.1016/j.gde.2015.11.002 [DOI] [PubMed] [Google Scholar]
- Gale AN, Sakhawala RM, Levitan A, Sharan R, Berman J, Timp W, Cunningham KW. 2020. Identification of essential genes and fluconazole susceptibility genes in Candida glabrata by profiling Hermes transposon insertions. G3 (Bethesda) 10: 3859–3870. 10.1534/g3.120.401595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangadharan S, Mularoni L, Fain-Thornton J, Wheelan SJ, Craig NL. 2010. DNA transposon Hermes inserts into DNA in nucleosome-free regions in vivo. Proc Natl Acad Sci 107: 21966–21972. 10.1073/pnas.1016382107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11: 4241–4257. 10.1091/mbc.11.12.4241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Hose J, Newton MA, Sardi M, Yong M, Wang Z. 2016. Further support for aneuploidy tolerance in wild yeast and effects of dosage compensation on gene copy-number evolution. eLife 5: e14409. 10.7554/eLife.14409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Nislow C. 2014. The yeast deletion collection: a decade of functional genomics. Genetics 197: 451–465. 10.1534/genetics.114.161620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio S, Liu Z, Thornton J, Butow RA. 2005. Retrograde response to mitochondrial dysfunction is separable from TOR1/2 regulation of retrograde gene expression. J Biol Chem 280: 42528–42535. 10.1074/jbc.M509187200 [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. 2007. Frozen competent yeast cells that can be transformed with high efficiency using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2: 1–4. 10.1038/nprot.2007.17 [DOI] [PubMed] [Google Scholar]
- Grech L, Jeffares DC, Sadée CY, Rodríguez-López M, Bitton DA, Hoti M, Biagosch C, Aravani D, Speekenbrink M, Illingworth CJR, et al. 2019. Fitness landscape of the fission yeast genome. Mol Biol Evol 36: 1612–1623. 10.1093/molbev/msz113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR, Sambrook J. 2016. Precipitation of DNA with ethanol. Cold Spring Harb Protoc 2016: pdb.prot093377. 10.1101/pdb.prot093377 [DOI] [PubMed] [Google Scholar]
- Gresham D, Desai MM, Tucker CM, Jenq HT, Pai DA, Ward A, DeSevo CG, Botstein D, Dunham MJ. 2008. The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet 4: e1000303. 10.1371/journal.pgen.1000303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Park JM, Cui B, Humes E, Gangadharan S, Hung S, FitzGerald PC, Hoe K-L, Grewal SIS, Craig NL, et al. 2013. Integration profiling of gene function with dense maps of transposon integration. Genetics 195: 599–609. 10.1534/genetics.113.152744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine E, Runcie DE, McGuigan K, Blows MW. 2018. Uneven distribution of mutational variance across the transcriptome of Drosophila serrata revealed by high-dimensional analysis of gene expression. Genetics 209: 1319–1328. 10.1534/genetics.118.300757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Gresham D. 2014. Molecular specificity, convergence and constraint shape adaptive evolution in nutrient-poor environments. PLoS Genet 10: e1004041. 10.1371/journal.pgen.1004041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hose J, Yong CM, Sardi M, Wang Z, Newton MA, Gasch AP. 2015. Dosage compensation can buffer copy-number variation in wild yeast. eLife 4: e05462. 10.7554/eLife.05462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hose J, Escalante LE, Clowers KJ, Dutcher HA, Robinson D, Bouriakov V, Coon JJ, Shishkova E, Gasch AP. 2020. The genetic basis of aneuploidy tolerance in wild yeast. eLife 9: e52063. 10.7554/eLife.52063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, Roberts CJ, Dai H, Jones AR, Meyer MR, Slade D, Burchard J, Dow S, Ward TR, Kidd MJ, et al. 2000. Widespread aneuploidy revealed by DNA microarray expression profiling. Nat Genet 25: 333–337. 10.1038/77116 [DOI] [PubMed] [Google Scholar]
- Innan H, Kondrashov F. 2010. The evolution of gene duplications: classifying and distinguishing between models. Nat Rev Genet 11: 97–108. 10.1038/nrg2689 [DOI] [PubMed] [Google Scholar]
- Jain N, Janning P, Neumann H. 2021. 14-3-3 protein Bmh1 triggers short-range compaction of mitotic chromosomes by recruiting sirtuin deacetylase Hst2. J Biol Chem 296: 100078. 10.1074/jbc.AC120.014758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazwinski SM, Kriete A. 2012. The yeast retrograde response as a model of intracellular signaling of mitochondrial dysfunction. Front Physiol 3: 139. 10.3389/fphys.2012.00139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Mason SP, Barabási AL, Oltvai ZN. 2001. Lethality and centrality in protein networks. Nature 411: 41–42. 10.1038/35075138 [DOI] [PubMed] [Google Scholar]
- Katju V, Bergthorsson U. 2019. Old trade, new tricks: insights into the spontaneous mutation process from the partnering of classical mutation accumulation experiments with high-throughput genomic approaches. Genome Biol Evol 11: 136–165. 10.1093/gbe/evy252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S, Cimini D. 2019. Aneuploidy and gene expression: Is there dosage compensation? Epigenomics 11: 1827–1837. 10.2217/epi-2019-0135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov FA. 2012. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc Biol Sci 279: 5048–5057. 10.1098/rspb.2012.1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad A, Flibotte S, Taylor J, Waterston RH, Moerman DG, Bergthorsson U, Katju V. 2018. Mutational and transcriptional landscape of spontaneous gene duplications and deletions in Caenorhabditis elegans. Proc Natl Acad Sci 115: 7386–7391. 10.1073/pnas.1801930115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A. 2016. Fast gene set enrichment analysis. bioRxiv 10.1101/060012 [DOI]
- Kumar R. 2017. An account of fungal 14-3-3 proteins. Eur J Cell Biol 96: 206–217. 10.1016/j.ejcb.2017.02.006 [DOI] [PubMed] [Google Scholar]
- Larrimore KE, Barattin-Voynova NS, Reid DW, Ng DTW. 2020. Aneuploidy-induced proteotoxic stress can be effectively tolerated without dosage compensation, genetic mutations, or stress responses. BMC Biol 18: 117. 10.1186/s12915-020-00852-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer S, Gresham D. 2019. An evolving view of copy number variants. Curr Genet 65: 1287–1295. 10.1007/s00294-019-00980-0 [DOI] [PubMed] [Google Scholar]
- Lauer S, Avecilla G, Spealman P, Sethia G, Brandt N, Levy SF, Gresham D. 2018. Single-cell copy number variant detection reveals the dynamics and diversity of adaptation. PLoS Biol 16: e3000069. 10.1371/journal.pbio.3000069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan A, Gale AN, Dallon EK, Kozan DW, Cunningham KW, Sharan R, Berman J. 2020. Comparing the utility of in vivo transposon mutagenesis approaches in yeast species to infer gene essentiality. Curr Genet 66: 1117–1134. 10.1007/s00294-020-01096-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26: 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder RA, Greco JP, Seidl F, Matsui T, Ehrenreich IM. 2017. The stress-inducible peroxidase TSA2 underlies a conditionally beneficial chromosomal duplication in Saccharomyces cerevisiae. G3 (Bethesda) 7: 3177–3184. 10.1534/g3.117.300069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang J. 2019. Yeast spontaneous mutation rate and spectrum vary with environment. Curr Biol 29: 1584–1591.e3. 10.1016/j.cub.2019.03.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Sekito T, Špírek M, Thornton J, Butow RA. 2003. Retrograde signaling is regulated by the dynamic interaction between Rtg2p and Mks1p. Mol Cell 12: 401–411. 10.1016/s1097-2765(03)00285-5 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Sung W, Morris K, Coffey N, Landry CR, Dopman EB, Dickinson WJ, Okamoto K, Kulkarni S, Hartl DL, et al. 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci 105: 9272–9277. 10.1073/pnas.0803466105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makanae K, Kintaka R, Makino T, Kitano H, Moriya H. 2013. Identification of dosage-sensitive genes in Saccharomyces cerevisiae using the genetic tug-of-war method. Genome Res 23: 300–311. 10.1101/gr.146662.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani R, St Onge RP, Hartman JL IV, Giaever G, Roth FP. 2008. Defining genetic interaction. Proc Natl Acad Sci 105: 3461–3466. 10.1073/pnas.0712255105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.j 17: 10–12. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. 2016. The Ensembl variant effect predictor. Genome Biol 17: 122. 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AH, Hatakeyama R, Kimmig P, Arter M, Peter M, Matos J, De Virgilio C, Kornmann B. 2017. Functional mapping of yeast genomes by saturated transposition. eLife 6: e23570. 10.7554/eLife.23570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya H. 2015. Quantitative nature of overexpression experiments. Mol Biol Cell 26: 3932–3939. 10.1091/mbc.E15-07-0512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenzner J, Trébulle P, Agostini F, Messner CB, Steger M, Lehmann A, Caudal E, Egger A-S, Amari F, Barthel N, et al. 2022. The natural diversity of the yeast proteome reveals chromosome-wide dosage compensation in aneuploids. bioRxiv 10.1101/2022.04.06.487392 [DOI]
- Myhre S, Lingjærde O-C, Hennessy BT, Aure MR, Carey MS, Alsner J, Tramm T, Overgaard J, Mills GB, Børresen-Dale A-L, et al. 2013. Influence of DNA copy number and mRNA levels on the expression of breast cancer related proteins. Mol Oncol 7: 704–718. 10.1016/j.molonc.2013.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S, Miller B, Barends M, Jaidee A, Patel J, Mayxay M, Newton P, Nosten F, Ferdig MT, Anderson TJC. 2008. Adaptive copy number evolution in malaria parasites. PLoS Genet 4: e1000243. 10.1371/journal.pgen.1000243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neymotin B, Athanasiadou R, Gresham D. 2014. Determination of in vivo RNA kinetics using RATE-seq. RNA 20: 1645–1652. 10.1261/rna.045104.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning K, Ng HK, Srihari S, Leong HW, Nesvizhskii AI. 2010. Examination of the relationship between essential genes in PPI network and hub proteins in reverse nearest neighbor topology. BMC Bioinformatics 11: 505. 10.1186/1471-2105-11-505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. 1970. Evolution by gene duplication. Springer, New York. 10.1007/978-3-642-86659-3 [DOI] [Google Scholar]
- Paulander W, Andersson DI, Maisnier-Patin S. 2010. Amplification of the gene for isoleucyl–tRNA synthetase facilitates adaptation to the fitness cost of mupirocin resistance in Salmonella enterica. Genetics 185: 305–312. 10.1534/genetics.109.113514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavani M, Bonaiuti P, Chiroli E, Gross F, Natali F, Macaluso F, Póti Á, Pasqualato S, Farkas Z, Pompei S, et al. 2021. Epistasis, aneuploidy, and functional mutations underlie evolution of resistance to induced microtubule depolymerization. EMBO J 40: e108225. 10.15252/embj.2021108225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. 2010. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468: 321–325. 10.1038/nature09529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payen C, Sunshine AB, Ong GT, Pogachar JL, Zhao W, Dunham MJ. 2016. High-throughput identification of adaptive mutations in experimentally evolved yeast populations. PLoS Genet 12: e1006339. 10.1371/journal.pgen.1006339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter J, De Chiara M, Friedrich A, Yue J-X, Pflieger D, Bergström A, Sigwalt A, Barre B, Freel K, Llored A, et al. 2018. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556: 339–344. 10.1038/s41586-018-0030-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pränting M, Andersson DI. 2011. Escape from growth restriction in small colony variants of Salmonella typhimurium by gene amplification and mutation. Mol Microbiol 79: 305–315. 10.1111/j.1365-2958.2010.07458.x [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancati G, Pavelka N, Fleharty B, Noll A, Trimble R, Walton K, Perera A, Staehling-Hampton K, Seidel CW, Li R. 2008. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 135: 879–893. 10.1016/j.cell.2008.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders J, Zahedi RP, Pfanner N, Meisinger C, Sickmann A. 2006. Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J Proteome Res 5: 1543–1554. 10.1021/pr050477f [DOI] [PubMed] [Google Scholar]
- Reinders J, Wagner K, Zahedi RP, Stojanovski D, Eyrich B, van der Laan M, Rehling P, Sickmann A, Pfanner N, Meisinger C. 2007. Profiling phosphoproteins of yeast mitochondria reveals a role of phosphorylation in assembly of the ATP synthase. Mol Cell Proteomics 6: 1896–1906. 10.1074/mcp.M700098-MCP200 [DOI] [PubMed] [Google Scholar]
- Rice AM, McLysaght A. 2017. Dosage-sensitive genes in evolution and disease. BMC Biol 15: 78. 10.1186/s12915-017-0418-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Place M, Hose J, Jochem A, Gasch AP. 2021. Natural variation in the consequences of gene overexpression and its implications for evolutionary trajectories. eLife 10: e70564. 10.7554/eLife.70564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca-Portoles A, Tait SWG. 2021. Mitochondrial quality control: from molecule to organelle. Cell Mol Life Sci 78: 3853–3866. 10.1007/s00018-021-03775-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez MR, Miller AW, Liachko I, Sunshine AB, Lynch B, Huang M, Alcantara E, DeSevo CG, Pai DA, Tucker CM, et al. 2017. Differential paralog divergence modulates genome evolution across yeast species. PLoS Genet 13: e1006585. 10.1371/journal.pgen.1006585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrider DR, Hahn MW. 2010. Gene copy-number polymorphism in nature. Proc Biol Sci 277: 3213–3221. 10.1098/rspb.2010.1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scopel EFC, Hose J, Bensasson D, Gasch AP. 2021. Genetic variation in aneuploidy prevalence and tolerance across Saccharomyces cerevisiae lineages. Genetics 217: iyab015. 10.1093/genetics/iyab015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal ES, Gritsenko V, Levitan A, Yadav B, Dror N, Steenwyk JL, Silberberg Y, Mielich K, Rokas A, Gow NAR, et al. 2018. Gene essentiality analyzed by in vivo transposon mutagenesis and machine learning in a stable haploid isolate of Candida albicans. mBio 9: e02048-18. 10.1128/mbio.02048-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki AM, Dulmage K, Cowen LE, Anderson JB, Berman J. 2009. Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet 5: e1000705. 10.1371/journal.pgen.1000705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Torres EM, Dunham MJ, Amon A. 2012. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci 109: 12644–12649. 10.1073/pnas.1209227109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, Snyder M, Oliver SG, Cyert M, Hughes TR, et al. 2006. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell 21: 319–330. 10.1016/j.molcel.2005.12.011 [DOI] [PubMed] [Google Scholar]
- Spealman P, Burrell J, Gresham D. 2020. Inverted duplicate DNA sequences increase translocation rates through sequencing nanopores resulting in reduced base calling accuracy. Nucleic Acids Res 48: 4940–4945. 10.1093/nar/gkaa206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spealman P, Avecilla G, Matthews J, Suresh I, Gresham D. 2022. Complex genomic rearrangements following selection in a glutamine-limited medium over hundreds of generations. Microbiol Resour Announc 11: e0072922. 10.1128/mra.00729-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spealman P, De T, Chuong JN, Gresham D. 2023. Best practices in microbial experimental evolution: using reporters and long-read sequencing to identify copy number variation in experimental evolution. J Mol Evol 91: 356–368. 10.1007/s00239-023-10102-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer M, Weissman JS, Kirschner MW. 2010. A general lack of compensation for gene dosage in yeast. Mol Syst Biol 6: 368. 10.1038/msb.2010.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunshine AB, Payen C, Ong GT, Liachko I, Tan KM, Dunham MJ. 2015. The fitness consequences of aneuploidy are driven by condition-dependent gene effects. PLoS Biol 13: e1002155. 10.1371/journal.pbio.1002155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhorst A, Sandikci A, Keller A, Whittaker CA, Dunham MJ, Amon A. 2020. The environmental stress response causes ribosome loss in aneuploid yeast cells. Proc Natl Acad Sci 117: 17031–17040. 10.1073/pnas.2005648117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd RT, Selmecki A. 2020. Expandable and reversible copy number amplification drives rapid adaptation to antifungal drugs. eLife 9: e58349. 10.7554/eLife.58349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. 2007. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916–924. 10.1126/science.1142210 [DOI] [PubMed] [Google Scholar]
- Torres EM, Springer M, Amon A. 2016. No current evidence for widespread dosage compensation in S. cerevisiae. eLife 5: e10996. 10.7554/eLife.10996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai H-J, Nelliat AR, Choudhury MI, Kucharavy A, Bradford WD, Cook ME, Kim J, Mair DB, Sun SX, Schatz MC, et al. 2019. Hypo-osmotic-like stress underlies general cellular defects of aneuploidy. Nature 570: 117–121. 10.1038/s41586-019-1187-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitia RA, Bottani S, Birchler JA. 2008. Cellular reactions to gene dosage imbalance: genomic, transcriptomic and proteomic effects. Trends Genet 24: 390–397. 10.1016/j.tig.2008.05.005 [DOI] [PubMed] [Google Scholar]
- Wagner A. 2005. Energy constraints on the evolution of gene expression. Mol Biol Evol 22: 1365–1374. 10.1093/molbev/msi126 [DOI] [PubMed] [Google Scholar]
- Wagner A. 2007. Energy costs constrain the evolution of gene expression. J Exp Zool B Mol Dev Evol 308B: 322–324. 10.1002/jez.b.21152 [DOI] [PubMed] [Google Scholar]
- Wiles AM, Cai H, Naider F, Becker JM. 2006. Nutrient regulation of oligopeptide transport in Saccharomyces cerevisiae. Microbiology 152: 3133–3145. 10.1099/mic.0.29055-0 [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901–906. 10.1126/science.285.5429.901 [DOI] [PubMed] [Google Scholar]
- Yona AH, Manor YS, Herbst RH, Romano GH, Mitchell A, Kupiec M, Pilpel Y, Dahan O. 2012. Chromosomal duplication is a transient evolutionary solution to stress. Proc Natl Acad Sci 109: 21010–21015. 10.1073/pnas.1211150109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Li F, Qin Y, Bo X, Wu Y, Wang S. 2010. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 26: 976–978. 10.1093/bioinformatics/btq064 [DOI] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, He Q-Y. 2012. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16: 284–287. 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YO, Sherlock G, Petrov DA. 2016. Whole genome analysis of 132 clinical Saccharomyces cerevisiae strains reveals extensive ploidy variation. G3 (Bethesda) 6: 2421–2434. 10.1534/g3.116.029397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Tsai H-J, Gordon MR, Li R. 2018. Cellular stress associated with aneuploidy. Dev Cell 44: 420–431. 10.1016/j.devcel.2018.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.